


 下载:
下载:





-
铬作为主要的工业原料被广泛应用于金属冶炼、材料加工、电镀、化工及制革等行业中[1-3]。如果大量未经处理的含铬废水排入环境,会对环境造成严重危害[4]。美国环保署早已将Cr(Ⅵ)纳入优先控制污染物清单,中国《生活饮用水卫生标准》中规定Cr(Ⅵ)浓度<0.05 mg·L−1。受天然溶出、铬渣堆放、含铬废水排放等不同污染源的影响,环境中铬污染浓度往往存在很大差异[5-11]。以铬渣堆放点为例,由于周边扩散条件和水流方向等地质条件的差异,最大Cr(Ⅵ)污染浓度超出了生活饮用水卫生标准的2 000倍[8, 12]。
将毒性强、迁移性好的Cr(Ⅵ)还原为毒性低的Cr(III)沉淀是目前广泛采用的Cr(Ⅵ)污染治理方法[13-14]。Cr(Ⅵ)主要还原途径包括生物还原和非生物还原,生物还原通过环境中耐铬微生物对Cr(Ⅵ)进行还原。由于Cr(Ⅵ)本身对微生物有一定毒性作用[15-16],还原Cr(Ⅵ)的微生物较少,且还原速率慢,使得生物还原Cr(Ⅵ)存在很多限制。非生物还原主要利用环境中的还原性物质,如Fe(Ⅱ)、有机质等,其特点是还原速率快,但消耗以后很难再生。在自然界中,含铁黏土矿物中的结构态铁含量可占到土壤和沉积物中铁含量的50%[17],广泛分布的异化铁还原菌可还原含铁黏土矿物中的铁,实现Fe(Ⅲ)-Fe(Ⅱ)的循环,该循环对环境中污染物代谢有着重要的作用[18]。此外,环境中广泛存在的天然有机质对不同生物还原过程中电子传递过程也有着重要的影响[17, 19-20]。
前期研究中,已发现电子传递体与含铁黏土矿物的添加对微生物还原0.8 mmol·L−1 Cr(Ⅵ)产生了明显的协同促进作用,这是由于电子传递体加速了微生物与含铁黏土矿物之间的电子传递,使黏土矿物中的Fe(Ⅲ)还原成结构态Fe(Ⅱ),并促进Cr(Ⅵ)的非生物还原[21-22],本研究则重点考察不同浓度的Cr(Ⅵ)条件下,电子传递体与含铁黏土矿物存在时对水体中Cr(Ⅵ)生物还原过程的协同促进作用。选取含铁量较高的黏土矿物绿脱石(NAu-2)和模式希瓦氏菌(Shewanella oneidensis MR-1)为代表,并选取蒽醌-2,6-二磺酸(AQDS)作为电子传递体,重点分析复杂环境体系中不同浓度条件下Cr(Ⅵ)迁移转化过程及机理,为地下水中不同污染程度的Cr(Ⅵ)环境污染治理修复提供参考。
全文HTML
-
希瓦氏菌(Shewanella oneidensis MR-1,MR-1)采用不含葡萄糖的胰蛋白胨大豆肉汤(TSB-D)培养基进行培养。对培养后的MR-1进行离心(相对离心力为3 500 g,10 min)并弃掉上清液,加入20 mmol·L−1的1,4-哌嗪二乙磺酸缓冲溶液(pH=7.0,简称PIPES,实验过程中均采用此缓冲溶液)旋混,再离心,重复上述操作3次,最后一次清洗采用无氧PIPES缓冲液(经高纯氮气曝气脱氧),在厌氧手套箱中进行,最后取重悬液稀释,进行浓度测定备用。整个实验过程均在室温(25 ℃)下进行。
