

-
酚类化合物是一类会对生物神经组织、生殖功能产生影响且可能诱发癌症的有机污染物[1],在煤气、焦化、炼油、制药、农药等行业具有广泛的应用[2]. 酚类化合物在生产或使用过程中排放进入水环境后,短期内不能降解而长时间的存在于水体中,在食物链的累积放大下对人体造成危害[3 − 4]. 国际上高度重视酚类化合物对环境带来的影响 [5],国内外将苯酚(Phol)、3-甲酚(m-Cresol)、2,4-二氯酚(2,4-DCP)、2,4,6-三氯酚(2,4,6-TCP)、五氯酚(PCP)、4-硝基酚(p-Nitrophenol)这6种酚类化合物设置为环境优先控制污染物[6 − 7], 五氯酚也因其本身具有高毒性和难降解等特点,被列入我国最新的新污染重点管控名单之内[8]. 国家地下水质量标准GB/T
14848 中明确规定一类地下水中苯酚、2,4,6-三氯酚和五氯酚的限值为50.0 ng·L−1. 随着国家《地下水管理条例》的颁布实施和“十四五”期间地下水质量监控工作的加强,建立快速高效、准确灵敏的地下水中痕量酚类化合物的分析方法为相关调查研究提供技术支撑,具有积极的现实意义.目前水中酚类化合物的提取净化方法主要有液液萃取法和固相萃取法等[9 − 10]. 其中,固相萃取具有操作简便快速、有机溶剂消耗量低、自动化程度高等优点,在前处理方法中使用较多[11]. 气相色谱法(GC)、液相色谱法(LC)、气相色谱质谱联用法(GC-MS)、液相色谱质谱联用法(LC-MS)等是酚类化合物常用的仪器分析方法[12 − 15]. 部分极性较强的酚类化合物在气相色谱中峰型不好,通常要进行衍生化处理[16 − 17],降低了分析效率. 高效液相色谱法不用衍生化反应可对酚类化合物直接分析,但紫外检测器与质谱相比,其定性能力较弱,很难避免假阳性的产生. 极性较大的硝基酚类和氯酚类等酚类化合物在现有方法中大部分是采用液相色谱-电喷雾电离(ESI)-质谱法检测[18 − 20],大气压化学电离(APCI)源相比于ESI源更适合中等极性以及弱极性,甚至部分非极性的化合物,这些化合物在ESI源下难以电离,但在更高温度的化学电离中能够产生单电荷离子. 苯酚和3-甲酚因极性相对较低,通常采用气相色谱质谱联用分析,液相色谱-质谱法分析3-甲酚以及苯酚的方法较少,目前极性和弱极性酚类化合物通常需要两套方法分开提取、净化和定量,极大降低了分析效率[12 − 13,15]. 亟待建立地下水中不同极性酚类化合物同时测定分析方法,提高分析效率,满足日益增长的调查监测需求.
本工作以Phol、2,4-DCP、2,4,6-TCP、PCP、m-Cresol、p-Nitrophenol等极性差异较大的6种代表性酚类化合物为研究对象,通过优化对比APCI源和ESI源对代表性酚类化合物的响应差异,结果表明,APCI源可以实现6种中等极性和极性酚的同时高灵敏度分析. 通过重点优化质谱参数和前处理条件,建立了固相萃取-高效液相色谱-大气压化学电离-质谱法同时分析地下水中6种不同极性酚类化合物的方法,为中等极性和极性酚类化合物的同时高灵敏分析提供了新思路,本方法在精密度、准确度好的同时也兼具方便高效,具有较好的应用价值.
-
高效液相色谱仪:岛津Nexera X2(日本岛津公司);三重四极杆串联质谱仪:SCIEX TRIPLE QUAD
5500 (美国SCIEX公司);固相萃取设备、氮吹浓缩仪(上海安谱实验公司);HLB固相萃取柱(500 mg·6 mL−1,美国Waters公司)、Kinetex C18色谱柱(100 mm×3.0 mm,2.6 μm,美国Phenomenex公司);旋转蒸发仪(瑞士BUCHI公司).甲醇中质量浓度为
1000 μg·mL−1的酚类化合物标准溶液(美国AccuStandard公司):Phol、2,4-DCP、2,4,6-TCP、PCP、m-Cresol、p-Nitrophenol;甲醇中质量浓度为1000 μg·mL−1的内标标准溶液(美国AccuStandard公司):对硝基酚-d4;色谱纯甲醇和乙腈(美国Thermo Fisher公司);优级纯乙酸和盐酸(国药集团化学试剂有限公司). -
色谱条件:流动相:A纯净水,B甲醇;梯度洗脱:0.0—4.0 min,50%—100% B;4.0—6.0 min,100% B;6.0—8.0 min,100%—50% B;8.0—10.0 min,50% B;流速:0.3 mL·min−1;柱温:30 ℃;进样体积:20 μL.
质谱条件:APCI源负离子模式电离;针电流(NC)3 mA;碰撞气(CAD)48 kPa;气帘气体(CUR)276 kPa;雾化气(GS1)276 kPa;离子源温度550 ºC;多重反应监测模式(MRM),其他质谱条件见表1.
-
首先活化SPE小柱,顺序加入10 mL甲醇以及10 mL纯净水. 水样经过滤(0.45 μm滤膜)后,取100 mL用盐酸调至pH为2,然后上样(流速约10 mL·min−1),使目标物在固相萃取柱上富集,之后用10 mL纯净水淋洗小柱,用氮气吹扫15 min,去除固相萃取柱中的水分,最后用14 mL的乙腈(含1%乙酸)进行洗脱,使用旋转蒸发仪对收集的全部洗脱液浓缩,加入1.00 mg·L−1内标溶液10.0 μL,用初始流动相定容至1.0 mL,混匀后进样分析.
-
ESI源是常用的电离源,样品先带电后形成喷雾电离,在极性化合物分析中应用广泛. APCI源是使样品先形成喷雾再经电晕针放电电离,其电离程度较ESI源强,通常认为它可以增加中等极性化合物离子化产率. 目前仍没有确切的准则选择判断电离源,化合物电离效果多依赖实验. 本研究依次将浓度为100 μg·L−1的6种代表性酚类标准溶液经质谱直接进样分析,重点调整碰撞能量和入口电压等参数,找出物质分子离子峰和两个信号较强的子离子峰,采用多重反应监测模式扫描分析. 结果表明,与ESI源相比,在使用APCI源时,2,4,6-三氯酚和2,4-二氯酚响应强度无明显差异,五氯酚降低50%, 4-硝基酚降低80%,响应强度无数量级差异. 但苯酚和3-甲酚在APCI源的响应强度与ESI源相比,提高了近100倍,可以满足同时高灵敏度分析的需要,最终选用APCI离子源定量,最佳质谱参数见表1.
-
据文献报道,采用C18填料色谱柱和一定比例的甲醇水可以实现酚类化合物较好的分离[21 − 22]. 本研究中6种酚类化合物的pKa值在4.92—10.09之间(见表1),其在水中存在离解平衡,为了使在色谱柱上保留较弱的离子态酚更好的吸附在色谱柱上,因此通常促进目标物向分子形式转换,所以在流动相中加入酸性添加剂降低流动相pH. 但随着酸性增强可能会抑制此类化合物的电离. 为此,本研究比较了水相为0.01mmol·L−1甲酸铵、0.003 mmol·L−1甲酸和超纯净水时目标物的质谱响应特点,通过结果得到,苯酚和3-甲酚质谱响应显著下降是因为流动相中添加甲酸或甲酸铵所引起的,原因可能是苯酚和3-甲酚酸性较弱,随着其酸性减弱,酸性环境对其离子化抑制越明显. 最终选择以甲醇和水作为流动相,此时6种酚类化合物的分离度和响应最好. 最佳流动相比例和梯度条件见表2.
-
选择水样的pH、洗脱试剂的种类、洗脱试剂的体积、浓缩方式这4个样品前处理条件参数进行优化,6种酚类化合物最佳固相萃取条件的选择依据为每种物质的回收率. 取100 mL水样,分别加入盐酸调节pH值,然后加入6种目标物的标准溶液,使质量浓度均为1.00 μg·L−1. 设置不同的前处理条件分别进行实验,获取最佳萃取条件.
-
水样加标后,用盐酸将6个平行水样的pH分别调节为1、2、3、4、5、6,按照“1.3”前处理方法实验步骤通过HLB固相萃取小柱,使用含1%乙酸的乙腈进行洗脱,经旋转蒸发仪浓缩后定容至1.0 mL,经上机检测后其结果如表3所示. 由表3可以看出,6种目标物的回收率会随着pH的上升而下降,最终选择水样的pH为2.
-
加标后的水样,用盐酸将调至pH为2,按照“1.3”前处理方法实验步骤通过HLB固相萃取小柱,选择甲醇、乙腈、二氯甲烷、含1%乙酸的乙腈作为洗脱溶剂,将洗脱液经旋转蒸发仪浓缩后定容上机,经上机检测后其结果如表4所示. 由表4可知,乙腈(含1%乙酸)洗脱效果最优,平均回收率最好,最终以乙腈(含1%乙酸)作为洗脱试剂.
-
加标后的水样,用盐酸将调至pH为2,按照“1.3”前处理方法实验步骤通过HLB固相萃取小柱,分别选择2.0、4.0、6.0、8.0、10.0、12.0、14.0、16.0 mL 含1%乙酸的乙腈对固相萃取柱进行洗脱,将洗脱液经旋转蒸发仪浓缩后定容上机,经上机检测后其结果如表5所示. 根据表5可以得出,当洗脱体积逐渐增加时,6种目标物的回收率也逐渐增大,并且当洗脱体积达到14.0 mL后,6种目标物的回收率均达到最大,因此最优的洗脱体积为14.0 mL.
-
将14.0 mL加入6种目标物的含1%乙酸的乙腈(物质的量浓度为5 μg·L−1),分别用氮吹浓缩仪和旋转蒸发仪浓缩后定容上机. 结果发现经氮吹浓缩后,6种酚类化合物的回收率为72.5%—87.8%;经过旋转蒸发仪浓缩后,6种酚类化合物的回收率为85.8%—108%,因此选用旋转蒸发仪进行浓缩.
