








-
近年来,药物和个人护理品(pharmaceutical and personal care products,PPCPs)在地下水、地表水和饮用水中被广泛检出,引起研究者们关注。双氯芬酸钠(diclofenac sodium,DCF)是一种典型的PPCPs,作为一种消炎止痛类药物已被广泛使用,因其具有难生物降解和生物积累性的特点,故给生态环境和人类健康带来极大的威胁。常规的处理技术无法有效地去除DCF,传统的生物处理技术对DCF的去除率只能达到30%左右[1]。因此,亟需寻找一种新型有效的处理方法去除水中的DCF。
近年来,基于硫酸根自由基(
SO⋅−4 )的高级氧化技术受到研究者们的广泛关注[2-3]。与·OH相比,SO⋅−4 具有氧化还原电位高、pH适用范围广及半衰期长等优点,有利于污染物的降解。过硫酸盐(persulfate,PS)可在紫外光、热、碱、过渡金属离子(Mn2+)和零价铁等活化下产生SO⋅−4 。然而,不同的方法具有各自的优点和缺点,如:热活化不产生二次污染,但在使用过程中会消耗很多能量;过渡金属离子可以在室温下活化PS,但是易受溶液pH影响而产生沉淀。零价铁可以在室温下活化PS产生SO⋅−4 ,其主要反应如式(1)和式(2)所示。零价铁的化学性质活泼,在制备和存储的过程中与氧气接触,会形成氧化膜覆盖在零价铁表面,从而影响其在反应过程中的活性。为了解决零价铁钝化问题,研究者们提出了一些改进方法,如利用纳米零价铁[4]、酸洗[5]、制备零价铁双金属[6]和氢气还原等[7]。这些方法可以在一定程度上改善零价铁去除污染物的活性,但是在实际应用方便仍然会存在一定的问题,如增加使用的成本、操作复杂等。有关磁场效应影响零价铁去除污染物的研究是近几年来新兴的研究方向,受到了研究者的广泛关注。KIM等[8]研究发现,在零价铁降解4-氯酚的过程中加入磁场时,可以提高4-氯酚的去除效率。研究者认为磁场可以加速零价铁的腐蚀作用,并促进氧气扩散到零价铁的表面,使其相互作用生成·OH降解污染物。一些研究对磁场强化零价铁降解污染物进行了一系列的探讨,证明了在不同的反应体系中,磁场均可以促进零价铁的腐蚀和Fe2+的溶出,可以不同程度地提高污染物的降解速率[9-13]。综上所述,磁场可以明显改善零价铁的反应活性,且操作简单、成本低、无二次污染。因此,将磁场与其他污染物处理技术相结合具有非常广泛的应用前景。由于零价铁是铁磁性物质,在磁场中磁化后离开磁场仍能保持剩磁,具有“磁记忆性”。因此,本研究利用零价铁的磁记效应来提高其反应活性,以DCF为模型污染物,采用预磁化零价铁活化PS体系对DCF进行降解,考察了零价铁投加量、PS投加量、pH等因素对DCF降解的影响,并探讨了DCF的降解机理,为DCF实际废水的降解提供了科学依据。
-
过硫酸盐(K2S2O8,PS)、氢氧化钠(NaOH)、硫酸(H2SO4)购于天津科密欧试剂有限公司;盐酸羟胺(NH2OH·HCl)、邻菲啰啉(C12H8N2.H2O)购于天津博迪化工有限公司;双氯芬酸钠(C14H10Cl2NNaO2)购于北京百灵威科技有限公司;甲醇(CH3OH)、5,5-二甲基-1-吡咯啉-氮-氧化物(DMPO)均为色谱纯并购于上海阿拉丁生化科技股份有限公司,实验用水为超纯水。
BS124S电子天平(赛多利斯科学仪器有限公司);PHS-3C型pH计(上海仪电科学仪器股份有限公司);VI-1501可见分光光度计(天津港东科技发展有限公司);D2004W搅拌器(上海司乐仪器有限公司);DH101-3BS型电热鼓风干燥箱(天津中环实验电炉有限公司);EMX-6/1电子自旋共振波谱仪(德国Bruker公司);FL2200液相色谱仪(浙江福立分析仪器有限公司)。
-
实验采用1 000 mL的烧杯为反应器,以2片圆形钕-铁-硼永久磁铁提供磁场,用特斯拉计测定并调整所需磁场强度。将零价铁置于磁场中磁化2 min,磁化过程中以机械搅拌器搅拌使零价铁均匀悬浮于烧杯中。磁化后,将调节好pH的DCF溶液加入反应器中,然后加入一定量的PS开始计时,每隔一定的时间取样,最后加入叔丁醇终止反应,过0.22 μm滤膜后待测。同时,在其他条件相同的情况下,以非磁化零价铁作对照组。实验在常温常压下进行。
-
DCF采用液相色谱法测定,流动相为甲醇∶水=75∶25(体积比),流速为1 mL·min−1,检测波长为278 nm,柱温为40 ℃,进样量为10 μL;铁离子浓度采用邻菲啰啉分光光度法测定;pH采用玻璃电极法测定;利用电子自选共振波谱法(electron spin resonance,ESR)测定体系中自由基产生情况。
-
DCF的去除率计算方法如式(3)所示。
DCF氧化分解的反应符合动力学一级反应的特征,拟一级动力学方程如式(4)所示。
式中:R为DCF的去除率;C0为DCF的初始浓度,mg·L–1;Ct为t时间的DCF浓度,mg·L–1;k为DCF降解的一级动力学速率常数,min−1。
-
实验对比了DCF在PS、零价铁、预磁化零价铁、Fe0/PS和Pre-Fe0/PS几种体系中的去除效果,结果如图1所示。在零价铁体系中,反应60 min仅有6.8%的DCF被去除;Pre-Fe0体系在60 min内可以去除9.8%的DCF;在PS体系中,反应60 min,DCF的去除率为29.3%。这3种体系对DCF去除率较低的原因是零价铁和预磁化零价铁在溶液中无法参与反应,对DCF有较低的去除可能是由于吸附作用,预磁化零价铁比表面积有所增加[14],对DCF的吸附作用比零价铁稍强。而PS体系中尽管其氧化还原电位较高[15],但仍无法有效降解DCF。当向零价铁体系中加入PS时,DCF的降解率可在60 min达到99%,说明零价铁可以有效活化PS氧化降解DCF。值得注意的是,Pre-Fe0/PS体系中,反应5 min时,DCF的降解率可达99.7%。前期研究[14]表明,预磁化可以加速体系中Fe2+溶出,因此,预磁化零价铁可以更快的催化PS产生更多的
SO⋅−4 ,使污染物更快降解。 -
体系中PS的浓度决定了产生
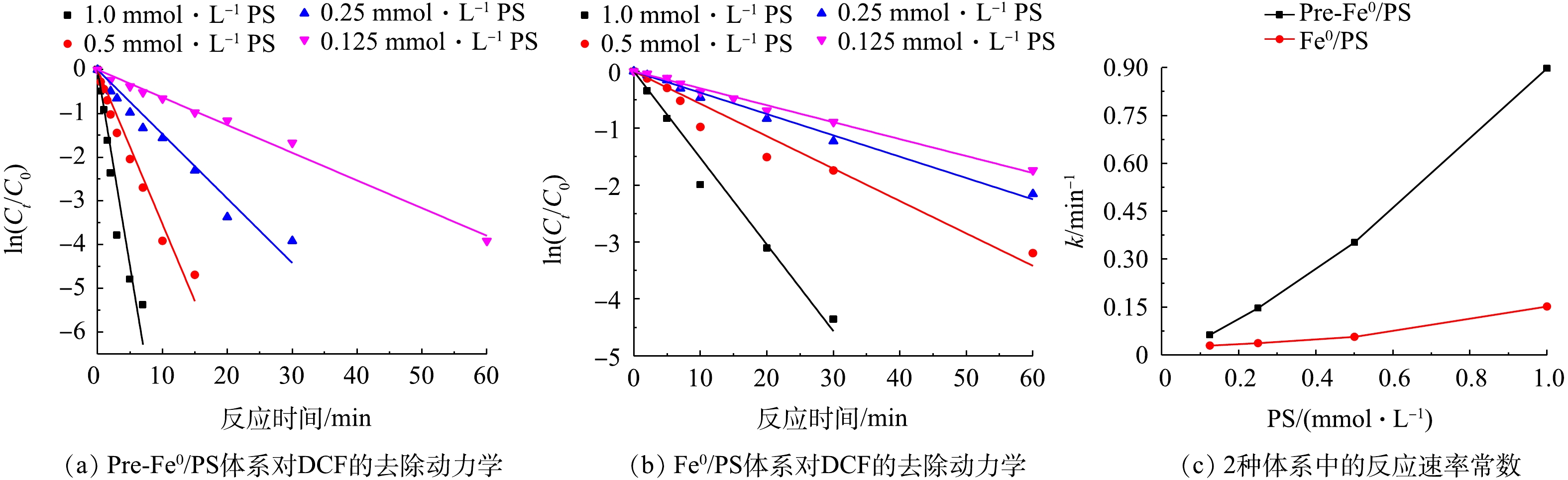
