


-
随着社会发展,水资源短缺问题日益严重. 在众多水处理技术中,多相光催化氧化技术具有操作简便、有机物矿化效率高、催化剂能循环利用等优点,在有机废水资源化利用中具有广阔的应用前景[1 − 2].
多相光催化氧化体系中,氧化剂主要有过氧化氢(H2O2)和过硫酸盐(PS)两大类[3 − 4],催化剂则通常是将过渡金属离子(Fe3+、Cu2+、Co2+等)负载到不同的载体上(活性炭、多孔材料等)制备得到[5 − 7]. MCM-41硅基介孔分子筛具有骨架结构稳定、合成简便、孔道结构规则、孔径尺寸可调节、比表面积大等优点,是多相光催化剂载体开发研究的热点[8 − 10]. 将铁、铜等过渡金属负载到MCM-41介孔分子筛表面或掺杂进骨架之中,可以制备高效活化H2O2或PS降解有机污染物的多相催化剂[11 − 14]. Deng等[13]发现,Fe/MCM-41介孔材料对H2O2具有很高的光催化活性,相应的催化氧化体系能在120 min内高效降解MB染料. Schlichter等[14]合成了对PS有高催化活性的铜基MCM-41多相催化剂,对应的催化氧化体系能在2 h内有效降解MO染料. 此外,MCM-41介孔分子筛表面的负电性能够在一定程度上抑制OH−对活性组分的沉淀反应,从而有望拓展多相光催化氧化体系的pH适用范围[15 − 16].
空间位阻效应是影响氧化剂作用于有机污染物的重要因素[17 − 18]. 例如,杨欢欢[17]进行了硫酸根自由基降解全氟羧酸化合物的DFT计算,发现正是空间位阻效应导致体积较大的硫酸根自由基只能进攻未被氟原子包裹的端基碳原子. Lee等[18]在研究PS活化机制和活性物种形成途径时发现,更大的空间位阻使过二硫酸盐(PDS)对有机物的反应性较过一硫酸盐(PMS)更低. 目前,在多相光催化氧化领域中,缺乏空间位阻效应影响催化剂活化氧化剂的研究. 过硫酸盐(PS)与H2O2分子结构相似,H2O2上的一个H原子被HSO3−取代即为过一硫酸盐(PMS,HSO5−),若两个H原子全被HSO3−取代便得到同样具有对称结构的过二硫酸盐(PDS,S2O82-)[19]. 因此,HSO5−和S2O82-相较于H2O2具备更大的分子尺寸. 介孔分子筛催化剂在活化分子尺寸相差较大的PDS和H2O2时,空间位阻效应的差异会更为显著,但目前关于介孔分子筛催化剂构型对氧化剂PDS和H2O2活化影响的研究鲜见报道.
基于此,本文选取Fe-MCM-41和Cu-MCM-41作为多相光催化剂,选取同样具有对称结构但分子尺寸相差较大的H2O2和PDS作为氧化剂,构建了中性条件下的多相光催化氧化降解金橙Ⅱ体系,分析了催化剂构型、孔道尺寸和电负性对Fe-MCM-41和Cu-MCM-41活化H2O2和PDS能力的影响,揭示了Fe-MCM-41和Cu-MCM-41光催化活化H2O2和PDS的构-效关系,为介孔分子筛光催化剂的开发利用提供理论依据.
-
SX2-4-10箱式电阻炉(上海实验仪器厂有限公司);PCX50C Discover多通道光反应系统(北京泊菲莱科技有限公司);UV-2300紫外分光光度计(中国上海天美科学仪器);Multi N/C 2100总有机碳分析仪(美国岛津分析仪器股份有限公司);Bruker D8 Advance型X射线衍射仪;麦克ASAP2460比表面积孔隙度分析仪;ZEISS GeminiSEM 300 扫描电子显微镜;Titan G260-300透射电子显微镜;Nano ZS90纳米粒度及zeta电位仪;电子顺磁共振波谱仪(EPR,EMX nano,Bruker 公司,德国).
-
金橙Ⅱ(生物染色剂)、正硅酸乙酯(化学纯)、L-组氨酸购于阿拉丁化学试剂有限公司;十六烷基三甲基溴化铵、过硫酸钠购于上海麦克林生化科技有限公司;氨水、30%H2O2、氢氧化钠、乙醇购于西陇化工有限公司;磷酸氢二钠、磷酸二氢钠、异丙醇、叔丁醇、九水合硝酸铁、三水合硝酸铜购于国药集团. 以上试剂如无特殊说明均为分析纯,试剂溶液均用超纯水配置.
-
Fe-MCM-41在室温下制备[8,12,20]:称取3.39 g九水合硝酸铁溶解于75 mL正硅酸乙酯(TEOS)和75 mL乙醇的混合液中,得到混合溶液A;称取25 g十六烷基三甲基溴化铵(CTAB)于1250 mL超纯水中,超声处理1 h使其完全溶解得到澄清液B;30 ℃条件下,将100 mL氨水缓慢加入澄清液B中,10 min后逐滴加入混合溶液A,滴加完成后持续搅拌16 h,得到凝胶C;将凝胶C于室温下静置老化24 h后过滤,产物用乙醇和超纯水反复离心洗涤至中性,80 ℃干燥至恒重;干燥后将产物研磨,放入马弗炉中于550 ℃煅烧6 h(升温速率5 ℃·min−1),自然冷却至室温后取出,充分研磨后得到Fe-MCM-41. 按照上述方法制备得到MCM-41(混合溶液A为75 mLTEOS+75 mL乙醇)和Cu-MCM-41(混合溶液A为1.77 g三水合硝酸铜+75 mLTEOS+75 mL乙醇).
-
样品晶型采用Bruker D8 Advance型X射线衍射仪分析;样品比表面积使用麦克ASAP2460比表面积孔隙度分析仪测定;样品形貌使用ZEISS GeminiSEM 300扫描电子显微镜观察;样品孔道情况通过Titan G260-300透射电子显微镜拍摄分析;样品在不同pH条件下的zeta电位使用Nano ZS90纳米粒度及zeta电位仪测定.
-
采用5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)作为·OH、SO4−·和O2−·等活性物质的捕获剂,2,2,6,6-四甲基哌啶(TEMP)作为1O2的捕获剂,使用电子顺磁共振波谱仪(EPR,EMX nano,Bruker 公司,德国)检测.
-
实验在PCX50C Discover多通道光催化反应系统中进行,恒温水浴控制反应温度为30 ℃;磁力搅拌系统转速设定为250 r·min−1;光通道底部光源为紫外灯(波长254 nm,功率5 W,光强59.6 lux). 反应条件:金橙Ⅱ浓度为0.2 mmol·L−1、pH值为6.8、催化剂用量为1 g·L−1、氧化剂浓度为10 mmol·L−1.
实验预先将催化剂和金橙Ⅱ溶液加入反应瓶中,无光条件下搅拌30 min达到吸附平衡;而后加入氧化剂,同时打开光源并开始计时,在设定的时刻取样、过滤(0.45 μm水系滤头)、测定.
-
金橙Ⅱ浓度通过分光光度计在484 nm处测得吸光度,再经过标准曲线换算得到;总有机碳含量(TOC)采用总有机碳分析仪测定,取样后按体积比1:1加入终止剂(0.1 mmol·L−1Na2SO3,0.1 mmol·L−1 KH2PO4,0.1 mmol·L−1 KI,0.05 mmol·L−1 NaOH)[21 − 22]以终止反应.
-
图1为MCM-41、Fe-MCM-41和Cu-MCM-41的X射线衍射图谱. 其中,图1a是样品在0.5°—10°小角区域的XRD图谱. MCM-41存在(100)、(110)和(200)的3个晶面的特征衍射峰,这说明合成的MCM-41具备有序六方介孔结构[12,23];与MCM-41相比,Fe-MCM-41的3个衍射峰位置和强度均没有明显变化,可见Fe掺杂后材料的介孔结构和有序性几乎没有变化;Cu-MCM-41的(100)晶面衍射峰强度明显降低,且(110)和(200)晶面的衍射峰基本消失,说明Cu的掺杂对材料的结构和有序性产生了较大的影响,但基本保留了MCM-41的介孔结构.
样品在10°—80°广角区域的XRD图谱如图1b所示. 3个样品均在23°左右有一个特征峰,即SiO2特征衍射峰,与样品主体为SiO2吻合;Fe-MCM-41没有发现铁氧化合物的衍射峰,表明铁元素高度分散在MCM-41骨架中;而Cu-MCM-41在2θ=35.5°、38.7°和48.7°处出现了CuO的特征峰,说明掺杂的铜元素既有部分分散在MCM-41骨架,还有部分以CuO的形式共存.
BET分析结果显示MCM-41、Fe-MCM-41、Cu-MCM-41的比表面积分别为1058 m2·g−1、1040 m2·g−1和951 m2·g−1. Fe-MCM-41比表面积相较于MCM-41几乎不变,进一步证明Fe的掺杂对MCM-41结构的影响很小;而CuO的形成则导致Cu-MCM-41的比表面积减小明显.
为进一步分析材料的形态,对MCM-41、Fe-MCM-41和Cu-MCM-41进行了扫描电镜(SEM)分析,结果如图2. 由图2可见,MCM-41呈现棒状或球状形态,Fe-MCM-41呈现椭球状或球状形态. Fe-MCM-41的Fe元素mapping图显示Fe元素均匀地掺杂到了分子筛骨架[24],这与XRD分析结果一致. 除椭球状和球状形态外,Cu-MCM-41还呈现了不规则纳米块状形态. Cu元素mapping图表明,纳米块状形态颗粒为铜的纳米颗粒或金属氧化物簇,结合广角XRD测试结果可以推断其为纳米CuO.
由以上表征可以推断,Fe-MCM-41为Fe元素均匀掺入MCM-41骨架内部的同晶替换型催化剂;Cu-MCM-41为Cu元素分布不均匀、部分以CuO形式结合在MCM-41骨架外的同晶替换和孤岛共存型催化剂.
-
图3为MCM-41、Fe-MCM-41和Cu-MCM-41的透射电镜(TEM)图. 在50 nm标尺图中可以看到,3种材料均在孔道方向上呈现出六方排列的蜂巢结构,说明经Fe或Cu金属掺杂后,MCM-41的有序介孔特征结构能被保留,这与小角XRD分析结果一致. 进一步放大观察尺度,在20 nm标尺图中对材料的孔径进行测量. 每种样品分别随机选取3个孔道测量孔径,取平均值得到其平均孔径. 根据比例尺测出MCM-41平均孔径为2.14 nm,Fe-MCM-41和Cu-MCM-41平均孔径均为2.02 nm. BET分析对应3个样品介孔平均孔径依次为3.06 nm、2.97 nm、2.71 nm,孔径大小集中在3 nm左右,这与文献报道一致[8,25]. 一定量的金属掺杂会导致MCM-41介孔分子筛的孔容孔径减小[26].
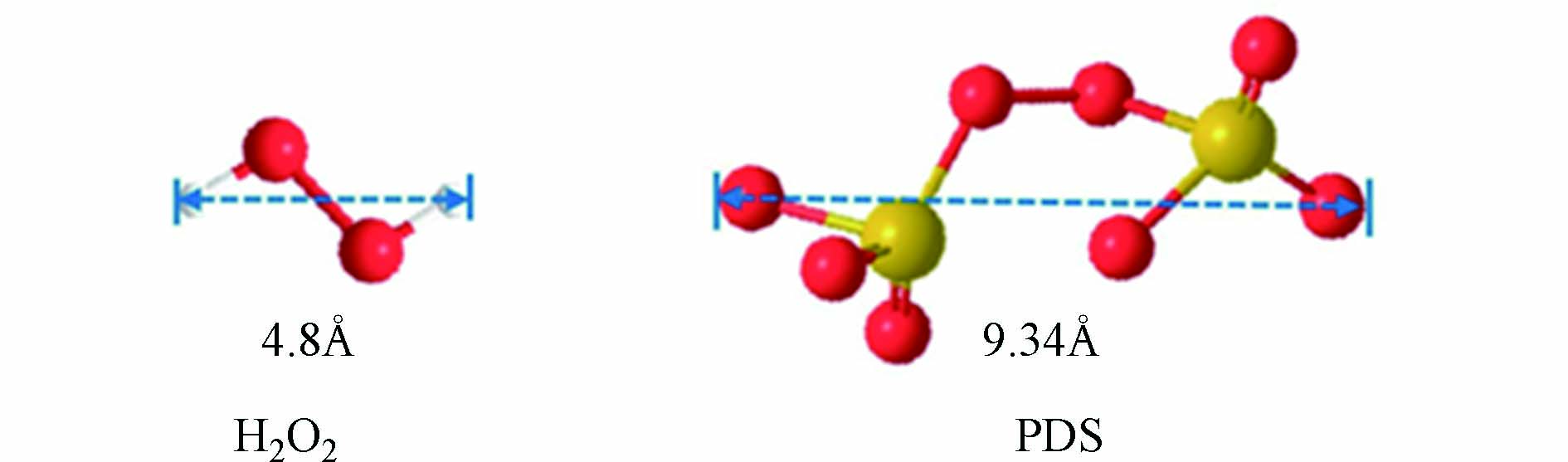
理想状态且不考虑水合影响时,氧化剂H2O2和PDS最长径分别为0.48 nm和0.934 nm,对应球体模型最大截面积分别为1.81nm2和6.85 nm2(图4); Fe-MCM-41和Cu-MCM-41的平均孔径均为2.02 nm,即其正六边形孔道边长为1.166 nm,孔道面积为35.32 nm2. PDS能够同时通过分子筛孔道截面的分子数(5个)远少于H2O2(19个),显然催化PDS的过程会受到更大的空间位阻影响.