-
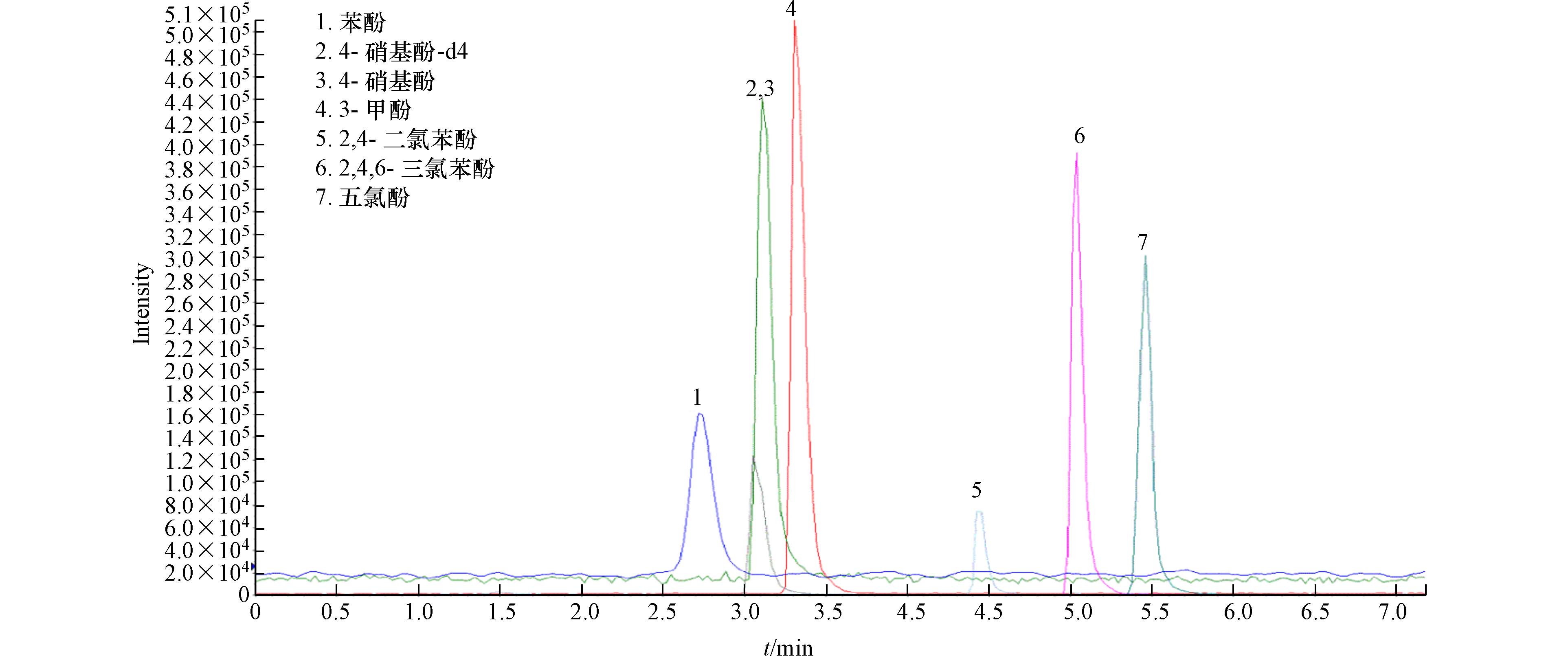
用初始流动相逐级稀释含6种目标物的混合标准溶液,配制成1.00、5.00、10.0、20.0、50.0、100 μg·L−1,加入内标后上机测定. 酚类化合物的总离子流图如图1所示. 横坐标为内标与目标物的质量浓度之比,纵坐标为内标与目标物的峰面积之比,创建标准曲线. 分析7个空白加标水样并求出其标准偏差s,带入HJ 168-2020[23]附录A.1 MDL=s×t(n-1,0.99)计算方法检出限. 6种目标物的线性范围等如表6所示,各组分的相关系数r均在0.998以上,6种目标物的方法检出限为0.005—0.050 μg·L−1,优于或与现有文献报道处在同一浓度水平[12,15,19],可以满足地下水中痕量酚类化合物的检测要求.
-
分6次各取100 mL地下水样品,加入6种目标物标准品,使加标量均为0.50 μg·L−1,结果如表7所示. 由表7可知,该样品共检出5种目标物,浓度在0.005—0.748 μg·L−1之间,该化工区的地下水样品的平均加标回收率为介于77.6%到106%之间,相对标准偏差(RSD)介于4.0%到8.0%之间,表明方法可以满足实际水样中痕量酚类化合物同时分析的需要,具有较强的实用价值.
-
为了深入认识6种酚类化合物在APCI源的质谱裂解行为,本研究对其可能的裂解途径进行了推断. 6种物质均是在负离子模式下获得最佳电离响应,苯酚的准分子离子峰 [M-H]−为m/z 93.0,推测裂解方式一种是失去1分子H2O后形成m/z 75.0的碎片离子,另一种是失去1分子CO,苯环重排为结构稳定的五元芳环,形成m/z 65.0的碎片离子. 4-硝基苯酚的准分子离子峰为 [M-H]− m/z 138.0,推测硝基与苯环间的C-N键断裂,形成m/z 46.1 NO2−碎片离子和m/z 92.0的苯酚负离子. 3-甲基酚的准分子离子峰为[M-H]− m/z 106.9,推测裂解方式一种是直接脱去1分子CH3,形成m/z 92.0的碎片离子,另一种是脱去甲基上的一个H,形成m/z 105.8的碎片离子. 2,4-二氯酚的准分子离子峰为[M-H]− m/z 160.9,推测其裂解方式为整体先脱去1分子HCl, 形成m/z 125.0的碎片离子,然后继续脱去1分子HCl,最后形成结构较为稳定的m/z 89.0的碎片离子. 2,4,6-三氯酚的准分子离子峰为[M-H]− m/z 195.0,推测裂解途径为先脱去1分子HCl, 形成m/z 159.0的碎片离子,然后该碎片再失去一个Cl−,形成m/z 35和m/z 124.0碎片离子. 五氯酚的准分子离子峰为[M-H]− m/z 264.8,推测裂解途径主要是脱Cl−并形成m/z 35.0或m/z 37.0 Cl离子碎片. 6种有机酚的分子式和裂解途径详见表8.
-
本研究建立了固相萃取-高效液相色谱-大气压化学电离源-串联质谱法同时分析地下水中6种不同极性酚类化合物分析测定方法. 方法仅采用100 mL样品,即可实现痕量酚类化合物的同时分析. 方法检出限在0.005—0.050 μg·L−1之间,实际样品加标回收率在77.6%—106%之间,相对标准偏差在4.0%—8.0%之间. 通过6种酚类化合物的质谱裂解规律的详细推断,增进了对该类化合物质谱裂解行为的认识. 将该方法在地下水中6种酚类化合物的同时分析中应用效果较好,结果表明该方法方便高效,准确度和精密度高,为中等极性和极性酚类化合物同时分析提供了新思路,具有较强的实用价值.
固相萃取-高效液相色谱-大气压化学电离-串联质谱法同时分析地下水中6种酚类化合物
Determination of six phenolic compounds in groundwater by solid phase extraction –high performance liquid chromatography-APCI-tandem mass spectrometry
-
摘要: 建立了固相萃取-高效液相色谱-大气压化学电离-串联质谱法(SPE-HPLC-APCI-MS/MS)同时分析地下水中6种不同极性酚类化合物的分析方法. 通过重点优化固相萃取、色谱条件和质谱参数等确定了最佳分析条件,详细解析了大气压化学电离源下6种酚的裂解规律. 方法采用100 mL酸化水样(pH=2),经HLB固相萃取柱富集净化和浓缩定容后上机检测,内标法定量. 结果显示:目标分析物线性范围在1—100.0 μg·L−1之间,相关系数r为
0.9978 —0.9996 ,方法检出限在0.005—0.050 μg·L−1之间. 方法加标回收率在77.6%—106%之间,相对标准偏差(RSD)介于4.0%到8.0%之间 (n=6). 方法在地下水实际样品分析中应用效果较好,方便高效,灵敏度高,精密度和准确度好,符合地下水中痕量酚类化合物分析的要求,为中等极性和极性酚类化合物同时分析提供新思路.-
关键词:
- 酚类化合物 /
- 地下水 /
- 高效液相色谱-质谱联用 /
- 固相萃取 /
- 大气压化学电离.
