


-
随着现代社会工业化的高速发展,天然水中砷的含量迅速增加[1]。水中砷污染已成为全社会亟待解决的重要环境问题之一[2]。目前,常用的除砷方法主要有混凝沉淀法、离子交换法、膜分离法、吸附法[3-6]等。混凝沉淀法中预氧化过程会增加后续污水处理的问题[3];离子交换法须采用特殊材料,工艺流程较繁琐[4];膜分离法易出现膜污染等问题,该法适用于处理量相对较少的砷污染水[5]。因此,吸附法在处理效率高、操作简单和适用性强等方面的优势越来越受到人们的重视[6]。
多壁碳纳米管(CNTs)具有比表面积大、孔隙结构丰富、表面易于被修饰等特点,近年来在制备复合吸附材料方面已有诸多的应用[7]。IBRAHIM等[8]研究了深共熔溶剂(DESs)功能化CNTs对水中甲基橙的吸附性能;李德云等[9]在研究改性CNTs吸附水中亚甲基蓝过程时提到Cu/CuO改性CNTs对亚甲基蓝的吸附效果优于原始CNTs;杨爱丽等[10]在研究臭氧氧化改性CNTs对铀的吸附去除时提到,含氧量的增加会显著提高改性CNTs对铀的去除率。
目前,对水中砷去除的研究多集中在吸附剂的氧化改性以及对As(Ⅴ)的吸附去除上,对As(Ⅲ)的削减效果分析和去除机制等方面的研究相对较少[11]。本研究从活化CNTs管壁接枝点位、负载高活性基团的酰胺类材料的角度出发,采用强酸氧化和酰胺化等方法制备了酰胺化/氧化碳纳米管-聚苯胺(NMCNTs-PANI),并探究该复合材料对水中As(Ⅲ)的吸附性能。
-
基准纯三氧化二砷(As2O3,99%);高锰酸钾(KMnO4)、硫酸(98%H2SO4)、氯化亚砜(SOCl2)、N-聚乙烯吡咯烷酮、三乙胺、乙二胺、N, N-二环己基碳二亚胺、聚苯胺(PANI)、丙酮均为分析纯。碳纳米管原粉(CNTs)购自苏州石墨烯科技有限公司,采用改性催化碳气相沉积法(CVD)制备。
-
真空干燥箱(DZF-6050,上海福絮仪器厂);磁力加热搅拌器(HJ-4,江苏新瑞仪器厂);pH计(pHS-25型,上海仪电公司);原子荧光光度计(AFS-933,美国Thermo Fisher Scientific公司);扫描电子显微镜(TM3030,日立公司);比表面孔径分布测定仪(TRISTAR3000,美国MICROMERITICS公司);X射线衍射仪(D2 PHASER,德国布鲁克AXS公司);傅里叶变换红外光谱仪(EQUINOX 55,美国赛默飞公司);拉曼光谱仪(LABRAM ARAMIS,法国HORIBA公司)。
实验采用恒温振荡器,实验装置及实物照片见图1。设置温度为25 ℃,转速为180 r·min−1,避光运行,振荡后静置,抽滤。
-
冰水浴中,称取1 g CNTs、3 g KMnO4加入到30 mL浓硫酸中,搅拌2 h;升温至40 ℃,搅拌反应4 h;置于70 ℃磁力搅拌器中搅拌0.3 h;加入适量FeCl3及FeSO4,溶解后抽滤,将所得固体烘干备用。取1 g上述烘干后的固体置于250 mL烧杯中,加入30 mL SOCl2、2 mL DMF和1 g N-聚乙烯吡咯烷酮,搅拌均匀,缓慢加入13 mL三乙胺、7 mL乙二胺,升温至80 ℃,搅拌0.5 h,加入适量N,N-二环己基碳二亚胺,超声0.5 h;置于摇床振荡,洗涤抽滤,干燥得到黑色固体[12-13]。取0.5 g上述黑色固体分散在水溶液中,与50 mL含有2 g PANI的丙酮溶液混合,置于60 ℃水浴锅反应1 h,超声0.5 h;移至摇床振荡20 h,洗涤抽滤,110 ℃真空干燥箱烘干,得到NMCNTs-PANI。
采用SEM观察表面特征;比表面积分析仪测定比表面积和孔径分布;X射线衍射仪(XRD)分析物相组成[14];傅里叶变换红外光谱仪(FT-IR)定性分析表面的化学键和官能团信息[15];拉曼光谱(Raman)分析材料的晶体结构[16]。
-
配置10 mg·L−1 As2O3溶液,取7份100 mL置于系列250 mL锥形瓶中,分别投加50 mg NCNTs/PANI,反应温度为25 ℃,设置不同的接触时间并置于振荡器中以180 r·min−1转速振荡,静置后过滤,用原子荧光光度计测定滤液中As(Ⅲ)浓度并计算吸附量。As(Ⅲ)初始浓度、初始pH、共存阴离子的考察过程与接触时间类似。溶液pH采用NaOH及HCl调节。
吸附量、去除率依次按式(1)和式(2)计算。
式中:q为NMCNTs-PANI对溶液中As(Ⅲ)的吸附量,mg·g−1;η为去除率;C0为As(Ⅲ)初始浓度,mg·L−1;C为滤液中As(Ⅲ)浓度,mg·L−1;V为溶液体积,L;m为NMCNTs-PANI投加量,g。
-
图2是改性前后CNTs的SEM图。从图2(a)中可以看出,CNTs原粉是相互缠绕在一起的管状结构,其管壁无序、混乱;图2(b)和图2(c)显示,NMCNTs-PANI管状结构部分消失,管间缺陷增大,并出现聚集现象。该现象可能是因为引入某种活性剂或CNTs表面功能化导致CNTs结构受到破坏[17]。
