


-
氮氧化物(NOx)是造成空气污染的主要污染物之一。在脱除NOx的应用研究中,目前研究较多的方法是选择性催化还原NOx(SCR),如以NH3为还原剂的方法(NH3-SCR)。但在20世纪90年代,IWAMOTO[1]和HELD等[2]发现,富氧条件下,Cu-ZSM-5催化剂可利用烃类物质选择性催化还原NO,烃类的SCR还原NO受到广泛关注。已有研究[3-6]对不同类型的催化剂对C2~C3的碳氢燃料的SCR脱硝特性进行了深入的探讨,在一定条件下取得了丰富的结果。
与其他烃类相比,作为天然气主要成分的甲烷储量丰富,价格低廉,远比其他烃类容易获得,因此,甲烷的选择性催化脱硝(CH4-SCR)具有显著的工程应用优势。CH4的碳氢键能较高,CH4的活化非常困难,在有O2条件下,易发生燃烧反应[7]。因此,对C2~C3烃具有催化活性的催化剂,对于CH4-SCR的反应活性却很低[8]。目前,一些研究[9-22]表明Co、In、Pd、Ga等离子具有一定的CH4-SCR催化活性。但是由表1可知,使用Co、In、Pd等作为活性金属的CH4-SCR催化效率较低。虽然COSTILLA等[13]使用离子交换法制得Pd-mordenite催化剂可在600 ℃达到90%脱硝效率,但是N2选择性较差,并且在有5%H2O的条件下,脱硝效率不超过60%。而GIL等[14]发现,经脱羟基处理后的镁碱沸石分子筛负载Co、In后(InCoFER),在0.25%H2O的条件下,450 ℃时达到97.5%的NO转化率,然而实验中加入的水蒸气的量过少,难以准确地评估InCoFER催化剂的抗水性能。由表1可知,镓作为活性金属,具有很高的甲烷催化活性。但是研究[18-22]发现,通过负载、溶剂、喷雾热解、共沉淀等方法制成的Ga2O3-Al2O3均受水蒸气影响较大,仅加入少量的水蒸汽,便会导致催化剂效率下降至30%。MIYAHARA等[19]研究发现,利用溶剂热法制备γ-Ga2O3-Al2O3的催化剂在2.5%H2O条件下,550 ℃仍具有50%的CH4催化NO活性;但在5%H2O条件下,溶剂法制备得到的γ-Ga2O3-Al2O3在500 ℃催化效率不足20%[20]。因此,选择使用镓基催化剂仍存在抗水差的问题。
已有研究[23]表明,金属铁在HC还原NO的反应中具有良好的抗水抗硫特性。负载了Fe的堇青石催化剂可在600 ℃达到97%的NO还原效率,通入2.1%水蒸气,仍保持60%以上的催化效率[24]。以柱撑黏土为载体负载铁离子的催化剂Fe/Ti-PILC[25]和Fe-PILC[26],在350 ℃可达到95%以上的脱硝效率,同时在10%水蒸气和0.2%的SO2下,400 ℃仍保持80%的催化效率。有研究[27]发现,采用Fe修饰的Fe-Ag/Al2O3/CM催化剂,可在500 ℃达到超过90%的脱硝效率,分别通入8%水蒸气和0.02%的SO2,脱硝效率基本无变化,有效地提高了Ag/Al2O3/CM 催化剂抵抗烟气中的SO2和H2O的能力。为改善Ga2O3-Al2O3催化活性,并提高其抗水能力,本研究采用Fe对镓基催化剂进行修饰,制备Fe/Ga2O3-Al2O3催化剂,对其CH4-SCR反应特性进行实验研究,并通过XRD、N2吸附脱附、XPS、H2-TPR、Py-IR等技术手段对催化剂的物理化学性质进行表征。
-
称取一定量的Ga(NO3)3·xH2O、Al(NO3)3·9H2O和Fe(NO3)3·9H2O,混合溶解在100 mL的去离子水中,得到盐溶液。根据研究[22],固定Ga与Al物质的量的比例为3∶7。另取5倍沉淀当量的氨水,溶入200 mL去离子水中,得到沉淀剂溶液。在室温条件下搅拌,往沉淀剂溶液中缓慢滴加金属盐溶液,在室温条件下,剧烈搅拌1 h。从溶液中离心得到前驱体,依次使用去离子水和无水乙醇各洗涤3次,在干燥箱中80 ℃条件下干燥12 h,然后在马弗炉内700 ℃、空气气氛条件下煅烧2 h,自然冷却到室温,得到xFe/Ga2O3-Al2O3催化剂(其中x为Fe所占金属离子物质的摩尔比)。
-
在程序控温固定床石英管微反应器上进行xFe/Ga2O3-Al2O3催化CH4选择性还原NO的测试,石英管内径为8 mm。催化剂压片、粉碎、过筛,至24~50目,将0.5 g催化剂放置于石英管固定床内。配气采用模拟烟气环境,模拟烟气组成为0.1%NO、0.2%CH4、1%O2、0.02%SO2、5%H2O,气体总流量为200 mL·min−1,其余气体由N2配平,反应的体积空速GHSV为16 000 h−1。实验开始之前,首先在N2氛围、300 ℃条件下对催化剂样品进行30 min的预处理,去除催化剂样品表面吸附的水蒸气和其他气体;待反应器与样品冷却至室温后,开始进行CH4-SCR反应实验,温度为200~600 ℃,各个温度稳定20 min后记录数据,反应器升温速率为5 ℃·min−1。反应后的NO、NO2、NOx通过烟气分析仪在线检测,CH4由气相色谱仪(GC-4000A)KB-Al2O3/Na2SO4毛细管柱氢火焰电离检测器(FID)检测,温度稳定后进行采样,每5 min采样1次。
NO转化率、CH4转化率、N2选择性计算方法见式(1)~式(3)。
式中:RNO为NO转化率;
RCH4 为CH4转化率;SN2 为N2选择性;cNOi为进口NO浓度;cNOo为出口NO浓度;cCH4i 为进口CH4浓度;cCH4o 为出口CH4浓度;cNO2o 为出口NO2浓度;cN2Oo 为出口N2O浓度。 -
催化剂的基础物理化学性质分别采用XRD、N2吸附-脱附、XPS、UV-vis、H2-TPR、Py-FTIR等进行表征。
使用18 kW转靶X射线衍射仪(D/max-2550VB+)进行催化剂物相表征,采用Cu Kα作为辐射源,5°~80°测试,扫描速率2 (°)·min−1,操作电压为40 kV,电流为30 mA。
使用全自动比表面积与孔隙度分析仪(ASAP 2460)进行介孔全分析测试,利用BET方法计算催化剂的比表面积,BJH方法计算催化剂脱附孔容、平均孔径以及孔径分布。
使用Thermo Fisher Scientific公司的ESCALAB 250 XI型号仪器测定催化剂的表面元素及其化学状态。
使用SHIMADZU公司的紫外可见近红外光谱仪(UV 3600)测试催化剂的吸收光谱,检测波长为200~800 nm。
H2-TPR在自组装的程序升温装置测试,在立式石英管中装填0.4 g催化剂,使用程序升温炉加热。实验前,300 ℃ N2氛围预处理30 min,冷却至室温,通入5%H2/95%N2的混合气进行催化剂的还原特性测试,升温速率5 ℃·min−1,尾气H2含量通过气相色谱仪热导检测器(TCD)测试,每5 min采样分析。
使用FT-IR Frontier型吡啶红外光谱仪(PE)测定催化剂表面的酸性位(Lewis酸和Brønsted)及含量。保持10−3 Pa的真空度,样品500 ℃预处理1 h。室温吸附吡啶,分别在40、150和300 ℃下进行测试。
-
由图1(a)可知,随着反应温度的增加,Ga2O3-Al2O3催化剂的CH4-SCR反应的NO转化率增大,在550 ℃时达到最大值81%。当反应温度继续升高后,NO转化率有所减小,这是因为高温促进了甲烷的燃烧反应[19],使得甲烷参与选择性还原NO的反应减弱。经铁修饰后的xFe/Ga2O3-Al2O3催化剂在350~500 ℃,CH4-SCR反应的NO转化率均高于Ga2O3-Al2O3催化剂,在500 ℃时NO转化率约为75%。反应温度超过500 ℃后,NO转化率有所下降,但仍保持在65%左右。随着铁含量的增加,NO转化率先增大后减小,如5Fe/Ga2O3-Al2O3的NO转化率高于2Fe/Ga2O3-Al2O3,然而当铁进一步增加后,如10Fe/Ga2O3-Al2O3,NO转化率反而降低。
