




-
在众多的污水处理方法中,活性污泥法受到人们的广泛关注,活性污泥法作为重要的处理污水方法之一,具有很多优势. 但是随着国内外对污水治理的日益重视和城市污水处理厂的不断建设,大量的剩余污泥作为活性污泥法处理污水的副产物排出[1]. 污泥因其含水率高、含有大量病原体和微生物等有害生物、重金属及有机物含量高等特点,容易对环境造成二次污染[2],污泥的有效处理处置是亟待解决的重要问题. 污泥脱水是常规的污泥处理方法,在污泥脱水之前需要经过一定的调理使其满足后续脱水要求,所以,选择合适的污泥调理方法对改善污泥脱水性能尤为重要.
过氧化钙(CaO2)作为一种热稳定性好的环境友好型材料,被广泛应用于农业种植、水产养殖、食品保存、医疗以及环境领域[3]. CaO2具有高能的过氧化物共价键,当CaO2与水接触时,能够缓慢释放过氧化氢(H2O2),同时还会生成羟基自由基、过氧化氢自由基等具有强氧化性的自由基(反应式见式(1—5))[4]. 近年来,因其具有稳定的氧化性,CaO2在污泥处理方面的应用成为一个新的研究热点. Wang 等研究发现,通过CaO2预处理污泥后,难降解有机物可以转化为可生物降解,促进污泥中可生物降解基质的水解和分解代谢,进而增强污泥厌氧消化效果[5]. 有研究表明,CaO2可以破解污泥EPS结构,释放污泥中的束缚水[6]. Wang等的研究表明,通过联合CaO2和微波预处理污泥,预处理后污泥的CST值相较于原泥下降52% [7]. 通过热处理与CaO2联合调理,可以提升污泥脱水性能[8].
除了直接使用CaO2对目标物进行氧化,对CaO2进行活化也是一种常用的技术[8]. 有研究认为,通过微波活化CaO2,能促进CaO2产生更多的HO·和·O2-[7]. 通过过渡金属(Fe2+/Fe3+和Ag+)活化CaO2分解是常用的活化方法[9]. 利用Fe2+活化CaO2可以形成类芬顿反应,但如果不进行pH调节, Fe2+易于被氧化成Fe3+,限制了芬顿反应的效率. 有研究指出,利用含铁矿物对H2O2进行活化可以克服这一缺陷[10]. 黄铁矿(FeS2)是一种常见的脉石矿物,与矿床中的有价矿物伴生,可通过常规浮选方法轻松处理[11]. 最近有研究发现,利用黄铁矿活化CaO2降解磺胺,相比常规的芬顿反应,磺胺的氧化效率从30%提升至80%,(主要反应见式(6—9))[12]. Zhou等研究表明利用黄铁矿活化CaO2处理邻苯二甲酸二乙酯(DEP),78%的DEP在24 h内被降解[13]. 这些结果说明,通过黄铁矿活化CaO2能有效促进HO·产生,但目前尚未发现关于利用黄铁矿活化过氧化钙调理污泥的研究,其对污泥脱水性能的影响及机理尚未清晰,因此本研究利用黄铁矿-CaO2作为一种新型的芬顿法对污泥进行调理,以期达到破解EPS从而释放结合水的效果,并通过EPS性质及污泥絮体性质变化探究其对污泥脱水性能的影响机理.
本研究对不同污泥样品进行EPS的提取,并对提取出来的EPS样品进行含量测定、三维荧光光谱检测,以表征调理前后污泥EPS性质变化. 同时对不同污泥样品的粒径分布进行检测,探究调理方法对污泥絮体团聚性能变化的影响.
-
本研究中污泥取自于广州市某污水处理厂二沉池,污泥取至实验室后,先过20目筛,去除大颗粒杂质和毛发,之后置于冰箱在4 ℃下保存. CaO2采购于上海麦克林生化科技有限公司. 黄铁矿采购于佛山市大昌顺材料科技有限公司,黄铁矿在使用之前对其进行研磨,并过100目筛,利用0.1 mol·L−1HNO3 洗去表面杂质及氧化层,干燥后备用[14].
-
为了探究不同调理条件对污泥脱水性能的影响,本研究对黄铁矿单独调理、CaO2单独调理以及两者复合调理污泥进行实验室规模的污泥脱水性能实验,250 mL的烧杯作为污泥调理容器,在调理容器中加入100 mL污泥样品进行实验. 利用重量法对污泥总固体(TS)进行测定[15]. 在黄铁矿单独调理实验中,设置6组不同黄铁矿调理剂量实验组,各组黄铁矿投加量分别为0、1、2、4、6 g·L−1. CaO2单独调理实验中,设置6组不同CaO2调理剂量实验组,各组CaO2投加量分别为10、30、50、80、100 mg·g−1 TS. 为了研究单独调理与复合调理以及不同复合调理方法之间的污泥脱水性能变化,设置了两组复合调理实验,第一组:CaO2投加量30 mg·g−1 TS,黄铁矿投加剂量1 g·L−1,第二组:CaO2投加量100 mg·g−1 TS,黄铁矿投加剂量1 g·L−1. 将单独调理和复合调理的实验组分别设置为A30、A100和B30、B100. 其中,A30为30 mg·g−1 TS CaO2单独调理,B30为30 mg·g−1 TS CaO2 +1 g·L−1黄铁矿复合调理,A100为100 mg·g−1 TS CaO2单独调理,B100为100 mg·g−1 TS CaO2 +1 g·L−1黄铁矿复合调理.
-
本研究中利用毛细吸水时间(CST)作为评价污泥脱水性能的指标. CST利用CST测定仪进行测定(HDFC-10A),利用测定后CST数据进行标准化CST(SCST)计算[16],计算公式如下:
其中,CSTa为调理后污泥样品的CST值,CST0为原泥的CST值.
-
在本研究中,EPS根据其存在形态分类为溶解性EPS(S-EPS)、松散束缚EPS(LB-EPS)和紧密束缚EPS(TB-EPS)[17],本研究采用一种改进的热提取方式对EPS进行提取,具体方法参照文献[18]. EPS中的多糖含量利用硫酸-蒽酮法测定,蛋白质含量利用福林酚法进行测定[19].
-
本研究中利用荧光光谱仪(Hitachi F-4600)对提取出的EPS进行3D-EEM的测定,光谱数据的发射波长(Em)以及激发波长(Ex)范围从220 nm到450 nm,采集间隔为10 nm. 光谱数据的利用5 nm的发射和激发狭缝带宽以及1500 nm·min−1的扫描速度进行收集.
-
本研究利用激光粒度仪(Mastersize 3000)对污泥絮体粒径分布及絮体粒径D50和D90值的测定. 其中,D50与D90分别定义为颗粒直径的第50和第90百分位数[20].