改性前后CNTs物理性质见表1。可以看出,CNTs原粉、NMCNTs-PANI和吸附后材料的孔径为3.58~13.84 nm,说明样品为介孔材料;与CNTs原粉相比,NMCNTs-PANI和吸附后材料的比表面积稍有减小,而孔容和平均孔径均大大增加。H2SO4和KMnO4均具有强氧化性,可以氧化掉CNTs上部分管状结构,导致CNTs局部坍塌;或者少部分孔道在反应过程中被堵塞,碳管难以拉伸,从而减小NMCNTs-PANI和吸附后材料的比表面积;同时,也有部分管内杂质被溶解,疏通了CNTs孔道结构;或者使一些内部封闭的孔道被打开,导致NMCNTs-PANI和吸附后材料的孔容和平均孔径均增大[18]。
图3是改性前后CNTs的XRD图谱。从图3可以看出,改性前后表征CNTs晶面(002)的衍射角在26.0°处位置没发生变化[14];NMCNTs-PANI和吸附后材料衍射峰的强度均小于CNTs原粉,说明CNTs改性后结晶度变差。此外,NMCNTs-PANI中体现PANI的衍射特征峰在15.3°和22.6°凸起,可能原因是PANI大部分以无定性态存在[19]。
图4是改性前后CNTs的红外光谱图。可以看出,与CNTs原粉相比,NMCNTs-PANI和吸附后材料在500~4 000 cm−1出现多个吸收峰。其中NMCNTs-PANI的红外光谱中1 134 cm−1是O—H的振动峰[20];1 192 cm−1是环氧基C—O—C引起的伸缩振动峰[21];1 701 cm−1处的吸收峰为羰基C=O伸缩振动[22];1 257 cm−1处的红外吸收峰归属于C—O的伸缩振动[23];1 400 cm−1处是C—N键的伸缩振动特征峰,1 654 cm−1处是C=N的弯曲振动峰[24],以上说明制备过程中引入了羧基、羟基、环氧基等功能性基团;3 278 cm−1处归属于N—H的伸缩振动峰,该峰和C=O、C—N表明酰胺基团负载在材料表面;786 cm−1处的特征峰归属于Fe—O键的伸缩振动[25],这表明铁离子成功接枝在材料表面。吸附后材料的FT-IR图出现了位于582 cm−1处的金属配位体振动(Metal-N)[25],可能原因是水中As(Ⅲ)吸附于NMCNTs-PANI表面。
图5是改性前后CNTs的Raman光谱图。可以看出,改性前后CNTs均出现了2个典型特征峰,其中D峰位于1 340 cm−1左右,该缺陷峰反映的是碳材料中C—C的无序振动,峰值反映出CNTs结构的缺陷性和混乱程度;在1 580 cm−1左右的G峰是碳结构sp2的特征峰,用以说明CNTs的结晶程度,2个峰的强度比值ID/IG可以衡量CNTs结构缺陷密度,比值越低,结晶越完整[16]。计算表明,NMNCTs-PANI和吸附后材料的ID/IG分别为1.05、1.06,均高于CNTs原粉ID/IG(1.03),说明复合材料结构的无序性增加。综上所述,改性后CNTs的结构发生了变化,但XRD及Raman分析结果一致表明,CNTs的晶体结构并没有被破坏。
-
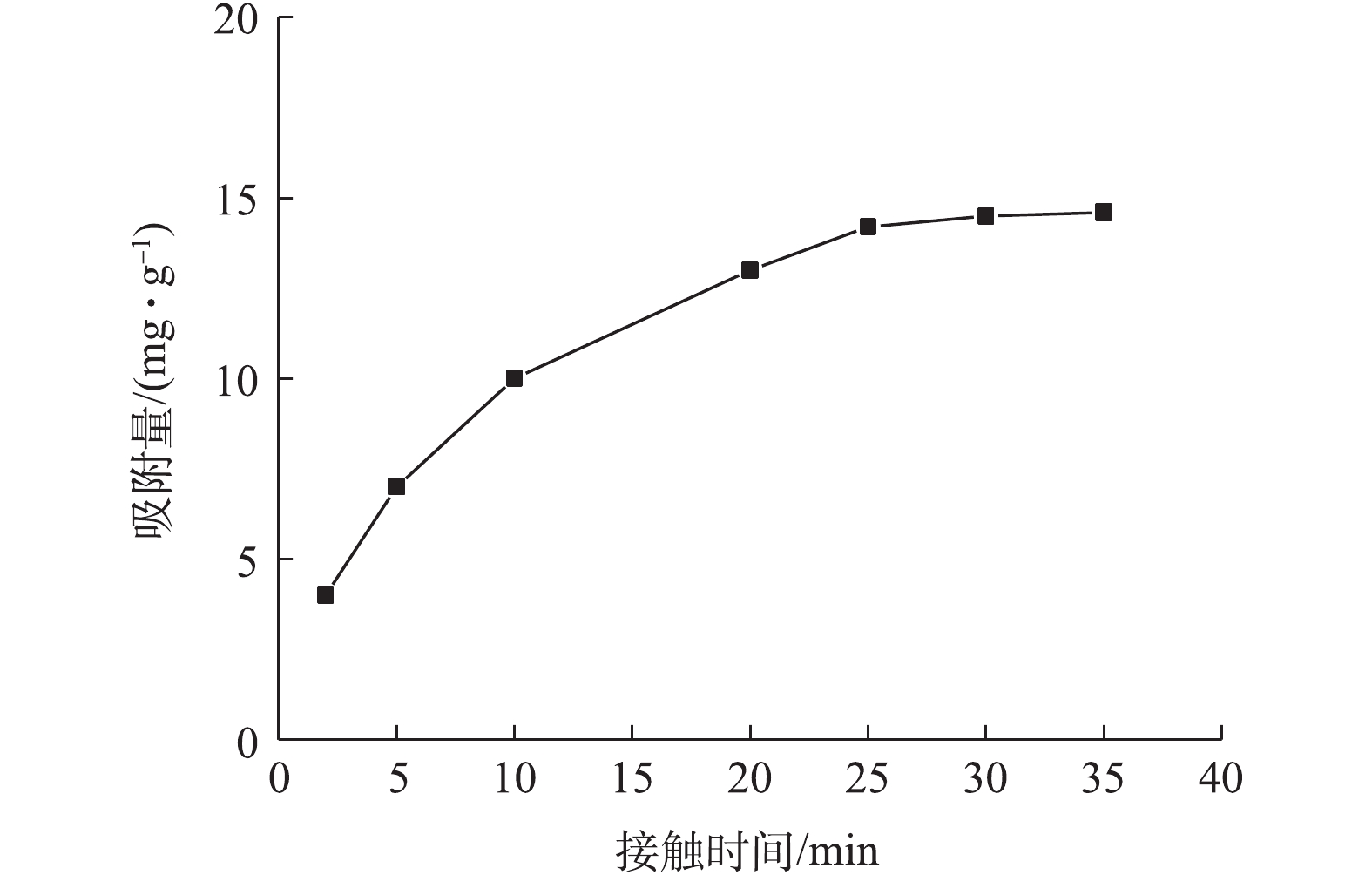
图6为接触时间对吸附量的影响结果。当接触时间从5 min延至20 min时,吸附量从4 mg·g−1快速上升到13 mg·g−1;随后吸附量缓慢增加;在30 min左右达到吸附平衡。这是由于初始阶段NMCNTs-PANI表面高能量的活性位点迅速被占领,吸附速率较高,此时溶液中大部分As(Ⅲ)占据了表面活性位点;后期由低能量的活性位点继续吸附,吸附速率随之降低。综合考虑吸附速率,接触时间选择30 min。
将数据代入准一级动力学方程和准二级动力学方程进行拟合,准一级动力学方程[26]和准二级动力学方程[27]见式(3)和式(4)。
式中:t为吸附过程某一时刻,min;qe为平衡吸附量,mg·g−1;qt为t时刻吸附量,mg·g−1;k1为一级动力学速率常数,min−1;k2为二级动力学速率常数,g·(mg·min)−1。
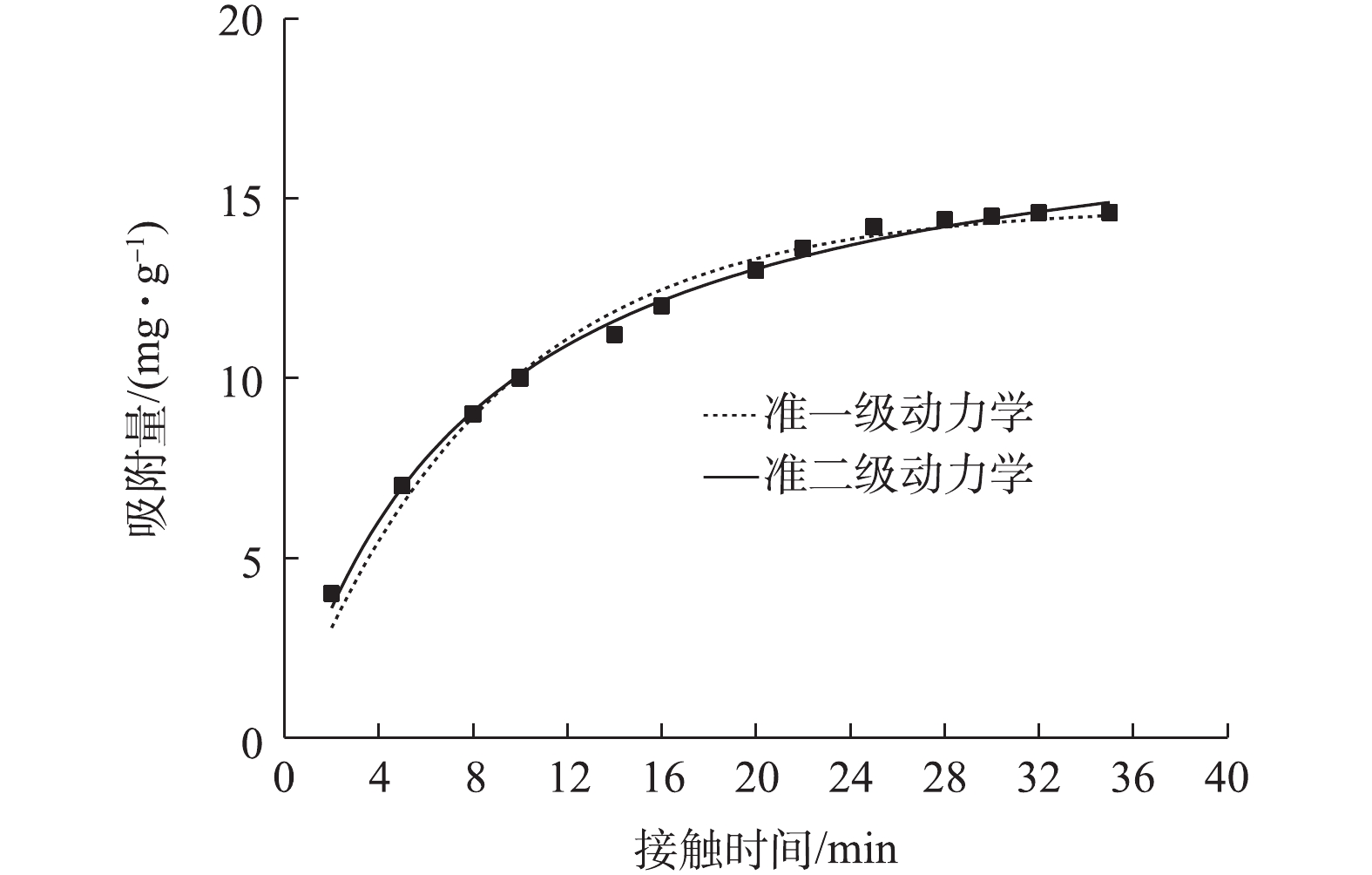
图7为采用不同动力学方程对动力学实验数据的拟合结果,拟合参数见表2。由图7可知,准一级动力学方程和准二级动力学方程均能较好描述NMCNTs-PANI吸附As(Ⅲ)的动力学过程。但表2显示准二级动力学方程R2为0.994,准一级动力学方程R2为0.983,故准二级动力学方程更为符合NMCNTs-PANI吸附As(Ⅲ)的动力学过程,即NMCNTs-PANI吸附水中As(Ⅲ)主要以化学吸附为主[28]。吸附过程中物理吸附主要是分子间作用力;化学吸附可能涉及离子交换或NMCNTs-PANI上活性基团与As(Ⅲ)形成化学键的吸附[28],尤其是改性材料表面所带有的酰胺基团,具有较高反应活性,可与As(Ⅲ)发生结合[29]。
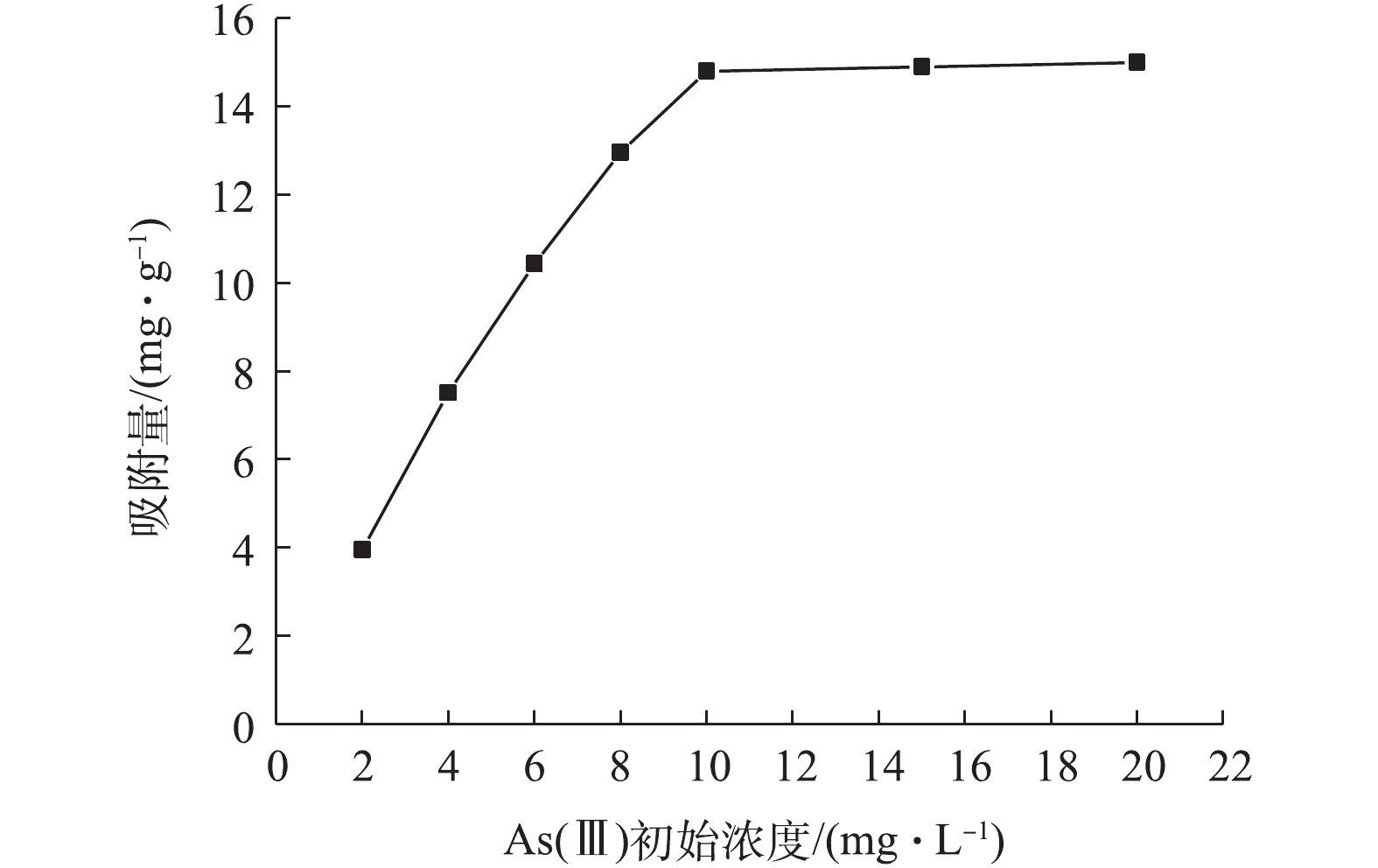
