





-
塑料作为人类生活中不可或缺的一种材料,被广泛运用于各个行业[1]. 随之而来的,塑料对环境的污染也成为了亟待解决的热点问题[2]. 近年来,微塑料(microplastics,MPs) ,即尺寸小于5 mm的塑料碎片[3- 4],由于其在水环境中的普遍分布、对生物体的生物影响以及不可降解的特性而备受关注[5-6]. 微塑料由于其体积小、密度低的特点很容易在水体中扩散[7],湖泊、河口、海滩等地区都是微塑料聚集的重点地区[8-12] ,甚至在北极地区都有关于微塑料的报道[13]. 与大中型塑料相比,微塑料具有粒径小、比表面积大、疏水性强等特点[14-15],更易于吸附污染物质[16-17],并且将污染物转移到食物链中[18],最后到达人类体内[19],重金属就是典型污染物其中之一[20-21].
重金属可以吸附到微塑料上[15, 22],其吸附量会受其自身物理化学特性影响,例如形态、比表面积、老化程度等[14-15, 23-26]. 并且塑料在制造过程中也会添加少量金属作为催化剂、颜料或者稳定剂[14]. 部分重金属作为一种可溶性有毒污染物[27],即使在低浓度下,也会对生物体造成伤害[25, 28]. 由于重金属离子和微塑料的毒性,重金属离子吸附到微塑料上可能会加剧两者对水生环境的潜在风险[23]. 重金属在微塑料上的吸附量也与其所处环境条件有关,例如pH值、盐度等[22]. Gao等通过室内实验与现场实验对比发现,现场实验中微塑料对重金属吸附量小于室内实验[29]. Holmes等发现,从海滩收集的微塑料颗粒中重金属浓度超过了当地河口沉积物中的浓度,这可能是由于塑料表面形成有机物及微生物,与重金属生成共沉淀导致的[30]. 真实水环境中微塑料对重金属吸附的影响因素远大于室内实验环境,这可能会导致两者的吸附量存在差异. 近年来,虽然关于微塑料对重金属吸附的研究越来越多,但是单个研究中,微塑料及涉及的重金属数量往往较少,考虑的影响因素也多局限在单一因素,较少有文章将多种微塑料/重金属、多种因素联系在一起进行综合比较. 此外,关于微塑料与重金属吸附作用的研究多集中于室内实验,也缺乏与真实水环境的对比.
本文以Web of Science核心数据库及Science Direct数据库作为数据源,对不同环境条件下不同微塑料对于不同重金属的吸附及其影响因素进行综合对比分析,并且还对室内实验结果与实际环境的差异进行研究,旨在探究微塑料吸附重金属的主要影响因素以及室内实验结果能否用来评估实际水环境中的微塑料对重金属的吸附水平.
-
本文搜集了2016—2022年期间发表的相关论文,数据库包括Web of Science(
https://www.webofscience.com/wos/alldb/basic-search ), Science Direct (https://www.sciencedirect.com/ ). 搜索关键词为“microplastics AND heavy metal AND adsorption” [31]. 参考《海水中微塑料的测定 傅里叶变换显微红外光谱法》(DB21/T 2751-2017),微塑料的搜索种类包括聚乙烯(polyethylene,PE)、聚丙烯(polypropylene, PP)、聚氯乙烯(polyvinyl chloride, PVC)、聚苯乙烯(polystyrene, PS)、聚氨酯(polyurethane, PU)、聚酰胺(nylon)、聚对苯二甲酸乙二脂(polyethylene terephthalate, PET);参考《海水水质标准》(GB3097-1997)和《地表水环境质量标准》(GB 3838-2002),重金属的搜索种类包括铜(Cu)、铅(Pb)、锌(Zn)、镉(Cd)、铬(Cr). 从文献中直接提取出吸附动力学及吸附等温线模型、微塑料粒径及老化程度. 不同微塑料及重金属的最大吸附量(Qmax )主要通过Langmuir等温线分析或者通过GetData Graph Digitizer对图中数据进行提取. 文中将1.5IQR内的数据认定为有效数据,超过1.5IQR则认定为异常值. -
当样本量大于3时,纳入统计分析. 使用SPSS 26.0对组间数据进行单因素方差分析(ANOVA),若P<0.05,则表示差异显著. 本文采用Origin 2022b进行数据描述性分析以及箱线图、弦图的制作.