SO⋅−4 的量,进而影响DCF的降解率。为了研究不同PS投加量对DCF的降解,实验考察了PS浓度分别为0.125、0.25、0.5、1.0 mmol·L−1时,Pre-Fe0/PS和Fe0/PS体系对DCF的降解情况,结果如图2所示。由图2可知,随着PS浓度的增加,2种体系对DCF的去除率都呈升高趋势,Pre-Fe0/PS体系对DCF的去除率和去除速率均大于Fe0/PS体系。当PS投加量为0.125、0.25、0.5和1.0 mmol·L−1时,在反应30 min后,Fe0/PS体系对DCF的去除率分别为58.5%、70.2%、75.8%和96.8%;在Pre-Fe0/PS体系中,PS投加量为0.125 mmol·L−1和0.25 mmol·L−1时,反应30 min后,DCF的去除率为81.9%和98.1%;当PS的投加量为0.5 mmol·L−1时,反应进行10 min时,DCF的去除率可达98.2%,继续增加PS的量为1.0 mmol·L−1时,Pre-Fe0/PS体系对DCF的去除率在5 min达99.3%。这是因为随着PS的量增加,会有更多的SO⋅−4 产生,故2种体系中DCF的去除速率均会升高。在Pre-Fe0/PS体系中,由于零价铁的腐蚀速率加快,会促进PS的分解加速产生SO⋅−4 ,进而使DCF的降解速率更快。利用拟一级动力学反应方程对2种体系在不同PS投加量时的实验结果进行拟合。当PS投加量由0.125 mmol·L−1增加到1.0 mmol·L−1时,Fe0/PS体系降解DCF的反应速率常数由0.029 min−1增加到0.152 min−1,在实验条件范围内,任一浓度过硫酸钾条件下,Pre-Fe0/PS体系降解DCF的反应速率常数均高于Fe0/PS体系,可从0.063 min−1提高到0.898 min−1。 -
零价铁在反应过程中释放的铁离子对
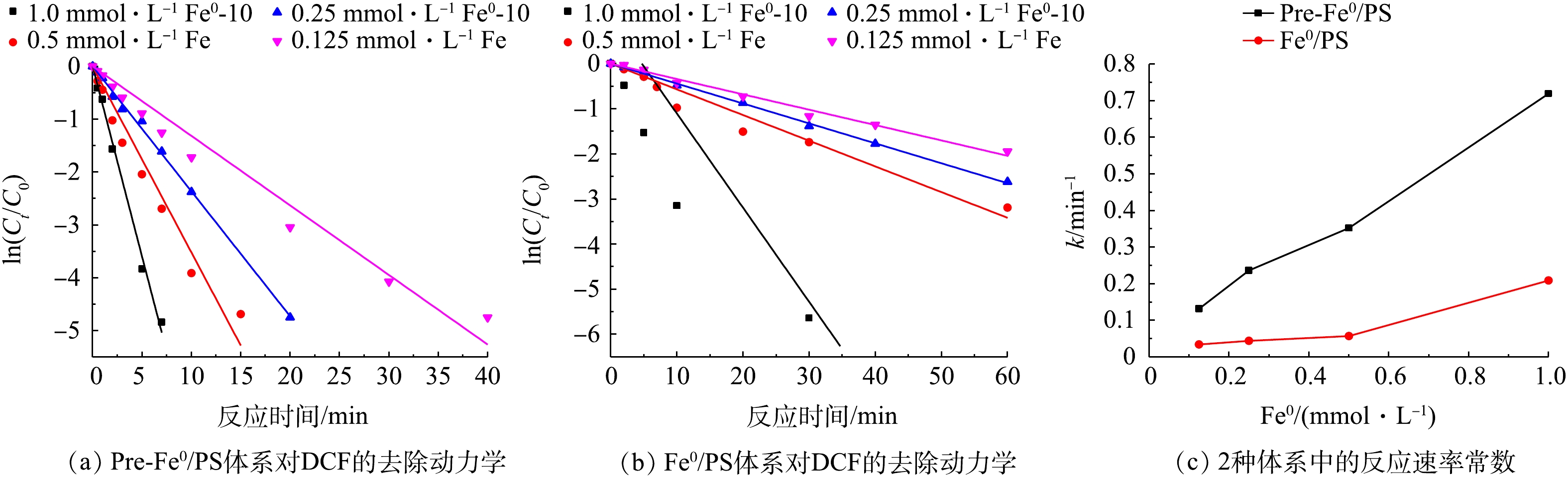
SO⋅−4 的产生起着非常重要的作用[16],因此,零价铁的浓度对污染物的降解有较大影响。为了研究不同零价铁投加量对DCF去除过程的影响,实验选取了0.125、0.25、0.5、1.0 mol·L−1零价铁投加量,在DCF初始浓度为20 mg·L−1,初始pH为7,PS投加量为0.5 mol·L−1时,同浓度的零价铁体系中DCF的反应速率如图3所示。可以看出,随着零价铁投加量的增加,2种体系对DCF的去除率有很大提升,且在相同零价铁投加量时,Pre-Fe0/PS体系的去除速率远大于化Fe0/PS体系。当零价铁投加量为0.125 mmol·L−1时,反应60 min后,Fe0/PS体系对DCF的去除率分别为80%;当零价铁投加量为1.0 mmol·L−1时,DCF在30 min的去除率可以达到99%。其原因是因为随着零价铁投加量的增加,体系中能产生更多的铁离子,进而活化PS产生SO⋅−4 ,最终加速DCF的去除。而Pre-Fe0/PS体系中零价铁投加量为0.125 mmol·L−1时,反应20 min时,对DCF的去除率为83.3%;随着零价铁投加量的增加,Pre-Fe0/PS体系对DCF的去除速率迅速增加,当零价铁投加量为0.25 mmol·L−1时,反应20 min后DCF的降解率接近100%。原因可能是因为在Pre-Fe0/PS体系中零价铁腐蚀速率较快,当零价铁为0.25 mmol·L−1时,溶出的铁离子能够在短时间内将体系中DCF完全去除,当零价铁的投加量继续增大时,对Pre-Fe0/PS体系的影响较小。2种体系的反应速率常数如图3(c)所示。2种体系对DCF降解的表观速率常数随零价铁投加量的增加而升高,在Pre-Fe0/PS体系中,反应速率常数由0.132 min−1增大到0.719 min−1;Fe0/PS体系由0.034 min−1增加到0.209 min−1。为了更好地表明零价铁投加量在Pre-Fe0/PS体系中对DCF去除率的影响,本研究增加了DCF浓度(40 mg·L−1),结果如图4所示。由图4可知,当零价铁浓度由0.125 mmol·L−1增加到0.5 mmol·L−1时,DCF的去除率随着零价铁投加量的增加而升高;当零价铁浓度继续增加,DCF去除率基本不变,这是因为当零价铁投加量过大时,体系中产生过多的铁离子会与
SO⋅−4 发生反应[17]。 -
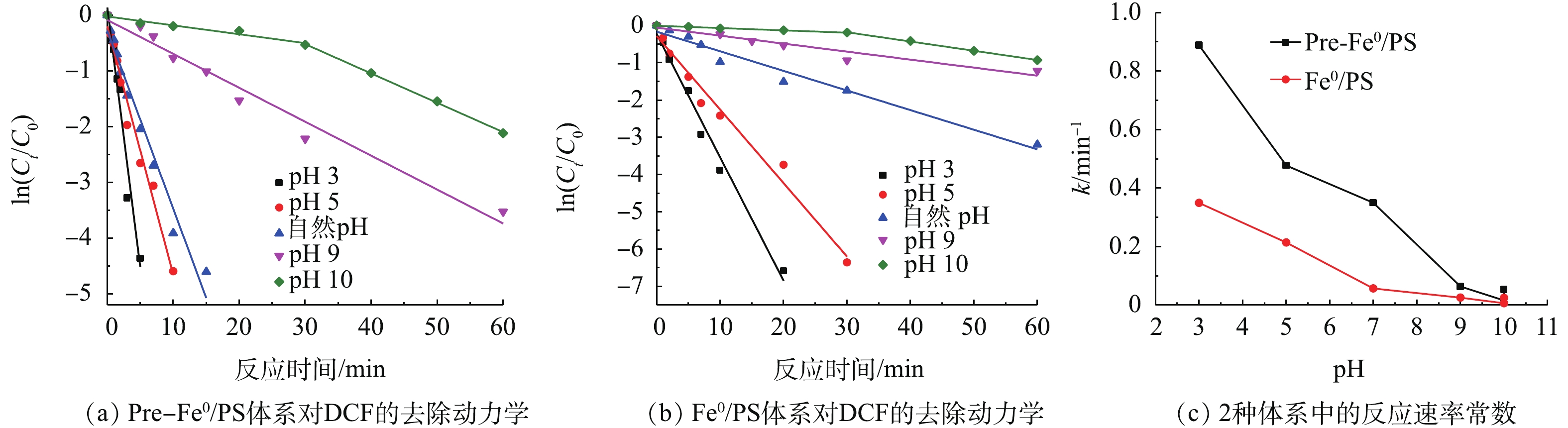
实验考察了当DCF初始浓度为20 mg·L−1,零价铁投加量为0.5 mmol·L−1,PS投加量为0.5 mmol·L−1,初始pH分别为3,5,7,9和10时DCF的降解效果,结果如图5所示。由图5可知,在Fe0/PS体系中,在初始pH为3~10时,DCF的去除率随初始pH值的升高而下降,特别是初始pH为10时下降尤为明显。当初始pH为3,反应20 min时,DCF的去除率为99%;初始pH为10时,DCF在60 min的去除率为50%左右。在Pre-Fe0/PS体系中,当初始pH为3、反应15 min时,对DCF的去除率可达100%;初始pH为10时,DCF在60 min时的降解率可达90.4%。
与Fe0/PS体系相比,在相同的pH下,Pre-Fe0/PS体系中DCF的去除率均有较大的提升,特别是在pH较低时,2种体系中DCF的降解较快。分析其原因可能是:零价铁在储存和运输过程中被氧化形成一层钝化膜覆盖其表面,当反应体系pH较低时,零价铁表面的氧化膜更容易被溶解[18]。因此,零价铁在体系pH较低时的腐蚀速率和反应活性较高,在反应过程中会产生更多的氢参与加成反应[19]。由图5(b)可知,随着体系初始pH的升高,Fe0/PS体系对DCF的去除率急剧下降;而Pre-Fe0/PS体系对DCF的去除率仍能保持在较高的水平,当pH为10时,对DCF的降解率在60 min时仍可达到90.4%,是Fe0/PS体系的2倍左右。
由图5(c)可知,2种体系的反应速率常数随初始pH的升高而迅速减小,Pre-Fe0/PS体系的反应速率常数是Fe0/PS体系的2.1~6.2倍,Pre-Fe0/PS体系对反应速率常数提升的倍数并没有因初始pH的升高而下降,其原因为当体系的初始pH较高时,零价铁在参与反应时会形成铁氧化物或铁氢氧化物钝化膜覆盖其表面阻止反应的进行。然而,目前有研究显示,预磁化可以加速零价铁的腐蚀,阻止钝化膜的形成[20],从而提高DCF的降解。因此,Pre-Fe0可以在一定程度上使该体系pH适用范围增大,减少其在应用过程中pH调节剂的使用,降低污染物的降解成本。
-
1)铁离子的产生。在Fe0/PS体系中,零价铁可以与体系中的氧气、水和H+反应生成Fe2+,活化PS生成
SO⋅−4 ,而本身被氧化为Fe3+,为了研究体系中零价铁、Fe2+和Fe3+的作用,实验测定了在近中性条件下,体系中亚铁离子和铁离子的变化。2种体系的反应过程中都没有测出Fe2+(测定方法的最低检测限为0.03 mg·L−1),这一现象与XIONG等[21]的研究结果吻合,即Fe2+的溶出是反应活化PS的限速步骤。实验研究了当DCF初始浓度为20 mg·L−1,零价铁投加量为0.5 mmol·L−1,PS投加量为0.5 mmol·L−1,自然初始pH下,体系中铁离子浓度和pH变化情况。图6为2种反应体系中总铁离子浓度的变化。由图6可见,Pre-Fe0/PS体系在反应过程中铁离子浓度高于Fe0/PS体系,说明在Pre-Fe0/PS体系中铁离子的快速溶出导致了DCF的降解效率的升高。此外,我们还测定了反应过程中体系pH的变化,随着反应的进行,2种体系的pH都呈降低的趋势。其原因可能是在反应过程中生成的Fe3+会发生水解作用产生H+(式(5)),另外,在部分