-
Fe-MCM-41和Cu-MCM-41在不同pH条件下的zeta电位如图5所示. Fe-MCM-41和Cu-MCM-41的零电荷点分别为2和3,在近中性(pH=6.8)的实验条件下两种材料表面均带负电荷. 而H2O2分子呈电中性,不受电性影响;PDS带负电,会与同样带负电荷的材料相互排斥.
-
Fe-MCM-41和Cu-MCM-41光催化H2O2和PDS降解金橙Ⅱ的结果如图6所示. 由图6可知,单独H2O2或PDS难以直接有效氧化降解金橙Ⅱ. 近中性和无光条件下,Fe-MCM-41和Cu-MCM-41均不能有效活化H2O2降解金橙Ⅱ;相同条件下,Fe-MCM-41和Cu-MCM-41能活化PDS降解金橙Ⅱ,反应120 min后二者脱色率分别为10%和15%.
紫外光照射下,不同体系中金橙Ⅱ脱色的一级动力学常数见表1. UV能够活化H2O2和PDS[21, 27],生成羟基自由基或硫酸根自由基使金橙Ⅱ脱色. 金橙Ⅱ在UV+H2O2和UV+PDS体系的脱色动力学常数分别为0.017 min−1和0.031 min−1,说明紫外光对PDS的活化效率远高于H2O2.
Fe-MCM-41光催化活化H2O2和PDS时,H2O2分子受空间位阻和电性影响微弱,能顺利通过Fe-MCM-41内部孔道并与活性组分充分接触而被高效活化;而PDS同时受到很强的空间位阻和电性排斥影响,难以高效通过催化剂孔道进入催化剂内部,同时催化剂会对紫外光造成遮挡,导致其不能被有效活化. 因此,UV+H2O2+Fe-MCM-41体系降解金橙Ⅱ的动力学常数为0.026 min−1,较UV+H2O2体系提升了52.9%;而UV+PDS+Fe-MCM-41体系降解金橙Ⅱ的动力学常数为0.028 min−1,较UV+PDS体系有所下降.
Cu-MCM-41构型为同晶替换和孤岛共存型,其光催化活化H2O2和PDS时会受到催化剂构型、空间位阻和电性的三重影响. H2O2分子虽然受空间位阻和电性影响较小,能高效通过Cu-MCM-41孔道与活性组分充分接触,但铜离子对H2O2的活化能力微弱[28],且催化剂会对紫外光造成遮挡,因此UV+H2O2+Cu-MCM-41体系中H2O2不能被有效活化;而PDS分子虽然受到较强的空间位阻和电性排斥干扰,不能高效通过孔道,但催化剂表面和孤岛组分CuO对PDS有较强的活化能力[29 − 30],所以Cu-MCM-41能高效活化PDS降解金橙Ⅱ. 由表1可见,UV+H2O2+Cu-MCM-41体系降解金橙Ⅱ的动力学常数仅为0.015 min−1,较UV+H2O2体系有所下降;而UV+PDS+Cu-MCM-41体系降解金橙Ⅱ的动力学常数达到了0.043 min−1,较UV+PDS体系提升了38.7%.
金橙Ⅱ在各体系中反应开始时和反应180 min时的矿化情况如图7所示,根据反应前后体系的TOC值可以计算不同体系反应180 min时金橙Ⅱ的矿化率. Fe-MCM-41和Cu-MCM-41对金橙Ⅱ有一定的吸附作用,使得反应开始时(暗吸附平衡)不同体系的TOC值略有差异. 近中性UV+H2O2体系反应180 min时,加入Fe-MCM-41可以将矿化率由43%提高至57%,而加入Cu-MCM-41会使矿化率降至40%;近中性UV+PDS体系反应180 min时,加入Cu-MCM-41可以将矿化率由48%提高至76%,而加入Fe-MCM-41会使矿化率降至45%.
上述结果说明,在近中性和紫外光条件下,Fe-MCM-41是活化H2O2的高效催化剂,而Cu-MCM-41是活化PDS的高效催化剂.
-
通过淬灭实验研究各体系活性基团对金橙Ⅱ降解过程的贡献,结果如图8所示. 对于UV+H2O2+Fe-MCM-41体系,以异丙醇作为淬灭剂进行了淬灭实验. 异丙醇是·OH良好的淬灭剂,其与·OH的反应速率高达1.9×1010 L·(mol·s)−1[31]. 加入过量异丙醇后体系中金橙Ⅱ的降解几乎被完全抑制,说明UV+H2O2+Fe-MCM-41体系中·OH是降解金橙Ⅱ最主要的活性基团.
对于UV+PDS+Cu-MCM-41体系,分别以L-组氨酸(1O2、SO4−·和·OH的淬灭剂)[32 − 33]、乙醇(SO4−·和·OH的淬灭剂)[34 − 35]和叔丁醇(·OH的淬灭剂)[34 − 35]作为淬灭剂进行淬灭实验. L-组氨酸是1O2的淬灭剂(反应速率为 3.2 × 107 L·(mol·s)−1),其也能与SO4−·(反应速率为 2.5 × 109 L·(mol·s)−1)和·OH(反应速率为 5.0 × 109 L·(mol·s)−1)发生反应[32 − 33]. 乙醇与SO4−·和·OH的反应速率分别为1.6× 107—7.8 × 107 L·(mol·s)−1和1.1× 109—2.8 × 109 L·(mol·s)−1,而叔丁醇与SO4−·和·OH的反应速率分别为4× 105—9.1 × 105 L·(mol·s)−1和3.6× 109—7.6 × 109 L·(mol·s)−1 [33 − 35]. 由图8b可见,UV+PDS+Cu-MCM-41体系中加入以上淬灭剂后,金橙Ⅱ的降解均受到一定程度的抑制. 其中,L-组氨酸抑制效果最为突出,金橙Ⅱ的降解速率常数降至0.002 min−1(基础k=0.043 min−1);其次是乙醇,金橙Ⅱ的降解速率常数降至0.014 min−1;抑制效果最弱的是叔丁醇,金橙Ⅱ的降解速率常数仍有0.041 min−1. 以上结果说明,在UV+PDS+Cu-MCM-41体系中,主要活性物种为1O2和SO4−·.
为进一步鉴定体系生成的自由基,采用EPR对活性物种进行了测定. 以水为溶剂检测1O2、SO4−·和·OH,以二甲基亚砜(DMSO)为溶剂检测O2−·[36],结果如图9所示. UV+H2O2+Fe-MCM-41体系仅观察到了DMPO-·OH(1:2:2:1)复合物特征峰信号[36 − 37],说明该体系主要的活性物种为·OH. UV+PDS+Cu-MCM-41体系中检测到了DMPO-·OH(1:2:2:1)和DMPO-SO4−·(1:1:1:1:1:1)[36 − 37],证明该体系反应过程中产生了·OH和SO4−·(·OH是由SO4−·与水反应生成[30,36]). 另外,EPR图谱中观察到了DMPO-O2−·信号[38],表明该体系反应过程中有O2−·生成. 同时TEMP-1O2(1:1:1)[33]特征峰信号出现证明该体系中确有1O2生成.
结合淬灭实验结果,可知UV+H2O2+Fe-MCM-41体系主要活性物种为·OH,UV+PDS+Cu-MCM-41体系主要活性物种为1O2和SO4−·.
-
基于以上分析,得到中性条件下Fe-MCM-41和Cu-MCM-41光催化H2O2和PDS的构-效关系,结果如图10所示. 对于Fe-MCM-41,首先紫外光将催化剂上的≡FeⅢ光解还原成≡FeⅡ(式1);H2O2受空间位阻和电性排斥影响小,能顺利通过催化剂内部孔道;催化剂表面和孔道内部的≡FeⅡ能与H2O2发生芬顿反应生成≡FeⅢ和·OH(式2)[21,39];催化剂上≡FeⅡ与≡FeⅢ的循环促进了H2O2持续分解生成·OH进而降解金橙Ⅱ;PDS由于受到空间位阻和电性排斥的影响,难以进入Fe-MCM-41孔道之中,加上催化剂对紫外光的遮挡,导致UV+PDS+Fe-MCM-41体系中PDS不能得到有效活化.
对于Cu-MCM-41,首先紫外光将同晶替换于骨架中的≡CuⅡ和孤岛CuO上的≡CuⅡ光解还原成≡CuⅠ(式3);PDS因为空间位阻和电性排斥难以进入催化剂孔道,但催化剂表面的≡CuⅠ和孤岛CuO上的≡CuⅠ可以快速活化S2O82-产生≡CuⅡ和SO4−·[40](式4);此外,过硫酸盐还可以进一步转化生成O2−·和1O2(式5-7)[33,36 − 37];体系中生成的1O2和SO4−·能高效氧化降解金橙Ⅱ;H2O2虽然能自由进出催化剂孔道,但铜离子活化H2O2的效果极差,且催化剂会对紫外光造成遮挡,因此在UV+H2O2+Cu-MCM-41体系中H2O2不能被有效活化.
-
(1)在室温下制备了Fe-MCM-41和Cu-MCM-41,表征结果证明Fe-MCM-41为同晶替换型催化剂,Cu-MCM-41为同晶替换和CuO孤岛共存型催化剂.
(2)中性条件下,H2O2受空间位阻和电性排斥影响小,能被Fe-MCM-41孔道内外铁离子高效光催化,但难以被Cu-MCM-41中同晶替换和孤岛的铜离子高效光催化;PDS由于受到空间位阻和电性排斥的影响,难以进入Fe-MCM-41和Cu-MCM-41孔道之中,但Cu-MCM-41表面和孤岛上的铜离子能高效光催化PDS.
(3)中性条件下,Fe-MCM-41能高效光催化H2O2产生·OH降解金橙Ⅱ,Cu-MCM-41则能高效光催化PDS产生1O2和SO4−·降解金橙Ⅱ. UV+H2O2体系中加入Fe-MCM-41,金橙Ⅱ的脱色速率提升了52.9%,反应180 min时体系矿化率从43%提高到了57%;UV+PDS体系中加入Cu-MCM-41,金橙Ⅱ的脱色速率提升了38.7%,反应180 min时体系矿化率从48%提高到了76%.