-
本研究采用的含铁黏土矿物为绿脱石(NAu-2),购自美国黏土矿物协会,其主要成分为
M+0.72 (Si7.55Al0.16Fe0.29)(Al0.34Fe3.54Mg0.05)O20(OH)4,其中M可能是Ca、Na或者K。NAu-2中的铁含量约为4.1 mmol·g−1,主要以结构态Fe(Ⅲ)的形式存在。将NAu-2研磨分散至0.5 mol·L−1 NaCl溶液中,进行搅拌、超声等操作,再通过离心的方法获得0.5~2 μm的黏土矿物颗粒,最后用超纯水反复清洗至上清液中无氯离子检出后,在60 ℃下烘干,用无氧PIPES缓冲液配制20 g·L−1的NAu-2储备液备用。优级纯重铬酸钾(K2Cr2O7)购自国药集团化学试剂有限公司。在厌氧手套箱中,将烘干后的K2Cr2O7溶于无氧水(经高纯氮气曝气脱氧)中,配制12.0 mmol·L−1 Cr(Ⅵ)储备液备用,使用前过0.22 μm无菌滤膜。PIPES、乳酸钠及AQDS购自Sigma-Aldrich,均配制为无氧储备液备用,使用前采用高压灭菌(121 ℃,15 min)或者过无菌滤膜的方式去除微生物。
-
本研究中主要采用批实验的方式考察NAu-2与AQDS对不同浓度Cr(Ⅵ)生物还原过程的影响。Cr(Ⅵ)浓度选取0.1、0.2、0.5、0.8、1.2和2.0 mmol·L−1,其他条件均一致。每种Cr(Ⅵ)浓度条件下均考察单独或同时添加NAu-2和AQDS对Cr(Ⅵ)生物还原过程的影响,并考察相关对照组及无微生物存在下的系统稳定性。每种Cr(Ⅵ)浓度下批实验具体添加方式见表1。整个实验过程均在厌氧手套箱中进行(25 ℃),氧气含量低于0.1 mg·L−1,Cr(Ⅵ)空白组以及实验组反应过程的有效容积为10 mL,缓冲体系为20 mmol·L−1 PIPES(pH 7.0),NAu-2浓度为2.0 g·L−1。AQDS作为电子传递体,浓度为0.1 mmol·L−1;MR-1浓度7×108 cell·mL−1;乳酸钠作为电子供体,为反应的进行提供充足的电子,其浓度为0.5 mmol·L−1。本实验采用无菌注射器在厌氧箱中进行取样并分析。
-
溶液中的Cr(Ⅵ)通过离心和过滤的方式获得上清液,稀释后采用二苯碳酰二肼分光光度法[23-24],在波长540 nm下测定。生物还原NAu-2后溶液中结构态Fe(Ⅱ)浓度采用HF-H2SO4进行消煮,并用1,10-菲啰啉显色的方法测定,测定波长为510 nm[25]。
-
Cr(Ⅵ)生物还原过程中浓度变化符合一级动力学方程(式(1))。
式中:k为一级动力学常数,h−1;CCr(Ⅵ)为反应过程中Cr(Ⅵ)的浓度,mg·L−1;t为反应时间,h。
微生物还原含铁黏土矿物(NAu-2)生成Fe(Ⅱ),采用零级动力学方程(式(2))进行分析。
式中:kFe(Ⅱ)为Fe(Ⅱ)生成的零级动力学常数,mmol·(L·h) −1;C0为还原过程未进行(t=0 h)时消煮后体系中Fe(Ⅱ)的总浓度,mmol·L−1;Ct为t时样品消煮后Fe(Ⅱ)总浓度,mmol·L−1;t为反应时间,h;CFe(Ⅱ)为Fe(Ⅱ)浓度,mmol·L−1。
