

 DownLoad:
DownLoad:






-
对苯二甲酸(TA)是一种石油化工产品,广泛应用于工业生产,在自然条件下不易降解且具有累积效应[1-2]。目前,含TA废水处理方法主要包括生化处理法和物化处理法,生化处理法有膜生物反应器法[3]、好氧生物法[4]以及厌氧生物法[5-6]等;物化处理法包括吸附法[7]、过滤分离法[8]以及催化臭氧氧化法[9]等。催化臭氧氧化法与其他方法相比,具有操作简单、反应时间短、降解效果高和成本低等优点[10-15]。催化臭氧氧化法可将TA高效氧化,但在非均相条件下,活性组分易团聚,活性位点不能充分利用,严重制约了催化活性。而载体表面性质会影响活性组分的分散状态,因此,寻找新型载体材料非常重要。
LOPES等[16]研究发现,(Mn,Cu)/TiO2-CeO2催化剂降解苯酚类污染物有很高的活性。王宇轩等[17]研究发现,CuO和活性炭催化过硫酸盐产生的
SO−4 ·降解活性艳红X-3B燃料显示出较好的活性和重复利用性。张坤等[18]研究发现,Y2O3/WO3降解综合废水,催化剂具有较高的催化活性。陈坦等[19]研究发现,CuO活性组分能很好地分散在HZSM-5载体上,达到提高芳烃收率的目的。SASIDHARAN等[20]研究发现,在H2O2水溶液作用下,TS-1与Ti-ZSM-22、Ti-β等硅酸盐相比,氧化醚生成内酯或羧酸实验性能最佳。刘艳芳等[21]研究发现,改变臭氧投加量和停留时间2个影响因素,优化Cu/Y-TS-1催化臭氧去除COD、DOC及NH3-N的效率,取得了较好的实验结果,但降解过程和降解机理有待进一步研究。