图1(b)和图1(c)给出了甲烷转化率与N2选择性的结果。随着反应温度的增加,CH4转化率增大。在600 ℃以后,CH4的转化率都达到100%,且xFe/Ga2O3-Al2O3催化剂的CH4转化率都高于Ga2O3-Al2O3催化剂。xFe/Ga2O3-Al2O3催化剂的N2选择性明显高于Ga2O3-Al2O3催化剂,N2选择性随着Fe的加入量得到提高,在450 ℃以后能保证100%的N2选择性。这说明Fe物种的引入,促进甲烷活化与NO反应,并抑制了N2O和NO2的形成,从而提高了N2选择性[28-29]。
-
化石燃料燃烧产生的实际烟气存在一定量的水蒸气与SO2,因此,须考虑水蒸气和SO2对HC-SCR反应的影响。水蒸气与SO2会大大降低Cu-ZSM-5分子筛的催化活性,并且导致结构破坏[30]。前期研究表明,金属铁/氧化铁[31-32]以及铁基催化剂[26]在使用烷烃催化还原NO时具有良好的抗水硫特性,用铁修饰银基催化剂改善了原有银基催化剂的抗水硫特性[27],因此,对xFe/Ga2O3-Al2O3的抗水抗硫特性也需要进行评估。在500 ℃分别进行水蒸气和SO2氛围下脱硝测试,实验结果如图2所示。由图2(a)可知,当反应气体中通入5%的水蒸气后,Ga2O3-Al2O3催化剂的NO转化率出现大幅度下降,降低了20%。这是由于水蒸气与NO或CH4在催化剂同一位置产生竞争吸附,从而影响了NO还原反应的活性位点,不利于NO的吸附物种的形成,也不利于CH4的吸附与活化[19]。当停止水蒸气的加入时,NO转化率即恢复到之前水平,说明水蒸气导致催化剂中毒是可逆的。当用铁进行催化剂修饰后,如5Fe/Ga2O3-Al2O3,在反应气体中通入5%的水蒸气后,NO转化率仅下降10%,仍能保持在60%以上的NO转化效率。切断H2O后,催化剂活性迅速恢复,说明铁的引入提高了催化剂抗水性能。图2(b)反映了引入0.02%SO2前、后Ga2O3-Al2O3与5Fe/Ga2O3-Al2O3的催化效果变化。可以看出,Ga2O3-Al2O3受SO2的抑制较为明显,而5Fe/Ga2O3-Al2O3受SO2的影响较小,在500 ℃引入0.02%SO2,仍能保持70%左右的效率;在切断SO2后,脱硝效率与引入SO2前相比率有略微的下降(<2%)。这说明Fe的引入能够提高了催化剂的抗硫能力。
-
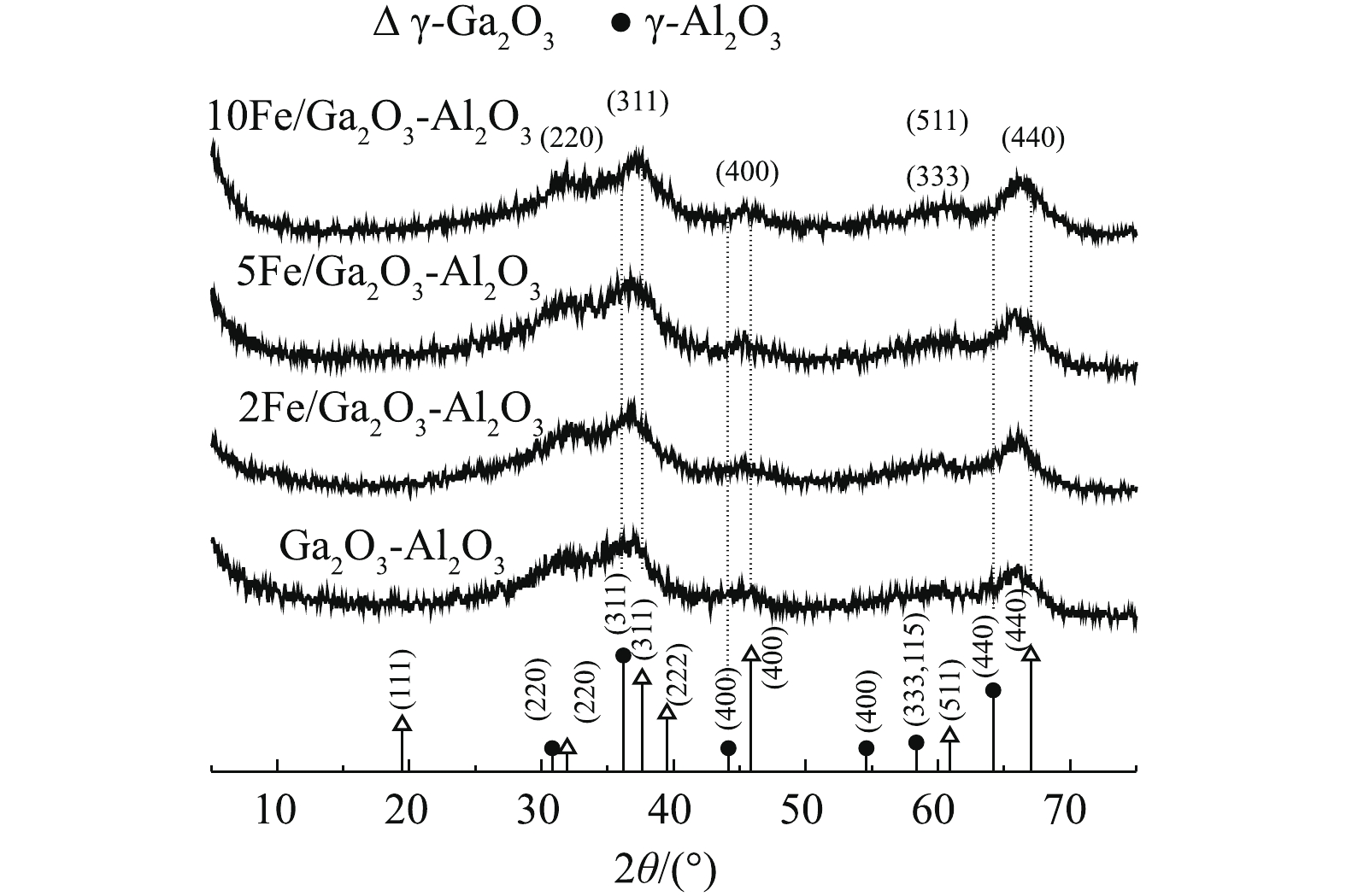
如图3所示,所有样品的衍射峰都非常宽,表明结晶度低,这种较低结晶度是大多数亚稳结构氧化铝的常见特征,同时也是纯的γ-Ga2O3多晶型的共同特征[33]。同时催化剂XRD谱图所示的宽度与低结晶度和高表面积有关[34]。催化剂XRD图谱与γ-Al2O3 (JCPDS#10-425)和γ-Ga2O3 (JCPDS#20-426)标准卡片进行比较,由图3可知,γ-Al2O3的d(400)晶面、d(440)晶面和d(311)晶面对应角度都向低角度偏移。Ga离子的半径为0.062 nm,Al离子对应的半径为0.051 nm,当Ga3+进入氧化铝的晶格中,取代Al3+位置,会使晶胞增大,导致γ-Al2O3特征峰向低角度偏移,说明催化剂均形成了尖晶石结构固溶体γ-Ga2O3-Al2O3[34],这是CH4催化还原NO的重要结构[35]。而加入Fe后,在XRD谱图中并没有观测到Fe2O3或其他形式的铁物质特征衍射峰,这说明铁物种可能高度分散,以无定形态存在,或者进入晶胞形成固溶体。引入过量Fe后,XRD图谱中γ-Ga2O3-Al2O3特征峰强度下降,可能是Fe在催化剂表面发生了团聚,从而影响了γ-Ga2O3-Al2O3结构,这可能是10Fe/Ga2O3-Al2O3的NO转化率下降的原因。
-
由表2可知,Ga2O3-Al2O3的比表面积、孔容和孔径分别为221 m2·g−1、0.582 cm3·g−1、8.2 nm。引入Fe后,2Fe/Ga2O3-Al2O3的比表面积基本无变化,孔容增加至0.643 cm3·g−1,孔径达到9.5 nm。进一步增加铁含量后,5Fe/Ga2O3-Al2O3的比表面积、孔容和孔径分别为213 m2·g−1、0.580 cm3·g−1、9.4 nm。当引入铁含量增加至10%,10Fe/Ga2O3-Al2O3的比表面积、孔容和孔径分别为217 m2·g−1、0.626 cm3·g−1、10.0 nm。说明共沉淀法引入Fe对催化剂比表面积影响小,但可以增大催化剂的孔径,而大孔径有利于降低水蒸气对催化活性的影响。
图4(a)为催化的N2吸附脱附脱附等温曲线,根据2015年IUPAC[36]更新分类可知,xFe/Ga2O3-Al2O3催化剂为Ⅳ(a)型,为介孔类吸附剂材料。在一定的相对压力下,吸附分支与脱附分支发生分离,形成明显的滞回环。合成的样品具有典型的介孔且具有较大孔径。在较低的相对压力区域,曲线向上微微凸起,主要是单分子层吸附作用,当压力足够大时,吸附质发生毛细凝结,使得吸附量急剧增加。随着相对压力进一步增加,吸附曲线趋于平稳,当p/p0接近1时,曲线继续上升,催化剂皆呈现H3型回滞环[37]。对于Ga2O3-Al2O3,在p/p0接近1时,等温吸附脱附曲线的滞后环出现平台,说明吸附已经达到饱和。由图4(a)可知,共沉淀方法引入铁物种,并没有影响到原有的孔隙结构。
由图4(b)可知,Fe的加入使得孔径增大,孔径分布朝更宽的区域分布。这说明Fe可以使催化剂表面孔隙结构变得疏松,从而增加了孔容与孔径。MASUDA等[22]研究发现,小孔径的Ga2O3-Al2O3催化剂在反应中更容易受到水蒸气的影响,而大孔径的催化剂的性能受水蒸气影响更小。引入铁物种后,催化剂的结构特性受影响较小,同时还增大了催化剂的孔径,这可能是其具有较好的抗水特性的原因。
-
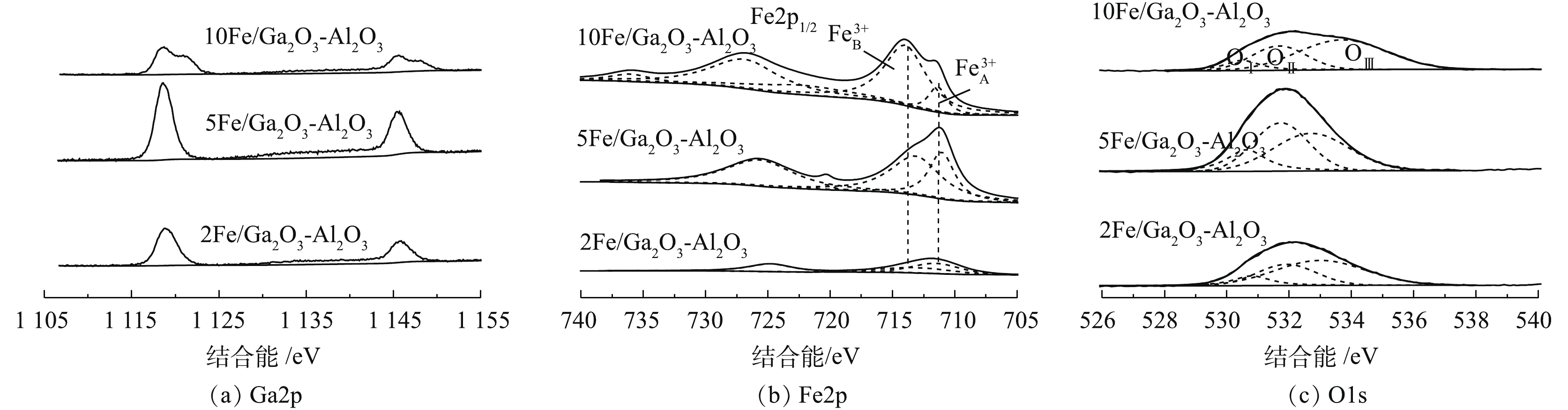
图5是 xFe/Ga2O3-Al2O3的XPS谱图。图5(a)为Ga2p的谱图,在1 118 eV和1 145 eV处附近,分别出现2个特征峰,峰间距为27 eV,分别对应于Ga2p3/2和Ga2p1/2自旋轨道,为+3价Ga特征峰,在1 118.7 eV处,Ga物种对应于四面体Ga物种[38]。而在镓铝固溶体结构中,为+3价Ga特征峰,离子会优先占据Al3+的四面体结构,处于四面体位置Ga3+具有更高的甲烷催化还原活性[39]。引入铁后,四面体位置Ga3+仍占主导地位。但引入过多铁后,10Fe/Ga2O3-Al2O3催化剂Ga2p谱图成不对称分布,说明Ga3+存在其他结构,因此,引入过量铁会影响Ga3+在Al3+的四面体位置分布,影响甲烷催化还原活性,这与XRD的分析结论相一致。图5(b)为Fe2p谱图,由Fe2p1/2峰和Fe2p3/2及其相应卫星峰组成,为+3价Fe特征峰[40]。通过分峰处理,Fe2p3/2可由位于711.0 eV附近的