当NAu-2与AQDS单独或共存时,在Cr(Ⅵ)生物还原过程产生的强化促进作用,采用强化系数(enhancement factor,EF)[18, 22]表示,计算方法见式(3)~式(5)。
式中:FEF,AQDS为AQDS强化系数;FEF,NAu-2为NAu-2强化系数;FEF,AQDS+NAu-2为AQDS+NAu-2强化系数。kcells为反应过程中只有微生物存在下还原过程的一级动力学常数;kcells+AQDS、kcells+NAu-2和kcells+NAu-2+AQDS分别为生物还原过程中单独/同时存在AQDS和NAu-2时还原过程的一级动力学常数。
当NAu-2与AQDS共同存在时,采用协同系数(synergy factor,SF)表示,计算方法见式(6)。
式中:FSF为协同系数;kcells+AQDS、kcells+NAu-2和kcells+NAu-2+AQDS含义同上。
当协同系数>1.0时,表示存在协同作用;当协同系数=1.0时,表示不存在协同作用;当协同系数<1.0时,表示存在抑制作用[26-29]。
1.1. 微生物的培养
1.2. 含铁黏土矿物与主要化学试剂
1.3. NAu-2与AQDS共存对不同浓度Cr(Ⅵ)生物还原过程的影响
1.4. 分析方法
1.5. 数据分析
-
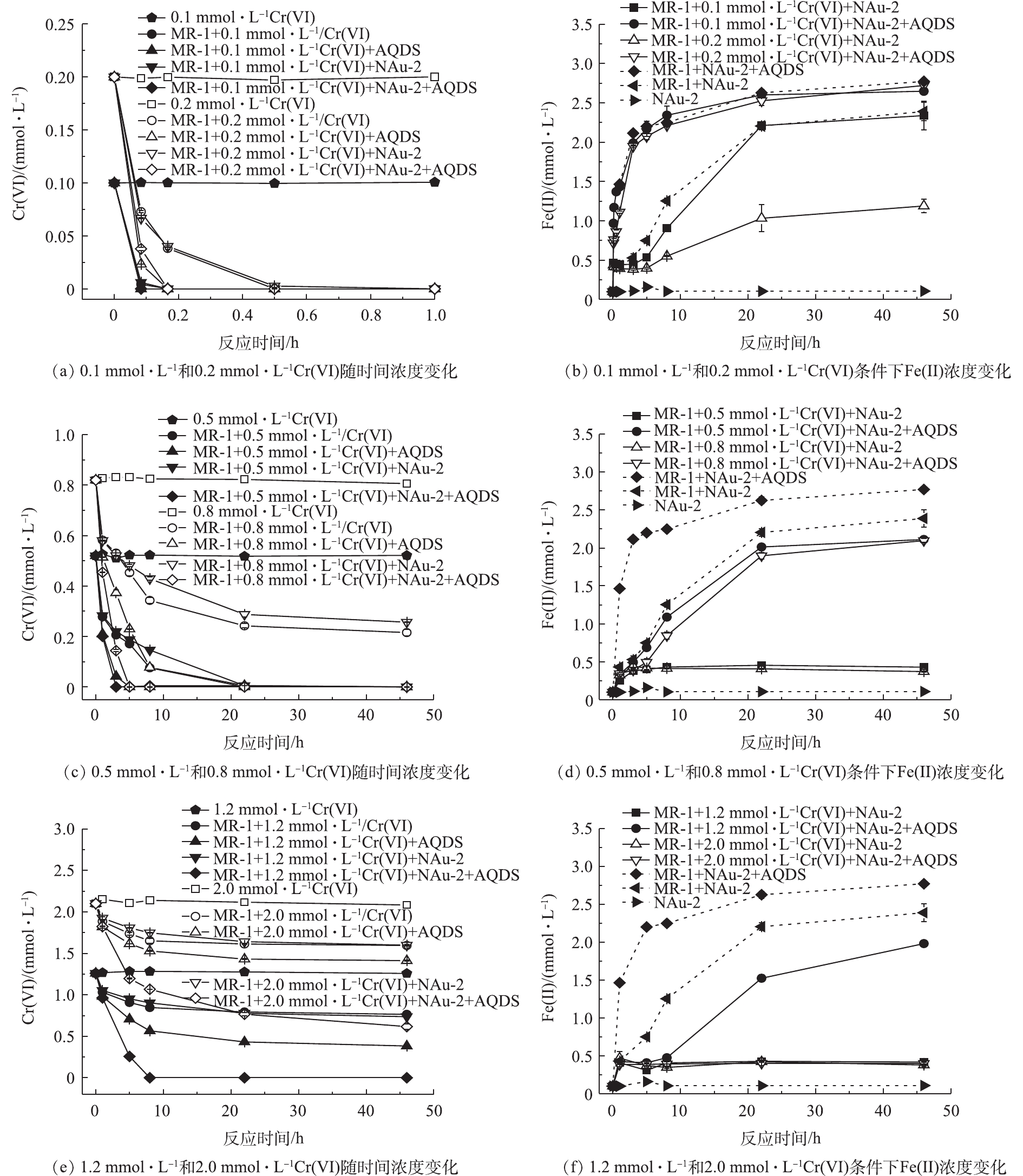
NAu-2与AQDS对不同浓度Cr(Ⅵ)生物还原过程的影响见图1。图1(a)、图1(c)和图1(e)反映了在不同浓度Cr(Ⅵ)条件下(0.1、0.2、0.5、0.8、1.2和2.0 mmol·L−1),AQDS与NAu-2单独/共存时对生物还原Cr(Ⅵ)的影响。当Cr(Ⅵ)初始浓度为0.1 mmol·L−1时(图1(a)),添加及未添加AQDS实验组中的Cr(Ⅵ)分别在5 min和10 min取样时降为0。随着Cr(Ⅵ)浓度升为0.2 mmol·L−1,MR-1单独还原 Cr(Ⅵ)实验组与添加NAu-2实验组中Cr(Ⅵ)浓度变化趋于一致。此时,两者一级动力学常数分别为(9.926±0.216) h−1和(8.622±0.976) h−1(表2),说明生物还原Cr(Ⅵ)这一过程中,NAu-2的单独添加并未起到明显促进作用。对于MR-1+Cr(Ⅵ)+AQDS和MR-1+Cr(Ⅵ)+AQDS+NAu-2的2组反应中,一级动力学常数分别为(25.787±0.071) h−1和(20.018±0.437) h−1,说明电子传递体(AQDS)对生物还原Cr(Ⅵ)起到明显的促进作用。当Cr(Ⅵ)初始浓度小于0.2 mmol·L−1时,微生物还原Cr(Ⅵ)的速率均较快,此时AQDS和NAu-2对这一过程的影响较小,无法计算一级动力学常数。此外,AQDS和NAu-2共存时,并未产生明显的促进作用。
