

 DownLoad:
DownLoad:









-
由于避蚊胺(DEET)对蚊子、苍蝇、跳蚤、蜱和其他叮咬昆虫有着显著的驱散效果,且具有成本低廉的优点,自20世纪50年代以来,被广泛的应用于军队和居民中[1]。据报道,美国平均每年要消耗1 800 t的避蚊胺[2]。随着DEET的广泛使用,其在全球的水环境中普遍可见,浓度为ng·L−1~μg·L−1[3]。例如,在美国54条河流中,其中74%的河流都可以检测到DEET,部分河流中DEET的浓度甚至高达1.1 μg·L−1[4];在中国5条主要河流流域的饮用水源中也检测到避蚊胺[5]。DEET具有毒性,在人鼻黏膜细胞中具有潜在的致癌性和遗传毒性[6],因此,对水中DEET的高效去除的研究变得极为重要。
近年来,有研究表明,传统的水处理过程对DEET几乎没有去除效果,ROSSNER等[7]研究表明,吸附剂种类会影响DEET去除率,但整体效果并不明显。而低温等离子体技术由于其具有高效率、优良的环境兼容性等优势,逐渐被广泛用于去除水体中的污染物[8]。在之前的研究中,湿壁介质阻挡放电(DBD)被应用于DEET的降解[9],虽然该技术将产生的等离子气体导入到溶液,但并没有充分的利用等离子气体中的臭氧,因此,单一的DBD等离子体的效果存在进一步的提高。
水中的臭氧可通过铁基材料催化,天然浮石是一种自然界中大量存在的多孔性火山岩,其成本低廉,是理想的天然原料。目前,对于浮石在水处理中应用的研究主要在于其吸附性能或作为一种催化剂载体[10-13]。同时为了进一步提高浮石的性能[14-15],通常负载铁。一些研究结果表明,羟基氧化铁能够催化臭氧,这是因为其表面的羟基官能团能促进臭氧分解并产生羟基自由基[16-17]。