本实验采用浸渍法制备CuO-Y2O3/TS-1臭氧催化剂,以TA为研究对象,构建非均相催化体系,通过考察臭氧通量、单金属与双金属催化剂、不同比例浸渍液浓度及初始溶液pH等因素对降解TA效果的影响,深入探讨了催化剂的循环稳定性及催化臭氧降解TA机理。本研究通过制备CuO-Y2O3/TS-1,实现高效降解TA的目标,并详细论述了对降解TA的机理,为降解工业废水中TA的研究和应用提供了参考。
全文HTML
-
硝酸钇(Y(NO3)3·6H2O)、硝酸铜(Cu(NO3)2·3H2O)、对苯二甲酸(TA)、碘化钾(KI)、无水乙醇均为分析纯;钛硅分子筛(TS-1);高纯氧气(O2,99.999%)。
-
采用蒸馏水反复冲洗TS-1去除杂质,阴干24 h,得到催化剂载体。采用浸渍法配制不同比例的Cu(NO3)2·3H2O和Y(NO3)3·6H2O浸渍液(Cu∶Y=0.5∶0.5、Cu∶Y=0.5∶1、Cu∶Y=0.5∶2、Cu∶Y=1∶0.5、Cu∶Y=2∶0.5、Cu∶Y=1∶1),取2 g粒径为40~60目的TS-1,放至浸渍液中,在温度50 ℃和转速30 r·min−1条件下,浸渍24 h,过滤除去浸渍液,得到TS-1,在马弗炉中105 ℃下,干燥2 h,煅烧3 h得到目标催化剂(升温速率为3 ℃·min−1,温度设定为550 ℃)。
-
X-射线衍射仪(XRD)采用Rigaku-D/max-2400型衍射仪,其中X射线源为Cu靶的Kα射线,扫描角度为5°~90°。X射线荧光光谱仪(XRF,AXIOS,荷兰)基于莫斯莱定律,以特征X射线为基础进行元素的定性、定量分析。利用傅里叶变换红外光谱仪(FT-IR,Bruker Tensor 27,德国)在DTGS检测器的光谱仪上记录FT-IR光谱,并用KBr颗粒测量样品。电子扫描电镜(SEM,S-4800-I,日本)测定催化剂外观形貌,电子加速3.0 kV,放大2×104倍。透射电子显微镜(TEM,JEM 2100,日本)测定催化剂结构。