当Cr(Ⅵ)初始浓度较高(0.5~2.0 mmol·L−1)时,NAu-2的单独添加对不同浓度Cr(Ⅵ)的还原均无明显促进作用。此时,kcells与kcells+NAu-2值相近,甚至添加NAu-2实验组中的k值略低(表2),且随着Cr(Ⅵ)初始浓度升高至0.8 mmol·L−1后,单独的微生物无法将Cr(Ⅵ)彻底还原。对于单独加入AQDS实验组,一级动力学常数kcells+AQDS均有明显升高,当浓度达到1.2 mmol·L−1和2.0 mmol·L−1后,实验组中单独添加AQDS时,Cr(Ⅵ)不能被彻底还原。Cr(Ⅵ)初始浓度为0.5~2.0 mmol·L−1时,AQDS和NAu-2的同时添加使Cr(Ⅵ)浓度降低最快,仅在最高浓度2.0 mmol·L−1时未被完全还原,且kcells+AQDS+NAu-2值均远高于kcells+AQDS和kcells+NAu-2的实验组(表2)。综上所述,当Cr(Ⅵ)浓度为0.8~2.0 mmol·L−1时,同时添加NAu-2和AQDS对生物还原Cr(Ⅵ)具有明显的促进作用。
-
图2反映了NAu-2和AQDS单独/共存时对生物还原不同浓度Cr(Ⅵ)的影响。通过引入强化系数,更好地对Cr(Ⅵ)生物还原过程进行评价。在不同初始浓度Cr(Ⅵ)条件下,生物还原Cr(Ⅵ)产生的强化系数见表3。与MR-1+Cr(Ⅵ)实验组相比,当强化系数>1.0时,说明起到明显的强化作用。由表3和图2可知,MR-1+Cr(Ⅵ)+NAu-2实验组强化系数均≤1.0,说明单独添加NAu-2对生物还原不同浓度Cr(Ⅵ)的过程均无强化作用,甚至产生一定程度的抑制,这是由于NAu-2生物还原过程与Cr(Ⅵ)生物还原过程存在一定的竞争关系[30]。MR-1+Cr(Ⅵ)+AQDS实验组与MR-1+NAu-2+AQDS组产生的强化系数分别为1.33~3.90和2.02~10.49,说明AQDS的加入与AQDS+NAu-2的同时加入对生物还原Cr(Ⅵ)均起到了明显的强化作用。
此外,对不同浓度Cr(Ⅵ)还原体系的协同促进作用进行分析,协同系数见表3。当Cr(Ⅵ)浓度为0.1 mmol·L−1时,反应太快,无法计算协同系数。当Cr(Ⅵ)浓度达到0.2 mmol·L−1和0.5 mmol·L−1时,协同系数分别为0.58和0.98,均小于1.0,说明当Cr(Ⅵ)浓度低于0.5 mmol·L−1时,同时加入AQDS和NAu-2,Cr(Ⅵ)生物还原过程并未表现出协同促进作用。随着反应体系中Cr(Ⅵ)浓度的继续升高(0.8~2.0 mmol·L−1),协同系数达到1.50~2.98,表现出明显的协同促进作用。
当Cr(Ⅵ)浓度为0.1 mmol·L−1时,AQDS与NAu-2单独/同时加入对还原速率并未影响,说明此时占主导作用的是单独微生物对Cr(Ⅵ)的还原。当Cr(Ⅵ)浓度为0.2~0.5 mmol·L−1时,单独添加AQDS对Cr(Ⅵ)生物还原过程均起到强化作用,但同时添加AQDS+NAu-2时,协同促进作用表现不明显,说明此浓度条件下AQDS对电子传递过程的促进作用占主导。当Cr(Ⅵ)浓度为0.8~2.0 mmol·L−1时,添加AQDS对Cr(Ⅵ)生物还原过程产生明显的强化作用,同时添加AQDS+NAu-2后,则表现出明显的协同促进作用,此时Cr(Ⅵ)还原过程中协同作用占主导。