Abstract: A method for simultaneous determination of six phenolic compounds in groundwater by solid phase extraction-high performance liquid chromatography-Atmospheric Pressure Chemical Ionization-tandem mass spectrometry(SPE-HPLC-APCI-MS/MS) was established. The optimum analytical conditions were determined by optimizing the solid-phase extraction conditions, chromatography conditions and mass spectrometry parameters. The mass spectrometric cleavage laws of six phenols were analyzed in detail. An aliquot of 100 mL acidified water sample (pH=2) was enriched with HLB solid-phase extraction column, and then determined by HPLC-MS/MS after concentration under gentle nitrogen flow and quantified by internal standard method. The results showed that the good linearity ranging from 0.10 μg·L−1 to 100.0 μg·L−1 with correlation coefficients of0.9978 —0.9996 were obtained.The detection limits of the method were between 0.005 μg·L−1 and 0.050 μg·L−1.The recoveries assessed by matrix addition were between 77.6%—106% with the relative standard deviations (RSD) in the range of 4.0%—8.0% (n = 6). It is proved that the proposed method showed simple operation, efficient, high sensitivity, good precision and accuracy, which is a new attempt and could meet the requirements for the rapid and simultaneous analysis of phenolic compounds of different polarity in groundwater.-
Key words:
- phenolic compounds /
- groundwater /
- HPLC-MS/MS /
- solid phase extraction /
- APCI
-
寻求和利用可再生绿色清洁能源替代化石燃料,是解决能源与环境危机的重要途径。微生物燃料电池(microbial fuel cell, MFC)作为一种集污水处理与生物产电于一体的新型技术,以产电细菌为主体,可将化学能转化为电能,同时去除水体中的污染物[1-2]。电极材料是影响MFC性能的关键因素之一,也是MFC产电微生物的附着载体和生长场所[3]。因此,找到一种可供微生物大量附着和生长的载体,同时具有良好导电性能的材料至关重要。
MFC电极多采用碳质材料,拥有良好的生物相容性、导电性和化学稳定性[4]。碳质材料一般包括石墨烯、碳毡、碳布、生物炭等。其中石墨烯电极机械强度较好,但其材料表面相对光滑,不利于微生物附着,因而导致胞外电子传递效率低[5-6];碳毡电极柔韧性良好,但其在MFC运行时,由于材质较厚,生物膜会妨碍底物由外向内的扩散,影响对污染物的降解效率;碳布电极表面粗糙但机械强度较差,不适于投入大规模的实际工程应用中[7]。相比于传统电极材料,生物炭材料具有来源广泛、成本低廉、电化学性能较好、比表面积高和孔隙结构多等优点。2018年CHEN等[8]大麻槿秸秆通过简单的碳化处理制成MFC阳极,其电流密度达到了32.5 A·m−2,是对照组石墨棒电极的3倍,由此可见生物炭作为MFC电极材料是具有一定优势的。
据2020年中国统计年鉴统计,我国核桃栽培面积为5.54×1010 m²,约1.3×109株[9]。每年有大量的废弃核桃壳产生,如何有效处理这些固体废物,实现减量化和资源化是环境领域的研究热点。采用高温裂解法制备生物炭,再通过化学活化,可使其表面结构相对于碳基材料的平面结构更为粗糙,更有效的提升活性表面积[10-13]。常见的生物炭化学活化剂包括ZnCl2、HPO4、KOH等,其中ZnCl2活化制备的活性炭具有产率高、过渡孔发达、价廉易得等优点[14],JIANG等通过ZnCl2活化甘蔗渣发现,锌离子浓度越高,比表面积越大[15]。
目前,以改性核桃壳作为电极材料的研究鲜有报道。因此,本研究主要以改性核桃壳作为生物炭基电极材料,通过不同温度的碳化、不同浓度的ZnCl2活化、不同比例的材料复合制成微生物燃料电池电极,通过表征分析,考察不同制备方法制备出的材料的性能差异,分析其在MFC中产电性能的差异,以及最佳条件MFC去除污染物的能力,为微生物燃料电池的发展方向提供参考。
1. 材料和方法
1.1 电极材料的制备
将市场上购买的核桃取果皮后粉碎,过40目分子筛后,置于石英舟中,再将其放入管式炉(OTF-1200 X),真空400 ℃炭化90 min后,得到黑色产物。称取一定量的黑色产物与氯化锌固体按质量比分别为5:1、5:3、5:5,置于烧杯中加去离子水刚好完全淹没,搅拌后,再将其置于105 ℃烘箱中烘干24 h。将烘干好的黑色产物放于管式炉中央,分别在400、600、800 ℃温度条件下真空煅烧2 h,反应结束后在真空保护下冷却至室温。煅烧好的样品先用10% HCl溶液洗涤,然后用去离子水洗涤,直至中性,最后将其置于105 ℃烘箱中烘干24 h,得到核桃壳碳化产物。
制备好的生物炭样品与聚苯胺和热熔胶按5:1:4和5:1:5质量比进行混合,然后将混合材料置于刚玉舟模具中压实,再放入200 ℃管式炉进行真空热熔,热熔30 min后,自然冷却至室温取出。制备后的电极材料样品尺寸为2 cm×3 cm×0.5(±0.05) cm。
1.2 表征与测试