-
由图1可见,单独投加CaO2之后,污泥SCST值随着CaO2的投加量的增加呈现先下降再上升的趋势,单独投加CaO2,投加量为30 mg·g−1 TS的实验组SCST值最低为0.61. 在投加剂量不高于80 mg·g−1 TS时,CaO2单独调理有利于提升脱水性能,但当CaO2投加量增加至100 mg·g−1 TS时,SCST值增加至1.39,说明过量的CaO2不仅不会提升污泥脱水性能,反而会使得原污泥脱水性能下降. 随着黄铁矿投加量增加,黄铁矿单独调理的SCST值也表现出先下降再上升的,最优黄铁矿单独调理剂量为1 g·L−1,SCST值为0.70. 但当投加量继续增加时,黄铁矿单独调理对污泥脱水性能的提升效果变弱,在投加量为6 g·L−1的单独调理下,SCST值为0.92,污泥脱水性能提升不明显. 这说明过量的过氧化钙投加,带来过强的氧化性能,会使得污泥的脱水性能下降,这一趋势与Chen等的研究结果相似,过强的氧化性可能会导致过量的EPS释放,降低污泥脱水性能[6]. 但在CaO2投加量为30 mg·g−1 TS复合调理时,虽然氧化性能更强,但污泥有更佳的脱水性能,SCST值下降至0.55,这说明利用黄铁矿活化过氧化钙对污泥进行复合调理能有效提升污泥的脱水性能.
-
不同结构的EPS对剩余污泥的脱水性能影响程度可能不同,Dai等认为S-EPS中有机物含量较高或LB-EPS中有机物含量较低,具有较好的脱水性能[21]. He等指出污泥脱水性与S-EPS中有机物浓度呈正相关,而与LB-EPS中生物聚合物含量呈负相关[22]. 剩余污泥脱水性能除了和EPS的组成结构有关,还与EPS的组成成分相关,Wei等研究发现,污泥脱水性能与EPS中蛋白质含量呈负相关性[23],而且蛋白质含量是决定污泥脱水性能的关键因素[24],为了进一步探究污泥调理过程中污泥性质的变化,本研究对提取出的EPS样品进行蛋白质和多糖含量的测定. CaO2调理后污泥EPS结构发生明显的变化(图2a),在30 mg·g−1 TS的CaO2投加量下,S-EPS蛋白质含量略有下降,而内层EPS(LB-EPS、TB-EPS)蛋白质含量增加,相较于单独调理,CaO2/黄铁矿复合调理由于其更强的氧化性能,在CaO2投加量为30 mg·g−1 TS时的复合调理污泥样品中,内层EPS蛋白质含量增加幅度更大. 当CaO2投加量增加至100 mg·g−1 TS后,所有层EPS中蛋白质含量均增加,与低CaO2投加量相似,复合调理因其更强的氧化性,内部EPS含量较单独调理增加更多. 调理后污泥的总EPS(T-EPS)蛋白质含量均增加,高剂量CaO2导致更多的蛋白质释放,而复合调理对蛋白质含量的提升高于单独调理.
调理前后EPS多糖含量的变化见图2b,随着CaO2投加量增加,内外层EPS多糖含量均增加. 值得注意的是,高CaO2投加剂量的复合调理样品中,S-EPS和LB-EPS的多糖含量较单独调理均下降. T-EPS中多糖的变化趋势与蛋白质不同,T-EPS中多糖含量随着氧化性能的增强表现出先增加后下降的趋势,这可能是低CaO2剂量调理下,EPS结构被破解,内层EPS释放至外层. 但在高剂量CaO2的复合调理下,多糖类物质可能被分解为更小的有机分子或直接被矿化,导致T-EPS中多糖含量下降.
有研究认为,LB-EPS中蛋白质/多糖比率(PN/PS)与脱水性有负相关性[25]. 本实验中,B30样品LB-EPS的PN/PS最小(图2c),且无论高剂量或低剂量,在同一剂量下复合调理得到的LB-EPS样品,其PN/PS值均小于单独调理. 但当用高剂量过氧化钙对污泥进行调理后,LB-EPS中的PN/PS上升,污泥脱水性能下降. 但本实验发现,高剂量的过氧化钙调理后虽然PN/PS上升,但仍然低于原泥,这与脱水性能变化不一致,这是因为污泥脱水性能的变化影响十分复杂,并不能只靠EPS中的PN/PS进行指示.
从EPS含量变化可以看出,使用CaO2单独调理以及CaO2/黄铁矿复合调理都可以改变EPS原有结构,破解EPS结构. 在同一CaO2投加量下,复合调理得到的EPS破解效果更加明显. 结合污泥脱水结果分析,污泥调理方法在一定范围内对EPS结构进行破解,可能有利于污泥脱水性能的提升,但对EPS结构的过度破解可能会使得大量有机质的释放,进而使得污泥脱水性能下降.
-
为了更深入地了解调理前后以及各调理方法对各层EPS的性质以及其含量的影响,本研究利用三维荧光光谱对各层EPS的有机成分进行表征,各样品EPS的三维荧光光谱见图3. 本研究中EPS的荧光光谱峰主要有两个,分别为A峰(Em/Ex:340 nm/225 nm)和B峰(Em/Ex:350 nm/280 nm). 根据Wen等提出的三维荧光光谱分区方法,A峰位于区域Ⅱ,归类为芳香类蛋白物质,B峰位于区域Ⅳ,归类为色氨酸和类蛋白物质[26].
A峰在原泥S-EPS中强度较低,但经过调理后,A峰强度上升,芳香类蛋白含量增加. 在A100中,S-EPS中的A峰出现最强的荧光强度,说明在此调理方法下内层EPS和胞内的芳香类蛋白向外释放,聚集在外层EPS中. 但经过氧化性更强的B100调理后,A峰强度下降,这可能是由于芳香类蛋白的分解导致含量下降. S-EPS中B峰的荧光强度在A30和B30调理下均下降,当CaO2投加量增加后,S-EPS的B峰强度增加,S-EPS中B峰最强峰强度出现在B100调理下. 原泥中LB-EPS中A峰和B峰强度稍强于S-EPS,经过预处理后污泥LB-EPS中A、B峰强度增加,且两峰强度的增加幅度明显大于S-EPS. 不同调理方法对LB-EPS的荧光光谱图影响与S-EPS相似,A、B峰在B100调理下均出现最强荧光强度. 原泥TB-EPS中的芳香类蛋白和色氨酸含量明显高于S-EPS和LB-EPS,这一结果与EPS含量一致. 不同调理手段下B峰强度在TB-EPS中的变化与在S-EPS、LB-EPS中的变化相似,B峰在A100调理下出现最大荧光强度,随后下降. 但与 S-EPS、LB-EPS 变化趋势不一致的是,TB-EPS 中 A 峰的最大荧光强度出现在 B30 调理下, 这一结果说明,芳香类蛋白比色氨酸更易于从胞内和内层 EPS 释放至胞外和外层 EPS.
荧光峰强度变化趋势可以说明,在一定条件下,随着调理方法的氧化性的增强,EPS中物质被分解,EPS结构破解程度增加,胞内物质向TB-EPS转移,同时TB-EPS中的物质向外层的LB-EPS和S-EPS转移. 当调理方法氧化性能过强,各层EPS中物质被分解甚至矿化,导致各层EPS中荧光峰强度下降,同时还发现,各层EPS中不同物质对于不同调理方法的变化趋势并不完全相同.
