


 下载:
下载:


-
根据2014年环境保护部国土资源部公布的《土壤污染状况调查公报》,全国土壤污染状况总体不容乐观,部分地区土壤污染较重,耕地土壤环境质量堪忧,工矿企业废弃地土壤环境问题突出。全国土壤总的点位超标率为16.1%,其中轻微、轻度、中度和重度污染比例分别为11.2%、2.3%、1.5%和1.1%[1]。土壤重金属主要来源是矿山开采与工农业生产等人为活动,尤其是金属矿产资源的开采、加工和利用过程[2-3]。土壤重金属污染不仅会导致粮食产量与品质下降,而且还会沿食物链富集,从而危害人体健康[4-6]。
目前,重金属污染土壤治理方法主要包括固化稳定[7]、植物修复[8]、微生物修复[9]和电动修复[10]等。其中,电动修复技术操作简单、效率高、费用低,具有较广泛的应用前景[11-14]。通过对污染土壤施加直流电场,重金属离子通过电迁移、电渗流、电泳等作用移动到电极附近区域,达到富集去除的目的[15]。电解液是影响修复效果的重要因素之一,受到了国内外学者广泛的研究和关注。
常用的电解液包括柠檬酸[16]、酒石酸、草酸[17]、醋酸[18]、EDTA[19]和琥珀酸等。其中,柠檬酸和酒石酸对金属的溶解能力更强,也更易于通过螯合作用将其去除。酒石酸对Cd2+的去除和柠檬酸无显著差异,而对Zn2+和Ni2+的去除效果要显著优于柠檬酸。另一方面,同柠檬酸相比,酒石酸作为电解液修复过程中土壤常量元素,损失更少[20]。利用酒石酸作为电解液,通过电动修复技术处理土壤中重金属复合污染,有助于丰富重金属复合污染土壤的修复技术,对我国采矿区附近污染土壤的修复具有潜在应用价值。