SO⋅−4 转化为·OH(式(6))的过程中也会产生H+,从而使体系pH下降。由于Pre-Fe0/PS体系中能产生更多的的Fe3+和SO⋅−4 ,因此,Pre-Fe0/PS体系中的pH下降较Fe0/PS体系更为明显。2)自由基的产生。自由基是降解污染物重要的活性物质,其在体系中的产生量决定了污染物的降解率。电子自旋共振波谱法(ESR)是测定短寿命自由基非常有效的手段,其信号可以半定量地反映自由基的产生量。由于自由基的寿命非常短暂,在水溶液中存在的时间小于10−4 s[22],实验过程中以5,5-二甲基-1-吡咯啉-氮-氧化物(DMPO)为捕获剂,生成寿命较长的自旋加合物进行测定。由图7可知,2种体系中均出现了DMPO-
SO−4 和DMPO-OH加合物的典型特征峰[23]。对比图7(a)和图7(b)可以看出,当反应条件相同时,Pre-Fe0/PS体系在任一取样时间点的加合物对应的峰高均大于Fe0/PS体系,即产生的SO⋅−4 和·OH量比Fe0/PS体系中多。由图7还可以看出,Pre-Fe0/PS体系在2 min时产生的SO⋅−4 和·OH比Fe0/PS体系在5 min时产生的量还要多,而且能在相当长的时间内保持较高的浓度水平,当取样时间为5 min时,DMPO-SO−4 和DMPO-OH加合物的信号峰仍然很强。然而,Fe0/PS体系中SO⋅−4 和·OH产生速度相对较慢,DMPO-SO−4 和DMPO-OH加合物的信号峰衰减较快。这一结果解释了Pre-Fe0/PS体系对DCF的去除率大于Fe0/PS体系的原因。 -
1)预磁化后的零价铁能够显著提升其对PS活化作用,进而提高其降解DCF的能力。
2) PS浓度、零价铁投加量及初始pH对Pre-Fe0/PS和Fe0/PS体系降解DCF均有较大影响。其中,在零价铁投加量为0.125~1.0 mmol·L−1、PS浓度为0.125~1.0 mmol·L−1条件中,反应速率常数均呈升高趋势,而DCF可在Fe0为 0.5 mmol·L−1,PS为0.5 mmol·L−1条件下几乎被完全去除;2种体系的反应速率常数随初始pH的升高而迅速减小,Pre-Fe0/PS体系的反应速率常数是Fe0/PS体系的2.1~6.2倍,在pH为6~8的条件下有利于反应进行。
3) Pre-Fe0/PS体系中铁离子溶出和pH下降趋势均比Fe0/PS体系快。
4) ESR结果表明,2种体系中都会产生
SO⋅−4 和·OH,且其对污染物的降解起主要作用,预磁化可以加速SO⋅−4 和·OH的产生,并能使其在较长的时间保持较高的浓度水平。
预磁化零价铁活化过硫酸盐体系降解双氯芬酸钠
Degradation of diclofenac sodium by premagnetized zero-valent iron-catalyzed persulfate
-
摘要: 双氯芬酸钠(diclofenac sodium,DCF)是一种常用的消炎止痛药,已在地下水、地表水和饮用水中被广泛检出,成为一种新型微量污染物,具有潜在危害,基于此,采用预磁化零价铁/过硫酸盐(Pre-Fe0/PS)和零价铁/过硫酸盐(Fe0/PS)2种体系对DCF进行降解。考察了过硫酸盐(PS)投加量、零价铁投加量、初始pH对2种体系降解DCF的影响,探究了2种体系中铁离子的产生情况和pH的变化,并利用ESR技术检测了体系中生成的自由基。结果表明,与Fe0/PS体系相比,在不同PS量(0.125~1.0 mmol·L−1)、Fe0量(0.125~1.0 mmol·L−1)和初始pH 3.0~10.0下,Pre-Fe0/PS体系对DCF的降解速率常数提高了2.1~6.2倍;Pre-Fe0/PS体系中会产生更多的铁离子,且在反应过程中pH下降更快;Pre-Fe0/PS体系比Fe0/PS体系产生更多的
SO⋅−4 和·OH,且能在较长的时间保持较高的浓度。Pre-Fe0/PS体系降解DCF可以适用更宽的pH范围,是DCF废水处理的有效途径。Abstract: Diclofenac sodium (DCF), a commonly used anti-inflammatory painkiller, has been widely detected in groundwater, surface water and drinking water. As an emerging pollutant, it can cause potential hazards. Based on this, the pre-magnetized Fe0 (pre-Fe0/PS) and Fe0/PS systems were used to degrade DCF. The effects of influencing operational parameters, including initial PS dosage, Fe0 dosage and pH, on the DCF degradation were investigated. The iron ions yield and pH changes in the both of systems were determined, and the generation of free radicals was also studied by using ESR. The results showed that pre-Fe0/PS process had 2.1~6.2 folds higher rate constant than Fe0/PS process for DCF degradation at different PS dosages (0.125~1.0 mmol·L−1), Fe0 dosages (0.125~1.0 mmol·L−1) and initial pH (3.0~10.0). More iron ions were generated and faster drop in pH occurred in pre-Fe0/PS process. EPR confirmed that stronger signals of DMPO-OH and DMPO-SO−4 adduct illustrated more and fasterSO⋅−4 and ·OH radicals produced in pre-Fe0/PS system than those of Pre-Fe0/PS system, and could keep relative high content for a long duration. The pre-Fe0/PS process presents wide pH range to degrade DCF, and it is a promising approach to remove DCF.-
Key words:
- pre-magnetization Fe0 /
- persulfate /
- diclofenac sodium /
