

 DownLoad:
DownLoad:







-
挥发性有机污染物(VOCs)是指在常温常压下沸点为50 ~ 260 ℃、饱和蒸汽压大于133.13 kPa的有机化合物。VOCs 通常来源于包装印刷、石油化工等行业及机动车尾气排放,可能导致雾霾和光化学烟雾的形成和臭氧层破坏,而且也给人体健康带来威胁,故需要对相关排放源进行治理。
光催化通过光源照射在半导体催化剂表面,引发电子从价带转移到导带,并与空气及水反应,生成羟基自由基等强氧化性基团;与污染物反应,从而达到强化降解污染物的目的[1-3]。通常,由于光催化材料的吸附能力通常较弱,因此,通过将光催化剂负载到吸附剂表面有着广阔的研究前景[4-7]。在基于真空紫外的光催化氧化(VUV-PCO)工艺中,O3通过185 nm真空紫外光辐射产生,可用于进一步增强污染物的氧化。但若未充分利用,出口处的高浓度O3同时也是二次污染物。活性炭(AC)拥有出色的吸附性能[8-9],具有一定的催化氧化作用,在减少O3污染排放的同时,可生成强氧化剂,增强对污染物的降解,并减少副产物的排放[10-11]。HUANG等[12]利用沸石良好的VOCs和臭氧吸收能力,以沸石作为载体制备了TiO2/ZSM-5催化剂,在提高苯吸收能力的同时,利用臭氧分解能力获得了较高的苯去除率。SHU等[13]制备0.1%Mn/40%TiO2/AC催化剂,在VUV-PCO工艺下,实现近86%的甲苯去除率,并可将O3催化转化为O(1D)和·OH高活性物种,辅助催化氧化。GUO等[14]和MATOS等[15]通过TiO2在活性炭上不同方式的负载,均得到了高效的气态甲苯去除率。采用溶胶-凝胶、浸渍法等原位负载或机械混合的方式负载活性炭[12],一方面发挥活性炭出色的吸附性能,另一方面也能发挥其与催化剂的协同效应,在增强降解性能的同时减少二次污染问题。
本研究采用共沉淀法制备了Ce掺杂ZnO纳米催化剂,通过机械混合的方式将其负载在活性炭上,通过考察负载活性炭对光催化性能(污染物去除率、矿化率和O3消耗量)的影响,获得了Ce-ZnO和粉末活性炭最佳负载比例,进而提出了可能的降解机理,并探究了活性炭对催化剂使用寿命及降解产物的影响,最后对该工艺的能耗进行了分析。本研究结果可为VUV-PCO体系下增强催化性能和稳定性,优化能量利用率提供参考。