但是,目前对于重金属复合污染土壤的电动修复过程研究较少,同时缺乏对其赋存形态变化的影响研究。因此,本研究根据安徽铜陵矿区金属复合污染土壤的特征,配制重金属(Zn2+、Cu2+、Pb2+、Mn2+、Cd2+)复合污染高岭土,研究不同修复时间下5种重金属离子的迁移和转化情况,探究不同浓度酒石酸电解液对电动修复效果的影响,分析修复前、后重金属离子的赋存状态的变化,为电动修复技术在重金属污染土壤修复工程实际的应用提供参考。
全文HTML
-
实验所用高岭土为化学纯,其余使用药品均为分析纯,实验用水为超纯水。使用金属硝酸盐配制重金属混合溶液,主要组成为1 500 mg·L−1 Cu2+、1 500 mg·L−1 Pb2+、1 500 mg·L−1 Zn2+、1 500 mg·L−1 Mn2+、150 mg·L−1 Cd2+。取400 mL重金属混合溶液加入600 g高岭土中,混合搅拌均匀后,静置4 d[2]。土壤中金属离子浓度为838.82 mg·kg−1 Cu2+、900.77 mg·kg−1 Pb2+、904.46 mg·kg−1 Zn2+、1 540.77 mg·kg−1 Mn2+、81.64 mg·kg−1 Cd2+,含水率为39%[21]。
-
实验装置如图1所示,主体材质为有机玻璃,分为土壤室与电解液室2个部分。装置总体尺寸为370 mm×100 mm×100 mm,其中两侧阴阳电极室内部尺寸均为50 mm×90 mm×95 mm,土壤室内部尺寸为250 mm×90 mm×95 mm;电极室与土壤室之间设置隔板,隔板布满直径5 mm的细密孔,隔板外包裹250目筛布,保证电解液可通过而土壤不能通过。采用φ5 mm×100 mm的石墨棒作为阴阳电极。