- degradation mechanism
-
聚乙烯醇(PVA)通过聚醋酸乙烯酯水解制得,是1种具有良好性能且可生物降解的水溶性聚合物[1]。PVA因其良好的水溶性,粘附性,耐磨性和强韧性等优良的性能,被广泛用于农用地膜,纺织上浆和食品包装等行业[2]。PVA具有较大的表面活性,因此,大量降解缓慢的PVA广泛存在于自然界中,造成水体和泥土富氧、生态环境危害和生物链的破坏[1, 3]。许多研究报道了处理PVA的物理和化学的方法,如吸附法[4]、化学凝固法[5]、超声波降解法[6]、膜过滤法[7]和催化氧化法[8]。但这些方法成本较高且可能造成二次污染,而生物降解法更加高效和经济,因此,筛选高效降解菌和优化降解方式对于减少PVA污染而言具有重要意义[9]。
自1973年首株能够降解PVA的假单胞菌(Pseudomonas O-3)[10]被分离出以来,大量关于PVA降解菌的研究被报道。从染料厂废水中分离出的鞘氨醇盒菌(Sphingopyxis sp. PVA3)在6 d的培养中对初始浓度为1 g·L−1 PVA的降解率达到90%[11]。粪产碱菌(Alcaligenes faecalis KK314)降解PVA的机理被详细报道,PVA上的羟基首先被氧化为β-羟基酮,然后被进一步水解为甲基酮类化合物和羧酸类化合物[12]。放线菌委内瑞拉链霉菌(Streptomyces venenzuelae GY1)能产生降解PVA的诱导型胞外酶[13]。地霉 (Geotrichum WF9101)对低分子质量的PVA具有降解的活性[14]。青霉菌(Penicillium sp.WSH02-21)能在12 d内将PVA降解完全,且能够分泌过氧化氢酶,防止氧化PVA过程中产生的过氧化氢对细胞产生损害[15]。共生降解也被广泛研究,其主要分为2种模式。首先是MORI等[16]报道的2种菌株共代谢,2株菌都不分泌生长因子。另外1种模式由SAKAZAWA等[17]提出。其中1株菌分泌生长因子,其他菌利用这种生长因子进行聚合物降解,例如假单胞菌(Pseudomonas sp.VM15C)和恶臭假单胞菌(Pseudomonas putida VM15A)[18],新鞘氨醇菌(Novosphingobium sp.) 和黄色杆菌(Xanthobacter flavus)等[19]。由于PVA降解菌广泛存在于自然界[20],因此,进一步筛选高效菌并研究降解特性就具有独特意义。本研究通过对聚乙烯醇材料堆肥,研究了材料表面的生物多样性变化,并且从经历了3年降解的PVA材料表面筛选到了能独立降解PVA的苏云金芽孢杆菌(Bacillus thuringiensis sp.),命名为DG01。对DG01进行了摇瓶降解实验和呼吸降解实验,并对其降解特性进行了研究。
1. 材料与方法
1.1 试剂
聚乙烯醇材料受赠于华南理工大学材料科学与工程学院;DNA提取试剂盒购于美国OMEGA公司;PVA2488(平均聚合度2 400±50,醇解度86.0%~90.0%)购于台湾长春化工有限公司;
1.2 堆肥试验
将材料切割成若干长方块(长3.5 cm,宽 2.0 cm,高 1.0 cm)。将长方块置于超净台中紫外灭菌45 min。用商业型堆肥对材料进行覆盖,覆盖深度为10 cm。将被肥料覆盖好的材料置于常温下避光保藏。在降解过程中,适量加水与翻动土壤,保证堆肥的含氧量和湿度。在降解1、2、3年时分别取样。每次取3个样,对材料表面细菌的多样性进行分析。
1.3 优势菌群分析
1.3.1 总DNA提取
在聚乙烯醇材料降解过程中,用DNA提取试剂盒提取材料表面微生物的DNA。将目标时间段的材料置于装有50 mL无菌生理盐水的250 mL三角烧瓶中,适当振荡后取出材料。按照试剂盒步骤提取DNA,提取液需−20 ℃保存。使用1%的琼脂糖进行电泳检查DNA样品是否合格。
1.3.2 PCR扩增及上机测序
选取通用引物(F338(5′-ACTCCTACGGGAGGCAGCA-3′)和R806(5′-GGACTACHVGGGTWTCTAAT-3′)),扩增细菌16S DNA片段的V3~V4区。PCR总体系为25 µL,包括25 µL premix,上下游引物各为0.5 µL,模板DNA 25 µL,ddH2O 23 µL。反应条件为,94 ℃预变性5分钟;94 ℃变性30 s;58 ℃退火45 s;72 ℃延伸60 s,循环25次;最后72 ℃延伸10 min。样品储存的温度为4 ℃。用1.2%凝胶琼脂糖做凝胶电泳检测,最后由广州基迪奥生物科技有限公司对其测序。
1.3.3 数据处理分析
数据预处理:基于序列末端相重合的部分,用FLASH将序列拼接起来。采用Usearch和Uchime除去非扩增区域序列和嵌合体。使用Prinseq 软件控制过滤去除编码序列以及部分低质量序列。读段与接头序列对比超过15 bp长度则认为接头污染。读段与接头序列超过9 bp长度则认为质量较低。
OTUs(operating taxonomic units)分类及Alpha多样性分析:使用Mothur平台数据对tag进行去冗余处理,挑选出Unique tag。在相似度97%的水平上对这些有效数据进行OTUs聚类分析,物种分析及稀释曲线分析。基于OTUs结果,用QIIME计算出多样性指数Chao、Ace和丰富度指数Shannon、Simpson。采用Bayesian算法对代表OTUs进行分类并选取最佳分类水平,比较微生物多样性的变化。
1.4 菌种
选取经历了3年降解的材料,从材料表面筛选出了可降解PVA的菌株DG01。经过16S鉴定,株菌与Bacillus thuringiensis BM-BT15426(CP020723.1)相似度达到100%,命名为DG01。
1.5 培养基
LB培养基(每升含量):蛋白胨 10.0 g,NaCl 5.0 g,葡萄糖 1.0 g,酵母膏粉 5.0 g,pH 7.0±0.2。
营养琼脂培养基(每升含量):蛋白胨 10.0 g,牛肉浸出粉 3.0 g,氯化钠 14.0 g,琼脂 14.0 g,pH 7.0±0.2。
PVA无机盐培养基(每升含量):PVA2488 3.0 g,酵母提取粉 1 g,NH4NO3 0.10 g,NaCl 0.02 g,MgSO4 0.05 g,CaCl2 0.05 g,FeSO4·7H2O 0.02 g,K2HPO4 1.60 g,KH2PO4 0.20 g,pH 7.0~7.2。
无机盐培养基(每升含量):酵母提取粉 1.0 g,NH4NO3 0.10 g,NaCl 0.02 g,MgSO4 0.05 g,CaCl2 0.05 g,FeSO4·7H2O 0.02 g,K2HPO4 1.60 g,KH2PO4 0.20 g,pH 7.0~7.2。
1.6 培养方法
从斜面上挑取适量菌株至100 mL(37 ℃,150 r·min−1)LB活化培养基中活化。活化16 h后,按无机盐培养基体积的5%接种菌液。