然而,纯浮石的催化效率比较有限,且羟基氧化铁的分离又比较困难,YUAN等[18]成功的制备了一种用于催化臭氧的材料α-FeOOH,提高了浮石的催化能力。其作用原理是:该负载材料通过羟基氧化铁催化了臭氧分解。DBD产生的等离子气体中含有较高浓度的臭氧,这些进入溶液的臭氧能否被该材料催化,尚未有相关的研究报道。
基于此,本文对湿壁介质阻挡放电(DBD)协同铁负载浮石降解水中DEET进行了研究。并对影响降解体系的因素进行了研究,分析浮石载羟基氧化铁在反应体系中的作用,并初步探究了DBD协同浮石载羟基氧化铁处理DEET的机制,以期为DEET降解的应用提供参考。
全文HTML
-
DEET购于上海阿拉丁试剂有限公司,通过将DEET溶解在超纯水中来模拟废水,实验中所用的浮石(pumice)产地为河北省行唐县。用于HPLC与LC-MS分析的试剂都是高效液相色谱级别。其他化学试剂均是分析纯。
天然浮石粉末都是经过300目标准检验筛(江苏省南京市雄晨筛网厂)的筛分后的颗粒,本研究通过将浮石浸渍在碱性的铁溶液中来制备浮石载羟基氧化铁(Fe-coated pumice)[14,18]。首先,将天然浮石粉末用蒸馏水超声清洗2次,然后在80 ℃下干燥12 h。其次,将经过上述预处理的浮石浸渍于0.1 mol·L−1的硝酸铁溶液中,用10 mol·L−1的氢氧化钠调节溶液pH到11,并室温下静置72 h。最后用超纯水冲洗直到pH和电导率维持稳定,并在80 ℃下烘干48 h,烘干后材料在真空下保存。
-
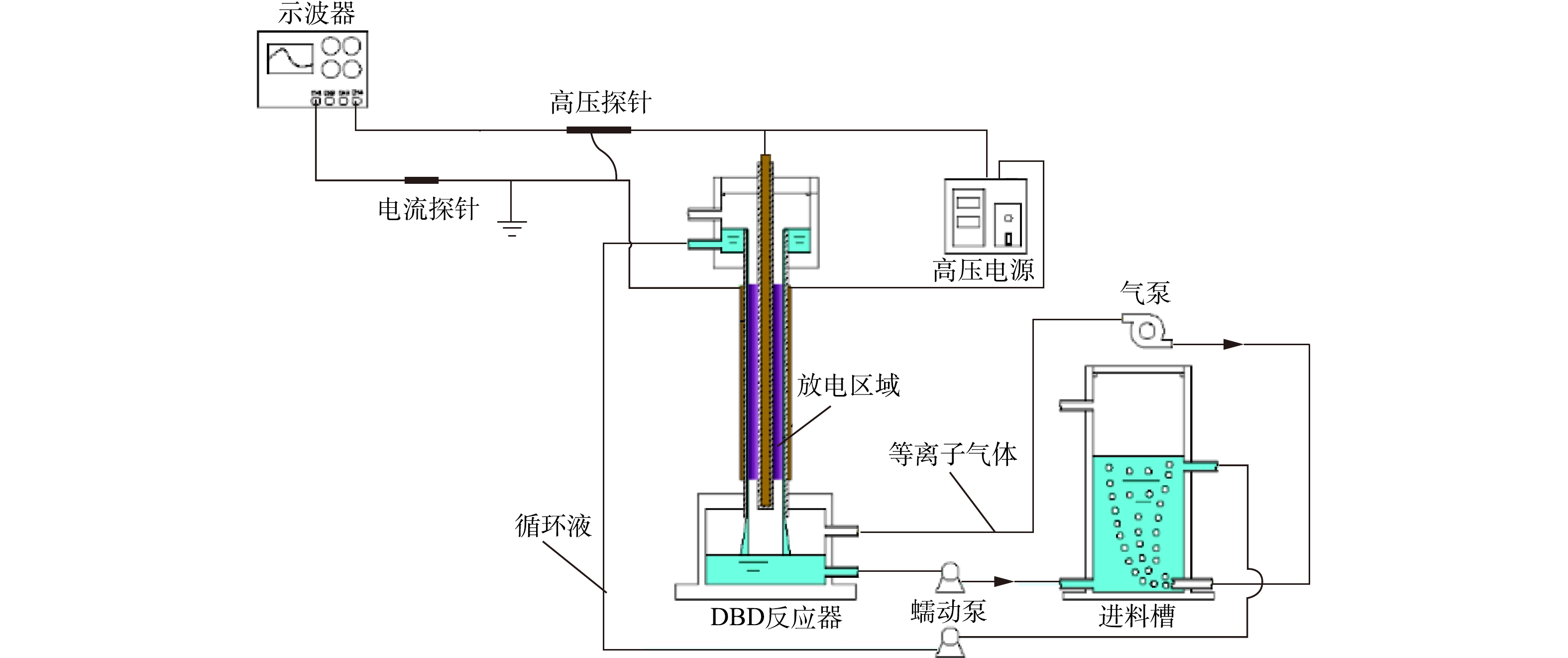