-
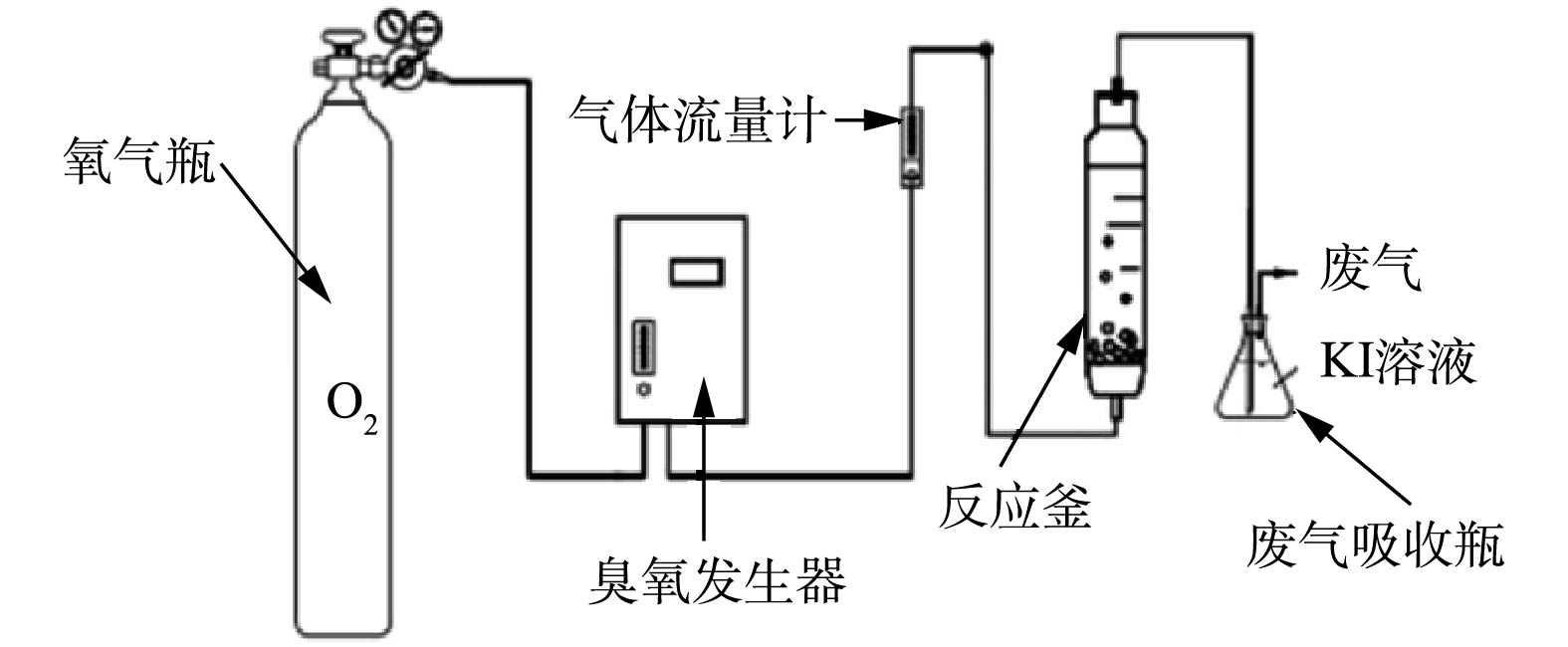
通过高效液相色谱法(HPLC)(Shimadzu LC-10 VP,日本)在反应过程中以规则的时间间隔测量水溶液中TA组分的浓度。实验装置如图1所示,氧气从气瓶进入臭氧发生器,在臭氧发生器产生臭氧,用气体流量计控制流量,使稳定的气流进入反应釜,反应后的尾气先通入废气吸收瓶,再排放到大气中。TA浓度为100 mg·L−1,臭氧通入量为6.3 mg·min−1,催化剂投加量为1.0 g,pH=9.0时,模拟废水量为250 mL。用高效液相色谱法测定TA组分的浓度,计算去除率。TA的去除率见式(1)。
式中:y为TA去除率;c进气为进气口TA浓度,μg·mL−1;c出气为进气口TA浓度,μg·mL−1。
1.1. 实验材料
1.2. 催化剂的制备
1.3. 表征技术
1.4. 催化活性评价
-
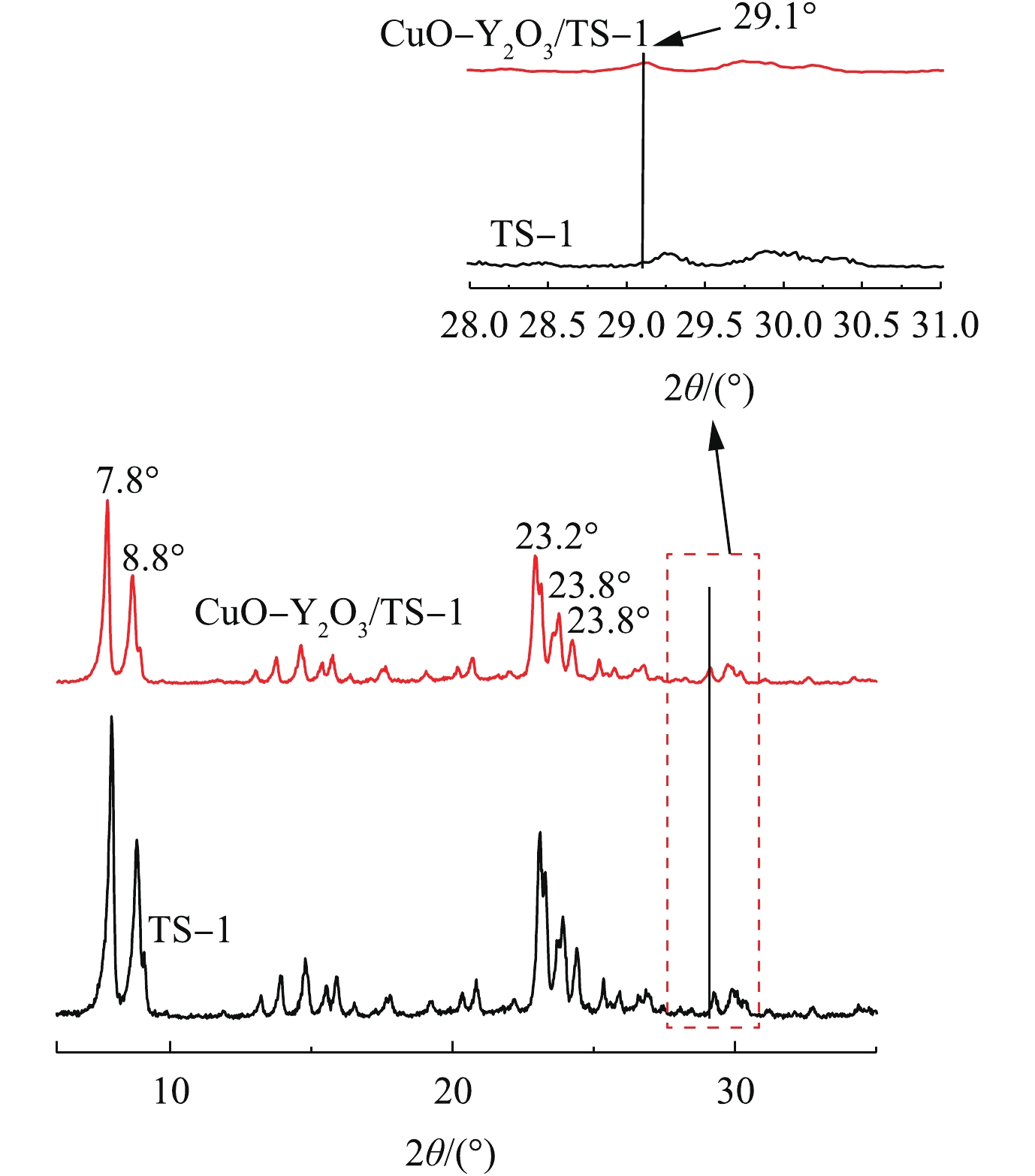
1) XRD表征。图2为TS-1和CuO-Y2O3/TS-1的XRD图。从图2(a)和图2(b)中可以得出,5个特征峰均是TS-1的峰且显示出较好的结晶度。图2(b)与图2(a)相比,7.8°、8.8°、23.2°、23.8°和24.3°处的5个特征峰的峰强增大,表明CuO、Y2O3的引入能够提高TS-1的结晶度。而从图2(b)中未发现CuO的特征峰,表明Cu2+半径小于Y3+半径,CuO进入到Y2O3里面;在29.1°处存在Y2O3的峰(标准卡片号#79-1256),且对比图2(a)与图2(b)发现峰向左偏移,由于CuO进入Y2O3里面,Cu2+与Y3+之间离子半径的差异很大,更多的氧进入Y2O3晶格中引起峰发生偏移。
