










-
高氯酸盐(perchlorate, ClO4−)来源和分布广泛,在一定环境条件下,可通过氯化物与大气臭氧发生光化学氧化反应自然生成[1 − 2]. 由于ClO4−被广泛应用于火箭推进剂、烟火生产制造、爆破、安全气囊和食品包装领域[3 − 5],在人为因素下将随工业废弃物处理和排放等途径暴露于大气、土壤、水体等公共环境中,这可能会产生大量的ClO4−有毒污染物,相较于自然来源,更容易导致ClO4−环境浓度超标.
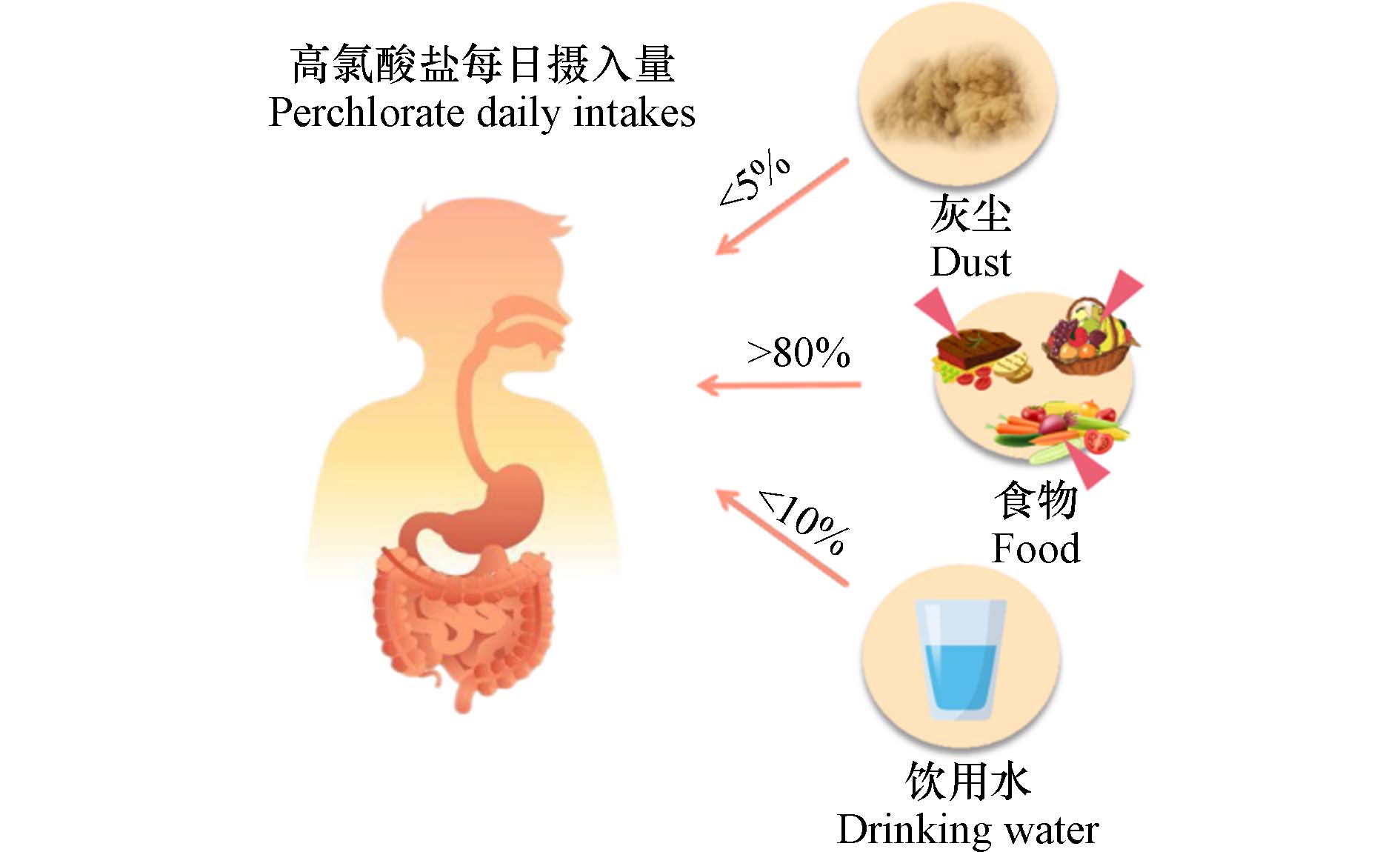
ClO4−中的Cl原子被4个氧原子呈正四面体结构包围,使其呈现化学惰性,具有极其稳定的物理化学性质. 此外,ClO4−易溶于水,在水中流动性强,扩散范围广,经水、土壤等途径被动植物吸收,并通过食物链的传递进入人体[6]. 总而言之,摄食和饮水是人类接触ClO4−的主要途径[7 − 8]. 此外,ClO4−还存在于室内外灰尘中[9],可能对人类健康构成直接威胁(图1).
ClO4−是一种具有持久性的有毒污染物质,通过与碘离子竞争性地进入哺乳动物和人体的甲状腺,阻碍甲状腺对碘离子的吸收,导致甲状腺激素失衡并扰乱甲状腺抗体水平[10]. 此外,它还能抑制三碘甲状腺原氨酸(T3)和甲状腺素(T4)的合成,影响人体的新陈代谢和生长发育[11],尤其是可能造成胎儿或婴儿的神经系统损伤和影响大脑组织的发育,引发智力缺陷、学习障碍等不良症状[12]. ClO4−污染及其造成的健康安全事件在世界各地相继发生,已成为一个与人类生活息息相关的全球性问题[13].
近年来,ClO4−领域的研究主要集中在ClO4−的检测、暴露风险评估以及其控制去除技术的研发与改进. 目前国际公认的ClO4−安全参考剂量(Reference Dose, RfD; RfD=0.7 µg·kg−1·d−1)由美国率先提出,后被美国环境保护署(Environment Protection Agency, EPA)采纳并作为官方推荐安全浓度限值[14]. 而其他大多数国家尚未确立ClO4−的限量标准,或是此类标准并未明确指出某些受ClO4−污染风险较高的检出源中的浓度限值. 鉴于此,本文对国际上较为典型的ClO4−的标准规定进行了直观地分类归纳,以期为各地相关法律法规的健全与完善提供一定参考. 针对世界各地出现的ClO4−污染情况,本文重点综述了ClO4−在饮用水、食品及室内外灰尘等主要暴露途径下的污染现状,进一步阐明其时空分布特征和迁移转化规律. 最后,对国内外饮用水中高氯酸盐的物理化学去除技术进行总结与分析,以推动ClO4−风险评估及污染防控工作的深入研究.
-
ClO4−浓度受饮食文化、工业化程度、环境治理水平等多种因素影响,在全球范围内ClO4−分布呈现出明显的地理性差异,因此,相关法律法规也因地制宜地对ClO4−浓度设定了不同的限值(表1).
EPA早在2005年就确定了食品中ClO4−的参考剂量RfD为0.7 µg·kg−1·d−1[14],折算到饮用水的当量水平为24.5 µg·L−1,2008年EPA将饮用水当量浓度更新至15 µg·L−1[15],这与法国食品、环境及劳动卫生署(French Agency for Food, Environmental and Occupational Health& Safety, ANSES)建议的饮用水中ClO4−的浓度限值一致[16]. 为了避免ClO4−区域分布差异对制订统一标准的影响,美国加州公共卫生部(California Department of Public Health, CDPH)[17]和马萨诸塞州环境保护部(Massachusetts Department of Environmental Protection, MDEP)[18]分别出台了相应的地方性环境法规.
2010年世界卫生组织(World Health Organization, WHO)将ClO4−的暂定每日最大耐受摄入量(provisional maximum tolerable daily intake, PMTDI)定为10 µg·kg−1·d−1体质量[19]. 欧洲食品安全局(European Food Safety Authority, EFSA)发布的每日可耐受摄入量(Tolerable Daily Intake, TDI)为0.3 µg·kg−1·d−1体质量[20]. 此外,EFSA还进一步提出了不同类别食品中ClO4−的临时参考水平(表2),主要包括水果、蔬菜、茶叶等.
多项研究表明,婴儿能通过摄食母乳、配方奶粉或其他婴儿食品等途径直接接触ClO4−[7,21 − 23],长期摄入可能导致甲状腺功能紊乱甚至影响婴儿生长发育. 因此,婴儿食品中ClO4−含量是否安全受到公众广泛关注. 但目前食品卫生安全领域的相关权威机构尚未明确规定婴儿食品中ClO4−浓度限值或对此制定法律法规.
-
居民生活饮用水主要有两大来源:市政自来水和瓶装/桶装水,其中自来水取自地表水或地下淡水,而瓶装/桶装水一般又分为纯净水、矿泉水和矿物质水(由纯净水经人工添加适量矿物质和灭菌处理加工而成). 目前已有多项研究报道了饮用水中ClO4−的存在(表3).
Erdemgil等[25]从土耳其5个城市采集的自来水中ClO4−的暴露水平虽均在安全限内,但值得注意的是开塞利市的ClO4−检测值明显高于其他4个城市,主要原因可能是自来水水源(地表水或地下水)的差异. 开塞利市当地气候干旱,选用地下水作为主要供水源,在高温环境中ClO4−的沉积速率远大于其在降水过程中的溶解速率[26],导致部分被土壤吸附或交换,最终ClO4−不断向地下水沉积富集. 有关智利地区的ClO4−暴露风险研究也显示出不同水源的ClO4−浓度差异,地下水ClO4−浓度(12.1 µg·L−1中值)超过地表水(中值1.8 µg·L−1)近10倍[27]. 由此可见,ClO4−在气候、地质和水文条件等因素影响下有逐渐向地下水沉积富集的趋势,使得各地区地下水ClO4−水平普遍高于地表水.
此外,水中ClO4−的含量受人类工业活动的影响较大. Alomirah等[28]发现,科威特艾哈迈迪省的自来水中ClO4−浓度最高为18.6 µg·L−1,超过中国成都市的暴露水平(最高浓度为1.61 µg·L−1)[29],这归因于该省是科威特的石油和天然气工业生产区;还有研究指出印度喀拉邦的地下水中ClO4−的平均浓度为773 µg·L−1,此检测值远超EPA建议的饮用水当量水平(15 µg·L−1)[30],原因在于取样点选取了当地高氯酸铵试验厂、航天中心等ClO4−生产及使用场所附近的水源. Kumarathilaka等[31]还分析了此类受污染程度较高的土壤或地下水中ClO4−与重金属/微量金属的影响关系,指出ClO4−可能会加速土壤矿物的溶解,以致土壤或地下水中重金属/微量金属浓度升高. Vigreux-Besret等[32]发现处理厂出水因含氯消毒剂的添加,其ClO4−浓度可能比集水区高0.15—0.5 µg·L−1. 因此,在选取饮用水水源时需充分考虑ClO4−的分布特征,同时保证可能被ClO4−影响的重金属含量等水质指标在标准限定范围内.
Lutter等[33]从经济成本的角度探讨了控制饮用水中ClO4−污染物的必要性. 由于公共饮用水系统中的ClO4−浓度处于相对安全水平,受ClO4−影响的高敏感人群占比较低,且通过降低饮用水中ClO4−水平以保障碘的正常吸收并非是最有效的途径,直接摄入微量碘补充剂足以满足人类健康需求[34]. 因此他认为降低饮用水中ClO4−浓度以控制ClO4−污染的水环境治理效益相对较低. 但“治标”还得“治本”,加强综合治理从水源中控制ClO4−污染至关重要.
近年来关于国内外饮用水中ClO4−暴露量的研究,绝大多数集中在自来水中污染物浓度检测层面,而对于矿物质水和纯净水中ClO4−的污染情况的认识仍处于空白,且不同水源的饮用水中其他离子与ClO4−之间的影响关系也有待进一步探索. 在饮用水ClO4−污染物治理方面,应优先加强环境中(特别是ClO4−生产和使用区)ClO4−污染监测与防治,严格管控相关企业的污染排放,以保障居民饮用水卫生安全.