-
取配制好的模拟土壤800 g均匀平铺于土壤室底部,向两侧电解液储存器中各加入500 mL电解液,实验过程中保持电解液液面与土壤土面平齐。开启直流电源,施加恒定直流电压,使土壤室内电压梯度为1 V·cm−1[22],并记录实验过程电流变化。采用完全随机实验设计,考察反应时间(12、24、48、72、96、120 h)和酒石酸浓度(0.05、0.1、0.2 mol·L−1)对土壤电动修复重金属修复效率的影响。如图1所示,实验结束后,将土壤均分为5份,标记为S1、S2、S3、S4、S5,测定土壤pH与重金属含量。以0.05 mol·L−1酒石酸为电解液电动修复120 h后土壤样品为代表,对金属赋存形式进行分析研究。
-
土壤样品采用电热板消解法进行预处理,然后利用火焰原子吸收光谱法测定金属离子浓度[23];采用BCR连续提取法测定金属赋存形态[24];取10 g土壤,按照土水质量比1∶2.5混合,搅拌静置后测量pH[25]。
重金属去除率计算如式(1)所示,重金属总去除率计算如式(2)所示。
式中:R为重金属去除率;Q和P分别为修复前后土壤重金属含量,mg·kg−1;R总为重金属总去除率;i=1, 2, 3, 4, 5代表5种金属。
1.1. 实验原料和装置
1.2. 实验方法和分析方法
-