2) SEM和TEM表征。由图3可以看出,图3(a)和图3(b)都有球状TS-1颗粒存在,图3(b)中TS-1负载CuO和Y2O3后,表面有一层涂层。由于CuO进入Y2O3内部,Y2O3负载到TS-1表面或内部,使TS-1晶格参数发生变化,晶粒生长过程中形貌结构也会发生差异,大量的晶粒形成紧密形貌结构的颗粒,表现出团聚现象。图3(c)~图3(f)均为CuO-Y2O3/TS-1的TEM图,从图3(d)和图3(f)可以看出,CuO-Y2O3没有晶格条纹且以氧化物的形式吸附在TS-1表面。其中图3(f)相比于图3(d),吸附在TS-1表面的CuO-Y2O3密度增大。由表1可以看出,在不同负载比例情况下,比表面积、孔容和孔径都发生了变化。主要由于TS-1少量负载CuO-Y2O3,浸渍过程也相当于TS-1的水洗过程,会将孔道中一些溶解物质带出,焙烧过程将一些结构不稳定的孔道破坏,焙烧高温会将一些挥发分带走,导致孔径增加;同时,加入的CuO-Y2O3会负载到TS-1的表面和孔道内,催化剂负载CuO-Y2O3后,比表面积增大,孔容减少。当大量负载CuO-Y2O3时,过量加入CuO-Y2O3,会使颗粒变大进而堵塞孔道,孔容孔径会变小,TS-1表面出现活性组分团聚,表面积降低。
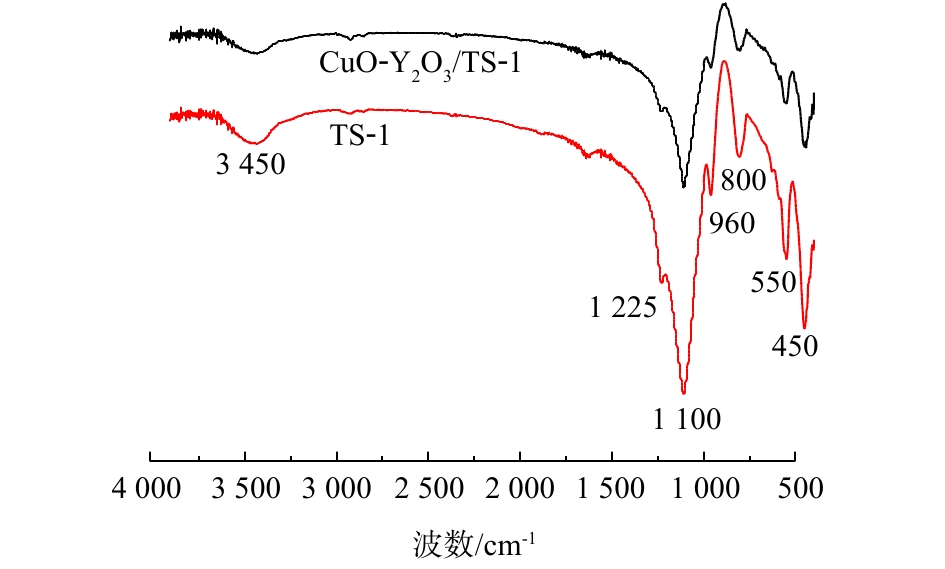
3) FT-IR表征。图4为TS-1和CuO-Y2O3/TS-1的FT-IR图。