-
植物不仅受天然水体、土壤等媒介中ClO4−环境浓度的影响,而且灌溉水和天然肥料中的ClO4−在一定程度上也能向外释放并在植物根系中积累[20,36 − 42]. 为评估ClO4−对植物源加工食品安全的潜在威胁,各国(地区)展开了一系列调查研究(表4).
Liao等[43]对京津冀地区植物源加工食品(包括谷物、豆制品、马铃薯制品、蔬菜产品、水果产品和食糖)中的ClO4−浓度进行测定,结果显示水果和蔬菜产品中的ClO4−检出水平最高,原因可能是其叶片面积较大,气孔数目多,在蒸腾作用下植物根部更易吸收水分,促进了ClO4−的运输. 就水果、蔬菜农产品而言,其对ClO4−的吸收速率可能受品种,气候条件以及竞争离子等因素影响而显现差异. Wang等[44]依据来自加拿大首都渥太华零售店的进口和国产食物样本,评估了加拿大人摄入水果和蔬菜可能接触到的ClO4−情况. 均由智利进口的两个不同品种的葡萄实验组的ClO4−含量存在差异,其中绿葡萄ClO4−的平均浓度为(45.5 ± 13.3)µg·kg−1,而无籽红提中ClO4−含量的平均值为(9.86 ± 15.1)µg·kg−1,产生这种差异的原因可能是受基因型以及生长和运输过程中环境因素的影响;值得注意的是,水果中哈密瓜的ClO4−暴露水平最高,其中产自危地马拉的哈密瓜的ClO4−平均含量高达(156 ± 99.5) µg·kg−1,已超过EFSA规定的临时参考水平(0.2 mg·kg−1). 另外,在中国武汉测定的蔬菜中ClO4−的结果显示,叶类蔬菜尤其是菠菜,相比于其他种类蔬菜(如黄瓜、胡萝卜等)更易吸收环境中的ClO4−[45],这一趋势与其他相关研究报道的结果一致[46 − 47]. Seyfferth等[38]还探究了两种气候条件下(“多云、潮湿、凉爽”:相对湿度=80%, 温度=18/15 ℃, 光量子通量密度=250 µmol·m−2·s−1;“晴朗、干燥、温暖”:相对湿度≤50%, 温度=28/18 ℃, 光量子通量密度=500 µmol·m−2·s−1)生菜在1.25 µg·L−1和10 µg·L−1ClO4−浓度体系中的污染物积累特征。研究表明,受气候影响,植物蒸腾速率相差2.0—2.7倍,导致生菜中ClO4−累积量呈现1.2—2.0倍的差异. 此外,NO3−已被证明能抑制大麦根对氯酸盐(ClO3−)的吸收,原因是这两种离子具有相同的转运机制[48],同理,ClO4−在植物中的跨膜运输效率可能也受NO3−等同类竞争离子的制约[49].
-
动物可在饮食过程中摄入ClO4−,食用动物加工食品时ClO4−可经食物链传递至人体内,对食品安全和人类健康构成直接威胁. 动物加工食品中肉类产品、蛋制品、水产品(包括海鲜)和乳制品中均存在可量化的ClO4−(表5).
Gan等[29]和Wang等[45]分别检测了中国成都市和武汉市动物食品样本中的ClO4−水平,发现鸡蛋中ClO4−的平均浓度(分别为15.3 µg·kg−1和15.86 µg·kg−1)远高于肉类,即使这两个地区肉类中ClO4−平均浓度相对不高,但却高出韩国肉类(0.60 µg·kg−1)[50]数倍. 值得注意的是,中国成都市的奶类中也检测出高浓度的ClO4−(均值:14.4 µg·kg−1)[29],这些高暴露水平的ClO4−可能来自喂养家畜的动物饲料和水源. 例如,Guruge等[51]比较了日本商业牛奶和现场直接采集的新鲜(生)牛奶中的ClO4−浓度,发现受畜牧业养殖过程中饲料差异的影响,商业牛奶中ClO4−的浓度明显更高. 此外,奶类中的ClO4−浓度或许还与家畜(牛/羊)的品种有关. Sungur等[52]测定了土耳其哈塔伊地区内牛奶、山羊奶、绵羊奶中ClO4−的平均含量,其中山羊奶(0.26 µg·kg−1)和牛奶(0.25 µg·kg−1)接近,绵羊奶检出水平最低(0.11 µg·kg−1). 同时,有关贝类的研究也表现了ClO4−的物种特异性积累. 位于中国南海海域内的深圳8种贝类(珠母贝、花蚬、近江牡蛎、华贵类栉孔扇贝、紫贻贝、方斑东风螺、杂色鲍和平蛤蜊)样本中,ClO4−平均浓度最高的是华贵类栉孔扇贝(14.0 µg·kg−1),其次是杂色鲍(11.6 µg·kg−1),方斑东风螺(2.33 µg·kg−1)最低[53],但对于物种差异导致ClO4−特异性积累的原因仍有待进一步研究.
-
人类除了通过摄食和饮用水途径接触ClO4−外,室内外灰尘也是ClO4−暴露的主要来源. 近年来,各国家及地区的灰尘样本中都检测出了一定浓度的ClO4−(表6).
Wan等[54]比较了中国、美国、印度等12个国家室内灰尘的ClO4−浓度,结果表明中国室内灰尘样本中的ClO4−浓度明显高于其他国家,其主要原因可能是中国人燃放烟花爆竹的节日习惯加剧了ClO4−污染. Gan等[55]在中国传统节日春节前后从中国北方地区采集了室外灰尘样本,部分采样点检出的ClO4−含量高达
5300 mg·kg−1,出现ClO4−浓度过高的原因可能是春节期间烟花爆竹的燃放产生的ClO4−残留物暴露于大气中,使得个别样品检测值偏高. Li等[9]发现在西藏某居住人口稀少的地区,灰尘中检出的ClO4−样本浓度值最低(0.01 mg·kg−1),而烟花生产区附近的ClO4−浓度高达815 mg·kg−1. 除上述烟火生产与燃放影响以外,人类生活习惯如开窗通风的频次也可能改变ClO4−水平[29]. Li等[9]还探究了季节差异对灰尘、土壤中ClO4−浓度的影响,结果表明中国人在冬季接触ClO4−的风险高于夏季,原因可能是夏季雨量充沛,灰尘中强水溶性的ClO4−被雨水溶解,转移到其他环境介质中. 室内外灰尘之间的ClO4−浓度在一定条件下存在相互影响关系[56]. Vella等[57]分析了马耳他国家同一地点收集的室内外灰尘之间ClO4−浓度的相关程度,结果表明两者具有强相关性,室外环境中负载ClO4−的大气颗粒可通过空气等媒介进入室内,影响室内ClO4−水平.综上可知,灰尘中ClO4−水平不仅受外部条件如人类活动、气候条件等因素的制约,室内外灰尘之间的ClO4−也会相互转移和累积,而室内外灰尘中ClO4−的来源及两者之间的转移机制有待进一步研究.
然而,据研究统计显示,室内灰尘对ClO4−日摄入的贡献不大(<5%)[29],摄食和饮水是中国人接触ClO4−的主要途径. 一般而言,大多数中国人的ClO4−日平均摄入量低于EPA的参考标准,但我国是世界上最大的烟花爆竹生产和消费国家,研究表明,烟花燃放后的水体和大气中存在高浓度ClO4−[58],故此标准未必能较好匹配或界定我国居民的ClO4−摄入量的安全限值. 即使该研究评估显示饮用水对ClO4−日摄入贡献率还不足10%,但由于ClO4−的水溶性极高(25 ℃时为200 g·L−1)[59],随地表水、地下水快速扩散,经食物链在动植物体内生物富集与积累,综上,究其原因在于饮用水水源为ClO4−污染途径的主要源头.
综上,ClO4−各暴露途径贡献率见图2.
-
如前所述,饮用水水源作为ClO4−污染途径的主要源头,如何有效控制其人群暴露风险是近年来研究工作的重点. 目前,处理饮用水中ClO4−污染物的方法包括物理法、化学法和生物法等,但由于生物法还原高氯酸盐对水质如pH值、温度和其他有机污染物非常敏感,电子供体的可获得性影响了ClO4−的生物降解的环境可持续性,且水体中各种病原生物的存在易造成处理工艺出水的二次污染,使得生物降解技术的应用受到限制[60],因此,本文重点综述了国内外饮用水中ClO4−的物理化学去除技术的研究进展,其中涉及的主要去除技术与原理如表7所示.
-
水处理过程中,常用颗粒活性炭(GAC)等吸附剂去除ClO4−. 一般来说,吸附效果受初始ClO4−浓度、溶液温度、pH值、接触时间和共存阴离子等多种因素的共同影响. 由于吸附剂的结构特征、表面性能直接制约着吸附材料的吸附能力,基于目前公认的离子交换、静电相互作用和表面络合的吸附机理,近年来大多数研究集中在新型吸附材料的探索以及吸附剂表面改性技术的研发与改进两大方面,以提高吸附法去除ClO4−的效果.
Krishnan等[61]采用纳米羟基磷灰石(nHA)及其磁性纳米复合材料(SPIONS@nHA)吸附实验室配水中的ClO4−,通过间歇吸附实验比较了不同吸附参数条件下两种材料对ClO4−的去除效果,nHA和SPIONS@nHA的最大吸附量分别为148.4 mg·g−1和305.8 mg·g−1,均高于目前使用广泛的GAC吸附剂. 此外,近中性pH值(pH=6—8)条件下ClO4−吸附同时发生PO43−的离子交换以及与正电荷吸附剂表面的静电相互作用,此时吸附效果最佳. 此外,季铵化改性磁性Mg/Al-层状双氢氧化合物(N8881Cl-LDH@Fe3O4)[62]、AIE效应超分子聚合物凝胶(PT-GEu)[63]和环氧氯丙烷交联壳聚糖水凝胶(ECH-CSBs)[64]等新型材料对水介质中的ClO4−均表现出良好的吸附性能,具有潜在的应用价值. 除吸附材料本身外,合成工艺对活性炭吸附剂的表面化学性质和多孔结构有显著影响,其中,表面改性可有效提高活性炭吸附容量. Rekha等[72]在Pluronic 123(P123)孔模板下利用KOH活化聚吡咯制备的氮掺杂活性炭对水中ClO4−的吸附容量高达587 mg·g−1,且材料可重复利用性能优良. Wang等[65]通过ZnO纳米颗粒调控生物炭的孔隙结构,用甜菜碱修饰生物炭的表面官能团,进而制备出了绿色季铵氮功能化介孔生物炭电极. 其增强机制为通过掺杂季胺氮基团引入额外的赝电容,改善电极的表面润湿性和电导率,从而加快双电层形成速率,提高了生物炭的电化学性能和电吸附能力.
需要注意的是,吸附过程只改变了ClO4−所在的位置,并没有将其还原降解成无毒的Cl−,因此,应充分考虑吸附剂解吸和避免产物可能引起的二次污染问题. 此外,新型吸附剂及其改性技术在市场上的竞争优势不仅取决于吸附材料的吸附性能,如吸附剂的吸附容量、化学稳定性、可重复使用性和可回收性等材料特性,材料成本的增加和高能耗的制备工艺等实际问题也不容忽视.