实验采用扫描电镜(捷克TESCAN MIRA LMS),通过磨成粉末过0.4 mm筛网制样,对生物炭基电极材料表面形貌进行表征;采用拉曼光谱(激光器波长532 nm,扫描范围50~4 000 cm−1)分析电极材料的石墨化程度;采用孔隙及比表面积分析仪(康塔4000 e,脱气温度120 ℃)分析电极材料的比表面积、孔体积和孔径;采用电化学工作站(CHI 660 e),通过LSV、EIS测试,分析电极材料的导电性能的差异;采用HACH高量程(20~1 500 mg·L−1)消解法测定COD;采用国标纳氏试剂比色法测定氨氮;采用电流电压数据采集器(KEYSIGHT 34972A)测定MFC产电性能、采集电流电压及功率密度。
1.3 MFC的构建
实验采用空气阴极单室MFC反应器,由有机玻璃制成,内径为10 cm,高度为14 cm,设置溢流堰用于出水,底部设置0.4 cm有机玻璃管用于进水。反应器阴阳极用尼龙螺栓固定,有效容积为377 mL。配制模拟废水(实验所用的去离子水为灭菌除氧后的水)用于MFC产电性能分析,每隔24 h进出水150 mL,其组成为1.356 g·L−1 C4H4Na2O4,0.15 g·L−1 (NH4)2SO4,0.253 5 g·L−1 KH2PO4,0.125 g·L−1 MgSO4·7H2O,0.125 g·L−1 NaCl,0.002 5 g·L−1 FeSO4·7H2O,0.002 g·L−1 MnSO4·H2O,1 mL·L−1 微量元素。其中微量元素包括1.5 g·L−1 FeC13·6H2O,0.02 g·L−1 CuC12·2H2O,0.18 g·L−1 KI,0.12 g·L−1 MnCl2·4H2O,0.01 g·L−1 ZnC12,0.06 g·L−1 Na2MoO4·2H2O,0.15 g·L−1 CoC12·6H2O,0.15 g·L−1 H3BO3,0.06 g·L−1 Na2MoO4·2H2O。
1.4 脱氮微生物接种
实验所用接种微生物为本研究组前期实验筛选出的异养硝化-好氧反硝化菌[16]。通过扩大培养,鉴定其异养硝化与好氧反硝化性能后接种至MFC反应器。具体过程如下:将菌种接种至LB液体培养基,在(30±2) ℃培养箱中培养24 h后,取菌种培养液与去离子水以1:9比例混合,用10 mL离心管离心去除上清液后加适量水摇匀,倒入好氧反硝化培养基,恒温振荡培养(160 min−1,30 ℃),每12 h测1次硝氮浓度和OD600;再取培养液置于异养硝化培养基,厌氧箱中恒温培养(30 ℃),每12 h测1次氨氮浓度和OD600。实验所用LB液体培养基含有3.0 g·L−1 牛肉膏、5.0 g·L−1 NaCl、10.0 g·L−1 蛋白胨(pH=7.0);好氧反硝化培养基(100 mL)含有1.356 g·L−1 C4H4 Na2O4、0.064 1 g·L−1 KNO3、0.253 5 g·L−1 KH2PO4、0.125 g·L−1 MgSO4·7H2O、0.125 g·L−1 NaCl、0.002 5 g·L−1 FeSO4·7H2O、0.002 g·L−1 MnSO4·H2O;异养硝化培养基(100 mL)含有1.356 g·L−1 C4H4Na2O4、0.253 5 g·L−1 KH2PO4、0.125 g·L−1 MgSO4·7H2O、0.125 g·L−1 NaCl、0.002 5 g·L−1 FeSO4·7H2O、0.002 g·L−1 MnSO4·H2O、0.15 g·L−1 (NH4)2SO4。
1.5 MFC启动
将目标菌株菌悬液接种至MFC反应器中,接种比例10%,上下电极间距4 cm。MFC置于恒温气候箱中,温度和湿度分别控制30 ℃、50%。进水pH为7.0±0.1,连接1 kΩ的外电阻,每5 min对MFC输出电流电压进行实时监控。在MFC电压达到稳定输出时,测量分析极化曲线和功率密度曲线(文中功率密度和电流密度以反应器有效容积为参比)。具体方法为:依次将外电阻由2 000 Ω调到100 Ω,每30 s记录1次MFC外阻的电流电压值,其中每更换一次电阻需等待3 min让电压值稳定。
2. 结果与讨论
2.1 生物炭/氯化锌质量比对电极材料性能的影响
为阐明ZnCl2用量对生物炭材料孔隙结构的影响,实验采用生物炭/氯化锌质量比分别为5:1、5:3、5:5的电极材料,在600 ℃煅烧后,通过比表面积和孔径分布来评估核桃壳生物炭的比表面积及相应孔径分布。由图1可知,ZnCl2活化后的生物炭孔结构的孔径主要集中在3.5 nm附近,在相对压力为0.1~1.0内出现较显著的滞后环,按照国际纯化学和应用化学联合会的定义,核桃壳生物炭是典型的Ⅰ型和Ⅱ型特性[17],说明核桃壳生物炭的结构属于尺寸较小的介孔结构。由表1可知,随着ZnCl2质量比的不断增加,改性核桃壳生物炭的比表面积由590 m2·g−1增加到883 m2·g−1,孔容由0.009 cm3·g−1逐渐增加到0.017 cm3·g−1。这说明随着ZnCl2用量的增加,活化后的核桃壳生物炭的比表面积也越大,可为微生物的生长提供更多的场所[18],做成MFC电极后其微生物负载量可得到提升,从而促进MFC的产电。
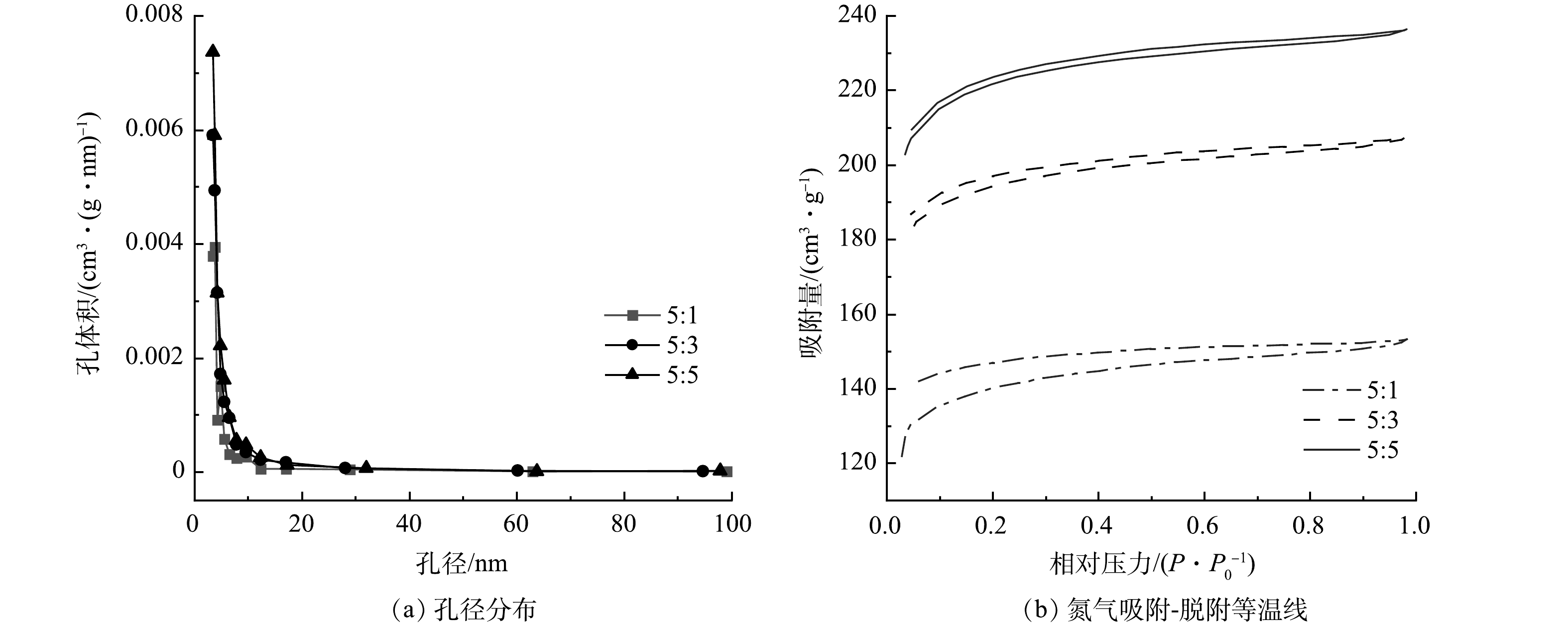 图 1 不同生物炭/氯化锌质量比条件下电极材料BET分析图Figure 1. BET analysis chart of electrode materials at different biochar/zinc chloride mass ratios表 1 BET测量时获得的比表面积、孔径和孔容Table 1. Specific surface area, pore size and pore volume determined by BET measurement
图 1 不同生物炭/氯化锌质量比条件下电极材料BET分析图Figure 1. BET analysis chart of electrode materials at different biochar/zinc chloride mass ratios表 1 BET测量时获得的比表面积、孔径和孔容Table 1. Specific surface area, pore size and pore volume determined by BET measurement生物炭/氯化锌质量比 比表面积/(m2·g−1) 孔径/nm 孔容/(cm3·g−1) 5:1 590 3.818 0.009 5:3 657 3.424 0.015 5:5 883 3.421 0.017 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.2 煅烧温度对电极材料性能的影响