图8为As(Ⅲ)初始浓度对吸附量的影响结果。由图8可知,As(Ⅲ)初始浓度从1 mg·L−1增加到8 mg·L−1时,吸附量从1.98 mg·g−1增加到12.96 mg·g−1;当As(Ⅲ)初始浓度达到10 mg·L−1以上时,NMCNTs-PANI对As(Ⅲ)吸附量几乎不再增加。这是因为随着As(Ⅲ)浓度的增加,NMCNTs-PANI与As(Ⅲ)之间的碰撞机率增大,导致吸附量不断增高;同时NMCNTs-PANI表面吸附位点数量有限,吸附位点被占据后不能继续吸附溶液中As(Ⅲ),NMCNTs-PANI逐渐达到吸附饱和状态。改性后CNTs表面官能团数量大大增加,水中As(Ⅲ)可以与羟基、羧基、酰胺键等官能团发生络合,极大地促进了NMCNTs-PANI对As(Ⅲ)的吸附。
将数据代入Langmuir等温线方程和Freundlich等温线方程进行拟合,前者常用于描述单分子层吸附;后则多用于描述复杂不均匀表面的多分子层吸附[30-31]。Langmuir方程[30]和Freundlich方程[31]见式(5)和式(6)。
式中:Ce为平衡浓度,mg·L−1;qe为平衡吸附量,mg·L−1;qm为饱和吸附量,mg·g−1;kL为Langmuir平衡常数,L·mg−1;kF为Freundlich常数mg·g−1;n为吸附强度。
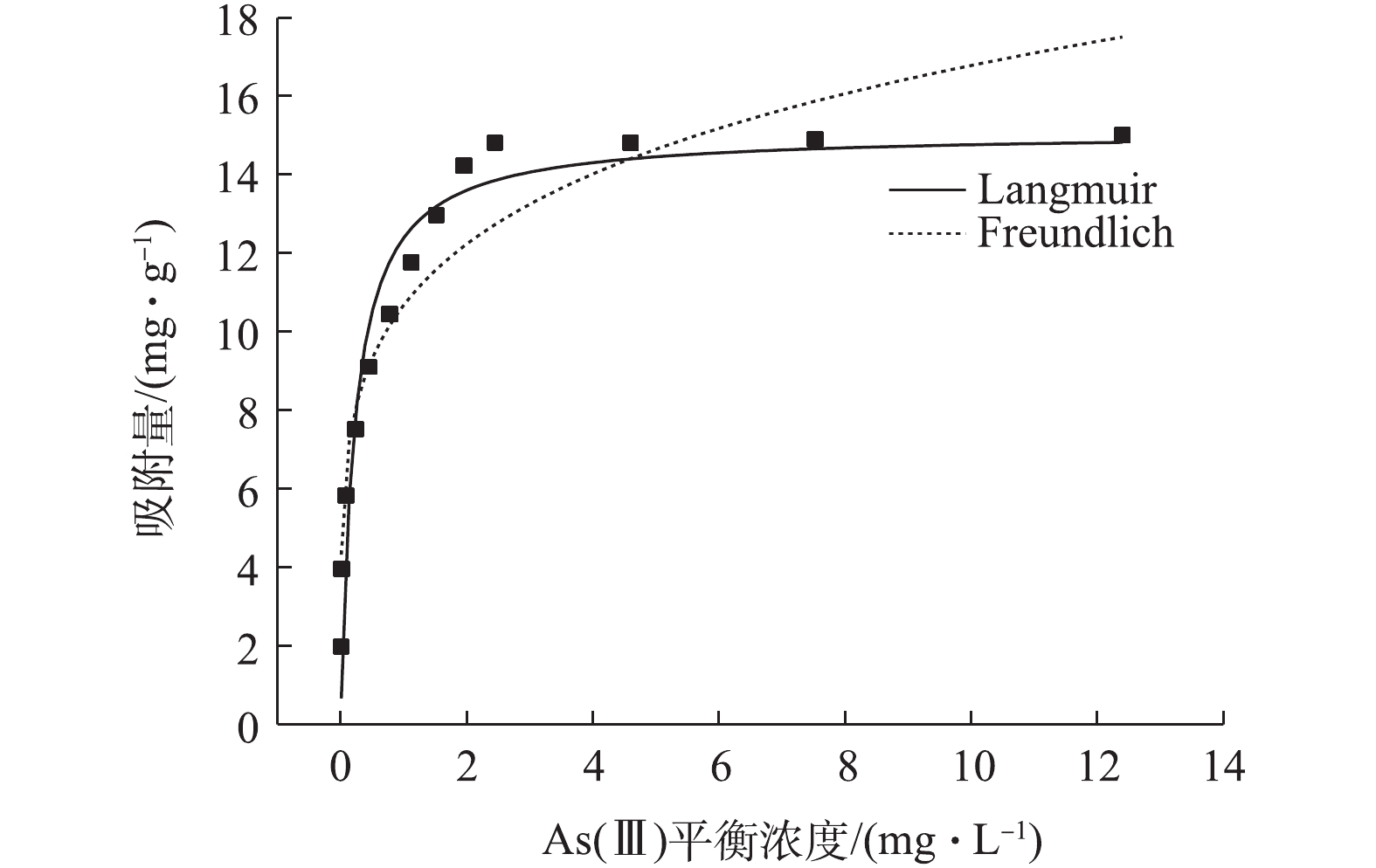
图9是利用Langmuir、Freundlich等温线对实验数据的拟合结果,拟合参数见表3。由图9可知,Langmuir、Freundlich等温线均能较好描述NMCNTs-PANI吸附水中As(Ⅲ)过程,但表3显示,前者R2大于后者,说明Langmuir等温线能更好地描述NMCNTs-PANI吸附水中As(Ⅲ)过程。实验中NMCNTs-PANI对10 mg·L−1 As(Ⅲ)饱和吸附量为14.80 mg·g−1。有研究发现,除部分研究[32]针对含As(Ⅲ)地下水利用生物催化合成施氏矿物对As(Ⅲ)的饱和吸附量为67.70 mg·g−1外,大多数研究[33-35]中吸附剂对水中As(Ⅲ)的饱和吸附量为1.78~10.90 mg·g−1,如Fe-Mn双氧化物改性的硅藻土(1.68 mg·g−1)、针铁矿(0.38 mg·g−1)和赤铁矿(0.26 mg·g−1)。因此,相比其他研究的结果,本研究所用吸附剂的吸附性能更优。
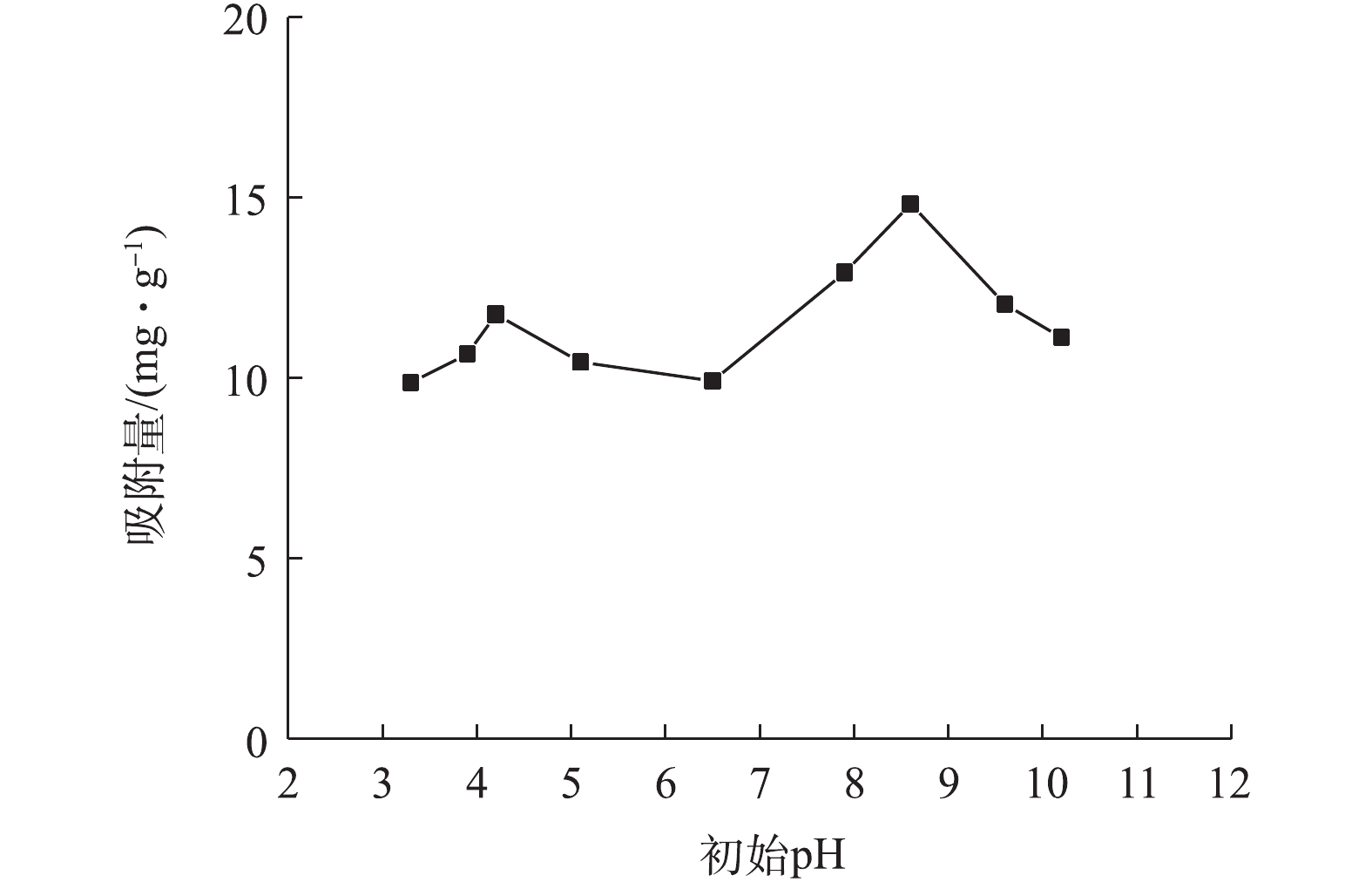
溶液pH可影响溶液中As(Ⅲ)的存在形态及吸附剂表面官能团的性能[36]。图10为初始pH对As(Ⅲ)吸附量的影响结果。由图10可知,在较低pH(3.3~4.2)条件下,As(Ⅲ)吸附量从9.87 mg·g−1升至11.76 mg·g−1;在pH为4.2~6.5的弱酸性环境中,As(Ⅲ)的吸附量下降至9.92 mg·g−1;当pH升高至6.5~8.6时,吸附量达到最大值,为14.82 mg·g−1,此时,去除率为74.1%;继续提高pH,吸附量则再次降低。有研究[37]表明,在溶液pH<9.2时,As(Ⅲ)主要以中性H3AsO3形态存在(
H3AsO3=H2AsO−3+ H+ ,pKa1=9.2);当pH>9.2 时,As(Ⅲ)则主要以阴离子形态H2AsO−3 存在。酸度较低条件下,中性状态存在的H3AsO3主要是通过与活性基团(羟基、羧基、酰胺基)的结合被吸附,且酸性基团的活性在该条件下会增强[38];弱酸性环境中,H3AsO3与活性基团的化学结合作用随着水中酸度降低而减少,吸附量出现下滑,故在pH=4.2出现较小的峰值;pH升高至碱性时,NMCNTs-PANI对As(Ⅲ)的去除主要归为以下2个原因:一方面,其通过与活性基团发生作用,此时碱性基团的活化增大了吸附作用;另一方面,As(Ⅲ)与吸附剂表面Fe离子发生配位络合作用,此过程主要依靠非离子H3AsO3和铁离子间强大的约束力[39],所以在pH=8.6时吸附量达到最高;随着pH继续上升,H3AsO3解离为H2AsO−3 ,吸附量出现大幅度降低,可能是因为吸附剂表面带负电荷的羧基、羟基和酰胺基团会与阴离子形态As(Ⅲ)发生静电斥力作用;且碱性条件下高浓度的OH−与H2AsO−3 激烈的竞争吸附剂表面的活性位点[35]。图11为共存阴离子对吸附量的影响结果。将Cl−、
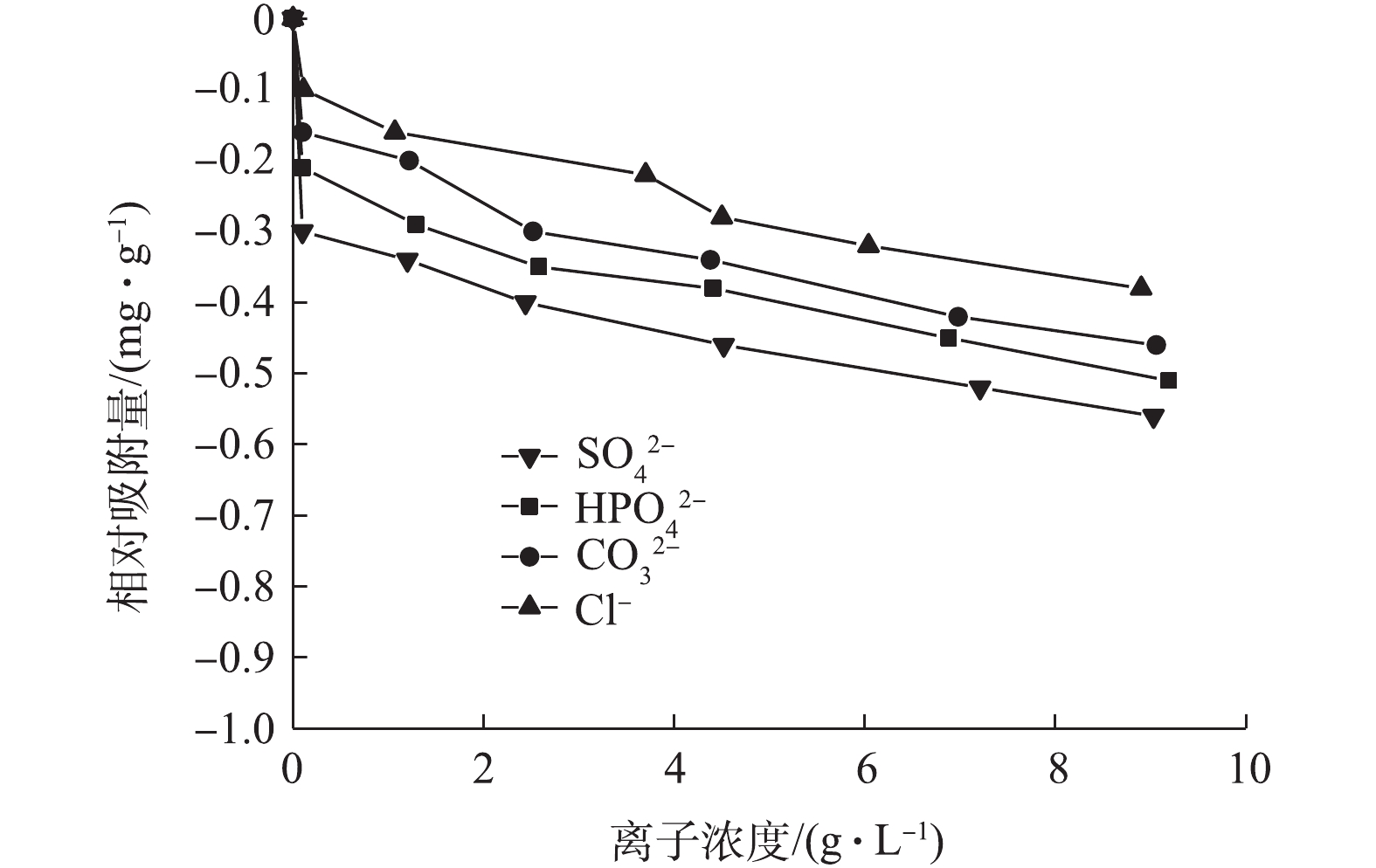
CO2−3 、HPO2−4 、SO2−4 的浓度分别设置为0.11~8.90、0.10~9.06、0.10~9.19、0.10~9.03 g·L−1。由图11可知,共存阴离子Cl−、CO2−3 、HPO2−4 、SO2−4 对吸附量几乎不产生影响,4种离子浓度分别在8.90、9.06、9.19、9.03 g·L−1时,吸附量也仅下降0.38~0.56 mg·g−1,说明NMCNTs-PANI对As(Ⅲ)的吸附具有选择性。有研究[40]发现,如果吸附机制为配位体交换和络合物形成,那么离子强度则对吸附无影响,进一步说明化学吸附是NMCNTs-PANI吸附As(Ⅲ)的主要机制。 -
1)实验结果显示,CNTs管状结构部分溶解,形成了孔容和平均孔径更大的多孔物质NMCNTs-PANI;且负载在NMCNTs-PANI表面的活性基团大大增加;吸附量随接触时间延长而增大,随As(Ⅲ)初始浓度增大而增大;初始pH对吸附量的影响较显著,NMCNTs-PANI对As(Ⅲ)吸附量在pH 8.6时达到最大;溶液中共存阴离子Cl−、