-
压力驱动膜过滤技术如纳滤(NF)、超滤(UF)、反渗透(RO)和电渗析(ED),被认为是去除ClO4−的有效工艺[73]. 针对水体中ClO4−污染防控的水净化膜技术,Li等[66]设计了一种聚偏氟乙烯-金属有机骨架复合超滤膜(PVA/Cu-iMOFs/PVDF-0.05),利用Cu-iMOFs的磺酸(R-SO3)配体与ClO4−之间的离子交换特性来捕获水中的ClO4−. 此外,反渗透膜主要用于地下水中ClO4−的去除,但由于污染物长期在膜表面或膜孔内的吸附、沉积,易造成膜孔堵塞,导致膜渗透流量下降,影响反渗透膜性能. 为减缓反渗透膜的污染,Yang等[67]在反渗透系统前增加了倒极电渗析工艺,ClO4−在电解过程中会生成盐酸,抑制氢氧化物的形成,从而减缓后期反渗透处理中污垢在膜上的积累. 此外,研究表明,倒极电渗析和反渗透一体化组合工艺(EDR + RO)中ClO4−的分离效果与工作电压呈正比,在40 V电压下,2.5 h内可去除高达95%的ClO4−. 经两级处理(EDR + RO)后,出水ClO4−浓度降至0.02 mg·L−1以下. 然而,由于RO工艺截留率易受初始ClO4−浓度影响,通常在处理低水平ClO4−污染的地下水时展现出良好的竞争力. 为了拓宽其适用范围,Russel等[74]在RO装置前串联了厌氧膜生物反应器(AFBR)和陶瓷微滤(MF)单元,以实现井水中ClO4−较高水平暴露条件下(15 mg·L−1)的水污染修复. 结果表明,约97%的ClO4−在AFBR单元被生物降解,RO膜只需负责吸附剩余部分(0.4 ± 0.35 mg·L−1),而MF的作用主要是控制AFBR出水中的菌落总数,对RO膜进水预处理,以减缓膜污染,保障AFBR-MF-RO生物-物理复合工艺能持续稳定发挥降解效能.
-
离子交换因其高效、操作方便和吸附容量高的特点已成为目前去除饮用水中ClO4−污染的主要途径. 近年来,多种阴离子交换树脂(不同基体或官能团)被研发用于水体中ClO4−的去除,其去除效率受树脂的交换能力、稳定性、选择性和再生能力等多种因素影响[31]. Zhu等[69]研究表明,在磁性离子交换树脂(MIEX)、Purolite A530E和Purolite A532E等 3种树脂中,由于不同树脂特定官能团和基体结构组成的差异,这两种Purolite树脂相较于MIEX树脂具有更优越的ClO4−选择性. 另外,ClO4−能自发地经历本体溶液迁移、边界层迁移、颗粒内迁移和本征吸附4个阶段被树脂化学吸附去除. 然而,天然水体中通常存在多种无机阴离子与ClO4−构成竞争关系,从而影响树脂吸附能力和ClO4−去除效果. Song等[70]探讨了共存离子(Cl−、SO42−、NO3−)对玉米秸秆改性磁性生物聚合物离子交换树脂(CS-MAB)去除水中ClO4−的影响,发现上述共存离子抑制CS-MAB去除ClO4−效果的影响程度排序为:SO42−>Cl−>NO3−. 此外,与MIEX树脂降解机理不同,CS-MAB降解ClO4−的整个过程中主要是以化学反应尤其是离子交换为主.
离子交换法去除ClO4−的技术限制主要是后期如何实现树脂中ClO4−的高效解吸,以保证树脂的可再生性和可持续利用. Faccini等[71]的研究表明,ClO4−在强碱性阴离子交换树脂中吸附和解吸的速率可能主要受化学吸附阶段限制. 此外,有研究指出,强碱性阴离子交换树脂似乎更适用于解决ClO4−浓度低于50 mg·L−1的水污染问题[75].
-
随着ClO4−在生产和使用过程中的排放,环境中的ClO4−含量逐渐上升,全球范围内饮用水、食品、室内外灰尘等介质中都能检测到ClO4−的存在. 对ClO4−的研究最早在美国引起了广泛关注,目前大多数国家对饮用水和食品中ClO4−的限量标准也通常基于美国及欧盟有关环保部门制订的法规或参考意见. 然而,由于ClO4−在不同地理区域的分布差异显著,此标准未必能适应当前形势下各国或地区对ClO4−安全限值摄入量的根本要求,亟待进一步完善. 在此基础上,应加强ClO4−在多介质环境中的监测并摸清其危害程度,以根据相应的人群暴露风险优先对特定污染源实行分类分级监管. 此外,为推动ClO4−环境化学行为的深入研究,针对ClO4−的分布、迁移与转化规律,提出如下建议:(1)加强ClO4−与多介质环境中共存阴离子的竞争机制研究;(2)明晰室内外灰尘中ClO4−的来源及两者之间的转移机制;(3)大气尘中的ClO4−是否可能成为土壤潜在污染源的问题有待进一步研究和解答.
饮用水水源是ClO4−污染途径的主要源头,为控制饮用水中ClO4−暴露风险,物理化学处理是去除饮用水中痕量ClO4−最常用的技术,包括吸附、膜过滤和离子交换. 在吸附法中,许多新型吸附剂及其表面改性复合材料均可经济高效地去除饮用水中的ClO4−,但吸附过程仅改变ClO4−所在的位置,并没有还原降解ClO4−并转化为Cl−,这可能导致ClO4−再次释放到环境中. 因此,应充分考虑吸附剂解吸和避免产物可能引起的二次污染问题. 与吸附法相似,离子交换也需实现树脂中ClO4−的高效解吸. 需要注意的是,选择性树脂是不可再生的,而非选择性树脂在可再生时会产生含高浓度ClO4−的废物流,今后研究可重点关注离子交换树脂与物化及生物降解技术的联合应用,解决其选择性和可再生性等应用难题. 此外,膜过滤技术易引起膜污染且成本较高,通常不适用于净化高浓度ClO4−的污染水体,但在膜过滤技术前增设预处理和膜污染防治单元或许是拓宽其应用前景的有效策略.
人群暴露高氯酸盐污染及其在饮用水中去除技术:综述
Population exposure to perchlorate contamination and its removal technologies in drinking water: A review
-
摘要: 高氯酸盐(perchlorate,ClO4−)是一种具有高水溶性、高度扩散性和持久性的有毒污染物质. 受自然和人为因素影响,ClO4−存在于大气、土壤、水体等公共环境中,并可通过食物链的传递进入人体. 此外,饮水和接触室内外灰尘也是人类暴露于ClO4−的主要途径. 因此,ClO4−的广泛存在会对人类健康构成直接或潜在威胁. 目前的研究主要集中在ClO4−的检测、暴露风险评估以及其控制去除技术的研发与改进. 大多数国家仍尚未确立ClO4−的限量标准,而是借鉴美国及欧盟有关环保部门制订的法规或参考意见. 鉴于此,本文对国际上有关ClO4−限量标准的规定进行了归纳分类,以期为形成更全面的环境健康和食品安全标准提供参考. 此外,还分别综述了ClO4−在不同暴露途径下(饮用水、食品及室内外灰尘等)的污染现状. 最后,讨论了物理化学方法去除水中ClO4−的内在机理与技术难点,由此展望相关技术的应用前景.Abstract: Perchlorate (ClO4−) is a toxic pollutant with high water solubility, high mobility and persistence. Perchlorate is present in atmosphere, soil, water, and other public environments due to natural and human factors, and can enter the human body through the food chain. In addition, drinking water and contacting with indoor and outdoor dust are also the main ways of human exposure to perchlorate. Hence, the presence of perchlorate poses a direct or potential threat to human health. The current researches mainly focus on the detection, assessment of exposure risks, as well as development and refinement of control technology of perchlorate. Most countries have not yet set up the guideline of perchlorate, but referred to the regulations or reference opinions present by the relevant environmental protection departments of the United States and the