Fe3+A 和713.0 eV左右的Fe3+B 共同组成,前者可对应游离态Fe3+物种,后者对应Fe2O3物种[41-42]。ZHANG等[43]研究发现,富氧条件下Fe2O3颗粒物种的存在促进甲烷参与完全氧化,导致了CH4选择性还原NO的反应减弱。由图5(b)可知,引入铁后,2Fe/Ga2O3-Al2O3催化剂出现Fe3+A 与Fe3+B 2种铁物种。提高引入铁量后,5Fe/Ga2O3-Al2O3催化剂主要以游离态Fe3+A 存在。继续增加铁量,10Fe/Ga2O3-Al2O3催化剂Fe3+B 比例上升。图5(b)显示,5Fe/Ga2O3-Al2O3催化剂具有高含量游离态Fe3+与低含量Fe2O3颗粒,这可能与其具有较好的活性有关,也与图1(a)和图1(b)中10Fe/Ga2O3-Al2O3比5Fe/Ga2O3-Al2O3具有更高的CH4转化率的同时,却具有更低的NO转化率的实验现象相一致。O物种对催化剂的活性具有重要的影响,因此,须对O1s进行分峰研究,探讨表面氧物种的种类与含量。图5(c)是催化剂的O1s XPS图谱,通过曲线拟合分析,可分成3个峰型。最低结合能峰OⅠ(529.3~529.7 eV)为晶格氧的能谱峰,结合能最高峰OⅢ (>533.0 eV)属于羟基与吸附水组成表面氧能谱峰,位于中间的OⅡ(531.5~531.8 eV)可归于催化剂表面吸附和弱结合氧物种[44-45]。在催化反应中,表面弱结合氧物种OⅡ具有高移动性,含量越高,催化活性越高[46]。由表3可知,5Fe/Ga2O3-Al2O3的OⅡ含量高,这与其具有高催化活性有关系。 -
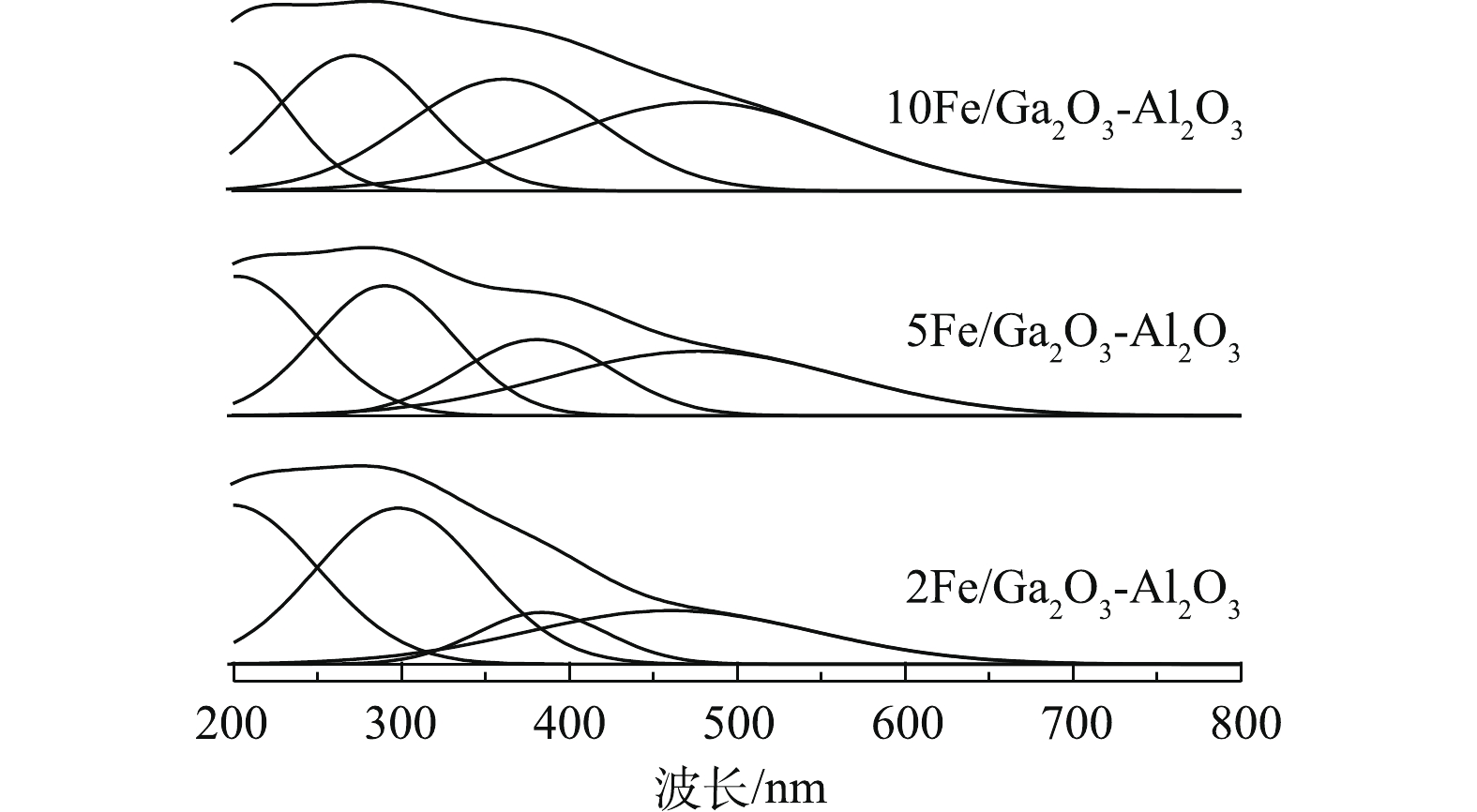
催化剂的化学组成与配位结构可以使用UV-vis光谱进行分析。Ga2O3是一种透明的宽禁带半导体材料,吸收波长<250 nm[47]。根据LI等[48]的研究,将铁的UV-vis吸收光谱分为3个峰,将300 nm以下归于游离态Fe3+,将300~400 nm的峰归属FexOy团聚物种,将400 nm以上的峰归属Fe2O3颗粒。对图6进行分峰处理,2Fe/Ga2O3-Al2O3中游离态Fe3+、FexOy团聚物、Fe2O3颗粒均有分布,且以游离态Fe3+为主要存在形式。进一步增加铁的含量,5Fe/Ga2O3-Al2O3催化剂中游离态Fe3+、FexOy团聚物种与Fe2O3颗粒含量有所增加,且以游离态Fe3+存在;继续增加铁,10Fe/Ga2O3-Al2O3催化剂显示具有更高的Fe2O3含量。研究认为,游离态Fe3+低温能促进甲烷活化成HCHO[43],而HCHO能参与NO还原反应[49],促进了NO转化,而Fe2O3颗粒会催化CH4的完全氧化[50]。因此,5Fe/Ga2O3-Al2O3的催化活性高可能与高含量游离态Fe3+有关,这与XPS中Fe2p的分析结果一致。
-
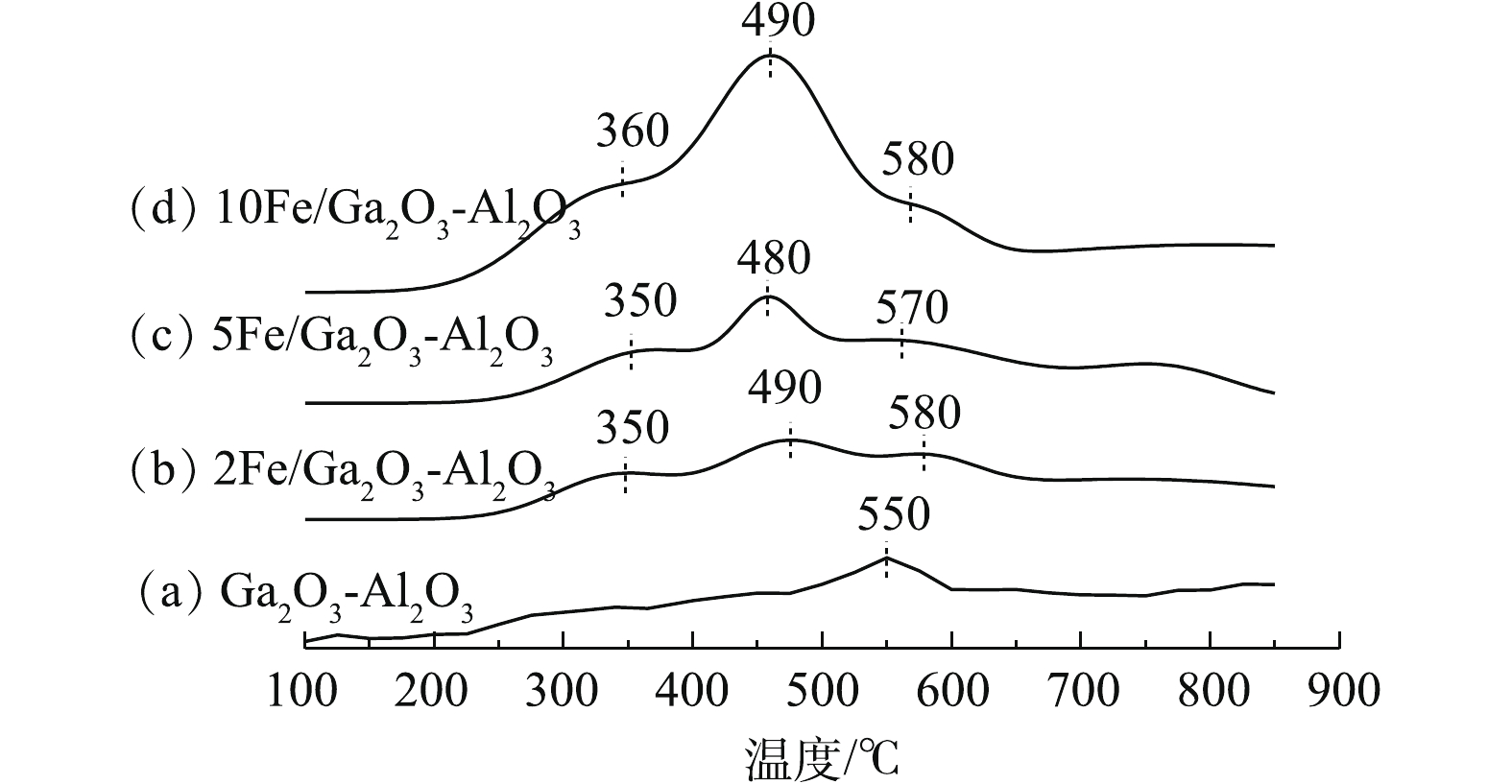
催化剂还原能力是选择性催化还原的重要参数,通过H2-TPR研究催化剂的还原性能。实验结果如图7所示,Ga2O3-Al2O3仅在550 ℃附近出现了一个较宽还原峰,这归属于Ga3+→Ga+还原[51-52]。引入Fe后,xFe/Ga2O3-Al2O3在350 ℃和500 ℃附近出现2个新的还原峰,这说明引入铁后,增强了催化剂在中高温时的还原能力,从而增强了中高温时的催化活性。根据研究,Fe催化剂的还原分为2步,将350 ℃附近还原峰归属于Fe3+、FexOy、Fe2O3中的Fe3+→Fe2+,500 ℃附近还原峰归属于Fe2+→Fe0还原。通过比较起始还原温度,发现5Fe/Ga2O3-Al2O3的还原峰与2Fe/Ga2O3-Al2O3和10Fe/Ga2O3-Al2O3相比,温度更低,因此,其具有更高的氧化能力和更好的氧移动性[46],这与XPS中O1s分析结果一致,原因可能是5Fe/Ga2O3-Al2O3具有更高催化反应活性。
-
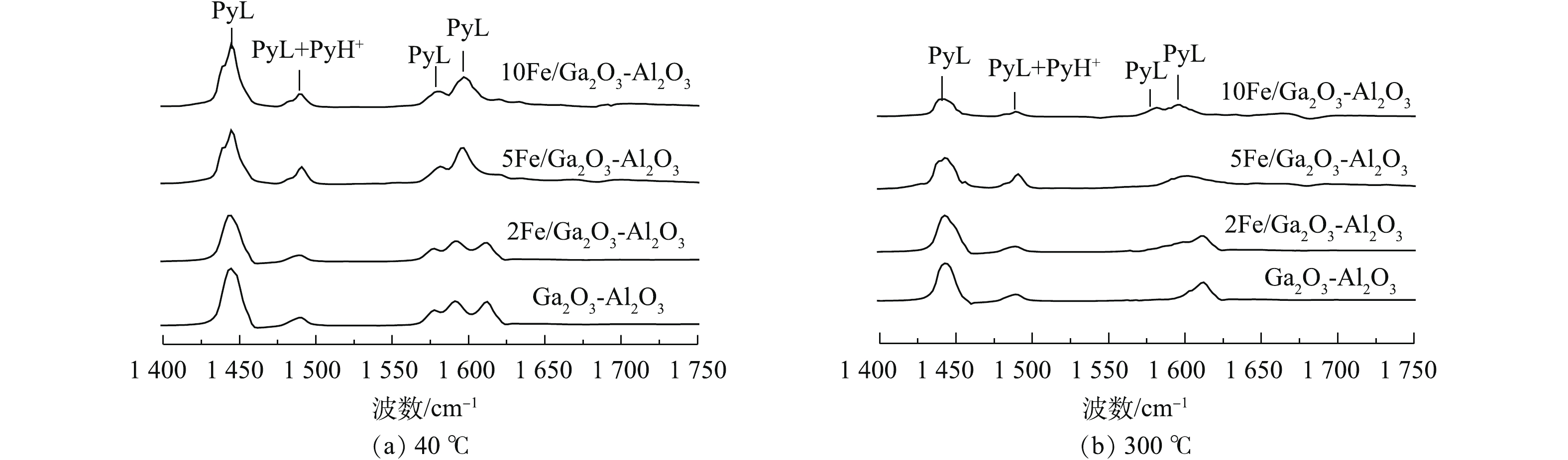
催化剂表面酸性中心一般采取吡啶吸附红外光谱进行分析。吡啶分子可被吸附在催化剂表面,利用在1 640~1 440 cm−1光谱上的差异,可以分析得到Lewis酸部位和Brønsted酸部位。图8为催化剂在室温下吸附饱和后,在40 ℃和300 ℃抽真空后的红外图谱。DATKA等[53]研究表明,波数1 440~1 460 cm−1和1 600~1 635 cm−1为L酸吸收峰,波数1 535~1 550 cm−1为Brønsted酸吸收峰。BARZETTI等[54]研究报道,在1 450 cm−1和1 590~1 620 cm−1的图谱对应Lewis酸,1 490 cm−1 和1 576 cm−1处吸收峰对应于Brønsted酸和Lewis酸。因此,1 445、1 576和1 600 cm−1处对应于Lewis酸,1 490 cm−1处出现的是Brønsted酸和Lewis酸共同峰。
在1 450 cm−1附近形成的L酸中心上出现强吸收峰,说明4组样品主要表现出Lewis酸性特征,催化剂含有少量的Brønsted酸性位,这可能是催化剂表面形成表面羟基,从而形成了B酸性位[55],B酸能够促进NO的氧化,形成重要反应中间体[56]。分别根据峰面积与对应消光系数计算酸量,结果见表4。与Ga2O3-Al2O3相比,xFe/Ga2O3-Al2O3催化剂样品在40 ℃具有更高的Lewis酸酸量,增加铁的引入量,L酸酸量增加,说明铁的引入的确可以促进Lewis的酸量生成。KANTCHEVA等[57]研究发现,CH4可以被Lewis酸吸附活化,并形成能够催化还原的中间体。因此,引入Fe后,催化剂样品会具有更高的甲烷转化率,这与图1(b)中甲烷转化率随引入铁量增加而增加的实验现象相一致。
-
1)采用共沉淀法制备了xFe/Ga2O3-Al2O3催化剂,研究了在富氧条件下的SCR-CH4脱硝特性。经铁修饰后的5Fe/Ga2O3-Al2O3比Ga2O3-Al2O3具有更高的催化活性和更高的N2选择性,在500 ℃、富氧条件下,达到76%的NO转化率和100%的N2选择性,且具有较好的抗烟气中的H2O和SO2的能力。
2)催化剂表征结果显示,加入铁后,引入反应活性物质游离态Fe3+,从而促进了甲烷活化。而当引入过量的Fe时,催化剂表面产生大量Fe2O3颗粒物,从而影响了CH4还原NO反应。共沉淀方法引入铁物种,在不影响原有孔隙结构的同时,提高了催化剂表面的Lewis酸量和氧化还原性能。因此,Fe修饰Ga2O3-Al2O3是提高Ga2O3-Al2O3催化剂的SCR-CH4脱硝性能的有效方法。
Fe/Ga2O3-Al2O3催化甲烷还原NO的性能
Performance of Fe/Ga2O3-Al2O3 catalysts on methane selective catalysis and NO reduction
-
摘要: 以甲烷为还原剂的选择性催化脱硝技术(SCR-CH4)是一种很有潜力的新的脱硝方法,但催化剂的催化活性比较低。为了提高催化剂的活性以及抗水能力,可使用Fe对Al2O3负载的Ga2O3催化剂进行改性。采用共沉淀法,制备了xFe/Ga2O3-Al2O3催化剂,在固定床反应器中测试其选择性催化CH4还原NO的性能。使用XRD、N2吸附脱附、XPS、H2-TPR、Py-IR等方法进行表征。结果表明:经过Fe改性后的催化剂提高了中高温的催化活性,提高了催化剂的N2选择性,并改善了催化剂的抗水特性;5Fe/Ga2O3-Al2O3催化剂在500 ℃、富氧条件下,达到76%的NO转化率和100%的N2选择性;在5%水蒸气条件下,5Fe/Ga2O3-Al2O3在500 ℃仍保持60%以上的NO转化率。N2吸附脱附结果显示,引入Fe后,催化剂保持了原有比表面积,并且大大增加了催化剂孔径,可提高催化剂抗水能力。XPS与UV-vis显示,5Fe/Ga2O3-Al2O3具有高含量的游离态Fe3+,可提高催化剂的中高温活性。H2-TPR结果显示,Fe的引入提高了催化剂氧化还原能力,增强了原有Ga2O3-Al2O3中高温的还原活性。Py-FT-IR结果显示,催化剂表面同时存在Lewis酸和Brønsted酸,铁的引入增加了催化剂表面的Lewis酸量。因此,Fe修饰Ga2O3-Al2O3是提高Ga2O3-Al2O3催化剂的SCR-CH4脱硝性能的有效方法。