CO2−3 、HPO2−4 、SO2−4 对吸附量的影响可忽略不计。2)动力学特征符合准一级动力学和准二级动力学方程;吸附等温线符合Langmuir模型,饱和吸附容量为14.80 mg·g−1。吸附动力学和等温线研究结果表明,NMCNTs-PANI吸附水中As(Ⅲ)的过程以化学吸附为主。
3)酰胺化/氧化碳纳米管-聚苯胺具有较佳的吸附性能,当微污染水中As(Ⅲ)浓度不超过饮用水标准50倍时,采用0.5 g·L−1 NMCNTs/PANI吸附处理可达到饮用水标准。此研究为解决饮用水中低浓度三价砷的污染问题提供了新方法。
酰胺化/氧化碳纳米管-聚苯胺吸附三价砷
Adsorption of As(Ⅲ) from aqueous solution by amidated/oxidized carbon nanotube-polyaniline
-
摘要: 为探讨改性碳纳米管(CNTs)对砷的吸附特性,采用化学修饰对CNTs进行了改性。将CNTs先后进行氧化和酰胺化处理,并与聚苯胺反应,得到酰胺化/氧化碳纳米管-聚苯胺(NMCNTs-PANI),利用SEM观察、比表面积测定、含氧含氮官能团和分子结构分析对改性前后CNTs进行了表征;研究了NMCNTs-PANI在不同反应体系对As(Ⅲ)的吸附效果。结果表明:NMCNTs-PANI总孔容和平均孔径均有所增加;表面含氧含氮基团增加;初始pH对吸附量影响较显著;共存阴离子对吸附量影响可忽略不计;吸附过程符合准一级动力学和准二级动力学方程,证实该过程主要以化学吸附为主;吸附等温线符合Langmuir模型。NMCNTs-PANI通过表面吸附-化学诱导作用可较好地去除水中As(Ⅲ),是一种优良的含砷污染水的吸附剂。Abstract: To promote the As(Ⅲ) adsorption capacity of carbon nanotubes (CNTs), the modification of CNTs was conducted in this study. Firstly, CNTs were modified with oxidation and amidation, followed by reaction with polyaniline to obtain amidated/oxidized carbon nanotube-polyaniline (NMCNTs-PANI). Both the modified and pristine carbon nanotubes were observed by scanning electron microscope to compare their morphological differences. Their specific surface, functional groups and molecular structure were also determined. The adsorption behavior of As(Ⅲ) on NMCNTs-PANI was further studied. The results show that the total pore volume and average pore size of NMCNTs-PANI increased, as well as the surface oxygen and nitrogen-containing groups. The initial pH had significant effects on the adsorption capacity, while the coexisting anions had a negligible effect on As(Ⅲ) adsorption. The kinetic data fitted well with the pseudo-first-order and pseudo-second-order kinetic model, which demonstrated that chemical adsorption was the main mechanism. The adsorption isotherms of NMCNTs-PANI fitted well with the Langmuir model. The good performance on As(Ⅲ) removal by NMCNTs-PANI was completed through surface adsorption-chemical induction actions. The high As(Ⅲ) removal efficiency of the NMCNTs-PANI makes it to be an excellent adsorbent for arsenic-contaminated water treatment.