European Union. Given this, our review compiled and classified the international standard regulations on perchlorate, with a view to providing a reference for the establishment of more comprehensive environmental health and food safety standards. In addition, the pollution status of perchlorate under different exposure ways (i.e., drinking water, food, as well as indoor and outdoor dust, etc.) was summarized. Finally, the intrinsic mechanism and technical difficulties of physical-chemical treatment technologies for removing ClO4− from water were discussed, and the application prospects of related technologies were prospected.
-
Key words:
- perchlorate /
- standard regulations /
- pollution status /
- physical-chemical treatment /
- human exposure.
-
石化企业是国民经济的支柱产业,给国民带来了极大的能源和经济利润,同时也属于重大的污染源。其中大气污染以无组织排放为主,污染物主要是种类繁多的挥发性有机污染物(VOCs)。挥发性有机污染物(VOCs)是一类常见的大气污染物,是臭氧和二次有机气溶胶的重要前体物[1-2],本身也具有毒理特性,对人体肝脏、血液健康等具有剧烈的生理毒害作用[3-4],部分污染物三氯甲烷、四氯乙烯、苯等对人体甚至有致癌、致畸、致突变作用[5],其污染物排放清单和污染物排放特征研究引起国内外学者的广泛关注。
储罐是石化企业最常用的生产装置,更是最主要的无组织排放源之一,据统计,我国每年约有千万吨级的VOCs从有机液体储罐挥发到大气中[6];美国2003年的一份调查显示,在美国18家石油公司各项污染源VOCs排放量中,储存过程中VOCs排放量约占总排放量的29%[7]。自储罐产生的无组织排放VOCs,一方面降低了油品质量、造成了资源的大量浪费,另一方面排放的大气污染物严重危害了人们的生命健康和生态环境,并带来了一系列的安全问题[8-10]。
目前世界各国自有其适用的储罐无组织排放VOCs定量方法,或是半经验半理论法,或是纯经验法,比如美国环保署EPA推荐方法、美国石油协会API经验法、日本资源能源厅方法等。我国亦于2015年出台了《石化行业VOCs污染源排查工作指南》,然而工作指南中的核算方法是以EPA推荐方法为基准的,相关参数也是基于美国储罐构造现状推导出的,而由于在原料品质、储罐设计标准和管理水平等方面的差异,并不能完全适用于我国。
扩散模式反推法,计算精准、需求污染源信息少、示踪气体扩散过程拟合效果好,是工业点源、面源污染物排放量核算的重要方法,在工业无组织排放源强特征研究中应用广泛[11-14]。然而传统的扩散模式反推法,多采用以电化学和气相色谱为主的定点采样分析方法,就其本身而言,具有采样分析过程复杂、耗费人力物力大、测量范围小、只能反映监测区域定时、定点、局部的监测结果,不能实现大范围区域环境监测,无法实现实时在线自动监测,不能满足及时、准确、全面反映环境质量动态和污染源动态变化的需求。
随着科技的进步,环境监测技术的发展,仪器分析以及计算机技术的广泛应用,各国科技工作者将遥感技术广泛应用于大气环境自动监测系统。本文以遥感FTIR技术为基础,提出了一种针对于石化罐区VOCs源强的核算方法:基于遥感FTIR的扩散模式反推法[15],旨在为我国石化罐区无组织排放VOCs核算方法的构建提供数据、技术支持。
1. 实验部分(Experimental section)
1.1 源强反演模型构建
如果已知污染源下风向某点位的浓度及影响排放的相关信息,根据大气扩散理论,则可以计算出污染源的排放量,这是传统扩散模式反推法的理论基础。与传统的扩散模式反推法相比,本文所建立的源强反演模型的创新点在于以遥感FTIR技术取代了传统的定点监测技术,摆脱了以电化学和气相色谱为代表的定点采样分析方法所带来的局限性问题,并同时保留了遥感FTIR监测技术与扩散模式反推法两者的优势。
FTIR是一种基于光的干涉原理基础上进行了傅里叶变换的红外光谱监测技术,近年来,取得了迅猛的发展[16]。遥感FTIR能够实现大范围的环境监测,具有更强的实用性,在环境监测中具有良好的应用前景[17],但在研究气体组分或颗粒物浓度时,遥感FTIR的测定结果只能获得光路积分浓度数据(path integrated concentrations),不能直接反应待测参数(如气体浓度)在空间的分布,应用受到了限制。
如果一个模型能够精准的模拟大气污染物的扩散过程,任何可以测量且与大气扩散模型及污染物排放量Q相关的指标都可以用来表征大气污染物排放量Q。光路积分浓度PICs即为一合适指标,既避免了遥感FTIR不能直接反应待测参数(如气体浓度)的难题,又发挥了光路积分浓度的优势,如避免了定点采样分析所带来数据波动性问题,可以进行大范围的现场监测、测量速度快、灵敏度高、反应大范围浓度变化等[18-19]。
扩散模式反推法成功的关键其一便在于选择合适的大气扩散模型,考虑到模型的工程适用性,选择工业上应用最为普遍且经过大量实验验证过的高斯模型。在工程应用中,多将石化罐区简化为面源进行处理,常用的面源模式计算方法主要有两种,即ATDL模式和虚点源模式后置法。相比更适用于城市尺度大范围计算的ATDL模式,后置虚点源法更适用于工业面源。
反演模型成功的关键其二在于光路积分浓度PICs与排放量Q之间联系的建立。由于高斯模式在数理上具有解析形式,极大的降低了PICs与Q之间关系建立的难度。即基于监测光路与主导风向垂直假设,将点式浓度沿监测光路积分,依托高斯大气扩散模型,则可构建公式(1):
stringUtils.convertMath(!{formula.content}) (1) 式中,
u σy σy0 σz σz0 CL Cb QP 1.2 实验方案
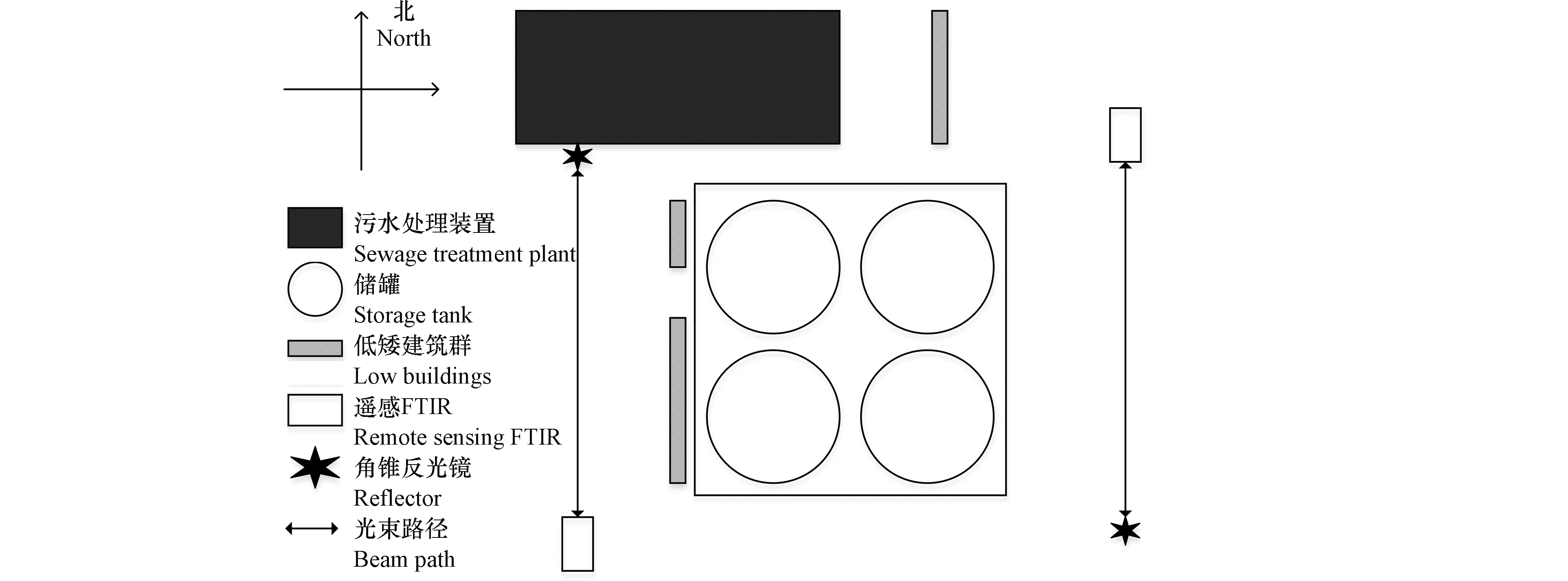
本研究所选取的20万m3石脑油实验罐区,位于我国北方地区某石化企业烯烃部东南角,罐区北侧和西侧周边有稀疏的建筑物,且高度普遍较低,西北角约500 m处有一污水处理装置,南侧和东侧为开阔荒野地带,无公路以及其它交通设施,车流量极少,其它污染源的影响较小,满足反演模型的应用条件。
在进行外场监测之前,进行了近一周的气象观测和现场勘查,以确定上下风向的监测点位,气象观测表明主导风向为西风,在该风向条件下,20万m3石脑油罐区的上下风向均有足够的开阔地,以布设OP-FTIR及辅助设备。
外场监测工作于2017年10月份持续进行了6 d,每天至少进行6 h上下风向连续不间断的光谱数据与气象数据同步采集,其中背景光谱数据采集在主导风向的上风向实施,VOCs排放光谱数据采集在主导风向的下风向进行,气象数据采集主要包括风向、风速、云量、温度、监测时间、大气压强等。外场监测布设详见图1。
在进行现场应用之前,已于我国北方某实验基地进行反演模型准确性验证及边界适用条件探索,研究表明,其监测点位的设置及数据采集分析处理应遵循以下原则:(1)背景监测点位应设置在主导风向的上风向,光路至污染源的监测距离应足够长,以避免污染源自身扩散对背景点位监测的干扰;(2)VOCs排放监测应设置在主导风向的下风向,监测距离需适中,以保证污染物扩散均匀,并满足所选用仪器设备的灵敏度需求,监测极限距离由监测设备与现场地形条件共同决定;(3)根据主导风向布置OP-FTIR主机与角锥反光镜,以保证主导风向与光束路径近似垂直,风向波动角度以小于15°为宜;(4)风速、大气稳定度、数据采集周期等分别以>1 m·s−1、C—E、15—60 min为宜,在本实验中以15 min为一个数据采集周期。
2. 结果与讨论(Results and discussion)
2.1 20万m3石脑油罐区周边VOCs浓度及化学组分分析
外场实验在2017年10月份开展,由于进行了6 d连续监测,风速、风向等气象条件也极为相近,具体数据采集状况如表1所示。