-
由图4a可以看出,经过调理后的污泥絮体粒径分布曲线均向左移动,同时图4b中看到原泥有最大的D90以及D50值,调理后污泥的D50以及D90均有明显的下降,说明调理后污泥的絮体粒径下降. 这是由于强氧化性的调理方法将EPS结构破解后,会使得污泥絮体分解,形成尺寸更小的絮体[27]. 随着调理方法的氧化性能增强,污泥的粒径分布曲线左移程度越大,且有更小的D50和D90值,可以认为氧化性能越强的调理方法能够更高效、更彻底地破坏原有污泥絮体结构,使得原有稳定的大颗粒絮体失稳进而形成众多小尺寸的絮体. 这一现象与Ling等研究结果一致,通过对污泥絮体的破解,可以有效地释放束缚水,提升污泥脱水性能[28]. 在本研究中,在同一CaO2投加量下,复合调理后的污泥样品相较于单独调理后的污泥样品有更小的粒径,这也再次说明本研究中复合调理有更高效的EPS破解性能,但高剂量的过氧化钙投加量可能会过度破解絮体结构,过度破解絮体使得絮体粒径下降可能会增加小颗粒污泥对过滤介质的堵塞作用,降低污泥的脱水性能[29].
-
本研究提出一种利用黄铁矿活化CaO2的污泥调理技术,结果表明,单独利用CaO2或者黄铁矿对污泥进行调理,随着CaO2或黄铁矿投加量的增加,污泥脱水性能呈现先上升后下降的趋势,在30 mg·g−1 TS CaO2和1 g·L−1黄铁矿的投加量下分别得到过氧化钙和黄铁矿的最优单独调理效果,同时发现,当CaO2和黄铁矿投加量为30 mg·g−1 TS和1g L−1时,复合调理后的污泥样品脱水性能优于单独调理. 但实现污泥脱水性能的提升需要对调理药剂投加量进行控制,过多的药剂投加可能会带来污泥脱水性能的下降.
黄铁矿活化过氧化钙(CaO2)调理提升污泥脱水性能
Improving sludge dewatering performance by conditioning of activated calcium peroxide (CaO2) with pyrite
-
摘要: 本研究提出了一种利用黄铁矿活化过氧化钙(CaO2)的污泥调理技术,考察了黄铁矿和CaO2单独/复合调理污泥对污泥脱水性能的影响. 本研究中结合胞外聚合物(EPS)性质变化(各层EPS含量、三维荧光光谱)以及污泥絮体结构变化(污泥絮体粒径)分析不同调理方法对污泥性质的影响. 结果表明,在黄铁矿和CaO2投加量分别为1 g·L−1和30 mg·g−1 TS调理条件下,污泥样品有最低的SCST值(0.55). 在研究中发现,通过破解EPS结构虽然能提升污泥脱水性能,但过度的EPS结构破解,会导致大量污泥内部有机物释放,絮体粒径大幅下降,反而降低污泥的脱水性能.Abstract: In this study, a sludge conditioning technology using pyrite activated calcium peroxide (CaO2) was proposed to improve sludge dewaterability, and the effect of pyrite and CaO2 alone/combined conditioning sludge on sludge dewatering performance was investigated. The effects of different conditioning methods on the properties of sludge were analyzed by combining the changes of extracellular polymeric substance (EPS) properties (EPS content and Three-Dimention fluorescence spectrum of each layer) and the structural changes of sludge floc (particle size of sludge floc). The results showed that the sludge samples had a lowest SCST value (0.55) when the dosage of pyrite and CaO2 was 1 g·L−1 and 30 mg·g−1TS, respectively. It was found that cracking EPS structure can improve the sludge dewatering performance, however, excessive decomposing EPS structure will lead to the release of a large number of organic compounds into the sludge and the particle size of flocs will decrease significantly, resulting in the poor sludge dewatering performance.
-
Key words:
- pyrite /
- calcium peroxide /
- sludge dewatering.
-
多环芳烃(polycyclic aromatic hydrocarbons,PAHs)是一类具有代表性的持久性有机污染物(persistent organic pollutants,POPs)[1],主要由化石燃料或生物质的不完全燃烧产生;其来源分为自然源(森林火灾和火山喷发)和人为源(垃圾焚烧、道路扬尘、石油精炼、交通运输等),其中人为源排放是导致PAHs含量剧增的主要原因[2]. 相较于其它有机污染物,PAHs具有种类多、浓度高、分布广、毒性作用显著等特点. 由于具有致癌、致畸、致突变的“三致”毒性[3],USEPA在1979年规定16种PAHs为优先控制污染物[4].
多环芳烃及其衍生物对人体的暴露途径主要包括吸入、摄入和皮肤接触. PAHs污染物作为外源性化学物质在进入人体后,首先被血液吸收,然后通过血液系统进行分布,将PAHs运输到靶器官[5]. 研究证明,多环芳烃可诱发多种疾病,如白内障、肾和肝损伤以及黄疸、肺癌、皮肤癌、膀胱癌等[6]. 近年来,PAHs衍生物也被发现具有致毒致癌效应[7],如含氧多环芳烃(oxygenated-polycyclic aromatic hydrocarbons,OPAHs)引发人体过敏性疾病和细胞凋亡[7]. 目前为止,关于人体PAHs、OPAHs的暴露水平的研究大多集中在外暴露中,依据环境中检测到的污染物浓度水平,运用模型公式和评估参数计算个体的污染物摄入量. 由于这些数据存在时间和空间上的差距,在人体暴露评估中可能存在局限性,不能反映出真实的暴露水平. 相比之下,内暴露通过检测人体体液或组织液的污染物浓度,其暴露水平更具有准确性、真实性. 通过研究人体内多环芳烃类污染物的暴露特征,有助于评估这些物质对人体的身体负担和潜在的毒性作用.
本研究选择天津市45名青年男性为研究对象,进行血浆样本的采集,对血浆中的PAHs、OPAHs的浓度水平及化学组成进行分析,并结合特征比值法和主成分分析方法解析目标物的来源,运用苯并[a]芘毒性当量(BaPeq)和致癌风险模型评估健康风险.
1. 材料与方法(Materials and methods)
1.1 样品采集与储存
本项研究共采集了天津45名青年男性的血液,参与人员的年龄在25—35岁之间,均在市区居住;无不良生活习惯,工作内容相似且无职业暴露风险. 将采集的静脉血置于含抗凝剂的2 mL EDTA采血管中,以3000 r·min−1离心10 min,取血浆于冻存管,并转移至-20 ℃冰箱中保存直至分析. 所有参与者都被详细告知研究目的,均签订知情同意书. 本研究符合卫生部人类医学伦理审查法和赫尔辛基宣言.
1.2 血浆样品前处理
将500 µL血浆转移至10 mL聚丙烯离心管中,加入1 mL醋酸铵溶液(1 mol·L−1)、0.25 mL甲酸溶液(1 mol·L−1)和1.2 mL超纯水使蛋白质变性. 超声处理10 min后,以4000 r·min−1离心5 min. 提取物的净化使用SPE柱进行,依次用5 mL正己烷、5 mL二氯甲烷、5 mL丙酮、5 mL甲醇和5 mL超纯水预先活化. 样品中加入40 µL替代标准品(Phe-D10、Chr-D12),经SPE柱提取后, 9 mL正己烷:二氯甲烷(V:V=1:1)混合溶剂进行目标物质的的洗脱. 安瓿瓶收集洗脱液,在温和的氮气流下吹至近干;然后使用500 µL正己烷进行复溶,样品移至进样瓶.