全文HTML
-
对二甲苯、Ce(NO3)3·6H2O、Na2CO3购于阿拉丁试剂有限公司,ZnSO4·7H2O购置于上海凌峰化工有限公司,活性炭购于溧阳市天顺活性炭有限公司,无水乙醇购于安徽安特食品股份有限公司。若无特别说明,所有药剂均为分析纯。
-
Ce-ZnO的制备:将4.34 g硝酸铈六水合物溶于100 mL超纯水中,并加至七水硫酸锌溶液中,制备摩尔比为0.96%的混合溶液,以300 r·min–1的搅拌速度搅拌3 h,从而获得Ce掺杂的ZnO复合物的前体物;用乙醇和超纯水利用超声波清洗机洗涤沉淀物3次至纯净,置于65 ℃烘箱中干燥后,在351 ℃下煅烧339 min,得到Ce-ZnO。
Ce-ZnO负载活性炭的制备:将Ce-ZnO和活性炭按不同比例加入无水乙醇,放至搅拌器中搅拌(100 r·min–1),然后将其放入超声波清洗器中,混合搅拌30 min;将混合物均匀涂覆在石英舟上,并将其置于65 ℃下干燥10 h。
-
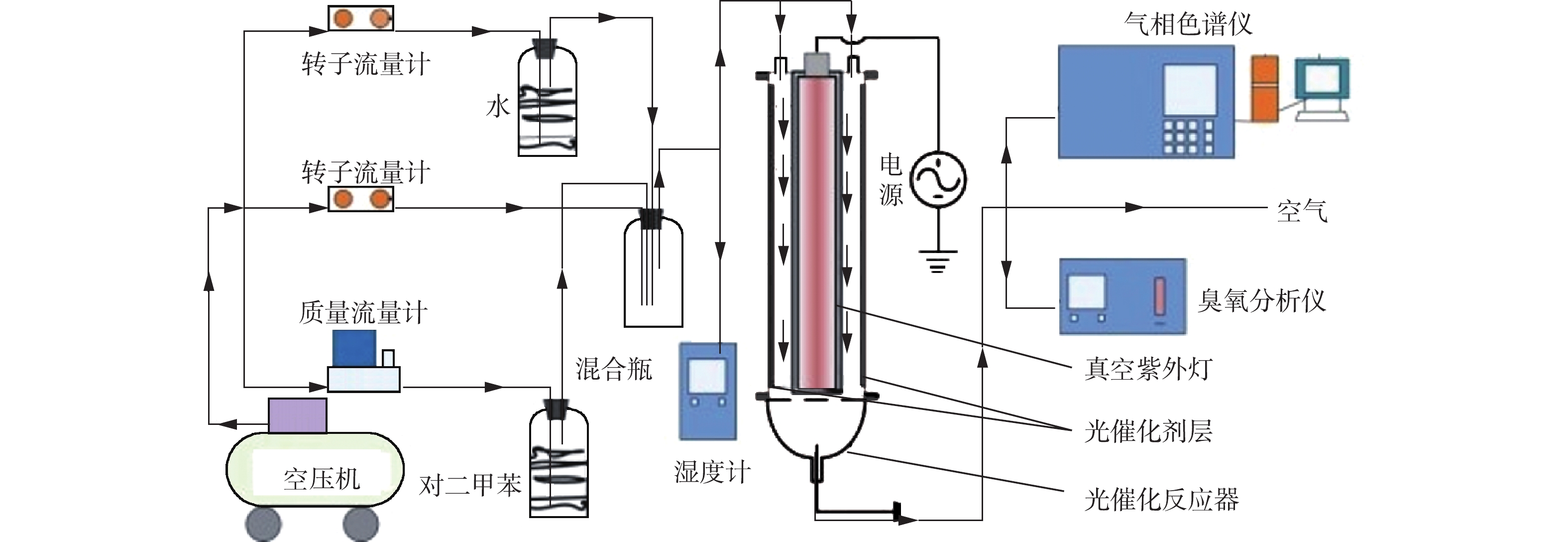
本研究中使用的性能测试系统见图1。测试系统主要由配气系统、光催化反应系统和检测系统3部分组成。通过配气系统产生不同浓度和湿度的对二甲苯废气。光催化反应系统由真空紫外灯管、光催化反应器和石英舟构成。光催化反应器包含圆形套管,内筒内放置紫外灯管,内筒(长44 cm)与外筒(长44 cm)夹层间放置涂覆有光催化剂的石英舟(长43 cm)。反应器的体积为600 cm3,长40 cm的紫外灯管(36 W,185 nm)置于反应器中,使VUV均匀辐射气流和光催化剂。实验中用无水乙醇将1 g催化剂均匀涂覆在石英舟上,涂覆面积为1 130 cm2。打开紫外灯的同时,连续通入对二甲苯混合气体,在一定时间内分别测定反应器出口处的对二甲苯浓度、二氧化碳浓度和臭氧浓度。实验中,通过调节配气比例和流量,控制对二甲苯浓度为150 mg·m–3,相对湿度为40%、停留时间为30 s。对二甲苯浓度和二氧化碳浓度由Agilent 6890气相色谱仪HP-Innowax型和HP-Plot-Q型毛细管柱定量分析,臭氧浓度利用德国德图(testo625)臭氧分析仪测定,中间产物由气相色谱-质谱联用(Agilent GC-7890,MS-5790)进行分析。