可以看出,图4出峰位置相同,450、550、800、960、1 100、1 225 cm−1均为TS-1骨架特征峰[22],550 cm−1和1 225 cm−1附近的峰归属于TS-1,1 110 cm−1处的吸收峰是Si—O四面体的不对称伸缩特征峰,8 00 cm−1和450 cm−1附近的吸收峰是[SiO4]四面体的Si—O弯曲振动峰,960 cm−1吸收峰断定为Si—O—Ti键的伸缩振动峰。图4中CuO-Y2O3/TS-1在450~1 225 cm−1处吸收峰变小,而谱带并未发生位移,这是由于CuO和Y2O3负载到TS-1表面或内部造成的。而3 045 cm−1峰归属为分子筛内部的硅羟基,它能通过氢键合水分子,该峰的强弱表示分子筛的吸水能力[23],CuO-Y2O3/TS-1的吸水能力比TS-1要弱,降低水对催化剂活性的影响,从而提高抗中毒性。
-
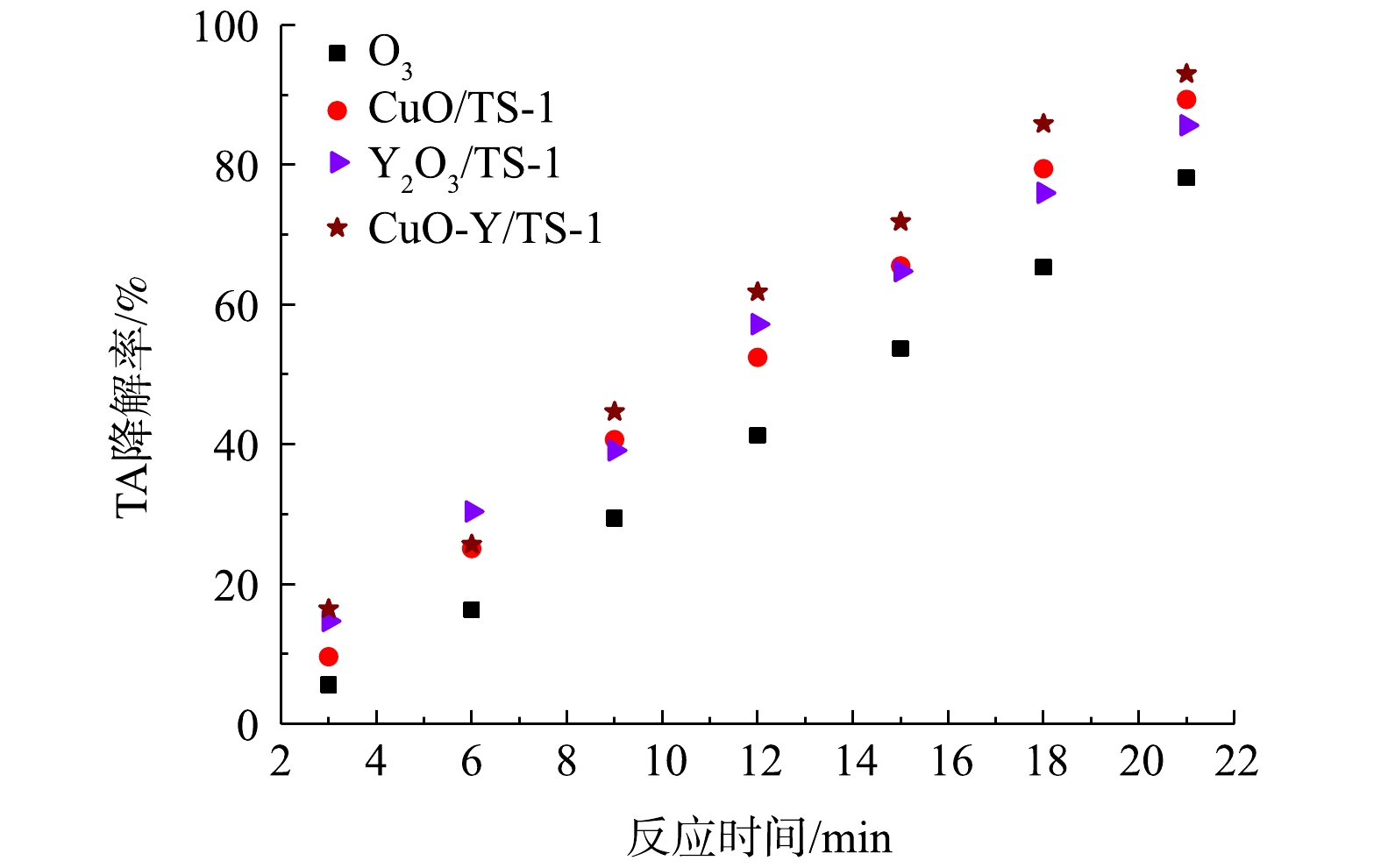
利用浸渍法制备催化剂,浸渍液浓度为0.5 mol·L−1,控制反应初始条件:pH为9.0,臭氧通量为3.6 mg·min−1,催化剂投加量为1.0 g。图5显示了TS-1负载金属催化剂降解TA的降解率。单独使用臭氧,催化降解效率较低,TS-1负载单金属催化剂的加入提高了降解效率,当反应时间为21 min时,CuO/TS-1的催化效率最高,降解率为91%。CuO-Y2O3/TS-1双金属催化剂降解TA的降解率最高,催化效率优于CuO/TS-1,9 min TA降解率为49%,21 min降解率为93%。活性顺序为CuO-Y2O3/TS-1>CuO/TS-1>Y2O3/TS-1>O3。钇负载TS-1上能提高Y2O3/TS-1的氧空位,从而提高臭氧降解TA的速率;铜负载TS-1上能提高CuO/TS-1的氧空位,Cu2+与臭氧反应,被还原成Cu+,Cu+与O2反应被氧化成Cu2+,Cu2+与Cu+之间相互转换,从而提高活性氧的迁移速率,显示出CuO/TS-1比Y2O3/TS-1降解TA的速率高[24]。