1.3 仪器分析
本研究选用岛津气相色谱-质谱联用仪(GC-MS)对多种PAHs及OPAHs进行定性和定量测定.
色谱参数设置:色谱柱型号为DB-5MS毛细管柱((SHIMADZU;30 m × 0.25 mm,0.25 μm),升温程序如下:初始温度为65 ℃,保持0.5 min;以15 ℃·min−1的升温速率升至130 ℃,保持0.5 min;再以9 ℃·min−1的速率升至220 ℃,保持1 min;然后以7 ℃·min−1的速率升至240 ℃,保持1.5 min;最后以15 ℃·min−1升温至320 ℃,保持5 min,升温程序的总时长为31.02 min. 进样口温度为270 ℃;进样量为5 µL. 采用脉冲不分流方式进样,载气为氦气,流速为1.4 mL·min−1.
质谱参数设置:质谱仪配置电子轰击电离源(EI),电离电压为70 eV;离子源温度230 ℃;传输线温度为270 ℃;溶剂延迟时间5 min;本研究采用选择离子监测模式(SIM)对目标组分进行检测,具体参数见表1.
表 1 目标组分的检测离子Table 1. The monitoring ions of target components目标组分 Target component 定量离子 Quantifier ion (m/z) 定性离子 Qualifier ion (m/z) 芴 Fluorene(Flo) 166 165.00—164.00 菲 Phenanthrene(Phe) 178 176.00—152.00 蒽 Anthracene(Ant) 178 176.00—179.00 荧蒽 Fluoranthene(Flu) 202 200.00—203.00 芘 Pyrene(Pyr) 202 200.00—201.00 二苯并(a,h)蒽 Dibenz[a,h]anthracene(DBahA) 278 276.00—279.00 苯并(g,h,i)芘 Benzo[ghi]perylene(BghiP) 276 274.00—138.00 氧芴 Dibenzofuran(Dibf) 168 139.00—169.00 10H-9-蒽酮 10H-9-Anthracenone(ATQ) 208 152.00—180.00 菲-9-醛 Phenanthrene-9-carboxaldehyde(Phe-9-Ald) 178 206.00—176.00 苯并蒽酮 7H-Benz[de]anthracen-7-one(BZO) 230 202.00—200.00 5,12-四并苯醌 5,12-Naphthacenedione(NCQ) 258 202.00—230.00 苯并[a]蒽-7,12-二酮 Benz(a)anthracene-7,12-dione(7,12-BaAO) 258 202.00—230.00 菲-D10 Phenanthrene-D10(Phe-D10) 188 184.00—160.00 䓛-D12 Chrysene-D12(Chr-D12) 240 236.00—241.00 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.4 质量控制与质量保证
为确保PAHs及其衍生物测定的准确性,样本分析前测定正己烷溶液检测仪器本底值,直至仪器本底值低于检出限后进行样品测定. 每5个样品中插入正己烷空白,空白样品中均未检测到目标物. 在样品中加入替代标准品,所有血样和空白的两种替代标准的平均回收率在87.6%—150.2%之间,基质加标的相对标准偏差(RSD)小于10%. 目标化合物的检出限(LOD)定义为3倍信噪比,方法检出限范围为0.40—1.80 ng·mL−1.
1.5 数据处理
采用SPSS 22.0统计软件进行基础数据的处理和主成分分析、Origin 8.0软件进行数据分析和绘图.
2. 结果与讨论(Results and discussion)
2.1 PAHs、OPAHs暴露水平
本研究通过对血浆样品中PAHs及OPAHs进行分析,共检测到7种PAHs、检出率范围为17.8%—80.0%(如表2所示),与Yang等报道的PAHs检出率范围相似[8]. 在7种单体PAHs中,Phe的检出率最高(80.0%),其次是DBahA(66.7%),Koukoulakis等对希腊成年人血清的研究中也表明了Phe具有较高的检出率[9]. 其他单体PAHs检出率均低于50%,其中BghiP的检出率最低(17.8%).
表 2 PAH和OPAHs在血浆中的分布情况Table 2. Concentrations of PAH and OPAHs in plasma分类Species 物质名称Compound 检出率/%Detection rate 平均浓度/(ng·mL−1)Average concentration 范围/(ng·mL−1)Range 标准偏差/(ng·mL−1) Standard deviation PAHs Phe 80.0 10.8 N.d.—51.7 10.4 DBahA 66.7 14.2 N.d.—36.1 4.8 BghiP 17.8 1.4 N.d.—3.4 1.2 Flo 33.3 5.4 N.d.—15.6 3.6 Ant 26.7 2.3 N.d.—4.4 1.0 Flu 31.1 4.4 N.d.—14.1 3.4 Pyr 33.3 8.1 N.d.—25.2 6.0 ∑PAHs 100 24.8 10.1—111.2 17.4 OPAHs Dibf 28.9 8.9 N.d.—21.7 6.6 ATO 44.4 27.1 N.d.—56.0 7.5 Phe-9-Ald 66.7 6.7 N.d.—7.0 0.2 BZO 66.7 4.3 N.d.—5.3 0.5 NCQ 66.7 6.3 N.d.—9.2 1.4 7,12-BaAO 66.7 8.7 N.d.—13.5 1.7 ∑OPAHs 95.6 31.9 N.d.—85.5 21.6 | Show TableDownLoad:
CSV
ΣPAHs的浓度范围为10.1—111.2 ng·mL−1,平均值为24.8 ng·mL−1 (表2). 在检测到的单体PAHs中,DBahA、Phe具有较高的浓度(分别为14.2 ng·mL−1、10.8 ng·mL−1),也是其中检出率最高的2个单体PAHs,浓度最低的是BghiP(1.4 ng·mL−1). Koukoulakis等在血清中的研究发现Phe等7种PAHs的浓度范围为1.95—62.2 ng·mL−1(病例组)和1.26—48.6 ng·mL−1(控制组)[9];Singh等[10]对印度儿童血清PAHs检测的结果显示Phe、Ant、Flu、Pyr浓度分别为9.0 ng·mL−1、3.6 ng·mL−1、6.0 ng·mL−1和9.0 ng·mL−1. 本研究中的PAHs浓度范围上述两项研究中浓度范围基本一致.
在血浆样品中OPAHs的检出率为95.6%,其中Phe-9-Ald、BZO、NCQ、7,12-BaAOs的检出率较高(均大于60%),而Dibf的检出率最低(28.9%). Σ6OPAHs的浓度范围为Nd—85.5 ng·mL−1,平均值为31.9 ng·mL−1,在检测到的单体中,ATO具有最高的浓度(27.1 ng·mL−1),浓度最低的是BZO(4.3 ng·mL−1). 目前,国内外关于OPAHs的研究报道主要集中在环境介质中,动物、人体中OPAHs的研究报道较少,尚不能提供数据上的参照.