中性条件Fe/Cu-MCM-41光催化活化H2O2/PDS的构-效关系
The structure-activity relationship of Fe/Cu-MCM-41 on photocatalytic activation of H2O2/PDS under neutral condition
-
摘要: 本文在室温下制备了Fe-MCM-41和Cu-MCM-41,利用XRD、BET、SEM和TEM等手段对催化剂进行了表征. 选取金橙Ⅱ为探针分子、H2O2和PDS作为氧化剂,构建了多相光催化氧化体系,研究中性条件下Fe-MCM-41和Cu-MCM-41光催化活化H2O2和PDS的构-效关系. 结果表明,Fe-MCM-41为同晶替换型催化剂,Cu-MCM-41为同晶替换和CuO孤岛共存型催化剂. 中性条件下,H2O2受空间位阻和电性排斥影响小,能被Fe-MCM-41孔道内外铁离子高效光催化产生·OH,但难以被Cu-MCM-41中同晶替换和孤岛的铜离子高效光催化;PDS由于受到空间位阻和电性排斥的影响,难以进入Fe-MCM-41和Cu-MCM-41孔道之中,但Cu-MCM-41表面和孤岛上的铜离子能高效光催化PDS产生1O2和SO4−·. UV+H2O2体系中加入Fe-MCM-41,金橙Ⅱ的脱色速率提升了52.9%,反应180 min时体系矿化率从43%提高到了57%;UV+PDS体系中加入Cu-MCM-41,金橙Ⅱ的脱色速率提升了38.7%,反应180 min时体系矿化率从48%提高到了76%. 本研究为介孔分子筛光催化剂的开发利用提供理论依据.Abstract: In this study, Fe-MCM-41 and Cu-MCM-41 were synthesized at room temperature and were characterized by XRD, BET, SEM and TEM techniques. In the heterogeneous photocatalytic oxidation system, Fe-MCM-41 and Cu-MCM-41, Orange Ⅱ, H2O2 and PDS were selected as catalysts, modal pollutant and oxidants, respectively. The structure-activity relationship of Fe-MCM-41 and Cu-MCM-41 on photocatalytic activation of H2O2 and PDS under neutral condition was discussed. The results showed that the configuration of Fe-MCM-41 was isomorphous substitution, and the configuration of Cu-MCM-41 was isomorphous substitution coexisting with CuO island. Under neutral condition, it was easy for H2O2 to enter the pore of Fe-MCM-41 or Cu-MCM-41 since H2O2 was less influenced by steric hindrance and electrical repulsion. ·OH could be efficiently generated through H2O2 photo-activation by iron ions inside and outside the pore of Fe-MCM-41, but it was difficult for H2O2 to be efficiently catalyzed by copper ions in Cu-MCM-41. It was hard for PDS to enter the pore of Fe-MCM-41 or Cu-MCM-41 due to the higher steric hindrance and electrical repulsion under neutral condition, but the copper ions on the surface of Cu-MCM-41 and CuO island could efficiently photo-activate PDS to produce 1O2 and SO4−·. When Fe-MCM-41 was added into UV+H2O2 system, the enhanced efficiency of Orange Ⅱ decolorization rate could reach 52.9%, and its mineralization increased from 43% to 57% at 180 min. When Cu-MCM-41 was added into UV+PDS system, the enhanced efficiency of Orange Ⅱ decolorization rate could reach 38.7%, and its mineralization increased from 48% to 76% at 180 min. This study provided a theoretical basis for the development and utilization of mesoporous molecular sieve photocatalysts.
-
Key words:
- MCM-41 /
- H2O2 /
- PDS /
- photocatalytic activation /
- structure-activity relationship.
-
近年来,我国一些发达地区的村落建设了分散式农村污水处理设施,并取得了较好的环境效益,但这些村落污水的治理,仍以COD、氨氮、总磷等污染物的降解为考核目标,而农村居民生活水平和医疗条件在不断提高,村落水环境中EDCs浓度水平也相应增加,其对水环境生态和人类健康危害日益严重[1],尤其是农村地区,EDCs通过灌溉形式,直接被稻、麦、瓜果等农作物吸收,进而进入食物链。EDCs是一种能扰乱生物体新陈代谢平衡的化学物质,主要分为天然产生(E1、E2、E3)及人工合成(EE2)[2]。据报道,各种环境基质中均检测到不同浓度的EDCs,水体中其质量浓度可低至10−6(1 μg·L−1)量级和10−9(1 ng·L−1)量级,而EDCs在极低浓度下就可引起水生生物的生殖发育障碍[3-4]。主要原因在于,EDCs与生物体内的雌激素受体结合而干扰生物内分泌系统正常代谢[5]。Legler等[6]研究发现,当自然水体中E2浓度达到1.0 ng·L−1时,可引起生物体内分泌紊乱。Cappiello等[7]发现不少猝死婴儿体内残留的EDCs含量相对普通新生婴儿较高;Clarke等[8]研究表明,妊娠期女性若接触过量EE2,则将増加母女患乳腺癌的风险。由此可见,当EDCs进入动物食物链,再经过层层传递,最终在人类体内积累,对人体健康损害威胁相应不断增大。