-
图1(b)、图1(d)和图1(f)反映了加入NAu-2后不同浓度Cr(Ⅵ)生物还原体系中Fe(Ⅱ)浓度的变化情况,同时也与单独NAu-2及微生物直接还原NAu-2的空白和对照实验组中Fe(Ⅱ)浓度进行对比分析。在单独NAu-2的空白实验中,Fe(Ⅱ)浓度一直稳定在0.1 mmol·L−1左右,说明体系本身不会对NAu-2还原过程有影响。MR-1直接还原NAu-2实验组中,Fe(Ⅱ)浓度随着反应进行逐渐升高,速率常数为0.10 mmol·(L·h)−1 (图3),在22 h后趋于稳定,最终达到2.4 mmol·L−1。与微生物直接还原NAu-2相比较,加入AQDS对NAu-2生物还原过程起到明显促进作用,Fe(Ⅱ)生成速率提高20%(图3),8 h后,Fe(Ⅱ)浓度增长趋于稳定,最终达到2.8 mmol·L−1。
在Cr(Ⅵ)浓度为0.1 mmol·L−1的实验组中(图1(b)),在0~22 h反应时间内,MR-1+0.1 mmol·L−1Cr(Ⅵ)+NAu-2实验组中Fe(Ⅱ)浓度上升曲线略低于MR-1+NAu-2组;22 h后,两者Fe(Ⅱ)浓度变化基本一致。而加入AQDS后,MR-1+0.1 mmol·L−1Cr(Ⅵ)+NAu-2+AQDS与MR-1+NAu-2+AQDS实验组中的Fe(Ⅱ)变化趋势基本重合,零级动力学常数分别为0.118 mmol·(L·h)−1和0.119 mmol·(L·h)−1,说明0.1 mmol·L−1Cr(Ⅵ)加入对NAu-2生物还原过程并无影响,且NAu-2加入对于0.1 mmol·(L·h)−1Cr(Ⅵ)还原过程也未产生影响(图1(a))。当Cr(Ⅵ)浓度升高至0.2 mmol·L−1时,MR-1+0.2 mmol·L−1 Cr(Ⅵ)+NAu-2+AQDS实验组中k0(0.115 mmol·(L·h)−1)与加入0.1 mmol·L−1及不加Cr(Ⅵ)时的实验组基本一致(k0分别为0.118 mmol·(L·h)−1和0.119 mmol·(L·h)−1,见图3),但MR-1+0.2 mmol·L−1 Cr(Ⅵ)+NAu-2实验组中Fe(Ⅱ)浓度上升速率明显降低,k0仅为0.047 mmol·(L·h)−1。当Cr(Ⅵ)浓度达到0.5~2.0 mmol·L−1时,MR-1+Cr(Ⅵ)+NAu-2实验组中Fe(Ⅱ)浓度均保持在0.4 mmol·L−1左右,随着反应的进行没有升高的趋势,k0仅为0.018~0.021 mmol·(L·h)−1 (见图3)。而MR-1+Cr(Ⅵ)+NAu-2+AQDS实验组中,随着Cr(Ⅵ)浓度升高(0.5~2.0 mmol·L−1),k0从0.091 mmol·(L·h)−1降至0.018 mmol·(L·h)−1,说明AQDS的加入促进了微生物与NAu-2之间电子传递效率,但随着Cr(Ⅵ)浓度的增加,仍会使Fe(Ⅱ)的生成速率降低。