1.1. 实验药品与仪器
1.2. 催化剂的制备
1.3. 吸附-光催化性能测试
-
许多研究[16-19]表明,Ce掺杂ZnO能够显著提高催化剂的光催化性能;但受制于其有限的活性位点,在较短的反应时间内,污染物无法与催化剂充分接触而被高效去除。活性炭作为一种有效的吸附材料,具有大比表面积和丰富的孔结构,通过表面碳原子与O3结合形成含氧基团,同时,其本身的催化活性也有一定的O3去除效果[20]。通过活性炭与Ce-ZnO的有效结合,有望获得较高的催化氧化性能。
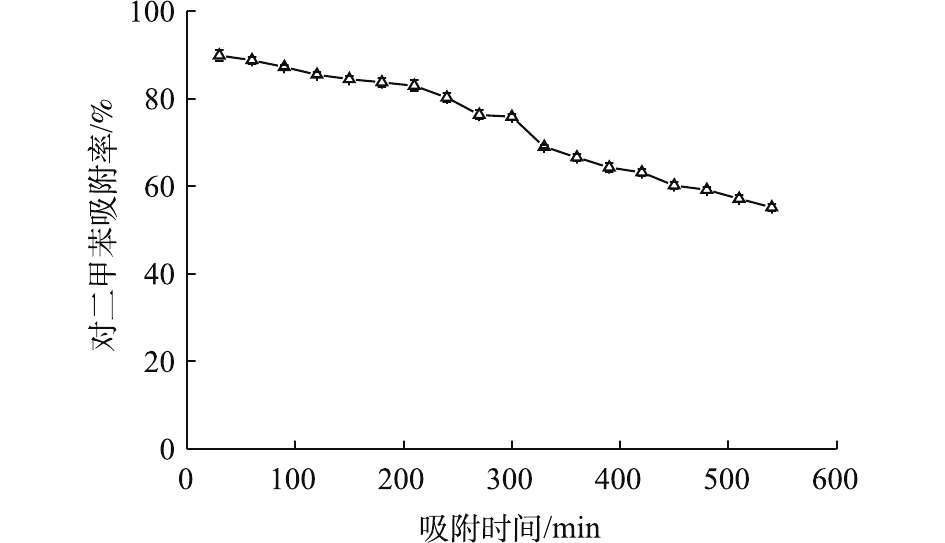
为考察Ce-ZnO/AC的吸附性能,在没有紫外光的条件下对催化剂进行了吸附稳定性测试(图2)。可以发现,复合材料对对二甲苯的吸附率持续下降。200 min后下降速率增加,并在9 h时降至60%以下。同时,测定了反应器出气中的二氧化碳,发现其浓度与进气中二氧化碳浓度相似。这说明在没有紫外光照射时,复合催化材料对对二甲苯只存在吸附作用。
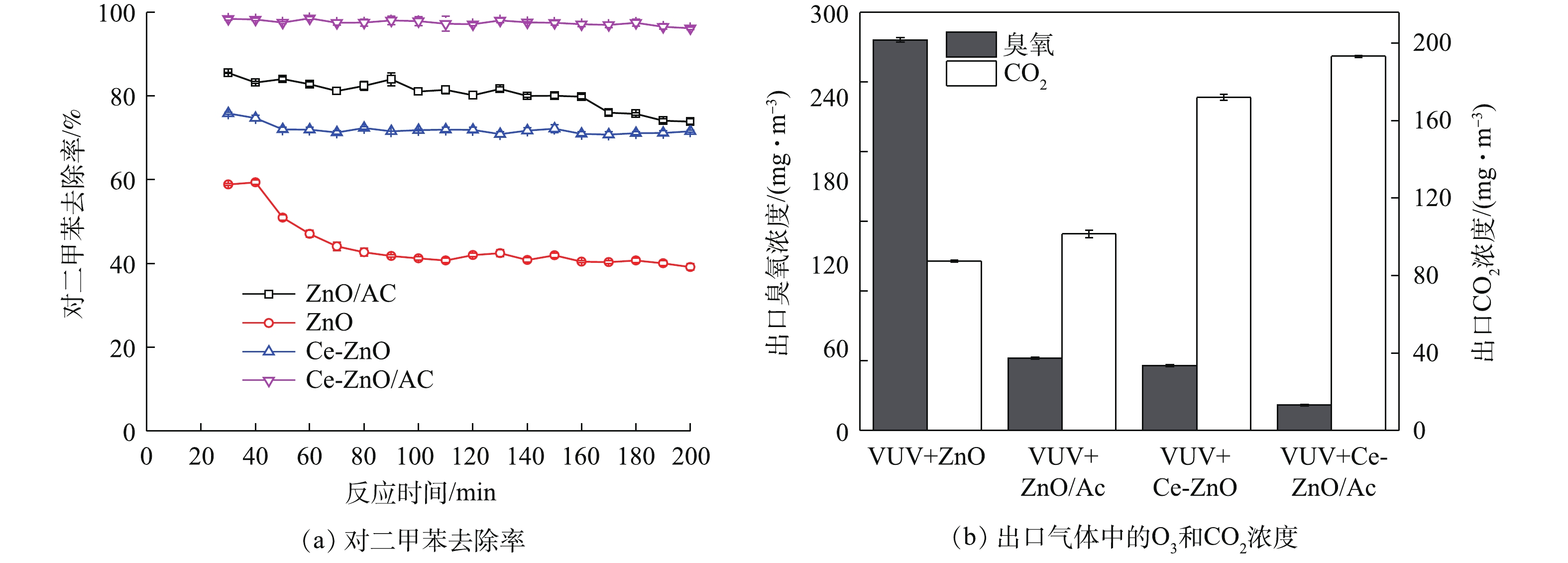
为验证复合催化剂的催化性能,考察不同工艺(VUV+ZnO、VUV+ZnO/AC、VUV+Ce-ZnO和VUV+Ce-ZnO/AC)对对二甲苯的去除效率,同时测定了反应器出口O3浓度和CO2浓度。如图3 (a)所示,单独使用ZnO作为光催化剂,120 min后对二甲苯的去除率从60%降低至40%左右,表明ZnO在反应过程中缓慢失活,催化稳定性较差。对照实验表明,ZnO失活后的降解效率来自真空紫外的贡献。当Ce掺杂到ZnO内部后,对二甲苯的去除率升高到70%以上,并且在120 min之后没有出现明显下降,表明Ce-ZnO具有良好的催化稳定性。ZnO负载活性炭时,对二甲苯的去除率在80%以上,并在160 min后开始有下降趋势;而当Ce-ZnO/AC放入反应器中时,对二甲苯的去除率进一步提高至98%以上,并在测试时段内保持稳定。这说明吸附-催化混合型催化剂具有优异的对二甲苯降解性能及稳定性。