与之前的研究[9]相同,实验装置如图1所示,主要包括同轴线筒式双介质阻挡等离子体反应器、等离子体电源和液体循环系统。圆筒反应器由2个内径分别为6 mm和25 mm的石英管组成。内外石英管厚度均为2 mm,内管插入直径为5 mm的铝杆,外管用长120 mm的铜弹簧紧密包裹。等离子体发生器的高压电极与铝棒连接,接地电极与铜弹簧连接,内外石英管的中间区域为放电区域,本研究使用的等离子体电源(CTP-2000 K,南京苏曼电子有限公司,中国)是一种高压交流电源。
-
如图1所示,向进料水箱中加入300 mL 20 mg·L−1的DEET溶液,并加入一定量的浮石载羟基氧化铁,DEET溶液通过蠕动泵循环输送,从进料水箱到反应池,然后在外石英管内壁形成一层遍及整个放电区域的湿壁流,同时生成的等离子气体通过气泵输送到DEET溶液中,待溶液在反应器内均匀循环流动时,开启等离子电源对水样进行处理,每隔3 min取一次样,使用H2SO4和NaOH调节pH。
DEET浓度分析采用高效液相色谱法(HPLC, Agilent 1200,USA),流动相为30%超纯水和70%的甲醇,流速为1 mL·min−1,柱温25 ℃,进样量为20 μL,色谱柱为安捷伦C18柱(4.6 mm×150 mm,5 μm),保留时间为3.41 min,检测波长为230 nm。
水溶液中的臭氧浓度采用靛蓝褪色法进行测定;催化剂溶出总铁离子(Fe2+/Fe3+)浓度采用石墨炉原子吸收光谱仪(Thermo Solaar M6, USA)配备空心阴极灯(Thermo Fe, USA)进行测定;样品材料中元素的组成使用ARL-9800型X射线荧光光谱进行测定分析;使用Swiss ARL X’TRA型X射线衍射仪测定样品存在的晶型结构进行物相分析;Hitachi S-3400N II型扫描电镜观察样品的表面形貌和微观结构;样品的官能团由USA NICOLET NEXUS 870型傅里叶红外光谱仪检测;样品的孔径分布和比表面积由ASAP 2020型比表面积分析仪测定。