1.7 Bacillus thuringiensis DG01降解PVA2488实验
1.7.1 摇瓶降解实验
按培养方法,将接种了DG01的PVA无机盐培养基置于37 ℃,150 r·min−1 摇床中培养。每组3个平行,每隔48 h取样,测定培养基中PVA的浓度、pH、活菌浓度。
活菌浓度的测定:根据GB 4789.2-2016对摇瓶降解实验的菌浓度进行测定。取1 mL的菌液,稀释至原菌液浓度的10−4、10−5、10−6倍。各取50 μl,涂布于营养琼脂培养基平板。将涂布好的平板置于37 ℃培养8 h左右,记录每个平板的菌落数。活菌浓度及其平均值按式(1)计算。
Ni=∑Cni×20 N=∑Nii (1) 式中:N为活菌浓度,CFU·mL−1;Ni为第i个样品的活菌浓度,CFU·mL−1;∑C为平板上的菌落之和;ni为稀释的倍数。
PVA浓度的测定:采用改良的finely法[21],取上清液通过96孔板测量PVA浓度和PVA降解率。取摇瓶培养后的发酵液,8 000 g离心10 min后,保留上清液。将碘化钾-碘溶液60 μl(碘1.27 g·L−1、碘化钾2.5 g·L−1),上清液40 μl和100 μl硼酸(浓度4 g·L−1)依次加入96孔板。在避光,4 ℃下反应15 min。用酶标仪检测波长为510 nm处的吸光度,通过标准曲线测定PVA的浓度,且以显色后的吸光度计算降解率。
降解率按照式(2)进行计算。
η=A0−A1A0−A×100% (2) 式中:η为PVA的降解率,%;A0为标准对照,降解前培养基的吸光度;A1为降解后培养基的吸光度;A为空白,无机盐培养基的吸光度。
1.7.2 以二氧化碳为指标的降解率的测定
根据EN ISO 14852-2004的方法搭建测定装置。在37 ℃下测试15 d。装置分为3部分,第1个部分吸收流入空气中的二氧化碳,由3个500 mL三角瓶组成,前2个瓶装200 mL的氢氧化钠溶液(0.05 mol·L−1,吸收二氧化碳),第3个瓶中装氢氧化钡溶液(0.012 5 mol·L−1),以检验二氧化碳是否除净。第2个部分由DG01利用PVA产生二氧化碳,由1个500 mL三角瓶组成,瓶子装有200 mL接种DG01后的PVA无机盐培养基。第3部分吸收由菌株产生的二氧化碳,由3个500 mL的三角烧瓶,分别装有200 mL 经过矫正的氢氧化钡溶液。气体流速控制在80 mL·min−1左右。用标准氢氧化钠(0.05 mol·L−1)矫正盐酸,然后用矫正后的盐酸滴定第3部分的氢氧化钡溶液来计算二氧化碳排放量。
以二氧化碳排放量为指标的降解率按照式(3)计算,
η1=(2C0V0−C1V1)M2TCO2×100% (3) 式中:η1为降解率,%;C0为装置第3部分氢氧化钡的浓度,mol·L−1;V0为装置中第3部分所含氢氧化钡的总体积,mL;C1为盐酸的浓度,mol·L−1;V1为滴定时消耗盐酸的总体积,mL;M为CO2的相对分子质量,g·mol−1;TCO2为理论上排放的总二氧化碳,在本装置中取值为1.198 8 g。
1.7.3 DG01降解PVA2488最适条件研究
最适降解温度:按培养方法将PVA无机盐培养基分别置于不同温度下(33、35、37、39、41、43、45 ℃),培养48 h,每组3个平行,测定降解前后的浓度,以此计算出PVA的降解率。考察最优降解温度。
最适酵母粉浓度:按培养方法将DG01接种于酵母粉浓度不同(0.2、0.5、0.8、1.1、1.4、1.7、2.0 g·L−1)的无机盐培养基中,培养48 h,每组3个平行,测定降解前后PVA浓度,以此来计算PVA的降解率。考察最优酵母粉浓度。
最适初始pH:配制初始pH不同的无机盐培养基(5.0、5.5、6.0、6.5、7.0、7.5、8.0)。按培养方法将DG01接种到配好的培养基中,培养48 h,每组3个平行,测定降解前后浓度,以此计算出PVA的降解率。考察最优初始pH。
2. 结果与讨论
2.1 聚乙烯醇材料表面的菌群分析
群落多样性分析:基于V3+V4区的测序,经过处理,最终从降解时间为1、2、3年的材料表面的样品中分别获得427,556和604个OTUs。降解1、2、3年样品的稀释曲线分别在600,550和400左右趋于平缓,说明测序覆盖率良好,基本覆盖样品的全物种。由表1可以看出,降解2年的Chao1指数达到最大值,Simpson指数微量增高。能利用聚乙烯醇材料生长的微生物渐渐汇集在材料表面,不能利用材料的微生物逐渐减少,导致多样性减少。堆肥带来大量的有机质以及营养物质,对汇集在材料表面的微生物有促进作用,使细菌的丰富度增加[22]。降解3年的Chao1指数大幅度下降,Simpson大幅度增高。材料被长期覆盖后,其表面的结构,营养成分以及通氧量发生了改变,限制了菌株的生长,导致物种多样性和丰富度都下降[23]。
表 1 不同降解时间的微生物群落丰富度和多样性指数Table 1. Community richness and diversity indices of microbes at different degradation time降解时间/年 Chao 1 Simpson OTUs/个 1 868 0.265 604 2 873 0.276 556 3 580 0.583 427 | Show Table DownLoad:
CSV
DownLoad:
CSV
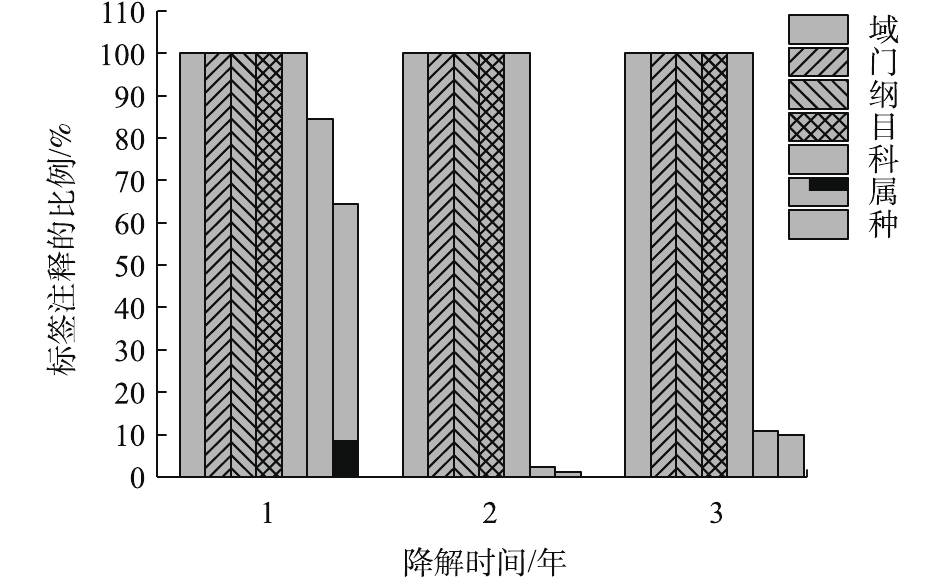
菌群结构变化及优势菌株分析:通过尽可能降低分类水平和提高tag的注释率,可获得最佳分类水平。分别在界、门、纲、目、科、属、种分类水平上统计tag序列数。结果如图1所示。在界,门,纲,目,科的分类水平上均有100%的tag序列能被注释。在分类水平属上,降解3年的样品有接近80%的tag能被注释,降解1年和2年样品的tag只有10%能被注释。因此,科为最佳分类水平。
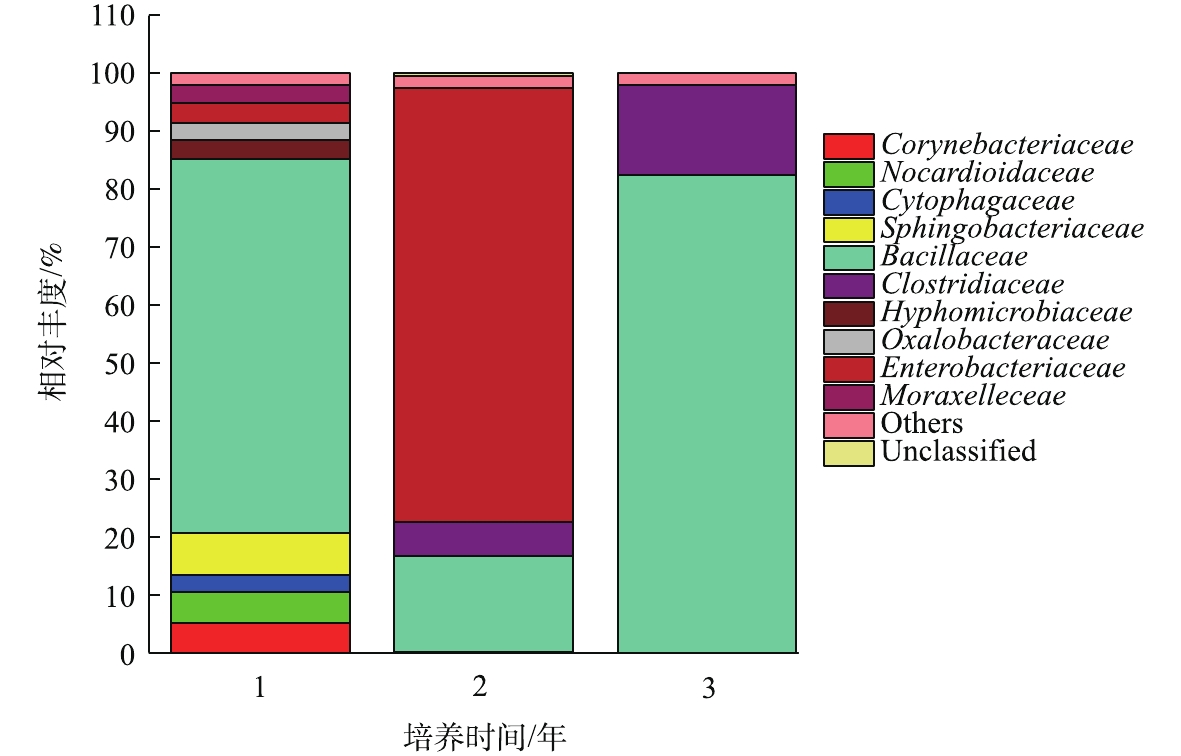
如图2所示,聚乙烯醇材料在堆肥中降解时,汇集在材料表面的菌群主要包括10个科(每个科的丰度在2%以上),分别是状杆菌科(Corynebacteriaceae)、诺卡氏菌科(Nocardioidaceae)、鞘氨醇杆菌科(Sphingobacteriaceae)、杆菌科(Bacillaceae)、细胞噬菌体科(Cytophagaceae)、梭状芽孢杆菌科(Clostridiaceae)、生丝微菌科(Hyphomicrobiaceae)、草杆菌科(Oxalobacteraceae)、肠杆菌科(Enterobacteriaceae)、摩拉克菌科(Moraxelleceae)。其中在3个时期都占主要成分是杆菌科(Bacillaceae),在降解的第1年和第3年都占绝大多数,相对丰度分别达到82.47%及64.62%。肠杆菌科(Enterobacteriaceae)主要在中前期被检测到,而在第3年几乎检测不到。梭状芽孢杆菌科(Clostridiaceae)随着时间增长其相对丰度也从5%缓慢增加到16%左右。其他7类菌株在前期丰度相对较少,在中后期几乎检测不出。在材料降解的第1年,第2年,第3年,Bacillaceae的丰度都明显较高,故认为其是优势菌。
