

-
选择性催化还原法(selective catalytic reduction, SCR)脱除烟气中NOx是目前工业上烟气脱硝首选技术, 广泛应用于燃煤电厂等固定污染源的烟气脱硝, 脱硝效率较高, 能够满足我国当前脱硝实际, 是目前最好的NOx烟气净化治理脱硝技术[1]. SCR技术的核心是催化剂. 目前, 国内外应用较成熟SCR催化剂为V2O5/TiO2系列催化剂[2-4], 该催化剂在较高的反应温度(>350 ℃)具有较高的催化活性和抗硫性能. 由于我国的脱硝工艺必须采用末端布置, 然而末端放置的工艺方式会使得烟气温度大大降低, 低于SCR催化剂所需的活性温度. 又因V2O5/TiO2系列催化剂重金属元素V毒性较大对环境和人体健康的毒副作用以及成本较高等, 中低温(<250 ℃)高活性SCR脱硝催化剂的研究开发受到国内外众多研究者的高度关注[5-7].
中低温NH3-SCR催化剂主要包括Mn、Fe、Cu、Ce、Cr等非钒基氧化物催化剂[8-12], 其中, Mn基氧化物催化剂由于其较高的低温NH3-SCR脱硝性能、环境友好、价格低廉的优点而备受关注. Peña等[13]比较了各种负载金属氧化物的催化剂, 发现TiO2负载V、Cr、Fe、Mn、Ni、Cu、Co在NH3-SCR过程中, 活性顺序为Mn>Cu>Cr>Co>Fe>V>Ni. 研究表明锰的氧化形态、制备方法等对锰系催化剂的催化性能影响显著[14-15]. 本课题组对原位生长法制备脱硝催化剂进行了较为深入地研究. Zhang等[9]以管状的埃洛石为载体, 采用原位生长法成功地制备出了MnOx/HNTs催化剂, 在36000 h−1下, 50—300 ℃之间NO转化率可保持在90%以上, 催化剂表现出的高活性与MnOx呈非晶态良好地分散在埃洛石纳米管的管内与管外有关, 埃洛石所具有的空间纳米结构限制了MnOx的生长, 使其趋于无定形态, 促进了活性位点的形成与分散. Zhang等[12]分别采用原位生长法与浸渍法制备了CeMn/TiO2催化剂, 探究了制备方法对以粉末状TiO2为载体所制备的催化剂脱硝性能的影响. 其中, 采用原位生长法所制备的样品表现出更好的催化活性, Ce(1.0)Mn/TiO2-SP在125—300 ℃之间, 27000 h−1下, NO转化率接近100%.
综上可知, 原位生长法制备的脱硝催化剂具有良好的催化活性, 且活性组分在载体表面呈非晶态, 分散性高. 然而, 原位生长法制备过程中MnOx的负载机制尚不清晰[9,11-12]. 为了明晰原位生长法制备过程中MnOx的负载机制, 阐明催化剂的催化活性机理. 本文以20—40目硅藻土基多孔陶瓷颗粒为载体, 制备了Mn基多孔陶瓷催化剂, 以传统浸渍法作为对比, 考察了不同制备方法Mn基催化剂的低温SCR脱硝性能, 探究了原位生长法制备催化剂过程中活性组分的负载机制.
-
埃洛石(HAL, 郑州金阳光陶瓷有限公司),主要化学组成(质量分数)为: 50.31% SiO2、38.27% Al2O3、7.59% SO3、1.22% K2O、1.12% Fe2O3; 硅藻土(DE, 国药集团化学试剂有限公司), 主要化学成分(质量分数)为: 88.80% SiO2、 2.98% Al2O3、2.06% Fe2O3、2.03% CO2、1.21% Na2O; 乙酸锰 Mn(CH3COO)2,高锰酸钾KMnO4, 聚乙烯醇(PVA, (CH2CHOH)n),石墨(Graphite, C), 均由国药集团化学试剂有限公司提供,纯度为AR;模拟烟气由南京上元工业气体厂提供.
-
称取6 g硅藻土粉末与4 g埃洛石混合, 再称取0.4 g淀粉, 采用湿法球磨(球:原料:酒精= 2:1:0.6)将上述粉末混合均匀, 110 ℃干燥24 h, 过200目筛, 滴加4% wt. PVA, 研磨,经红外压片机4 MPa压片成型, 再经管式炉1000 ℃煅烧2 h, 得到硅藻土基多孔陶瓷. 将所得多孔陶瓷破碎后过20目筛, 作为催化剂载体, 并标记为DE-HAL.
-
原位生长法: 称取5 g上述制备的硅藻土基多孔陶瓷, 称取化学计量的Mn(Ac)2溶解于一定量的去离子水中, 用以作为锰的前驱体. 将多孔陶瓷等体积浸渍于Mn(Ac)2溶液中, 静置24 h后加入KMnO4溶液, 搅拌后静置24 h, 多次洗涤抽滤至滤液澄清无色以达到除去K+的目的, 于鼓风干燥箱中110 ℃干燥24 h, 即得所需催化剂, 标记为Mn(8)/DE-HAL(SP)(催化剂中Mn元素质量分数为8%). 重复上述制备步骤, 但催化剂不经过洗涤抽滤, 干燥后在300 ℃下煅烧3 h, 标记为Mn(8)/DE-HAL(SP-300).
浸渍法: 称取5 g制备的硅藻土基多孔陶瓷, 称取化学计量的Mn(Ac)2溶解于一定量的去离子水中, 将多孔陶瓷等体积浸渍于Mn(Ac)2溶液中, 静置24 h后于烘箱中110 ℃干燥24 h, 再置于马弗炉中, 空气氛围, 300 ℃煅烧3 h, 标记为Mn(8)/DE-HAL(IP-300).
-
催化剂的脱硝活性评价测试装置为长80 cm, 内径15 mm的竖式石英玻璃常压固定床反应器, 活性评价装置主要由模拟烟气(1000 mg·L−1 NO, 1000 mg·L−1 NH3, 6%vol H2O, 3%vol O2, N2为平衡气混合而成, 总流量为350 mL·min−1), 固定床反应器和分析检测装置的3部分组成. 反应空速为52000 h−1, 反应温度为100—300 ℃. 采用MRU OPTIMA7烟气分析仪在线测量反应器进出口NOx浓度,准确度为 ±3%。采用MGA6plus分析仪测定反应器出口N2O浓度,准确度为 ±2%。采用GT-1000-NH3分析仪测定NH3浓度,准确度为 ±1%.
催化剂脱硝活性以NO转化率和N2的选择性衡算,计算公式为:
-
采用美国康塔NOVA 2200e型比表面积及孔径分析仪分析样品的比表面积和孔径孔容. 采用荷兰帕纳科PANalytical X-Pert PRO MPD型X射线衍射仪对多孔陶瓷和催化剂的物相与晶型进行表征,Cu Kα射线, 操作电压为40 kV, 操作电流为40 mA, 扫描速度为4(o)·min−1, 扫描区间范围2θ=5°—70°. 采用美国Thermo ESCALAB250Xi型X射线光电子能谱仪对催化剂进行XPS测试, Al Kα射线, 管电压为1486.6 eV. 采用德国卡尔蔡司Gemini 500型热场发射扫描电子显微镜观察多孔陶瓷与催化剂的表面微观结构.采用全自动多功能吸附仪(TP-5080D)进行H2的程序升温还原(H2-TPR)实验.采用Micromeritics Autochem-II仪器对催化剂进行NH3/NO程序升温脱附(NH3/NO-TPD)实验,利用质谱(Hiden QIC-200)对脱附过程中出口气体浓度进行实时监测。.
-
图1为不同制备方法催化剂的脱硝活性-温度曲线. 由图1可以看出, Mn/DE-HAL(SP)具有最优异的催化活性, 在100—250 ℃之间NO转化率高于90%, 225 ℃之后催化活性虽然随温度的升高而下降, 但在整个测试温度段, NO转化率不低于80%. Mn/DE-HAL(SP-300)在100 ℃时NO转化率约为38%, 150—200 ℃之间NO转化率约为70%, 该样品催化活性较差可能是由于催化剂中存在大量K元素, 研究表明, 碱金属会占据催化剂的部分酸性位点, 降低催化剂对NH3的吸附和活化能力[16], 从而导致催化剂催化活性较差. 此外, 煅烧也是降低样品NO转化率的原因之一. Mn/DE-HAL(IP-300)低温下催化活性较差, 150 ℃之后NO转化率显著增加, 在225—300 ℃之间催化活性较为稳定, NO转化率约为80%. 下文将通过表征分析对催化剂的理化性质进行进一步探究.
-
N2的选择性为Mn基SCR催化剂的重要性质之一,其实质上反映了反应系统中目的反应与副反应反应速率的竞争关系,选择性越高越有利于催化反应的进行. 图2为不同制备方法催化剂N2选择性曲线图. 由图2可以看出,在整个温度区间范围内, Mn/DE-HAL(SP)催化剂N2选择性均在95%左右,说明在100—300 ℃温度范围内,Mn/DE-HAL(SP)催化剂稳定性较好且具有高N2选择性.
-
为了更直观地研究催化剂的形貌, 对样品进行了SEM表征. 图3为催化剂的SEM与Mapping图. 由图3(c)可以看出, 原位生长法所制备的催化剂表面MnOx呈絮状, 由图3(f)可以看出采用浸渍法所制备的样品表面的MnOx为颗粒状, 两种方法所制备的催化剂表面的活性组分形貌差异显著. 由图3(b)和图3(e)的Mapping图可以清晰地观察到Mn(8)/DE-HAL(SP)表面的活性组分的分散情况与Mn(8)/DE-HAL(IP-300)差别并不明显. 可见, 制备方法影响载体表面MnOx的形貌, 而对活性组分在载体表面的分散性没有太大影响. 对比两种不同制备方法催化剂形貌, 结果表明, 催化剂表面活性组分的分散性并非是影响催化剂脱硝性能的决定性因素. 图3(g)和(h)分别为原位生长法制备的催化剂的断裂面形貌图与Mapping图, 由图3(h)可以看出, MnOx并未进入颗粒内部; 图3(i)和图3(j)分别为浸渍法制备的催化剂的断裂面形貌图与Mapping图, 由图3(j)可以看出, MnOx同时负载在了颗粒内部. 该结果表明, 原位生长法所制备的催化剂, MnOx仅附着在陶瓷颗粒表面, 而浸渍法所制备的样品, MnOx进入了陶瓷颗粒内部. 下面将进一步探究出现这一现象的原因.
两种方法制备催化剂的第一步均是将载体浸渍于Mn(Ac)2溶液中, 浸渍法经过干燥煅烧后, 颗粒表面与内部均生成了MnOx, 而原位生长法在加入KMnO4进行化学反应, 再经过洗涤抽滤后颗粒内部已观察不到MnOx的存在. 现采用原位生长法制备催化剂, 不经水洗抽滤, 干燥后经300 ℃煅烧3 h, 探究是否是洗涤抽滤除去了颗粒内部的Mn(Ac)2.
图4为原位生长法制备Mn(8)/DE-HAL(SP-300)催化剂的SEM与Mapping图. 图4(a)为催化剂外表面的SEM图, 图4(d)为催化剂内部断裂面的SEM图. 由图4(b)和(c)可以看出, MnOx分散在催化剂的外表面, 由于干燥前未经过洗涤抽滤, 使得K大量存在于样品表面. 由图4(e)和(f)可以看出, 催化剂内部仍然有K, 但并未观察到MnOx的存在. 该结果表明, 采用原位生长法制备的样品内部的活性组分并非被洗涤除去. KMnO4不仅与样品表面的Mn(Ac)2反应, 位于多孔陶瓷颗粒内部的Mn2+也会迁移到样品表面与KMnO4反应生成MnOx. 这意味着, 原位生长法制备的催化剂, 活性组分富集在载体表面, 结合催化剂的催化活性分析可知, 这些负载在载体表面的MnOx在SCR反应中发挥着重要作用.