如图3 (b)所示,在单独VUV体系中,反应器出气中O3浓度为3 002.5 mg·m−3。当使用ZnO作为催化剂时,O3浓度降至340 mg·m–3,同时产生150 mg·m–3的CO2。当ZnO负载活性炭后,出气中O3浓度迅速降低至60 mg·m−3,但CO2生成量提升幅度较小。这说明,虽然活性炭具有较好的污染物富集能力[21-22],但富集到活性炭表面的对二甲苯不能被催化剂彻底矿化,因而CO2没有大幅提升。同时,活性炭具有一定的臭氧分解能力[23-25],使得出气中O3浓度大幅下降。当Ce-ZnO作为催化剂时,O3分解加快,CO2生成量达到170 mg·m−3。当Ce-ZnO与活性炭混合之后,出气中O3浓度进一步降低至20 mg·m−3,同时对二甲苯的矿化率进一步提高,CO2产生量达到190 mg·m−3。这些结果说明,活性炭的加入不仅对光催化氧化反应起促进作用,提高对二甲苯的降解效率,而且在臭氧的分解中也起到积极作用。
-
虽然吸附-催化复合材料对对二甲苯具有良好的降解效果,但当催化剂在活性炭上的负载量过高时,部分催化剂将占据活性炭表面的微孔,抑制活性炭的吸附性能;当负载量过低时,光催化剂就无法发挥较大的作用,从而降低催化氧化的效果。因此,催化剂在活性炭上的负载比例尤为重要。因此,实验考察了Ce-ZnO与活性炭的质量比分别为1∶4、2∶4、3∶4和4∶4的催化性能,实验结果如图4(a)所示。当光催化剂与活性炭比例为1∶2时,对二甲苯去除率最高,且出口的O3浓度最低,为20 mg·m–3左右,CO2生成量达到175 mg·m–3。此后随着催化剂比例的增加,降解性能和矿化率都有所下降,出口气体中的臭氧残留较多。当催化剂负载比例较低时,在反应初始阶段,吸附催化性能较高,去除效果较好;但随着时间的推移,去除率缓慢下降。这是因为低负载比例下催化剂的降解性能有限,而活性炭的吸附作用占主导地位。当催化剂负载比例较高时,活性炭表面的催化剂含量相对较高,初始阶段的降解效果较好。但由于活性炭内部的部分孔径被催化剂所占据,无法与对二甲苯充分接触,发挥不了其富集作用,导致降解效率迅速降低。