2.2 组分特征分析
血浆PAHs和OPAHs的分子组成从3环到6环均有分布,其中3环为Phe、Flo、Ant和ATO、Phe-9-Ald,占总量的56%,4环为Flu、Pyr和BZO、NCQ、7,12-BaAO,占总量的29%,5环和6环分别为DBahA和BghiP,分别占13.1%和1.3%(图1a). 血浆中检测到的PAHs和OPAHs成分主要以低-中分子量物质为主,Yin等在脐带血清的研究中发现了低分子量多环芳烃的比例高于高分子量多环芳烃[11],来自印度不同地区的多环芳烃监测研究也报告了儿童血清中低-中分子量多环芳烃多于高分子量多环芳烃[10],低-中-高分子量多环芳烃在人体中的分布呈现一致性. 该分布状况的形成与高分子量物质易与大颗粒物结合富集在地面,低中分子量物质易与较小的颗粒物结合悬浮在空气中有关,通过呼吸进入人体的低中环分子量物质较多,是导致血浆中PAHs和OPAHs以低中分子含量为主的重要因素之一.
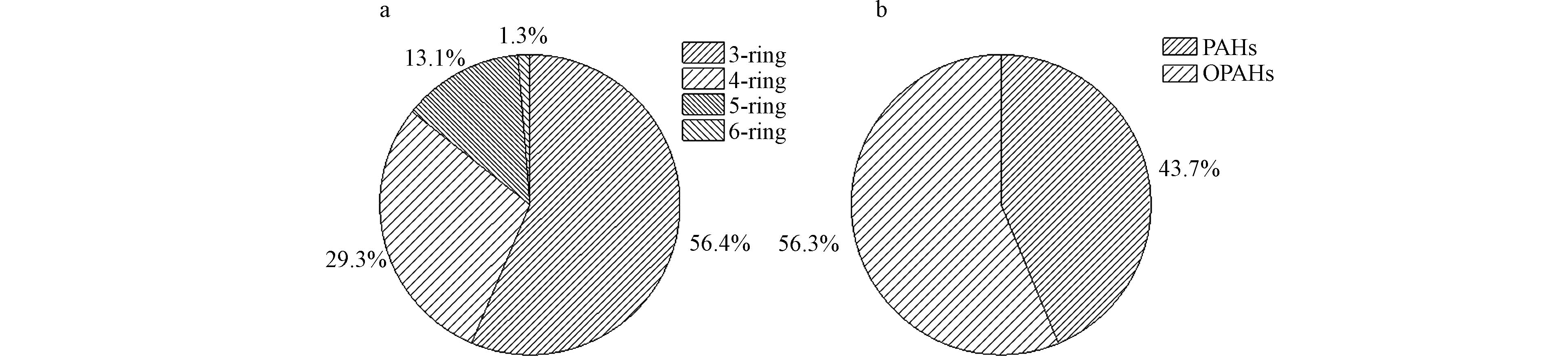 图 1 血浆中PAHs和OPAHs分布比例Figure 1. The distribution ratio of PAHs and OPAHsa 不同环数 Different ring numbers;b 浓度水平Concentration level
图 1 血浆中PAHs和OPAHs分布比例Figure 1. The distribution ratio of PAHs and OPAHsa 不同环数 Different ring numbers;b 浓度水平Concentration level在血浆样品中检测到的OPAHs浓度高于PAHs,分别占比56%和44%(图1b). 由于目前在人体内暴露中检测OPAHs的研究有限,目前在一些环境介质中的研究中,发现PAHs衍生物低于PAHs浓度;Bandowe等的研究报告称,乌兹别克斯坦一处工业区的土壤中多环芳烃衍生物(MPAHs,OPAH)的含量低于PAHs的含量[12];Wang等在北京大气颗粒中的研究中发现OPAHs的浓度低于PAHs[13],在环境中的调查与本研究结果不同. 据报道,多环芳烃衍生物与其母体PAHs相似,可以直接从岩源或热源中产生[14],此外,PAHs衍生物还可以通过PAHs在人体内的代谢而逐渐生成[15],这种转化可能是OPAHs的浓度高于PAHs的原因. 人体中出现OPAHs浓度较高的情况应引起重视,在有限的研究中表明OPAHs的毒性可能与PAHs的毒性相同甚至更高. OPAHs在细胞水平上的毒性作用能显著降低人脐静脉内皮细胞(HUVECs)一氧化氮(NO)的生成,存在潜在的内皮损伤效应,可成为人类细胞高致突变的诱变剂[16].
2.3 PAHs来源解析
2.3.1 特征值分析
由于不同的排放源会产生特定的多环芳烃标志物,PAHs特征比值法常被用来判别多环芳烃类污染物的可能来源. 本研究选用以往研究中广泛使用的4种特征值应用于本研究血清中PAHs的来源解析[9, 17-18]. 各类成因对应污染源中PAHs特征比值如表3所示,在本研究中通过特征值的分析,表明石油源和化石燃料的不完全燃烧是PAHs产生的的重要来源.
表 3 各类成因对应污染源中PAHs的特征比值Table 3. Various causes correspond to the diagnostics ratios of PAHs in pollution sources特征比值Diagnostics ratios 石油类来源Petroleum source 化石燃料的不完全燃烧类来源Incomplete combustion of fossil fuels 本研究This study 荧蒽/芘 Flu/Pyr 0—1 >1 0.55 菲/蒽 Phe/Ant >10 0—10 5.8 荧蒽/(荧蒽+芘) Flu/(Flu+Pyr) 0—0.4 0.4—0.5a,>0.5b 0.33 蒽/(蒽+菲) Ant/ (Phe+Ant) <0.1 >0.1 0.15 注:a. 主要在石油类产品的不完全燃烧过程中形成Mainly formed in the incomplete combustion process of petroleum products;b. 主要在木材、煤炭和草类的不完全燃烧过程中形成Mainly formed in the incomplete combustion process of wood, coal and grass | Show TableDownLoad:
CSV
2.3.2 主成分分析
为进一步研究PAHs的来源情况,采用主成分分析法定量分析血浆中PAHs类物质的来源,提取初始特征值大于1的因子,2个主因子的累计方差贡献率为79.7%,7种PAHs主成分因子载荷如表4所示.