为阐明热处理温度对生物炭材料分子结构的影响规律,控制生物炭/氯化锌质量比为5:5,分别在400、600、800 ℃热处理条件下,对制备的生物炭进行拉曼光谱分析,实验结果如图2所示。由图2可见,核桃壳生物炭在1 316 cm−1和1 586 cm−1处有2个显著的拉曼峰,分别为炭材料的特征D峰和G峰[19-20]。其中D峰主要是芳香环之间的C—C结构,为环数大于6环的芳香环结构,是由炭材料缺陷引起;G峰与炭材料的C=C键Sp2杂化有关。D峰与G峰的强度比(ID/IG)可以在一定程度反映材料的缺陷程度,ID/IG值越高,代表材料的无序率越高;ID/IG值越低,说明材料的石墨化程度越高,导电性能越好[21]。根据拉曼光谱图Gauss拟合曲线方法可以得到ID/IG。由图2可以看出,随着热解温度的增加,ID/IG比值变小。这说明材料的石墨化程度增加,导电性能越好,制作的MFC电极性能越好。
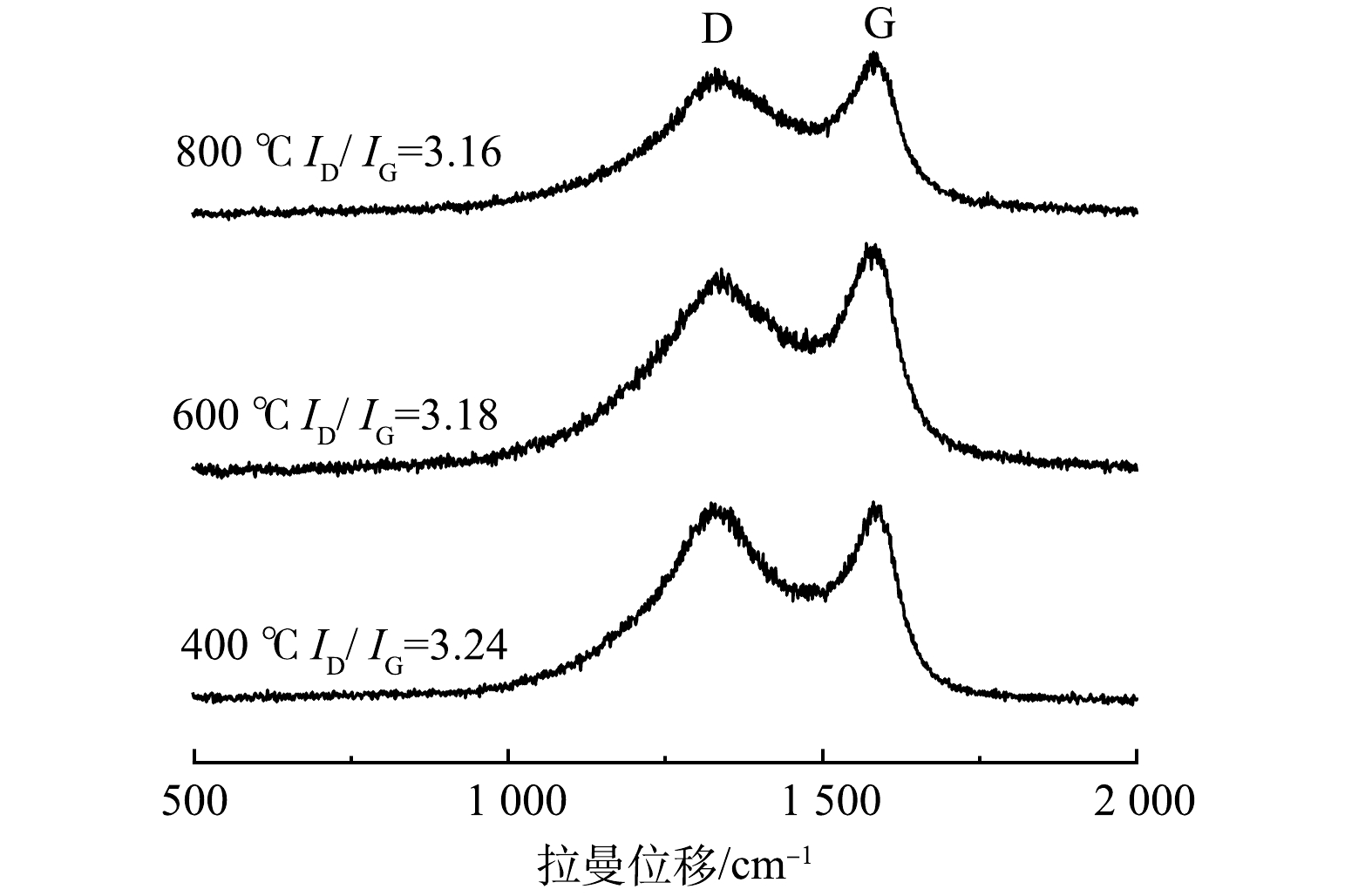 图 2 不同煅烧温度条件下改性核桃壳生物炭拉曼光谱分析Figure 2. Raman spectra analysis of modified walnut shell biochar at different calcination temperatures
图 2 不同煅烧温度条件下改性核桃壳生物炭拉曼光谱分析Figure 2. Raman spectra analysis of modified walnut shell biochar at different calcination temperatures2.3 生物炭/聚苯胺/热熔胶复合比例对电极材料性能的影响
聚苯胺与热熔胶比例也是考察MFC电极制作过程的因素之一。图3为在真空煅烧温度为600 ℃、生物炭与氯化锌活化质量比为5:3的条件下,生物炭/聚苯胺/热熔胶比例分别为5:1:4和5:1:5所制成的MFC复合电极的扫描电镜图。
 图 3 不同生物炭/聚苯胺/热熔胶复合比例条件下扫描电镜分析Figure 3. SEM analysis at different composite ratios of biochar/polyaniline/hot melt adhesive
图 3 不同生物炭/聚苯胺/热熔胶复合比例条件下扫描电镜分析Figure 3. SEM analysis at different composite ratios of biochar/polyaniline/hot melt adhesive由于当生物炭/聚苯胺/热熔胶的比例为5:1:1和5:1:2时,经过煅烧的生物炭电极几乎不成型,依旧保持粉末状态;当生物炭/聚苯胺/热熔胶添加比例为5:1:3时,经过煅烧的生物炭电极机械强度低,易碎,因此这些复合比例均不能到达作为MFC电极材料的要求,故实验选用的生物炭/聚苯胺/热熔胶比例为5:1:4和5:1:5。由图3可以看出,随着热熔胶中聚乙烯粉末和导电态聚苯胺的加入,其对生物炭的表面起到了修饰作用,但未对生物炭的多孔结构产生明显的影响。
2.4 生物炭/聚苯胺/热熔胶复合电极电化学表征
图4反映了电解池三电极体系中生物炭/聚苯胺/热熔胶复合电极的电化学性能。以1 cm2的铂片作为辅助电极,以Ag/AgCl作为参比电极,将制备得到BPP 5:1:4和BPP 5:1:5的复合电极分别连接在电极夹上作为工作电极。所有的电化学性能测试均是在1 mmol·L−1 铁氰化钾混合溶液(0.1 mol·L−1 KCl)中完成。
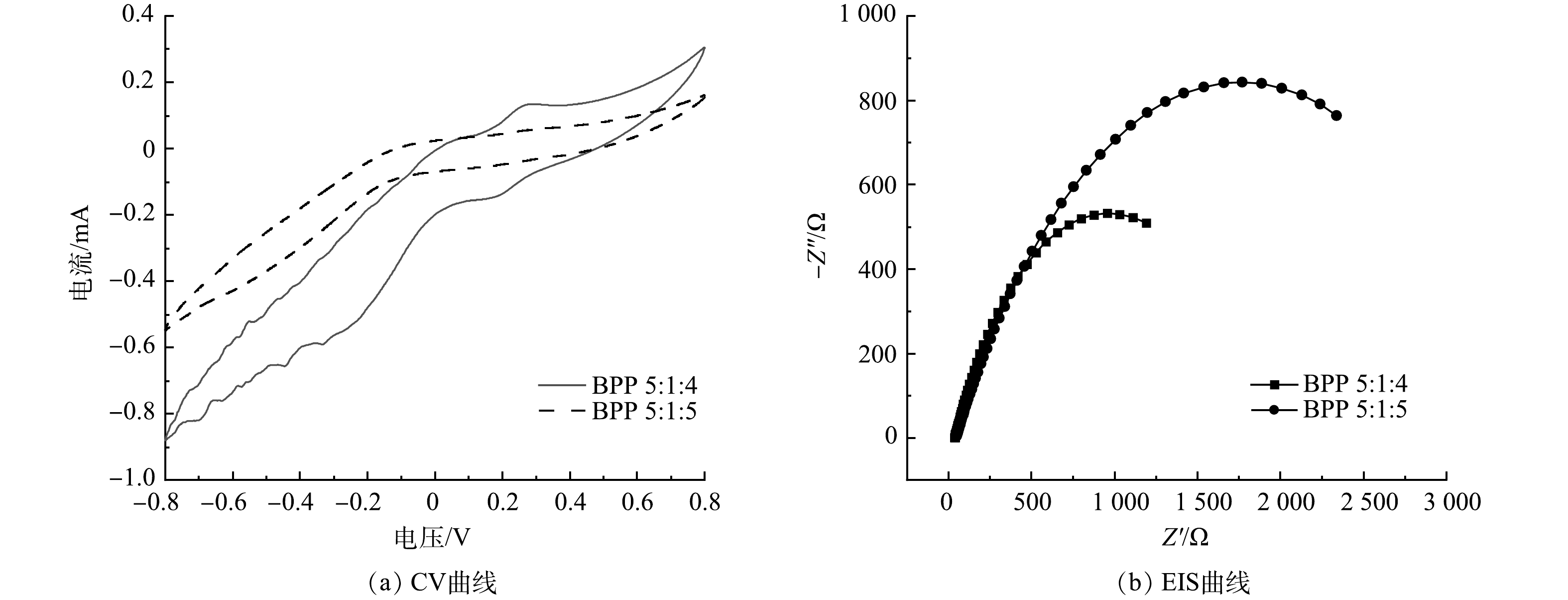 图 4 不同生物炭/聚苯胺/热熔胶复合比例电极电化学分析Figure 4. Electrochemical analysis of electrodes with different composite ratios of biochar/polyaniline/hot melt adhesive
图 4 不同生物炭/聚苯胺/热熔胶复合比例电极电化学分析Figure 4. Electrochemical analysis of electrodes with different composite ratios of biochar/polyaniline/hot melt adhesive如图4(a)所示,BPP 5:1:4材料在-0.8~0.8 V内的电流为0.305~-0.879 9 mA,且CV曲线有2对微弱的氧化还原峰;而BPP 5:1:5材料的电流为0.154~-0.546 mA,CV曲线没有氧化还原峰。材料氧化还原峰越多越明显,材料的电子传递能力越好[22],因此,BPP 5:1:4材料比BPP 5:1:5材料具有更良好的电子传递能力,更易促进氧化还原反应。
交流阻抗(EIS)曲线如图4(b)所示,其中,正弦信号频率为0.01~105 Hz,交流振幅为0.006。MFC中EIS的表征大多用于分析欧姆内阻和扩散内阻,由于低频区对扩散内阻的表征存在较大偏差,所以在数据拟合过程中未将低频区部分纳入拟合范围[23]。本次拟合使用软件Zview2,等效电路模型中RΩ为欧姆内阻,Rct为电荷转移内阻,电荷转移内阻与一个双电层电容并联,但因弥散效应的存在,该电容偏离理想双电层电容器,因而在本次拟合电路中使用常相位角原件代替传统双电层电容器。根据拟合结果,BPP 5: 1: 4欧姆内阻为37.17 Ω,电荷转移内阻为1 854 Ω,BPP 5:1:5欧姆内阻为46.54 Ω,电荷转移内阻为343 Ω。总的来看,Rct均大于RΩ,说明MFC系统内组主要受Rct控制。就Rct而言,BPP 5:1:4小于BPP 5:1:5,Rct主要反映电活性微生物与电极之间电子传递过程的内阻[24],Rct越小,其电子传递速率越快,因此,BPP 5:1:4生物电化学活性优于BPP 5:1:5。
2.5 菌株硝化反硝化能力的分析
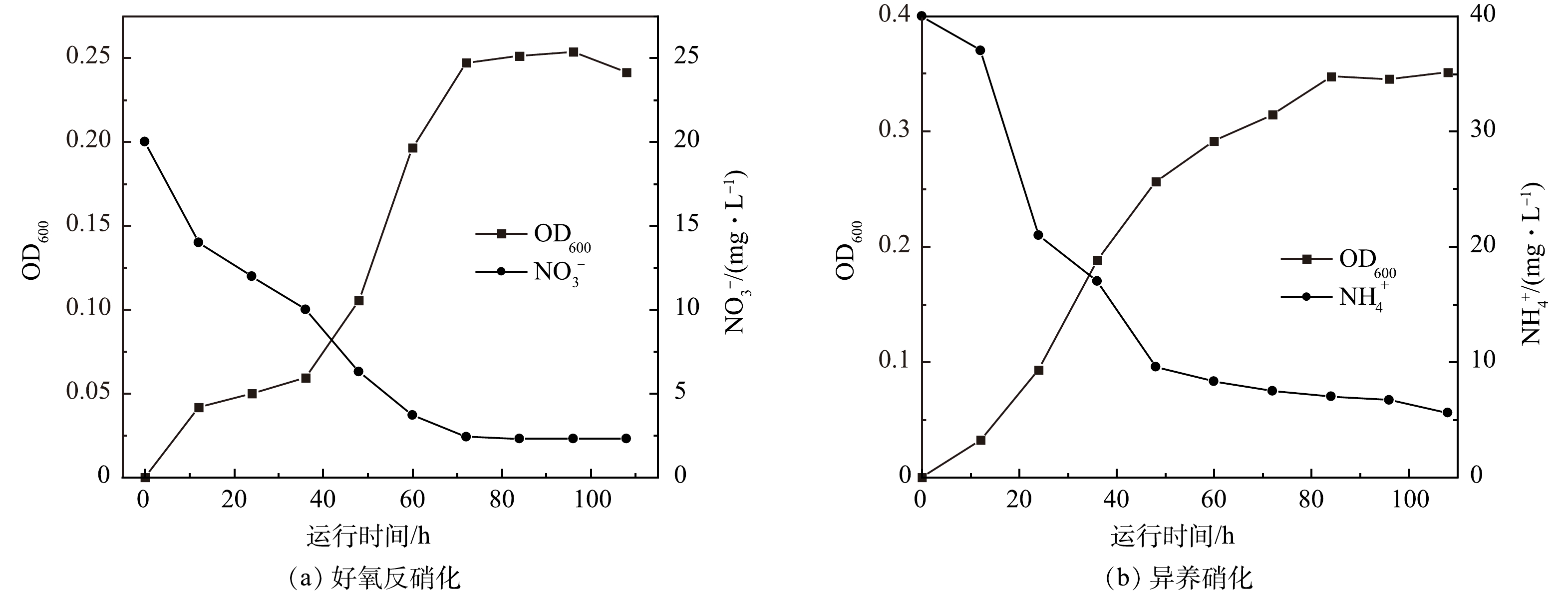
本研究组前期筛选出的异养硝化-好氧反硝化菌[16],通过扩大培养后,接种到异养硝化与好氧反硝化培养基中。由图5可见,在好氧反硝化、异养硝化培养基中菌株不断进行自我繁殖,并消耗培养基中的硝酸根与氨氮。这说明实验接种的微生物具有好氧反硝化与异养硝化能力,后续实验将采用此细菌做为MFC的产电菌。
2.6 改性核桃壳生物炭电极MFC产电性能
实验构建单室MFC反应器,将脱氮菌株接种至MFC反应器中,连接不同条件下制备的改性核桃壳基生物炭电极材料,每5 min对MFC输出电流电压进行实时监控,其产电性能、功率密度与极化曲线如图6所示。
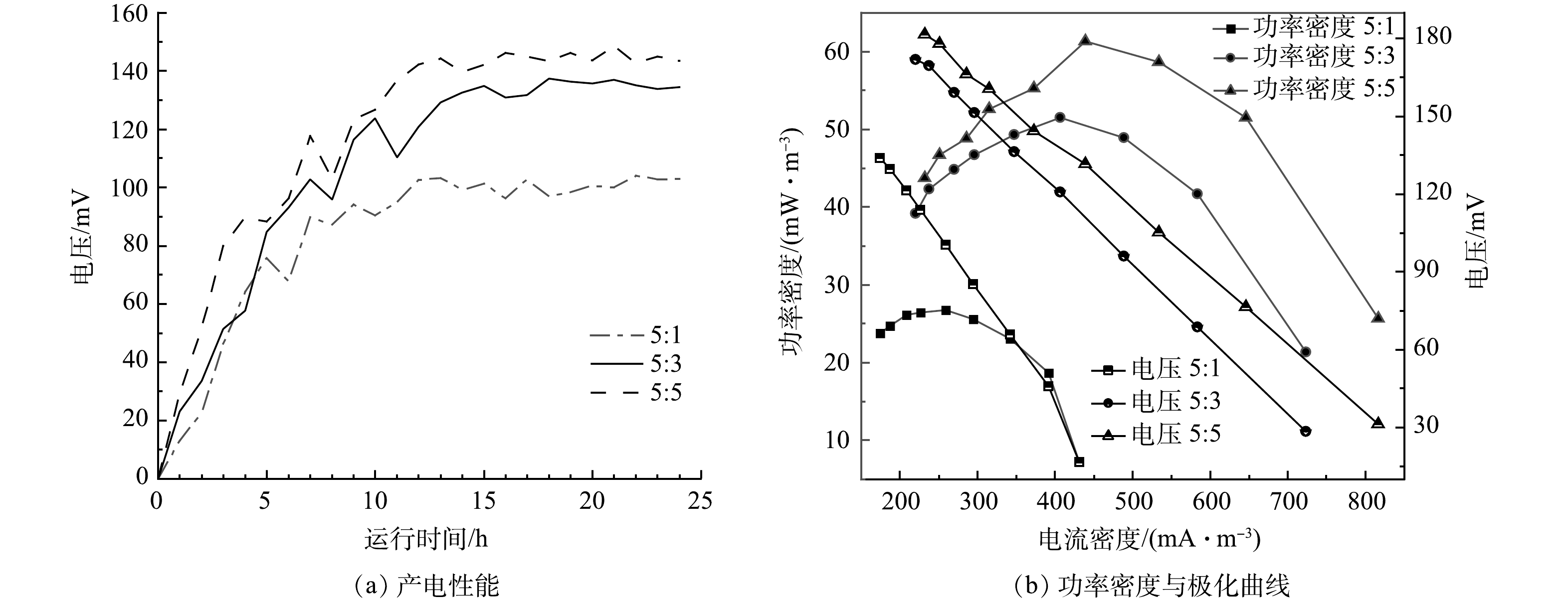 图 6 不同生物炭/氯化锌质量比条件下制备的电极材料Figure 6. Electrode materials prepared at different biochar/zinc chloride mass ratios