此外,在考虑负载比例时,既要充分利用O3的氧化性能,又要防止出口气体中O3浓度过高。由图4(b)可以看出,光催化剂与活性炭的比例为1∶2时,对二甲苯的转化率更高,矿化率更高,O3的排放量也更少。这说明在该比例下,催化剂充分利用了O3的氧化性能,污染物降解率有所提高。
-
与传统的PCO工艺相比,VUV-PCO体系具有更多的降解途径。VUV(λ=185 nm) 辐射产生的高能光子可以使氧和水蒸气直接解离[26],反应见式(1)和式(2)。Ce-ZnO催化剂带隙激发产生的激发态导带电子和价带空穴,与吸附的电子供体得到受体发生电子转移反应[27-28],反应见式(3))。VUV辐射还将产生O3,并在后续降解过程中发挥重要作用[26-29],反应见式(4)~式(6)。
VUV辐射产生的O3不仅具有一定的氧化作用,O3作为强氧化剂和良好的电子受体还可有效防止电子-空穴对的复合,从而延长光催化作用中空穴的寿命。同时,O3能在Ce-ZnO催化剂的作下,分解产生激发态的氧原子O(1D)和羟基自由基(·OH)等自由基[12, 26-30]。负载的活性炭可以将O3吸附至其表面,捕获电子形成臭氧化物离子(
O⋅−3 ),然后形成·OH[13, 30-31],反应见式(7)~式(9)。O3分子也可以通过VUV辐射分解为O (1D) 和一个氧分子[32],反应见式(10)。以上反应过程中产生的激发态氧原子和自由基等均将增强VOC的降解效果。O3与活性炭之间存在的化学相互作用将生成羧基等含氧官能团。在催化剂负载过量的情况下,活性炭上活性位点被催化剂部分占据,此时官能团的迅速积累会导致活性炭表面更少的活性位点参与O3的分解(2个O3分子与表面位点相互作用以产生3个O2分子)[33-34]。而当负载量过少的情况下,虽然活性炭表面空余的活性位点相对增加,但此时Ce-ZnO的含量也相对较低,对吸附物质的催化氧化能力降低,协同效果不明显。
在VUV-PCO体系中,Ce-ZnO/AC作为催化剂时对二甲苯的降解效率最优。由图3(a)可以看出,未负载活性炭时,VUV + Ce-ZnO对对二甲苯降解率约为70%,其中单独VUV作用时的降解率为40%,而活性炭的加入可将其提升至99%,降解效率提高了29%。同时,活性炭的加入能够产生更多的二氧化碳。如图3(b)所示,与未负载的Ce-ZnO催化剂相比,复合催化剂在降解过程中的矿化率提升了11.8%。