CuO-Y2O3降解速率高的原因是CuO、Y2O3掺杂到TS-1内部,能提高TS-1的氧空位,反应的活性位点为Cu2+,Cu2+与Cu+之间相互转换,进一步提高了反应速率,晶格氧失去电子被氧化成氧气,原晶格氧变成氧空穴,富氧状态下,氧空穴被还原成晶格氧,氧空穴与晶格氧循环转变是催化还原的关键步骤[25]。氧在反应过程中起2个作用:1)促进氧迁移,提高反应速率;2)促进Cu2+与Cu+之间相互转换,提高反应效率。
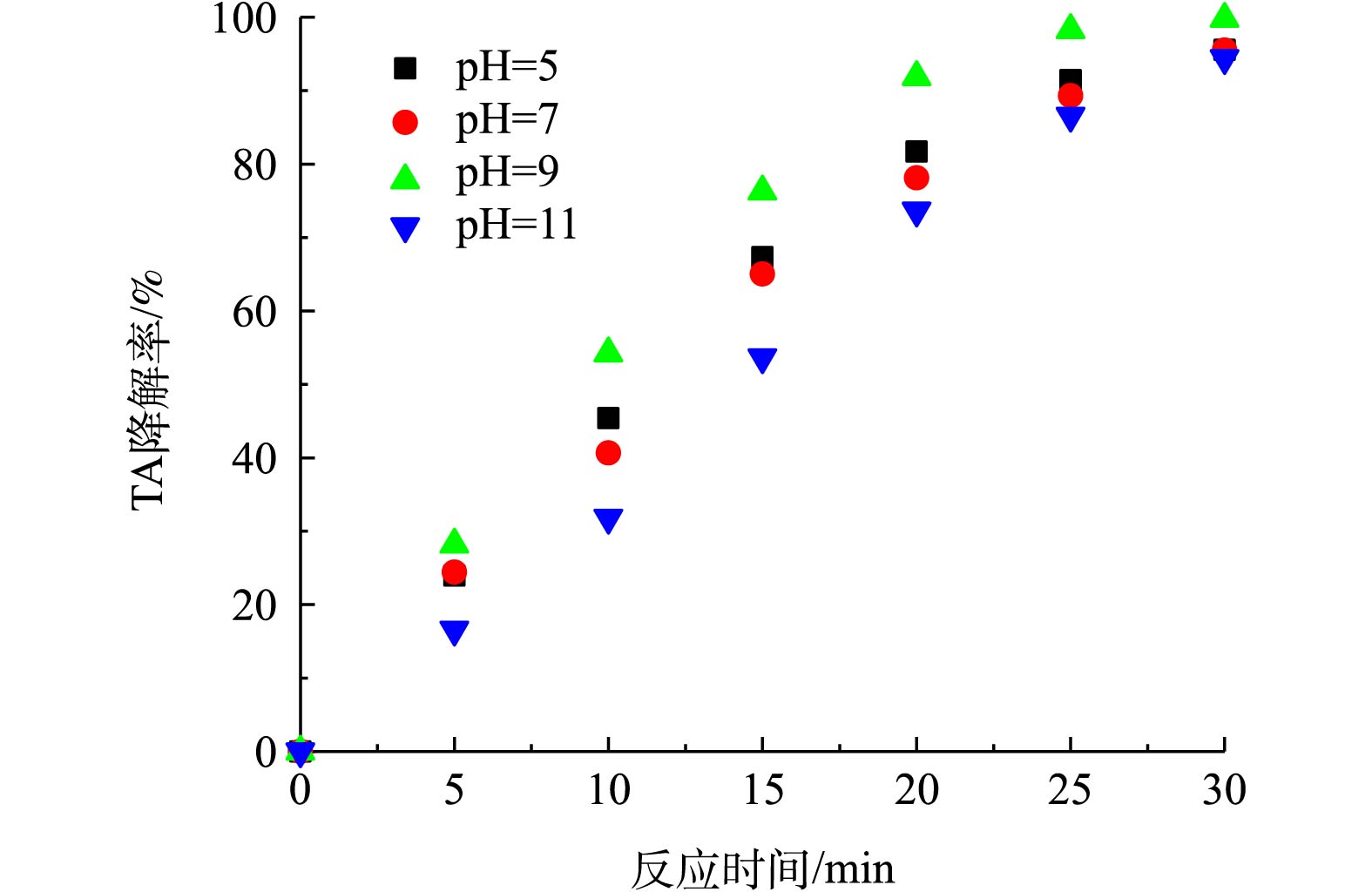
如图6所示,在pH = 9.0时,TA降解率最高。臭氧氧化属于自由基反应,反应[26-28]见式(2)~式(6)。
碱性介质反应见式(7)~式(10)。
臭氧在弱碱性条件下,易产生·HO2和·OH等活性自由基。·OH氧化能力比臭氧高,更易攻击有机分子,形成中间体和自由基,从而提高臭氧氧化的效率。臭氧与废水中的OH−反应,产生·OH,再氧化有机废水。
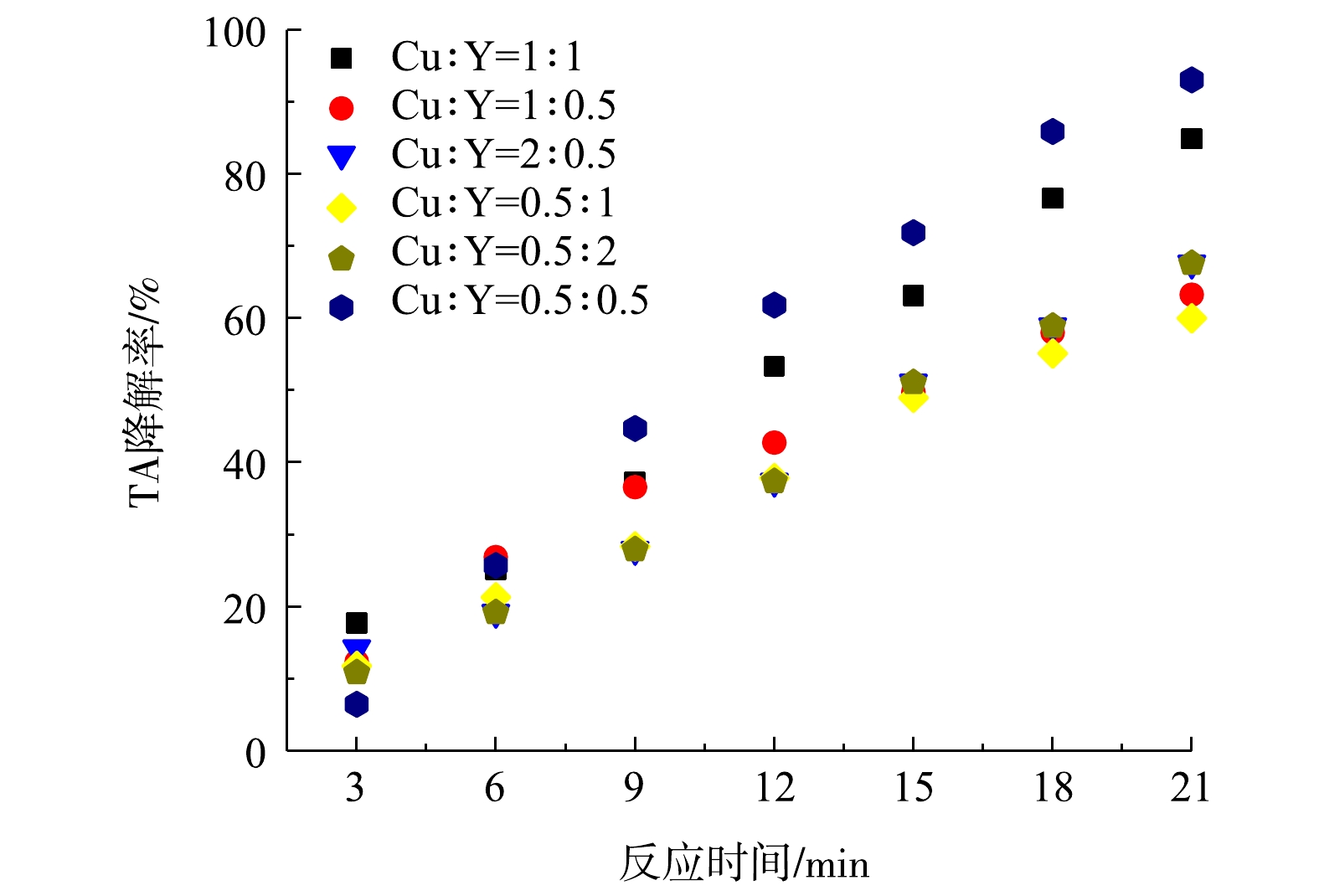
由图7可以看出,不同浓度比例Cu(NO3)2·H2O和Y(NO3)3·6H2O负载到TS-1上,对TA降解效率顺序为Cu∶Y(0.5∶0.5)>Cu∶Y(1∶1)>Cu∶Y(0.5∶2)>Cu∶Y(2∶0.5)>Cu∶Y(1∶0.5)>Cu∶Y(0.5∶1)。Cu与Y都是过渡金属,加入后可以提高TS-1的氧空位,Cu2+与Cu+之间相互转换,从而提高氧化还原速率;Cu∶Y=0.5∶0.5时,CuO与Y2O3摩尔比最大为19.58,氧空位、Cu2+与Cu+之间相互转换速率与其内部空间表面积达到最优,从而对TA降解效率达到最高。