1.1. 实验材料
1.2. 实验装置
1.3. 实验方法
-
浮石和浮石载羟基氧化铁催化剂通过SEM、XRF、XRD、FT-IR和BET等技术进行了表征分析。图2是浮石和浮石载羟基氧化铁的SEM图。由图2可知,纯浮石呈现为块状结构,表面比较光滑,宽度在20 μm左右。当引入羟基氧化铁后,浮石的块状结构遭受破坏,可能是被高浓度氢氧化钠腐蚀所致,且表面变得粗糙,有许多不规则分布的孔道,与纯浮石结构明显不同。
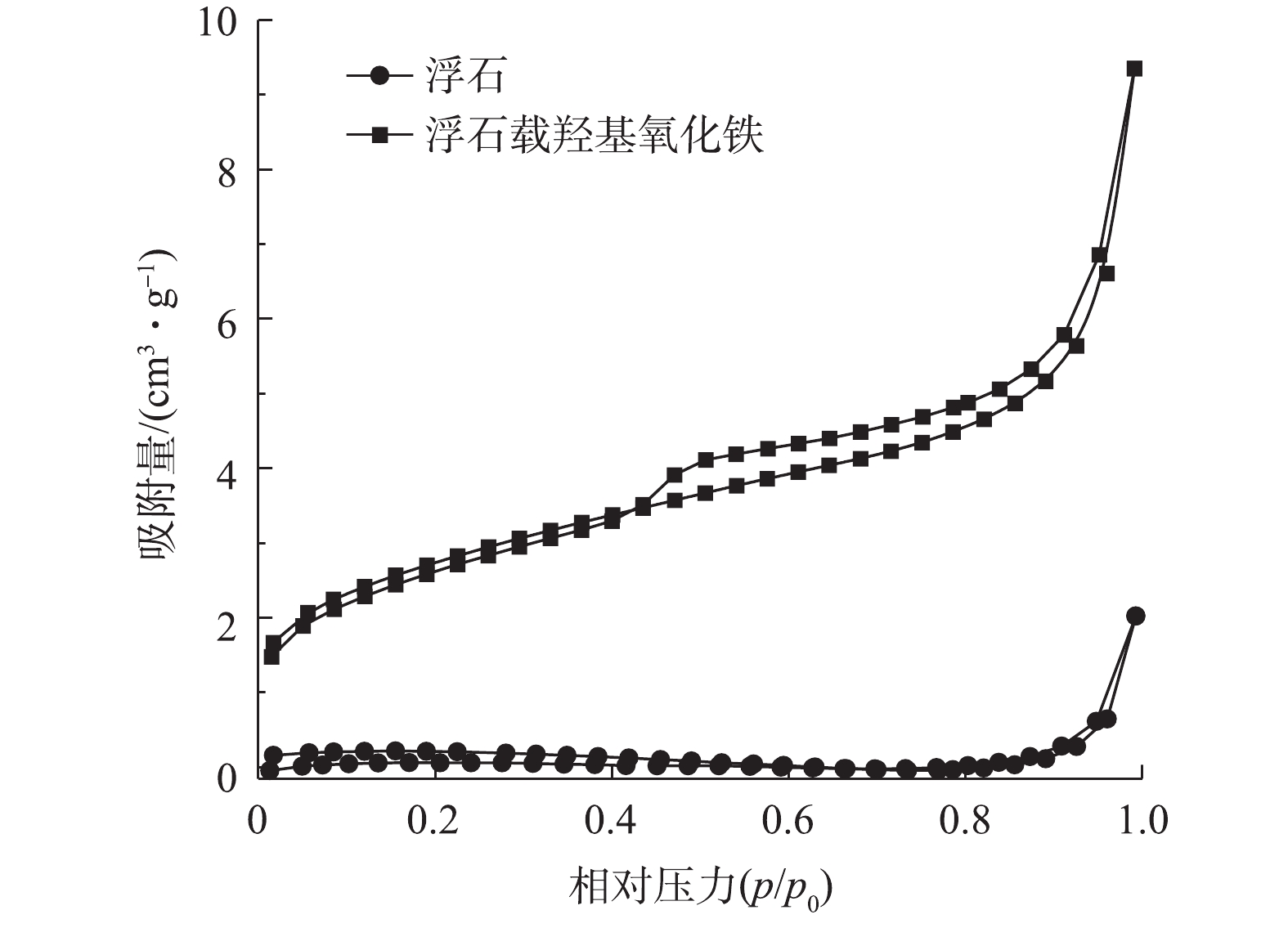
为了进一步研究样品的表面形貌和孔容,对样品进行BET分析,浮石和浮石载羟基氧化铁催化剂的物理吸附等温线如图3所示。浮石及浮石载羟基氧化铁的吸附等温线呈现出具有磁滞回线的Ⅳ型特点,这说明其均是介孔材料。表1展示了浮石和浮石载羟基氧化铁的比表面积和孔容,相比于纯浮石,浮石载羟基氧化铁的比表面积由0.56 m2·g−1提高到9.56 m2·g−1,孔容变大3.66倍。而浮石载羟基氧化铁的作用主要是催化臭氧氧化,是一种表面反应[19-20]。增加的比表面积和孔容也为臭氧提供更多的活性位点,从而提高其催化臭氧氧化效率。
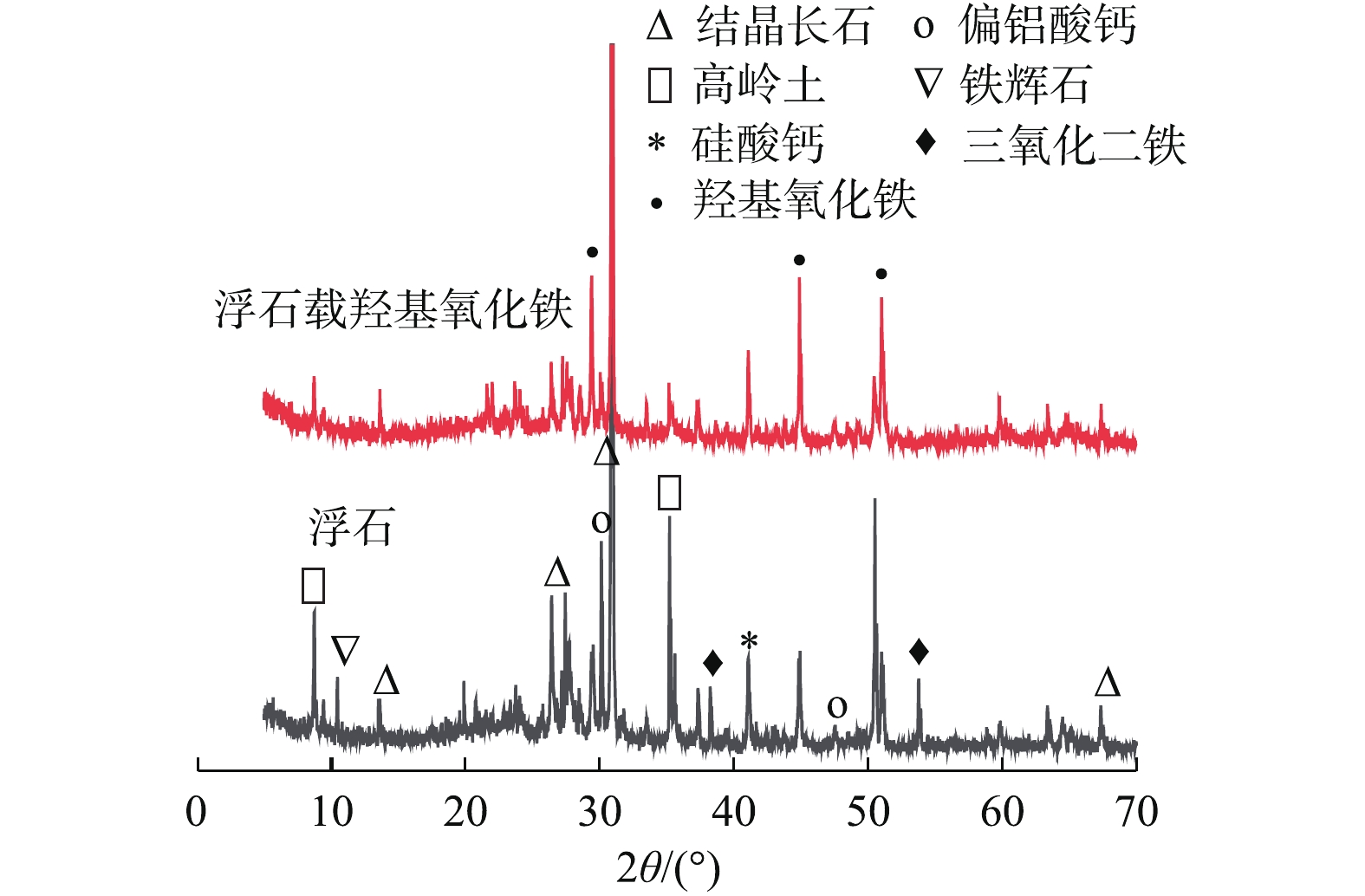
XRF的表征结果表明浮石主要由O(35.1%)、Si(17.9%)、Ca(11.7%)、Mg(5.9%)、Al(4.5%)和Fe(1.8%)构成。与之相比,浮石载羟基氧化铁含有的元素包括O(35.1%)、Si(17.4%)、Ca(11.2%)、Mg(5.8%)、Al(4.3%) 和Fe(3.3%)。浮石与浮石载羟基氧化铁的XRD的表征结果如图4所示。浮石的主要物相为结晶长石和硅铝酸盐。而浮石载羟基氧化铁催化剂与纯浮石却不相同,铁的负载导致浮石表面生成了与铁元素有关的α-FeOOH晶体[18]。