表 4 主成分分析因子载荷矩阵Table 4. Factor loading matrix of principal component analysis变量Variables 主因子1 Factor 1 主因子2 Factor 2 Flo −0.929 0.301 Phe −0.881 0.390 Ant −0.897 0.265 Flu −0.862 0.329 Pyr −0.899 0.317 DBahA 0.845 0.275 BghiP 0.262 0.379 Dibf −0.790 0.154 ATO 0.601 0.443 Phe-9-Ald 0.939 0.241 BZO 0.923 0.254 NCQ 0.894 0.258 7,12-BaAO 0.908 0.290 解释方差变量% 70.2 9.5 累计方差贡献率% 70.2 79.7 | Show TableDownLoad:
CSV
一般认为低分子量的PAHs主要来源于石油的泄漏、化石燃料和生物质的不完全燃烧,而高温燃烧过程主要形成高分子量的PAHs;如在PAHs的来源分析中Pyr、Phe等低中分子量主要与煤燃烧有关,DBahA、BghiP是汽车排放的重要化合物[19-21]. 主因子1具有最高的方差贡献率(70.2%),在主因子1的单体PAHs中只有DBahA具有很高的载荷,表明PAHs的来源是交通燃油排放;主因子2的方差贡献率为9.5%,在单体PAHs中Phe具有最高载荷,其次是Pyr、Flu等低分子量PAHs也具有较高载荷,表明燃煤是主因子2的主要来源. 本研究通过主成分分析表明交通燃油排放是主要污染源,而燃煤来源为PAHs的第二污染源. 其分析结果与特征比值法的结果相互吻合. 综上所述石油源和化石燃料的燃烧是PAHs产生的重要来源,通过对PAHs来源的解析有助于更好地了解排放途径和不同来源的贡献,为未来的风险管理提供科学依据.
有关OPAHs来源途径较多,即可来自于一次排放,也可来自于二次生成. 目前关于OPAHs的具体来源途径尚未有明确的判别方法,可供参考的研究资料有限,尚需进行进一步的研究工作,以期揭示OPAHs具体来源途径及其影响因素.
2.4 健康风险评估
2.4.1 每日总摄入量和毒性当量
依据药代动力学模型从血浆中多环芳烃含量推算每日总摄入量(TEDIs)如公式(1)所示,当血浆中的多环芳烃的浓度不在随时间变化时,即
dc/dt vdcdt=TEDIs×BW×A×f−b×V×C (1) 式中,C为血浆中单体多环芳烃的浓度;V为人体的血量(L),一般认为是男性血量为体重的8%;BW为体重(kg);A(无量纲)为Pyr摄入剂量的吸收速率为0.90;f(无量纲)是Pyr吸收剂量分布在血液中的比例为0.052;b代表Pyr的消除速率常数为0.068;式中这些参数参考Haddad[22]和Viau[23]的研究结果.
以血浆中Pyr浓度为基础,基于公式(1)计算血浆中Pyr的每日总摄入量. 其TEDI的范围为0.296—2.928 μg·kg−1·d−1 bw,平均浓度为0.935 μg·kg−1·d−1 bw,见表5.
表 5 Pyr的每日总摄入量(TEDI,μg·kg−1·d−1 bw)Table 5. Total estimated daily intake of Pyr in plasma (TEDI, μg·kg−1·d−1 bw)目标物Compounds 百分位数Percentiles 平均值Average 范围 Range 25% 50% 75% Pyr 0.487 0.777 1.156 0.935 0.296—2.928 | Show TableDownLoad:
CSV
由于每种单体PAHs具有不同程度的毒性,国内外的研究中通常采用苯并[a]芘当量浓度(BaP equivalent, BaPeq)来评估得到其他单体PAHs的毒性,各单体PAHs的BaPeq毒性当量计算公式如(2)所示。
BaPeqi=TEDIi×TEFi (2) 式中,BaPeqi为各PAHs单体的BaP当量毒性,TEFi为对应PAHs单体的毒性当量因子(Pyr的毒性当量因子为0.001).
2.4.2 非致癌健康风险和致癌风险
(1)非致癌系数(HQ)是每日摄入量(TEDI)与美国环保局提供每日参考剂量(RfD)的比值,若HQ>1,则说明存在非致癌风险;HQ<1,则表明不存在致癌风险[24]. Pyr的参考剂量为30 μg·kg−1-bw·d−1,计算公式如(3)所示.
HQ=TEDIRfD (3) (2)PAHs暴露的致癌风险的计算,如公式(4)所示。
CR=CSF×BaPeq (4) 式中,CSF为致癌斜率因子,本研究选用BaP的口服致癌斜率因子[8][7.3 (kg·d)·mg−1]. 根据美国环保署规定,CR<10−6时,引发的致癌风险不明显;CR在10−4和10−6之间时,表明具有潜在的致癌风险;CR>10−4时,表明存在很高的致癌风险[25].
分别计算了HQ和CR值(图2),以评估血浆中Pyr内暴露的非致癌和致癌风险. 血浆中Pyr的HQ值范围为0.072—0.712,中位数为0.189,表明人体血浆中的多环芳烃不存在非致癌风险. 然而,当考虑致癌风险时,基于苯并[a]芘当量浓度的致癌风险应引起足够关注,血浆中CR值的范围为2.16×10−6—2.14×10−5,中位数为5.67×10−6,所用个体的CR值均超过了1×10−6,表明血浆中PAHs存在潜在的致癌风险.
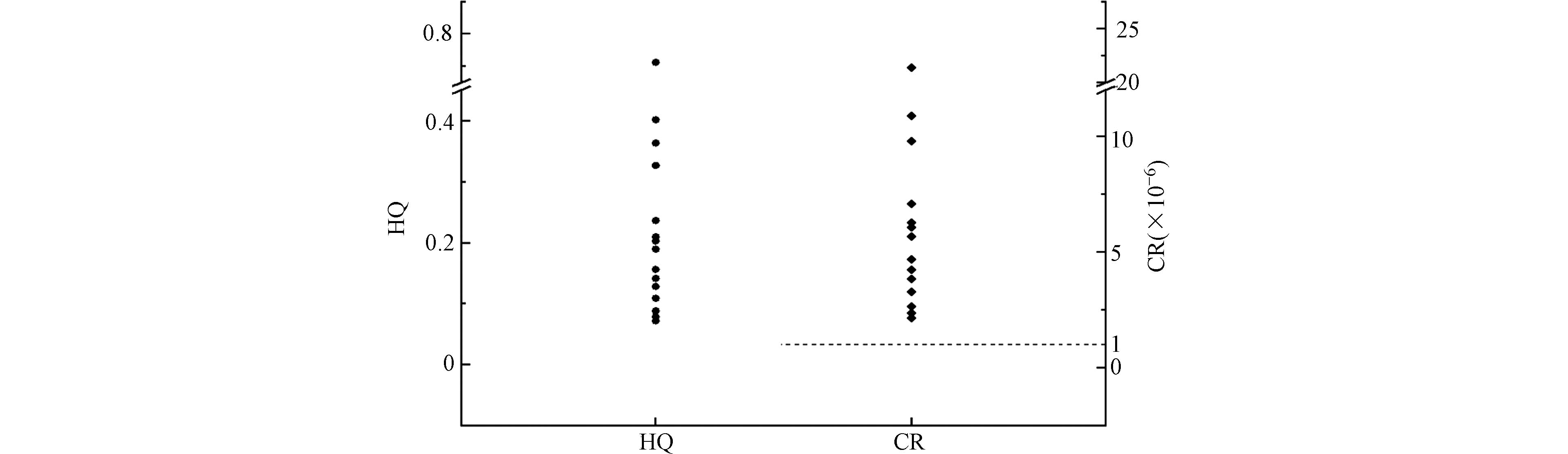 图 2 血浆样本芘的个体危险系数(HQ)和癌症风险(CR)Figure 2. Individual hazard quotient (HQ) and cancer risk (CR) of Pyr in serum
图 2 血浆样本芘的个体危险系数(HQ)和癌症风险(CR)Figure 2. Individual hazard quotient (HQ) and cancer risk (CR) of Pyr in serum3. 结论(Conclusion)
在天津市45名青年人群血浆中共检测出7种PAHs和6种OPAHs,ΣPAHs和∑OPAHs平均浓度分别为24.8 ng·mL−1和31.9 ng·mL−1. 其单体化合物中ATO、DBahA的相对含量较高,对人群健康的潜在危害较大,应重点关注. 且PAHs和OPAHs组成特征以低中环物质为主,占总质量浓度的85%. 石油源和化石燃料的燃烧为人群血浆中PAHs的重要来源. 通过致癌风险评估,表明血浆中的PAHs存在潜在的致癌风险,后续应加大对人群内暴露的全面监测和长期健康风险的评估.