催化剂活性和寿命是反映催化剂性能的重要指标。催化剂用去离子水及无水乙醇交替洗涤,50 ℃烘干,以备下一次循环使用。在最优条件下,对催化剂进行寿命研究,统计5次循环实验结果。结果表明,模拟废液中的TA的去除率由99.8%降低到了98.2%,去除率降低1.6%左右,表明该催化剂活性稳定性良好。
-
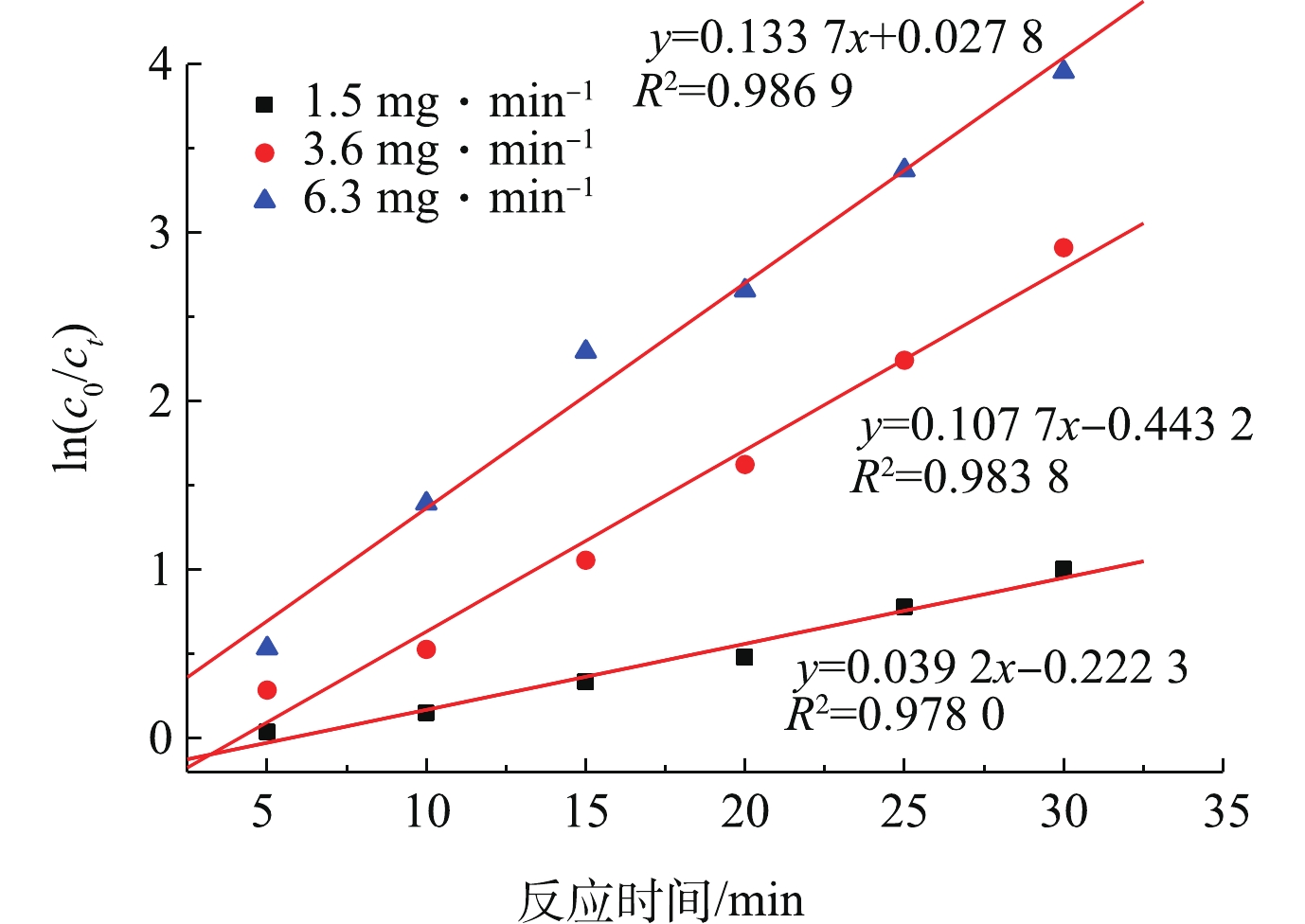
温度、pH、催化剂投加量保持不变,在实验中,有3种不同的臭氧通量,实验结果符合一级反应动力学方程,反应动力学曲线见图8。线性方程的表达式[29]见式(11)。
式中:Kt为表观反应速率常数;c0为初始浓度,mg·L−1;ct为瞬时浓度,mg·L−1。
如图8所示,臭氧通量对催化剂降解TA的顺序为6.3 mg·min−1>3.6 mg·min−1>1.5 mg·min−1。在3种臭氧通量情况下,通量为6.3 mg·min−1时,催化剂降解TA最好。由于TS-1负载过渡金属,提高的氧空位是一定的,臭氧量影响Cu2+与Cu+之间相互转换速率,当臭氧通量在6.3 mg·min−1时,氧空位、Cu2+与Cu+之间相互转换速率达到最优。各曲线的可决系数接近于1,3组均符合一级动力学反应。同时,随着臭氧通量的增加,反应速率常数从0.039 2增加到0.133 7,这说明臭氧浓度的增大加速了TS-1分子筛臭氧催化氧化处理模拟对TA废水的反应速率。
-
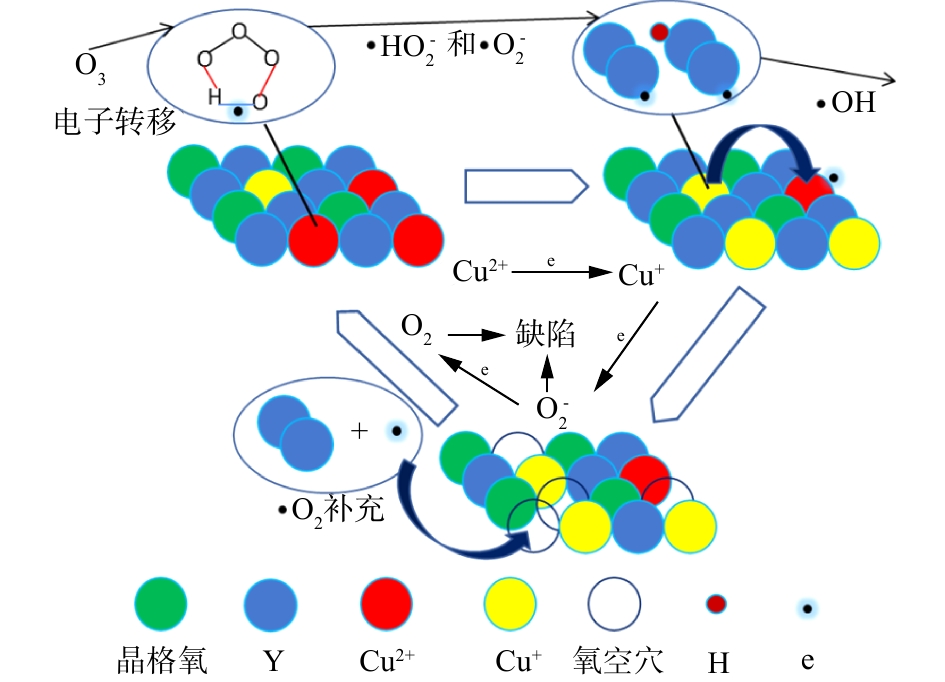