国内外众多学者研究表明,耕作型稻田复合生态系统通过“微生物-稻田湿地”耦合的复合系统对村落污水中的有机污染物进行生物降解,其主要依靠水稻复杂的根系及其附着的生物膜协同净化作用,不但可以达到净化村落污水的效果,还可以产生水稻增肥的效益[9]。但存在类固醇类激素(EDCs)环境污染及生态危害问题。现阶段,人为去除环境中雌激素类污染物主要通过吸附[10]、光催化氧化[11]、生物降解[12]的3种途径使EDCs在环境中迁移、降解。阳春等[10]研究表明,污泥对雌激素的吸附主要来自于污泥中的活性成分,而被生物表面所吸附的雌激素才能被生物降解。光解与氧化作用是EDCs真正的分解过程,因为它不可逆的改变了分子结构,强烈影响其在环境中的归趋,但光降解类固醇雌激素极易受pH的影响,近期有一些研究表明类固醇雌激素光氧化降解后产生的代谢产物仍具有雌激素活性[11]。生物降解通过微生物新陈代谢和自身与周围环境进行物质交换,达到将污染物去除或转化为无害无毒的物质[12],日益受到人们的重视。
本文针对村落污水中的类固醇类激素(EDCs)环境污染及生态危害问题,构建耕作型稻田湿地[13],并以本课题组筛选的农药降解菌HD为EDCs生物强化降解菌[14],考察其对EDCs的降解效能,以期为村落水环境中的EDCs降解机制及环境生态影响评价提供参考。
1. 材料与方法(Materials and methods)
1.1 试验装置与进水水质
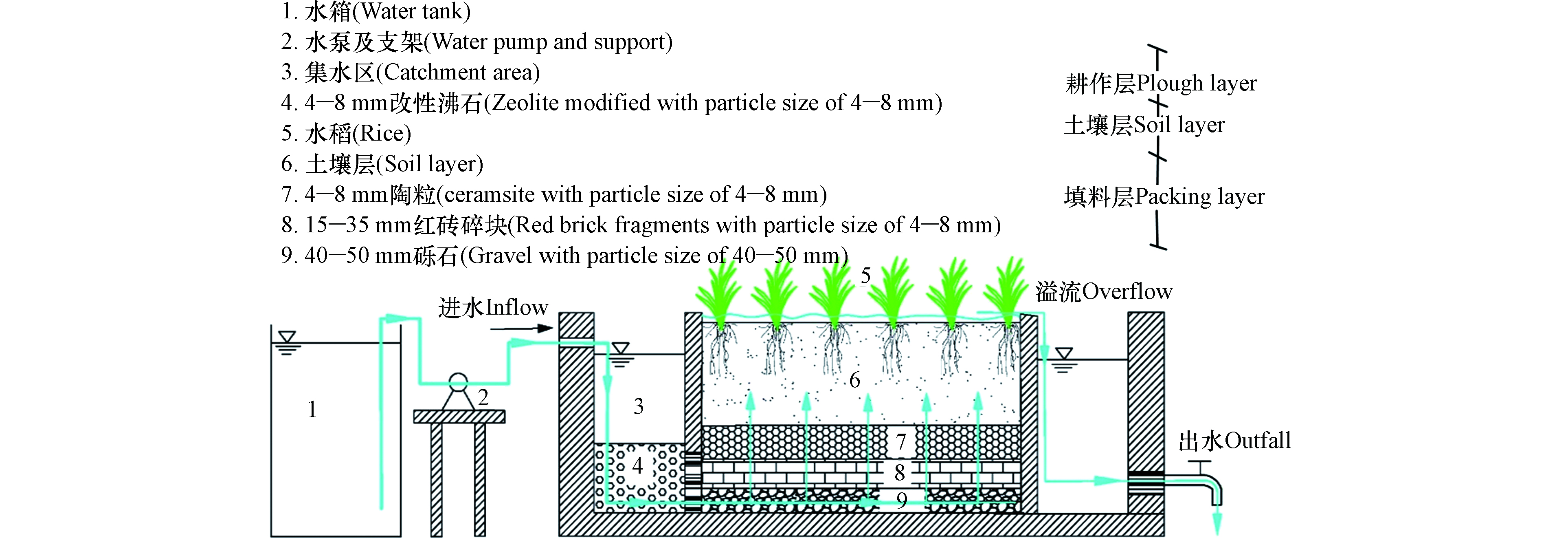
耕作型稻田湿地试验装置分2组,A组为空白对照组,B组为HD菌剂强化组。试验装置如图1所示,整个耕作型稻田湿地装置采用PP板(L×B×H=1 m×0.4 m×0.65 m),由集水区、湿地处理区和出水区3部分所组成,集水区沸石[15]层厚度200 mm,湿地填料层厚度300 mm,自下而上由40—50 mm砾石、15—35 mm红砖碎块、4—8 mm陶粒组成,孔隙率约30.8%;试验进水通过恒流水泵抽入集水区,经集水区的沸石层有效拦截后,再进入稻田湿地。装置内的土壤,取自常州洛阳镇薛家河周边稻田表层15—20 cm处土壤,所取土壤为该地区连年种植水稻土,装置内土壤层厚度为20 cm。耕作层厚度10 cm。水稻秧苗取自常州市洛阳镇薛家河周边水稻田中,种植密度为45株·m−2。
菌剂投加:A、B二组均采用自然进水法生物膜培养,当镜检可见填料上有褐色生物膜和原生动物及COD降解率超过60%时,认为生物挂膜成功。此时在B组中投加HD菌剂,配制100 mL的基础液体培养基(氯化钠:0.5 g,七水合硫酸镁:0.5 g,七水合硫酸亚铁:0.002 g,硫酸铵:1.5 g,氯化钙:0.04 g,磷酸氢二钾:1.5 g,磷酸二氢钾:1.5 g,蒸馏水:1 L,琼脂粉:20 g,pH:7.2—7.4),按照体积分数2.5%的比例接入菌种HD,培养48 h,将菌液混合后,再按照1%的投加比例混入进水中,随进水进入B组耕作型稻田湿地系统,连续投加2周,进行菌剂强化挂膜。A组不投菌,在正常条件下进行生物挂膜,为试验对照组。
农药降解菌HD筛选自南京某废弃农药厂的土壤中,是一株具有降解2,4-二氯苯酚能力的菌,现保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏编号:CGMCC No. 15123。经理化特性和分子鉴定判断属于摩式假单胞菌属(Pseudomonas mosselii)。
试验进水水质如表1所示。在生活污水的基础上添加少量内分泌干扰物,模拟进水内分泌干扰物浓度波动,如表2所示。
表 1 试验进水水质Table 1. Test water quality指标Index CODcr/(mg·L−1) 总磷/(mg L−1)TP 氨氮/(mg·L−1) NH+4 总氮/(mg·L−1)TN pH 范围 87—156 2.23—5.69 8.75—16.34 10.86—18.33 7.39—7.84 | Show Table DownLoad:
CSV
表 2 进水内分泌干扰物浓度(μg·L-1)Table 2. Concentration of endocrine disruptors in water inlet(μg·L-1)
DownLoad:
CSV
表 2 进水内分泌干扰物浓度(μg·L-1)Table 2. Concentration of endocrine disruptors in water inlet(μg·L-1)类固醇Steroid estrogen E1 Estrone E2 Estradiol EE2 17-α-ethinylestradiol E3 Estriol 原水浓度 10.13—15.25 0.79—1.1 0.88—1.82 6.31—9.58 模拟进水浓度 38.69—60.13 9.86—14.32 10.89—13.21 33.28—54.36 | Show TableDownLoad:
CSV
1.2 试验仪器与试剂
表 3 主要试验试剂Table 3. Main experimental reagents药品名称Drug names 分子式Molecular formula 规格Specification 生产单位Production unit E1 C18H22O2 — 阿拉丁 E2 C18H24O2 — 阿拉丁 E3 C18H24O3 — 阿拉丁 EE2 C20H24O3 — 阿拉丁 BSTFA C8H18F3NOSi2 — 阿拉丁 吡啶 C5H5N AR 永华化学科技(江苏) 丙酮 CH3COCH3 AR 国药 正己烷 C6H14 AR 江苏强盛功能化学 二氯甲烷 CH2Cl2 AR 永华化学科技(江苏) 雄烷 C19H23 — 北京谱析科技有限公司 | Show TableDownLoad:
CSV
表 4 试验主要仪器Table 4. Experimental main instruments仪器设备Instrument and equipment 型号Model number 生产单位Production unit 多用途高速离心机 SORVALL Thermo electron corporation 行星式球磨机 QM-1SP2 南京大学仪器厂 超声波细胞粉碎机 JY96-Ⅱ 宁波新芝生物科技股份有限公司 气质联用 Trace ISQLT 美国赛默飞科技有限公司 | Show TableDownLoad:
CSV
1.3 水样预处理
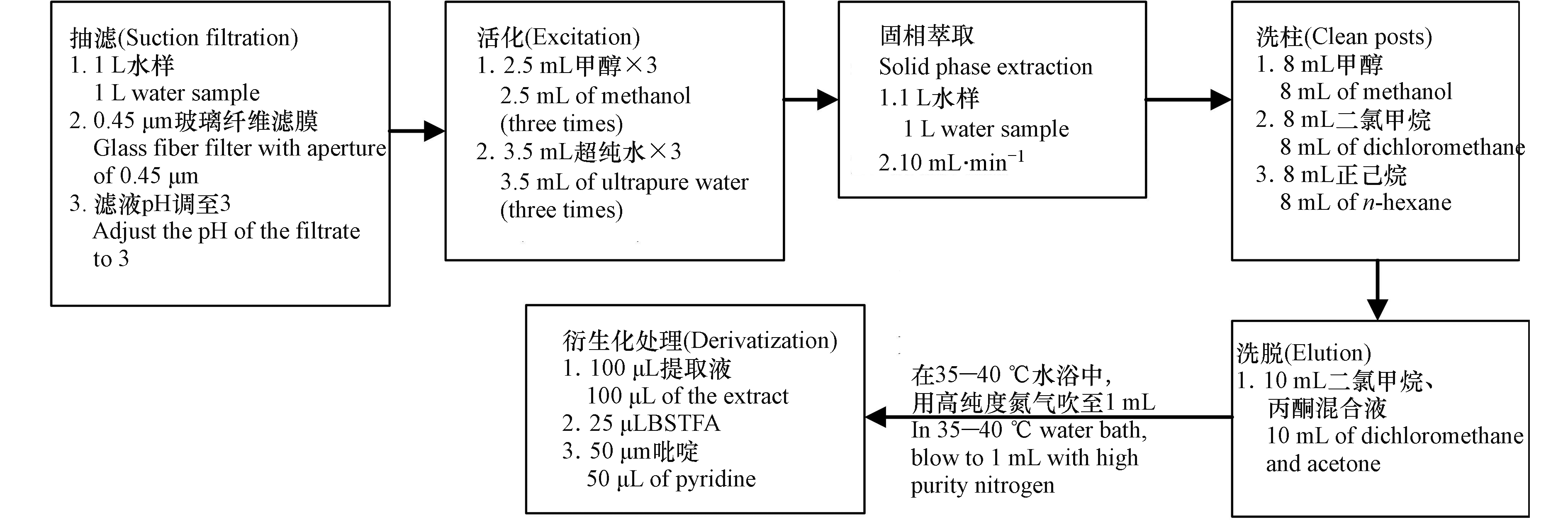
采集1 L水样,GF/F(0.45 μm)滤膜抽滤,滤液用99%的浓硫酸调节至pH3以下。
固相萃取:利用Simon Acti-Carb SPE柱进行固相萃取,首先活化SPE,分别加入2.5 mL甲醇3次,3.5 mL超纯水3次,控制流速在10 mL min−1,进行萃取,待水样萃取完,再分别加入8 mL甲醇、8 mL二氯甲烷、8 mL正己烷进行洗柱,最后用10 mL的二氯甲烷和丙酮的混合溶液淋洗,收集淋洗液,在35—40 ℃的水浴中,用高纯度氮气吹至1 mL,放入冰箱待用,具体步骤见图2。
1.4 土样预处理