2.2 Bcaillus thuringiensis DG01降解特性实验
从经历了3年降解的聚乙烯材料表面筛出了能降解聚乙烯醇的菌株。经16S rRNA分子生物学鉴定,该菌株属于在降解过程中占优势的杆菌科(Bacillaceae),命名为Bacillus thuringiensis DG01。能降解PVA的Bacillus thuringiensis鲜有报道。
2.2.1 无机盐培养基下DG01形态SEM观察
对在PVA无机盐培养基里培养8 d的菌株进行电镜观察,结果如图3所示,DG01呈椭圆杆状长度和宽度各约为1.30 μm和0.64 μm。不同于在活化培养基的大小约1.4 μm×3.2 μm(图片没有给出),微生物形态变化可能与吸收PVA有关。HU[24]发现Spingopyxis sp.113P3在吸收PVA时,膜结构发生改变,以利于微生物吸收培养基中PVA。
 图 3 菌株Bacillus thuringiensis DG01的扫描电镜图Figure 3. SEM image of strain Bacillus thuringiensis DG01
图 3 菌株Bacillus thuringiensis DG01的扫描电镜图Figure 3. SEM image of strain Bacillus thuringiensis DG012.2.1 DG01降解PVA2488实验
摇瓶降解实验。Bacillus thuringiensis DG01降解PVA2488的过程如图4所示,活菌浓度在48 h内进入稳定期并达到最大值,在随后的6 d内维持不变。平衡期DG01的pH维持在8.20左右,降解环境维持稳定。对培养基中PVA浓度的下降趋势进行了动力学分析,结果表明PVA浓度下降趋势与一级动力学模型吻合良好,降解速率常数k和相关系数R2分别为0.195 d−1和0.984[25]。
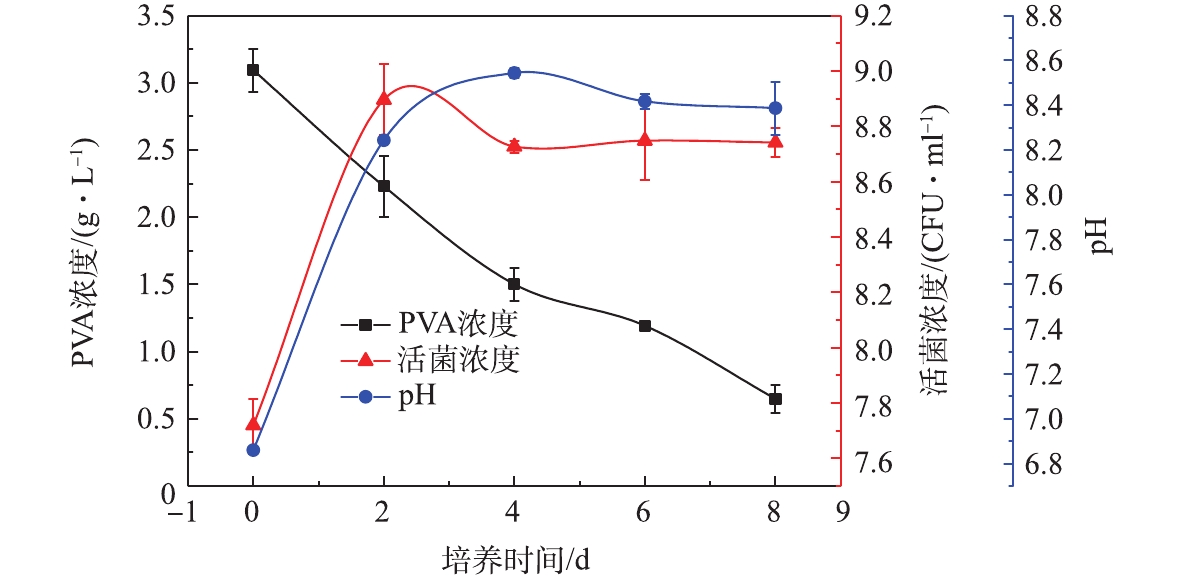 图 4 Bacillus thuringiensis DG01生长过程中PVA2488浓度、活菌浓度、pH的变化Figure 4. Changes in PVA2488 concentration, living bacteria concentration,pH during Bacillus thuringiensis DG01growth
图 4 Bacillus thuringiensis DG01生长过程中PVA2488浓度、活菌浓度、pH的变化Figure 4. Changes in PVA2488 concentration, living bacteria concentration,pH during Bacillus thuringiensis DG01growth图5表明,DG01在前2 d降解率比较高,随后每天的降解率急剧下降。第1 天和第2 天的降解率之和达到18.04%,在后13 d内每天的降解率均较低。降解趋势符合一级动力学模型,相关系数R2为0.983 5。由图4和图5可知,以PVA浓度为指标表示的降解率与以二氧化碳排放量为指标表示的降解率不相符,后者的降解滞后于前者,说明细胞吸收的PVA并没有立即全部转化为CO2,降解时产生了中间体。目前普遍认为PVA的降解主要分为2步。首先,PVA上的羟基被氧化,形成单酮和β–二羰基结构;然后,这些单酮和β–二羰基被进一步水解,PVA分子链断开,生成羧酸和甲基酮类物质。通过重复以上2步反应,PVA分子链不断变短,形成短链羧酸类物质,这些物质最终进入微生物的中心代谢途径,成为微生物的能源物质和细胞生长可利用的碳源[3,26]。通过对比降解前后PVA2488的红外光谱图,发现有新的羧基产生。凝胶排阻色谱图表明降解前后PVA2488的分子质量有所减小。故认为在PVA2488在降解过程中,可能产生了分子链相对较短的羧酸类物质。
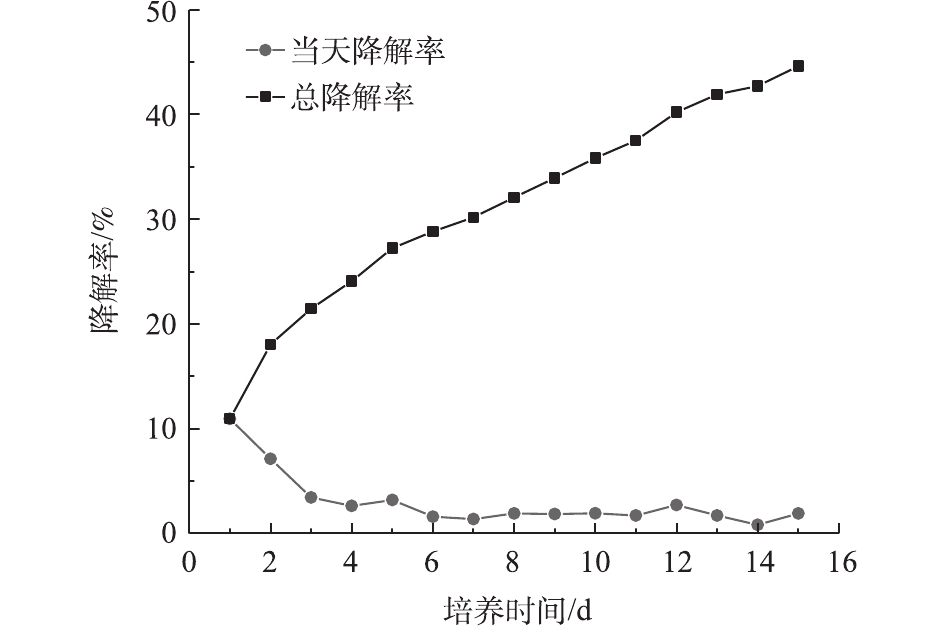 图 5 以二氧化碳排放量为指标的PVA降解率随时间的变化Figure 5. The relationship between PVA degradation rate and culture time with CO2 emission as indicator
图 5 以二氧化碳排放量为指标的PVA降解率随时间的变化Figure 5. The relationship between PVA degradation rate and culture time with CO2 emission as indicator2.2.2 DG01降解PVA2488最适条件研究
分别对温度、初始pH和初始酵母粉浓度进行了单因素优化,结果如图6所示。当培养温度为41 ℃ 时,PVA2488降解率最高,可达到40.65%;当初始酵母粉浓度为1.4 g·L−1时,PVA2488降解率最高,可达到34.36%;当初始pH为7.0时,PVA2488降解率最高,可达到25.14%。因此,选择最优培养温度为41 ℃,最优初始酵母粉浓度为1.4 g·L−1,最优初始pH为7.0。
 图 6 Bacillus thuringiensis DG01菌株降解PVA2488的条件优化Figure 6. Optimization of PVA2488 degradation conditions with Bacillus thuringiensis DG01
图 6 Bacillus thuringiensis DG01菌株降解PVA2488的条件优化Figure 6. Optimization of PVA2488 degradation conditions with Bacillus thuringiensis DG01按培养方法,在酵母粉浓度为1.4 g·L−1,初始pH为7.0的PVA培养基中接种DG01。在温度为41 ℃的条件下培养48 h,测量3个平行样降解前后培养基中PVA浓度,计算其降解率。结果表明在最适条件下,DG01对初始浓度为3 g·L−1 PVA的降解率可达到45.21%,降解了1.356 g·L−1的PVA,降解率提高了2.10倍。优化后PVA降解速率为0.678 g·(L·d)−1,高于Stenotrophomonas rhizophila QL-P4[26]和Sphingopyxis sp.PVA3[11],其降解速率在PVA初始浓度为1 g·L−1时分别为0.350 g·(L·d)−1和0.125 g·(L·d)−1。也高于Pseudomonas sp.strain VM15C[18]和Penicillium sp.WSH02-21[15], 其降解速率在PVA初始浓度为5 g·L−1时分别为0.500 g·(L·d)−1和0.417 g·(L·d)−1。由此可见,DG01有相对较高的降解速率,有潜在的应用前景。