图 6 不同生物炭/氯化锌质量比条件下制备的电极材料Figure 6. Electrode materials prepared at different biochar/zinc chloride mass ratios在真空煅烧温度为600 ℃,BPP为5:1:4的条件下,考察了不同浓度氯化锌对产电性能的影响,结果如图6(a)和图6(b)所示。当生物炭/氯化锌质量比为5:1时,MFC电极最大输出电压为0.103 V,随外电阻由大到小变化,反应器极化曲线电压由133 mV降至16 mV,最大体积功率为26 mW·m−3,电流密度为259 mW·m−3;当生物炭/氯化锌质量比为5:3时,最大输出电压为0.137 V,随外电阻由大到小变化,反应器极化曲线电压由171 mV降至28 mV,最大体积功率为51 mW·m−3,电流密度为406 mA·m−3;当生物炭/氯化锌质量比为5:5时,MFC电极最大输出电压为0.148 V,随外电阻由大到小变化,反应器极化曲线电压由181 mV降低至31 mV,最大功率密度为61 mW·m−3,电流密度为438 mA·m−3。根据极化曲线斜率可以得出MFC电极电阻,斜率越小MFC的内阻越大[25]。随着ZnCl2质量比的增加,MFC的产电能力增加,内阻逐渐减小,电极材料的产电性能越好。当生物炭/氯化锌质量比从5:3提升到5:5,MFC的产电性能提升不大,可能原因是ZnCl2对生物炭的造孔能力几乎达到饱和[26]。
在BPP为5:1:4,生物炭/氯化锌比为5:3条件下,考察了煅烧温度对电极性能的影响,结果如图7(a)和图7(b)所示。在400 ℃煅烧条件下,MFC电极最大输出电压为0.096 V,随外电阻由大到小变化,反应器极化曲线电压由119 mV降低至17 mV,最大功率密度为22 mW·m−3,电流密度为238 mA·m−3;在600 ℃煅烧条件下,最大输出电压为0.137 V,随外电阻由大到小变化,反应器极化曲线电压由171 mV降低至28 mV,最大体积功率为51 mW·m−3,电流密度为406 mA·m−3;在800 ℃煅烧条件下,MFC电极最大输出电压为0.143 V,随外电阻由大到小变化,反应器极化曲线电压由181 mV降低至29 mV,最大功率密度约为57 mW·m−3,此时的电流密度为436 mA·m−3。随着煅烧温度的增加,MFC的产电能力增加,内阻逐渐减小,电极材料的产电性能越好。结合图2可知,这是由于材料石墨化程度的增加,导致MFC的内阻减小。由图7可见,在600 ℃和800 ℃条件下,制备的电极性能相差不大。由此可见,当煅烧温度达到一定程度,MFC的产电性能提升不大,可能的原因是温度的增加破坏了部分生物炭的微孔和大孔,虽然生物炭石墨化程度增加,但是微生物的负载量减少[27]。
 图 7 不同煅烧温度条件下制备的电极材料Figure 7. Electrode materials prepared at different calcination temperatures
图 7 不同煅烧温度条件下制备的电极材料Figure 7. Electrode materials prepared at different calcination temperatures在生物炭/氯化锌比为5:3,真空煅烧温度为600 ℃条件下,考察了不同材料复合情况对电极产电性能的影响,结果如图8(a)和图8(f)所示。当生物炭/聚苯胺/热熔胶复合比例为5:1:4时,最大输出电压为0.137 V,随外电阻由大到小变化,反应器极化曲线电压由171 mV降至28 mV,最大体积功率为51 mW·m−3,电流密度为406 mA·m−3;当生物炭/聚苯胺/热熔胶复合比例为5:1:5时,最大输出电压为0.077 V,随外电阻由大到小变化,反应器极化曲线电压由100 mV降至13mV,最大体积功率为16 mW·m−3,电流密度为176 mA·m−3。由此可见,生物炭的含量对复合材料有显著影响。结合图3可知,在保证材料成型的前提下,生物炭含量越高,MFC的内阻越小,材料的产电性能越好[28]。
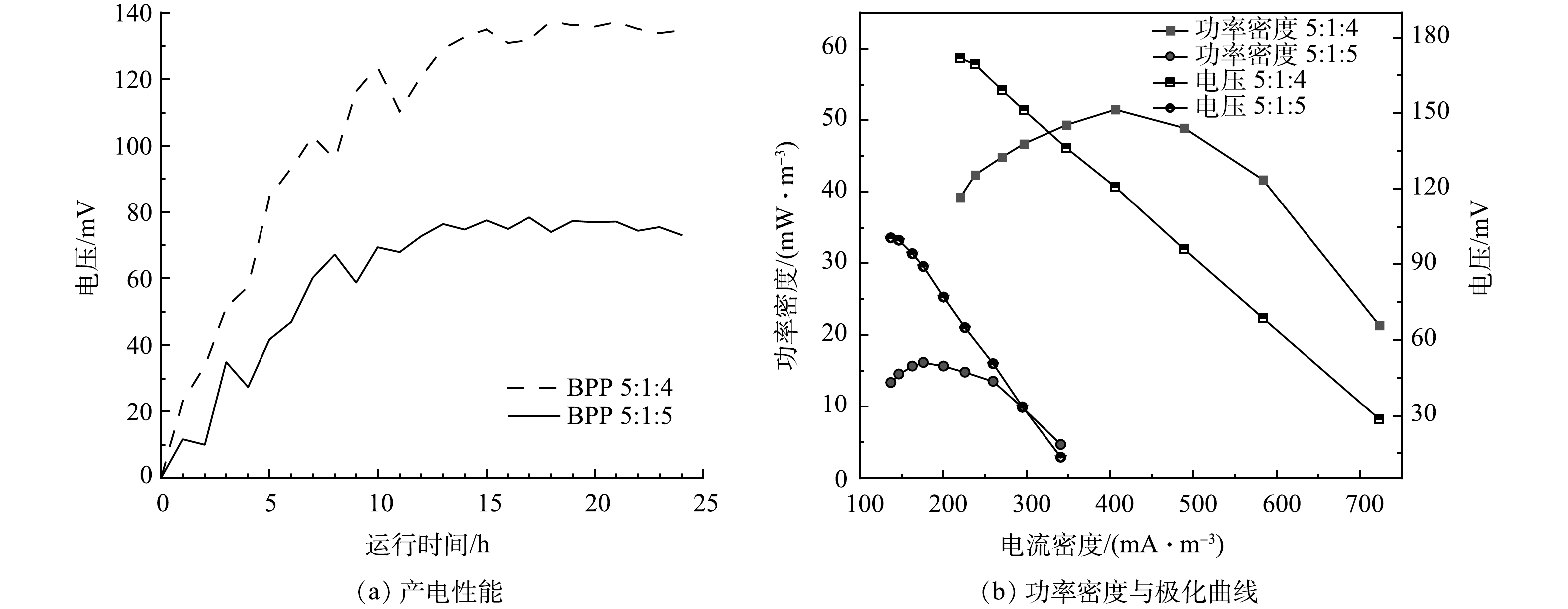 图 8 不同BPP条件下制备的电极材料Figure 8. Electrode materials prepared at different BPP conditions
图 8 不同BPP条件下制备的电极材料Figure 8. Electrode materials prepared at different BPP conditions2.7 改性核桃壳生物炭MFC对污染物的去除性能
根据以上结果,确认电极制备的最佳条件为BPP 5:1:4、煅烧温度600 ℃、生物炭/氯化锌比5:3时,即节约了生产成本,又达到最大产电量的90%,为减少对环境的污染,选用此种方法制备的电极材料用来探讨改性核桃壳生物炭电极材料用于MFC反应器降解污染物的长期效果,结果如图9所示。可以看出,随着时间的推移,MFC中出水COD由685 mg·L−1降至100 mg·L−1。第1天时,出水COD大幅下降,这说明微生物在反应初期消耗废水中大量有机物用于增殖。随着微生物增殖所需能量减少,有机物需求也逐渐减少,最终COD稳定去除率为85%。出水氨氮质量浓度由38 mg·L−1降低至4.5 mg·L−1,在第4天达到稳定,改性核桃壳生物炭MFC对氨氮的去除率最终达到88%。水体中的硝氮在7 d内先上升后下降,可以看出硝化菌株产生的硝态氮会被好氧反硝化菌株利用,MFC具有较好的脱硝态氮能力。
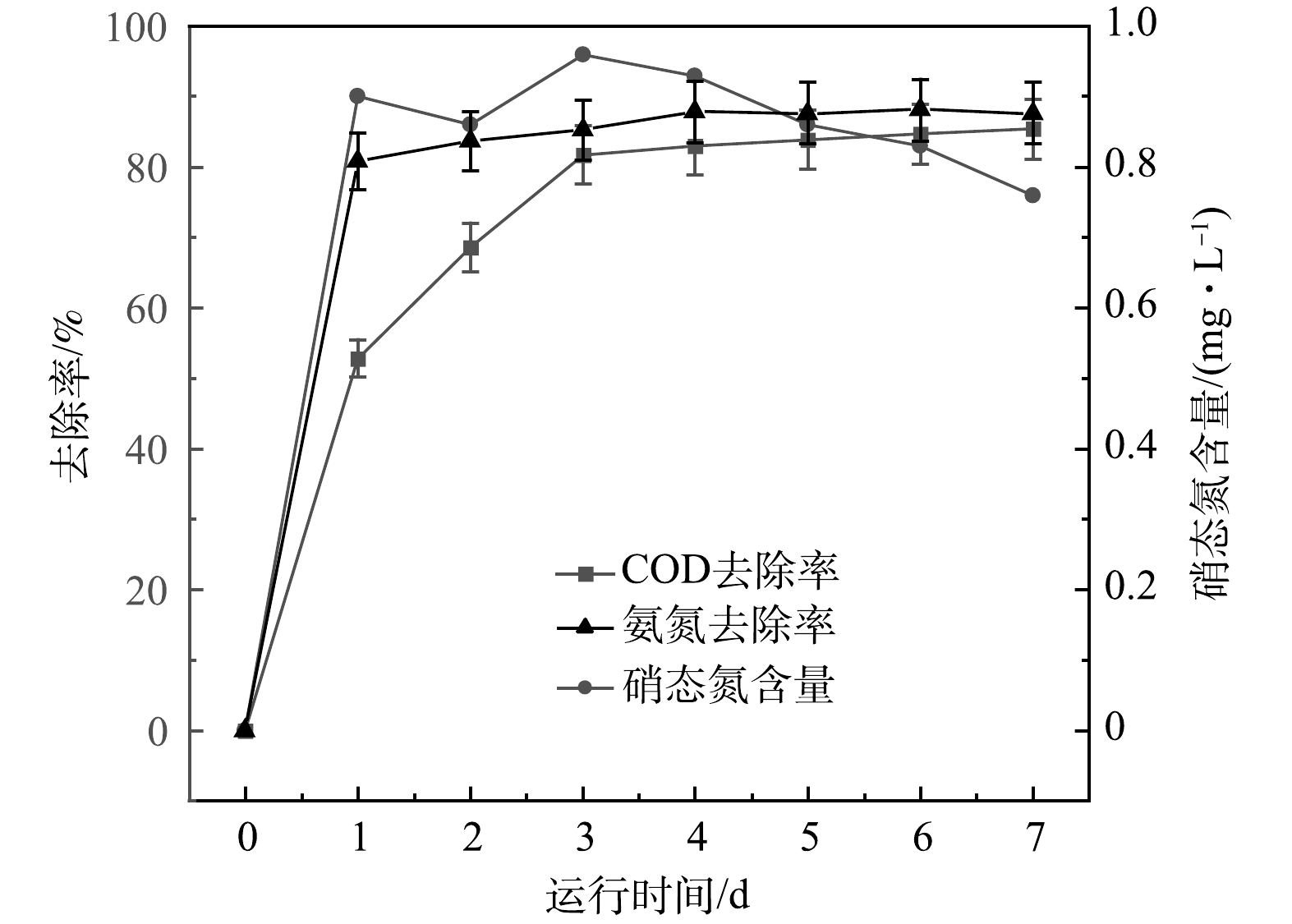 图 9 改性核桃壳生物炭MFC处理模拟废水中污染物的去除效果Figure 9. Removal effect of pollutants in simulated wastewater treated by modified walnut shell biochar MFC
图 9 改性核桃壳生物炭MFC处理模拟废水中污染物的去除效果Figure 9. Removal effect of pollutants in simulated wastewater treated by modified walnut shell biochar MFC3. 结论
1) MFC电极的最佳制备条件:活化时生物炭/氯化锌质量比5:3,真空煅烧温度为600 ℃,生物炭/聚苯胺/热熔胶复合电极比例为5:1:4,应用于MFC中最大的体积功率密度可达51 mW·m−3,对模拟废水中COD和氨氮的去除率分别为85%和88%。
2)相较于传统生物炭电极在水体中易碎,通过复合聚苯胺与热熔胶来制作生物炭电极可以在模拟废水中稳定运行。
3)用核桃壳生物炭通过简单的过程制备的MFC电极,为成本低廉,绿色清洁,操作简单的MFC发展方向提供了新的选择。
-
表 1 六种酚类化合基本信息和质谱参数
Table 1. Basic information and MS parameters of 6 phenolic compounds