-
关键词:
- 选择性催化还原 /
- NO /
- CH4 /
- Fe/Ga2O3-Al2O3催化剂
Abstract: The selective catalytic denitration with methane reductant (SCR-CH4) is a very promising alternative method, however, the current reported catalysts showed low catalytic reactivity for SCR-CH4. In order to improve the catalytic reactivity and the water resistance of the catalysts, Fe was used to modify the Ga2O3 catalysts supported on Al2O3. The xFe/Ga2O3-Al2O3 catalysts were prepared by co-precipitation method, and their catalytic performance on methane selective catalysis and NO reduction was tested in a fixed bed reactor. XRD, N2 adsorption desorption, XPS, H2-TPR, Py-IR, etc were used to characterize the xFe/Ga2O3-Al2O3 catalysts. The results showed that the catalysts modified by Fe improved the catalytic activity at medium and high temperature, their N2 selectivity, and their tolerance for water presented in the feed gas. At 500 ℃ and oxygen-rich conditions, the 5Fe/Ga2O3-Al2O3 catalyst could achieve 76% NO conversation and 100% N2 selectivity. Under 5% water vapor conditions, 5Fe/Ga2O3-Al2O3 still maintained over 60% NO conversation at 500 ℃. The results of N2 adsorption and desorption showed that the original specific surface area was maintained for the Fe-doped catalysts, and their pore size increased significantly, which improved their water-resistance ability. XPS and UV-vis detection showed that 5Fe/Ga2O3-Al2O3 had a high content of free Fe3+, which contributed to the medium-high temperature activity. The H2-TPR results showed that the introduction of Fe elevated the redox capacity of the catalysts and enhanced the medium-high temperature reduction activity of the original Ga2O3-Al2O3. Py-FT-IR results showed that both Lewis acid and Brønsted acid existed on the surface of the catalysts, and the introduction of Fe raised the content of the Lewis acid. Therefore, Fe modification of Ga2O3-Al2O3 is an effective method to improve the performance of SCR-CH4 of Ga2O3-Al2O3.-
Key words:
- selective catalytic reduction /
- NO /
- CH4 /
- Fe/Ga2O3-Al2O3 catalyst
-
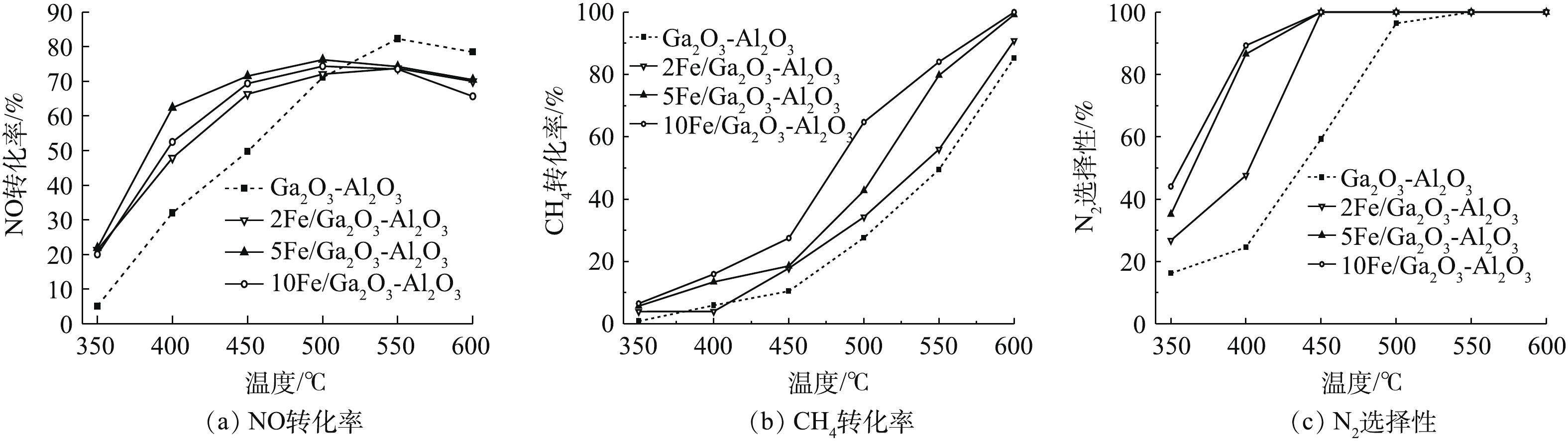
图 1 催化剂的NO转化率、CH4转化率、N2选择性
Figure 1. NO conversion to N2, CH4 conversion, N2 selectivity of the catalysts
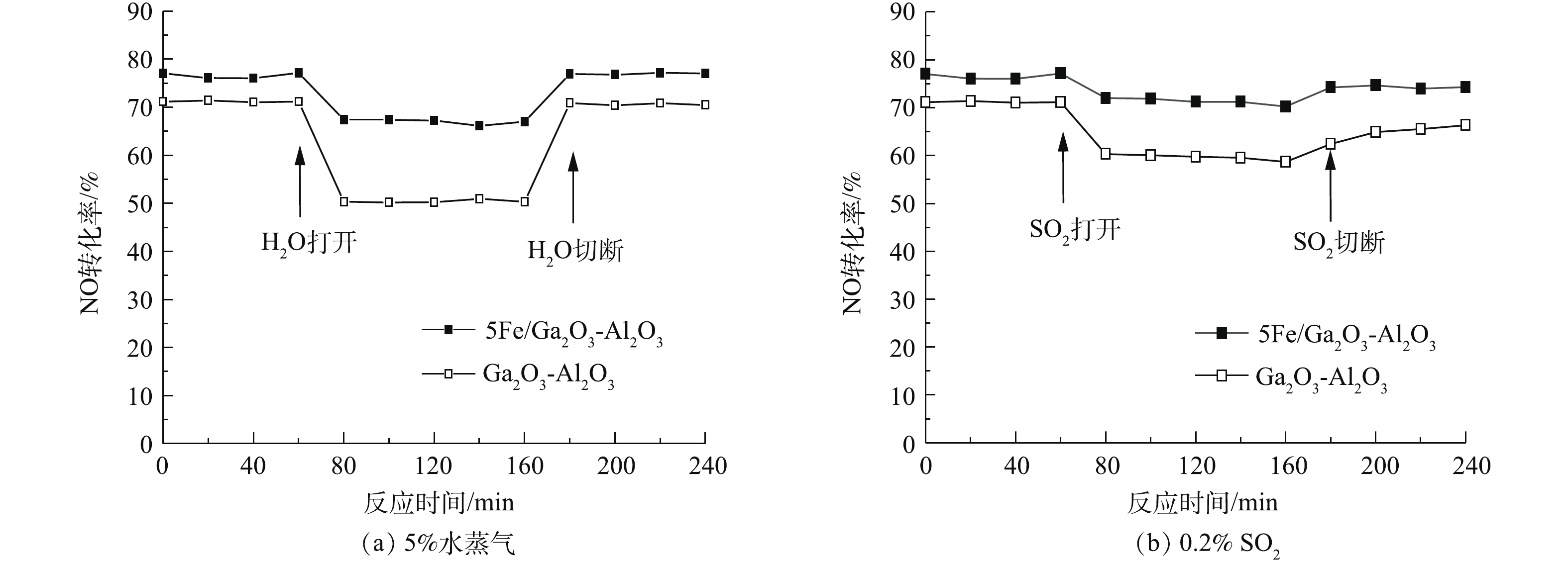
图 2 500 ℃时水蒸气和SO2分别对Ga2O3-Al2O3和5Fe/Ga2O3-Al2O3催化剂活性的影响
Figure 2. Influences of water vapor and SO2 on NO conversion of Ga2O3-Al2O3 and 5Fe/Ga2O3-Al2O3 at 500 ℃
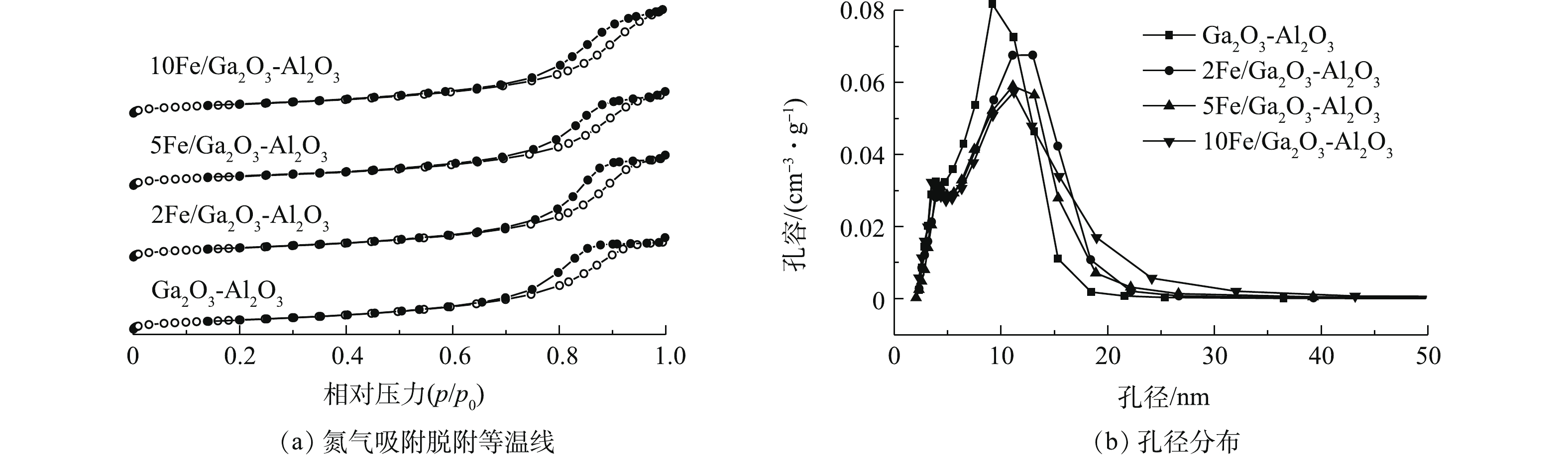
图 4 催化剂的氮气吸附脱附等温线和孔径分布
Figure 4. N2 adsorption/desorption isotherms and BJH pore size distribution of the catalysts
表 1 甲烷选择还原NO催化剂
Table 1. Catalysts for selective catalytic reduction of NO with methane
催化剂 反应工况 NO转化率/% 温度/℃ 来源 Co-ZSM-5 0.2%NO+0.2%CH4+2%O2 50 500 [9] Co-ZSM-5 0.082%NO+0.07%CH4+2.5%O2 50 400 [10] Co-ZSM-5 0.5%NO+0.2%CH4+3%O2 70 400 [11] Co, H-mordenite 0.4%NO+0.4%CH4+2%O2 60 550 [12] Pd-mordenite 0.101%NO+0.33%CH4+4.1%O2 90 600 [13] InCoFER 0.1%NO+0.2%CH4+4%O2+0.25%H2O 97.5 450 [14] Pd-MOR 0.1%NO+0.1%CH4+7%O2 25 500 [15] Ce/Pd-MOR 0.1%NO+0.1%CH4+7%O2 35 500 [15] Ga-H-ZSM-5 0.161%NO+0.1%CH4+2.5%O2 34 500 [16] Ga/H-ZSM-5 0.1%NO+0.1%CH4+10%O2 90 500 [17] Ga2O3/Al2O3 0.1%NO+0.1%CH4+6.7%O2 70 550 [18] γ-Ga2O3-Al2O3(ST) 0.1%NO+0.1%CH4+6.7%O2 90 550 [19-20] γ-Ga2O3-Al2O3(SP) 0.1%NO+0.2%CH4+6.7%O2 70 550 [21] γ-Ga2O3-Al2O3(CP) 0.1%NO+0.1%CH4+6.7%O2 85 550 [22]  下载: 导出CSV