3. 结 论
1) 通过高通量测序,分析了聚乙烯醇复合材料降解过程中材料表面种群结构的变化,并找到了优势种群Bacillaceae,为堆肥降解PVA的研究提供了基础。在接下来的研究中将对堆肥中降解PVA菌群的功能进行分析。
2) 从降解了3年的材料中筛选出属于优势种群Bacillaceae的菌株Bacillus thuringiensis DG01。分别以培养基中PVA浓度和CO2排放量为指标研究降解趋势。2种指标下的降解趋势都符合一次降解动力学模型,且培养基中被微生物吸收的PVA没有完全转化为CO2。进一步研究发现,PVA在降解过程中产生了羧酸类物质,为研究该菌株降解PVA的机理做出了指导。在接下来的研究中将进一步详细探讨该菌株降解PVA的过程。
3) 对DG01降解PVA2488的条件进行了优化,结果表明其最适温度,初始pH和酵母粉浓度分别为41 ℃,7.0和1.4 g·L−1。在最佳条件下,3 g·L−1 的PVA在24 h的降解率为45.21%,提高了2.10倍。为该菌株降解PVA的实际应用打下了坚实基础。
-
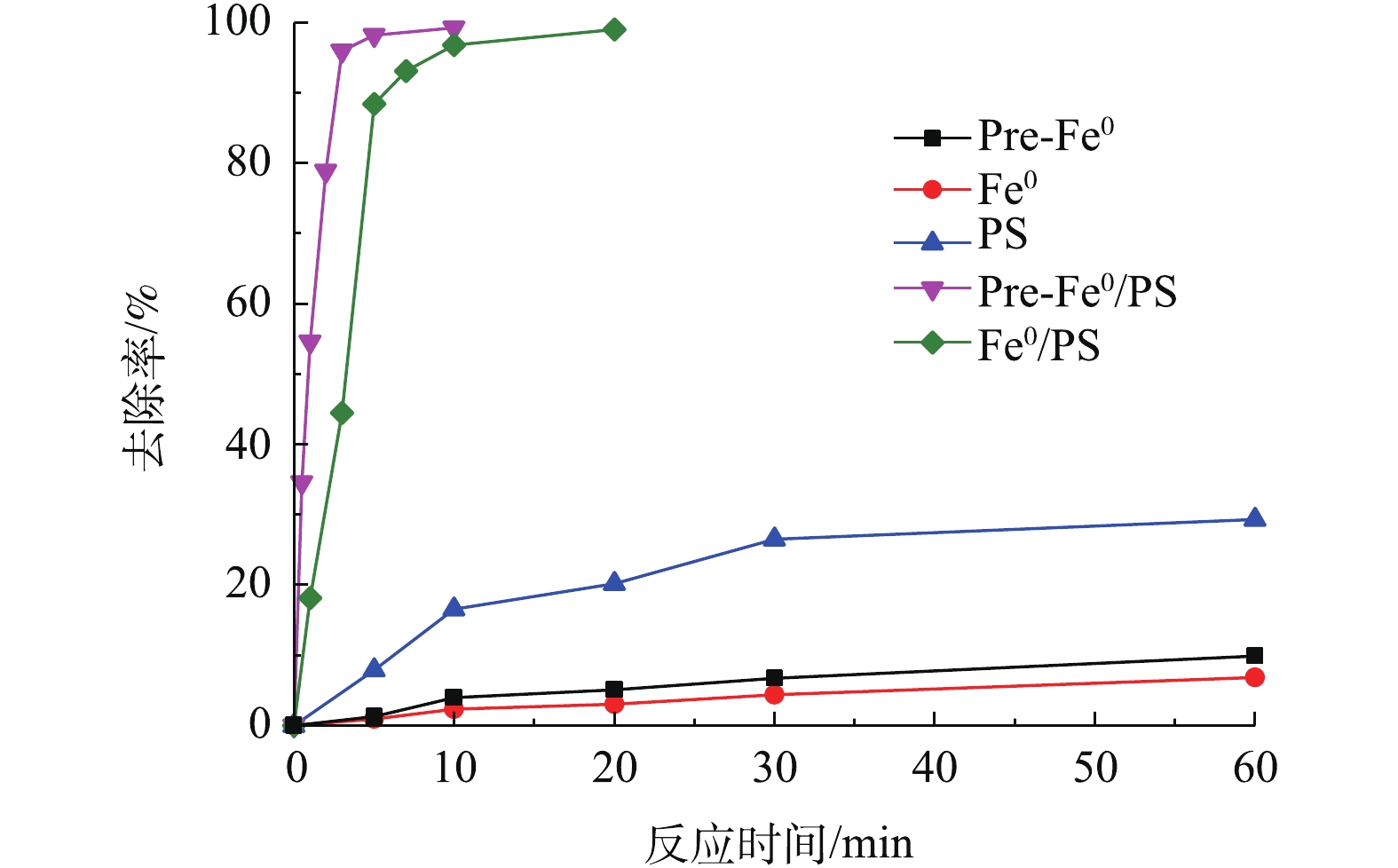
图 1 不同体系对DCF去除率的影响
Figure 1. Removal efficiency of diclofenac sodium by different system
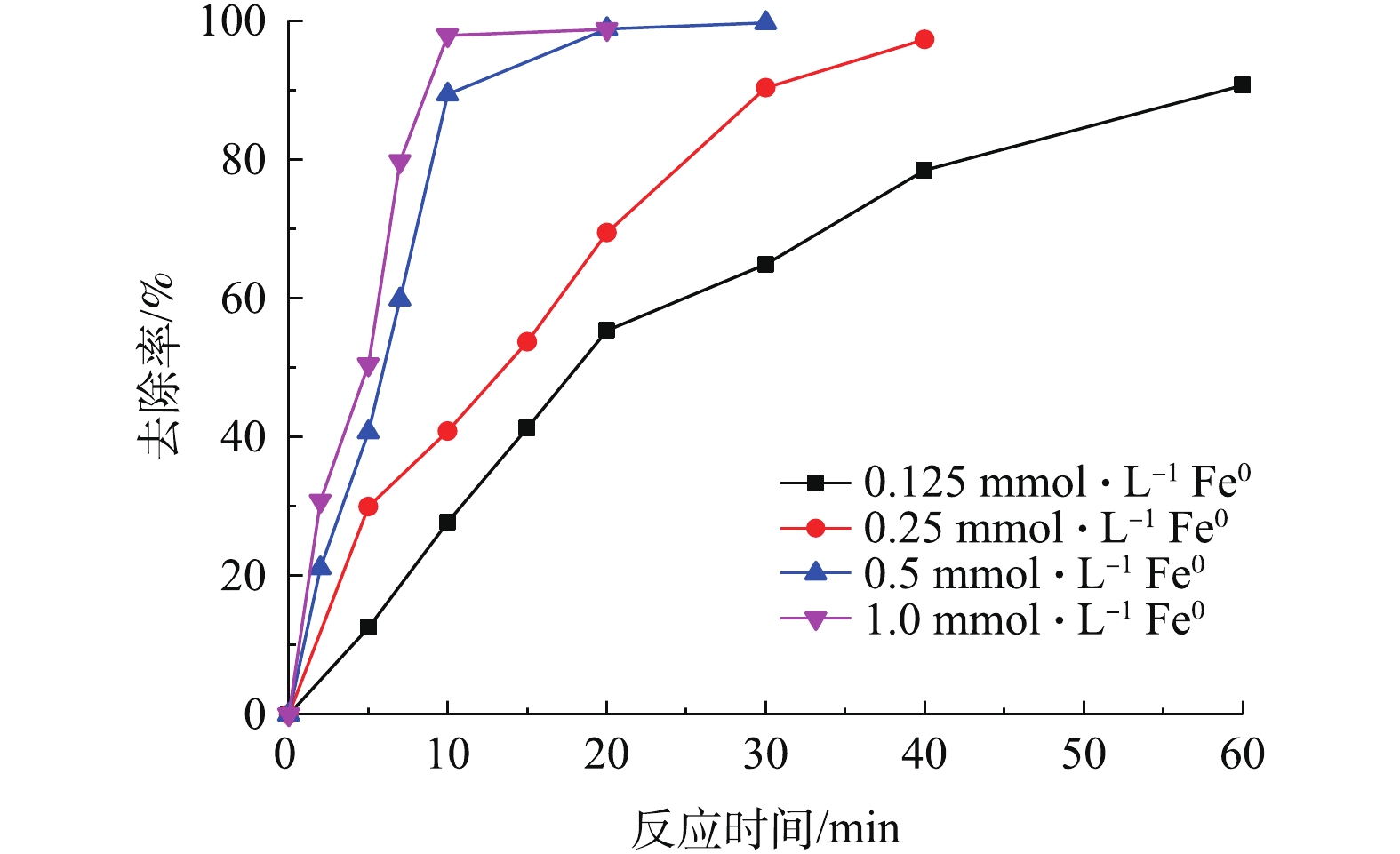
图 4 零价铁投加量对Pre-Fe0/PS体系降解DCF的影响
Figure 4. Influence of dosage of Fe0 on DCF degradation by Pre-Fe0/PS process
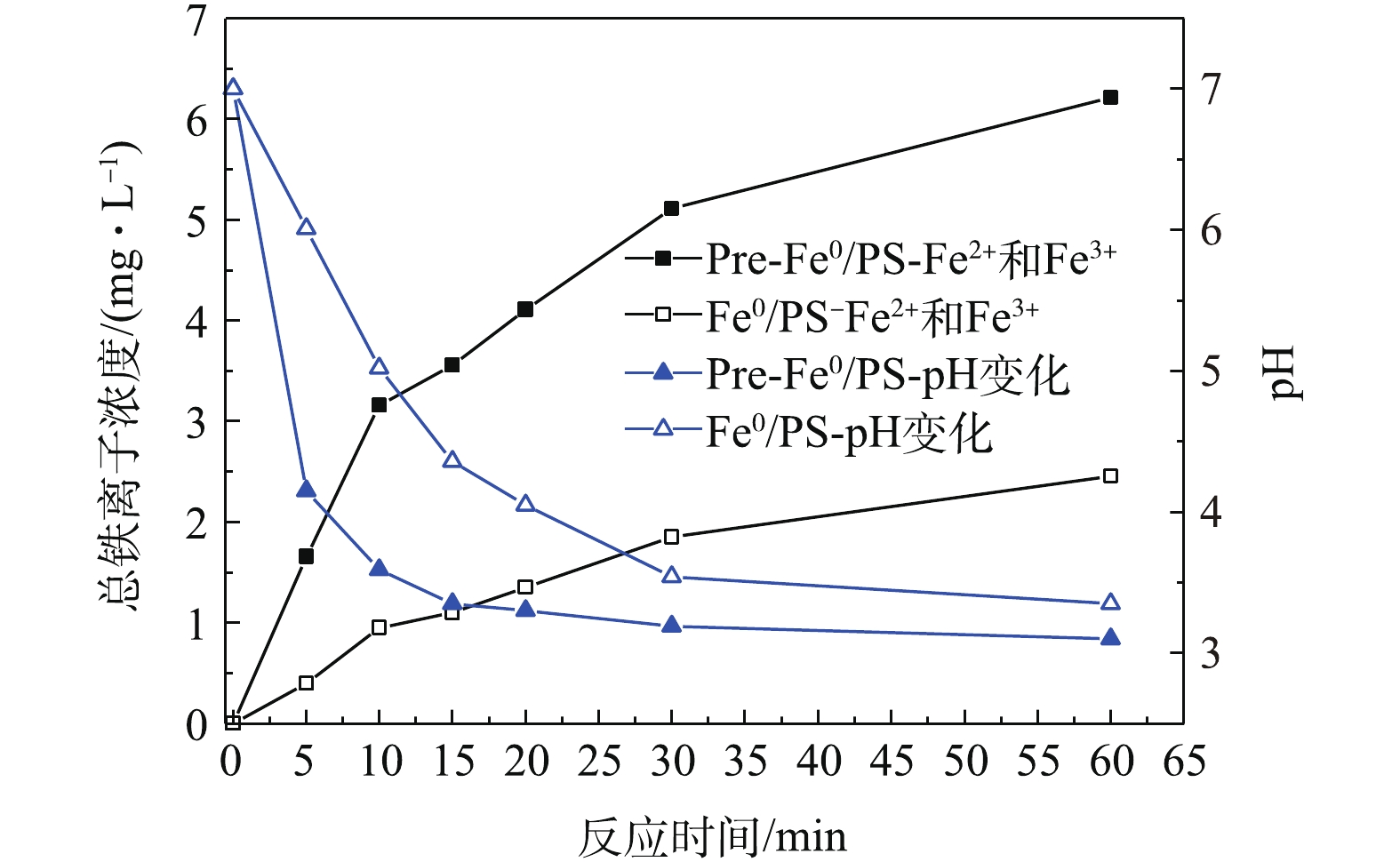
图 6 反应过程中2种体系中铁离子浓度和pH变化
Figure 6. Changes of total iron concentration and pH in Fe0/PS and pre-Fe0/PS process.
-
[1] ZHANG Y J, GEISSEN S U, GAL C. Carbamazepine and diclofenac: Removal in wastewater treatment plants and occurrence in water bodies[J]. Chemosphere, 2008, 73(8): 1151-1161. doi: 10.1016/j.chemosphere.2008.07.086 [2] AHMAD M, TEEL A L, WATTS R J. Mechanism of persulfate activation by phenols[J]. Environmental Science & Technology, 2013, 47(11): 5864-5871. [3] WANG J L, WANG S Z. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 2018, 334: 1502-1517. doi: 10.1016/j.cej.2017.11.059 [4] DONG H, DENG J, XIE Y, et al. Stabilization of nanoscale zero-valent iron (nZVI) with modified biochar for Cr(VI) removal from aqueous solution[J]. Journal of Hazardous Materials, 2017, 332: 79-86. doi: 10.1016/j.jhazmat.2017.03.002 [5] LAI K C K, LO I M C. Removal of chromium (VI) by acid-washed zero-valent iron under various groundwater geochemistry conditions[J]. Environmental Science & Technology, 2008, 42(4): 1238-1244. [6] SONG X Z, SHI Q, WANG H, et al. Preparation of Pd-Fe/graphene catalysts by photocatalytic reduction with enhanced electrochemical oxidation-reduction properties for chlorophenols[J]. Applied Catalysis B: Environmental, 2017, 203: 442-451. doi: 10.1016/j.apcatb.2016.10.036 [7] LIN C J, LO S L. Effects of iron surface pretreatment on sorption and reduction kinetics of trichloroethylene in a closed batch system[J]. Water Research, 2005, 39(6): 1037-1046. doi: 10.1016/j.watres.2004.06.035 [8] KIM D H, KIM J, CHOI W. Effect of magnetic field on the zero valent iron induced oxidation reaction[J]. Journal of Hazardous Materials, 2011, 192(2): 928-931. doi: 10.1016/j.jhazmat.2011.05.075 [9] XU H Y, SUN Y K, LI J X, et al. Aging of zerovalent iron in synthetic groundwater: X-ray photoelectron spectroscopy depth profiling characterization and depassivation with uniform magnetic field[J]. Environmental Science & Technology, 2016, 50(15): 8214-8222. [10] FENG P, GUAN X H, SUN Y K, et al. Weak magnetic field accelerates chromate removal by zero-valent