-
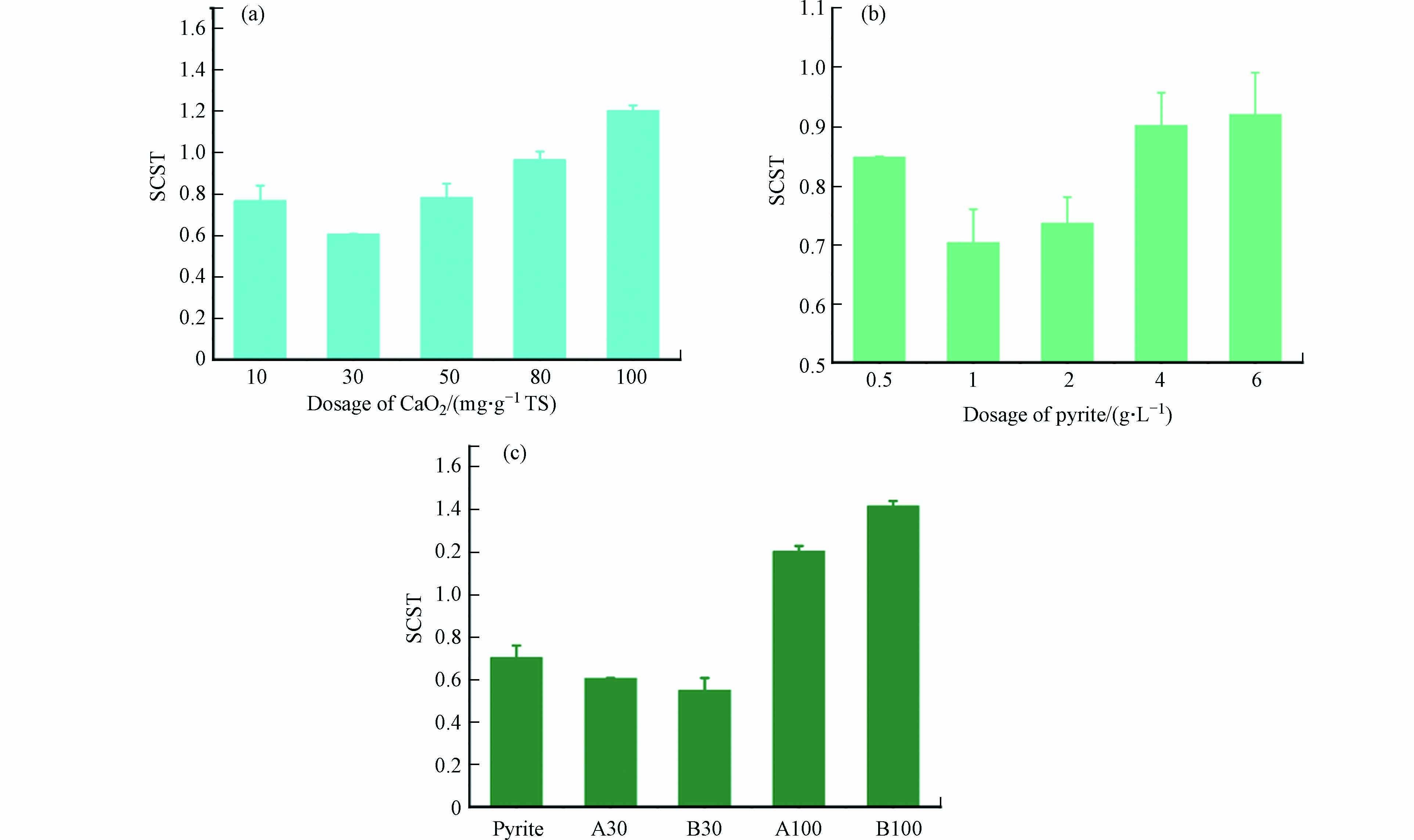
图 1 过氧化钙(a)、黄铁矿(b) 单独调理和复合调理(c)污泥样品的SCST值
Figure 1. SCST values of calcium peroxide (a), pyrite (b) single conditioning and composite conditioning (c) sludge samples

图 2 原泥以及调理后污泥EPS中蛋白质含量(a),多糖(b)蛋白质-多糖含量比率(c)变化
Figure 2. The concentration of protein (a), polysaccharide (b) and the ratio of protein to polysaccharide (c) in EPS of raw sludge and conditioned sludge

图 3 原泥(a)、A30(b)、B30(c)、A100(d)、B100(e)EPS样品三维荧光光谱图
Figure 3. 3D-EEM spectra of EPS samples of raw sludge(a); A30(b); B30(c); A100(d); B100(e)
-
[1] CHRISTENSEN M L, KEIDING K, NIELSEN P H, et al. Dewatering in biological wastewater treatment: A review[J]. Water Research, 2015, 82: 14-24. doi: 10.1016/j.watres.2015.04.019 [2] 汤连生, 罗珍贵, 张龙舰, 等. 污泥脱水研究现状与新认识[J]. 水处理技术, 2016, 42(6): 12-17. doi: 10.16796/j.cnki.1000-3770.2016.06.003 TANG L S, LUO Z G, ZHANG L J, et al. Research status and new views of sludge dewatering[J]. Technology of Water Treatment, 2016, 42(6): 12-17 (in Chinese). doi: 10.16796/j.cnki.1000-3770.2016.06.003
[3] MA Y, ZHANG B T, ZHAO L X, et al. Study on the generation mechanism of reactive oxygen species on calcium peroxide by chemiluminescence and UV-visible spectra[J]. Luminescence, 2007, 22(6): 575-580. doi: 10.1002/bio.1003 [4] XU Q X, HUANG Q S, WEI W, et al. Improving the treatment of waste activated sludge using calcium peroxide[J]. Water Research, 2020, 187: 116440. doi: 10.1016/j.watres.2020.116440 [5] WANG C, WEI W, DAI X H, et al. Calcium peroxide significantly enhances volatile solids destruction in aerobic sludge digestion through improving sludge biodegradability[J]. Bioresource Technology, 2022, 346: 126655. doi: 10.1016/j.biortech.2021.126655 [6] CHEN Z, ZHANG W J, WANG D S, et al. Enhancement of waste activated sludge dewaterability using calcium peroxide pre-oxidation and chemical re-flocculation[J]. Water Research, 2016, 103: 170-181. doi: 10.1016/j.watres.2016.07.018 [7] WANG J, SHANG K K, DA L J, et al. Synergetic effect of combined CaO2 and microwave treatment on waste active sludge dewaterability and organic contaminants’ removal[J]. Frontiers in Environmental Science, 2021, 9: 734277. doi: 10.3389/fenvs.2021.734277 [8] ZHOU Y B, FANG X B, WANG T H, et al. Chelating agents enhanced CaO2 oxidation of bisphenol A catalyzed by Fe3+ and reuse of ferric sludge as a source of catalyst[J]. Chemical Engineering Journal, 2017, 313: 638-645. doi: 10.1016/j.cej.2016.09.111 [9] ZHANG X, GU X G, LU S G, et al. Degradation of trichloroethylene in aqueous solution by calcium peroxide activated with ferrous ion[J]. Journal of Hazardous Materials, 2015, 284: 253-260. doi: 10.1016/j.jhazmat.2014.11.030 [10] CHEN H, ZHANG Z L, YANG Z L, et al. Heterogeneous Fenton-like catalytic degradation of 2, 4-dichlorophenoxyacetic acid in water with FeS[J]. Chemical Engineering Journal, 2015, 273: 481-489. doi: 10.1016/j.cej.2015.03.079 [11] WANG L P, CHANG Y Z, LI A M. Hydrothermal carbonization for energy-efficient processing of sewage sludge: A review[J]. Renewable and Sustainable Energy Reviews, 2019, 108: 423-440. doi: 10.1016/j.rser.2019.04.011 [12] KIM J G, KIM H B, JEONG W G, et al. Enhanced-oxidation of sulfanilamide in groundwater using combination of calcium peroxide and pyrite[J]. Journal of Hazardous Materials, 2021, 419: 126514. doi: 10.1016/j.jhazmat.2021.126514 [13] ZHOU Y, HUANG M, WANG X L, et al. Efficient transformation of diethyl phthalate using calcium peroxide activated by pyrite[J]. Chemosphere, 2020, 253: 126662. doi: 10.1016/j.chemosphere.2020.126662 [14] LI Y, CHEN J M, ZHONG J, et al. Acceleration of traces of Fe3+-activated peroxymonosulfate by natural pyrite: A novel cocatalyst for improving Fenton-like processes[J]. Chemical Engineering Journal, 2022, 435: 134893. doi: 10.1016/j.cej.2022.134893 [15] DAI Z H, LIU