稻田土壤采集后,自然风干,去除杂石杂草,用球磨机在400 r·min−1的条件下充分研磨,过20目筛网,混匀后放入冰箱待用。预处理时,称取1 g的样品,放入10 mL的离心管中,加入5 mL溶剂(甲醇∶丙酮=1∶1),超声波萃取10 min,随后以5000 r·min−1转速离心10 min,收集上清液至25 mL离心管中。重复以上步骤2次,合并上清液后,经高纯度氮气吹至1 mL后,定容至20 mL。最后按照图2水样预处理的方法,进行固相萃取。
1.5 衍生化处理
环境中类固醇类EDCs的含量相对较低,且由于这类化学物质都含有—OH官能团,极性较强,若直接采用气质联用测定,在测定过程中容易被色谱柱所吸附,从而造成实测浓度小于实际浓度,因此为降低该类物质的极性,提高其热稳定性,增加了衍生化步骤,即在正常衍生化的步骤中,又增添了吡啶,可以有效降低EE2的衍生产物转变为E1的衍生产物。黄成等[16]对某制药厂污水样品中E1、E2、E3和EE2衍生化后使用气相色谱/质谱法(GC-MS)分析表明,4种目标化合物的加标回收率达到(94.0%±2.9%)—(101.0%±3.8%),说明GC-MS法可应用于污水中雌激素化合物定量检测。
衍生化方法是在1.5 mL色谱进样瓶中加入100 μL的混合标液,通入高纯度氮气将其缓慢吹干,接着在进样瓶中加入25 μL BSTFA和50 μL吡啶,待其反应一定时间后吹干,最后加入V(二氯甲烷)∶V(正己烷)=1∶4的进样溶剂和10 μL 0.01 g·L−1的内标,取1 μL注入GC-MS分析,在空白污水样品中添加100,300、500 ng·L−1 等3个浓度水平的目标物,测定回收率。衍生化的具体反映结构变化如下:

1.6 EDCs的测定条件
试验中E1、E2、EE2、E3选用气质联用进行测定,色谱柱为TG-5MS(30 m×0.25 mm×0.25 μm),气相条件如下:
GC:以氦气为载气,流速1 mL·min−1;不分流方式进样,进样口温度280 ℃,进样体积1 μL;柱初始温度为50 ℃,保持2 min,以12 ℃·min−1程序升温至260 ℃,保持8 min,再以3 ℃·min−1升温至280 ℃,保持5 min;
MS:接口温度280 ℃,传输线温度300 ℃,离子源为EI源,温度250 ℃,电子轰击能量70 eV,溶剂延迟时间12 min,以全扫描模式定性,扫描范围50—600 m/z,以选择离子扫描模式定量;
根据其衍生产物的特征碎片离子分布特征来确定目标产物的实际浓度,衍生产物实际参数如表5所示。
表 5 衍生产物的相应参数Table 5. corresponding parameters of derivative products衍生产物Derivative product 保留时间/min Retention time 特征碎片离子(m/z)Characteristic fragment ion 线性回归方程Equation of linear regression TMS-E1 24.28 342、327、285 Y=(9.43×108)x+(1.02×109);R2=0.91 di-TMS-E2 25.43 416、401、285 Y=(1.08×107))x+(7.55×106);R2=0.91 di-TMS-EE2 27.03 440、425、285 Y=(1.90×107))x+(3.92×106);R2=0.92 Tri-TMS-E3 28.43 504、489、285 Y=(1.14×106)x+(2.38×106);R2=0.90 注:x为目标产物的实际浓度,单位mg·L−1,Y为色谱峰面积. Note:x is the actual concentration of the target product, unit: mg·L−1, Y is the peak area. | Show TableDownLoad:
CSV
2. 结果与讨论(Results and discussion)
2.1 耕作型稻田湿地去除EDCs效能分析
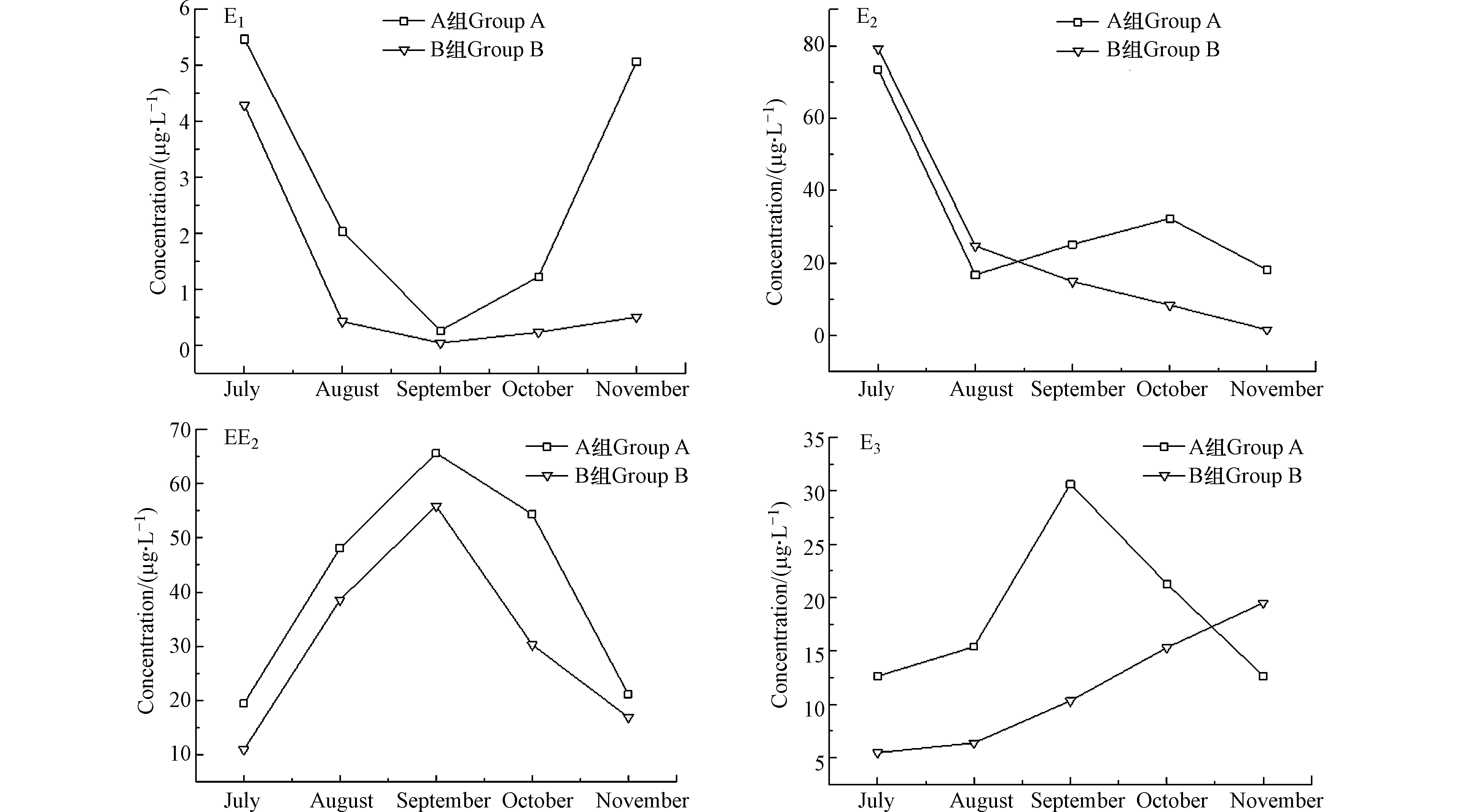
图3是2018年7月至11月,水稻生长过程中,耕作型稻田湿地对EDCs中E1、E2、EE2、E3的去除效果图。从图3可以看出,生活污水中EDCs通过耕作型稻田湿地吸附降解后,A组的E1、E2、EE2、E3的平均去除率为80.1%、72.4%、68.3%、51.4%,B组的平均去除率为82.6%、73.4%、75.6%、60.5%,除了E3外,其余各目标污染物的去除率均在65%以上,Baronti[17]在试验过程中发现E2在生物降解过程中,一部分的E2较易氧化转化成E1,遵循典型的醇氧化为酮原则,而E1通过水和作用又转化为E3,随着中间产物的不断产生,进而导致了出水E3浓度偏高。B组投加HD菌后,可能是假单胞菌HD诱导产生了羟基酶[18],相比A组E1、E2去除率未发生明显变化,EE2和E3去除率增高7%—9%,Huang等[19]发现,在BPA(双酚A)降解的河流底泥中,假单胞菌和鞘单胞菌占据了原有细菌群落的73%,由此可见,向湿地中投加假单胞菌HD有助于提高EDCs的去除率。
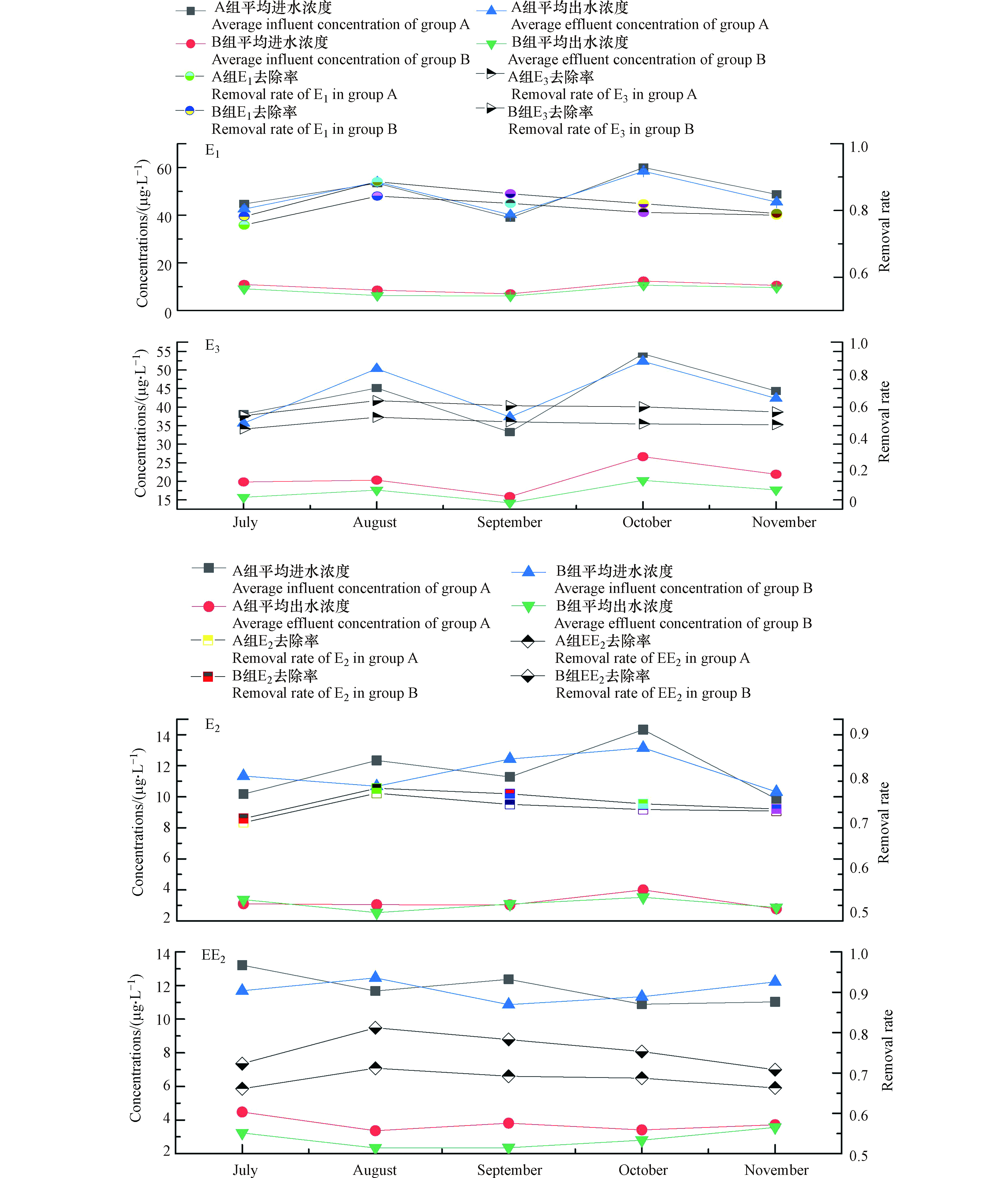 图 3 耕作型人工湿地对E1、E3、E2、EE2的去除效果Figure 3. Removal effect of cultivated constructed wetland on E1, E3, E2 and EE2
图 3 耕作型人工湿地对E1、E3、E2、EE2的去除效果Figure 3. Removal effect of cultivated constructed wetland on E1, E3, E2 and EE2湿地经过5个月的连续运行后,投菌组B中的EDCs残留量明显低于未投菌组A。由图3中的实测数据,经计算B组湿地出水中E1、E2、E3和EE2含量相比A组分别降低约26.6%、17.1%、30.3%、13.3%。试验时间跨秋夏两季,夏季EDCs的去除效果要明显优于秋季,这主要是因为夏季正值水稻生长期,水稻根系对EDCs有一定吸收作用,夏季温度较高,微生物活性较强,微生物新陈代谢较为旺盛,运行至秋季时,温度降低,水稻收割,微生物活性逐渐降低,EDCs的降解率并未大幅下降,说明人工湿地中填料层及土壤层对EDCs的去除起到了一定作用。
2.2 HD菌剂强化后耕作型稻田湿地各生物单元对EDCs去除分析
图4为耕作型稻田湿地经HD菌剂强化后EDCs浓度变化趋势图,沿程对E1、E2、E3和EE2的总去除率分别为88.6%、76.3%、64.8%和81.2%。3个生物单元填料层、土壤层、耕作层对E1去除率分别为16.2%、75.1%、28.2%,对E2去除率分别为14.5%、61.8%、18.7%,对E3去除率分别为20.6%、49.8%、10.4%,对EE2去除率分别为8.9%、43.4%、-8.1%,显然土壤层对EDCs的去除贡献占比相对最大,主要是因为土壤中微生物种群丰度要远高于填料层孔隙中生物膜上的微生物种群丰度,同时,土壤中还有分布广泛的水稻根系,通过其根系的泌氧作用,刺激微生物活性,促进微生物新陈代谢,进而加强了微生物降解能力,而填料层的填料作为微生物的代谢场所[20],其主要作用有二:一是直接吸附EDCs,二是作为微生物的载体,在其表面形成微生物集聚(生物膜),为生物膜微生物吸附、吸收、降解EDCs提供平台。其中E1、E2、E3的降解沿程越往后,目标污染物浓度越低,但EE2在经过土壤层的降解后,其浓度却略有提升,一方面,据Ascenzo等[21]研究显示,EE2的生物降解只发生在好氧段,因为EE2拥有乙炔基,从空间结构上来看,这对基团基质和受体的结合有阻碍作用,使得酶活性表达受阻,且在厌氧阶段,会使其由结合态变为游离态,导致EE2不易被降解,另一方面,Li等[22]研究发现,EE2在厌氧条件下更有利用吸附,耕作层分布有植物根系,植物根系的泌氧作用使得填料层吸附作用相比土壤层较弱。同时在降解过程中E3的含量在进水时略低于E1,但随着沿程越往后E3出水浓度高于E1,因为E2在生物降解过程中,一部分的E2较易氧化转化成E1,而E1通过水和作用又转化为E3,随着中间产物的不断产生,进而导致出水E3浓度偏高。