-
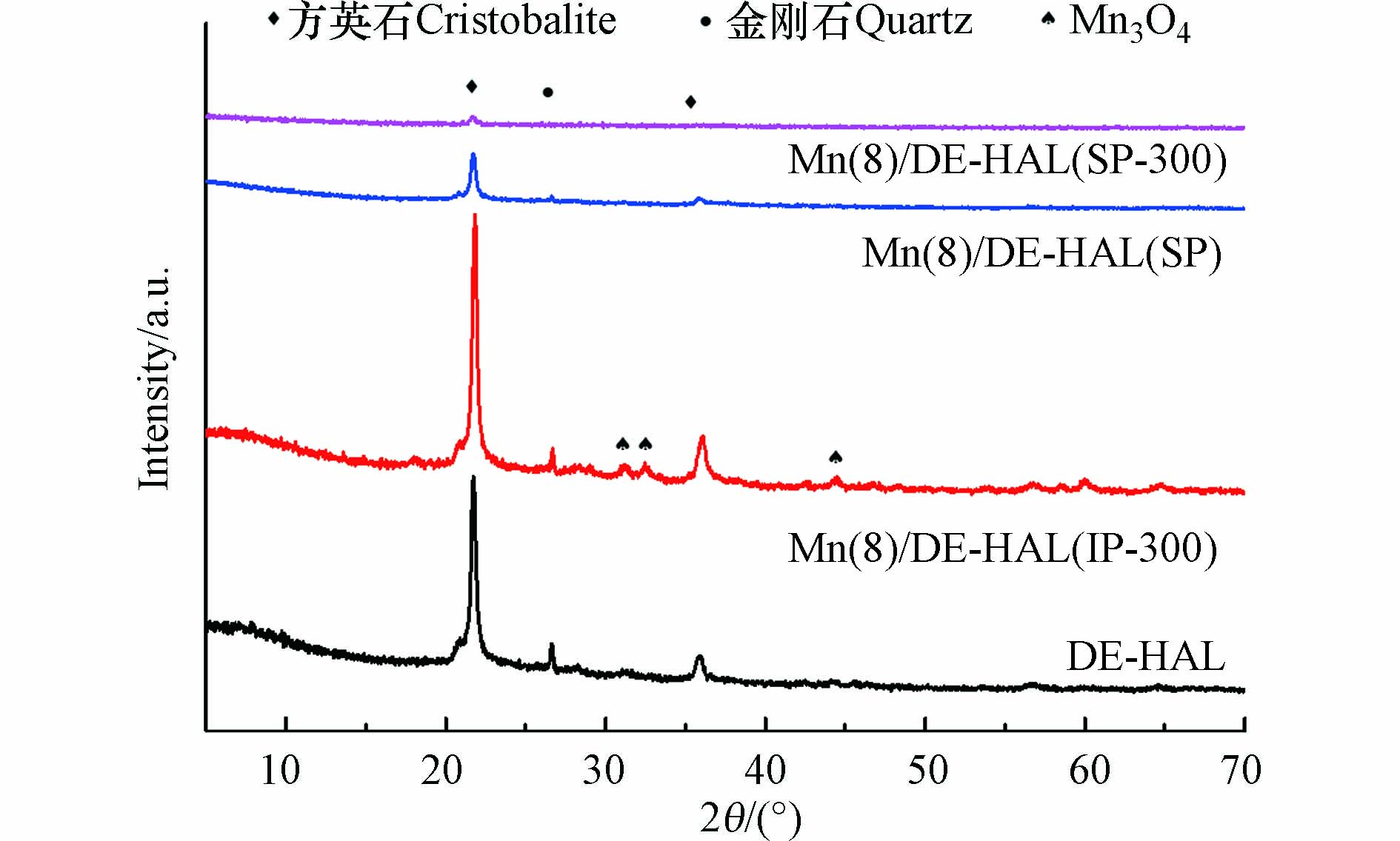
图5为多孔陶瓷和催化剂的XRD图谱. 如图5所示, DE-HAL在21.66°、35.87°以及31.05°处的峰归属于方石英的特征衍射峰(PDF#39-1425), 26.62°处的峰归属于石英的特征衍射峰(PDF#46-1045).
Mn/DE-HAL(SP)的石英峰与方石英峰均显著减弱, 出现这一现象原因一方面是因为MnOx包覆在载体表面, 削弱了其原有的衍射峰, 另一方面, 由于样品的特殊性, 使得筛选出来的样品MnOx含量较多. 此外, 样品中并未观察到MnOx的衍射峰, 这主要是由于活性组分在载体表面呈无定形态分布. Mn/DE-HAL(IP-300)中除了石英峰与方石英峰外, 在31.03°、32.44°以及44.37°处还可观察到Mn3O4的特征衍射峰(PDF#18-0803). 催化剂表面活性组分结晶度越低, 越具有较多的表面活性点位, 有利于提升催化剂的催化活性, 这可能是Mn/DE-HAL(SP)表现出更好的催化活性的重要原因.
-
比表面积和孔结构是评价催化剂优良的重要指标, 总体而言, 较大的比表面积能够为催化剂提供更多的活性位点, 使活性组分MnOx得到更好的分散, 更有利于反应气体的吸附, 从而提高催化活性. 表1为样品的孔结构与BET数据, 其比表面积大小顺序为: Mn(8)/DE-HAL(SP) > Mn(8)/DE-HAL(SP-300) > Mn(8)/DE-HAL(IP-300) > DE-HAL. 可以看到, 与Mn(8)/DE-HAL(SP)相比, Mn(8)/DE-HAL(SP-300)与Mn(8)/DE-HAL(IP-300)的孔容较小, 这可能是由于煅烧使得活性组分堵塞了部分小孔. 此外, 负载MnOx后, 所有样品的孔径均有不同程度的减小, 这与活性组分和载体结合后, 形成了丰富的孔道结构有关. 相较于DE-HAL多孔陶瓷, 负载MnOx后, 样品的比表面积显著增加, 主要是由于MnOx成功负载后提升了样品的比表面积, 这意味着催化剂的比表面积主要是由MnOx贡献的. 结合SEM与Mapping分析, Mn/DE-HAL(SP)的活性组分主要分散在载体表面, Mn(8)/DE-HAL(IP-300)的活性组分分散在载体表面与内部孔隙中, 即MnOx对Mn/DE-HAL(SP)比表面积的贡献集中在载体表面, 对Mn(8)/DE-HAL(IP-300)比表面积的提升作用于载体表面与内部, 而表面的活性组分更容易参与到SCR反应中, 催化剂表面具有更多的反应活性位点, 有利于提升催化剂的脱硝性能, 这是Mn(8)/DE-HAL(SP)具有良好的催化活性的重要原因.
-
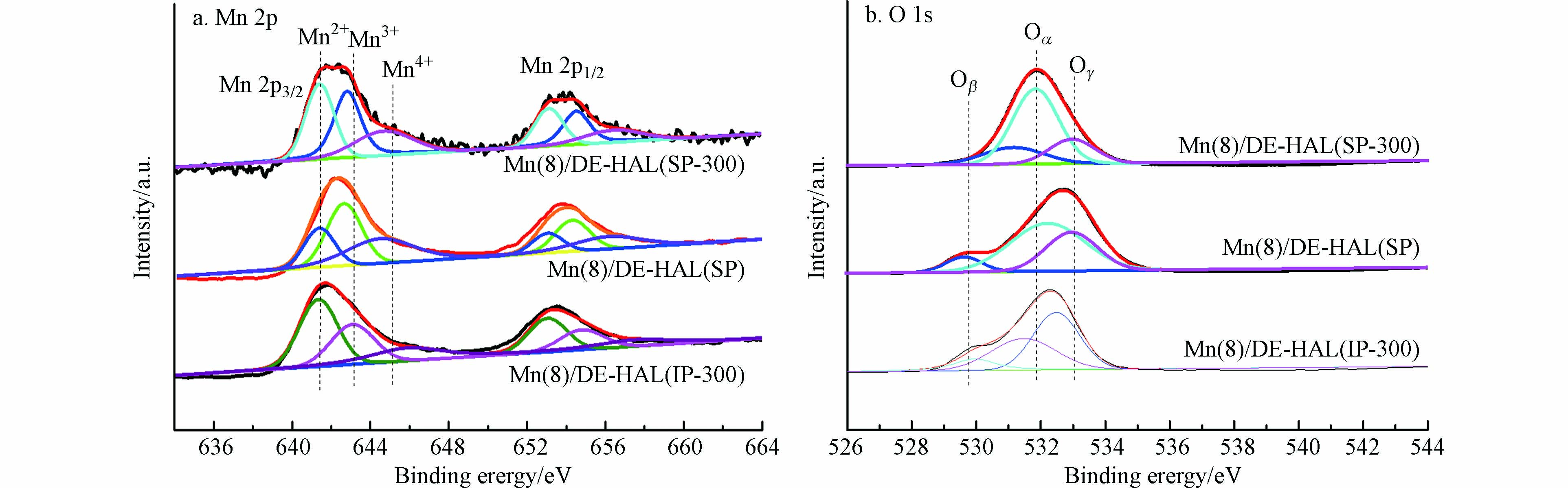
图6为采用两种方法所制备的催化剂XPS图谱, 表2为催化剂的XPS分析结果. 图6(a)中644.4—645 eV、642.2—642.7 eV和 640.9—641.4 eV的3个峰分别归属于Mn4+、Mn3+和Mn2+ [17-18]. Mn4+是Mn基催化剂在NH3-SCR反应中最活跃的物种, Mn4+易于吸附NH3并生成Mn4+-NH3, 而Mn3+有利于吸附NO并生成Mn3+-NO[19]. 研究表明, Mn4+-NH3可以与Mn3+-NO结合生成中间产物NH4NO3[19], 与NO反应生成NH4NO2会进一步分解成N2和H2O[20], 从而促进NH3-SCR反应的进行. 图6(b)中530.1—530.2 eV, 531.7—532.4 eV和533.5—534.4 eV的 3个峰分别归属于表面晶格氧(Oβ)、化学吸附氧(Oα)和吸附的水物种(Oγ)[21-22]. Oα在NH3-SCR反应中比Oβ更活泼, 因为其具有更高的迁移率[23], 有利于将NO氧化成NO2, 促进快速SCR反应的进行[24]. 与Mn(8)/DE-HAL(SP)相比, Mn(8)/DE-HAL(SP-300)的Mn4+比例降低了14.53%, 意味着碱金属的存在降低了Mn4+浓度; Oα的比例降低了11.58%, 研究表明, 碱金属会与表面氧中心形成强键[25], 抑制催化剂表面化学吸附氧的形成[26]. 相较于Mn(8)/DE-HAL(SP), Mn(8)/DE-HAL(IP-300)中Mn4+和Oα的比例分别降低了17.10%和9.54%. 该结果表明, 原位生长法可以提升催化剂表面Mn4+和Oα的浓度, 更高的Mn4+和Oα的相对百分比含量, 有利于促进SCR反应的进行, 提升样品的催化活性.