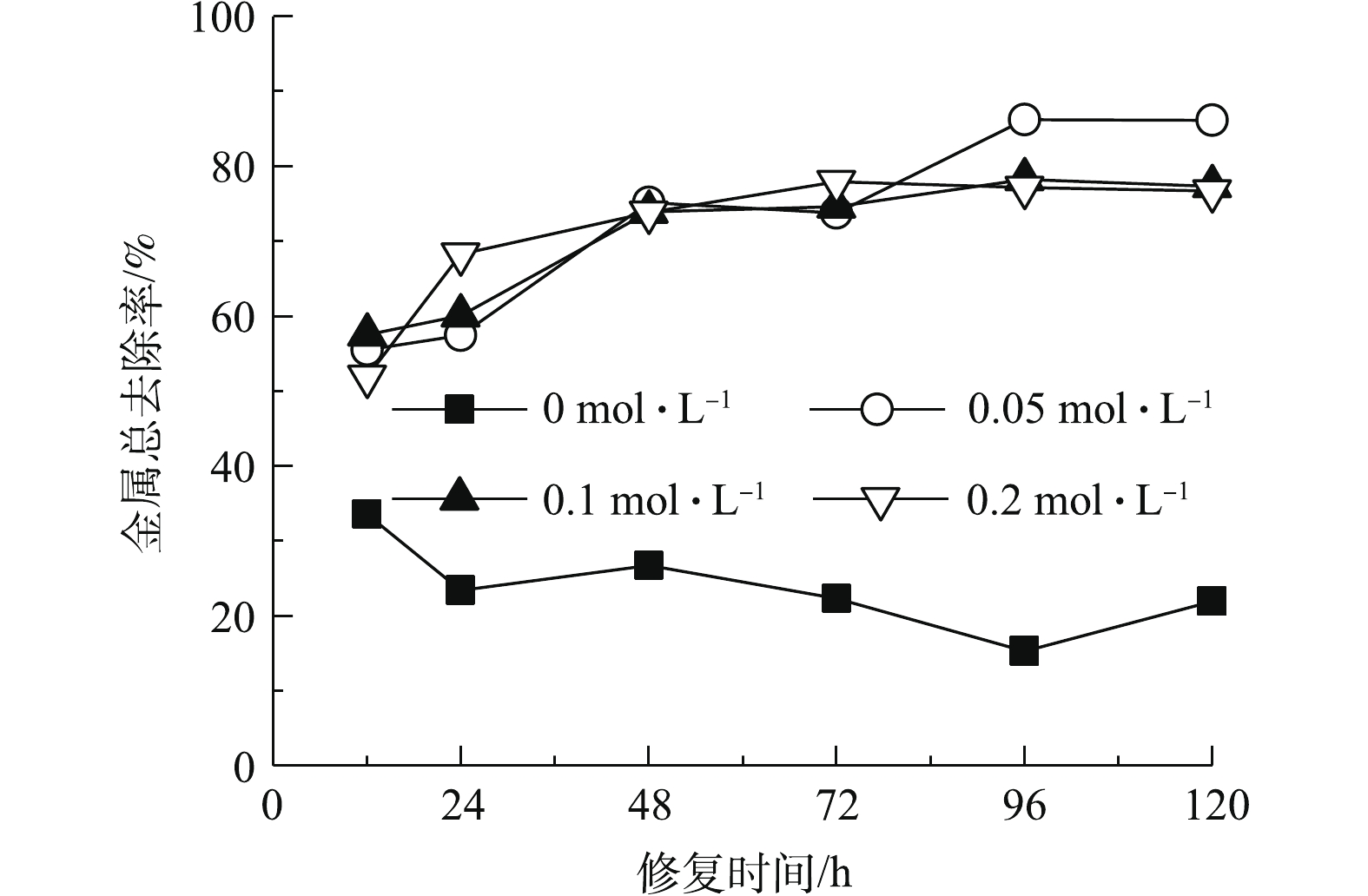
如图2所示,以纯水为电解液时,重金属总去除率较低。这主要是由于,高岭土对金属离子具有吸附性[26],限制了其迁移能力;同时阴极区产生的OH−与金属结合形成氢氧化物沉淀,进一步降低了金属移动能力。对照组在修复时间为12 h时金属去除率最高,延长修复时间导致去除率下降。这主要是由于,修复时间为12 h时,靠近阴阳电解室的土壤与纯水接触,金属离子主要通过扩散进入阴阳极电解液;提高作用时间,在电迁移作用下,阳极电解液中金属阳离子重新进入土壤室向阴极侧迁移,反而导致金属总去除率降低。
与对照相比,添加酒石酸明显提高了重金属总去除率。酒石酸作为电解液能够降低土壤pH,减少金属氢氧化物的生成。酒石酸也能够与金属离子发生配位作用,降低土壤吸附作用,增强其迁移能力[17]。酒石酸浓度为0.05 mol·L−1时,金属总去除率高于0.1 mol·L−1和0.2 mol·L−1。这主要是由于,酒石酸浓度提高使得体系内呈电中性的金属-有机酸化合物含量升高,从而限制了金属的电迁移[3]。
-
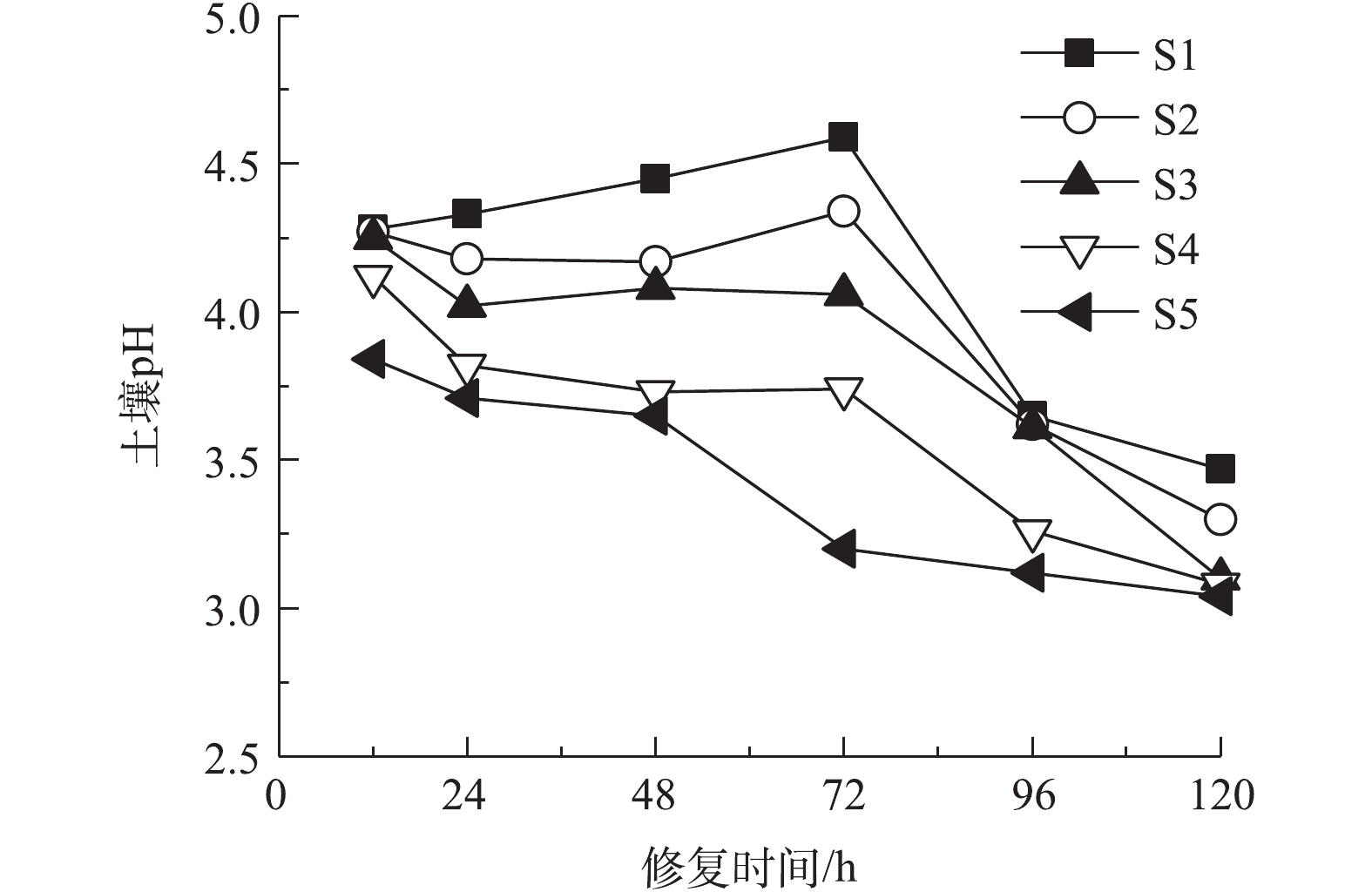
以0.05 mol·L−1酒石酸作为电解液不同时间电动修复后各区域土壤pH分布如图3所示。电动修复反应中,电解室发生水的电解反应,使阳极电解液pH下降,阴极电解液pH升高,H+、OH−在电场力作用下发生迁移,在土壤室内形成酸性带与碱性迁移带[27]。本体系土壤初始pH为5.74,修复过程中土壤pH均处于酸性环境。