-
共搜集到文献200篇,文献筛选流程见图1. 文献数量逐年递增(图2),说明该研究领域的研究热度也在逐年递增. 搜集到的文献中,室内实验文献共33篇,微塑料种类涉及PE、PET、PP、PVC、PS,重金属涉及Cu、Pb、Zn、Cd、Cr. 关于PS的论文有17篇, Pb的论文有19篇,说明PS与Pb是被研究的最多的微塑料和重金属,涉及Zn的研究最少,仅检索出4篇论文. 微塑料老化及粒径是关于微塑料对重金属吸附考虑最多的两个的因素,其中搜集到提及微塑料老化的文献有13篇,及微塑料粒径的文献共有33篇,粒径范围主要集中在0—2 mm. 关于真实水环境中微塑料吸附重金属的文献仅检索出6篇,远少于室内实验研究.
-
由图3(a)可以看出,对于Cu、Pb、Cr等3种重金属,PS对它们的亲和力最强,其次是PVC、PE、PP与PET. PS特有的苯环结构可能是导致其对重金属吸附能力强的主要原因[32]. PVC和PP中都含有钛,但是PVC的吸附性能高于PP,这说明微塑料对重金属的吸附量不仅仅受单一的特性影响,而是所有这些特性的组合决定了微塑料与重金属的吸附作用[14]. PET在生产过程中会添加保护其免受老化和生物定殖的添加剂,这会导致其对于Pb的吸附量最弱 [14]. 而不同微塑料对于Cd的吸附量由大到小的顺序为,PVC>PP>PS>PE. 与其它微塑料相比,PVC可以为Cd提供更多的吸附位点[32]. Cd在PE上的吸附机制归因于范德华力,而PS则为π-π相互作用. 吸附机制的不同可能是导致其吸附量差异的主要原因[33].
由图3(b)可以看出,针对不同重金属,PE、PS、PVC对Pb吸附量最大,PP对Pb的吸附量也相对较大,主要是因为在这些重金属中,Pb的水合离子半径最小[34],而对于等电荷金属的静电吸附,吸附亲和力与离子半径呈负相关. 尽管Cu的水合离子半径与Cd相似,但PS对Cu的吸附量比Cd高. 这一结果可以用Cu比Cd更强的络合能力来解释[34]. 而PVC的结晶度较小,更有利于其对Cd的吸附[32]. 对于PP,其对重金属的吸附量的大小顺序为Cd>Pb>Cu>Cr. 重金属在PP颗粒上的吸附主要集中在表面,此时PP对不同重金属的吸附机理可能是导致吸附量产生差异的主要原因[35].
微塑料对重金属吸附量差异主要与微塑料官能团、结构,重金属水合离子半径等因素有关. 由图3(c)可以看出,PS较其它微塑料更易吸附重金属,而Pb则更容易被微塑料所吸附.