下载: 导出CSV
表 2 不同催化剂的织构特性
Table 2. Textural properties of different catalysts
催化剂 比表面积/(m2·g−1) 孔容/(cm3·g−1) 孔径/nm Ga2O3-Al2O3 221 0.582 8.2 2Fe/Ga2O3-Al2O3 221 0.643 9.5 5Fe/Ga2O3-Al2O3 213 0.580 9.4 10Fe/Ga2O3-Al2O3 217 0.626 10.0
下载: 导出CSV
表 3 xFe/Ga2O3-Al2O3的表面组成(原子分数)
Table 3. Surface composition of xFe/Ga2O3-Al2O3 (atomic fraction)
% 催化剂 Ga Fe OⅠ OⅡ OⅢ 2Fe/Ga2O3-Al2O3 14.66 1.83 7.46 18.47 33.26 5Fe/Ga2O3-Al2O3 15.95 2.19 10.44 23.74 25.47 10Fe/Ga2O3-Al2O3 13.03 3.71 6.85 18.51 36.35
下载: 导出CSV
表 4 催化剂的B酸和L酸含量
Table 4. Brønsted and Lewis acid content of catalysts
μmol·g−1 催化剂 40 ℃ 170 ℃ 300 ℃ B酸 L酸 B酸 L酸 B酸 L酸 Ga2O3-Al2O3 3.88 608.85 2.14 374.83 0 181.66 2Fe/Ga2O3-Al2O3 2.43 813.51 1.18 250.31 0 162.40 5Fe/Ga2O3-Al2O3 4.15 835.59 2.23 298.28 0 100.71 10Fe/Ga2O3-Al2O3 3.48 976.56 1.28 356.35 0 172.97
下载: 导出CSV
-
[1] IWAMOTO M. Selective reduction of NO by lower hydrocarbons in the presence of O2 and SO2 over cupper ion-exchanged zeolites[J]. Shokubai, 1990, 32: 430-433. [2] HELD W, KÖNIG A, RICHTER T, et al. Catalytic NOx reduction in net oxidizing exhaust gas[J]. SAE Transactions, 1990, 99: 209-216. [3] YUAN M H, DENG W Y, DONG S L, et al. Montmorillonite based porous clay heterostructures modified with Fe as catalysts for selective catalytic reduction of NO with propylene[J]. Chemical Engineering Journal, 2018, 353: 839-848. doi: 10.1016/j.cej.2018.07.201 [4] CHAIEB T, DELANNOY L, LOUIS C, et al. On the origin of the optimum loading of Ag on Al2O3 in the C3H6-SCR of NOx[J]. Applied Catalysis B: Environmental, 2013, 142-143: 780-784. doi: 10.1016/j.apcatb.2013.06.010 [5] KOMVOKIS V G, ILIOPOULOU E F, VASALOS I A, et al. Development of optimized Cu-ZSM-5 deNOx catalytic materials both for HC-SCR applications and as FCC catalytic additives[J]. Applied Catalysis A: General, 2007, 325(2): 345-352. doi: 10.1016/j.apcata.2007.02.035 [6] PAN H, SU Q F, CHEN J, et al. Promotion of Ag/H-BEA by Mn for lean NO reduction with propane at low temperature[J]. Environmental Science & Technology, 2009, 43(24): 9348-9353. [7] OHTSUKA H, TABATA T. Influence of Si/Al ratio on the activity and durability of Pd-ZSM-5 catalysts for nitrogen oxide reduction by methane[J]. Applied Catalysis B: Environmental, 2000, 26(4): 275-284. doi: 10.1016/S0926-3373(00)00127-2 [8] 张涛, 任丽丽, 林励吾. 甲烷选择催化还原NO研究进展[J]. 催化学报, 2004, 25(1): 75-83. doi: 10.3321/j.issn:0253-9837.2004.01.017 [9] BELLMANN A, ATIA H, BENTRUP U, et al. Mechanism of the selective reduction of NOx by methane over Co-ZSM-5[J]. Applied Catalysis B: Environmental, 2018, 230: 184-193. doi: 10.1016/j.apcatb.2018.02.051 [10] LI Y, ARMOR J N. Catalytic reduction of nitrogen oxides with methane in the presence of excess oxygen[J]. Applied Catalysis B: Environmental, 1992, 1(4): L31-L40. doi: 10.1016/0926-3373(92)80050-A [11] LI Y J, BATTAVIO P J, ARMOR J N. Effect of water vapor on the selective reduction of NO by methane over cobalt-exchanged ZSM-5[J]. Journal