化合物Compound 保留时间/mintR 相对分子量Relative molecular mass pka 母离子Parent Ion 子离子daughter ion 去簇电压/VDP 碰撞能量/VCE 入口电压/VEP 碰撞室出口电压/VCXP 苯酚 2.74 94.1 9.89 93.0 65.0* −60 −28 −10 −14 75.0 −60 −28 −10 −14 4-硝基酚 3.11 139.1 7.08 138.0 46.1* −70 −48 −10 −14 92.0 −60 −26 −10 −14 3-甲酚 3.33 108.1 10.09 106.9 92.0* −90 −27 −10 −14 105.8 −90 −27 −10 −14 2,4-二氯酚 4.45 163.0 7.68 160.9 125.0* −80 −44 −10 −14 89.0 −80 −21 −10 −14 2,4,6-三氯酚 5.04 197.5 7.41 195.0 35.0* −60 −50 −10 −14 159.0 −60 −30 −10 −14 五氯酚 5.67 266.3 4.92 264.8 34.9* −110 −59 −10 −14 37.0 −92 −59 −10 −14 4-硝基酚-d4 3.09 143.1 — 141.9 45.9* −70 −48 −10 −14 96.0 −60 −26 −10 −14 注: *是二级质谱定量子离子,剩余为定性子离子.
下载: 导出CSV
表 2 最佳流动相比例和梯度洗脱条件
Table 2. The Optimal mobile phase proportion and gradient elution conditions
时间/mintR 流动相A/%Mobile phase A 流动相B/%Mobile phase B 0.00 70 30 7.0 30 70 9.0 30 70 15.0 20 80 22.0 70 30 30.0 70 30 A:纯净水Pure water. B:甲醇 Methanol.
下载: 导出CSV
表 3 不同pH条件下萃取6种目标物的回收率
Table 3. Recoveries of 6 target substances using different pH
化合物Compound 回收率/%Recovery pH=1 pH=2 pH=3 pH=4 pH=5 pH=6 苯酚 86.7 85.2 77.4 75.1 76.6 75.1 4-硝基酚 106 107 99.9 102 96 97.0 3-甲酚 76.8 76.4 74.9 72.0 70.7 69.3 2,4-二氯酚 90.9 92.1 87.9 85.6 89.2 84.2 2,4,6-三氯酚 78.1 79.5 79.6 78.0 80.2 78.9 五氯酚 92.0 87.7 87.4 81.9 73.1 71.1
下载: 导出CSV
表 4 使用不同洗脱溶剂时6种目标物的回收率
Table 4. Recoveries of 6 target substances using different elution solvents
化合物Compound 回收率/%Recovery 二氯甲烷 甲醇 乙腈 乙腈(含1%乙酸) 苯酚 56.7 81.4 82.0 85.2 4-硝基酚 67.5 87.7 94.6 107 3-甲酚 52.8 69.9 69.4 76.4 2,4-二氯苯酚 57.9 78.9 81.7 92.1 2,4,6-三氯苯酚 62.1 73.7 78.4 79.5 五氯酚 60.0 74.2 77.4 87.7
下载: 导出CSV
表 5 使用不同洗脱体积时6种目标物的回收率
Table 5. Recoveries of 6 target substances using different elution solvents
化合物Compound 回收率/%Recovery 2 mL 4 mL 6 mL 8mL 10 mL 12 mL 14 mL 16 mL 苯酚 16.7 63.2 86.3 87.0 88.5 86.4 87.7 87.9 4-硝基酚 1.6 23.8 61.7 95.8 105 106 105 106 3-甲酚 11.3 49.1 75.2 76.9 77.6 78.1 77.2 76.4 2,4-二氯苯酚 0.0 8.2 32.2 61.2 83.3 90.6 92.0 91.4 2,4,6-三氯苯酚 0.4 13.7 38.2 70.4 78.1 79.6 80.0 80.2 五氯酚 0.0 0.7 9.8 27.9 50.5 74.2 92.0 90.1
下载: 导出CSV
表 6 线性范围、标准曲线与方法检出限
Table 6. The Linear range, standard curve and method detection limit
化合物Compound 线性范围/(μg·L−1)Linearity range 线性方程Linearity equation 相关系数r 检出限/(μg·L−1)LOD 苯酚 1—100 y=0.604x+ 0.0323 0.9996 0.050 4-硝基酚 1—100 y=13.4x+0.311 0.9992 0.050 3-甲酚 1—100 y=0.810x+ 0.0212 0.9995 0.005 2,4-二氯苯酚 1—100 y= 0.0267 x+6.21×10−40.9991 0.010 2,4,6-三氯苯酚 1—100 y=0.683x+ 0.00243 0.9996 0.005 五氯酚 1—100 y= 0.0883 x+0.00334 0.9987 0.005
下载: 导出CSV
表 7 地下水样品分析精密度与准确度试验结果
Table 7. The results of precision and accuracy for analysis of groundwater
化合物Compound 本底值/(μg·L−1)Background values 加标量/(μg·L−1)Spiked value 测定值/(μg·L−1)Measured value 相对标准偏差/%RSD 平均加标回收率/%The average spiked recovery 苯酚 0.748 0.500 1.279 4.0 106 4-硝基酚 0.282 0.500 0.738 7.0 91.4 3-甲酚 0.040 0.500 0.428 4.2 77.6 2,4-二氯酚 ND* 0.500 0.470 5.1 94.0 2,4,6-三氯酚 0.005 0.500 0.470 6.6 92.9 五氯酚 0.238 0.500 0.661 8.0 84.6 注:ND,未检出 not detected.