-
(1)微塑料老化 微塑料老化会改变微塑料表面性质而影响其对重金属的吸附行为,老化方式与老化程度的不同也对微塑料自身官能团、结构产生影响[36-38]. 老化微塑料对重金属的吸附量往往大于新鲜微塑料(图4). Hadiuzzaman等通过光降解的方式对PE进行人工老化,发现老化PE与新鲜PE相比,生成了新的含氧官能团,导致老化PE对Pb吸附量比新鲜PE高[36]. Li等发现老化后的PE比表面积增加,同时生成新的官能团,对Cr的吸附量比新鲜PE高[37]. 微塑料老化后,其表面存在更多的孔洞或裂缝,导致与重金属接触的表面积更大,吸收污染物的量提高[38]. Liu等通过对比PE与PP的老化过程,发现两者官能团生成顺序不同,这表明不同微塑料的老化机制存在差异[39]. 结合图3(a)发现,其实验结果中PE对于Cd的吸附数据偏高,该实验在PE老化时采用了长江水振荡老化18个月的方式,老化时间较长. 这表明老化微塑料对重金属的吸附量随着老化时间的延长而增加[38, 40]. Lin等通过FTIR观察到PP光化学老化过程中,羟基、羰基和乙烯基的形成,由此判断官能团的变化可以用来评价PP的老化程度[41]. Fan通过实验发现老化PP的zeta电位由老化前的-5.36mV下降至-9.52 mV,这也进一步促进了PP对Pb的吸附[42],但是在其实验结果中,老化PP对Pb吸附量达到1126 μg·g−1,远高于新鲜PP对Pb吸附量,通过实验条件对比发现,该实验中新鲜微塑料的吸附量在正常范围内,因此推断其老化方式是导致吸附量偏离正常值的主要原因[42]. Liu等也发现在老化过程中,与化学氧化作用相比,光老化起主要作用,这表明老化程度确实与老化方式有关[39]. PS塑料老化后吸附量中值略大于新鲜PS,但是总体吸附量与新鲜PS差异并不显著. Xi等实验发现,老化PS与原始PS相比,含氧官能团增多,对重金属吸附量增强[43]. 由此可以推测对于PS塑料来说,老化程度会影响其对重金属的吸附量,但是影响程度较小.
自然环境中的微塑料老化增大了微塑料对于重金属的吸附作用,进而强化了微塑料作为重金属污染物的载体作用,从而加大了微塑料和重金属对生态环境的联合风险[44- 45].
(2)微塑料粒径 不同粒径微塑料的比表面积不同,进而对重金属的吸附量也不同[29, 46]. 由图5可以看出,微塑料对重金属的吸附量总体趋势随粒径的增加而减小. Wang等通过对比3种不同粒径的PE对Cd的吸附量发现,粒径是决定Cd吸附到PE上的主要因素,较小的颗粒具有更大的比表面积和更多的吸附点位[46],从而导致粒径较小的微塑料对重金属吸附量更大. 而Zhou等研究中PE的粒径虽然大于Wang等采用的微塑料,但是由于其取自商业材料,表面较为粗糙,导致其吸附量更大[34]. Gao等比较了不同粒径的PP与重金属之间的亲和性,结果表明,随着PP粒径的增大,Pb、Cu、Cd在PP上的吸附量显著降低[29]. Shen等探究了PP对Pb吸附量,虽然粒径最小,但是其吸附量小于其它粒径微塑料,这可能是吸附实验中微塑料初始浓度太低所导致的[47]. 而当粒径小于0.5 mm时,PS对Pb的吸附量随粒径变化较为明显,通过条件对比发现,除了粒径,pH值与重金属初始浓度的不同也对两组微塑料对重金属的吸附量有一定的影响,进一步导致了吸附量的剧烈变化. 值得指出的是,在微塑料粒径较小时表现出的较大变动(图5b, 5c, 5f),本文由于样本量所限还无法确认并揭示其准确原因,但这一现象值得进一步探究. 粒径越小的微塑料越容易成为环境中重金属迁移的载体,被生物摄食并在体内积聚,进而表现出更高的毒性[48-50].