of Catalysis, 1993, 142(2): 561-571. doi: 10.1006/jcat.1993.1231 [12] LÓNYI F, SOLT H E, PÁSZTI Z, et al. Mechanism of NO-SCR by methane over Co,H-ZSM-5 and Co,H-mordenite catalysts[J]. Applied Catalysis B: Environmental, 2014, 150-151: 218-229. doi: 10.1016/j.apcatb.2013.12.024 [13] COSTILLA I O, SANCHEZ M D, VOLPE M A, et al. Ce effect on the selective catalytic reduction of NO with CH4 on Pd-mordenite in the presence of O2 and H2O[J]. Catalysis Today, 2011, 172(1): 84-89. doi: 10.1016/j.cattod.2011.03.025 [14] GIL B, JANAS J, WŁOCH E, et al. The influence of the initial acidity of HFER on the status of Co species and catalytic performance of CoFER and InCoFER in CH4-SCR-NO[J]. Catalysis Today, 2008, 137(2): 174-178. doi: 10.1016/j.cattod.2008.01.004 [15] MENDES A N, ZHOLOBENKO V L, THIBAULT-STARZYK F, et al. On the enhancing effect of Ce in Pd-MOR catalysts for NOx CH4-SCR: A structure-reactivity study[J]. Applied Catalysis B: Environmental, 2016, 195: 121-131. doi: 10.1016/j.apcatb.2016.05.004 [16] LI Y J, ARMOR J N. Selective Catalytic reduction of NO with methane on gallium catalysts[J]. Journal of Catalysis, 1994, 145(1): 1-9. doi: 10.1006/jcat.1994.1001 [17] KIKUCHI E, YOGO K. Selective catalytic reduction of nitrogen monoxide by methane on zeolite catalysts in an oxygen-rich atmosphere[J]. Catalysis Today, 1994, 22(1): 73-86. doi: 10.1016/0920-5861(94)80093-6 [18] SHIMIZU K, SATSUMA A, HATTORI T. Selective catalytic reduction of NO by hydrocarbons on Ga2O3/Al2O3 catalysts[J]. Applied Catalysis B: Environmental, 1998, 16(4): 319-326. doi: 10.1016/S0926-3373(97)00088-X [19] MIYAHARA Y, TAKAHASHI M, MASUDA T, et al. Selective catalytic reduction of NO with C1~C3 reductants over solvothermally prepared Ga2O3-Al2O3 catalysts: Effects of water vapor and hydrocarbon uptake[J]. Applied Catalysis B: Environmental, 2008, 84(1): 289-296. doi: 10.1016/j.apcatb.2008.04.005 [20] TAKAHASHI M, INOUE N, NAKATANI T, et al. Selective catalytic reduction of NO with methane on γ-Ga2O3-Al2O3 solid solutions prepared by the glycothermal method[J]. Applied Catalysis B: Environmental, 2006, 65(1): 142-149. doi: 10.1016/j.apcatb.2006.01.007 [21] WATANABE T, MIKI Y, MASUDA T, et al. Performance of γ-Ga2O3-Al2O3 solid solutions prepared by spray pyrolysis for CH4-SCR of NO[J]. Applied Catalysis A: General, 2011, 396(1): 140-147. doi: 10.1016/j.apcata.2011.02.005 [22] MASUDA T, WATANABE T, MIYAHARA Y, et al. Synthesis of Ga2O3-Al2O3 catalysts by a coprecipitation method for CH4-SCR of NO[J]. Topics in Catalysis, 2009, 52(6/7): 699-706. [23] 周皞, 苏亚欣, 戚越舟, 等. 水蒸气对甲烷在金属铁表面还原NO行为的影响[J]. 燃料化学学报, 2014, 42(11): 1378-1386. doi: 10.3969/j.issn.0253-2409.2014.11.016 [24] ZHOU H, SU Y X, LIAO W Y, et al. NO reduction by propane over monolithic cordierite-based Fe/Al2O3 catalyst: Reaction mechanism and effect of H2O/SO2[J]. Fuel, 2016, 182: 352-360. doi: 10.1016/j.fuel.2016.05.116 [25] 董士林, 苏亚欣, 刘欣, 等. Fe/Ti-PILC用于C3H6选择性催化还原NO的研究[J]. 燃料化学学报, 2018, 46(10): 1231-1239. doi: 10.3969/j.issn.0253-2409.2018.10.011 [26] 李前程, 苏亚欣, 董士林, 等. Fe-PILC在贫燃条件下催化丙烯选择性还原NO[J]. 燃料化学学报, 2018, 46(10): 1240-1248. doi: 10.3969/j.issn.0253-2409.2018.10.012 [27] 杨溪, 苏亚欣, 钱文燕, 等. Fe-Ag/Al2O3 催化丙烯还原NO的实验研究[J]. 