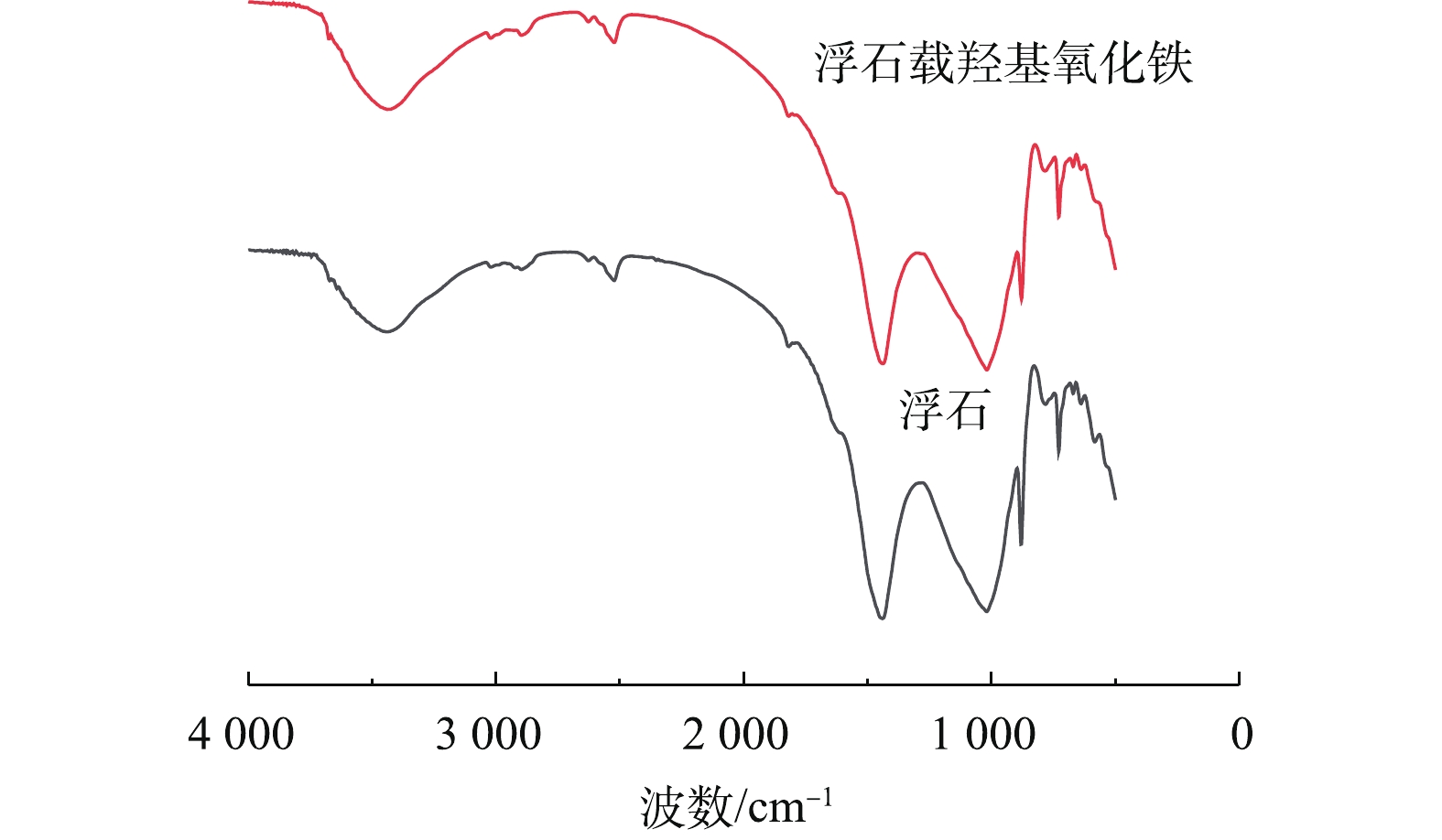
表面羟基在红外光谱中常于3 200~3 650 cm−1处出现峰值。由图5可知,浮石载羟基氧化铁和浮石在3 450 cm−1处均有吸收峰,主要为水分子和表面羟基(Si—OH、Ca—OH、Mg—OH或Fe—OH)的拉伸运动[18],而1 620 cm−1处的波峰对应于羟基的弯曲模式[21]。浮石载羟基氧化铁表面羟基官能团峰高的增加,表明α-FeOOH含量的增加。1 020 cm−1处附近的峰代表Si—O—Al键的伸缩振动[15],880 cm−1处的峰可能是由于Al—OH的振动[22],Fe—OH键的弯曲振动和Fe—O键的伸缩振动导致催化剂在500~800 cm−1出现吸收峰[23],这些结果与以前的研究结果[18]相一致。
-
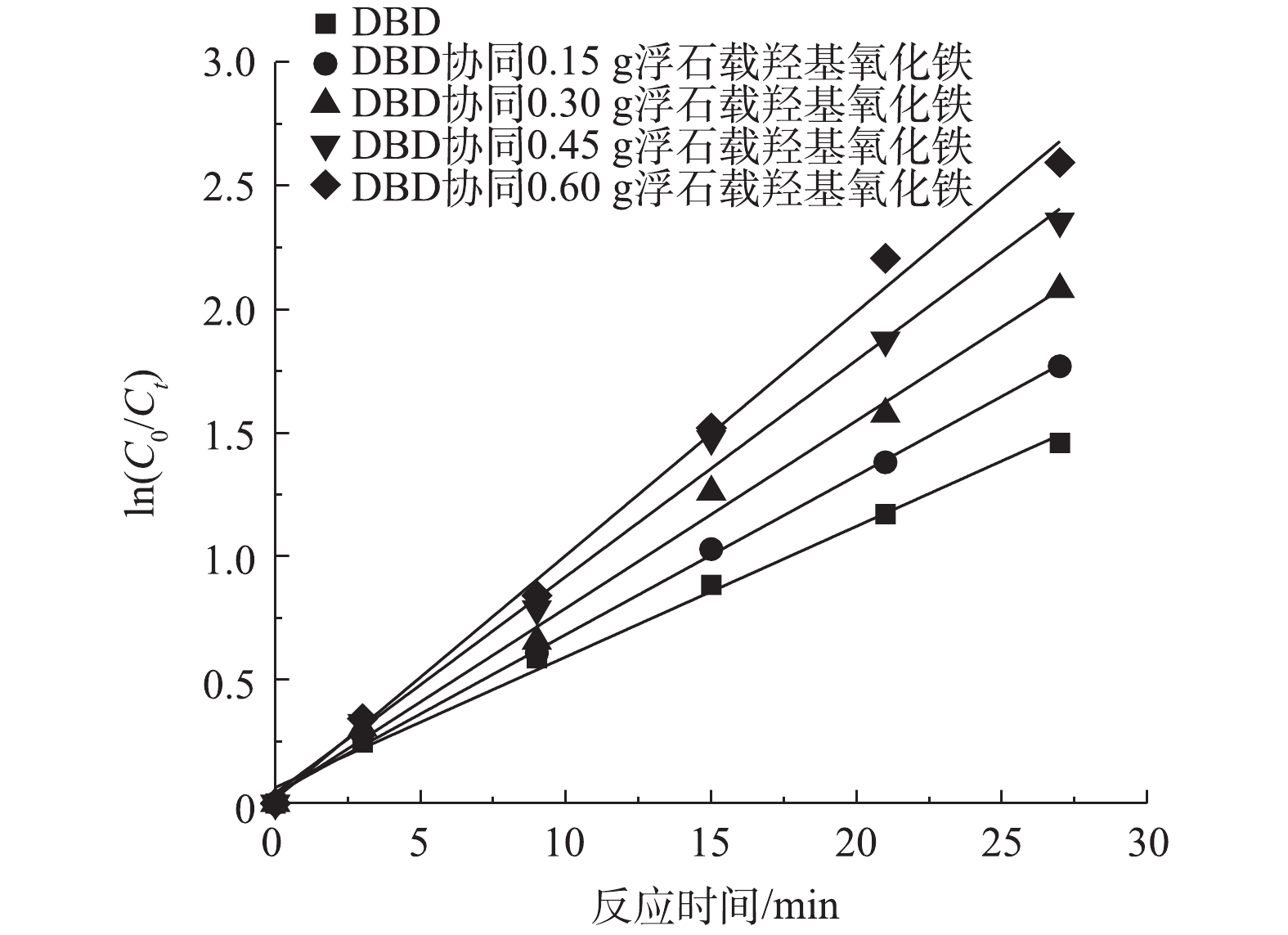
催化剂的投加量会影响DEET的去除速率和效果。DBD协同不同量的浮石载羟基氧化铁降解DEET的速率对比如图6所示,DEET的去除速率随着催化剂投加量的增加而上升,无催化剂投加的情况下,DBD降解DEET的一级动力学反应速率常数为0.053 min−1,而当浮石载羟基氧化铁投加量增加到0.45 g时,DBD协同浮石载羟基氧化铁的一级动力学反应速率常数为0.086 min−1,这可能是因为催化剂量的增加,提高了表面反应的位点数目,加速臭氧分解形成·OH。当催化剂添加量达到0.6 g时,DBD协同浮石载羟基氧化铁的一级动力学反应速率常数为0.098 min−1,DEET的去除速率只有轻微的提升,这可能是由于当持续增加催化剂添加量时,臭氧吸附位点富余,催化剂的利用率变低。