下载: 导出CSV
表 8 六种有机酚的分子式和裂解途径
Table 8. Molecular formula and cleavage pathway of six organic phenols
化合物Compound 分子式Molecular formulas 裂解途径Fragmentation pathway 苯酚 C6H5OH 
4-硝基酚 C6H5NO3 3-甲基酚 C7H8O 2,4-二氯酚 C6H4Cl2O 2,4,6-三氯酚 C6H3Cl3O 五氯酚 C6HCl5O
下载: 导出CSV
-
[1] NASSIRI M, ZAHEDI M M, POURMORTAZAVI S M, et al. Optimization of dispersive liquid-liquid microextraction for preconcentration and spectrophotometric determination of phenols in Chabahar Bay seawater after derivatization with 4-aminoantipyrine[J]. Marine Pollution Bulletin, 2014, 86(1/2): 512-517. [2] LIU W T, XIE M Y, HAO X T, et al. Rapid synergistic cloud point extraction for simultaneous determination of five polar phenols in environmental water samples via high performance liquid chromatography with fluorescence detection[J]. Microchemical Journal, 2021, 164: 105963. doi: 10.1016/j.microc.2021.105963 [3] McLELLAN I, CARVALHO M, PEREIRA C S, et al. The environmental behaviour of polychlorinated phenols and its relevance to cork forest ecosystems: A review[J]. Journal of Environmental Monitoring, 2007, 9(10): 1055-1063. doi: 10.1039/b701436h [4] WU Y L, HU B, HOU Y L. Headspace single drop and hollow fiber liquid phase microextractions for HPLC determination of phenols[J]. Journal of Separation Science, 2008, 31(21): 3772-3781. doi: 10.1002/jssc.200800365 [5] XING H Z, WANG X, CHEN X F, et al. Accelerated solvent extraction combined with dispersive liquid-liquid microextraction before gas chromatography with mass spectrometry for the sensitive determination of phenols in soil samples[J]. Journal of Separation Science, 2015, 38(8): 1419-1425. doi: 10.1002/jssc.201500022 [6] HUANG X J, QIU N N, YUAN D X. Development and validation of stir bar sorptive extraction of polar phenols in water followed by HPLC separation in poly(vinylpyrrolididone-divinylbenzene) monolith[J]. Journal of Separation Science, 2009, 32(9): 1407-1414. doi: 10.1002/jssc.200800708 [7] 周文敏, 傅德黔, 孙宗光. 中国水中优先控制污染物黑名单的确定[J]. 环境科学研究, 1991, 4(6): 9-12. ZHOU W M, FU D Q, SUN Z G. Determination of black list of China’s priority pollutants in water[J]. Research of Environmental Sciences, 1991, 4(6): 9-12 (in Chinese).
[8] 杨淑贞, 韩晓冬, 陈伟. 五氯酚对生物体的毒性研究进展[J]. 环境与健康杂志, 2005, 22(5): 396-398. YANG S Z, HAN X D, CHEN W. Advances in the toxicity of pentacholrophenol on organism[J]. Journal of Environment and Health, 2005, 22(5): 396-398 (in Chinese).
[9] DOLATTO R G, MESSERSCHMIDT I, FRAGA PEREIRA B, et al. Preconcentration of polar phenolic compounds from water samples and soil extract by liquid-phase microextraction and determination via liquid chromatography with ultraviolet detection[J]. Talanta, 2016, 148: 292-300. doi: 10.1016/j.talanta.2015.11.004 [10] ABOLGHASEMI M M, PARASTARI S, YOUSEFI V. Polypyrrole-montmorillonite nanocomposite as sorbent for solid-phase microextraction of phenolic compounds in water[J]. Journal of Separation Science, 2014, 37(23): 3526-3532. doi: 10.1002/jssc.201400602 [11] LU W H, WANG X Y, WU X Q, et al. Multi-template imprinted polymers for simultaneous selective solid-phase extraction of six phenolic compounds in water samples followed by determination using capillary electrophoresis[J]. Journal of Chromatography A, 2017, 1483: 30-39. doi: 10.1016/j.chroma.2016.12.069 [12] MASI O H, GULICK JR W M. An optimized gas chromatographic determination of priority pollutant phenols[J]. Journal of High Resolution Chromatography, 1987, 10(12): 647-649. doi: 10.1002/jhrc.1240101205 [13] PROESTOS C, SERELI D, KOMAITIS M. Determination of phenolic compounds in aromatic plants by RP-HPLC and GC-MS[J]. Food Chemistry, 2006, 95(1): 44-52. doi: 10.1016/j.foodchem.2004.12.016 [14] SALCEDO G M, KUPSKI L, DEGANG L, et al. Determination of fifteen phenols in wastewater from petroleum refinery samples using a dispersive liquid—Liquid microextraction and liquid chromatography with a photodiode array detector[J]. Microchemical Journal, 2019, 146: 722-728. doi: 10.1016/j.microc.2019.01.075 [15] PAN S D, CHEN X H, SHEN H Y, et al. Retracted: Rapid and effective sample cleanup based on graphene oxide-encapsulated core-shell magnetic microspheres for determination of fifteen trace environmental phenols in seafood by liquid chromatography-tandem mass spectrometry[J]. Analytica Chimica Acta, 2019, 1046: 208. doi: 10.1016/j.aca.2018.10.049 [16] 李娟, 王荟. 水中多种酚类化合物衍生化方法研究[J]. 环境监控与预警, 2014, 6(5): 23-25,28. LI J, WANG H. Derivative methods of phenolic compounds in water[J]. Environmental Monitoring and Forewarning, 2014, 6(5): 23-25,28 (in Chinese).
[17] 张莉, 桂建业, 张永涛, 等. 改性聚合物萃取-五氟苄基衍生化-气相色谱-质谱法测定水中酚类化合物[J]. 理化检验-化学分册, 2013, 49(10): 1155-1158. ZHANG L, GUI J Y, ZHANG Y T, et al. GC-MS determination of phenols in water after extraction with modified polymer and derivatization with pentafluorobenzyl bromide[J]. Physical Testing and Chemical Analysis (Part B:Chemical Analysis), 2013, 49(10): 1155-1158 (in Chinese).
[18] 唐雪, 李强, 郝红元, 等. 超高效液相色谱/三重四极杆质谱-直接进样分析地表水中硝基酚类化合物[J]. 环境化学, 2020, 39(3): 831-834. TANG X, LI Q, HAO H Y, et al. Simultaneous determination of nitrophenols in surface water by ultra high performance liquid chromatography-tandem mass spectrometry[J]. Environmental Chemistry, 2020, 39(3): 831-834 (in Chinese).
[19] 罗碧容, 钱蜀, 潘乐丹, 等. 高效液相色谱-串联质谱法同时测定水中18种酚类污染物[J]. 分析测试学报, 2016, 35(8): 974-980. LUO B R, QIAN S, PAN L D, et al. Simultaneous determination of 18 phenols pollutants in water samples by high performance liquid chromatography-tandem mass spectrometry[J]. Journal of Instrumental Analysis, 2016, 35(8): 974-980 (in Chinese).
[20] 杨秋红, 程小艳, 杨坪, 等. 固相萃取-高效液相色谱串联质谱法同时检测地表水中的2, 4-二氯酚、2, 4, 6-三氯酚和五氯酚[J]. 分析化学, 2011, 39(8): 1208-1212. YANG Q H, CHENG X Y, YANG P, et al. Simultaneous determination of 2, 4-dichlorophenol, 2, 4, 6-trichlorophenol and pentachlorophenol in surface water by high performance liquid chromatography-tandem mass spectrometry with solid phase extraction[J]. Chinese Journal of Analytical Chemistry, 2011, 39(8): 1208-1212 (in Chinese).
[21] 范云场, 胡正良, 陈梅兰等. 离子液体液-液萃取-高效液相色谱测定水中酚类化合物[J]. 分析化学, 2008, 36(9): 1157-1161. FAN Y C, HU Z L, CHEN M L, et al. Analysis of Phenolic Compounds by lonic Liquid Based Liquid-Liquid Extraction Coupled with High Performance Liquid Chromatography[J]. Chinese Journal of Analytical Chemistry, 2008, 36(9): 1157-1161. (in Chinese)
[22] 宣栋梁, 黎源倩. 高效液相色谱法测定环境水中的酚类化合物[J]. 中国公共卫生, 2002, 18(9): 1102-1104. XUAN D L, LI Y Q. Determination of Phenols in Environmental Water by Solid Phase Extraction and High Performance Liquid Chromatography[J]. Chinese Journal of Public Health, 2002, 18(9): 1102-1104. (in Chinese)
[23] 中华人民共和国环境保护部. 环境监测 分析方法标准制修订技术导则: HJ 168—2010[S]. 北京: 中国环境科学出版社, 2008. Ministry of Environmental Protection of the People's Republic of China. Environmental monitoring-Technical guideline on drawing and revising analytical method standards: HJ 168—2010[S]. Beijing: China Environmental Science Press, 2008(in Chinese).
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 543
- HTML全文浏览数: 543
- PDF下载数: 15
- 施引文献: 0