根据XRD、SEM、FT-IR和XRF等的表征结果,可推测催化臭氧降解TA机理。分析催化剂表面吸附的表面羟基与水中臭氧发生相互作用,可推测出催化剂活性位点吸附水中的水分子,使其分解为H+和OH−,形成表面羟基结构。水中溶解的臭氧分子会由于吸附等原因与催化剂表面的羟基相连,形成一个五元环的结构,催化剂表面的Cu2+离子通过表面羟基结构传递电子到五元环中,破坏五元环结构,使之分解为·HO2和·O2 2种活性物质,这2种活性物质会引发链式反应,诱发产生具有更强氧化性能的·OH,同时Cu2+得到电子,转化为Cu+离子。催化剂恢复活性过程是由于Cu+还原催化剂中的晶格氧,氧化为Cu2+,被还原的晶格氧被消耗,产生的氧空位则会由水中的活性物质补充进来,整个反应过程的机理[30-33]见图9。
2.1. 催化剂表征
2.2. 催化剂活性
2.3. 降解动力学
2.4. 催化臭氧降解TA机理
-
1)本研究采用浸渍法制备CuO-Y2O3/TS-1催化剂,通过构建非均相催化臭氧氧化降解TA(100 mg·L−1)体系考察其催化性能。与单金属催化剂及单独臭氧反应相比,催化臭氧降解TA具有明显优势。
2)当选择Cu(NO3)2·3H2O和Y(NO3)3·6H2O浸渍液浓度均为0.5 mol·L−1、浸渍时间为24 h、焙烧温度为550 ℃、焙烧时间为2h时,制备得到的催化剂催化效果最好。臭氧通入量为6.3 mg·min−1、催化剂投加量为1.0 g、pH=9.0时,反应30 min,TA降解率高达99.8%。经5次重复实验后,TA去除率仍然保持在98.2%,表明催化剂具有较强的稳定性。
3)通过对催化臭氧降解机理分析,在CuO-Y2O3/TS-1催化臭氧氧化反应过程中,铜和钇的加入能增加氧空穴。同时Cu2+和Cu+之间相互转化可促进氧的迁移速率,从而能快速生成小分子物质、H2O和CO2,大大提高了废水的可生化性能。
4)催化臭氧化处理TA实验符合一级反应动力学方程。