-
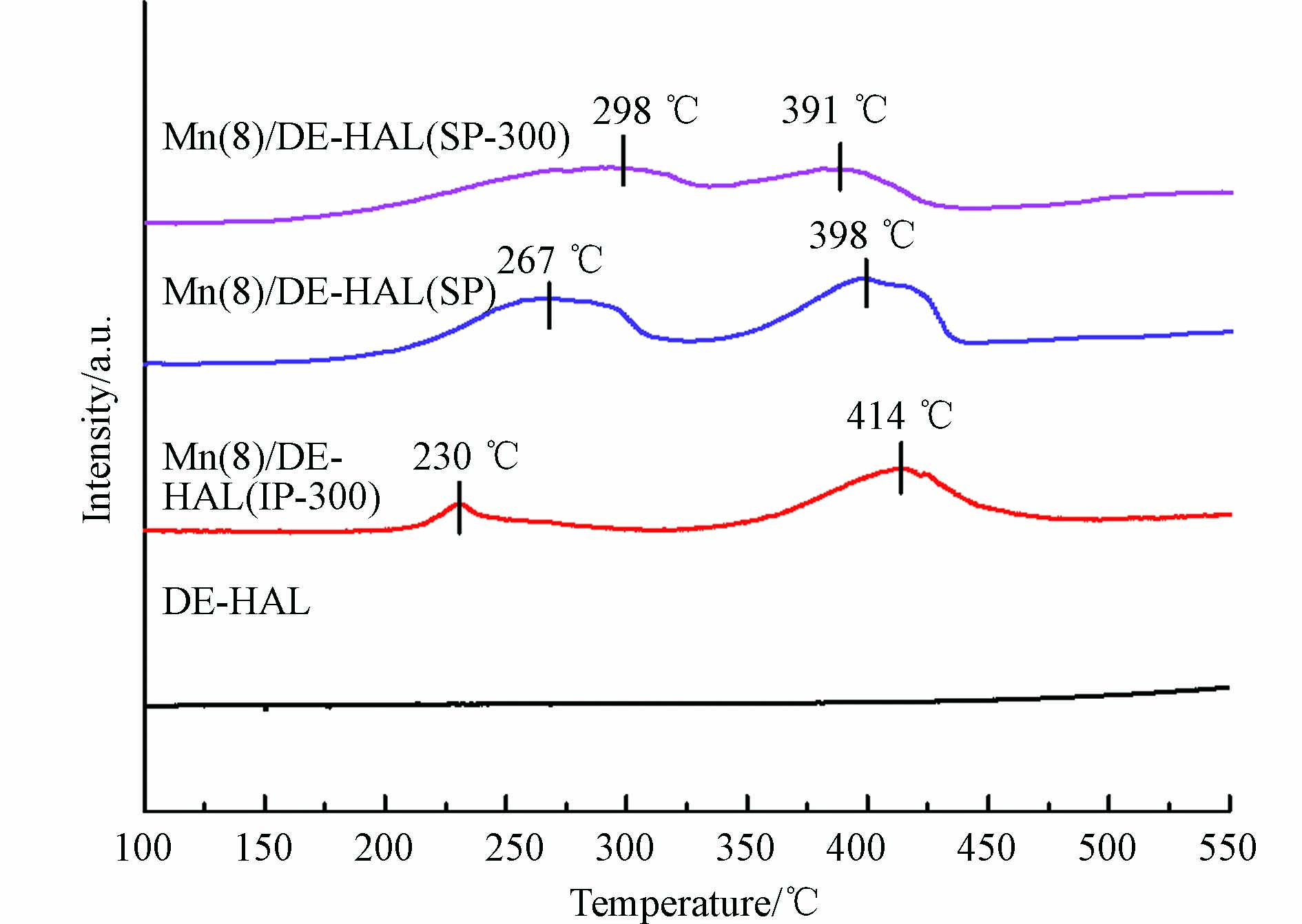
通过H2-TPR对催化剂氧化还原能力进行测试, 结果如图7所示, 未负载活性组分的陶瓷在100—550 ℃之间没有明显的还原峰, 而Mn(8)/DE-HAL(SP)在267 ℃和398 ℃处出现了2个还原峰, Mn(8)/DE-HAL(SP-300)在298 ℃和391 ℃处出现了两个还原峰, Mn(8)/DE-HAL(IP-300)的2个还原峰分别出现在230 ℃和414 ℃处, 催化剂在100—550 ℃之间的2个还原峰分别归因于MnO2到Mn2O3的还原和Mn2O3到MnO的还原[27]. 虽然Mn(8)/DE-HAL(IP-300)第1个还原峰温度最低, 但其峰面积较小, 而H2消耗峰的面积在某种程度上可以反应出催化剂表面活性物种的数量, 可以看出, Mn(8)/DE-HAL(IP-300)表面Mn4+浓度相对较低, 这与XPS结果是一致的. 原位生长法所制备的样品, 活性组分虽然仅分散在载体表面, 但却表现出更强的还原性, 这意味着载体表面的活性组分对催化剂的氧化还原能力有重要影响. 在低温区域, Mn(8)/DE-HAL(SP)的还原峰比Mn(8)/DE-HAL(SP-300)高, 且前者的H2消耗量也比后者大, 意味着Mn(8)/DE-HAL(SP)的氧化还原能力强于Mn(8)/DE-HAL(SP-300), 该结果表明, K的存在降低了催化剂的氧化还原能力.
-
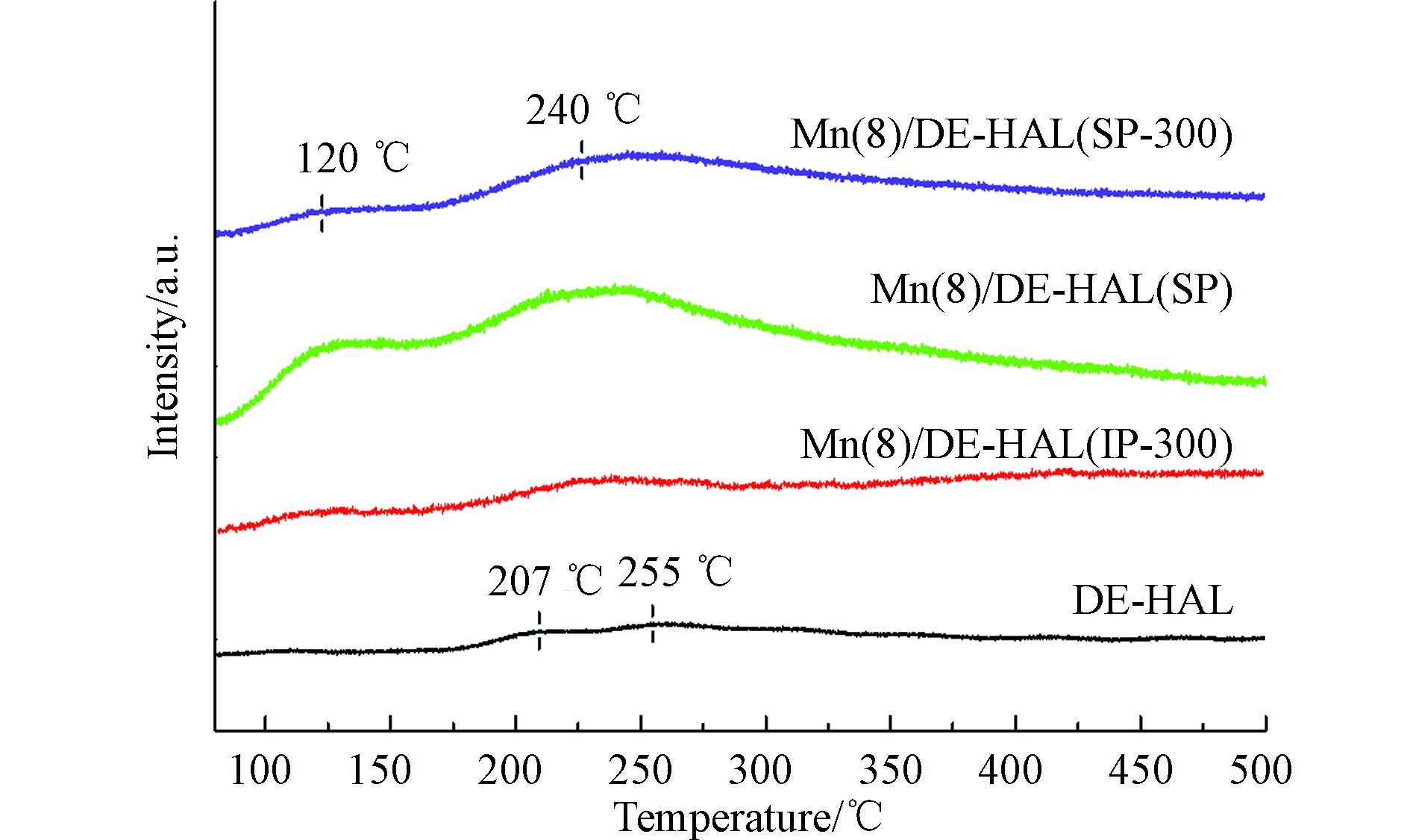
图8为催化剂的NH3-TPD图谱. 研究表明, 在NH3-TPD中, 200 ℃以下的峰可归因于NH3在弱酸位点上物理和弱化学吸附的连续解吸, 200—450 ℃范围内的峰可归因于中弱酸位点[19]. 酸性位点的数量与分布情况是催化剂脱硝性能的重要影响因素, 是保证NH3吸附和活化的前提[23], 其中, 位于100—350 ℃的酸性位点数量影响催化剂低温下的脱硝性能[28].
由图8可观察到, 未负载活性组分的陶瓷分别在207 ℃和255 ℃处出现了脱附峰, 3个催化剂分别在120 ℃和240 ℃处出现了脱附峰, 三者峰面积相差较大, 研究表明, NH3-TPD的峰面积与酸性位点的含量呈正比[29]. 对样品的峰面积进行拟合计算, 结果列于表3, 由计算结果知, 样品的峰面积大小顺序为Mn(8)/DE-HAL(SP) > Mn(8)/DE-HAL(IP-300) > Mn(8)/DE-HAL(SP-300). 研究表明, 碱金属会占据部分酸性位点, 降低催化剂对NH3的吸附与活化能力[16], 这是Mn(8)/DE-HAL(SP-300)的NH3脱附峰面积较小的重要原因. 采用原位生长法制备的催化剂, 活性组分富集在陶瓷颗粒表面, 使得样品表面含有大量酸性位点, 催化剂表面酸性位点越多, 吸附在其表面的氨种类就越多, 越有利于SCR反应.