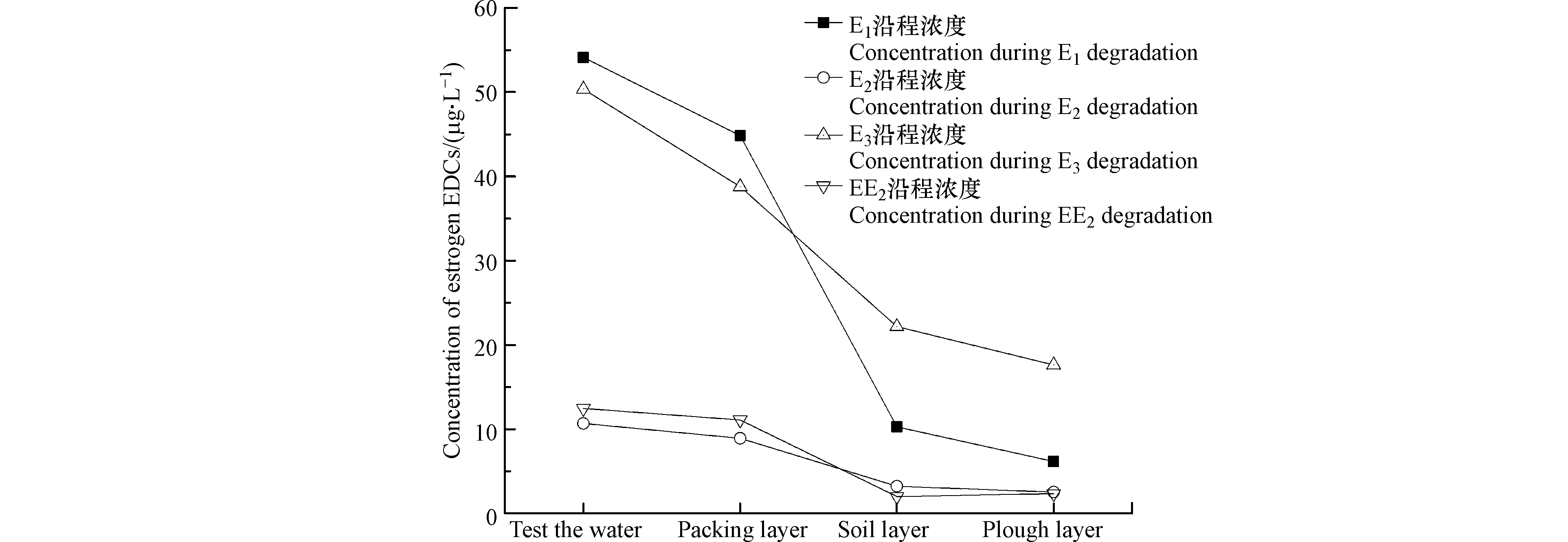 图 4 耕作型湿地沿程EDCs浓度变化Figure 4. Changes of EDCs concentration along the course of cultivated wetland
图 4 耕作型湿地沿程EDCs浓度变化Figure 4. Changes of EDCs concentration along the course of cultivated wetland2.3 耕作型稻田湿地水芹种植期土壤中残留EDCs的变化特征
类固醇类EDCs多半有亲脂疏水的特性,易被土壤所吸附,一般类固醇类EDCs在环境中的半衰期为5—25 d不等,土壤中残留的EDCs会在一定时期,通过迁移、浸出到水体中,所以稻田湿地水体中EDCs含量通常偏高[23]。图5反应了耕作型稻田湿地净化过程中,土壤中残留EDCs含量的变化趋势,可看出投菌组B中E1、E2、EE2残留量明显小于A组,可见稻田湿地微生物对EDCs进行生物降解时,反硝化假单胞菌[24]使得反硝化细菌种群丰度提升,有利于E1的生物降解,说明HD菌促进了土壤层中羟基酶的产生,提高了其对EDCs的去除效率。从图5可看出,EE2浓度是先增后减,可能是由于EE2较为稳定不易降解。Ternes等[25]研究EE2的生物降解特性,发现1 mg·L−1的EE2经24 h后几乎无降解。10月水稻收割后,新种植了水芹,据Schroeder等[26]研究发现水芹在24 h内便能通过茎部富集近0.9 ng·L−1的EE2,所以后期EE2浓度又显下降趋势。B组中E3浓度一直呈上升趋势,任海燕等[27]研究发现,对EE2降解中间产物进行质谱分析推测,EE2在降解过程中首先被氧化为E1,后经过一系列生物催化作用生成2-羟基-2,4-二烯-戊酸和2-羟基-2,4-二烯-1,6-己二酸两种中间代谢产物,E1通过水合作转化为E3,又E2在生物降解过程中部分较易被氧化转化为E1,E1通过水合作用又转化为E3,故而导致土壤中E3浓度不断增高。湿地运行初期E1、E2浓度分别增加了85%、25%,EE2、E3浓度下降了60%、30%,这主要是因为生活污水中EDCs的构成主要以天然雌激素中的E1、E2为主,且其主要为结合态形式,极性更强,更易被土壤所吸收[28],且溶解性有机物的共轭物在经细菌酶分解时,亦会产生E1的衍生产物[29],进而导致E1浓度提高。虽然土壤层对于EDCs的降解效果较好,但初期由于土壤中生物种群尚未稳定,所以导致E1、E2的处理效果受限。
3. 结论(Conclusions)
(1)未投菌的A组对雌酮(E1)、雌二醇(E2)、雌三醇(E3)及17α-乙炔基雌二醇(EE2)的平均去除率分别为80.1%、72.4%、51.4%、68.3%,投加HD菌剂强化的B组平均去除率分别为82.6%、73.4%、60.5%、75.6%,A、B两组耕作型稻田湿地对E3去除率相比E1、E2、EE2最低。E2在生物降解过程中较易氧化转化成E1,而E1通过水和作用又转化为E3,随着中间产物的不断产生,出水E3浓度增大。A、B两组耕作型稻田湿地中,除E3外E1、E2、EE2去除率均在65%以上,EE2的生物降解只发生在好氧段,EE2拥有乙炔基,从空间结构上来看,这对基团基质和受体的结合有阻碍作用,使得酶活性表达受阻,且在厌氧阶段,会使其由结合态变为游离态,致使EE2不易被降解。
(2)投加HD菌后,耕作型稻田湿地对E1、E2、E3和EE2的总去除率分别为88.6%、76.3%、64.8%和81.2%。耕作层、土壤层、填料层3个生物单元对EDCs(E1、E2、E3、EE2)去除效果均有提升,其中:对E1去除率分别为16.2%、75.1%、28.2%;对E2去除率分别为14.5%、61.8%、18.7%;对E3去除率分别为20.6%、49.8%、10.4%;对EE2去除率分别为8.9%、43.4%、−8.1%。HD菌对耕作型稻田湿地中微生物群落产生影响,诱导产生的羟基酶有利于降解EDCs,从而提高了EDCs的去除效率。耕作型稻田湿地经HD菌剂强化后,3个生物单元中明显土壤层对EDCs去除贡献相对较大,土壤层微生物种群丰度更大,且土壤层中还分布有广泛的水稻根系,不仅为微生物代谢提供营养物质和氧气,还进一步提高了其生物分解能力。
(3)湿地运行初期,耕作型稻田背景土壤中残留的EDCs含量相对较高,经过5个月的连续运行后,土壤中EDCs含量明显下降,投菌组B中的EDCs残留量要明显低于未投菌组A,B组湿地出水中E1、E2、E3和EE2含量相比A组可分别降低约26.6%、17.1%、30.3%、13.3%。表明HD菌剂能强化耕作型稻田湿地土壤中EDCs的降解。本研究对于村落水环境中的EDCs降解机制及环境生态影响评价有一定的参考价值。
-
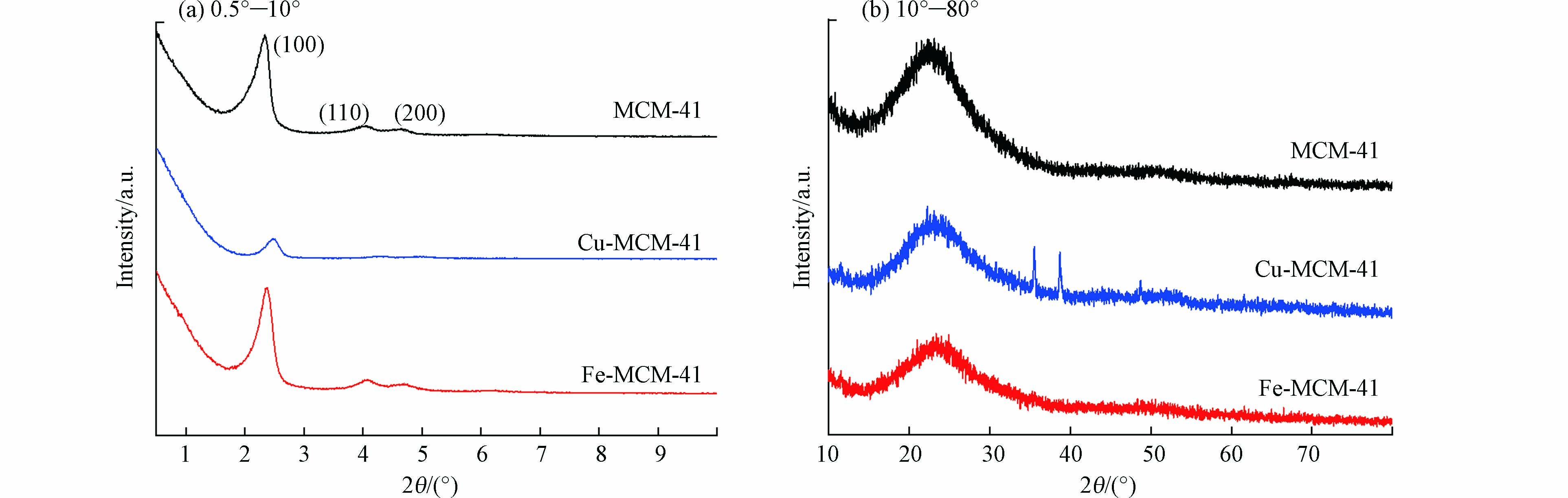
图 1 MCM-41、Fe-MCM-41和Cu-MCM-41的XRD图谱
Figure 1. XRD patterns of MCM-41,Fe-MCM-41 and Cu-MCM-41

图 2 MCM-41(a)、Fe-MCM-41(b)和Cu-MCM-41(c)的SEM图及Fe-MCM-41(d)和Cu-MCM-41(e)的mapping照片
Figure 2. SEM images of MCM-41(a),Fe-MCM-41(b),Cu-MCM-41(c)and mapping images of Fe-MCM-41(d)and Cu-MCM-41(e)

图 3 MCM-41(a、d),Fe-MCM-41(b、e),Cu-MCM-41(c、f)的TEM照片
Figure 3. TEM images of MCM-41(a, d),Fe-MCM-41(b, e) and Cu-MCM-41(c, f)
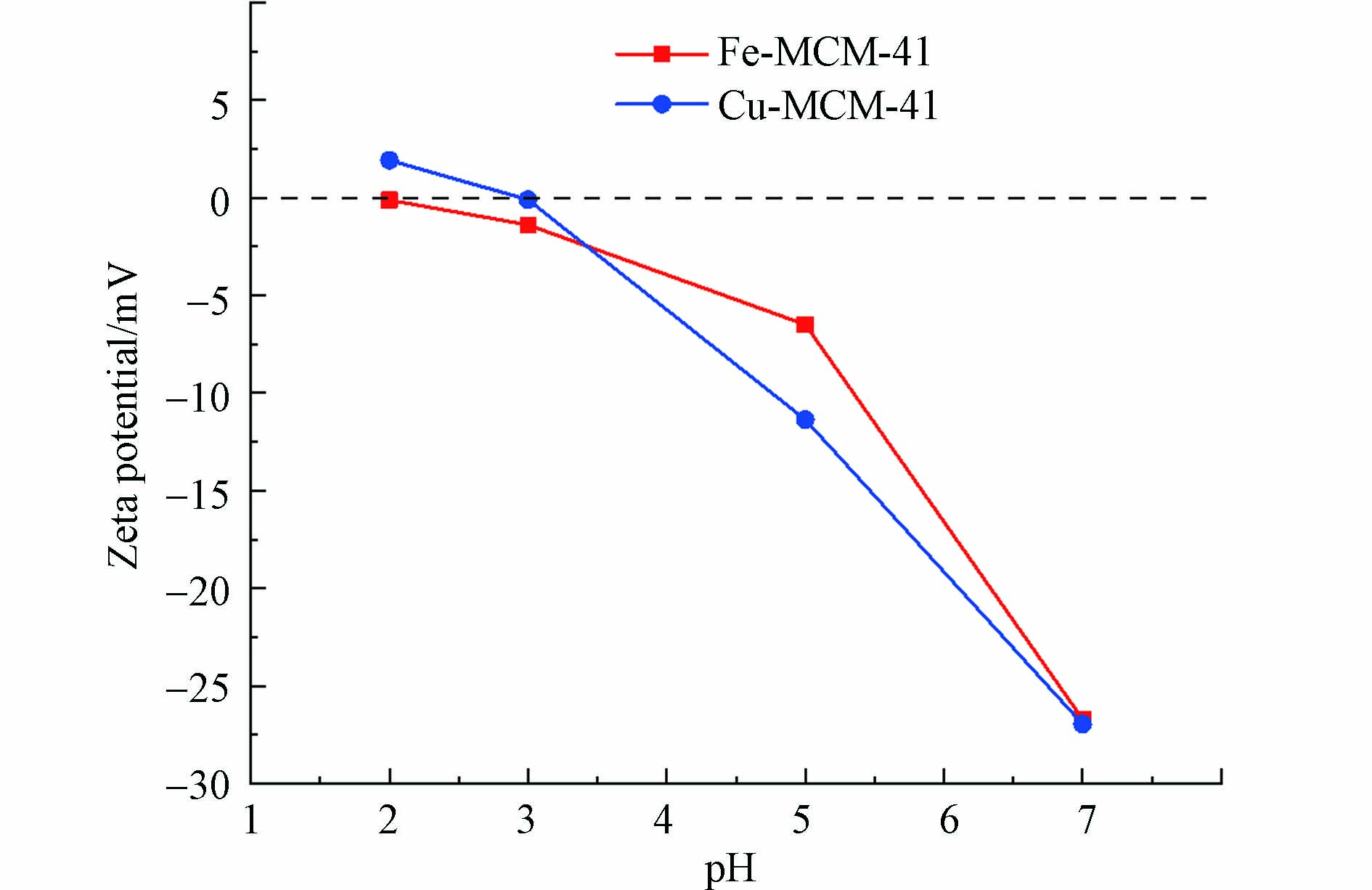
图 5 Fe-MCM-41和Cu-MCM-41在不同pH下的zeta电位
Figure 5. Zeta potential of Fe-MCM-41 and Cu-MCM-41 at different pH values
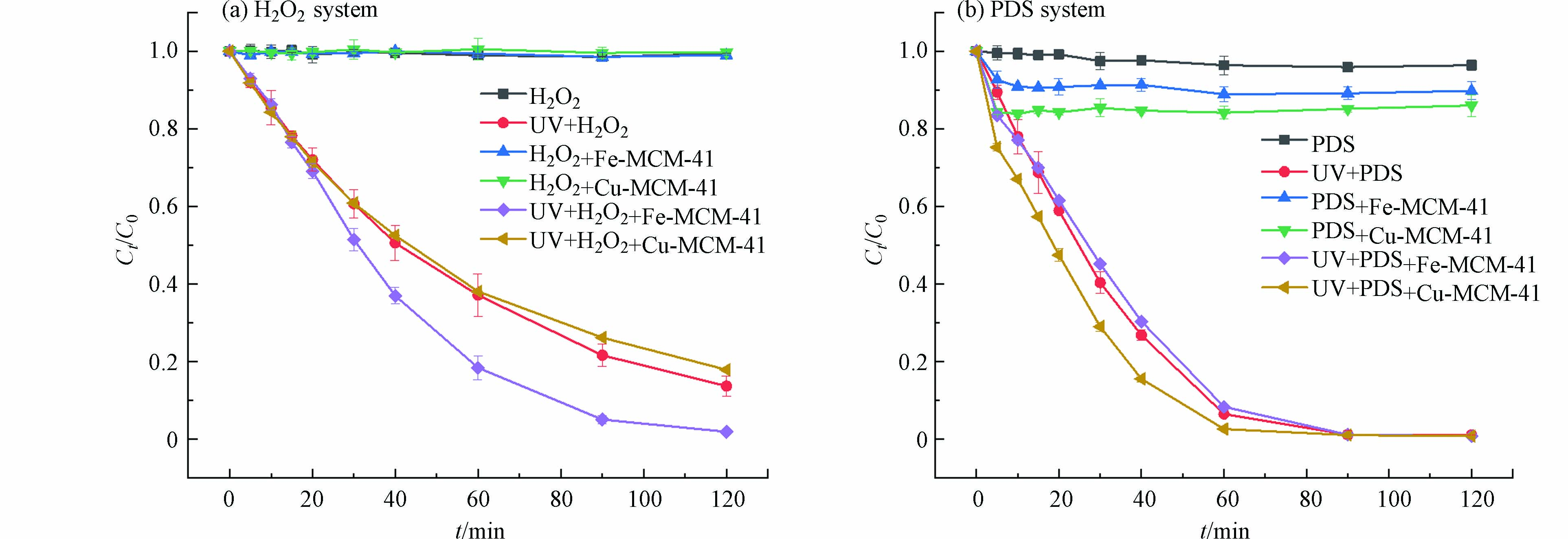
图 6 不同体系下金橙Ⅱ的脱色情况([Orange Ⅱ]=0.2 mmol∙L−1, pH=6.8, T=30 ℃, 催化剂=1 g·L−1, 紫外灯功率=5 W, H2O2或PDS =10 mmol·L−1)
Figure 6. Discoloration of Orange Ⅱ under different systems