(3)盐度 盐度对微塑料对重金属吸附的影响较为显著. 当盐度从5增加到35时,PS和PET颗粒对Cu的吸附量呈小幅下降趋势. 盐度的增加会破坏吸附剂的电荷平衡,中和吸附剂的表面电荷,从而抑制静电作用[51]. Barus等研究了PS在不同盐度下对Pb、Zn和Cu等3种金属的吸附量,其吸附量均随盐度的增加而降低[22]. 当盐度增加时,微塑料不仅会与重金属相互作用,还会与其它离子相互作用. 海水中Na、Ca、Mg等离子的浓度增加,导致电解质离子的竞争效应[15, 40]. 随着离子强度增加到一定水平,特别是较高浓度的Na离子可能会激烈竞争微塑料表面的阳离子交换位点,进一步抑制重金属和微塑料相互作用,导致吸附量下降. 此外,Ho等发现,在一定盐度条件下,Ca对Pb的抑制作用远大于Na,可能的原因是Ca和Pb具有相同的价态,因此阻止了Pb与微塑料有限活性位点的结合[52].
(4)pH值 微塑料对重金属的吸附会受pH影响. Holmes等研究发现,PE对Cd、Co、Ni和Pb的吸附量随着pH值的增加而增加,但对Cr的吸附量降低[30]. 随着pH从2.0增加到6.0,Pb在不同微塑料上的吸附均增加. 在Pb吸附到微塑料上的过程中,溶液 pH 影响溶液中 Pb物种的分布和微塑料的表面性质. 该现象可归因于带负电的微塑料与带正电的Pb之间的静电吸引力. pH值的增加会加速重金属在微塑料中的吸附过程[15, 24]. 值得注意的是由于气候变化的影响,海水pH值总体呈现酸化趋势 [53-54],海水酸化总体上不利于微塑料对于重金属的吸附,一定程度上削弱了微塑料对于重金属的携带和运输作用.
-
微塑料对重金属的吸附动力学模拟模型主要有Pseudo-first order model(PFO)模型、Pseudo-second order(PSO)模型,而PSO模型往往有更好的模拟结果[33, 48, 55](表1),这表明吸附过程不仅包括污染物吸附到微塑料上的不同位点,还涉及多重吸附,传质和离子扩散等不同阶段[59]. 通过绘制
qt 与t1/2 的关系图可以发现,重金属在微塑料上的吸附是一个多阶段的过程[26, 33]. 在吸附的第一阶段,微塑料表面暴露出外部活性位点,吸附主要以液膜扩散为主,吸附速度快. 到了第二阶段,重金属从液膜缓慢扩散到微塑料孔隙中,颗粒内扩散在这一阶段起关键作用[48]. 最后,通过小孔扩散的第三阶段,随后达到吸附/解吸动态平衡状态. 因此,这意味着颗粒内扩散和最终平衡过程是重金属吸附到微塑料上的限速步骤.Freundlich模型、Langmuir模型与Brunauer-Emmett-Teller(BET)模型,都能较好地拟合微塑料与重金属吸附等温线. Freundlich模型表明吸附过程发生在微塑料的异质表面,涉及单层和多层吸附[21, 38],往往为物理吸附,重金属通过弱键吸附在微塑料上,并在微塑料表面形成一层薄膜[60]. 该模型适用于较多微塑料与重金属吸附场景,可以很好的拟合PVC对Pb的吸附[33],同时也适用于PE对Cr的吸附[56]. Langmuir等温线模型通常用于描述单层吸附. 该模型假设吸附剂表面是均匀的,并且表面上的所有吸附位点具有相同的吸附物亲和力[61]. Godoy同时使用Freundlich与Langmuir模型对5种不同的微塑料与重金属进行拟合,结果Langmuir模型具有更好的相关系数. BET等温线模型是从Langmuir等温线模型扩展得出的多层吸附系统的一种特殊形式[61]. 该模型多用于液固平衡系统,能较好的拟合PS与PE对Pb的吸附过程[33].