-
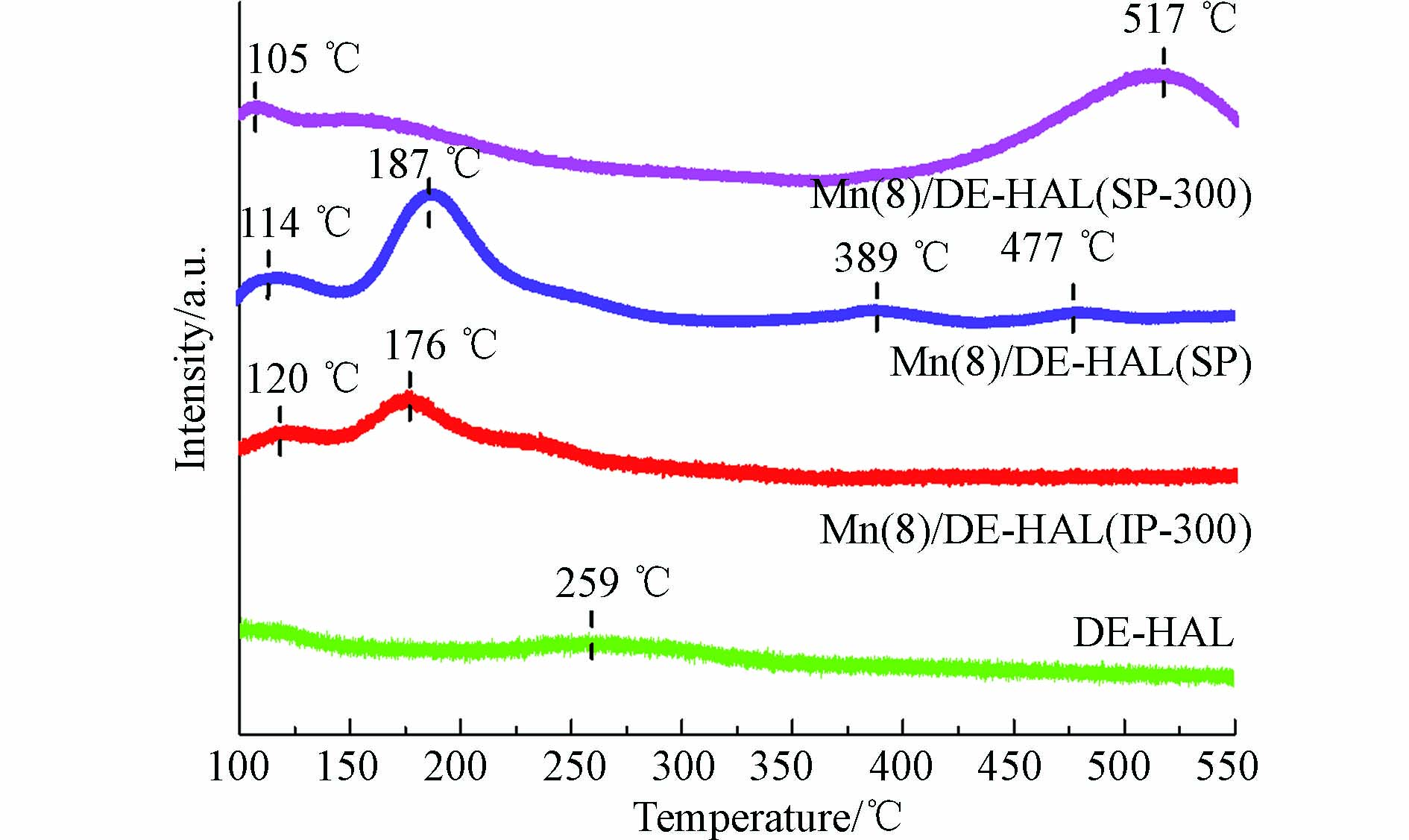
为了探明NO在催化剂表面的吸脱附行为与温度的关系, 对DE-HAL多孔陶瓷以及催化剂进行了NO-TPD测试. 如图9所示. DE-HAL仅在259 ℃有1个不明显的峰, Mn(8)/DE-HAL(IP-300)的脱附峰出现在120 ℃和176 ℃处. Mn(8)/DE-HAL(SP)分别在114 ℃和187 ℃处出现了2个脱附峰, 在389 ℃和477 ℃处出现了2个脱附峰. Mn(8)/DE-HAL(SP-300)分别在105 ℃个517 ℃处出现了NO的脱附峰. 研究表明, 200 ℃以下的脱附峰是由于催化剂表面弱配位的单齿亚硝酸盐物种的受热分解, 200 ℃以上的峰则是由于强配位的桥式亚硝酸盐和双齿硝酸盐的受热分解[30-31]. 在3个催化剂中, Mn(8)/DE-HAL(SP)在200 ℃以下的脱附峰面积最大, 表明其表面存在较多的亚硝酸盐, 这些物种有利于促进SCR反应的进行. 与Mn(8)/DE-HAL(SP)相比, Mn(8)/DE-HAL(SP-300)的脱附峰向更高温度转移, 这可能是由于K的存在改变了催化剂表面的化学环境, 影响了桥式亚硝酸盐和双齿硝酸盐分解温度.
-
图10为催化剂制备过程中MnOx的负载机理图.
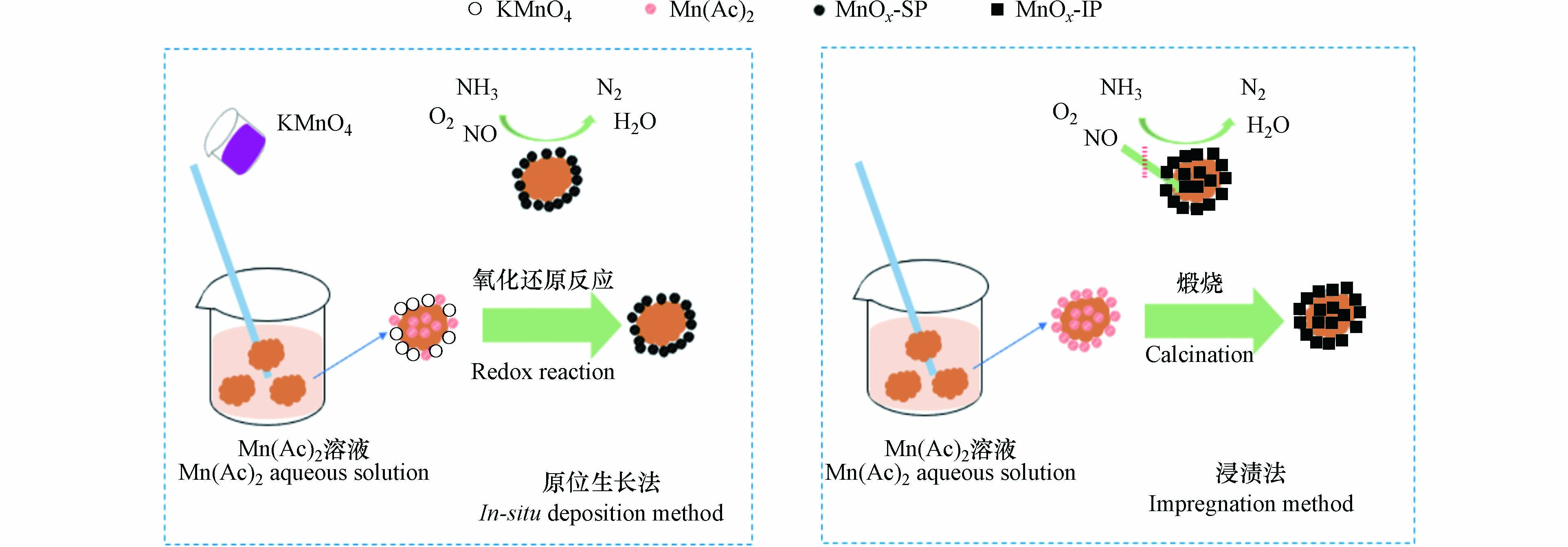
两种方法制备MnOx的原理虽不相同,但第一步均是将多孔陶瓷载体浸渍于Mn(Ac)2溶液中, 在这个过程中, Mn(Ac)2会分布于多孔陶瓷的表面以及内部孔隙中. 随后,原位生长法制备催化剂的过程中会往溶液中加入化学计量的KMnO4, 部分KMnO4与多孔陶瓷表面的Mn(Ac)2反应被消耗, 未被消耗的部分会将载体内部的Mn(Ac)2吸引到表面进行氧化还原反应, 所生成的MnOx均聚集在多孔陶瓷表面. 而浸渍法则是通过煅烧使Mn(Ac)2分解生成MnOx, Mn(Ac)2在分解过程中迁移较少. 最终, 所生成的活性组分分布于多孔陶瓷表面与内部孔隙中. 分散在载体表面的MnOx更容易与混合气体接触, 从而参与SCR反应, 而由于传质阻力, 混合气体不易进入到载体内部, 这意味着载体内部的活性组分对催化剂的催化活性贡献较小. 结合前面的数据与表征分析可知, 20—40目多孔陶瓷载体表面的活性组分在SCR反应中具有重要作用,原位生长法使活性组分富集在载体表面, 这一方面提升了催化剂的催化活性, 另一方面也提高了MnOx的利用率.
-
原位生长法制备催化剂的过程中, MnOx最终只存在于多孔陶瓷颗粒表面, 浸渍于载体内部的Mn(Ac)2并非随着水洗抽滤被除去, 而是迁移到载体表面与KMnO4发生反应被消耗完全; 采用浸渍法制备的催化剂, 活性组分分散在载体表面与多孔陶瓷内部. 分布在20—40目多孔陶瓷颗粒载体表面的活性组分更容易与混合反应气体接触, 在SCR反应中发挥着重要作用, 而载体内部的活性组分由于传质阻力, 不易参与到SCR反应中. 原位生长法使活性组分聚集到载体表面, 既提升了催化剂的脱硝能力, 又提高了MnOx的利用率.
锰氧化物负载硅藻土-埃洛石多孔陶瓷制备低温SCR脱硝催化剂
Preparation of low temperature SCR denitration catalyst by MnOx supported diatomite - halloysite porous ceramics
-
摘要: 分别采用原位生长法和浸渍法(IP)制备了Mn/DE-HAL催化剂,考察了不同制备方法对催化剂性能的影响,探究了原位生长法制备过程中MnOx的负载机制. 研究发现,在100—300 ℃之间,Mn(8)/DE-HAL(SP)表现出了更优异的催化活性. 表征结果表明,Mn(8)/DE-HAL(SP)的活性组分仅分散在载体表面,而Mn(8)/DE-HAL(IP-300)的活性组分分布于载体表面与内部. 这是因为原位生长法制备催化剂的过程中,载体表面的Mn(Ac)2与KMnO4反应被消耗完后,载体内部的Mn(Ac)2也会迁移到表面与之反应,而浸渍法制备催化剂的过程中Mn(Ac)2迁移较少. 负载在载体表面的活性组分由于更容易与混合气体接触从而进行化学反应,在SCR反应中发挥着重要作用. 原位生长法使MnOx富集到载体表面,既提升了催化剂的催化活性,又提高了MnOx的利用率.Abstract: Mn/DE-HAL catalysts were prepared by in-situ growth method and impregnation method (IP), respectively. The effects of different preparation methods on the performance of the catalysts were investigated, and the loading mechanism of MnOx in the preparation process of in-situ growth method was explored. It was found that Mn(8)/DE-HAL(SP) exhibited high SCR activity at temperature of 100—300 ℃. The characterization results showed that the active components of Mn(8)/DEHAL(SP) were only distributed on the surface of the support, while the active components of Mn(8)/DEHAL(IP-300) were distributed on the surface and inside of the support. This was due to the fact that during the process of preparing catalyst by in-situ growth method, when the Mn(Ac)2 on the surface of the carrier was consumed due to the reaction with KMnO4, the Mn(Ac)2 inside the carrier would also migrate to the surface to react with it; However, for the process of preparing catalysts by impregnation, Mn(Ac)2 inside the support could rarely migrate to the surface of the support. The active components loaded on the surface of the carrier played an important role in SCR reactions because they were easier to contact with mixed gases for chemical reactions. The in-situ growth method enriched MnOx onto the surface of the carrier, which not only improved the catalytic activity of the catalyst, but also improved the utilization rate of MnOx.
-
Key words:
- MnOx /
- in situ growth /
- low temperature SCR.
-
选择性催化还原法(selective catalytic reduction, SCR)脱除烟气中NOx是目前工业上烟气脱硝首选技术, 广泛应用于燃煤电厂等固定污染源的烟气脱硝, 脱硝效率较高, 能够满足我国当前脱硝实际, 是目前最好的NOx烟气净化治理脱硝技术[1]. SCR技术的核心是催化剂. 目前, 国内外应用较成熟SCR催化剂为V2O5/TiO2系列催化剂[2-4], 该催化剂在较高的反应温度(>350 ℃)具有较高的催化活性和抗硫性能. 由于我国的脱硝工艺必须采用末端布置, 然而末端放置的工艺方式会使得烟气温度大大降低, 低于SCR催化剂所需的活性温度. 又因V2O5/TiO2系列催化剂重金属元素V毒性较大对环境和人体健康的毒副作用以及成本较高等, 中低温(<250 ℃)高活性SCR脱硝催化剂的研究开发受到国内外众多研究者的高度关注[5-7].