修复12 h时,电解反应时间较短,阴极产生OH−较少,电解液pH变化较小。阴阳极酸性电解液进入土壤室,使S1~S5整个区域土壤pH均下降,且pH从S1区域到S5区域逐渐降低。延长通电时间,阴极电解室中产生大量OH−,阴极电解液pH升高,OH−向阳极侧迁移,S1区域的H+不断被中和,使土壤pH不断上升,在72 h时达到最大值;同时阳极电解液产生大量H+,且不断向阴极侧迁移,使S3区域到S5区域,土壤pH持续降低。修复时间达到96 h和120 h后,整个土壤室中含有大量H+,5个土壤区域pH大幅降低,S5~S1区域土壤pH为3.0~3.5,且从S1区域到S5区域呈下降趋势。
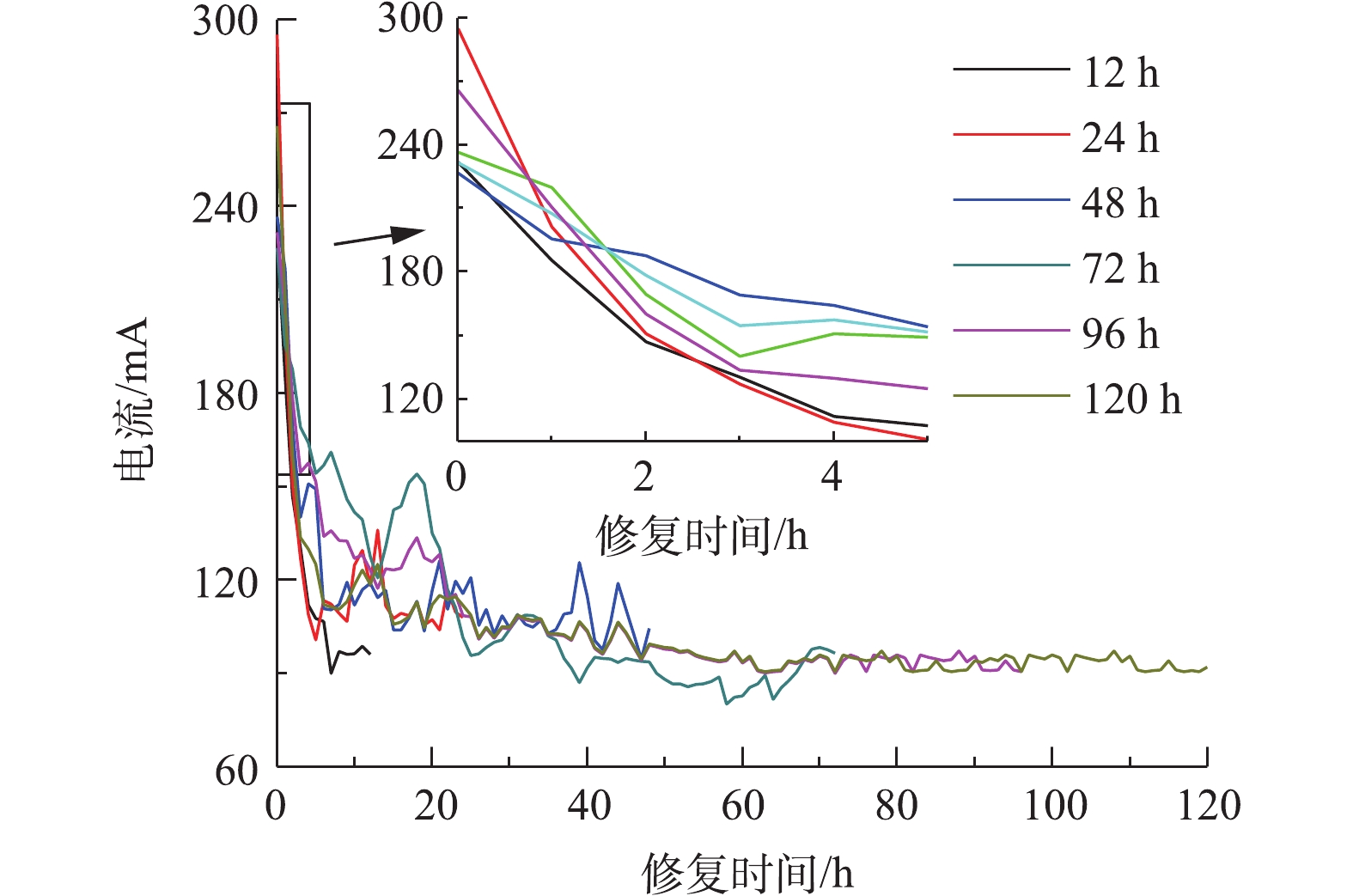
体系内电流大小能够反映自由离子数量的多少,以电流变化判断可溶性离子的迁移情况。以0.05 mol·L−1酒石酸作为电解液不同时间电动修复实验过程中电流变化如图4所示,在开始反应4 h内,电流快速下降,之后缓慢下降,最终趋于稳定。反应初始,体系内离子多,电流较大,为230~290 mA;反应开始后4 h内,在扩散作用下,离子浓度快速下降,电流快速减小至120 mA;随实验时间延长,体系内离子浓度缓慢减小,电流缓慢下降,直至趋于平稳为90~100 mA,H+和酒石酸根离子为土壤中主要的导电离子。
-
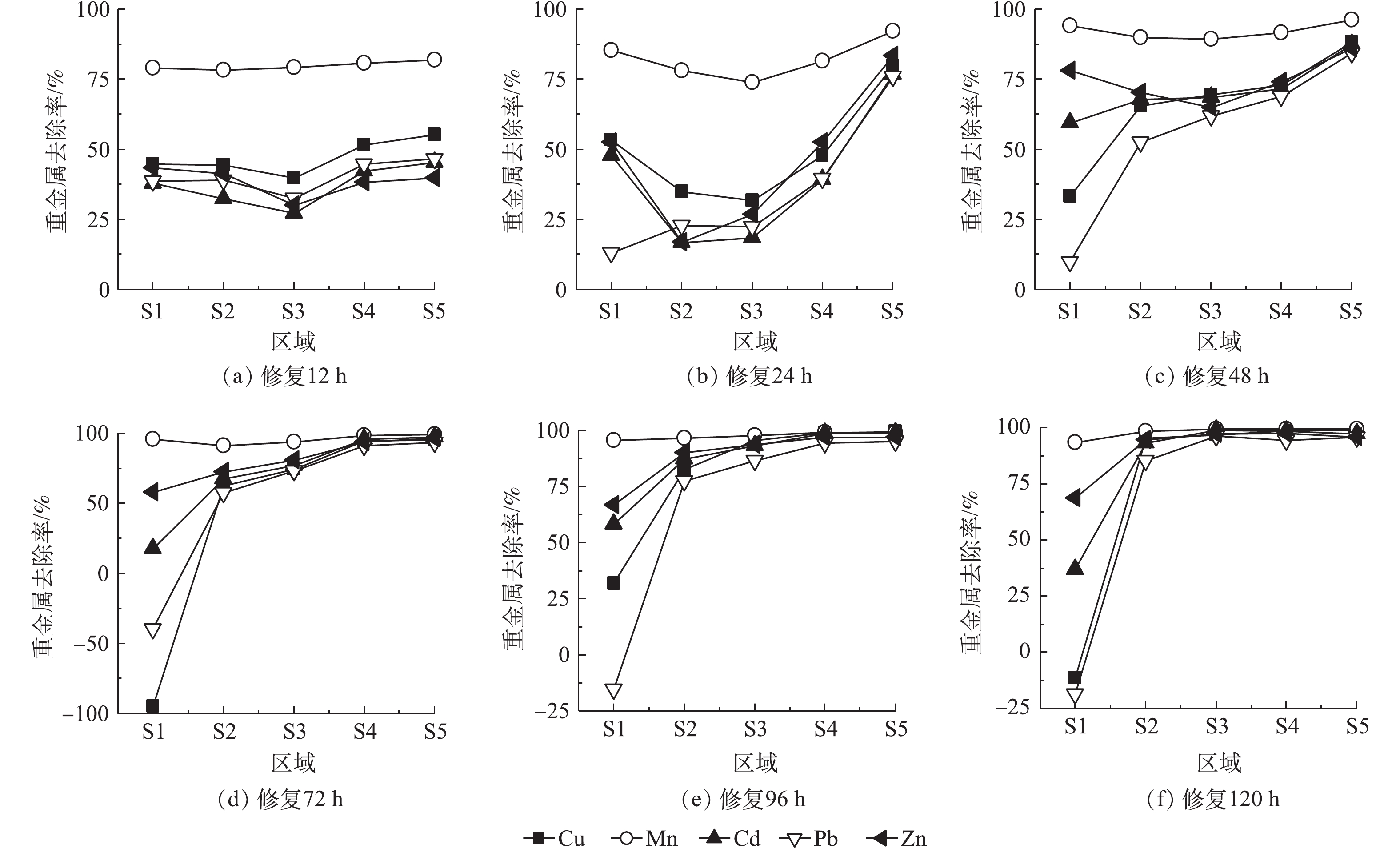