-
由图6可知,真实水环境中和室内实验条件下微塑料均呈现出对重金属的富集作用,但真实水环境中微塑料对重金属的吸附量小于室内实验中微塑料对重金属的吸附. 造成这一结果的原因可能有以下几点:
第一,真实水环境中微塑料及重金属浓度与室内实验条件有较大差异. Wang等研究发现,随着微塑料浓度的增加,微塑料对重金属的吸附量显著增加,当微塑料浓度从1.0 g·L−1增加到5.0 g·L−1时,微塑料对Cu的吸附量从40.3 µg·g−1增加到了55.1 µg·g-1[25]. 但是随着微塑料浓度的持续增加,微塑料表面的空白吸附点数量增加,导致微塑料的单位吸附容量下降[55]. 室内实验中,微塑料与重金属浓度通常维持在在10—100 g·L−1、1—20 mg·L−1之间,而真实水环境中微塑料与重金属浓度要远低于该区间. 室内实验中微塑料与重金属的浓度范围跨度较大,这也导致了微塑料对重金属的吸附量数据离散程度大于真实水环境. 同时,真实水环境中微塑料对重金属的吸附行为并不明显,这也可能是真实水环境中重金属环境浓度较低导致.
第二,环境中存在悬浮颗粒物. 除了微塑料,重金属还会与水中的其它颗粒物共存,并且它们对重金属有较高的亲和力[62]. Qi等比较了悬浮颗粒物、沉积物以及微塑料对Pb的吸附作用,结果表明,在中性条件下,这三者之间对Pb都存在竞争性吸附[63]. 悬浮颗粒物与沉积物对重金属的吸附量大于微塑料[64—65],这说明微塑料在水环境中作为重金属载体的功能与悬浮颗粒物、沉积物相比较弱,很难对环境产生重大影响[66].
第三,真实水环境中有多种重金属离子共存. Gao发现,Cu、Pb、Cd共存的混合溶液的吸附容量低于单一金属溶液的吸附容量. 由于3种金属离子的存在,金属离子在微塑料表面的吸附是竞争的[29]. 微塑料可以从Cu和Cd共存的溶液中选择性吸附Cu和Cd,但重金属离子会在微塑料表面竞争活性吸附位点. Shen通过共存离子实验也发现,Cu的存在会干扰Pb的吸附[47]. 同时,当混合重金属溶液浓度较低时,共存体系的存在对吸附行为影响较小. 随着重金属溶液浓度的增加,微塑料对重金属的吸附才开始受到抑制[35].
第四,真实水环境中溶液介质与室内实验环境有差异. 不同环境水体也会对吸附产生影响,在废水和灌溉水中,有机物的存在增加了塑料对Pb和Cr的吸附. Fu等发现以雨水为溶液介质,微塑料对重金属的吸附量高于纯水,这主要是由于雨水中存在有机物,会影响吸附过程,提高吸附效率[67]. 而海水中盐度和pH值的共同作用有时会增加微塑料对重金属的吸附,有时会减少微塑料对重金属的吸附,这可能是由于阳离子之间对微塑料吸附位点的竞争所致[14].
-
(1)不同微塑料对不同重金属的吸附量不同,微塑料官能团、结构,重金属水合离子半径等是影响其吸附量的主要因素. 其中,PS较其它微塑料更易吸附重金属,而Pb则更容易被微塑料吸附. 另外,微塑料老化程度、粒径大小以及盐度、pH值等因素也都会影响微塑料对重金属吸附.
(2)在一般情况下,重金属与微塑料的吸附动力学模型可以用PSO模型进行很好的解释,而吸附等温线模型需要由微塑料与重金属类型共同决定.
(3)真实水环境中微塑料颗粒对重金属的吸附量小于室内实验结果,其主要原因为:微塑料与悬浮颗粒物以及各种重金属之间均存在竞争吸附,不同溶液介质pH值、盐度、有机物均存在差异,室内实验中微塑料与重金属的浓度设置远高于实际环境.