中低温NH3-SCR催化剂主要包括Mn、Fe、Cu、Ce、Cr等非钒基氧化物催化剂[8-12], 其中, Mn基氧化物催化剂由于其较高的低温NH3-SCR脱硝性能、环境友好、价格低廉的优点而备受关注. Peña等[13]比较了各种负载金属氧化物的催化剂, 发现TiO2负载V、Cr、Fe、Mn、Ni、Cu、Co在NH3-SCR过程中, 活性顺序为Mn>Cu>Cr>Co>Fe>V>Ni. 研究表明锰的氧化形态、制备方法等对锰系催化剂的催化性能影响显著[14-15]. 本课题组对原位生长法制备脱硝催化剂进行了较为深入地研究. Zhang等[9]以管状的埃洛石为载体, 采用原位生长法成功地制备出了MnOx/HNTs催化剂, 在36000 h−1下, 50—300 ℃之间NO转化率可保持在90%以上, 催化剂表现出的高活性与MnOx呈非晶态良好地分散在埃洛石纳米管的管内与管外有关, 埃洛石所具有的空间纳米结构限制了MnOx的生长, 使其趋于无定形态, 促进了活性位点的形成与分散. Zhang等[12]分别采用原位生长法与浸渍法制备了CeMn/TiO2催化剂, 探究了制备方法对以粉末状TiO2为载体所制备的催化剂脱硝性能的影响. 其中, 采用原位生长法所制备的样品表现出更好的催化活性, Ce(1.0)Mn/TiO2-SP在125—300 ℃之间, 27000 h−1下, NO转化率接近100%.
综上可知, 原位生长法制备的脱硝催化剂具有良好的催化活性, 且活性组分在载体表面呈非晶态, 分散性高. 然而, 原位生长法制备过程中MnOx的负载机制尚不清晰[9,11-12]. 为了明晰原位生长法制备过程中MnOx的负载机制, 阐明催化剂的催化活性机理. 本文以20—40目硅藻土基多孔陶瓷颗粒为载体, 制备了Mn基多孔陶瓷催化剂, 以传统浸渍法作为对比, 考察了不同制备方法Mn基催化剂的低温SCR脱硝性能, 探究了原位生长法制备催化剂过程中活性组分的负载机制.
1. 实验部分(Experimental section)
1.1 材料与试剂
埃洛石(HAL, 郑州金阳光陶瓷有限公司),主要化学组成(质量分数)为: 50.31% SiO2、38.27% Al2O3、7.59% SO3、1.22% K2O、1.12% Fe2O3; 硅藻土(DE, 国药集团化学试剂有限公司), 主要化学成分(质量分数)为: 88.80% SiO2、 2.98% Al2O3、2.06% Fe2O3、2.03% CO2、1.21% Na2O; 乙酸锰 Mn(CH3COO)2,高锰酸钾KMnO4, 聚乙烯醇(PVA, (CH2CHOH)n),石墨(Graphite, C), 均由国药集团化学试剂有限公司提供,纯度为AR;模拟烟气由南京上元工业气体厂提供.
1.2 催化剂的制备
1.2.1 硅藻土基多孔陶瓷的制备
称取6 g硅藻土粉末与4 g埃洛石混合, 再称取0.4 g淀粉, 采用湿法球磨(球:原料:酒精= 2:1:0.6)将上述粉末混合均匀, 110 ℃干燥24 h, 过200目筛, 滴加4% wt. PVA, 研磨,经红外压片机4 MPa压片成型, 再经管式炉1000 ℃煅烧2 h, 得到硅藻土基多孔陶瓷. 将所得多孔陶瓷破碎后过20目筛, 作为催化剂载体, 并标记为DE-HAL.
1.2.2 Mn基硅藻土多孔陶瓷催化剂的制备
原位生长法: 称取5 g上述制备的硅藻土基多孔陶瓷, 称取化学计量的Mn(Ac)2溶解于一定量的去离子水中, 用以作为锰的前驱体. 将多孔陶瓷等体积浸渍于Mn(Ac)2溶液中, 静置24 h后加入KMnO4溶液, 搅拌后静置24 h, 多次洗涤抽滤至滤液澄清无色以达到除去K+的目的, 于鼓风干燥箱中110 ℃干燥24 h, 即得所需催化剂, 标记为Mn(8)/DE-HAL(SP)(催化剂中Mn元素质量分数为8%). 重复上述制备步骤, 但催化剂不经过洗涤抽滤, 干燥后在300 ℃下煅烧3 h, 标记为Mn(8)/DE-HAL(SP-300).
浸渍法: 称取5 g制备的硅藻土基多孔陶瓷, 称取化学计量的Mn(Ac)2溶解于一定量的去离子水中, 将多孔陶瓷等体积浸渍于Mn(Ac)2溶液中, 静置24 h后于烘箱中110 ℃干燥24 h, 再置于马弗炉中, 空气氛围, 300 ℃煅烧3 h, 标记为Mn(8)/DE-HAL(IP-300).
1.3 催化剂活性测试
催化剂的脱硝活性评价测试装置为长80 cm, 内径15 mm的竖式石英玻璃常压固定床反应器, 活性评价装置主要由模拟烟气(1000 mg·L−1 NO, 1000 mg·L−1 NH3, 6%vol H2O, 3%vol O2, N2为平衡气混合而成, 总流量为350 mL·min−1), 固定床反应器和分析检测装置的3部分组成. 反应空速为52000 h−1, 反应温度为100—300 ℃. 采用MRU OPTIMA7烟气分析仪在线测量反应器进出口NOx浓度,准确度为 ±3%。采用MGA6plus分析仪测定反应器出口N2O浓度,准确度为 ±2%。采用GT-1000-NH3分析仪测定NH3浓度,准确度为 ±1%.
催化剂脱硝活性以NO转化率和N2的选择性衡算,计算公式为:
NOconversion=CinNO−CoutNOCinNO×100% (1) N2selectivity=[NOin]+[NH3]in−[NO2]out−2[N2O]out[NOin+[NH3]in]×100% (2) 1.4 催化剂表征
采用美国康塔NOVA 2200e型比表面积及孔径分析仪分析样品的比表面积和孔径孔容. 采用荷兰帕纳科PANalytical X-Pert PRO MPD型X射线衍射仪对多孔陶瓷和催化剂的物相与晶型进行表征,Cu Kα射线, 操作电压为40 kV, 操作电流为40 mA, 扫描速度为4(o)·min−1, 扫描区间范围2θ=5°—70°. 采用美国Thermo ESCALAB250Xi型X射线光电子能谱仪对催化剂进行XPS测试, Al Kα射线, 管电压为1486.6 eV. 采用德国卡尔蔡司Gemini 500型热场发射扫描电子显微镜观察多孔陶瓷与催化剂的表面微观结构.采用全自动多功能吸附仪(TP-5080D)进行H2的程序升温还原(H2-TPR)实验.采用Micromeritics Autochem-II仪器对催化剂进行NH3/NO程序升温脱附(NH3/NO-TPD)实验,利用质谱(Hiden QIC-200)对脱附过程中出口气体浓度进行实时监测。.
2. 结果与讨论(Results and discussion)
2.1 催化剂活性评价
2.1.1 不同制备方法催化剂的脱硝活性
图1为不同制备方法催化剂的脱硝活性-温度曲线. 由图1可以看出, Mn/DE-HAL(SP)具有最优异的催化活性, 在100—250 ℃之间NO转化率高于90%, 225 ℃之后催化活性虽然随温度的升高而下降, 但在整个测试温度段, NO转化率不低于80%. Mn/DE-HAL(SP-300)在100 ℃时NO转化率约为38%, 150—200 ℃之间NO转化率约为70%, 该样品催化活性较差可能是由于催化剂中存在大量K元素, 研究表明, 碱金属会占据催化剂的部分酸性位点, 降低催化剂对NH3的吸附和活化能力[16], 从而导致催化剂催化活性较差. 此外, 煅烧也是降低样品NO转化率的原因之一. Mn/DE-HAL(IP-300)低温下催化活性较差, 150 ℃之后NO转化率显著增加, 在225—300 ℃之间催化活性较为稳定, NO转化率约为80%. 下文将通过表征分析对催化剂的理化性质进行进一步探究.
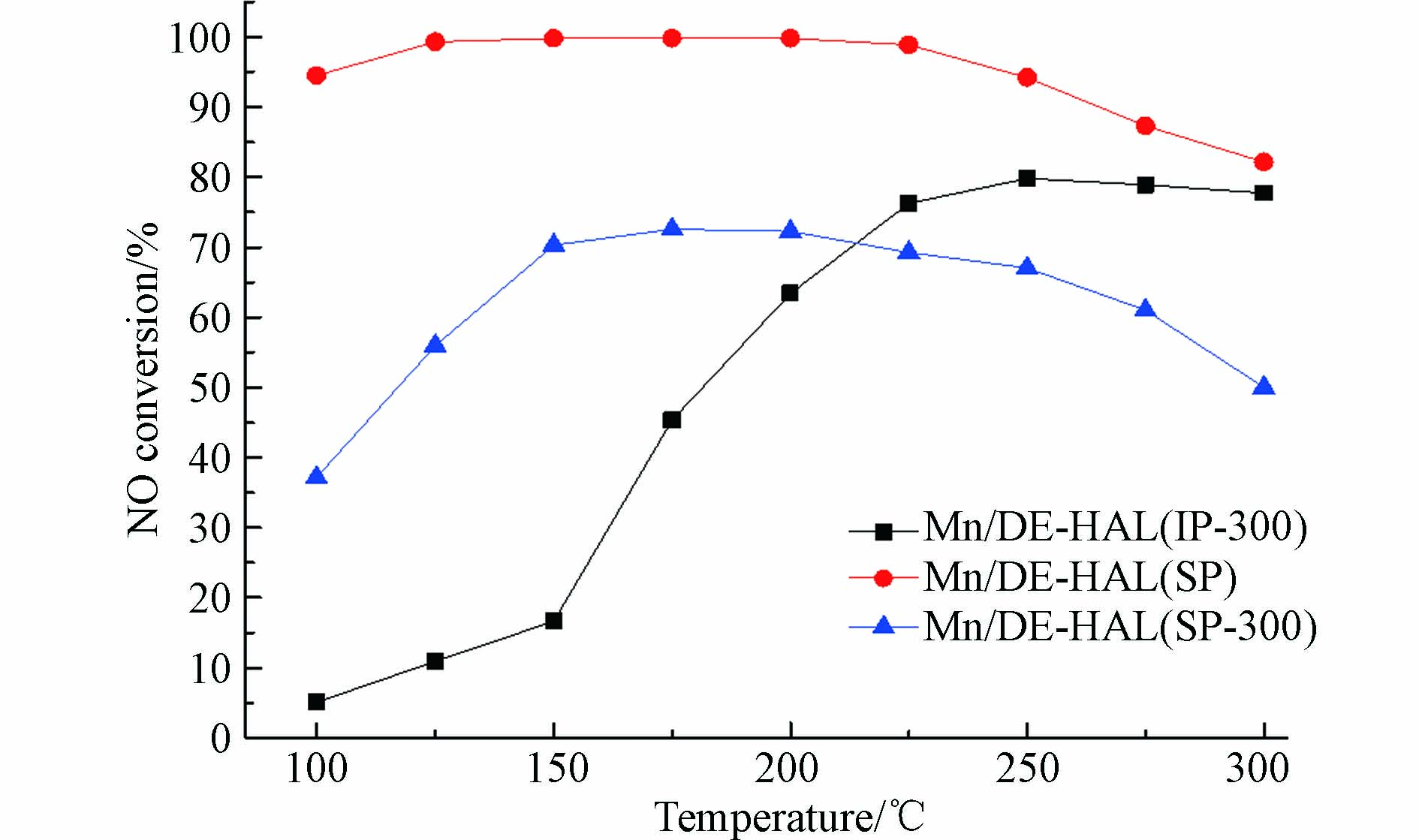 图 1 不同制备方法催化剂的脱硝活性Figure 1. Denitration activity of catalysts prepared by different methods反应条件: [NO] = [NH3] = 1000 mg·L−1, [H2O] = 6 %vol, [O2] = 3 %vol, N2平衡气, 总流速350 mL·min−1, 空速52000 h−1.