-
Key words:
- trivalent arsenic /
- modified carbon nanotube /
- polyaniline(PANI) /
- adsorption
-
随着现代社会工业化的高速发展,天然水中砷的含量迅速增加[1]。水中砷污染已成为全社会亟待解决的重要环境问题之一[2]。目前,常用的除砷方法主要有混凝沉淀法、离子交换法、膜分离法、吸附法[3-6]等。混凝沉淀法中预氧化过程会增加后续污水处理的问题[3];离子交换法须采用特殊材料,工艺流程较繁琐[4];膜分离法易出现膜污染等问题,该法适用于处理量相对较少的砷污染水[5]。因此,吸附法在处理效率高、操作简单和适用性强等方面的优势越来越受到人们的重视[6]。
多壁碳纳米管(CNTs)具有比表面积大、孔隙结构丰富、表面易于被修饰等特点,近年来在制备复合吸附材料方面已有诸多的应用[7]。IBRAHIM等[8]研究了深共熔溶剂(DESs)功能化CNTs对水中甲基橙的吸附性能;李德云等[9]在研究改性CNTs吸附水中亚甲基蓝过程时提到Cu/CuO改性CNTs对亚甲基蓝的吸附效果优于原始CNTs;杨爱丽等[10]在研究臭氧氧化改性CNTs对铀的吸附去除时提到,含氧量的增加会显著提高改性CNTs对铀的去除率。
目前,对水中砷去除的研究多集中在吸附剂的氧化改性以及对As(Ⅴ)的吸附去除上,对As(Ⅲ)的削减效果分析和去除机制等方面的研究相对较少[11]。本研究从活化CNTs管壁接枝点位、负载高活性基团的酰胺类材料的角度出发,采用强酸氧化和酰胺化等方法制备了酰胺化/氧化碳纳米管-聚苯胺(NMCNTs-PANI),并探究该复合材料对水中As(Ⅲ)的吸附性能。
1. 材料与方法
1.1 实验原料
基准纯三氧化二砷(As2O3,99%);高锰酸钾(KMnO4)、硫酸(98%H2SO4)、氯化亚砜(SOCl2)、N-聚乙烯吡咯烷酮、三乙胺、乙二胺、N, N-二环己基碳二亚胺、聚苯胺(PANI)、丙酮均为分析纯。碳纳米管原粉(CNTs)购自苏州石墨烯科技有限公司,采用改性催化碳气相沉积法(CVD)制备。
1.2 实验装置
真空干燥箱(DZF-6050,上海福絮仪器厂);磁力加热搅拌器(HJ-4,江苏新瑞仪器厂);pH计(pHS-25型,上海仪电公司);原子荧光光度计(AFS-933,美国Thermo Fisher Scientific公司);扫描电子显微镜(TM3030,日立公司);比表面孔径分布测定仪(TRISTAR3000,美国MICROMERITICS公司);X射线衍射仪(D2 PHASER,德国布鲁克AXS公司);傅里叶变换红外光谱仪(EQUINOX 55,美国赛默飞公司);拉曼光谱仪(LABRAM ARAMIS,法国HORIBA公司)。
实验采用恒温振荡器,实验装置及实物照片见图1。设置温度为25 ℃,转速为180 r·min−1,避光运行,振荡后静置,抽滤。
1.3 实验方法
冰水浴中,称取1 g CNTs、3 g KMnO4加入到30 mL浓硫酸中,搅拌2 h;升温至40 ℃,搅拌反应4 h;置于70 ℃磁力搅拌器中搅拌0.3 h;加入适量FeCl3及FeSO4,溶解后抽滤,将所得固体烘干备用。取1 g上述烘干后的固体置于250 mL烧杯中,加入30 mL SOCl2、2 mL DMF和1 g N-聚乙烯吡咯烷酮,搅拌均匀,缓慢加入13 mL三乙胺、7 mL乙二胺,升温至80 ℃,搅拌0.5 h,加入适量N,N-二环己基碳二亚胺,超声0.5 h;置于摇床振荡,洗涤抽滤,干燥得到黑色固体[12-13]。取0.5 g上述黑色固体分散在水溶液中,与50 mL含有2 g PANI的丙酮溶液混合,置于60 ℃水浴锅反应1 h,超声0.5 h;移至摇床振荡20 h,洗涤抽滤,110 ℃真空干燥箱烘干,得到NMCNTs-PANI。
采用SEM观察表面特征;比表面积分析仪测定比表面积和孔径分布;X射线衍射仪(XRD)分析物相组成[14];傅里叶变换红外光谱仪(FT-IR)定性分析表面的化学键和官能团信息[15];拉曼光谱(Raman)分析材料的晶体结构[16]。
1.4 分析方法
配置10 mg·L−1 As2O3溶液,取7份100 mL置于系列250 mL锥形瓶中,分别投加50 mg NCNTs/PANI,反应温度为25 ℃,设置不同的接触时间并置于振荡器中以180 r·min−1转速振荡,静置后过滤,用原子荧光光度计测定滤液中As(Ⅲ)浓度并计算吸附量。As(Ⅲ)初始浓度、初始pH、共存阴离子的考察过程与接触时间类似。溶液pH采用NaOH及HCl调节。
吸附量、去除率依次按式(1)和式(2)计算。
q=(C0−C)VC0 (1) η=(C0−CC0)×100% (2) 式中:q为NMCNTs-PANI对溶液中As(Ⅲ)的吸附量,mg·g−1;η为去除率;C0为As(Ⅲ)初始浓度,mg·L−1;C为滤液中As(Ⅲ)浓度,mg·L−1;V为溶液体积,L;m为NMCNTs-PANI投加量,g。
2. 结果与讨论
2.1 对NMCNTs-PANI的表征
图2是改性前后CNTs的SEM图。从图2(a)中可以看出,CNTs原粉是相互缠绕在一起的管状结构,其管壁无序、混乱;图2(b)和图2(c)显示,NMCNTs-PANI管状结构部分消失,管间缺陷增大,并出现聚集现象。该现象可能是因为引入某种活性剂或CNTs表面功能化导致CNTs结构受到破坏[17]。
 图 2 CNTs原粉、NMCNTs-PANI和吸附后材料的SEM图Figure 2. SEM images of CNTs, NMCNTs-PANI and post-adsorption composite
图 2 CNTs原粉、NMCNTs-PANI和吸附后材料的SEM图Figure 2. SEM images of CNTs, NMCNTs-PANI and post-adsorption composite改性前后CNTs物理性质见表1。可以看出,CNTs原粉、NMCNTs-PANI和吸附后材料的孔径为3.58~13.84 nm,说明样品为介孔材料;与CNTs原粉相比,NMCNTs-PANI和吸附后材料的比表面积稍有减小,而孔容和平均孔径均大大增加。H2SO4和KMnO4均具有强氧化性,可以氧化掉CNTs上部分管状结构,导致CNTs局部坍塌;或者少部分孔道在反应过程中被堵塞,碳管难以拉伸,从而减小NMCNTs-PANI和吸附后材料的比表面积;同时,也有部分管内杂质被溶解,疏通了CNTs孔道结构;或者使一些内部封闭的孔道被打开,导致NMCNTs-PANI和吸附后材料的孔容和平均孔径均增大[18]。
表 1 CNTs原粉、NMCNTs-PANI和吸附后材料的物理性质Table 1. Physical characteristics of CNTs, NMCNTs-PANI and post-adsorption composite改性前后的材料 比表面积/(m2·g−1) 孔容/(cm3·g−1) 平均孔径/nm CNTs原粉 156.59 0.58 3.58 NMCNTs-PANI 134.97 0.99 13.84 吸附后材料 120.80 0.87 12.85 | Show Table DownLoad:
CSV
DownLoad:
CSV
图3是改性前后CNTs的XRD图谱。从图3可以看出,改性前后表征CNTs晶面(002)的衍射角在26.0°处位置没发生变化[14];NMCNTs-PANI和吸附后材料衍射峰的强度均小于CNTs原粉,说明CNTs改性后结晶度变差。此外,NMCNTs-PANI中体现PANI的衍射特征峰在15.3°和22.6°凸起,可能原因是PANI大部分以无定性态存在[19]。
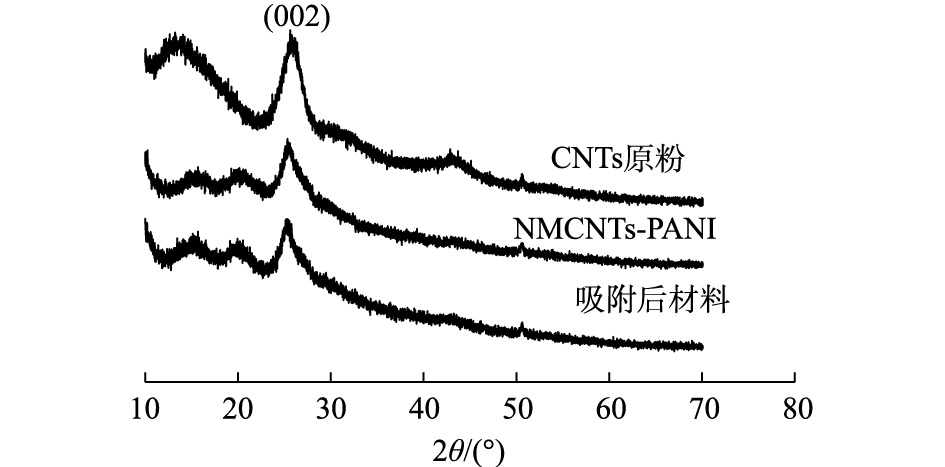 图 3 CNTs原粉、NMCNTs-PANI和吸附后材料的XRD图Figure 3. X-ray diffraction patterns of CNTs, NMCNTs-PANI and post-adsorption composite
图 3 CNTs原粉、NMCNTs-PANI和吸附后材料的XRD图Figure 3. X-ray diffraction patterns of CNTs, NMCNTs-PANI and post-adsorption composite图4是改性前后CNTs的红外光谱图。可以看出,与CNTs原粉相比,NMCNTs-PANI和吸附后材料在500~4 000 cm−1出现多个吸收峰。其中NMCNTs-PANI的红外光谱中1 134 cm−1是O—H的振动峰[20];1 192 cm−1是环氧基C—O—C引起的伸缩振动峰[21];1 701 cm−1处的吸收峰为羰基C=O伸缩振动[22];1 257 cm−1处的红外吸收峰归属于C—O的伸缩振动[23];1 400 cm−1处是C—N键的伸缩振动特征峰,1 654 cm−1处是C=N的弯曲振动峰[24],以上说明制备过程中引入了羧基、羟基、环氧基等功能性基团;3 278 cm−1处归属于N—H的伸缩振动峰,该峰和C=O、C—N表明酰胺基团负载在材料表面;786 cm−1处的特征峰归属于Fe—O键的伸缩振动[25],这表明铁离子成功接枝在材料表面。吸附后材料的FT-IR图出现了位于582 cm−1处的金属配位体振动(Metal-N)[25],可能原因是水中As(Ⅲ)吸附于NMCNTs-PANI表面。
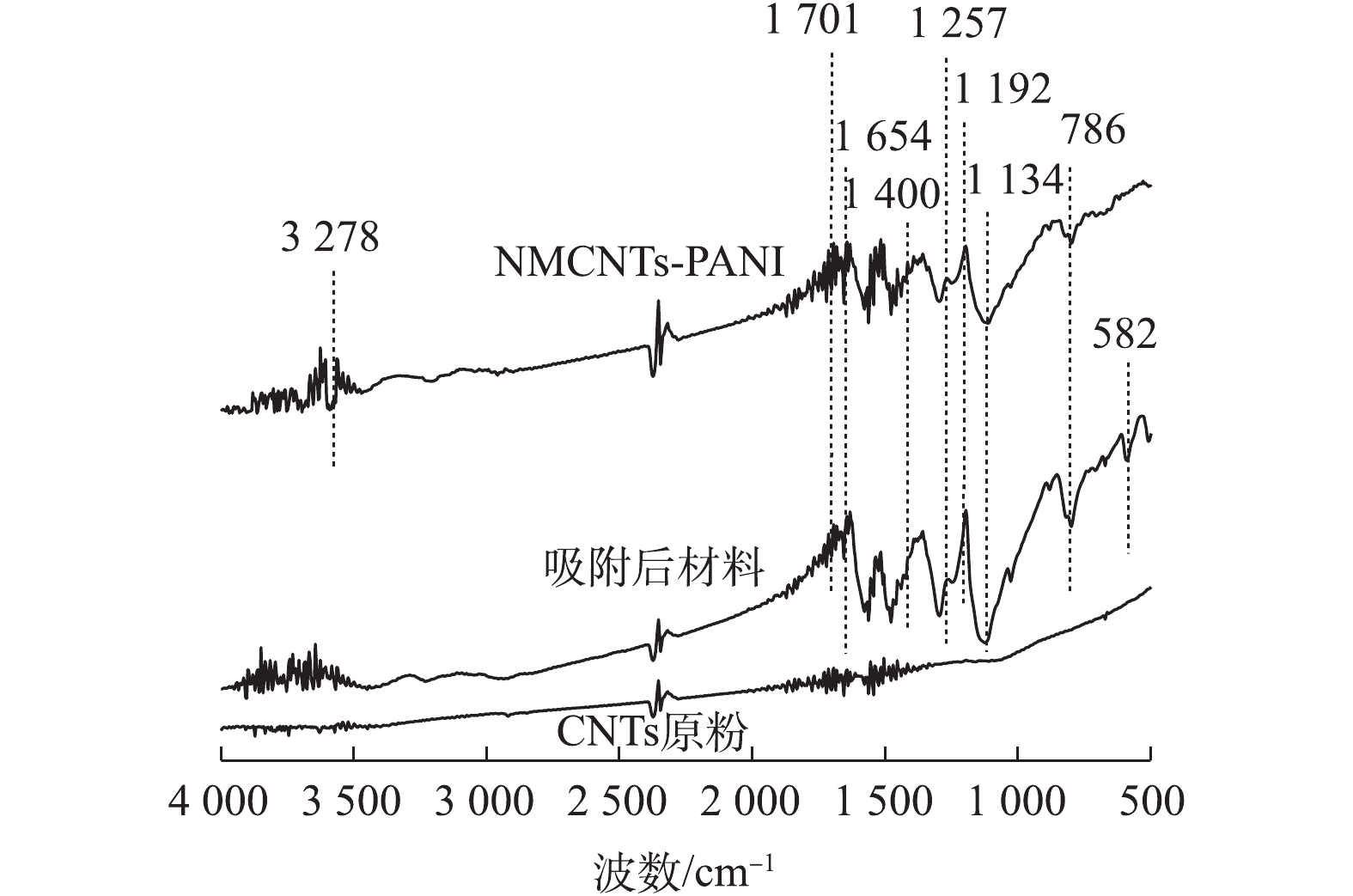 图 4 CNTs原粉、NMCNTs-PANI和吸附后材料的FT-IR图Figure 4. FT-IR spectra of CNTs, NMCNTs-PANI and post-adsorption composite