复合催化剂的吸附催化机理可总结如下:1) 185 nm VUV产生高活性物质(如羟基自由基和O3)并对污染物产生降解作用;2)残留的对二甲苯及大量中间产物通过吸附在活性炭和Ce-ZnO表面延长接触时间,并通过光催化氧化和臭氧辅助氧化进一步降解;3)活性炭本身的催化氧化性能在分解臭氧减少二次污染的同时,产生的高活性物质也能进一步促进污染物的转化。活性炭的强吸附性能和Ce-ZnO的催化氧化性能共同形成了复合催化剂的高催化活性,两者的协同作用导致了对二甲苯的高效降解。
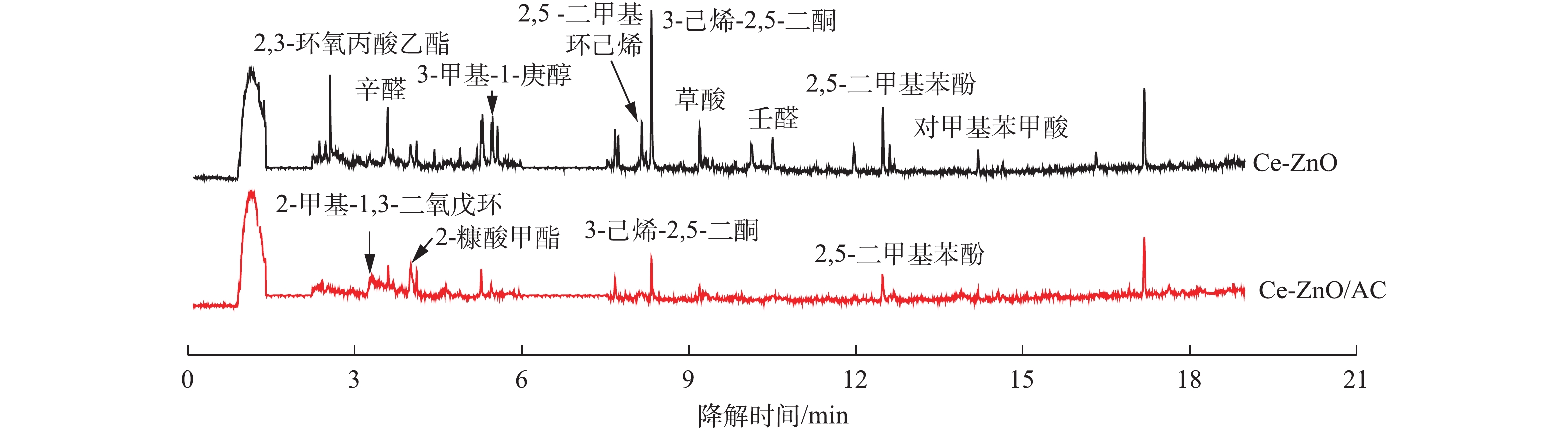
利用气相色谱-质谱联用(GC-MS)对反应器出口尾气中的成分进行检测,考察活性炭的负载对对二甲苯在VUV-PCO体系下中间产物生成的影响。如图5所示,尾气中除了未参与反应的对二甲苯外,还有大量的降解中间产物。当采用Ce-ZnO/AC时,中间产物中部分大分子物质(如2,3-环氧丙酸乙酯、3-甲基-1-庚醇、壬醛和2,5 -二甲基环己烯等)几乎被完全去除,一些小分子物质的种类也有所减少。说明活性炭的存在能够减少难降解中间产物的生成,促进对二甲苯向小分子物质和二氧化碳转变,验证了矿化率的增加。
-
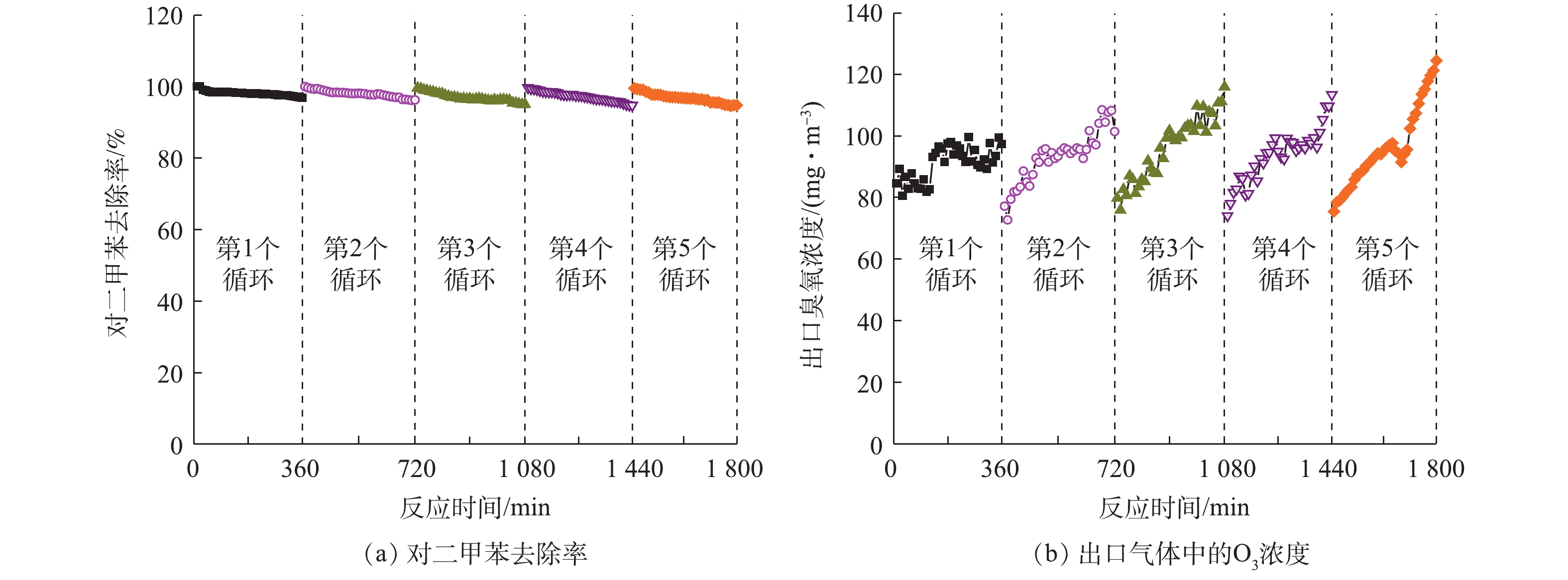
许多研究探讨了催化剂的稳定性,如Pt-ZnO-HAP[35]、Si-GO/ZnO[36]和胶体Au-CeO[37]催化剂在降解气态苯时,分别在400、1 260、300 min后均保持了较好的催化活性。为评价本研究制备催化剂的稳定性,将Ce-ZnO/AC (Ce-ZnO∶AC=1∶2) 放入光催化反应器中连续运行6 h。由图6 (a)可以看出,污染物的去除率在6 h内从99%降低到97%,虽然活性炭的负载可促进中间产物的进一步转化,但仍有部分小分子有机物积聚在催化剂表面,导致其降解效率略有下降[38]。在停留时间为60 s,相对湿度为50%时,采用VUV对Ce-ZnO/AC进行再生,3 h后催化剂可恢复其原始催化活性,并且该操作可进行5个循环以上。再生过程中,VUV产生的O3等活性物质可以氧化去除催化剂表面覆盖的有机中间产物,恢复位点活性。由图6 (b)可以看出,反应器出口O3的浓度变化幅度较小,这进一步证明了该复合材料具有稳定的O3分解能力。这些结果表明,制备的负载活性炭的Ce-ZnO催化剂具有较好的稳定性,这对其潜在的工业应用具有重要意义。