图5为0.05 mol·L−1酒石酸作为电解液,不同修复时间Cu、Mn、Cd、Pb、Zn等金属在土壤室不同区域迁移情况和去除率。修复时间为12 h时,所有区域5种金属含量均下降,Mn去除率为78%~81%,Cu、Cd、Pb、Zn去除率分别为39%~55%、27%~45%、32%~46%、30%~43%。靠近阴、阳极电解液侧土壤金属去除率略高于中部。这主要是由于,土壤含水率较高,电解液进入土壤区域,由于稀释与自由扩散作用,游离态的金属离子向阴阳两极电解液中扩散。随后在电场力作用下,进入阳极液的金属离子会重新进入S5区域并向S1方向迁移。通电24 h后,靠近阳极的S5区域金属去除率明显上升,均达到75%以上,其余区域去除率变化较小;随着修复过程的进行,金属向阴极侧逐渐迁移。48 h时,S2~S5区域5种金属去除率均上升达到50%以上。72 h后,5种金属S5区域的去除率均接近100%,其中S1区域中Cu、Zn、Pb、Cd含量最高,甚至Pb、Zn含量高于修复前,分别达到修复前含量的1.5倍和2倍。原因是,S1区域土壤pH较高,不利于高岭土吸附的金属离子的解吸,且金属离子形成了氢氧化物沉淀。96 h时,S2~S5区域5种金属含量继续下降,去除率持续上升。此时,S1区域pH大幅下降,Cu、Pb从高岭土上解吸,氢氧化物沉淀溶解后进入阴极电解液,Cu去除率上升至31.81%,Pb富集量下降至原来的1.25倍。实验时间为120 h时,S2~S5区域中除S2区域中Pb去除率为85.42%,其余去除率均达到93%以上。此时,金属Cu、Mn、Cd、Pb、Zn的总去除率分别为75.7%、98.1%、85.1%、70.8%、90.9%,土壤中金属污染物得到较好的去除。