图 4 CNTs原粉、NMCNTs-PANI和吸附后材料的FT-IR图Figure 4. FT-IR spectra of CNTs, NMCNTs-PANI and post-adsorption composite图5是改性前后CNTs的Raman光谱图。可以看出,改性前后CNTs均出现了2个典型特征峰,其中D峰位于1 340 cm−1左右,该缺陷峰反映的是碳材料中C—C的无序振动,峰值反映出CNTs结构的缺陷性和混乱程度;在1 580 cm−1左右的G峰是碳结构sp2的特征峰,用以说明CNTs的结晶程度,2个峰的强度比值ID/IG可以衡量CNTs结构缺陷密度,比值越低,结晶越完整[16]。计算表明,NMNCTs-PANI和吸附后材料的ID/IG分别为1.05、1.06,均高于CNTs原粉ID/IG(1.03),说明复合材料结构的无序性增加。综上所述,改性后CNTs的结构发生了变化,但XRD及Raman分析结果一致表明,CNTs的晶体结构并没有被破坏。
 图 5 CNTs原粉、NMCNTs-PANI和吸附后材料的Raman光谱图Figure 5. Raman spectra of CNTs, NMCNTs-PANI and post-adsorption composite
图 5 CNTs原粉、NMCNTs-PANI和吸附后材料的Raman光谱图Figure 5. Raman spectra of CNTs, NMCNTs-PANI and post-adsorption composite2.2 静态吸附实验
图6为接触时间对吸附量的影响结果。当接触时间从5 min延至20 min时,吸附量从4 mg·g−1快速上升到13 mg·g−1;随后吸附量缓慢增加;在30 min左右达到吸附平衡。这是由于初始阶段NMCNTs-PANI表面高能量的活性位点迅速被占领,吸附速率较高,此时溶液中大部分As(Ⅲ)占据了表面活性位点;后期由低能量的活性位点继续吸附,吸附速率随之降低。综合考虑吸附速率,接触时间选择30 min。
将数据代入准一级动力学方程和准二级动力学方程进行拟合,准一级动力学方程[26]和准二级动力学方程[27]见式(3)和式(4)。
qt=qe[1−exp(−k1t)] (3) qt=q2ek2t1+qek2t (4) 式中:t为吸附过程某一时刻,min;qe为平衡吸附量,mg·g−1;qt为t时刻吸附量,mg·g−1;k1为一级动力学速率常数,min−1;k2为二级动力学速率常数,g·(mg·min)−1。
图7为采用不同动力学方程对动力学实验数据的拟合结果,拟合参数见表2。由图7可知,准一级动力学方程和准二级动力学方程均能较好描述NMCNTs-PANI吸附As(Ⅲ)的动力学过程。但表2显示准二级动力学方程R2为0.994,准一级动力学方程R2为0.983,故准二级动力学方程更为符合NMCNTs-PANI吸附As(Ⅲ)的动力学过程,即NMCNTs-PANI吸附水中As(Ⅲ)主要以化学吸附为主[28]。吸附过程中物理吸附主要是分子间作用力;化学吸附可能涉及离子交换或NMCNTs-PANI上活性基团与As(Ⅲ)形成化学键的吸附[28],尤其是改性材料表面所带有的酰胺基团,具有较高反应活性,可与As(Ⅲ)发生结合[29]。
表 2 不同动力学模型拟合参数Table 2. Fitting parameters for various kinetic modelsqe,exp/(mg·g−1) 准一级动力学方程 准二级动力学方程 k1 qe,cal R2 k2 qe,cal R2 14.50 0.12 14.78 0.983 0.006 18.37 0.994 注:qe,exp和qe,cal依次为平衡吸附量实验值和拟合值。 | Show TableDownLoad:
CSV
图8为As(Ⅲ)初始浓度对吸附量的影响结果。由图8可知,As(Ⅲ)初始浓度从1 mg·L−1增加到8 mg·L−1时,吸附量从1.98 mg·g−1增加到12.96 mg·g−1;当As(Ⅲ)初始浓度达到10 mg·L−1以上时,NMCNTs-PANI对As(Ⅲ)吸附量几乎不再增加。这是因为随着As(Ⅲ)浓度的增加,NMCNTs-PANI与As(Ⅲ)之间的碰撞机率增大,导致吸附量不断增高;同时NMCNTs-PANI表面吸附位点数量有限,吸附位点被占据后不能继续吸附溶液中As(Ⅲ),NMCNTs-PANI逐渐达到吸附饱和状态。改性后CNTs表面官能团数量大大增加,水中As(Ⅲ)可以与羟基、羧基、酰胺键等官能团发生络合,极大地促进了NMCNTs-PANI对As(Ⅲ)的吸附。
将数据代入Langmuir等温线方程和Freundlich等温线方程进行拟合,前者常用于描述单分子层吸附;后则多用于描述复杂不均匀表面的多分子层吸附[30-31]。Langmuir方程[30]和Freundlich方程[31]见式(5)和式(6)。
qe=qmkLCe1+kLCe (5) qe=kFC1/ne (6) 式中:Ce为平衡浓度,mg·L−1;qe为平衡吸附量,mg·L−1;qm为饱和吸附量,mg·g−1;kL为Langmuir平衡常数,L·mg−1;kF为Freundlich常数mg·g−1;n为吸附强度。
图9是利用Langmuir、Freundlich等温线对实验数据的拟合结果,拟合参数见表3。由图9可知,Langmuir、Freundlich等温线均能较好描述NMCNTs-PANI吸附水中As(Ⅲ)过程,但表3显示,前者R2大于后者,说明Langmuir等温线能更好地描述NMCNTs-PANI吸附水中As(Ⅲ)过程。实验中NMCNTs-PANI对10 mg·L−1 As(Ⅲ)饱和吸附量为14.80 mg·g−1。有研究发现,除部分研究[32]针对含As(Ⅲ)地下水利用生物催化合成施氏矿物对As(Ⅲ)的饱和吸附量为67.70 mg·g−1外,大多数研究[33-35]中吸附剂对水中As(Ⅲ)的饱和吸附量为1.78~10.90 mg·g−1,如Fe-Mn双氧化物改性的硅藻土(1.68 mg·g−1)、针铁矿(0.38 mg·g−1)和赤铁矿(0.26 mg·g−1)。因此,相比其他研究的结果,本研究所用吸附剂的吸附性能更优。
表 3 不同吸附等温线主要拟合参数Table 3. Main fitting parameters for various adsorption isothermsLangmuir Freundlich qm/(mg·g−1) kL R2 kF n−1 R2 15.08 4.60 0.927 10.68 0.20 0.885 | Show TableDownLoad:
CSV
溶液pH可影响溶液中As(Ⅲ)的存在形态及吸附剂表面官能团的性能[36]。图10为初始pH对As(Ⅲ)吸附量的影响结果。由图10可知,在较低pH(3.3~4.2)条件下,As(Ⅲ)吸附量从9.87 mg·g−1升至11.76 mg·g−1;在pH为4.2~6.5的弱酸性环境中,As(Ⅲ)的吸附量下降至9.92 mg·g−1;当pH升高至6.5~8.6时,吸附量达到最大值,为14.82 mg·g−1,此时,去除率为74.1%;继续提高pH,吸附量则再次降低。有研究[37]表明,在溶液pH<9.2时,As(Ⅲ)主要以中性H3AsO3形态存在(
H3AsO3=H2AsO−3+ H+ ,pKa1=9.2);当pH>9.2 时,As(Ⅲ)则主要以阴离子形态H2AsO−3 存在。酸度较低条件下,中性状态存在的H3AsO3主要是通过与活性基团(羟基、羧基、酰胺基)的结合被吸附,且酸性基团的活性在该条件下会增强[38];弱酸性环境中,H3AsO3与活性基团的化学结合作用随着水中酸度降低而减少,吸附量出现下滑,故在pH=4.2出现较小的峰值;pH升高至碱性时,NMCNTs-PANI对As(Ⅲ)的去除主要归为以下2个原因:一方面,其通过与活性基团发生作用,此时碱性基团的活化增大了吸附作用;另一方面,As(Ⅲ)与吸附剂表面Fe离子发生配位络合作用,此过程主要依靠非离子H3AsO3和铁离子间强大的约束力[39],所以在pH=8.6时吸附量达到最高;随着pH继续上升,H3AsO3解离为H2AsO−3 ,吸附量出现大幅度降低,可能是因为吸附剂表面带负电荷的羧基、羟基和酰胺基团会与阴离子形态As(Ⅲ)发生静电斥力作用;且碱性条件下高浓度的OH−与H2AsO−3 激烈的竞争吸附剂表面的活性位点[35]。图11为共存阴离子对吸附量的影响结果。将Cl−、
CO2−3 、HPO2−4 、SO2−4 的浓度分别设置为0.11~8.90、0.10~9.06、0.10~9.19、0.10~9.03 g·L−1。由图11可知,共存阴离子Cl−、CO2−3 、HPO2−4 、SO2−4 对吸附量几乎不产生影响,4种离子浓度分别在8.90、9.06、9.19、9.03 g·L−1时,吸附量也仅下降0.38~0.56 mg·g−1,说明NMCNTs-PANI对As(Ⅲ)的吸附具有选择性。有研究[40]发现,如果吸附机制为配位体交换和络合物形成,那么离子强度则对吸附无影响,进一步说明化学吸附是NMCNTs-PANI吸附As(Ⅲ)的主要机制。3. 结论
1)实验结果显示,CNTs管状结构部分溶解,形成了孔容和平均孔径更大的多孔物质NMCNTs-PANI;且负载在NMCNTs-PANI表面的活性基团大大增加;吸附量随接触时间延长而增大,随As(Ⅲ)初始浓度增大而增大;初始pH对吸附量的影响较显著,NMCNTs-PANI对As(Ⅲ)吸附量在pH 8.6时达到最大;溶液中共存阴离子Cl−、
CO2−3 、HPO2−4 、SO2−4 对吸附量的影响可忽略不计。2)动力学特征符合准一级动力学和准二级动力学方程;吸附等温线符合Langmuir模型,饱和吸附容量为14.80 mg·g−1。吸附动力学和等温线研究结果表明,NMCNTs-PANI吸附水中As(Ⅲ)的过程以化学吸附为主。
3)酰胺化/氧化碳纳米管-聚苯胺具有较佳的吸附性能,当微污染水中As(Ⅲ)浓度不超过饮用水标准50倍时,采用0.5 g·L−1 NMCNTs/PANI吸附处理可达到饮用水标准。此研究为解决饮用水中低浓度三价砷的污染问题提供了新方法。
-
图 2 CNTs原粉、NMCNTs-PANI和吸附后材料的SEM图
Figure 2. SEM images of CNTs, NMCNTs-PANI and post-adsorption composite
图 3 CNTs原粉、NMCNTs-PANI和吸附后材料的XRD图