表 1 现场数据采集详细状况Table 1. Detail conditions of VOCs date acquisition实验编号Experiment number 监测日期Monitoring date 风向Wind direction 风速/(m·s−1)Wind speed 大气稳定度Atmospheric stability 光程长度/mOptical path length 1 2017.10.10 西 3.9—5.7 C,C—D 205 2 2017.10.11 西 3.7—6.9 C,C—D 205 3 2017.10.12 西 4.1—6.8 C,C—D 204 4 2017.10.14 西 4.1—6.5 C,C—D 205 5 2017.10.16 西、西-西北 3.9—6.3 C,C—D 205 6 2017.10.17 西、西-西南 4.1—6.9 C,C—D 205 C,弱不稳定。C, Less stable. D,中性;D, Neutral. | Show Table DownLoad:
CSV
DownLoad:
CSV
经过6 d的外场实验,共计采集光谱及气象数据76组,通过遥感FTIR监测仪对光谱数据进行分析,共计检出14种VOCs化合物,包括7种烷烃、3种烯烃、1种苯系物以及3种醛醚酚等其它化合物,将其浓度之和定义为VOCs。根据监测时间,将监测数据编为实验1—6,其背景点与受体点VOCs浓度如图2所示,实验5、实验6受体点VOCs浓度虽有一定波动,但整体上14种VOCs化合物的光路积分浓度均比较稳定,标准偏差约为0.31,说明20万m3石脑油罐区VOCs无组织排放稳定,适用于源强反演模型进行源强反演,此外受体点与背景点VOCs浓度存在明显差异,显示了罐区无组织排放VOCs对周边环境的显著影响,针对于实验5、实验6受体点VOCs排放浓度的差异性,后续将结合核算结果作进一步分析。
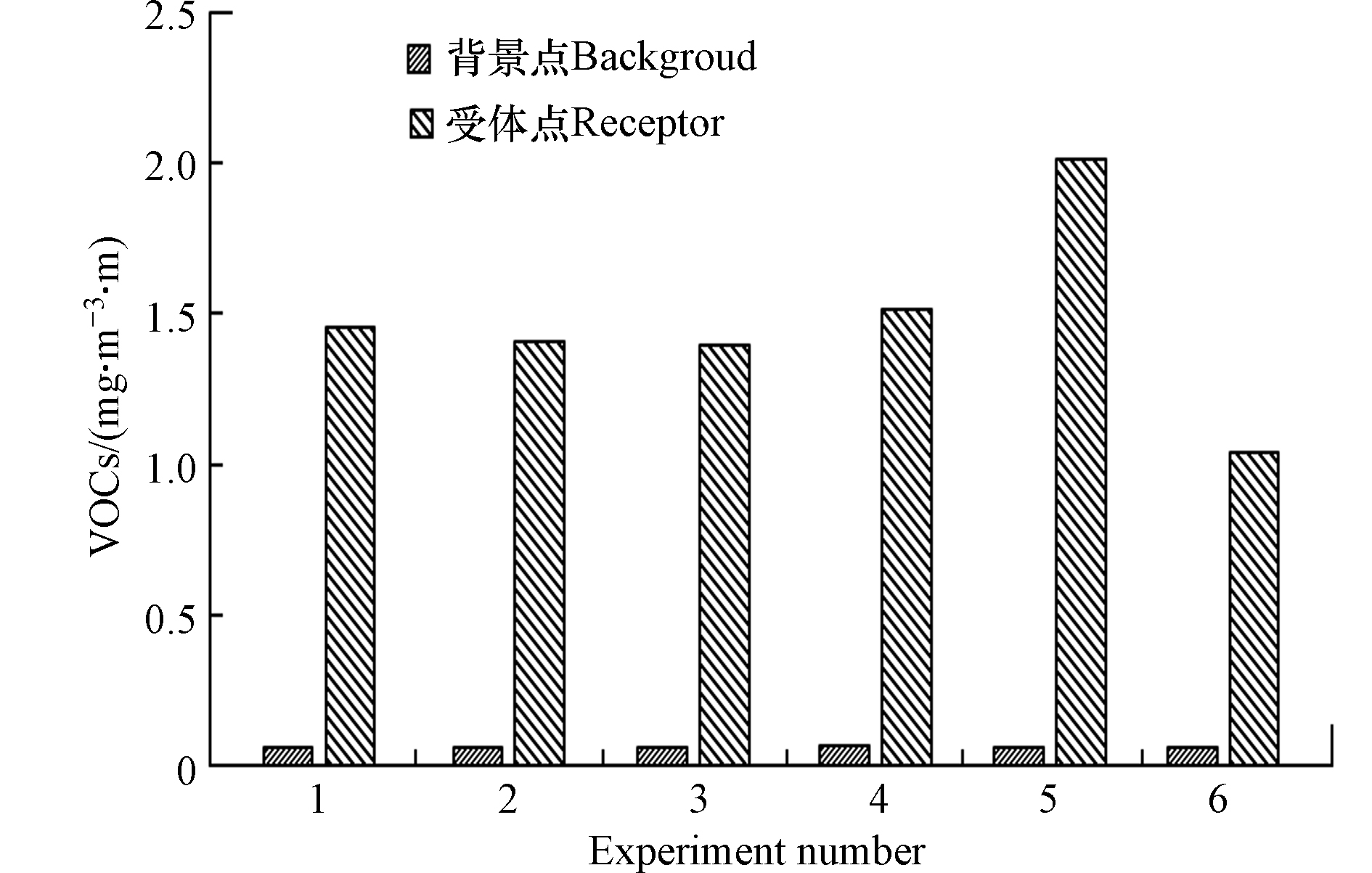 图 2 监测时段背景点与受体点VOCs浓度值Figure 2. VOCs concentration in receptors and background during monitoring period
图 2 监测时段背景点与受体点VOCs浓度值Figure 2. VOCs concentration in receptors and background during monitoring period2.2 20万m3石脑油罐区VOCs排放成分谱
受体点和背景点不止在VOCs浓度方面有着明显的区别,更在污染物成分上有着显著差异。在背景点中乙烯、乙烷所占比例最大,特别是乙烯占总体积浓度的30%,而在受体点中,乙烯的体积浓度百分比明显降低,仅占总体积浓度的8.48%,乙烷、庚烷的浓度明显增高,约占总体积浓度的50%。受体点和背景点之间VOCs的浓度差异以及污染物化学成分上的明显差异表明,20万m3石脑油罐区VOCs排放量大,对局地大气环境影响明显,可以使用源强反演模型进行VOCs排放量反演。
将剔除了背景点浓度的受体点VOCs作为该20万m3石脑油罐区所做的贡献,并以之来组成20万m3石脑油罐区VOCs排放成分谱图,其中14种主要的化合物体积浓度百分比如图3所示,20万m3石脑油罐区排放的VOCs主要成分为乙烯、乙烷、丙烷、异丁烷、己烷、庚烷、甲基叔丁基醚,其体积浓度百分比分别为8.48%、22.73%、10.35%、8.10%、7.11%、24.22%、6.90%。
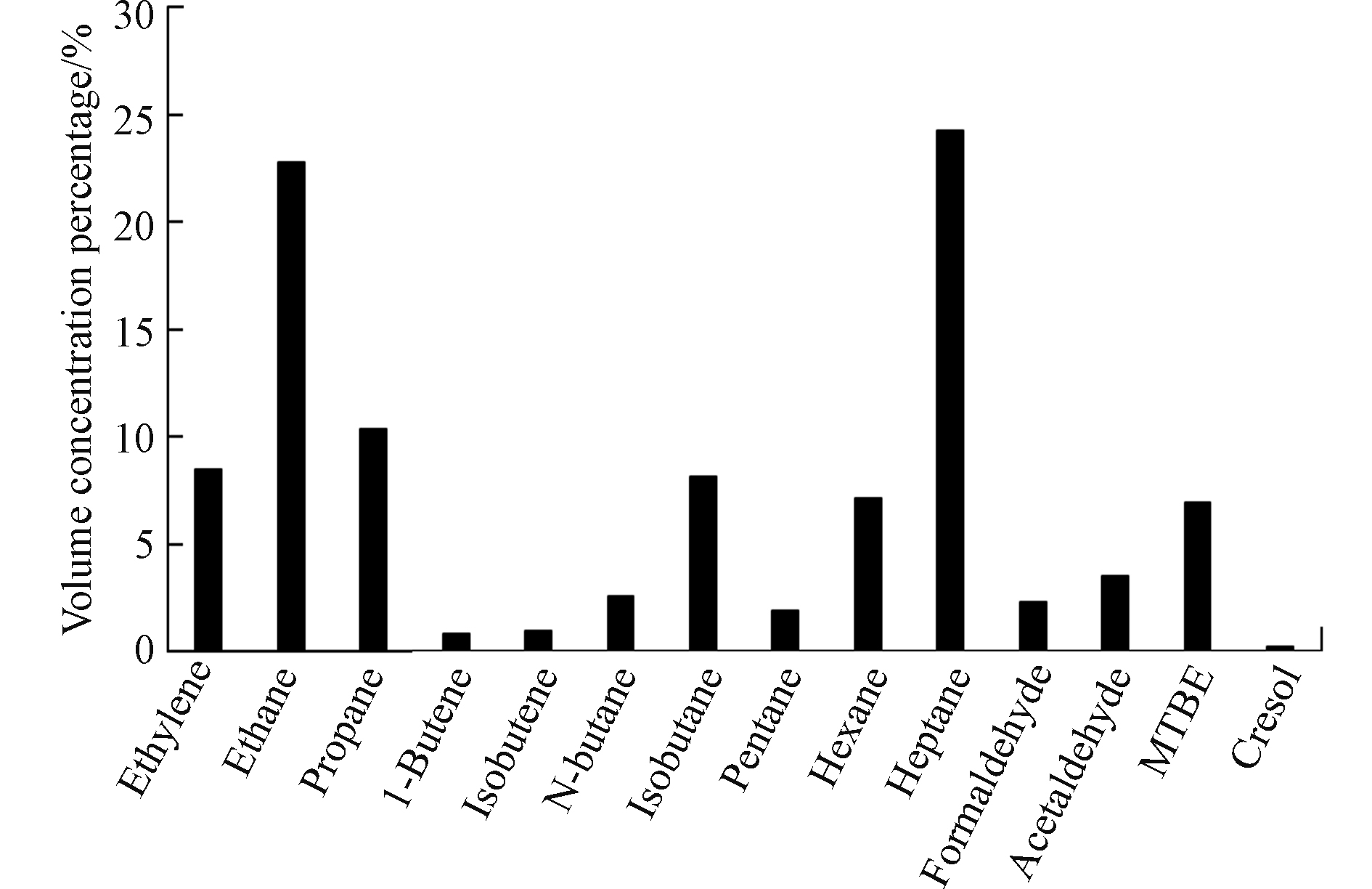 图 3 20万m3石脑油罐区VOCs成分谱Figure 3. Chemical profile of VOCs emitted from 200 thousand cubic meters tank area
图 3 20万m3石脑油罐区VOCs成分谱Figure 3. Chemical profile of VOCs emitted from 200 thousand cubic meters tank area2.3 20万m3石脑油罐区VOCs排放源强反演
表2以10月10日实验1为例进行说明,依据本文提出的源强反演模型对14种主要污染物进行源强反演,以14种污染物之和作为VOCs的主要代表物质,进而计算20万m3石脑油罐区VOCs的排放量Q,其中∆C代表两者的浓度差值。
表 2 实验1中 14种VOCs化合物推算结果Table 2. Calculation case of 14 species at experiment-1污染物Compounds ∆C/(mg·m−3·m) Q/(g·s−1) 乙烯 0.0490 0.0140 乙烷 0.1569 0.0460 丙烷 0.1117 0.0347 正丁烯 0.0127 0.0054 异丁烯 0.0064 0.0033 正丁烷 0.0467 0.0155 异丁烷 0.0930 0.0267 戊烷 0.0169 0.0051 己烷 0.1397 0.0414 庚烷 0.5929 0.1843 甲醛 0.0093 0.0027 乙醛 0.0298 0.0089 甲基叔丁基醚 0.1270 0.0370 甲苯酚 0.0004 0.0001 | Show TableDownLoad:
CSV
本研究共计进行了6组实验,获得了76组实验数据,将6组实验的VOCs源强反演数值总结至图4。如图4所示,前4组实验VOCs浓度基本保持稳定,在1.38 mg·m−3·m上下波动,而实验5、实验6 VOCs浓度存在一定差异,分别为1.95 mg·m−3·m和0.98 mg·m−3·m,同时反映到20万m3石脑油罐区VOCs年排放量上也有相似的规律,以前4组实验稳定数据作为20万m3石脑油罐区的核算基准,可得其排放量为0.38—0.42 g·s−1,均值为0.41 g·s−1,而实验5与实验6的核算结果分别为0.55 g·s−1和0.26 g·s−1。
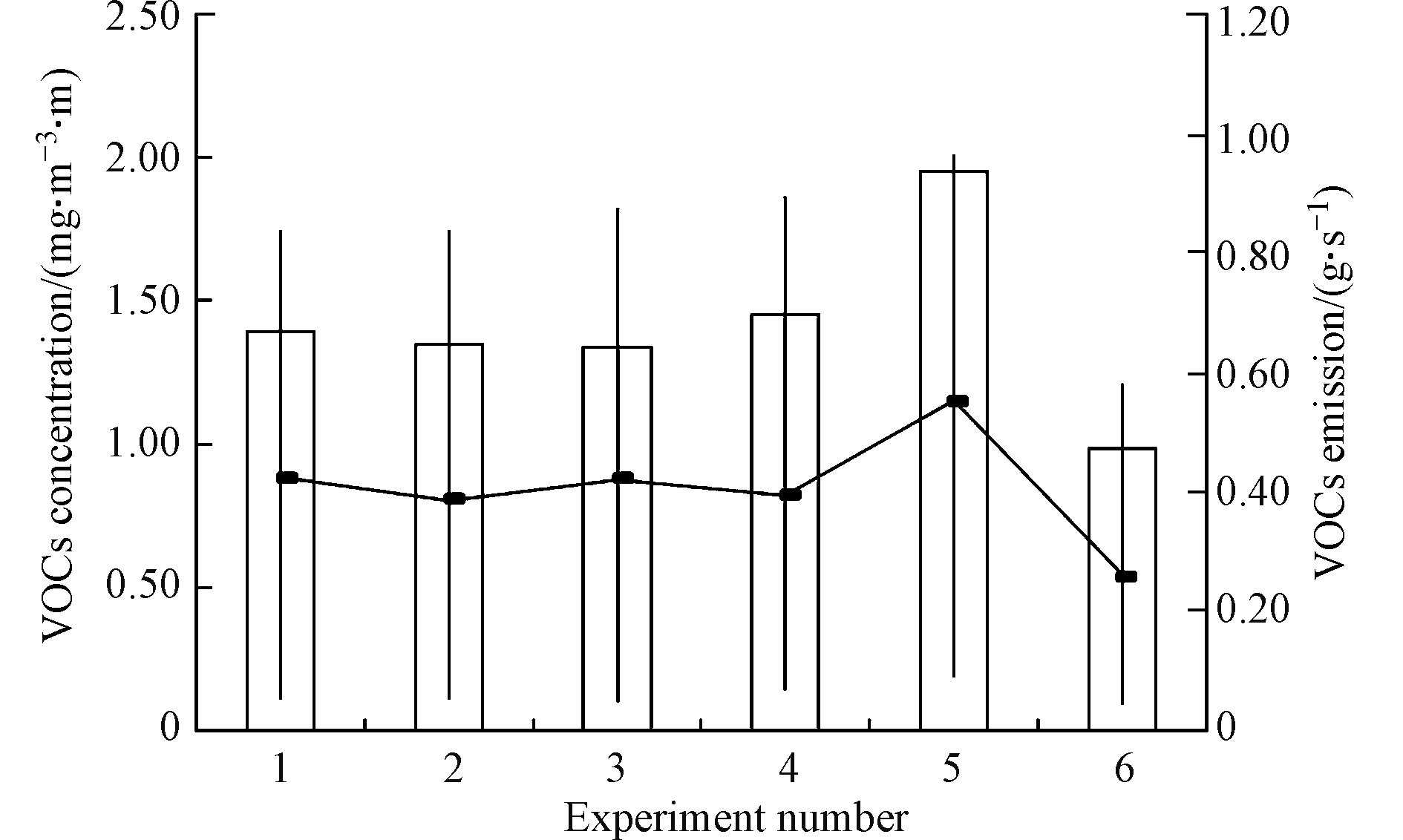 图 4 20万m3石脑油罐区VOCs排放量反演结果Figure 4. Estimate of annual VOCs emission amount by the 200 thousand cubic meters tank area
图 4 20万m3石脑油罐区VOCs排放量反演结果Figure 4. Estimate of annual VOCs emission amount by the 200 thousand cubic meters tank area对6组实验数据中的异常结果进行分析,实验5,污染物VOCs浓度明显高于其它实验,经与企业工作人员协商,罐区运行状态并无异常,与其它监测时段运行状态一致,下风向受体点污染物VOCs浓度偏高是受到西北侧污水处理单元的影响,而导致监测浓度并不仅仅来源于20万m3石脑油罐区,而是多源的叠加,监测数据明显异常,进而导致源强反演结果明显过大,实验6,参考采集的气象数据,实验6主导风向成西-西南风,而20万m3石脑油罐区西南侧为空旷地带,导致监测数据有所减小,亦使得数据的采集不符合反演模型的适用要求,反算结果误差较大。
2.4 经验公式法计算罐区排放VOCs
虽然通过源强反演模型对20万m3石脑油罐区无组织排放VOCs进行了源强反演,源强反演结果稳定,但是为了比较反演结果的准确性,仍需要与其它核算方法进行比较分析。
当下国内外主要的石化罐区无组织排放核算办法可分成两种,一种是半经验半理论方法,主要包括美国EPA推荐方法、中国《石油库节能设计导则》方法、中国石油化工(CPCC)估算法等,另一种是纯经验方法,主要包括美国石油协会(API)经验方法、欧盟排放系数方法[20]等。
我国亦于2015年出台了《石化行业VOCs污染源排查工作指南》[21],然而工作指南中的核算方法是以EPA推荐方法为基准的,对于罐区的无组织排放VOCs核算亦是如此,相关参数也是基于美国储罐构造现状推导出的,而由于在原料品质、储罐设计标准和管理水平等方面的差异,并不能完全适用于我国。尽管如此,工作指南中的核算方法,仍然是我国目前最主要的VOCs核算方法。如图5所示,模型反演结果与工作指南核算结果有一定差距,其中工作指南核算结果为0.65 t·month−1,计算结果来源于所研究炼化企业根据工作指南的自身核算,而源强反演核算结果为1.07 t·month−1,反演核算结果约比工作指南核算结果高45%。
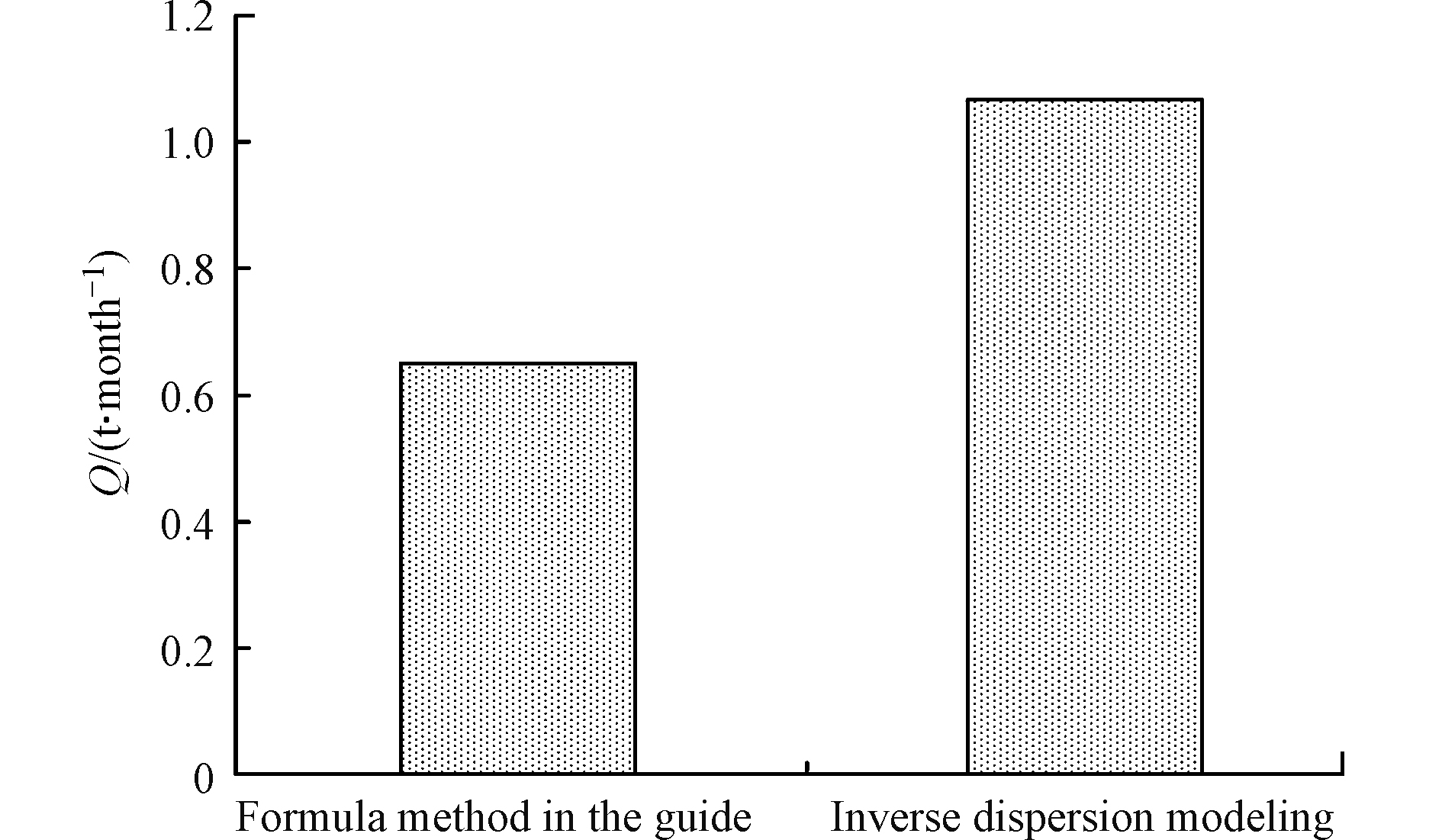 图 5 20万m3石脑油罐区10月份VOCs排放量估算结果Figure 5. Estimate of VOCs emission by the 200 thousand cubic meters tank area in October