图 1 不同制备方法催化剂的脱硝活性Figure 1. Denitration activity of catalysts prepared by different methods反应条件: [NO] = [NH3] = 1000 mg·L−1, [H2O] = 6 %vol, [O2] = 3 %vol, N2平衡气, 总流速350 mL·min−1, 空速52000 h−1.2.1.2 N2选择性
N2的选择性为Mn基SCR催化剂的重要性质之一,其实质上反映了反应系统中目的反应与副反应反应速率的竞争关系,选择性越高越有利于催化反应的进行. 图2为不同制备方法催化剂N2选择性曲线图. 由图2可以看出,在整个温度区间范围内, Mn/DE-HAL(SP)催化剂N2选择性均在95%左右,说明在100—300 ℃温度范围内,Mn/DE-HAL(SP)催化剂稳定性较好且具有高N2选择性.
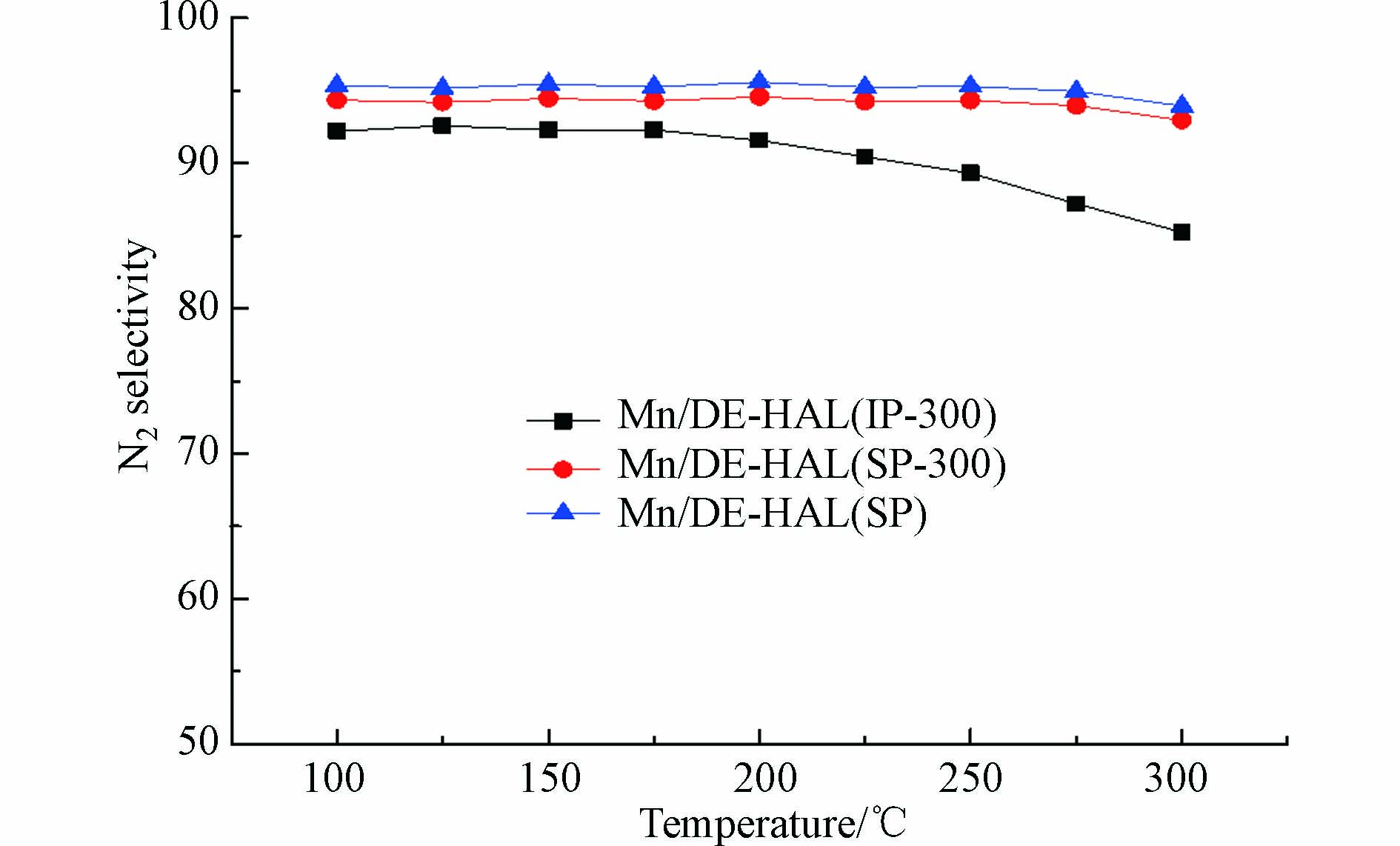 图 2 不同制备方法催化剂的N2选择性Figure 2. N2 selectivity of catalysts prepared by different methods
图 2 不同制备方法催化剂的N2选择性Figure 2. N2 selectivity of catalysts prepared by different methods2.2 催化剂表征
2.2.1 SEM分析
为了更直观地研究催化剂的形貌, 对样品进行了SEM表征. 图3为催化剂的SEM与Mapping图. 由图3(c)可以看出, 原位生长法所制备的催化剂表面MnOx呈絮状, 由图3(f)可以看出采用浸渍法所制备的样品表面的MnOx为颗粒状, 两种方法所制备的催化剂表面的活性组分形貌差异显著. 由图3(b)和图3(e)的Mapping图可以清晰地观察到Mn(8)/DE-HAL(SP)表面的活性组分的分散情况与Mn(8)/DE-HAL(IP-300)差别并不明显. 可见, 制备方法影响载体表面MnOx的形貌, 而对活性组分在载体表面的分散性没有太大影响. 对比两种不同制备方法催化剂形貌, 结果表明, 催化剂表面活性组分的分散性并非是影响催化剂脱硝性能的决定性因素. 图3(g)和(h)分别为原位生长法制备的催化剂的断裂面形貌图与Mapping图, 由图3(h)可以看出, MnOx并未进入颗粒内部; 图3(i)和图3(j)分别为浸渍法制备的催化剂的断裂面形貌图与Mapping图, 由图3(j)可以看出, MnOx同时负载在了颗粒内部. 该结果表明, 原位生长法所制备的催化剂, MnOx仅附着在陶瓷颗粒表面, 而浸渍法所制备的样品, MnOx进入了陶瓷颗粒内部. 下面将进一步探究出现这一现象的原因.
 图 3 不同制备方法催化剂的SEM和mapping图Figure 3. SEM and mapping images of catalysts prepared by different methods(a)、(c) Mn(8)/DE-HAL(SP); (d)、(f) Mn(8)/DE-HAL(IP-300); (b) Mn(8)/DE-HAL(SP)mapping; (e) Mn(8)/DE-HAL(IP-300) mapping; (g) Mn(8)/DE-HAL(SP) 断裂面; (i) Mn(8)/DE-HAL(IP-300) 断裂面; (h) Mn(8)/DE-HAL(SP) mapping断裂面; (j) Mn(8)/DE-HAL(IP-300) mapping断裂面.
图 3 不同制备方法催化剂的SEM和mapping图Figure 3. SEM and mapping images of catalysts prepared by different methods(a)、(c) Mn(8)/DE-HAL(SP); (d)、(f) Mn(8)/DE-HAL(IP-300); (b) Mn(8)/DE-HAL(SP)mapping; (e) Mn(8)/DE-HAL(IP-300) mapping; (g) Mn(8)/DE-HAL(SP) 断裂面; (i) Mn(8)/DE-HAL(IP-300) 断裂面; (h) Mn(8)/DE-HAL(SP) mapping断裂面; (j) Mn(8)/DE-HAL(IP-300) mapping断裂面.两种方法制备催化剂的第一步均是将载体浸渍于Mn(Ac)2溶液中, 浸渍法经过干燥煅烧后, 颗粒表面与内部均生成了MnOx, 而原位生长法在加入KMnO4进行化学反应, 再经过洗涤抽滤后颗粒内部已观察不到MnOx的存在. 现采用原位生长法制备催化剂, 不经水洗抽滤, 干燥后经300 ℃煅烧3 h, 探究是否是洗涤抽滤除去了颗粒内部的Mn(Ac)2.
图4为原位生长法制备Mn(8)/DE-HAL(SP-300)催化剂的SEM与Mapping图. 图4(a)为催化剂外表面的SEM图, 图4(d)为催化剂内部断裂面的SEM图. 由图4(b)和(c)可以看出, MnOx分散在催化剂的外表面, 由于干燥前未经过洗涤抽滤, 使得K大量存在于样品表面. 由图4(e)和(f)可以看出, 催化剂内部仍然有K, 但并未观察到MnOx的存在. 该结果表明, 采用原位生长法制备的样品内部的活性组分并非被洗涤除去. KMnO4不仅与样品表面的Mn(Ac)2反应, 位于多孔陶瓷颗粒内部的Mn2+也会迁移到样品表面与KMnO4反应生成MnOx. 这意味着, 原位生长法制备的催化剂, 活性组分富集在载体表面, 结合催化剂的催化活性分析可知, 这些负载在载体表面的MnOx在SCR反应中发挥着重要作用.
 图 4 原位生长法制备催化剂的SEM和mapping图(a) Mn(8)/DE-HAL(SP-300); (b)、(c) mapping of Mn(8)/DE-HAL(SP-300); (d) fracture surface of Mn(8)/DE-HAL(SP-300); (e)、(f) mapping of fracture surface of Mn(8)/DE-HAL(SP-300)Figure 4. SEM and mapping images of catalysts prepared by in-situ growth method
图 4 原位生长法制备催化剂的SEM和mapping图(a) Mn(8)/DE-HAL(SP-300); (b)、(c) mapping of Mn(8)/DE-HAL(SP-300); (d) fracture surface of Mn(8)/DE-HAL(SP-300); (e)、(f) mapping of fracture surface of Mn(8)/DE-HAL(SP-300)Figure 4. SEM and mapping images of catalysts prepared by in-situ growth method2.2.2 XRD分析
图5为多孔陶瓷和催化剂的XRD图谱. 如图5所示, DE-HAL在21.66°、35.87°以及31.05°处的峰归属于方石英的特征衍射峰(PDF#39-1425), 26.62°处的峰归属于石英的特征衍射峰(PDF#46-1045).
Mn/DE-HAL(SP)的石英峰与方石英峰均显著减弱, 出现这一现象原因一方面是因为MnOx包覆在载体表面, 削弱了其原有的衍射峰, 另一方面, 由于样品的特殊性, 使得筛选出来的样品MnOx含量较多. 此外, 样品中并未观察到MnOx的衍射峰, 这主要是由于活性组分在载体表面呈无定形态分布. Mn/DE-HAL(IP-300)中除了石英峰与方石英峰外, 在31.03°、32.44°以及44.37°处还可观察到Mn3O4的特征衍射峰(PDF#18-0803). 催化剂表面活性组分结晶度越低, 越具有较多的表面活性点位, 有利于提升催化剂的催化活性, 这可能是Mn/DE-HAL(SP)表现出更好的催化活性的重要原因.