L, DUAN H R, et al. Improving sludge dewaterability by free nitrous acid and lysozyme pretreatment: Performances and mechanisms[J]. Science of the Total Environment, 2023, 855: 158648. doi: 10.1016/j.scitotenv.2022.158648 [16] GUO J Y, JIA X J, GAO Q F. Insight into the improvement of dewatering performance of waste activated sludge and the corresponding mechanism by biochar-activated persulfate oxidation[J]. Science of the Total Environment, 2020, 744: 140912. doi: 10.1016/j.scitotenv.2020.140912 [17] NIU M Q, ZHANG W J, WANG D S, et al. Correlation of physicochemical properties and sludge dewaterability under chemical conditioning using inorganic coagulants[J]. Bioresource Technology, 2013, 144: 337-343. doi: 10.1016/j.biortech.2013.06.126 [18] LI X Y, YANG S F. Influence of loosely bound extracellular polymeric substances (EPS) on the flocculation, sedimentation and dewaterability of activated sludge[J]. Water Research, 2007, 41(5): 1022-1030. doi: 10.1016/j.watres.2006.06.037 [19] McDONALD C E, CHEN L L. The Lowry modification of the Folin reagent for determination of proteinase activity[J]. Analytical Biochemistry, 1965, 10(1): 175-177. doi: 10.1016/0003-2697(65)90255-1 [20] CHEN D Y, YANG J. Effects of explosive explosion shockwave pretreatment on sludge dewaterability[J]. Bioresource Technology, 2012, 119: 35-40. doi: 10.1016/j.biortech.2012.05.129 [21] DAI Q X, MA L P, REN N Q, et al. Investigation on extracellular polymeric substances, sludge flocs morphology, bound water release and dewatering performance of sewage sludge under pretreatment with modified phosphogypsum[J]. Water Research, 2018, 142: 337-346. doi: 10.1016/j.watres.2018.06.009 [22] HE D Q, WANG L F, JIANG H, et al. A Fenton-like process for the enhanced activated sludge dewatering[J]. Chemical Engineering Journal, 2015, 272: 128-134. doi: 10.1016/j.cej.2015.03.034 [23] WEI H, GAO B Q, REN J, et al. Coagulation/flocculation in dewatering of sludge: A review[J]. Water Research, 2018, 143: 608-631. doi: 10.1016/j.watres.2018.07.029 [24] LIU J, YANG Q, WANG D B, et al. Enhanced dewaterability of waste activated sludge by Fe(Ⅱ)-activated peroxymonosulfate oxidation[J]. Bioresource Technology, 2016, 206: 134-140. doi: 10.1016/j.biortech.2016.01.088 [25] GUO Z Y, MA L P, DAI Q X, et al. Role of extracellular polymeric substances in sludge dewatering under modified corn-core powder and sludge-based biochar pretreatments[J]. Ecotoxicology and Environmental Safety, 2020, 202: 110882. doi: 10.1016/j.ecoenv.2020.110882 [26] CHEN W, WESTERHOFF P, LEENHEER J A, et al. Fluorescence excitation–emission matrix regional integration to quantify spectra for dissolved organic matter[J]. Environmental Science & Technology, 2003, 37(24): 5701-5710. [27] XIAO K K, PEI K Y, WANG H, et al. Citric acid assisted Fenton-like process for enhanced dewaterability of waste activated sludge with in-situ generation of hydrogen peroxide[J]. Water Research, 2018, 140: 232-242. doi: 10.1016/j.watres.2018.04.051 [28] LING X, DENG J, YE C, et al. Fe(Ⅱ)-activated sodium percarbonate for improving sludge dewaterability: Experimental and theoretical investigation combined with the evaluation of subsequent utilization[J]. Science of the Total Environment, 2021, 799: 149382. doi: 10.1016/j.scitotenv.2021.149382 [29] LI Y F, ZHU Y Q, WANG D B, et al. Fe(Ⅱ) catalyzing sodium percarbonate facilitates the dewaterability of waste activated sludge: Performance, mechanism, and implication[J]. Water Research, 2020, 174: 115626. doi: 10.1016/j.watres.2020.115626 期刊类型引用(2)
1. 任瑛,李广科,桑楠. 氧化多环芳烃对大麦幼苗生长及抗氧化生理响应的影响. 环境化学. 2024(11): 3658-3664 .  本站查看
本站查看
2. 杨姝萍,钟芝清,黄子杰,罗宽能. 菜籽油中16种PAHs测定的不确定度评定. 粮油与饲料科技. 2024(10): 218-220 . 百度学术
其他类型引用(3)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 1619
- HTML全文浏览数: 1619
- PDF下载数: 45
- 施引文献: 5