Figure 3. X-ray diffraction patterns of CNTs, NMCNTs-PANI and post-adsorption composite
图 4 CNTs原粉、NMCNTs-PANI和吸附后材料的FT-IR图
Figure 4. FT-IR spectra of CNTs, NMCNTs-PANI and post-adsorption composite
图 5 CNTs原粉、NMCNTs-PANI和吸附后材料的Raman光谱图
Figure 5. Raman spectra of CNTs, NMCNTs-PANI and post-adsorption composite
表 1 CNTs原粉、NMCNTs-PANI和吸附后材料的物理性质
Table 1. Physical characteristics of CNTs, NMCNTs-PANI and post-adsorption composite
改性前后的材料 比表面积/(m2·g−1) 孔容/(cm3·g−1) 平均孔径/nm CNTs原粉 156.59 0.58 3.58 NMCNTs-PANI 134.97 0.99 13.84 吸附后材料 120.80 0.87 12.85
下载: 导出CSV
表 2 不同动力学模型拟合参数
Table 2. Fitting parameters for various kinetic models
qe,exp/(mg·g−1) 准一级动力学方程 准二级动力学方程 k1 qe,cal R2 k2 qe,cal R2 14.50 0.12 14.78 0.983 0.006 18.37 0.994 注:qe,exp和qe,cal依次为平衡吸附量实验值和拟合值。
下载: 导出CSV
表 3 不同吸附等温线主要拟合参数
Table 3. Main fitting parameters for various adsorption isotherms
Langmuir Freundlich qm/(mg·g−1) kL R2 kF n−1 R2 15.08 4.60 0.927 10.68 0.20 0.885
下载: 导出CSV
-
[1] ALAM M, ALLINSON G, STAGNITTI F, et al. Arsenic contamination in Bangladesh groundwater: A major environmental and social disaster[J]. International Journal of Environmental Health Research, 2002, 12(3): 236-253. [2] 邓安琪, 董兆敏, 高群, 等. 中国地下水砷健康风险评价[J]. 中国环境科学, 2017, 37(9): 3556-3365. doi: 10.3969/j.issn.1000-6923.2017.09.044 [3] YAN Q, GUI Y, ZHOU N, et al. Treatment of arsenic-containing mineral processing wastewater by coagulation and sedimentation process[J]. Chinese Journal of Environmental Engineering, 2014, 8(9): 3683-3688. [4] ORTEGA A, OLIVA I, CONTRERAS K, et al. Arsenic removal from water by hybrid electro-regenerated anion exchange resin/electrodialysis process[J]. Separation and Purification Technology, 2017, 184: 319-326. [5] ZHU N, ZHANG J, TANG J, et al. Arsenic removal by periphytic biofilm and its application combined with biochar[J]. Bioresource Technology, 2018, 248: 49-55. [6] SARI A, TUZEN M. Biosorption of As(III) and As(V) from aqueous solution by macrofungus(Inonotus hispidus) biomass: Equilibrium and kinetic studies[J]. Journalof Hazardous Materials, 2009, 164(2/3): 1372-1378. [7] ZHENG X, GAO H, DING A, et al. Self-assembly of polymer-functionalized carbon nanotubes induced by polymer microphase separation[J]. Chinese Journal of Organic Chemistry, 2013, 33(7): 1509-1513. [8] IBRAHIM R, ELSHAFIE A, HIN L, et al. A clean approach for functionalized carbon nanotubes by deep eutectic solvents and their performance in the adsorption of methyl orange from aqueous solution[J]. Journal of Environmental Management, 2019, 235: 521-534. [9] 李德云, 刘龙飞, 李成亮, 等. Cu/CuO改性碳纳米管对亚甲基蓝的吸附特征[J]. 农业环境科学学报, 2018, 37(10): 2289-2296. doi: 10.11654/jaes.2017-1749 [10] 杨爱丽, 武俊红, 张业新. 臭氧氧化改性碳纳米管对铀的吸附性能[J]. 核化学与放射化学, 2018, 40(4): 267-272. doi: 10.7538/hhx.2018.YX.2017036 [11] 严群, 余洋, 周娜娜. 饮用水中砷处理技术的研究进展[J]. 有色金属科学与工程, 2012, 3(3): 74-79. [12] CHIANG I, BRINSON B, SMALLEY R, et al. Purification and characterization of single-wall carbon nanotubes[J]. Journal of Physical Chemistry B, 2001, 105(6): 1157-1161. [13] LIU Z, SHEN Z, ZHU T, et al. Organizing single-walled carbon nanotubes on gold using a wet chemical self-assembling technique[J]. Langmuir, 2000, 16(8): 3569-3373. [14] 杨春霞, 赵文彬. 多壁碳纳米管的表面修饰及分散性[J]. 黑龙江科技大学学报, 2018, 28(3): 286-291. doi: 10.3969/j.issn.2095-7262.2018.03.010 [15] ZHOU Y, HE Y, XIANG Y, et al. Single and simultaneous adsorption of pefloxacin and Cu(II) ions from aqueous solutions by oxidized multiwalled carbon nanotube[J]. Science of the Total Environment, 2019, 646: 29-36. [16] KIM M, LEE C, JANG J. Fabrication of highly flexible, scalable, and highperformance supercapacitors using polyaniline/reduced graphene oxide film with enhanced electrical conductivity and crystallinity[J]. Advanced Functional Materials, 2014, 24(17): 2489-2499. [17] BEHABTU N, YOUNG C, TSENTALOVICH D, et al. Strong, light, multifunctional fibers of carbon nanotubes with ultrahigh conductivity[J]. Science, 2013, 339(6116): 182-186. [18] 刘苛. 磁性多壁碳纳米管吸附去除水中Cu(Ⅱ)和亚甲基蓝的研究[D]. 长沙: 湖南大学, 2015. [19] 高珍珍, 佟浩, 陈建慧. 聚苯胺共价接枝碳纳米管复合材料的制备及其超电容性能的研究[J]. 