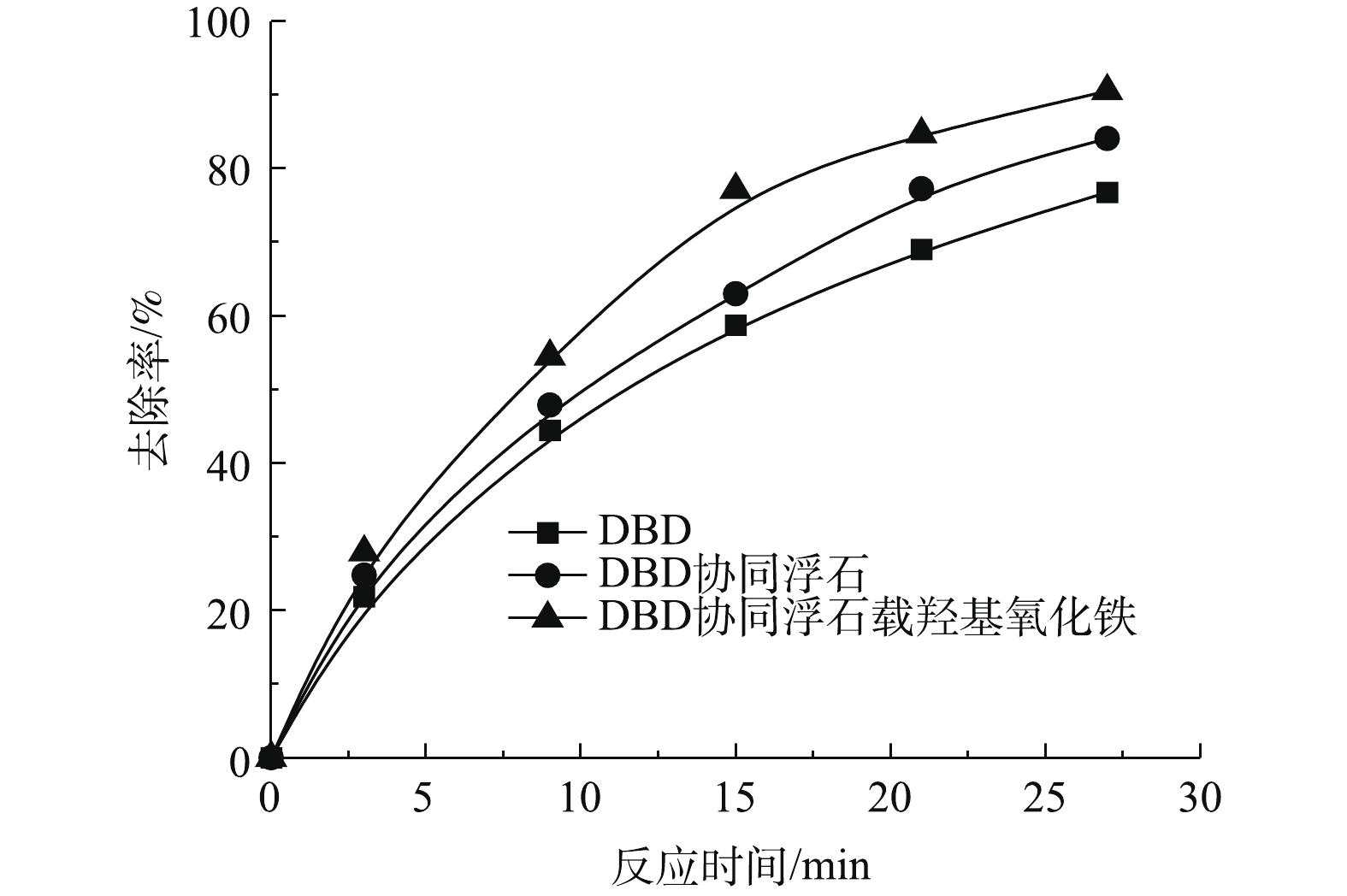
图7为DBD分别协同浮石、浮石载羟基氧化铁降解DEET的去除率随时间变化曲线,浮石和浮石载羟基氧化铁的添加均提高了DEET的去除率,这可能是浮石和浮石载羟基氧化铁催化DBD中产生的臭氧气体分解得到的·OH导致。同时浮石载羟基氧化铁具有更好的催化性能,该实验结果与前述的催化剂表征相匹配(更大的比表面积为臭氧提供更多的活性位点、更高的表面羟基含量)。为了去除催化剂对DEET吸附的影响,使用浮石和浮石载羟基氧化铁对DEET进行30 min的暗吸附实验,结果表明,浮石和浮石载羟基氧化铁吸附对DEET的去除率分别为3.82%和3.34%。
-
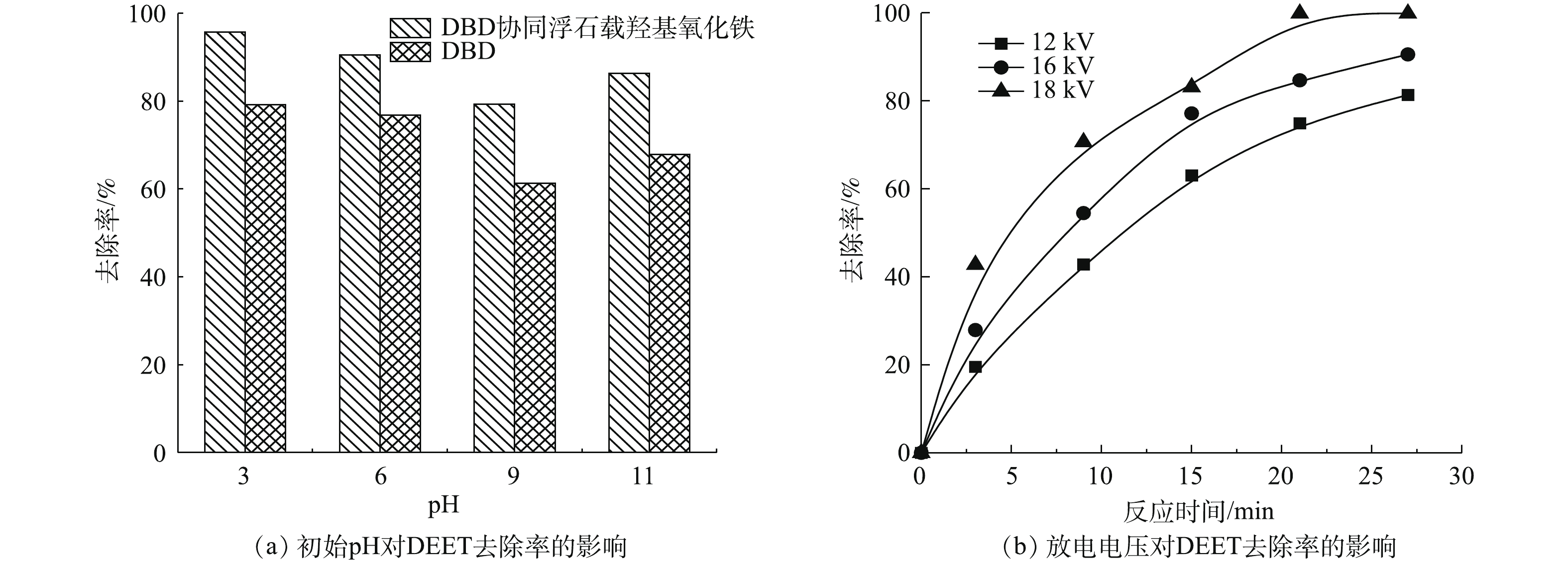
图8反映了pH和放电电压对DEET去除率的影响。如图8(a)所示,DEET的去除率随着pH由3增加到9而降低,其原因可能是等离子放电在pH较低时能产生更多的·OH[24]。另一方面,随着OH−浓度的增加,一些·OH可以与OH−结合形成水,从而降低DEET的去除率[25]。而当pH 为9~11时,DEET的去除率反而提升可能是因为臭氧分解产生·OH速率随着溶液中碱性的增强而变快[26-27]。由图8(a)可知,在pH为3~11时,浮石载羟基氧化铁均能提供稳定的催化性能。
如图8(b)所示,放电电压增加也表明放电功率的增加,随着放电电压的提高,DEET的去除率也有明显增加。放电电压为18 kV时DEET的去除率为放电电压为12 kV的1.23倍。等离子体中的自由电子通过碰撞使氧和水分发生强电离产生大量的活性物种(·O、O3和·OH等)[28]。当放电电压增加时,电子在放电区域会获得更多的能量,从而增加碰撞的概率,形成更多的活性物种,进而提高DEET的去除率。