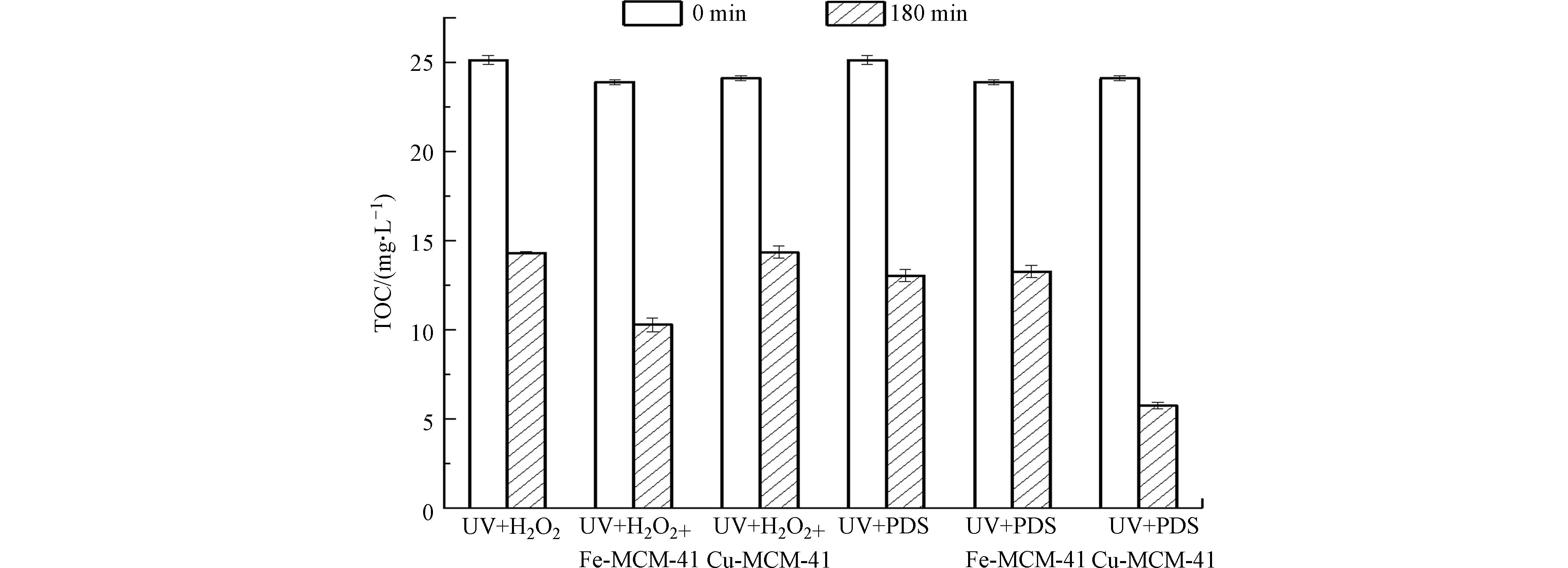
图 7 反应前后不同体系反应液的TOC值
Figure 7. TOC values of reactive liquids in different systems before and after the reaction
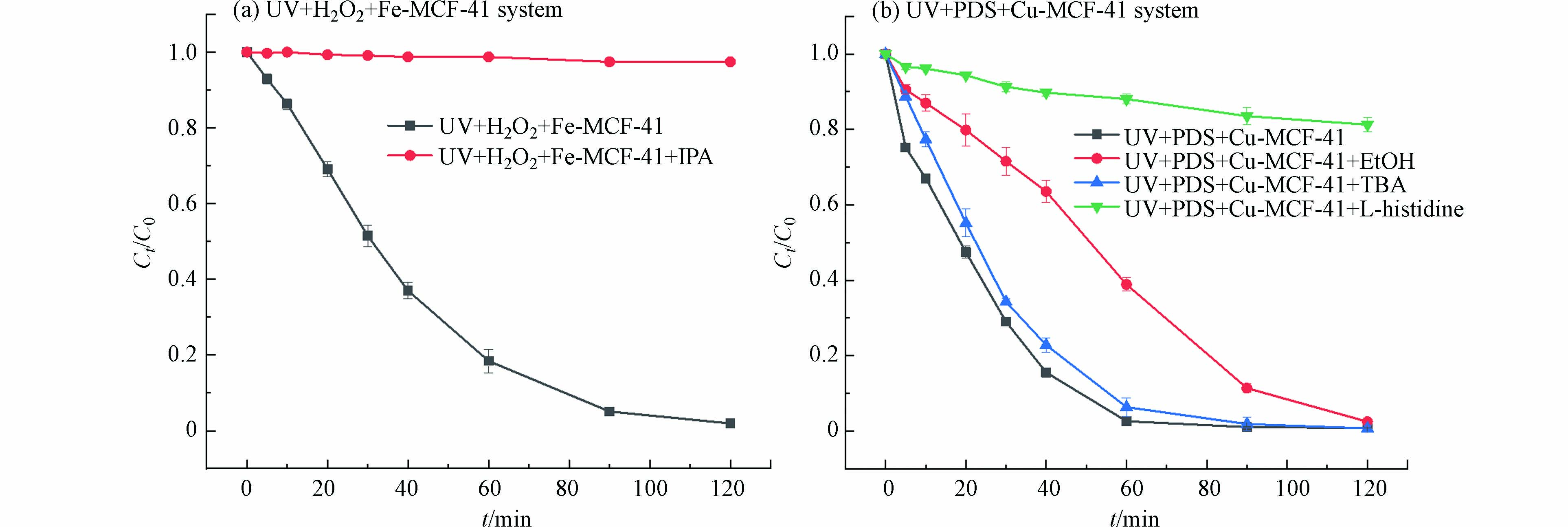
图 8 不同体系中淬灭剂对金橙Ⅱ脱色的影响
Figure 8. Effect of quencher on the degradation of Orange Ⅱ under different systems
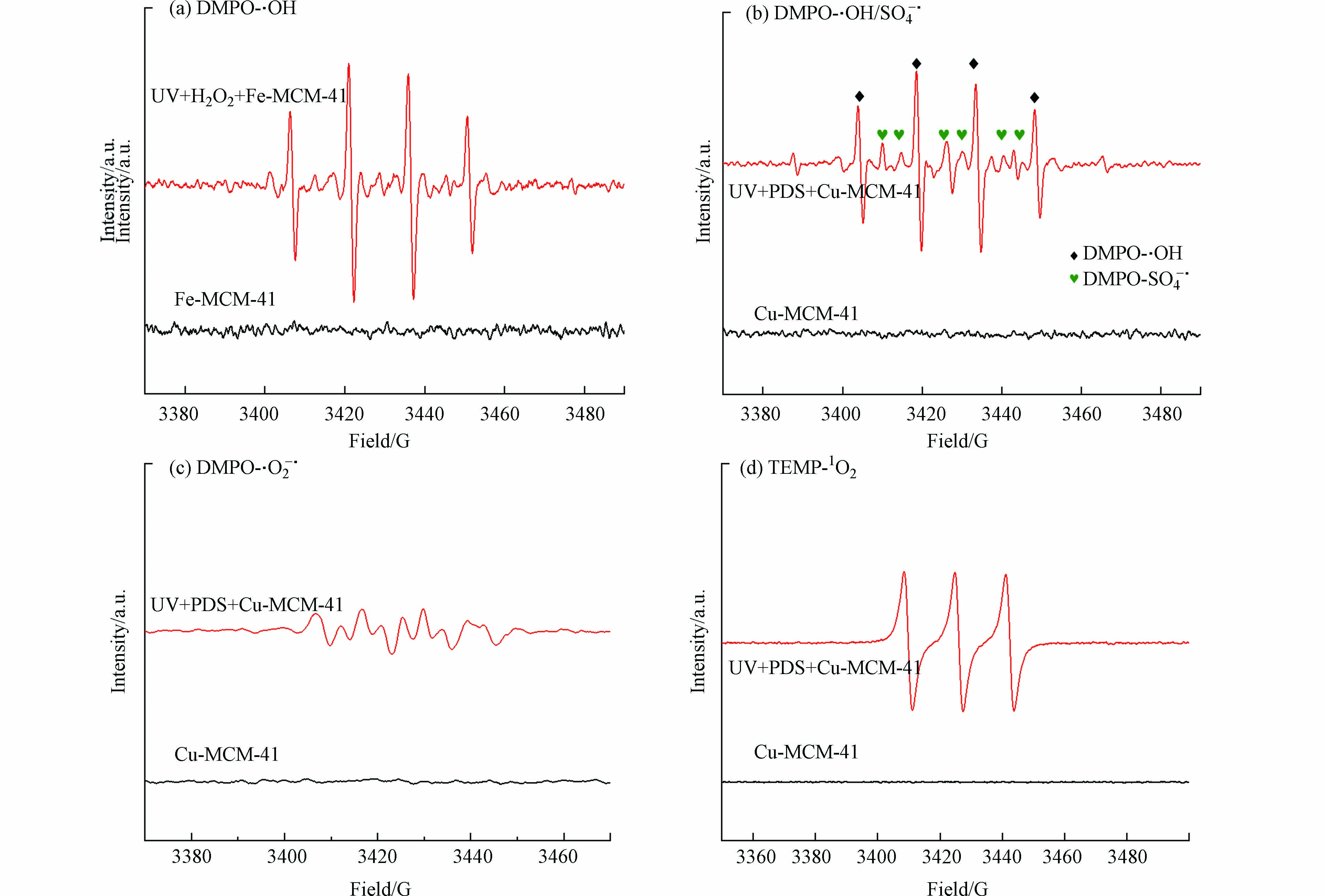
图 9 DMPO-·OH/SO4−·/O2−·和TEMP-1O2的EPR图谱
Figure 9. EPR spectra of DMPO-·OH/SO4−·/O2−· and TEMP-1O2
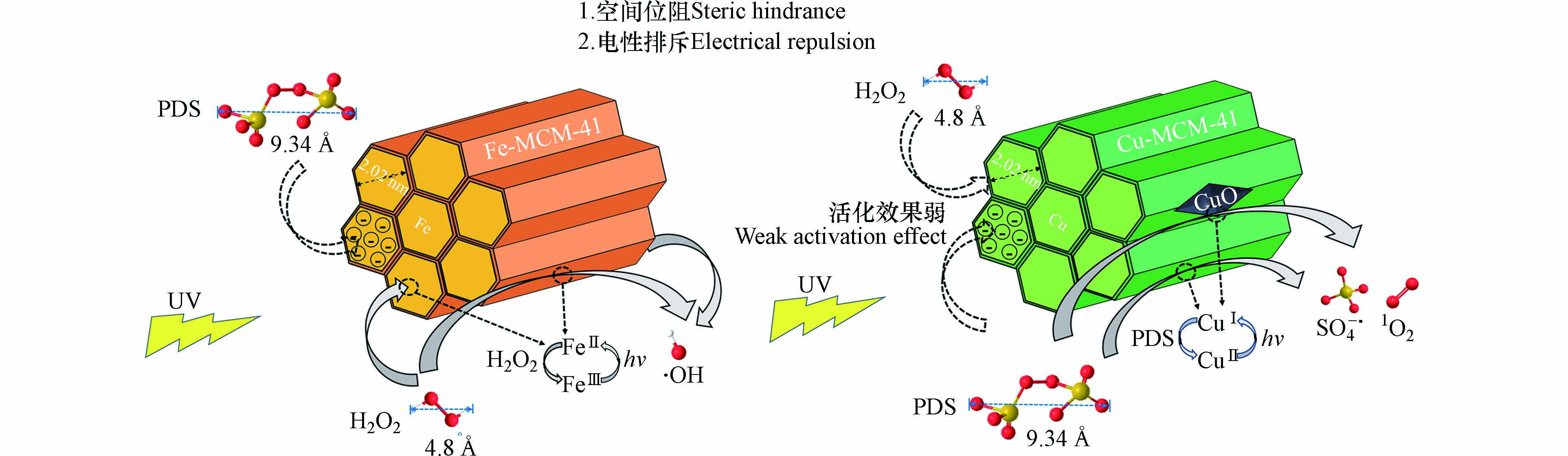
图 10 Fe-MCM-41和Cu-MCM-41光催化活化H2O2和PDS的构-效关系
Figure 10. Structure-activity relationship of Fe-MCM-41 and Cu-MCM-41 on photocatalytic activation of H2O2 and PDS
表 1 不同体系中金橙Ⅱ脱色的一级动力学常数
Table 1. The first order kinetics of Orange Ⅱ degradation under different systems
反应体系Reaction system k/min−1 R2 UV+H2O2 0.017 1.000 UV+H2O2+Fe-MCM-41 0.026 0.981 UV+H2O2+Cu-MCM-41 0.015 0.997 UV+PDS 0.031 0.991 UV+PDS+Fe-MCM-41 0.028 0.993 UV+PDS+Cu-MCM-41 0.043 0.992
下载: 导出CSV
-
[1] SARAVANAN A, DEIVAYANAI V C, KUMAR P S, et al. A detailed review on advanced oxidation process in treatment of wastewater: Mechanism, challenges and future outlook[J]. Chemosphere, 2022, 308: 136524. doi: 10.1016/j.chemosphere.2022.136524 [2] WANG L Y, LUO D, YANG J P, et al. Metal-organic frameworks-derived catalysts for contaminant degradation in persulfate-based advanced oxidation processes[J]. Journal of Cleaner Production, 2022, 375: 134118. doi: 10.1016/j.jclepro.2022.134118 [3] 钟欣, 吴迪, 张凯欣, 等. 光助Fe/BiOCl活化过硫酸盐降解橙黄Ⅱ[J]. 环境化学, 2019, 38(12): 2860-2868. doi: 10.7524/j.issn.0254-6108.2019070806 ZHONG X, WU D, ZHANG K X, et al. Photo-assisted activation of persulfate by using Fe/BiOCl for the degradation of azo dye Orange Ⅱ[J]. Environmental Chemistry, 2019, 38(12): 2860-2868 (in Chinese). doi: 10.7524/j.issn.0254-6108.2019070806
[4] 张艺伟, 卫培垚, 陈建新, 等. 可见光下苯醌类化合物诱导磷酸铁芬顿反应的铁离子源汇机制[J]. 环境化学, 2023, 42(2): 635-645. doi: 10.7524/j.issn.0254-6108.2021102005 ZHANG Y W, WEI P Y, CHEN J X, et al. Iron-ion source and sink mechanism for Fenton reaction based on iron phosphate induced by benzoquinones under visible light[J]. Environmental Chemistry, 2023, 42(2): 635-645 (in Chinese). doi: 10.7524/j.issn.0254-6108.2021102005
[5] ZHAO J J, SUN Y J, ZHANG Y, et al. Heterogeneous activation of persulfate by activated carbon supported iron for efficient amoxicillin degradation[J]. Environmental Technology & Innovation, 2021, 21: 101259. [6] LAM F L Y, YIP A C K, HU X J. Copper/MCM-41 as a highly stable and pH-insensitive heterogeneous photo-Fenton-like catalytic material for the abatement of organic wastewater[J]. Industrial & Engineering Chemistry Research, 2007, 46(10): 3328-3333. [7] HUANG T Y, CHEN J B, WANG Z M, et al. Excellent performance of cobalt-impregnated activated carbon in peroxymonosulfate activation for acid orange 7 oxidation[J]. Environmental Science and Pollution Research, 2017, 24(10): 9651-9661. doi: 10.1007/s11356-017-8648-7 [8] 孙文静, 王亚旻, 卫皇曌, 等. Fe-MCM-41催化臭氧氧化间甲酚废水[J]. 环境科学, 2015, 36(4): 1345-1351. SUN W J, WANG Y M, WEI H Z, et al. Degradation of m-cresol with Fe-MCM-41 in catalytic ozonation[J]. Environmental Science, 2015, 36(4): 1345-1351 (in Chinese).