5种金属去除率差异较大,Mn在所有区域的去除率均达到90%以上,其余4种金属去除效果为Zn>Cd>Cu>Pb。这主要是由于,高岭土对不同金属的吸附与解吸附能力差异所致。有研究报道,高岭土对于Cd2+、Cu2+、Pb2+ 3种金属的吸附能力为Pb2+>Cd2+>Cu2+,解吸能力为Cd2+>Cu2+>Pb2+[28],这与本实验重金属去除率顺序相同,说明土壤对金属的吸附解吸性能也是影响着电动修复中金属离子去除效果的重要因素。
-
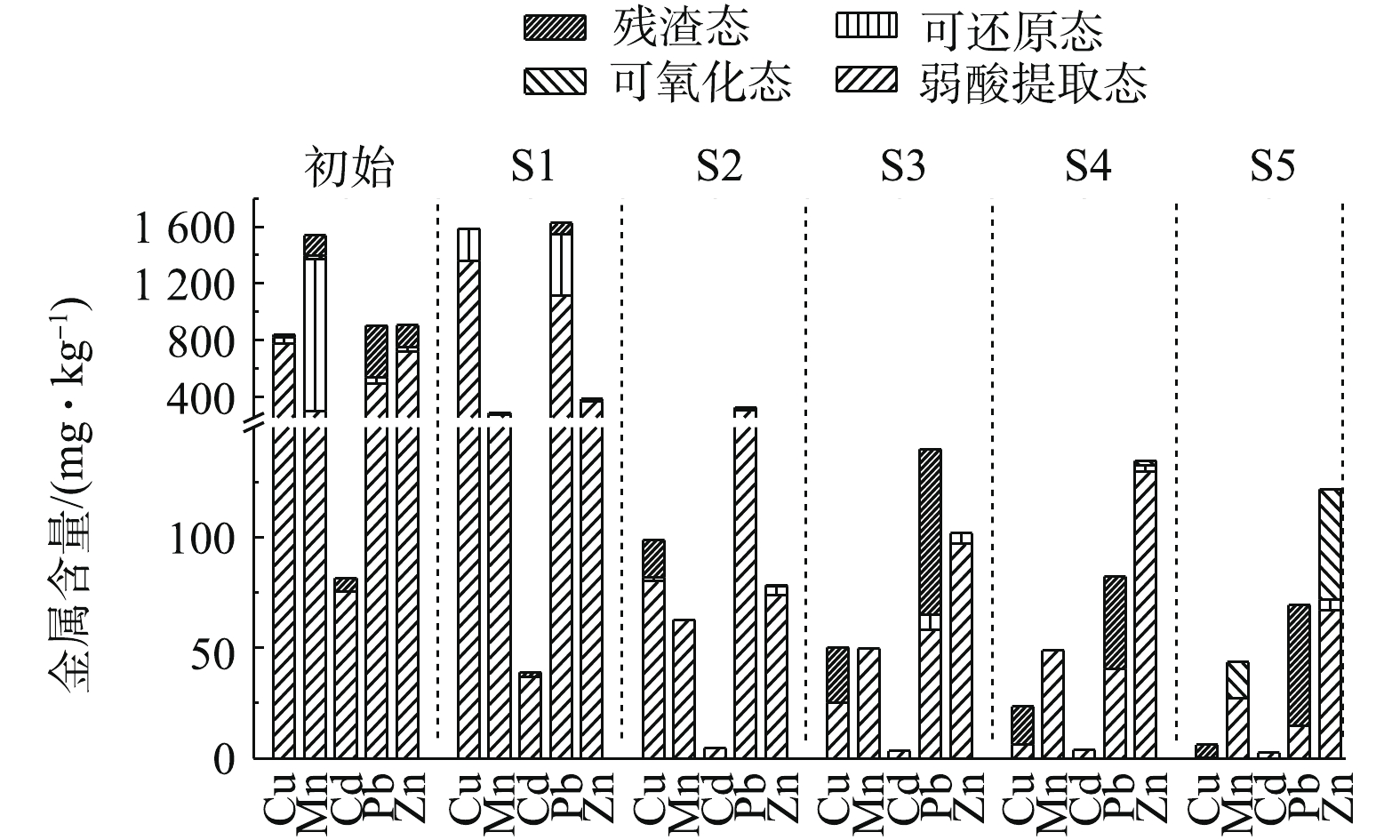
土壤中金属元素赋存形态将影响环境中植物、微生物的生理生化特性,同时影响电动修复过程中金属迁移转化情况。图6为采用0.05 mol·L−1酒石酸为电解液,电动修复120 h前、后土壤金属赋存形态的变化。修复前土壤中Cu存在形式主要是弱酸提取态、可还原态与残渣态。修复后Cu在S1区域中富集,存在形态为弱酸提取态与可还原态。从S1~S5可还原态与弱酸提取态含量下降,残渣态含量上升,S4和S5区域内可还原态含量为0 mg·kg−1,在S5区域中残渣态含量为5.8 mg·kg−1,占总含量的94.7%,说明电动修复中弱酸提取态的Cu迁移率较大,且添加酒石酸可促进残渣态Cu向弱酸提取态Cu转化,这与现有研究结果[29]相一致。