-
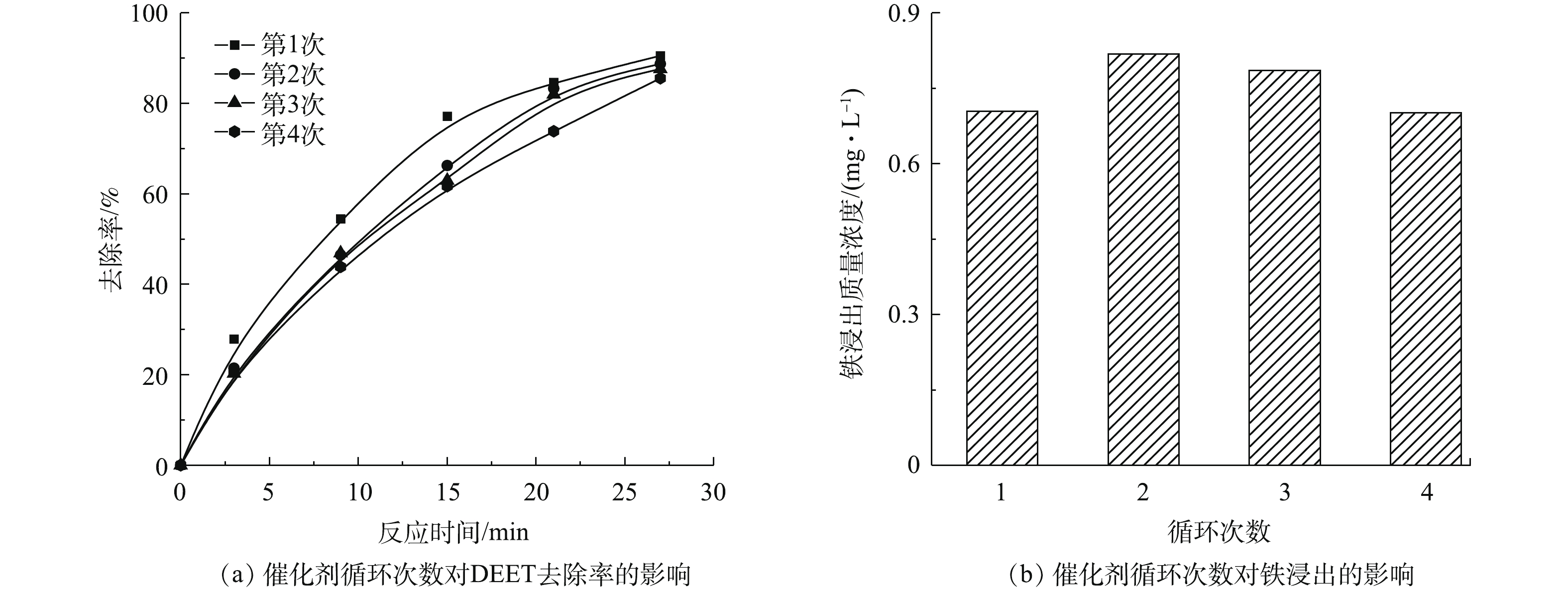
为了研究浮石载羟基氧化铁的稳定性,在相同的条件下对催化剂进行4次循环实验。每次循环后的样品经过离心收集后,用超纯水洗涤、干燥以备下次使用。如图9所示,材料经过4次循环后,DEET的去除率仅有轻微的降低。同时为了进一步检测材料的稳定性,使用石墨炉原子吸收光谱仪测定每次反应液中溶出铁的浓度,每0.45 g催化剂反应一次铁的损失量在0.21~0.24 mg,相比于前述表征中催化剂中铁的含量占比较小,由此可见,合成的浮石载羟基氧化铁具有良好的催化性能,且具有较好的重复使用稳定性。
-
使用异丙醇(IPA)作为羟基自由基的捕获剂。同时为了推测浮石载羟基氧化铁在体系中的作用,设计3种反应器运行模式:M1仅将放电区域产生的等离子气体通过气泵输送到进料水槽中,不存在液循环进入放电区域;M2存在液循环进入放电区域,但气泵停止工作;M3为上述实验方法中所述。
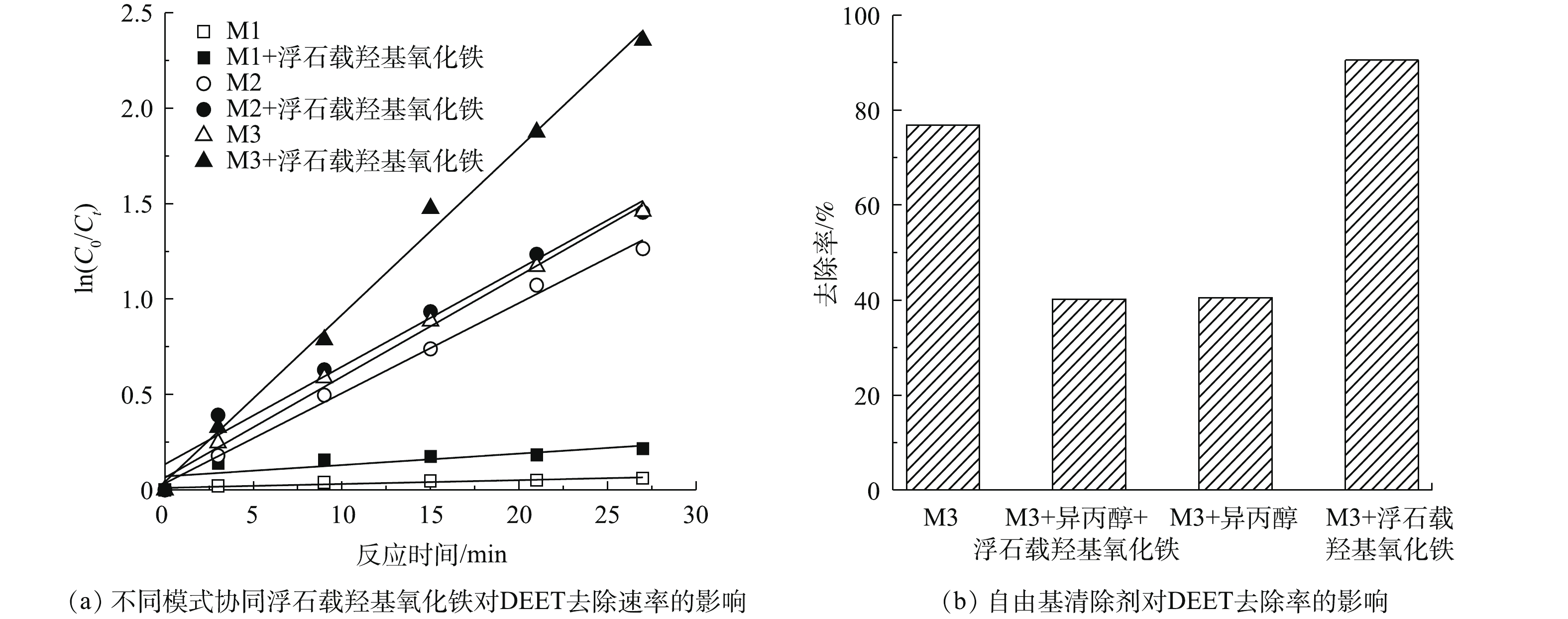
在之前的研究中[9],M1中臭氧浓度最高为7.125 μmol·L−1,M3中臭氧浓度略低于M1,为4.312 μmol·L−1,而M2中臭氧浓度很低,仅有1.416 μmol·L−1。图10(a)为不同模式下协同浮石载羟基氧化铁降解DEET的去除速率对比。相比于单独的介质阻挡放电,M2协同浮石载羟基氧化铁的一级动力学反应速率几乎没有提升。而M1协同浮石载羟基氧化铁的一级动力学反应速率常数提高1.94倍。由此可知,相比于DBD,协同体系处理DEET的速率提升可能主要归因于浮石载羟基氧化铁的催化臭氧氧化作用。