燃料化学学报, 2017, 45(11): 1365-1375. doi: 10.3969/j.issn.0253-2409.2017.11.012 [28] YUAN M H, SU Y X, DENG W Y, et al. Porous clay heterostructures (PCHs) modified with copper ferrite spinel as catalyst for SCR of NO with C3H6[J]. Chemical Engineering Journal, 2019, 375: 122091. doi: 10.1016/j.cej.2019.122091 [29] MRAD R, COUSIN R, POUPIN C, et al. Propene oxidation and NO reduction over MgCu-Al(Fe) mixed oxides derived from hydrotalcite-like compounds[J]. Catalysis Today, 2015, 257: 98-103. doi: 10.1016/j.cattod.2015.02.020 [30] FENG X B, KEITH H W. FeZSM-5: A durable SCR catalyst for NOx removal from combustion streams[J]. Journal of Catalysis, 1997, 166(2): 368-376. doi: 10.1006/jcat.1997.1530 [31] 苏亚欣, 任立铭, 苏阿龙, 等. 甲烷在金属铁及氧化铁表面还原NO的研究[J]. 燃料化学学报, 2013, 41(11): 1393-1400. [32] 苏亚欣, 邓文义, 苏阿龙. 甲烷在氧化铁表面还原NO的特性与反应机理研究[J]. 燃料化学学报, 2013, 41(9): 1129-1135. doi: 10.3969/j.issn.0253-2409.2013.09.016 [33] AREÁN C O, BELLAN A L, MENTRUIT M P, et al. Preparation and characterization of mesoporous γ-Ga2O3[J]. Microporous and Mesoporous Materials, 2000, 40(1): 35-42. doi: 10.1016/S1387-1811(00)00240-7 [34] AREÁN C O, DELGADO M R, MONTOUILLOUT V, et al. Synthesis and characterization of spinel-type gallia-alumina solid solutions[J]. Zeitschrift für Anorganische und Allgemeine Chemie, 2005, 631(11): 2121-2126. doi: 10.1002/zaac.200570027 [35] HANEDA M, KINTAICHI Y, SHIMADA H, et al. Selective reduction of NO with propene over Ga2O3-Al2O3: Effect of sol-gel method on the catalytic performance[J]. Journal of Catalysis, 2000, 192(1): 137-148. doi: 10.1006/jcat.2000.2831 [36] THOMMES M, KANEKO K, NEIMARK A V, et al. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report)[J]. Pure and Applied Chemistry, 2015, 87(9/10): 1051-1069. doi: 10.1515/pac-2014-1117 [37] WATANABE T, MIKI Y, MASUDA T, et al. Pore structure of γ-Ga2O3-Al2O3 particles prepared by spray pyrolysis[J]. Microporous and Mesoporous Materials, 2011, 145(1): 131-140. doi: 10.1016/j.micromeso.2011.05.002 [38] 高俊华, 刘平, 吉可明, 等. GaZSM-5分子筛的合成、表征及其在甲醇转化制烃(MTH)反应中的催化性能[J]. 燃料化学学报, 2018, 46(4): 465-472. doi: 10.3969/j.issn.0253-2409.2018.04.012 [39] MIYAHARA Y, WATANABE T, MASUDA T, et al. Evaluation of catalytic activity of Ga2O3-Al2O3 solid solutions for CH4-SCR by UV-vis spectra after adsorption of C3H6 as a probe[J]. Journal of Catalysis, 2008, 259(1): 36-42. doi: 10.1016/j.jcat.2008.07.007 [40] WANDELT K. Photoemission studies of adsorbed oxygen and oxide layers[J]. Surface Science Reports, 1982, 2(1): 1-121. doi: 10.1016/0167-5729(82)90003-6 [41] YANG S, GUO Y, YAN N, et al. Remarkable effect of the incorporation of titanium on the catalytic activity and SO2 poisoning resistance of magnetic Mn-Fe spinel for elemental mercury capture[J]. Applied Catalysis B: Environmental, 2011, 101(3): 698-708. doi: 10.1016/j.apcatb.2010.11.012 [42] LIU Y M, XU J, HE L, et al. Facile synthesis of Fe-loaded mesoporous silica by a combined detemplation−incorporation process through Fenton’s chemistry[J]. The Journal of Physical Chemistry C, 2008, 112(42): 16575-16583. doi: 10.1021/jp802202v [43] ZHANG Q H, LI Y, AN D L, et al. Catalytic behavior and kinetic features of FeOx/SBA-15 catalyst for selective oxidation of methane by oxygen[J]. Applied Catalysis A: General, 2009, 356(1): 103-111. doi: 10.1016/j.apcata.2008.12.031 [44] TIAN T F, ZHAN M C, WANG W D, et al. Surface properties and catalytic performance in methane combustion of La0.7Sr0.3Fe1−yGayO3−δ perovskite-type oxides[J]. Catalysis Communications, 2009, 10(5): 513-517. doi: 10.1016/j.catcom.2008.10.028 [45] 乐向晖, 张栖, 付名利, 等. SO2对La0.8K0.2Cu0.05Mn0.95O3钙钛矿催化剂氧化碳烟的影响[J]. 