修复前土壤中Mn的赋存形式主要为弱酸提取态、可还原态、可氧化态与残渣态。修复后S1~S5中残渣态Mn含量减小;S2~S4中全部以弱酸提取态存在;在靠近阴、阳极的S1、S5区域,分别存在少量的可还原态与可氧化态。在S1和S5区域内,Mn发生了氧化还原反应,使其形态发生改变。酒石酸促进了残渣态Mn向弱酸提取态Mn转化。修复前土壤中Cd的赋存形式主要为弱酸提取态和残渣态。修复后S1中Cd仍以弱酸提取态与残渣态存在,弱酸提取态占比为94.3%,但在S2~S5中全部转变为弱酸提取态Cd,由此可见,酒石酸促进了残渣态Cd向弱酸提取态Cd转化。修复前土壤中Pb、Zn的赋存形式主要为弱酸提取态、可还原态和残渣态。修复后在靠近阴极的S1区域,存在少量的可还原态Pb与Zn。S1~S5中弱酸提取态Pb、Zn含量逐渐下降;残渣态Pb比例逐渐增大。从金属赋存形态变化趋势可知,电动修复过程中弱酸提取态金属去除率高于残渣态金属,酒石酸可促进残渣态金属向弱酸提取态转化。
2.1. 金属总去除率
2.2. 土壤pH和电流的变化
2.3. 修复时间对金属去除率的影响
2.4. 金属赋存形态变化
-
1)酒石酸作为电解液时,其电离产生的H+可以提供酸性条件促进土壤中金属离子的溶解,抑制金属氢氧化物沉淀的生成,且酒石酸与金属离子的螯合作用可以促进土壤颗粒表面金属的解吸,改变金属存在形态,有利于电动修复。
2)电压梯度为1 V·cm−1,以0.05 mol·L−1酒石酸为电解液电动修复120 h后,土壤修复效果最好,重金属总去除率达到86.15%,Cu、Mn、Cd、Pb、Zn去除率分别为75.67%、98.11%、85.1%、70.75%、90.9%。
3)弱酸提取态金属较容易迁移,残渣态较稳定,酒石酸可促进残渣态金属向弱酸提取态转化。