2.2.3 比表面和孔结构分析
比表面积和孔结构是评价催化剂优良的重要指标, 总体而言, 较大的比表面积能够为催化剂提供更多的活性位点, 使活性组分MnOx得到更好的分散, 更有利于反应气体的吸附, 从而提高催化活性. 表1为样品的孔结构与BET数据, 其比表面积大小顺序为: Mn(8)/DE-HAL(SP) > Mn(8)/DE-HAL(SP-300) > Mn(8)/DE-HAL(IP-300) > DE-HAL. 可以看到, 与Mn(8)/DE-HAL(SP)相比, Mn(8)/DE-HAL(SP-300)与Mn(8)/DE-HAL(IP-300)的孔容较小, 这可能是由于煅烧使得活性组分堵塞了部分小孔. 此外, 负载MnOx后, 所有样品的孔径均有不同程度的减小, 这与活性组分和载体结合后, 形成了丰富的孔道结构有关. 相较于DE-HAL多孔陶瓷, 负载MnOx后, 样品的比表面积显著增加, 主要是由于MnOx成功负载后提升了样品的比表面积, 这意味着催化剂的比表面积主要是由MnOx贡献的. 结合SEM与Mapping分析, Mn/DE-HAL(SP)的活性组分主要分散在载体表面, Mn(8)/DE-HAL(IP-300)的活性组分分散在载体表面与内部孔隙中, 即MnOx对Mn/DE-HAL(SP)比表面积的贡献集中在载体表面, 对Mn(8)/DE-HAL(IP-300)比表面积的提升作用于载体表面与内部, 而表面的活性组分更容易参与到SCR反应中, 催化剂表面具有更多的反应活性位点, 有利于提升催化剂的脱硝性能, 这是Mn(8)/DE-HAL(SP)具有良好的催化活性的重要原因.
表 1 样品的孔隙结构和BET数据Table 1. Pore structure and BET data of samples样品Sample 比表面积/(m2·g−1)Specific surface area 孔容/(cm3·g−1)Pore volume 孔径/nmPore diameter DE-HAL 4 0.02 179.47 Mn(8)/DE-HAL(IP-300) 13 0.03 81.32 Mn(8)/DE-HAL(SP) 32 0.05 48.75 Mn(8)/DE-HAL(SP-300) 14 0.04 7.66 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.2.4 XPS分析
图6为采用两种方法所制备的催化剂XPS图谱, 表2为催化剂的XPS分析结果. 图6(a)中644.4—645 eV、642.2—642.7 eV和 640.9—641.4 eV的3个峰分别归属于Mn4+、Mn3+和Mn2+ [17-18]. Mn4+是Mn基催化剂在NH3-SCR反应中最活跃的物种, Mn4+易于吸附NH3并生成Mn4+-NH3, 而Mn3+有利于吸附NO并生成Mn3+-NO[19]. 研究表明, Mn4+-NH3可以与Mn3+-NO结合生成中间产物NH4NO3[19], 与NO反应生成NH4NO2会进一步分解成N2和H2O[20], 从而促进NH3-SCR反应的进行. 图6(b)中530.1—530.2 eV, 531.7—532.4 eV和533.5—534.4 eV的 3个峰分别归属于表面晶格氧(Oβ)、化学吸附氧(Oα)和吸附的水物种(Oγ)[21-22]. Oα在NH3-SCR反应中比Oβ更活泼, 因为其具有更高的迁移率[23], 有利于将NO氧化成NO2, 促进快速SCR反应的进行[24]. 与Mn(8)/DE-HAL(SP)相比, Mn(8)/DE-HAL(SP-300)的Mn4+比例降低了14.53%, 意味着碱金属的存在降低了Mn4+浓度; Oα的比例降低了11.58%, 研究表明, 碱金属会与表面氧中心形成强键[25], 抑制催化剂表面化学吸附氧的形成[26]. 相较于Mn(8)/DE-HAL(SP), Mn(8)/DE-HAL(IP-300)中Mn4+和Oα的比例分别降低了17.10%和9.54%. 该结果表明, 原位生长法可以提升催化剂表面Mn4+和Oα的浓度, 更高的Mn4+和Oα的相对百分比含量, 有利于促进SCR反应的进行, 提升样品的催化活性.
表 2 催化剂的XPS分析结果Table 2. XPS results of catalysts样品 Sample 表面原子浓度/% Surface atomic concentration Mn4+/Mn3+ Oα/(Oα+Oβ) Mn(8)/DE-HAL(SP) 83.46 86.70 Mn(8)/DE-HAL(SP-300) 66.36 77.16 Mn(8)/DE-HAL(IP-300) 68.93 75.12 | Show TableDownLoad:
CSV
2.2.5 H2-TPR分析
通过H2-TPR对催化剂氧化还原能力进行测试, 结果如图7所示, 未负载活性组分的陶瓷在100—550 ℃之间没有明显的还原峰, 而Mn(8)/DE-HAL(SP)在267 ℃和398 ℃处出现了2个还原峰, Mn(8)/DE-HAL(SP-300)在298 ℃和391 ℃处出现了两个还原峰, Mn(8)/DE-HAL(IP-300)的2个还原峰分别出现在230 ℃和414 ℃处, 催化剂在100—550 ℃之间的2个还原峰分别归因于MnO2到Mn2O3的还原和Mn2O3到MnO的还原[27]. 虽然Mn(8)/DE-HAL(IP-300)第1个还原峰温度最低, 但其峰面积较小, 而H2消耗峰的面积在某种程度上可以反应出催化剂表面活性物种的数量, 可以看出, Mn(8)/DE-HAL(IP-300)表面Mn4+浓度相对较低, 这与XPS结果是一致的. 原位生长法所制备的样品, 活性组分虽然仅分散在载体表面, 但却表现出更强的还原性, 这意味着载体表面的活性组分对催化剂的氧化还原能力有重要影响. 在低温区域, Mn(8)/DE-HAL(SP)的还原峰比Mn(8)/DE-HAL(SP-300)高, 且前者的H2消耗量也比后者大, 意味着Mn(8)/DE-HAL(SP)的氧化还原能力强于Mn(8)/DE-HAL(SP-300), 该结果表明, K的存在降低了催化剂的氧化还原能力.
2.2.6 NH3-TPD分析
图8为催化剂的NH3-TPD图谱. 研究表明, 在NH3-TPD中, 200 ℃以下的峰可归因于NH3在弱酸位点上物理和弱化学吸附的连续解吸, 200—450 ℃范围内的峰可归因于中弱酸位点[19]. 酸性位点的数量与分布情况是催化剂脱硝性能的重要影响因素, 是保证NH3吸附和活化的前提[23], 其中, 位于100—350 ℃的酸性位点数量影响催化剂低温下的脱硝性能[28].
由图8可观察到, 未负载活性组分的陶瓷分别在207 ℃和255 ℃处出现了脱附峰, 3个催化剂分别在120 ℃和240 ℃处出现了脱附峰, 三者峰面积相差较大, 研究表明, NH3-TPD的峰面积与酸性位点的含量呈正比[29]. 对样品的峰面积进行拟合计算, 结果列于表3, 由计算结果知, 样品的峰面积大小顺序为Mn(8)/DE-HAL(SP) > Mn(8)/DE-HAL(IP-300) > Mn(8)/DE-HAL(SP-300). 研究表明, 碱金属会占据部分酸性位点, 降低催化剂对NH3的吸附与活化能力[16], 这是Mn(8)/DE-HAL(SP-300)的NH3脱附峰面积较小的重要原因. 采用原位生长法制备的催化剂, 活性组分富集在陶瓷颗粒表面, 使得样品表面含有大量酸性位点, 催化剂表面酸性位点越多, 吸附在其表面的氨种类就越多, 越有利于SCR反应.
表 3 样品的NH3-TPD积分结果Table 3. NH3-TPD integration of samples样品Sample Mn(8)/DE-HAL(SP) Mn(8)/DE-HAL(SP-300) Mn(8)/DE-HAL(IP-300) DE-HAL NH3-TPD /(eV·s) 1.18×10−7 1.09×10−7 1.14×10−7 1.06×10−7 | Show TableDownLoad:
CSV
2.2.7 NO-TPD分析
为了探明NO在催化剂表面的吸脱附行为与温度的关系, 对DE-HAL多孔陶瓷以及催化剂进行了NO-TPD测试. 如图9所示. DE-HAL仅在259 ℃有1个不明显的峰, Mn(8)/DE-HAL(IP-300)的脱附峰出现在120 ℃和176 ℃处. Mn(8)/DE-HAL(SP)分别在114 ℃和187 ℃处出现了2个脱附峰, 在389 ℃和477 ℃处出现了2个脱附峰. Mn(8)/DE-HAL(SP-300)分别在105 ℃个517 ℃处出现了NO的脱附峰. 研究表明, 200 ℃以下的脱附峰是由于催化剂表面弱配位的单齿亚硝酸盐物种的受热分解, 200 ℃以上的峰则是由于强配位的桥式亚硝酸盐和双齿硝酸盐的受热分解[30-31]. 在3个催化剂中, Mn(8)/DE-HAL(SP)在200 ℃以下的脱附峰面积最大, 表明其表面存在较多的亚硝酸盐, 这些物种有利于促进SCR反应的进行. 与Mn(8)/DE-HAL(SP)相比, Mn(8)/DE-HAL(SP-300)的脱附峰向更高温度转移, 这可能是由于K的存在改变了催化剂表面的化学环境, 影响了桥式亚硝酸盐和双齿硝酸盐分解温度.
2.3 催化剂制备过程机理
图10为催化剂制备过程中MnOx的负载机理图.
两种方法制备MnOx的原理虽不相同,但第一步均是将多孔陶瓷载体浸渍于Mn(Ac)2溶液中, 在这个过程中, Mn(Ac)2会分布于多孔陶瓷的表面以及内部孔隙中. 随后,原位生长法制备催化剂的过程中会往溶液中加入化学计量的KMnO4, 部分KMnO4与多孔陶瓷表面的Mn(Ac)2反应被消耗, 未被消耗的部分会将载体内部的Mn(Ac)2吸引到表面进行氧化还原反应, 所生成的MnOx均聚集在多孔陶瓷表面. 而浸渍法则是通过煅烧使Mn(Ac)2分解生成MnOx, Mn(Ac)2在分解过程中迁移较少. 最终, 所生成的活性组分分布于多孔陶瓷表面与内部孔隙中. 分散在载体表面的MnOx更容易与混合气体接触, 从而参与SCR反应, 而由于传质阻力, 混合气体不易进入到载体内部, 这意味着载体内部的活性组分对催化剂的催化活性贡献较小. 结合前面的数据与表征分析可知, 20—40目多孔陶瓷载体表面的活性组分在SCR反应中具有重要作用,原位生长法使活性组分富集在载体表面, 这一方面提升了催化剂的催化活性, 另一方面也提高了MnOx的利用率.
3. 结论(Conclusion)
原位生长法制备催化剂的过程中, MnOx最终只存在于多孔陶瓷颗粒表面, 浸渍于载体内部的Mn(Ac)2并非随着水洗抽滤被除去, 而是迁移到载体表面与KMnO4发生反应被消耗完全; 采用浸渍法制备的催化剂, 活性组分分散在载体表面与多孔陶瓷内部. 分布在20—40目多孔陶瓷颗粒载体表面的活性组分更容易与混合反应气体接触, 在SCR反应中发挥着重要作用, 而载体内部的活性组分由于传质阻力, 不易参与到SCR反应中. 原位生长法使活性组分聚集到载体表面, 既提升了催化剂的脱硝能力, 又提高了MnOx的利用率.