图 5 20万m3石脑油罐区10月份VOCs排放量估算结果Figure 5. Estimate of VOCs emission by the 200 thousand cubic meters tank area in October综合考虑指南核算方法与源强反演核算方法,从两个角度对图5的结果进行分析,指南核算方法以EPA核算方法为依托,EPA推荐的核算方法,综合考虑了各种储罐和储存物料,计算过程考虑了各种影响因素,并具有缜密的公式推导和实验基础,然而针对于储罐无组织排放的VOCs核算自然是以美国的自身状况为基础,其气候条件、罐体特征、管理方法、控制措施等与我国存在一定的区别,使得该方法在美国适用良好,在我国进行的石化现场核算必然存在一定差距,国外的许多核算方法在我国石化企业直接使用,会造成较大的误差,不宜直接使用。
基于遥感FTIR-扩散模式反推的源强反演方法,前期通过模拟气体释放实验,已验证该反演模型反演结果稳定可接受,其中模拟误差<15%,然而在现场的实际应用中还存在来自现场监测环境、气象条件以及储罐自身运行状态等因素的影响,以此评估储罐VOCs排放状况,误差范围会进一步扩大,不过,45%的差距仍是难以接受,分析可能与温度等气象条件有关,现场监测的数据采集是在晴朗的白天进行,并以此为基础推算10月份的VOCs排放量,没有考虑到夜晚低温带来的低排放量状况,而导致整体核算结果偏高。
3. 结论(Conclusion)
针对于石化罐区无组织排放VOCs,建立了同时具备遥感FTIR和扩散模式反推法两者优势的源强反演模型,摆脱了遥感FTIR在不能直接反映待测参数(如气体浓度)在空间的分布方面的限制,拓宽了遥感FTIR技术的应用范围,亦避免了传统扩散模式反推法在定点采样分析方面的缺陷。
为了更详尽地检验源强反演模型的实际应用状况,同时为我国石油炼制行业储罐区VOCs排放特征提供参考,为工业面源VOCs排放量计算方法的构建提供数据支撑,在我国北方某石化企业20万m3石脑油罐区进行了现场应用实验。研究表明:(1)监测时段内20万m3石脑油罐区VOCs无组织排放稳定,受体点与背景点在VOCs浓度及化学组分上存在明显差异,20万m3石脑油罐区VOCs排放量大,对局地大气环境存在明显影响;(2)以扣除了背景浓度的受体浓度VOCs作为20万m3石脑油罐区所在的贡献,获得了20万m3石脑油罐区VOCs排放成分谱图;(3)针对20万m3石脑油罐区,分别以源强反演模型和指南公式法对其10月份VOCs排放量进行计算,结果分别为1.07 t·month−1、0.65 t·month−1,两者存在一定差异,分析主要原因是指南公式法在我国应用的不适用以及监测时段的温度条件所致。
虽构建了针对于罐区无组织排放VOCs的源强反演模型,也通过模拟实验检验了该模型核算结果的准确性,但现场应用条件的复杂性,打破了模拟实验的反演误差,为了准确确定污染源VOCs的排放状况,需进行长期的现场监测,综合考虑环境温度及装置的运行状态,并将排放量与之建立数量关系,以量化反演模型的准确性。
-
表 3 各国(地区)饮用水中ClO4−检出情况
Table 3. Perchlorate pollution in indoor and outdoor dust in various countries (regions)
国家(地区)Countries(regions) 检测对象Detection objects 检出浓度/(µg·L−1)Detection concentration 参考文献References 中国 成都 自来水 0.57—1.61(均值=0.86,中值=0.83, DN=51) [29] 矿泉水 0.15—2.84(均值=1.03,中值=0.75, DN=12) 智利 地下水 中值=12.1, DN=6 [27] 地表水 中值=1.8, DN=6 土耳其 伊斯坦布尔 自来水 0.04—0.09(中值=0.08, DN=60) [25] 安卡拉 0.07—0.21(中值=0.07, DN=35) 萨卡利亚 中值=0.04, DN=20 伊斯帕尔塔 0.02—0.07(中值=0.03, DN=15) 开塞利 0.23—0.31(中值=0.25, DN=15) 科威特 艾哈迈迪 自来水 0.01—18.6(均值=1.24) [28,35] 印度 喀拉拉邦 地下水 均值=773, DN=160 [30] 地表水 均值=79.41, DN=10 注:DN, 样本数. data number.
下载: 导出CSV
表 4 各国(地区)植物源加工食品中ClO4−检出情况
Table 4. Perchlorate contamination in plant-source processed foods in various countries (regions)
国家(地区)Countries(regions) 检测对象Detection objects 检出浓度/(µg·kg−1)Detection concentration 参考文献References 中国 北京, 天津, 河北 谷物 均值=6.2, DN=60 [43] 豆制品 均值=7.7, DN=60 马铃薯制品 均值=7.6, DN=60 蔬菜产品 均值=11.2, DN=60 水果产品 均值=10.4, DN=60 食糖 均值=2.0, DN=60 中国 武汉 菠菜 均值=140.2 ± 102.4, DN=15 [45] 生菜 均值=29.28 ± 66.37, DN=15 黄瓜 均值=7.77 ± 4.55, DN=15 胡萝卜 均值=8.73 ± 4.75, DN=15 加拿大 商品 原产国 [44] 菠菜 美国 均值=133 ± 24.9, DN=6 哈密瓜 哥斯达黎加 均值=0.61 ± 1.13, DN=6 哈密瓜 危地马拉 均值=156 ± 99.5, DN=6 绿葡萄 智利 均值=45.5 ± 13.3, DN=6 无籽红提 智利 均值=9.86 ± 15.1, DN=6 注:DN, 样本数. data number.
下载: 导出CSV
表 5 各国(地区)动物加工食品中ClO4−污染情况
Table 5. Perchlorate contamination in animal processed foods in various countries (regions)
国家(地区)Countries(regions) 检测对象Detection objects 检出浓度/(µg·kg−1)Detection concentration 参考文献References 中国 北京, 天津, 河北 肉类 均值=3.8, DN=60 [43] 蛋制品 均值=3.1, DN=60 水产品 均值=8.0, DN=60 乳制品 均值=3.7, DN=60 中国 成都 肉类 猪肉 ND—13.8(均值=4.60, DN=20) [29] 牛肉 ND—28.8(均值=5.61, DN=20) 淡水鱼 鲫鱼 ND—15.7(均值=4.45, DN=20) 鲤鱼 ND—16.0(均值=4.48, DN=20) 草鱼 1.40—11.7(均值=4.43, DN=20) 海鲜 带鱼 3.17—8.80(均值=5.94, DN=5) 螳螂虾 6.89—15.8(均值=12.1, DN=5) 扇贝 4.40—8.25(均值=6.34, DN=5) 蛋制品 蛋 LOQ—22.5(均值=15.3, DN=20) 乳制品 奶 9.38—25.2(均值=14.4, DN=20) 中国 武汉 肉类 猪肉 ND—14.48(均值=2.99, DN=15) [45] 牛肉 0.89—10.68(均值=3.72, DN=15) 蛋制品 蛋 ND—59.31(均值=15.86, DN=15) 韩国 肉类 猪肉 均值=0.32, DN=20 [50] 牛肉 均值=0.62, DN=20 鱼类和贝类 均值=0.95, DN=100 土耳其 哈塔伊 奶 牛奶 均值=0.25, DN=3 [52] 山羊奶 均值=0.26, DN=3 绵羊奶 均值=0.11, DN=3 中国 深圳 贝类 珠母贝 均值=5.99, DN=16 [53] 花蚬 均值=5.64, DN=15 近江牡蛎 均值=7.40, DN=42 华贵类栉孔扇贝 均值=14.0, DN=31 紫贻贝 均值=4.45, DN=13 方斑东风螺 均值=2.33, DN=11 杂色鲍 均值=11.6, DN=27 平蛤蜊 均值=4.22, DN=23 注:DN, 样本数. data number; ND, 未检出. not detected; LOQ, 定量极限. limit of quantification.