化学学报, 2014, 72(11): 1175-1181. [20] HU X J, LIU Y G, WANG H, et al. Removal of Cu(II) ions from aqueous solution using sulfonated magnetic graphene oxide composite[J]. Separation and Purification Technology, 2013, 108: 189-195. [21] WANG H, LIU Y G, ZENG G M, et al. Grafting of beta-cyclodextrin to magnetic graphene oxide via ethylenediamine and application for Cr(VI) removal[J]. Carbohydrate Polymers, 2014, 113: 166-173. [22] BOURLINOS A, GOUMIS D, PETRIDIS D, et al. Graphite oxide: Chemical reduction to graphite and surface modification with primary aliphatic amines and amino acids[J]. Langmuir, 2003, 19(15): 6050-6055. [23] CHIN S, IYER K, RASTON C. Fabrication of carbon nano-tubes decorated with ultra fine superparamagnetic nano-particles under continuous flow conditions[J]. Lab on a Chip, 2008, 8: 439-442. [24] LEE H Y, VOGEL W, CHU P J. Nanostructure and surface composition of Pt and Ru binary catalysts on ppolyaniline-functionalized carbon nanotubes[J]. Langmuir, 2011, 27(23): 14654-14661. doi: 10.1021/la202169j [25] STANKOVICH S, PINER R D, NGUYEN S T, et al. Synthesis and exfoliation of isocyanate-treated graphene oxide nanoplatelets[J]. Carbon, 2006, 44(15): 3342-3347. doi: 10.1016/j.carbon.2006.06.004 [26] DOGAN M, KARAOGLU M, ALKAN M. Adsorption kinetics of maxilon yellow 4GL and maxilon red GRL dyes on kaolinite[J]. Journal of Hazardous Materials, 2009, 165(1/2/3): 1142-1151. [27] IDREES M, BATOOL S, KALSOOM T, et al. Animal manure-derived biochars produced via fast pyrolysis for the removal of divalent copper from aqueous media[J]. Journal of Environmental Management, 2018, 213: 109-118. [28] TAN L, LIU Q, JING X, et al. Removal of uranium(VI) ions from aqueous solution by magnetic cobalt ferrite/multiwalled carbon nanotubes composites[J]. Chemical Engineering Journal, 2015, 273: 307-315. doi: 10.1016/j.cej.2015.01.110 [29] CHENG Z, LIU P, GUO B, et al. Surface activation of carbon paper with potassium dichromate lotion and application as a supercapacitor[J]. Applied Surface Science, 2015, 349: 833-838. doi: 10.1016/j.apsusc.2015.05.086 [30] CEGLOWSKI M, SCHROEDER G. Preparation of porous resin with Schiff base chelating groups for removal of heavy metal ions from aqueous solutions[J]. Chemical Engineering Journal, 2015, 263: 402-411. doi: 10.1016/j.cej.2014.11.047 [31] ZHANG X, JIAO C, WANG J, et al. Removal of uranium(VI) from aqueous solutions by magnetic Schiff base: Kinetic and thermodynamic investigation[J]. Chemical Engineering Journal, 2012, 198: 412-419. [32] 陈福星. 施氏矿物对污染水体中六价铬及三价砷的吸附去除研究[D]. 南京: 南京农业大学, 2006. [33] CHANG F, QU J H, LIU H J, et al. Fe-Mn binary oxide incorporated into diatomite as an adsorbent for arsenite removal: Preparation and evaluation[J]. Journal of Colloid and Interface Science, 2009, 338(2): 353-358. [34] WU Y, FENG S, LI B, et al. The characteristics of Escherichia coli adsorption of arsenic(III) from aqueous solution[J]. World Journal of Microbiology & Biotechnology, 2010, 26(2): 249-256. [35] SARI A, ULUOZLU O, TUZEN M. Equilibrium, thermodynamic and kinetic investigations on biosorption of arsenic from aqueous solution by algae (Maugeotia genuflexa) biomass[J]. Chemical Engineering Journal, 2011, 167(1): 155-161. [36] RAHAMN M, ISLAM M. Effects of pH on isotherms modeling for Cu(II) ions adsorption using maple wood sawdust[J]. Chemical Engineering Journal, 2009, 149(1/2/3): 273-280. [37] SHARMA V, SOHN M. Aquatic arsenic: Toxicity, speciation, transformations, and remediation[J]. Environment International, 2009, 35(4): 743-759. [38] TEIXEIRA M C, CIMINELLI V S T. Development of a biosorbent for arsenite: Structural modeling based on X-ray spectroscopy[J]. Environmental Science & Technology, 2005, 39(3): 895-900. [39] RAHAMAN M, BASU A, ISLAM M. The removal of As(III) and As(V) from aqueous solutions by waste materials[J]. Bioresource Technology, 2008, 99(8): 2815-2823. [40] WANG Z, XING M, FANG W, et al. One-step synthesis of magnetite core/zirconia shell nanocomposite for high efficiency removal of phosphate from water[J]. Applied Surface Science, 2016, 366: 67-77. 期刊类型引用(5)
1. 石嘉浩,保琼莉,储玉檀,孙红羽,黄益宗. 铁基石灰质强化钝化材料的制备及其对Sb的钝化效果. 环境科学. 2025(02): 1118-1129 .  百度学术
百度学术
2. 朱晓丽,林姝欢,张星,张子夜,马晓杰. 生物炭固定化解有机磷菌对Pb~(2+)的吸附行为研究. 环境科学学报. 2023(03): 116-126 . 百度学术
3. 张欣禄,孙天杭,姜伟,张海韵,沈晓芳. 氨基化碳纳米管对水中新烟碱类农药的吸附研究. 功能材料. 2023(08): 8207-8216 . 百度学术
4. 马明明,张明明. 联苯胺分子印迹聚合物对苯胺的吸附性研究. 化学研究与应用. 2022(07): 1479-1485 . 百度学术
5. 侯嫔,杨晓瑜,霍燕龙,张南南,刘昊,郭建辉,董慧峪,强志民. 超声氧化多壁碳纳米管对水中Ni(Ⅱ)的吸附效能. 环境工程学报. 2021(07): 2256-2264 . 本站查看
其他类型引用(5)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3913
- HTML全文浏览数: 3913
- PDF下载数: 74
- 施引文献: 10