iron[J]. Journal of Environmental Sciences, 2015, 31: 175-183. [11] GUAN X H, JIANG X, QIAO J L, et al. Decomplexation and subsequent reductive removal of EDTA-chelated Cu(Ⅱ) by zero-valent iron coupled with a weak magnetic field: Performances and mechanisms[J]. Journal of Hazardous Materials, 2015, 300: 688-694. doi: 10.1016/j.jhazmat.2015.07.070 [12] JIANG X, QIAO J L, LO I M C, et al. Enhanced paramagnetic Cu2+ ions removal by coupling a weak magnetic field with zero valent iron[J]. Journal of Hazardous Materials, 2015, 283: 880-887. doi: 10.1016/j.jhazmat.2014.10.044 [13] LI J L, BAO H L, XIONG X M, et al. Effective Sb(V) immobilization from water by zero-valent iron with weak magnetic field[J]. Separation and Purification Technology, 2015, 151: 276-283. doi: 10.1016/j.seppur.2015.07.056 [14] LI X, ZHOU M, PAN Y, et al. Pre-magnetized Fe0/persulfate for notably enhanced degradation and dechlorination of 2,4-dichlorophenol[J]. Chemical Engineering Journal, 2017, 307: 1092-1104. doi: 10.1016/j.cej.2016.08.140 [15] SHU H Y, CHANG M C, HUANG S W. UV irradiation catalyzed persulfate advanced oxidation process for decolorization of acid blue 113 wastewater[J]. Desalination and Water Treatment, 2014, 54(4/5): 1013-1021. [16] SINGH P, RAIZADA P, KUMARI S, et al. Solar-Fenton removal of malachite green with novel Fe0-activated carbon nanocomposite[J]. Applied Catalysis A: General, 2014, 476: 9-18. [17] LIANG C, BRUELL C J, MARLEY M C, et al. Persulfate oxidation for in situ remediation of TCE. I. Activated by ferrous ion with and without a persulfate-thiosulfate redox couple[J]. Chemosphere, 2004, 55(9): 1213-1223. doi: 10.1016/j.chemosphere.2004.01.029 [18] NURMI J T, TRATNYEK P G, SARATHY V, et al. Characterization and properties of metallic iron nanoparticles: Spectroscopy, electrochemistry, and kinetics[J]. Environmental Science & Technology, 2005, 39(5): 1221-1230. [19] LIU Y Q, LOWRYG V. Effect of particle age (Fe0 content) and solution pH on NZVI reactivity: H2 evolution and TCE dechlorination[J]. Environmental Science & Technology, 2006, 40(19): 6085-6090. [20] SUEPTITZ R, KOZA J, UHLEMANN M, et al. Magnetic field effect on the anodic behaviour of a ferromagnetic electrode in acidic solutions[J]. Electrochimica Acta, 2009, 54(8): 2229-2233. [21] XIONG X M, SUN B, ZHANG J, et al. Activating persulfate by Fe0 coupling with weak magnetic field: Performance and mechanism[J]. Water Research, 2014, 62: 53-62. doi: 10.1016/j.watres.2014.05.042 [22] 韩鹤友, 何志柯, 曾云鹗. 钌(II)-邻菲咯啉偶合化学发光法测定Fenton反应产生的羟自由基[J]. 分析化学, 1999, 27(8): 890-893. doi: 10.3321/j.issn:0253-3820.1999.08.005 [23] MA J H, MA W H, SONG W J, et al. Fenton degradation of organic pollutants in the presence of low-molecular-weight organic acids: Cooperative effect of quinone and visible light[J]. Environmental Science & Technology, 2006, 40(2): 618-624. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4480
- HTML全文浏览数: 4480
- PDF下载数: 80
- 施引文献: 0