下载: 导出CSV
表 6 各国(地区)室内外灰尘中ClO4−污染情况
Table 6. Perchlorate pollution in indoor and outdoor dust in various countries (regions)
国家(地区)Countries(regions) 检测对象Detection objects 检出浓度/(mg·kg−1)Detection concentration 季节Season 参考文献References 中国 华北地区 室外灰尘 0.22—215(中值=5.01) 夏季 [9] 华南地区 0.01—921(中值=13.0) 全国范围 中值=8.1 北方地区 室内灰尘 中值=15.2 南方地区 中值=9.79 全国范围 中值=11.4 全国范围 土壤 均值=0.33, 中值=0.05 中国 北方地区 室外灰尘 0.132— 5300 (中值=37.4)春季2—3月(春节期间) [55] 南方地区 0.270— 3700 (中值=48.7)中国 天津 室内灰尘 0.72—119(均值=30.1, DN=54) 夏季 [29] 成都 0.11—38.8(均值=7.91, DN=51) 夏季 希腊 室内灰尘 0.08—4.67(中值=0.37, DN=30) — [54] 罗马尼亚 0.03—1.83(中值=0.16, DN=23) 美国 0.03—1.18(中值=0.41, DN=30) 印度 室内灰尘 0.04—19.1(中值=0.14, DN=30) — [54] 中国 0.88—60.7(中值=4.25, DN=30) 马耳他 室内灰尘 0.790—53(中值=7.8, DN=37) 秋季 [57] 注:DN, 样本数. data number; 灰尘中检测的ClO4−浓度指摄入、吸入和皮肤接触的暴露量.
下载: 导出CSV
表 7 饮用水中ClO4−的主要物理化学去除技术与原理
Table 7. Main physicochemical removal technologies and mechanism of perchlorate in drinking water
去除技术Removal technologies 材料/工艺Materials/Processes 实现途径Pathways 机理Mechanism 参考文献References 吸附 磁性纳米复合材料(SPIONS@nHA) ClO4−与吸附剂表面(FeOH2+-ClO4−,≡Ca-OH2+-ClO4−)的静电引力;PO43−与ClO4−交换 离子交换静电相互作用 [61] 季铵化改性磁性Mg/Al-层状双氢氧化合物(N8881Cl-LDH@Fe3O4) ClO4−与[N+(CH8CH17)3Cl−]中的Cl−发生离子交换 离子交换 [62] AIE效应超分子聚合物凝胶(PT-GEu) Eu3+与PT-G中的酰胺键配位并发生电子转移,随后ClO4−与Eu3+竞争配位 竞争配位 [63] 环氧氯丙烷交联壳聚糖水凝胶(ECH-CSBs) ECH-CSB上的质子化-NH3+位点在酸性条件下通过静电相互作用吸附ClO4− 静电相互作用 [64] 甜菜碱功能化介孔生物炭电极(BK-Z15%N) 电场引发的瞬时表面吸附在吸附过程中起主导作用,掺杂季胺氮基团引入额外的赝电容 电吸附 [65] 膜过滤 聚偏氟乙烯-金属骨架超滤膜(PVA/Cu-iMOFs/PVDF) Cu-iMOFs的磺酸(R-SO3)配体与ClO4−的可逆离子交换 离子交换 [66] 电渗析反渗透复合工艺(EDR+RO) 在EDR试验中,ClO4−电解产生HCl和Cl−,采用NZVI和纳米Fe0/Al0还原高浓度ClO4− 电解Fe还原 [67] 聚烯丙基胺盐酸盐(PAH)和聚丙烯酸(PAA)改性纳滤(NF)膜 NF膜的表面电荷随双层数变化不显著,改性NF膜的离子排斥机制中尺寸排除占主导地位 尺寸排除机制 [68] 离子交换 磁性离子交换树脂(MIEX)、Purolite A530E和Purolite A532E 3种树脂 吸附过程可分为4个连续的步骤:本体溶液迁移、边界层迁移、颗粒内迁移和本征吸附 化学吸附 [69] 玉米秸秆改性磁性生物聚合物树脂(CS-MAB) 物理吸附;ClO4−与CS-MAB的含Cl基团发生离子交换反应 物理吸附离子交换 [70] 强碱性阴离子交换树脂(SBA) 吸附解吸速率限制阶段可能是化学吸附 化学吸附 [71]
下载: 导出CSV
-
[1] CHANG W H, CHEN P H, HERIANTO S, et al. Aggregating exposures and toxicity equivalence approach into an integrated probabilistic dietary risk assessment for perchlorate, nitrate, and thiocyanate: Results from the National food monitoring study and National Food Consumption Database[J]. Environmental Research, 2022, 211: 112989. doi: 10.1016/j.envres.2022.112989 [2] TRUMPOLT C W, CRAIN M, CULLISON G D, et al. Perchlorate: Sources, uses, and occurrences in the environment[J]. Remediation Journal, 2005, 16(1): 65-89. doi: 10.1002/rem.20071 [3] COUNCIL N R. Health implications of perchlorate ingestion[M]. Washington D C : National Academies Press, 2005. [4] MAFFINI M V, TRASANDE L, NELTNER T G. Perchlorate and diet: Human exposures, risks, and mitigation strategies[J]. Current Environmental Health Reports, 2016, 3(2): 107-117. doi: 10.1007/s40572-016-0090-3 [5] TRASANDE L, SHAFFER R M, SATHYANARAYANA S, et al. Food additives and child health[J]. Pediatrics, 2018, 142(2): e20181408. doi: 10.1542/peds.2018-1408 [6] 许建红, 高乃云, 刘祖文, 等. 去除饮用水中高氯酸盐的研究新进展[J]. 水处理技术, 2011, 37(9): 28-32. XU J H, GAO N Y, LIU Z W, et al. The new development of perchlorate removal in the drinking water[J]. Technology of Water Treatment, 2011, 37(9): 28-32 (in Chinese).
[7] LI M H, XIAO M H, XIAO Q R, et al. Perchlorate and chlorate in breast milk, infant formulas, baby supplementary food and the implications for infant exposure[J]. Environment International, 2022, 158: 106939. doi: 10.1016/j.envint.2021.106939 [8] ZHANG L, FANG C R, LIU L P, et al. A case-control study of urinary levels of iodine, perchlorate and thiocyanate and risk of papillary thyroid cancer[J]. Environment International, 2018, 120: 388-393. doi: 10.1016/j.envint.2018.08.024 [9] LI Y W, LIAO R Y, GAN Z W, et al. Seasonal variation and exposure risks of perchlorate in soil, indoor dust, and outdoor dust in China[J]. Archives of Environmental Contamination and Toxicology, 2018, 75(3): 367-376. doi: 10.1007/s00244-018-0526-x [10] WANG H R, JIANG Y S, SONG J Y, et al. The risk of perchlorate and iodine on the incidence of thyroid tumors and nodular goiter: A case-control study in southeastern China[J]. Environmental Health, 2022, 21(1): 4. doi: 10.1186/s12940-021-00818-8 [11] 吴春笃, 李顺, 许小红, 等. 高氯酸盐的环境毒理学效应及其机制的研究进展[J]. 环境与健康杂志, 2013, 30(1): 85-89. WU C D, LI S, XU X H, et al. Environmental toxicological effect and mechanism of perchlorate[J]. Journal of Environment and Health, 2013, 30(1): 85-89 (in Chinese).
[12] BELLANGER M, DEMENEIX B, GRANDJEAN P, et al. Response to the letter by middlebeek and veuger[J]. The Journal of Clinical Endocrinology & Metabolism, 2015, 100(6): L54-L55. [13] CALDERÓN R, GODOY F, ESCUDEY M, et al. A review of perchlorate (ClO4−) occurrence in fruits and vegetables[J]. Environmental Monitoring and Assessment, 2017, 189(2): 82. doi: 10.1007/s10661-017-5793-x [14] U. S. Environmental Protection Agency (USEPA). Perchlorate (ClO4−) and perchlorate salts[EB/OL].2005, [2021-2-12]. [15] BRANDHUBER P, CLARK S, MORLEY K. A review of perchlorate occurrence in public drinking water systems[J]. Journal AWWA, 2009, 101(11): 63-73. doi: 10.1002/j.1551-8833.2009.tb09991.x [16] French Agency for Food, Environmental and Occupational Health& Safety (ANSES). Opinion on the presence of perchlorate in infant formula and in drinking water in France[EB/OL]. [2014-4-8]. [17] California Department of Public Health (CDPH). Comparison of MCLs and PHGs for regulated contaminants in drinking water[EB/OL].2007, [2014-7-5]. [18] U. S. Environmental Protection Agency (USEPA). EPA sets feference dose for perchlorate[EB/OL].2005, [2014-7-15]. [19] FAO J, NG J. Joint FAO/WHO expert committee on food additives seventy-second meeting: Summary and conclusions[R]. World Health Organization Technical Report Series, 2010. [20] BENFORD D, CECCATELLI S, COTTRILL B, et al. Scientific Opinion on the risks to public health related to the presence of perchlorate in food, in particular fruits and vegetables[J]. EFSA Journal, 2014, 12(10): 3869. doi: 10.2903/j.efsa.2014.3869 [21] PANSERI S, NOBILE M, ARIOLI F, et al. Occurrence of perchlorate, chlorate and polar herbicides in different baby food commodities[J]. Food Chemistry, 2020, 330: 127205. doi: 10.1016/j.foodchem.2020.127205 [22] CALDERÓN R, JARA C, ALBORNOZ F, et al. Accumulation and distribution of perchlorate in spinach and chard growing under greenhouse: Implications for food safety in baby foods commodities[J]. Food Chemistry, 2022, 370: 131101. doi: 10.1016/j.foodchem.2021.131101 [23] HAKME E, HERRMANN S S, POULSEN M E. Chlorate and perchlorate residues in food products on the Danish market[J]. Food Additives & Contaminants. Part A, 2022, 39(3): 551-559. [24] European Commission(EC). Commission regulation(EU) 2020/685 of 20 May 2020 amending Regulation (EC)No 1881/2006 as regards maximum levels of perchlorate incertain foods[EB/OL].2020, [2021-2-12]. [25] ERDEMGIL Y, GÖZET T, CAN Ö, et al. Perchlorate levels found in tap water collected from several cities in Turkey[J]. Environmental Monitoring and Assessment, 2016, 188(3): 158. doi: 10.1007/s10661-016-5161-2 [26] URBANSKY E T, COLLETTE T W. Survey of fertilizers and related materials for perchlorate (ClO4−)[M]. National Risk Management Research Laboratory, Office of Research and Development, US Environmental Protection Agency, 2001. [27] CALDERÓN R, PALMA P, ARANCIBIA-MIRANDA N, et al. Occurrence, distribution and dynamics of perchlorate in soil, water, fertilizers, vegetables and fruits and associated human exposure in Chile[J]. Environmental Geochemistry and Health, 2022, 44(2): 527-535. doi: 10.1007/s10653-020-00680-6 [28] ALOMIRAH H F, AL-ZENKI S F, ALASWAD M C, et al. Widespread occurrence of perchlorate in water, foodstuffs and human urine collected from Kuwait and its contribution to human exposure[J]. Food Additives & Contaminants. Part A, 2016, 33(6): 1016-1025. [29] GAN Z W, PI L, LI Y W, et al. Occurrence and exposure evaluation of perchlorate in indoor dust and diverse food from Chengdu, China[J]. Science of the Total Environment, 2015, 536: 288-294. doi: 10.1016/j.scitotenv.2015.07.057 [30] NADARAJA A V, PUTHIYAVEETTIL P G, BHASKARAN K. Surveillance of perchlorate in ground water, surface water and bottled water in Kerala, India[J]. Journal of Environmental Health Science and Engineering, 2015, 13(1): 56. doi: 10.1186/s40201-015-0213-z [31] KUMARATHILAKA P, OZE C, INDRARATNE S P, et al. Perchlorate as an emerging contaminant in soil, water and food[J]. Chemosphere, 2016, 150: 667-677. doi: 10.1016/j.chemosphere.2016.01.109 [32] VIGREUX-BESRET C, MAHÉ A, LEDOUX G, et al. Perchlorate: Water and infant formulae contamination in France and risk assessment in infants[J]. Food Additives & Contaminants. Part A, Chemistry, Analysis, Control, Exposure & Risk Assessment, 2015, 32(7): 1148-1155. [33] LUTTER R. An upper-bound assessment of the benefits of reducing perchlorate in drinking water[J]. Risk Analysis, 2014, 34(10): 1944-1956. doi: 10.1111/risa.12261 [34] LEWANDOWSKI T A, PETERSON M K, CHARNLEY G. Iodine supplementation and drinking-water perchlorate mitigation[J]. Food and Chemical Toxicology, 2015, 80: 261-270. doi: 10.1016/j.fct.2015.03.014 [35] ALOMIRAH H F, AL-ZENKI S F, ALASWAD M C, et al. Widespread occurrence of perchlorate in water, foodstuffs and human urine collected from Kuwait and its contribution to human exposure[J]. Food Additives & Contaminants. Part