[9] ALAMGHOLILOO H, NAZARI S, ASGARI E, et al. Facile fabrication of Z-scheme TiO2/ZnO@MCM-41 heterojunctions nanostructures for photodegradation and bioactivity performance[J]. Journal of Molecular Liquids, 2022, 364: 119990. doi: 10.1016/j.molliq.2022.119990 [10] LIU D P, LIN M X, CHEN W R, et al. Enhancing catalytic ozonation activity of MCM-41 via one-step incorporating fluorine and iron: The interfacial reaction induced by hydrophobic sites and Lewis acid sites[J]. Chemosphere, 2022, 292: 133544. doi: 10.1016/j.chemosphere.2022.133544 [11] SCHLICHTER S, DENNEHY M, ALVAREZ M. Activation of peroxymonosulfate and persulfate by metal loaded mesoporous catalysts for orange G dye degradation[J]. Environmental Processes, 2019, 6(4): 805-818. doi: 10.1007/s40710-019-00389-4 [12] SUN X W, XU D Y, DAI P, et al. Efficient degradation of methyl orange in water via both radical and non-radical pathways using Fe-Co bimetal-doped MCM-41 as peroxymonosulfate activator[J]. Chemical Engineering Journal, 2020, 402: 125881. doi: 10.1016/j.cej.2020.125881 [13] DENG Y X, XU X D, WANG R, et al. Characterization and photocatalytic evaluation of Fe-loaded mesoporous MCM-41 prepared using iron and silicon sources extracted from iron ore tailing[J]. Waste and Biomass Valorization, 2020, 11(4): 1491-1498. doi: 10.1007/s12649-018-0460-1 [14] SCHLICHTER S, SAPAG K, DENNEHY M, et al. Metal-based mesoporous materials and their application as catalysts for the degradation of methyl orange azo dye[J]. Journal of Environmental Chemical Engineering, 2017, 5(5): 5207-5214. doi: 10.1016/j.jece.2017.09.039 [15] MUTO S, IMAI H. Relationship between mesostructures and pH conditions for the formation of silica-cationic surfactant complexes[J]. Microporous and Mesoporous Materials, 2006, 95(1/2/3): 200-205. [16] 范钧朝, 陈爱因, 陈诗, 等. 过渡金属Fe、Co、Ni介孔分子筛MCM-41催化剂的制备及其氧化性能[J]. 环境化学, 2016, 35(6): 1116-1124. doi: 10.7524/j.issn.0254-6108.2016.06.2015102201 FAN J Z, CHEN A Y, CHEN S, et al. Synthesis of Fe, Co, Ni loaded MCM-41 mesoporous molecular sieves and their catalytic oxidation performance[J]. Environmental Chemistry, 2016, 35(6): 1116-1124 (in Chinese). doi: 10.7524/j.issn.0254-6108.2016.06.2015102201
[17] 杨欢欢. 全氟羧酸化合物降解机理的DFT研究[D]. 镇江: 江苏科技大学, 2019. YANG H H. DFT study on the degradation mechanism of perfluorocarboxylic acid compounds[D]. Zhenjiang: Jiangsu University of Science and Technology, 2019 (in Chinese).
[18] LEE J, von GUNTEN U, KIM J H. Persulfate-based advanced oxidation: Critical assessment of opportunities and roadblocks[J]. Environmental Science & Technology, 2020, 54(6): 3064-3081. [19] 刘路明, 高志敏, 邓兆雄, 等. 过硫酸盐的活化及其在氧化降解水中抗生素的机理和应用[J]. 环境化学, 2022, 41(5): 1702-1717. doi: 10.7524/j.issn.0254-6108.2021010601 LIU L M, GAO Z M, DENG Z X, et al. Activation of persulfate and its mechanism and application in oxidative degradation of antibiotics in water[J]. Environmental Chemistry, 2022, 41(5): 1702-1717 (in Chinese). doi: 10.7524/j.issn.0254-6108.2021010601
[20] MELÉNDEZ-ORTIZ H I, PERERA-MERCADO Y, MERCADO-SILVA J A, et al. Functionalization with amine-containing organosilane of mesoporous silica MCM-41 and MCM-48 obtained at room temperature[J]. Ceramics International, 2014, 40(7): 9701-9707. doi: 10.1016/j.ceramint.2014.02.051 [21] 苗笑增, 戴慧旺, 陈建新, 等. 草酸根对α-FeOOH多相UV-Fenton催化能力的增效实验[J]. 环境科学, 2018, 39(3): 1202-1211. MIAO X Z, DAI H W, CHEN J X, et al. Experiment to enhance catalytic activity of α-FeOOH in heterogeneous UV-Fenton system by addition of oxalate[J]. Environmental Science, 2018, 39(3): 1202-1211 (in Chinese).
[22] FENG J Y, HU X J, YUE P L. Discoloration and mineralization of Orange II using different heterogeneous catalysts containing Fe: A comparative study[J]. Environmental Science & Technology, 2004, 38(21): 5773-5778. [23] GRÜN M, UNGER K K, MATSUMOTO A, et al. Novel pathways for the preparation of mesoporous MCM-41 materials: Control of porosity and morphology[J]. Microporous and Mesoporous Materials, 1999, 27(2/3): 207-216. [24] SHEN S H, CHEN J, KOODALI R T, et al. Activation of MCM-41 mesoporous silica by transition-metal incorporation for photocatalytic hydrogen production[J]. Applied Catalysis B: Environmental, 2014, 150/151: 138-146. doi: 10.1016/j.apcatb.2013.12.014 [25] 张安超, 张洪良, 宋军, 等. Mn-Co/MCM-41吸附剂表征及脱除烟气中单质汞研究[J]. 中国环境科学, 2015, 35(5): 1319-1327. ZHANG A C, ZHANG H L, SONG J, et al. Characterization and performance of Mn-Co/MCM-41 for elemental mercury removal from simulated flue gas[J]. China Environmental Science, 2015, 35(5): 1319-1327 (in Chinese).
[26] LAN B Y, HUANG R H, LI L S, et al. Catalytic ozonation of p-chlorobenzoic acid in aqueous solution using Fe-MCM-41 as catalyst[J]. Chemical Engineering Journal, 2013, 219: 346-354. doi: 10.1016/j.cej.2012.12.083 [27] SONG T H, LI G Q, HU R H, et al. Degradation of antibiotics via UV-activated peroxodisulfate or peroxymonosulfate: A review[J]. Catalysts, 2022, 12(9): 1025. doi: 10.3390/catal12091025 [28] 龙学军. 降解染料废水类Fenton新体系构建及机理研究[D]. 武汉: 武汉大学, 2015. LONG X J. A study on the construction and mechanism of the new Fenton-like system for degradation of dye waste water[D]. Wuhan: Wuhan University, 2015 (in Chinese).
[29] ZHANG T, CHEN Y, WANG Y R, et al. Efficient peroxydisulfate activation process not relying on sulfate radical generation for water pollutant degradation[J]. Environmental Science & Technology, 2014, 48(10): 5868-5875. [30] 韩仪, 黄明杰, 周涛, 等. 氧化铜活化过硫酸盐的界面反应机理[J]. 环境化学, 2020, 39(3): 735-744. doi: 10.7524/j.issn.0254-6108.2019110101 HAN Y, HUANG M J, ZHOU T, et al. Interfacial reaction mechanism of copper oxide activating persulfate[J]. Environmental Chemistry, 2020, 39(3): 735-744 (in Chinese). doi: 10.7524/j.issn.0254-6108.2019110101
[31] 苏海英, 王盈霏, 王枫亮, 等. g-C3N4/TiO2复合材料光催化降解布洛芬的机制[J]. 中国环境科学, 2017, 37(1): 195-202. SU H Y, WANG Y F, WANG F L, et al. Preparation of g-C3N4/TiO2 composites and the mechanism research of the photocatalysis degradation of ibuprofen[J]. China Environmental Science, 2017, 37(1): 195-202 (in Chinese).
[32] SUN H Q, KWAN C, SUVOROVA A, et al. Catalytic oxidation of organic pollutants on pristine and surface nitrogen-modified carbon nanotubes with sulfate radicals[J]. Applied Catalysis B: Environmental, 2014, 154/155: 134-141. doi: 10.1016/j.apcatb.2014.02.012 [33] WANG Q, ZHOU D M, LIN K F, et al. Carbon nitride-based cuprous catalysts induced nonradical-led oxidation by peroxydisulfate: Role of cuprous and dissolved oxygen[J]. Chemical Engineering Journal, 2021, 419: 129667. doi: 10.1016/j.cej.2021.129667 [34] LEE H, LEE H J, SEO J, et al. Activation of oxygen and hydrogen peroxide by copper(II) coupled with hydroxylamine for oxidation of organic contaminants[J]. Environmental Science & Technology, 2016, 50(15): 8231-8238. [35] 杨奕飞, 杨天学, 吴代赦, 等. 改性沼渣生物质炭活化过硫酸盐降解酚类性能[J]. 中国环境科学, 2022, 42(5): 2153-2160. YANG Y F, YANG T X, WU D S, et al. Study on the performance of modified biogas residue biomass charcoal to activate persulfate to degrade phenols[J]. China Environmental Science, 2022, 42(5): 2153-2160 (in Chinese).
[36] 雷倩, 许路, 艾伟, 等. CDs-BOC复合催化剂可见光下活化过硫酸盐降解典型PPCPs[J]. 环境科学, 2021, 42(6): 2885-2895. LEI Q, XU L, AI W, et al. CDs-BOC nanophotocatalyst activating persulfate under visible light for the efficient degradation of typical PPCPs[J]. Environmental Science, 2021, 42(6): 2885-2895 (in Chinese).
[37] KHAN A, ZHANG K K, SUN P, et al. High performance of the A-Mn2O3 nanocatalyst for persulfate activation: Degradation process of organic contaminants via singlet oxygen[J]. Journal of Colloid and Interface Science, 2021, 584: 885-899. doi: 10.1016/j.jcis.2020.10.021 [38] LI Y, LI L, CHEN Z X, et al. Carbonate-activated hydrogen peroxide oxidation process for azo dye decolorization: Process, kinetics, and mechanisms[J]. Chemosphere, 2018, 192: 372-378. doi: 10.1016/j.chemosphere.2017.10.126 [39] MAEZONO T, TOKUMURA M, SEKINE M, et al. Hydroxyl radical concentration profile in photo-Fenton oxidation process: Generation and consumption of hydroxyl radicals during the discoloration of azo-dye Orange Ⅱ[J]. Chemosphere, 2011, 82(10): 1422-1430. doi: 10.1016/j.chemosphere.2010.11.052 [40] ZHOU P, ZHANG J, LIANG J, et al. Activation of persulfate/copper by hydroxylamine via accelerating the cupric/cuprous redox couple[J]. Water Science and Technology2016, 73(3): 493-500. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1515
- HTML全文浏览数: 1515
- PDF下载数: 85
- 施引文献: 0