-
在厌氧环境中,微生物还原Cr(Ⅵ)污染物主要有4种途径(图4)。第1种途径,微生物直接还原Cr(Ⅵ);第2种途径,微生物还原的电子传递体间接还原Cr(Ⅵ);第3种途径,微生物还原含铁矿物中Fe(Ⅲ),产生的Fe(Ⅱ)非生物还原Cr(Ⅵ);第4种途径,Fe(Ⅲ)矿物与电子传递体共存下协同还原Cr(Ⅵ)[22]。在本研究中,不同浓度条件下主要通过第1种、第2种和第4种途径还原Cr(Ⅵ)。由于Cr(Ⅵ)对微生物具有毒性,因此,不同浓度Cr(Ⅵ)对微生物还原Fe(Ⅲ)的过程也有重要影响。
在低浓度Cr(Ⅵ)(≤0.5 mmol·L−1)条件下,第1种和第2种还原途径占主导地位,即微生物快速将Cr(Ⅵ)还原并达到平衡,AQDS在还原过程中起到明显电子传递作用。在中浓度Cr(Ⅵ)(0.8~1.2 mmol·L−1)条件下,AQDS与NAu-2共存时的协同促进作用(第4种)占主导地位,可极大提高Cr(Ⅵ)还原效率。在高浓度Cr(Ⅵ)条件下(2.0 mmol·L−1),仍然存在明显的协同促进作用,但较高浓度Cr(Ⅵ)对微生物毒性作用较强,进而对微生物参与的第1种,第2种和第4种途径产生一定的抑制。
综上所述,当地下水环境体系受到Cr(Ⅵ)污染后,可利用环境中广泛存在的铁还原微生物对Cr(Ⅵ)污染浓度较低的区域进行修复,通过促进微生物电子传递过程提高Cr(Ⅵ)修复效率。随着Cr(Ⅵ)污染浓度的升高,可结合污染场地的环境条件,利用微生物还原与非生物还原相结合的方式协同还原Cr(Ⅵ),进而实现Cr(Ⅵ)污染场地高效修复。
2.1. NAu-2与AQDS对不同浓度Cr(Ⅵ)生物还原过程的影响
2.2. NAu-2与AQDS共存对不同浓度Cr(Ⅵ)生物还原过程的促进作用分析
2.3. 不同浓度Cr(Ⅵ)生物还原过程中Fe(Ⅱ)浓度变化情况分析
2.4. 不同浓度Cr(Ⅵ)生物还原途径及机理分析
-
1)单独添加AQDS可以加速生物还原过程的电子传递,对Cr(Ⅵ)生物还原过程产生明显的强化作用。
2)单独添加NAu-2时,对不同浓度Cr(Ⅵ)的生物还原过程均无促进作用,甚至会产生一定抑制;但不同浓度Cr(Ⅵ)对NAu-2生物还原过程有着重要影响,当Cr(Ⅵ)浓度为0.1 mmol·L−1时,Fe(Ⅱ)生成速率基本没有变化,但随着Cr(Ⅵ)浓度升高,Fe(Ⅱ)生成速率受到明显抑制,k0迅速从0.10 mmol·(L·h)−1降低至0.019 mmol·(L·h)−1。
3)同时加入AQDS和NAu-2时,对低浓度Cr(Ⅵ)(0.1~0.5 mmol·L−1)的生物还原过程产生明显的强化作用。随着Cr(Ⅵ)浓度升高至0.8~2.0 mmol·L−1,产生强化作用的同时还表现出明显的协同促进作用,协同系数达到1.50~2.98。