A, Chemistry, Analysis, Control, Exposure & Risk Assessment, 2016, 33(6): 1016-1025. [36] PARKER D R. Perchlorate in the environment: The emerging emphasis on natural occurrence[J]. Environmental Chemistry, 2009, 6(1): 10. doi: 10.1071/EN09001 [37] SEYFFERTH A L, PARKER D R. Determination of low levels of perchlorate in lettuce and spinach using ion chromatography-electrospray ionization mass spectrometry (IC-ESI-MS)[J]. Journal of Agricultural and Food Chemistry, 2006, 54(6): 2012-2017. doi: 10.1021/jf052897v [38] SEYFFERTH A L, PARKER D R. Effects of genotype and transpiration rate on the uptake and accumulation of perchlorate (ClO4−) in lettuce[J]. Environmental Science & Technology, 2007, 41(9): 3361-3367. [39] SEYFFERTH A L, PARKER D R. Uptake and fate of perchlorate in higher plants[J]. Advances in Agronomy, 2008, 99: 101-123. [40] SANCHEZ C A, CRUMP K S, KRIEGER R I, et al. Perchlorate and nitrate in leafy vegetables of North America[J]. Environmental Science & Technology, 2005, 39(24): 9391-9397. [41] SANCHEZ C A, KRIEGER R I, KHANDAKER N, et al. Accumulation and perchlorate exposure potential of lettuce produced in the Lower Colorado River region[J]. Journal of Agricultural and Food Chemistry, 2005, 53(13): 5479-5486. doi: 10.1021/jf050380d [42] SANCHEZ C A, KRIEGER R I, KHANDAKER N R, et al. Potential perchlorate exposure from Citrus sp. irrigated with contaminated water[J]. Analytica Chimica Acta, 2006, 567(1): 33-38. doi: 10.1016/j.aca.2006.02.013 [43] LIAO Z Y, CAO D L, GAO Z B, et al. Occurrence of perchlorate in processed foods manufactured in China[J]. Food Control, 2020, 107: 106813. doi: 10.1016/j.foodcont.2019.106813 [44] WANG Z W, FORSYTH D, LAU B P Y, et al. Estimated dietary exposure of Canadians to perchlorate through the consumption of fruits and vegetables available in Ottawa markets[J]. Journal of Agricultural and Food Chemistry, 2009, 57(19): 9250-9255. doi: 10.1021/jf901910x [45] WANG Y J, DONG J J, CHEN M Y, et al. Dietary exposure and risk assessment of perchlorate in diverse food from Wuhan, China[J]. Food Chemistry, 2021, 358: 129881. doi: 10.1016/j.foodchem.2021.129881 [46] VEJDOVSZKY K, GROSSGUT R, UNTERLUGGAUER H, et al. Risk assessment of dietary exposure to perchlorate for the Austrian population[J]. Food Additives & Contaminants. Part A, 2018, 35(4): 623-631. [47] CHANG W H, CHEN H L, LEE C C. Dietary exposure assessment to perchlorate in the Taiwanese population: A risk assessment based on the probabilistic approach[J]. Environmental Pollution, 2020, 267: 115486. doi: 10.1016/j.envpol.2020.115486 [48] TAN K, ANDERSON T A, JACKSON W A. Uptake and exudation behavior of perchlorate in smartweed[J]. International Journal of Phytoremediation, 2006, 8(1): 13-24. doi: 10.1080/15226510500506922 [49] LIANG Y B, ZHOU L, ZHANG X Z, et al. Uptake, accumulation, translocation, and subcellular distribution of perchlorate in tea (Camellia sinensis L. ) plants[J]. Journal of Agricultural and Food Chemistry, 2021, 69(16): 4655-4662. doi: 10.1021/acs.jafc.1c01270 [50] LEE J W, OH S H, OH J E. Monitoring of perchlorate in diverse foods and its estimated dietary exposure for Korea populations[J]. Journal of Hazardous Materials, 2012, 243: 52-58. doi: 10.1016/j.jhazmat.2012.09.037 [51] GURUGE K S, WU Q, KANNAN K. Occurrence and exposure assessment of perchlorate, iodide and nitrate ions from dairy milk and water in Japan and Sri Lanka[J]. Journal of Environmental Monitoring, 2011, 13(8): 2312-2320. doi: 10.1039/c1em10327j [52] SUNGUR Ş, ATAN M M. Determination of nitrate, nitrite and perchlorate anions in meat, milk and their products consumed in Hatay region in Turkey[J]. Food Additives & Contaminants. Part B, 2013, 6(1): 6-10. [53] CHEN Y N, ZHU Z, ZHAO Y, et al. Perchlorate in shellfish from South China Sea and implications for human exposure[J]. Marine Pollution Bulletin, 2021, 170: 112672. doi: 10.1016/j.marpolbul.2021.112672 [54] WAN Y J, WU Q, ABUALNAJA K O, et al. Occurrence of perchlorate in indoor dust from the United States and eleven other countries: Implications for human exposure[J]. Environment International, 2015, 75: 166-171. doi: 10.1016/j.envint.2014.11.005 [55] GAN Z W, SUN H W, WANG R N, et al. Occurrence and exposure evaluation of perchlorate in outdoor dust and soil in mainland China[J]. Science of the Total Environment, 2014, 470/471: 99-106. doi: 10.1016/j.scitotenv.2013.09.067 [56] ZHANG T, CHEN X J, WANG D, et al. Perchlorate in indoor dust and human urine in China: Contribution of indoor dust to total daily intake[J]. Environmental Science & Technology, 2015, 49(4): 2443-2450. [57] VELLA A J, CHIRCOP C, MICALLEF T, et al. Perchlorate in dust fall and indoor dust in Malta: An effect of fireworks[J]. Science of the Total Environment, 2015, 521/522: 46-51. doi: 10.1016/j.scitotenv.2015.03.071 [58] SHI Y L, ZHANG N, GAO J M, et al. Effect of fireworks display on perchlorate in air aerosols during the Spring Festival[J]. Atmospheric Environment, 2011, 45(6): 1323-1327. doi: 10.1016/j.atmosenv.2010.11.056 [59] 闻自强, 郑雯静, 沈昊宇, 等. 高氯酸盐的危害、水污染现状与去除技术研究进展[J]. 环境化学, 2019, 38(1): 209-216. doi: 10.7524/j.issn.0254-6108.2018022702 WEN Z Q, ZHENG W J, SHEN H Y, et al. Research progress on the hazards, water pollution status and removal technique of perchlorate[J]. Environmental Chemistry, 2019, 38(1): 209-216 (in Chinese). doi: 10.7524/j.issn.0254-6108.2018022702
[60] MA H Z, BONNIE N A, YU M A, et al. Biological treatment of ammonium perchlorate-contaminated wastewater: A review[J]. Journal of Water Reuse and Desalination, 2016, 6(1): 82-107. doi: 10.2166/wrd.2015.016 [61] KRISHNAN G R, PRABHAKARAN K, GEORGE B K. Biogenic magnetic nano hydroxyapatite: Sustainable adsorbent for the removal of perchlorate from water at near-neutral pH[J]. Journal of Environmental Chemical Engineering, 2021, 9(6): 106316. doi: 10.1016/j.jece.2021.106316 [62] MENG Z L, FAN J X, CUI X Y, et al. Removal of perchlorate from aqueous solution using quaternary ammonium modified magnetic Mg/Al-layered double hydroxide[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2022, 647: 129111. doi: 10.1016/j.colsurfa.2022.129111 [63] ZHANG Q, ZHANG Y M, YAO H, et al. Supramolecular AIE polymer-based rare earth metallogels for the selective detection and high efficiency removal of cyanide and perchlorate[J]. Polymer Chemistry, 2021, 12(13): 2001-2008. doi: 10.1039/D0PY01630F [64] YU X L, ZHANG J, ZHENG Y. Perchlorate adsorption onto epichlorohydrin crosslinked chitosan hydrogel beads[J]. Science of the Total Environment, 2021, 761: 143236. doi: 10.1016/j.scitotenv.2020.143236 [65] WANG B, ZHAI Y B, HU T J, et al. Green quaternary ammonium nitrogen functionalized mesoporous biochar for sustainable electro-adsorption of perchlorate[J]. Chemical Engineering Journal, 2021, 419: 129585. doi: 10.1016/j.cej.2021.129585 [66] LI T, REN Y, ZHAI S, et al. Integrating cationic metal-organic frameworks with ultrafiltration membrane for selective removal of perchlorate from Water[J]. Journal of Hazardous Materials, 2020, 381: 120961. doi: 10.1016/j.jhazmat.2019.120961 [67] YANG B M, LI J M, YOU Z Y, et al. Using integrated electrodialysis and RO hybrid system to remediate and reclaim perchlorate-contaminated groundwater[J]. Desalination, 2020, 480: 114377. doi: 10.1016/j.desal.2020.114377 [68] SANYAL O, SOMMERFELD A N, LEE I. Design of ultrathin nanostructured polyelectrolyte-based membranes with high perchlorate rejection and high permeability[J]. Separation and Purification Technology, 2015, 145: 113-119. doi: 10.1016/j.seppur.2015.03.011 [69] ZHU Y P, GAO N Y, WANG Q F, et al. Adsorption of perchlorate from aqueous solutions by anion exchange resins: Effects of resin properties and solution chemistry[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2015, 468: 114-121. [70] SONG W, GAO B Y, GUO Y, et al. Effective adsorption/desorption of perchlorate from water using corn stalk based modified magnetic biopolymer ion exchange resin[J]. Microporous and Mesoporous Materials, 2017, 252: 59-68. doi: 10.1016/j.micromeso.2017.06.019 [71] FACCINI J, EBRAHIMI S, ROBERTS D J. Regeneration of a perchlorate-exhausted highly selective ion exchange resin: Kinetics study of adsorption and desorption processes[J]. Separation and Purification Technology, 2016, 158: 266-274. doi: 10.1016/j.seppur.2015.12.019 [72] REKHA KRISHNAN G, PRABHAKARAN K, GEORGE B K. N-doped activated carbon with hierarchical pores for the efficient removal of perchlorate from water[J]. Microporous and Mesoporous Materials, 2021, 315: 110892. doi: 10.1016/j.micromeso.2021.110892 [73] YOON J, AMY G, CHUNG J, et al. Removal of toxic ions (chromate, arsenate, and perchlorate) using reverse osmosis, nanofiltration, and ultrafiltration membranes[J]. Chemosphere, 2009, 77(2): 228-235. doi: 10.1016/j.chemosphere.2009.07.028 [74] RUSSEL J G, THULASIRAMAN V, NAIR R R, et al. A novel bio-physical approach for perchlorate contaminated well water treatment[J]. Environmental Advances, 2021, 4: 100058. doi: 10.1016/j.envadv.2021.100058 [75] DARRACQ G, BARON J, JOYEUX M. Kinetic and isotherm studies on perchlorate sorption by ion-exchange resins in drinking water treatment[J]. Journal of Water Process Engineering, 2014, 3: 123-131. doi: 10.1016/j.jwpe.2014.06.002 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2456
- HTML全文浏览数: 2456
- PDF下载数: 36
- 施引文献: 0