-
吸附-催化复合催化剂的催化活化需要利用紫外光,因此,催化剂的选择须考虑能量的利用情况。以降解单位污染物所需能量为指标,对不同工艺在降解过程中能耗的利用情况进行分析,计算过程见式(11)。
式中:Cinlet为对二甲苯进口浓度,mg·m–3;Coutlet为对二甲苯出口浓度,mg·m–3;Q为进气流量,L·min–3;P为紫外灯管功率,W;E为降解单位所需能量,Wh·mg–1。
不同处理工艺的能耗情况差异较大。在相同的紫外光源照射下,仅使用VUV处理对二甲苯时,降解1 mg对二甲苯需要(12.400 ± 0.512 4) Wh的能量;加入ZnO后,所需能量为(9.300 ± 0.131 8) Wh;使用Ce-ZnO催化剂时,需要(5.300 ± 0.393 4) Wh能量;当使用活性炭作为载体的复合催化剂时,所需能量降至(3.900 ± 0.212 8) Wh。相对于单独VUV,使用了吸附-催化复合材料后的能量利用率提升了近3倍。
2.1. 吸附-催化复合材料性能测试
2.2. 负载比例的影响
2.3. VUV辐射下对二甲苯的催化氧化
2.4. 催化剂稳定性考察
2.5. 能耗分析
-
1) Ce-ZnO负载于活性炭上对对二甲苯的降解具有良好的促进作用。负载活性炭时,催化剂对二甲苯的去除率可达99%,相对未负载前矿化率提高了11.8%,同时提高了对真空紫外光解产生臭氧的利用效率,减少了出口气体中臭氧浓度。
2)当Ce-ZnO与活性炭的负载比例为1∶2时,能够使吸附-催化降解的协同效果达到最大化。此时,复合催化剂具有较高的稳定性,能够在原位再生恢复其原始催化活性,在5次循环后对二甲苯的去除率仍可达到94.8%。
3) VUV、光催化氧化和活性炭促进的臭氧辅助氧化的协同作用导致对二甲苯的高效去除。中间产物的分析表明,活性炭的负载能够促进对二甲苯转化为小分子物质,减少难降解中间产物的比例。
4)吸附-催化降解工艺的能量利用效率较高,相对单独催化工艺能耗节省了近1/3,仅为单独VUV工艺的1/4。