-
图 1 不同制备方法催化剂的脱硝活性
Figure 1. Denitration activity of catalysts prepared by different methods
图 2 不同制备方法催化剂的N2选择性
Figure 2. N2 selectivity of catalysts prepared by different methods
图 3 不同制备方法催化剂的SEM和mapping图
Figure 3. SEM and mapping images of catalysts prepared by different methods
图 4 原位生长法制备催化剂的SEM和mapping图(a) Mn(8)/DE-HAL(SP-300); (b)、(c) mapping of Mn(8)/DE-HAL(SP-300); (d) fracture surface of Mn(8)/DE-HAL(SP-300); (e)、(f) mapping of fracture surface of Mn(8)/DE-HAL(SP-300)
Figure 4. SEM and mapping images of catalysts prepared by in-situ growth method
表 1 样品的孔隙结构和BET数据
Table 1. Pore structure and BET data of samples
样品Sample 比表面积/(m2·g−1)Specific surface area 孔容/(cm3·g−1)Pore volume 孔径/nmPore diameter DE-HAL 4 0.02 179.47 Mn(8)/DE-HAL(IP-300) 13 0.03 81.32 Mn(8)/DE-HAL(SP) 32 0.05 48.75 Mn(8)/DE-HAL(SP-300) 14 0.04 7.66
下载: 导出CSV
表 2 催化剂的XPS分析结果
Table 2. XPS results of catalysts
样品 Sample 表面原子浓度/% Surface atomic concentration Mn4+/Mn3+ Oα/(Oα+Oβ) Mn(8)/DE-HAL(SP) 83.46 86.70 Mn(8)/DE-HAL(SP-300) 66.36 77.16 Mn(8)/DE-HAL(IP-300) 68.93 75.12
下载: 导出CSV
表 3 样品的NH3-TPD积分结果
Table 3. NH3-TPD integration of samples
样品Sample Mn(8)/DE-HAL(SP) Mn(8)/DE-HAL(SP-300) Mn(8)/DE-HAL(IP-300) DE-HAL NH3-TPD /(eV·s) 1.18×10−7 1.09×10−7 1.14×10−7 1.06×10−7
下载: 导出CSV
-
[1] 张先龙, 胡晓芮, 刘仕雯, 等. 锰基累托石低温NH3-SCR催化剂的制备方法 [J]. 环境化学, 2022, 41(3): 1043-1051. doi: 10.7524/j.issn.0254-6108.2020110905 ZHANG X L, HU X R, LIU S W, et al. The preparation method of manganese-based rectorite low-temperature NH3-SCR catalyst [J]. Environmental Chemistry, 2022, 41(3): 1043-1051(in Chinese). doi: 10.7524/j.issn.0254-6108.2020110905
[2] TOPSOE N Y, DUMESIC J A, TOPSOE H. Vanadia-titania catalysts for selective catalytic reduction of nitric-oxide by ammonia: I. I. Studies of Active Sites and Formulation of Catalytic Cycles [J]. Journal of catalysis, 1995, 151(1): 241-252. doi: 10.1006/jcat.1995.1025 [3] TOPSOE N, TOPSØE H, DUMESIC J. Vanadia/titania catalysts for selective catalytic reduction (SCR) of nitric-oxide by ammonia: I. combined temperature-programmed in situ FTIR and on-line mass-spectroscopy studies [J]. Journal of Catalysis, 1995, 151: 226-240. doi: 10.1006/jcat.1995.1024 [4] KOBAYASHI M, HAGI M. V2O5-WO3/TiO2-SiO2-SO42− catalysts: Influence of active components and supports on activities in the selective catalytic reduction of NO by NH3 and in the oxidation of SO2 [J]. Applied Catalysis B:Environmental, 2006, 63(1/2): 104-113. [5] DAMMA D, ETTIREDDY P R, REDDY B M, et al. A review of low temperature NH3-SCR for removal of NOx [J]. Catalysts, 2019, 9(4): 349. doi: 10.3390/catal9040349 [6] ZHAO Q, CHEN B B, LI J, et al. Insights into the structure-activity relationships of highly efficient CoMn oxides for the low temperature NH3-SCR of NOx [J]. Applied Catalysis B:Environmental, 2020, 277: 119215. doi: 10.1016/j.apcatb.2020.119215 [7] SHI Y R, TANG X L, YI H H, et al. Controlled synthesis of spinel-type mesoporous Mn–Co rods for SCR of NOx with NH3 at low temperature [J]. Industrial & Engineering Chemistry Research, 2019, 58(9): 3606-3617. [8] 刘福东, 单文坡, 石晓燕, 等. 用于NH3选择性催化还原NO的非钒基催化剂研究进展 [J]. 催化学报, 2011, 32(7): 1113-1128. LIU F D, SHAN W P, SHI X Y, et al. Research progress in vanadium-free catalysts for the selective catalytic reduction of NO with NH3 [J]. Chinese Journal of Catalysis, 2011, 32(7): 1113-1128(in Chinese).
[9] ZHANG X L, WANG P M, WU X, et al. Application of MnOx/HNTs catalysts in low-temperature NO reduction with NH3 [J]. Catalysis Communications, 2016, 83: 18-21. doi: 10.1016/j.catcom.2016.05.003 [10] ZHANG X L, WU Q, DIAO Q C, et al. Performance study for NH3-SCR at low temperature based on different methods of Mnx/SEP catalyst [J]. Chemical Engineering Journal, 2019, 370: 364-371. doi: 10.1016/j.cej.2019.03.065 [11] WU X P, SHI Q, XU Y Q, et al. Synthesis and catalytic performances of Manganese oxides-loaded porous halloysite/carbon composites for the selective catalytic reduction of NO with NH3 [J]. Applied Clay Science, 2020, 185: 105200. doi: 10.1016/j.clay.2019.105200 [12] ZHANG X L, ZHANG X C, YANG X J, et al. CeMn/TiO2 catalysts prepared by different methods for enhanced low-temperature NH3-SCR catalytic performance [J]. Chemical Engineering Science, 2021, 238: 116588. doi: 10.1016/j.ces.2021.116588 [13] PEÑA D A, UPHADE B S, SMIRNIOTIS P G. TiO2-supported metal oxide catalysts for low-temperature selective catalytic reduction of NO with NH3: I. Evaluation and characterization of first row transition metals [J]. Journal of Catalysis, 2004, 221(2): 421-431. doi: 10.1016/j.jcat.2003.09.003 [14] GONG P J, XIE J L, FANG D, et al. Effects of surface physicochemical properties on NH3-SCR activity of MnO2 catalysts with different crystal structures [J]. Chinese Journal of Catalysis, 2017, 38(11): 1925-1934. doi: 10.1016/S1872-2067(17)62922-X [15] LIU Y, YANG W J, ZHANG P Y, et al. Nitric acid-treated birnessite-type MnO2: An efficient and hydrophobic material for humid ozone decomposition [J]. Applied Surface Science, 2018, 442: 640-649. doi: 10.1016/j.apsusc.2018.02.204 [16] ZHU N, SHAN W P, SHAN Y L, et al. Effects of alkali and alkaline earth metals on Cu-SSZ-39 catalyst for the selective catalytic reduction of NOx with NH3 [J]. Chemical Engineering Journal, 2020, 388: 124250. doi: 10.1016/j.cej.2020.124250 [17] FRANCE L J, YANG Q, LI W, et al. Ceria modified FeMnOx—Enhanced performance and sulphur resistance for low-temperature SCR of NOx [J]. Applied Catalysis B:Environmental, 2017, 206: 203-215. doi: 10.1016/j.apcatb.2017.01.019 [18] GAO G, SHI J W, FAN Z Y, et al. MnM2O4 microspheres (M=Co, Cu, Ni) for selective catalytic reduction of NO with NH3: Comparative study on catalytic activity and reaction mechanism via in-situ diffuse reflectance infrared Fourier transform spectroscopy [J]. Chemical Engineering Journal, 2017, 325: 91-100. doi: 10.1016/j.cej.2017.05.059 [19] SHI X K, GUO J X, SHEN T, et al. Enhancement of Ce doped La–Mn oxides for the selective catalytic reduction of NOx with NH3 and SO2 and/or H2O resistance [J]. Chemical Engineering Journal, 2021, 421: 129995. doi: 10.1016/j.cej.2021.129995 [20] JIA B H, GUO J X, SHU S, et al. Effects of different Zr/Ti ratios on NH3–SCR over MnOx/ZryTi1-yO2: Characterization and reaction mechanism [J]. Molecular Catalysis, 2017, 443: 25-37. doi: 10.1016/j.mcat.2017.09.019 [21] GAO L, LI C T, LI S H, et al. Superior performance and resistance to SO2 and H2O over CoOx-modified MnOx/biomass activated carbons for simultaneous Hg0 and NO removal [J]. Chemical Engineering Journal, 2019, 371: 781-795. doi: 10.1016/j.cej.2019.04.104 [22] FAN J, NING P, WANG Y C, et al. Significant promoting effect of Ce or La on the hydrothermal stability of Cu-SAPO-34 catalyst for NH3-SCR reaction [J]. Chemical Engineering Journal, 2019, 369: 908-919. doi: 10.1016/j.cej.2019.03.049 [23] HUANG X S, DONG F, ZHANG G D, et al. A strategy for constructing highly efficient yolk-shell Ce@Mn@TiOx catalyst with dual active sites for low-temperature selective catalytic reduction of NO with NH3 [J]. Chemical Engineering Journal, 2021, 419: 129572. doi: 10.1016/j.cej.2021.129572 [24] YAO X J, MA K L, ZOU W X, et al. Influence of preparation methods on the physicochemical properties and catalytic performance of MnOx-CeO2 catalysts for NH3-SCR at low temperature [J]. Chinese Journal of Catalysis, 2017, 38(1): 146-159. doi: 10.1016/S1872-2067(16)62572-X [25] FANG D, HE F, XIE J L. Characterization and performance of common alkali metals and alkaline earth metals loaded Mn/TiO2 catalysts for NOx removal with NH3 [J]. Journal of the Energy Institute, 2019, 92(2): 319-331. doi: 10.1016/j.joei.2018.01.004 [26] LI C X, CHENG J, YE Q, et al. Poisoning effects of alkali and alkaline earth metal doping on selective catalytic reduction of NO with NH3 over the Nb-Ce/Zr-PILC catalysts [J]. Catalysts, 2021, 11(3): 329. doi: 10.3390/catal11030329 [27] FAN Z Y, SHI J W, GAO C, et al. Gd-modified MnOx for the selective catalytic reduction of NO by NH3: The promoting effect of Gd on the catalytic performance and sulfur resistance [J]. Chemical Engineering Journal, 2018, 348: 820-830. doi: 10.1016/j.cej.2018.05.038 [28] LI X D, HAN Z T, SHI Q D, et al. Characterization of WMnCeTiOx catalysts prepared by different methods for the selective reduction of NO with NH3 [J]. New Journal of Chemistry, 2021, 45(41): 19456-19466. doi: 10.1039/D1NJ03891E [29] SHI Y R, YI H H, GAO F Y, et al. Evolution mechanism of transition metal in NH3-SCR reaction over Mn-based bimetallic oxide catalysts: Structure-activity relationships [J]. Journal of Hazardous Materials, 2021, 413: 125361. doi: 10.1016/j.jhazmat.2021.125361 [30] MA L, CHENG Y S, CAVATAIO G, et al. in situ DRIFTS and temperature-programmed technology study on NH3-SCR of NOx over Cu-SSZ-13 and Cu-SAPO-34 catalysts [J]. Applied Catalysis B:Environmental, 2014, 156/157: 428-437. doi: 10.1016/j.apcatb.2014.03.048 [31] LIAN Z H, LIU F D, HE H, et al. Manganese–niobium mixed oxide catalyst for the selective catalytic reduction of NOx with NH3 at low temperatures [J]. Chemical Engineering Journal, 2014, 250: 390-398. doi: 10.1016/j.cej.2014.03.065 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1591
- HTML全文浏览数: 1591
- PDF下载数: 47
- 施引文献: 0