由图10(b)可知,在反应体系中,·OH是主要的活性物种,在纯M3体系内,·OH对DEET的去除率约36%,但在M3体系中加入了浮石载羟基氧化铁后,该复合体系中·OH对DEET的去除率约50%,由此可见,浮石载羟基化铁的加入为体系中引入了更多的·OH。
DBD中引入浮石载羟基氧化铁提高了DEET的去除率,说明体系存在明显的协同效应,而协同效应的产生主要因为浮石载羟基氧化铁的表面羟基官能团起着催化臭氧的作用,其与臭氧反应产生HO3·和氧气[17],HO3·再分解产生·OH和氧气,同时,等离子放电过程还会在体系中产生H2O2,羟基氧化铁还能诱发类芬顿反应从而形成·OH [29],进一步加速DEET的降解。所涉及的反应过程如式(1)~式(5)所示。
2.1. 催化剂的表征分析
2.2. 催化剂投加量对DEET去除性能的影响
2.3. 初始pH和放电电压对DEET去除率的影响
2.4. 催化剂的稳定性
2.5. DBD与浮石载羟基氧化铁的协同机制
-
1)湿壁介质阻挡放电协同浮石载羟基氧化铁对DEET有着较高的去除效果和一定的协同作用,同时浮石载羟基氧化铁在初始pH为3~11时均有稳定的催化性能。协同体系去除DEET的一级动力学常数是单独等离子体系的1.62倍。在27 min实验后,协同作用去除率提高了13.74%。
2)在一定范围内,增加浮石载羟基氧化铁的投加量,可以提升DEET的去除率。
3)加入羟基自由基清除剂可显著降低DEET的去除率,且湿壁介质阻挡放电协同浮石载羟基氧化铁体系中,DEET的去除率要高于单一的湿壁介质阻挡放电体系。
4)浮石载羟基氧化铁在协同体系中的主要作用是催化作用,其表面羟基官能团能促进等离子气中的臭氧最终分解成·OH,从而提高DEET的去除率。