无机化学学报, 2009, 25(7): 1170-1176. doi: 10.3321/j.issn:1001-4861.2009.07.006 [46] 叶青, 王瑞璞, 徐柏庆. 柠檬酸溶胶-凝胶法制备的Ce1-xZrxO2: 结构及其氧移动性[J]. 物理化学学报, 2006, 22(1): 33-37. doi: 10.3866/PKU.WHXB20060107 [47] GUO D Y, WU Z P, LI P G, et al. Fabrication of β-Ga2O3 thin films and solar-blind photodetectors by laser MBE technology[J]. Optical Materials Express, 2014, 4(5): 1067-1076. doi: 10.1364/OME.4.001067 [48] LI L D, SHEN Q, LI J J, et al. Iron-exchanged FAU zeolites: Preparation, characterization and catalytic properties for N2O decomposition[J]. Applied Catalysis A: General, 2008, 344(1): 131-141. doi: 10.1016/j.apcata.2008.04.011 [49] CAPELA S, CATALÃO R, RIBEIRO M F, et al. Methanol interaction with NO2: An attempt to identify intermediate compounds in CH4-SCR of NO with Co/Pd-HFER catalyst[J]. Catalysis Today, 2008, 137(2): 157-161. doi: 10.1016/j.cattod.2007.11.048 [50] FIERRO G, MORETTI G, FERRARIS G, et al. A Mössbauer and structural investigation of Fe-ZSM-5 catalysts: Influence of Fe oxide nanoparticles size on the catalytic behaviour for the NO-SCR by C3H8[J]. Applied Catalysis B: Environmental, 2011, 102(1): 215-223. doi: 10.1016/j.apcatb.2010.12.001 [51] SHAO C T, LANG W Z, YAN X, et al. Catalytic performance of gallium oxide based-catalysts for the propane dehydrogenation reaction: Effects of support and loading amount[J]. RSC Advances, 2017, 7(8): 4710-4723. doi: 10.1039/C6RA27204E [52] EL-MALKI E-M, VAN SANTEN R A, SACHTLER W M H. Introduction of Zn, Ga, and Fe into HZSM-5 cavities by sublimation: Identification of acid sites[J]. The Journal of Physical Chemistry B, 1999, 103(22): 4611-4622. doi: 10.1021/jp990116l [53] DATKA J, TUREK A M, JEHNG J M, et al. Acidic properties of supported niobium oxide catalysts: An infrared spectroscopy investigation[J]. Journal of Catalysis, 1992, 135(1): 186-199. doi: 10.1016/0021-9517(92)90279-Q [54] BARZETTI T, SELLI E, MOSCOTTI D, et al. Pyridine and ammonia as probes for FTIR analysis of solid acid catalysts[J]. Journal of the Chemical Society, Faraday Transactions, 1996, 92(8): 1401-1407. doi: 10.1039/ft9969201401 [55] 吴越. 取代硫酸、氢氟酸等液体酸催化剂的途径[J]. 化学进展, 1998, 10(2): 158-171. doi: 10.3321/j.issn:1005-281X.1998.02.007 [56] LÓNYI F, SOLT H E, VALYON J, et al. The SCR of NO with methane over In,H- and Co,In,H-ZSM-5 catalysts: The promotional effect of cobalt[J]. Applied Catalysis B: Environmental, 2012, 117-118: 212-223. doi: 10.1016/j.apcatb.2012.01.022 [57] KANTCHEVA M, VAKKASOGLU A S. Cobalt supported on zirconia and sulfated zirconia I: FT-IR spectroscopic characterization of the NOx species formed upon NO adsorption and NO/O2 coadsorption[J]. Journal of Catalysis, 2004, 223(2): 352-363. doi: 10.1016/j.jcat.2004.02.007 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 8699
- HTML全文浏览数: 8699
- PDF下载数: 65
- 施引文献: 0