水环境中微塑料对重金属的吸附及其影响因素
Review on the adsorption of heavy metals with microplastics in water environment and its influencing factors
-
摘要: 微塑料(microplastics,MPs)其自身是一种新兴污染物,且还可作为载体吸附重金属等其它污染物,对生态环境造成威胁. 本文以Web of Science核心数据库及Science Direct数据库作为数据源,搜集了2016—2022年间发表的有关微塑料对重金属吸附的文献,分析了不同环境条件下微塑料对重金属的吸附及其影响影响因素,还比较了室内实验结果与实际环境的差异. 结果表明,不同微塑料对重金属吸附量不同,其差异主要与微塑料官能团、结构,重金属水合离子半径等因素有关. 其中,聚苯乙烯(polystyrene,PS)较其它微塑料更易吸附重金属,而铅(Pb)则更易被微塑料吸附. 另外,微塑料老化程度、粒径大小以及盐度、pH值等因素也都会影响微塑料对重金属吸附. 研究还发现,真实水环境中微塑料对重金属的吸附量总体上小于室内实验结果,也就是说,室内实验往往高估了微塑料对重金属的吸附.Abstract: Microplastics (MPs) are an emerging pollutant that can be used as a vector to adsorb and transport heavy metals and other pollutants, posing a threat to the environment. In this paper, the core database of Web of Science and Science Direct database were used as data sources to collect the literature on the adsorption of heavy metals with MPs published from 2016 to 2022, and analyze the metal adsorption of MPs and the influencing factors. The difference between the lab experiments and the actual environment was also studied. The results showed that the difference of metal adsorption capacity for various MPs was mainly related to the functional groups, crystallinity, and the radius of heavy metal hydration ions. Polystyrene (PS) can adsorb more heavy metals than the other MPs, and lead (Pb) can be adsorbed more easily by MPs. In addition, the aging degree and particle size of MPs, and the pH and salinity of environmental conditions may also greatly affect the metal adsorption of MPs. It was also observed that the metal adsorption capacity of MPs in real water environment was smaller than the results from lab experiments. This means that lab experiments may often overestimate the metal adsorption capacity of MPs.
-
Key words:
- microplastics /
- heavy metals /
- adsorption /
- influence factor /
- laboratory experiment /
- real environment.
-
塑料作为人类生活中不可或缺的一种材料,被广泛运用于各个行业[1]. 随之而来的,塑料对环境的污染也成为了亟待解决的热点问题[2]. 近年来,微塑料(microplastics,MPs) ,即尺寸小于5 mm的塑料碎片[3- 4],由于其在水环境中的普遍分布、对生物体的生物影响以及不可降解的特性而备受关注[5-6]. 微塑料由于其体积小、密度低的特点很容易在水体中扩散[7],湖泊、河口、海滩等地区都是微塑料聚集的重点地区[8-12] ,甚至在北极地区都有关于微塑料的报道[13]. 与大中型塑料相比,微塑料具有粒径小、比表面积大、疏水性强等特点[14-15],更易于吸附污染物质[16-17],并且将污染物转移到食物链中[18],最后到达人类体内[19],重金属就是典型污染物其中之一[20-21].
重金属可以吸附到微塑料上[15, 22],其吸附量会受其自身物理化学特性影响,例如形态、比表面积、老化程度等[14-15, 23-26]. 并且塑料在制造过程中也会添加少量金属作为催化剂、颜料或者稳定剂[14]. 部分重金属作为一种可溶性有毒污染物[27],即使在低浓度下,也会对生物体造成伤害[25, 28]. 由于重金属离子和微塑料的毒性,重金属离子吸附到微塑料上可能会加剧两者对水生环境的潜在风险[23]. 重金属在微塑料上的吸附量也与其所处环境条件有关,例如pH值、盐度等[22]. Gao等通过室内实验与现场实验对比发现,现场实验中微塑料对重金属吸附量小于室内实验[29]. Holmes等发现,从海滩收集的微塑料颗粒中重金属浓度超过了当地河口沉积物中的浓度,这可能是由于塑料表面形成有机物及微生物,与重金属生成共沉淀导致的[30]. 真实水环境中微塑料对重金属吸附的影响因素远大于室内实验环境,这可能会导致两者的吸附量存在差异. 近年来,虽然关于微塑料对重金属吸附的研究越来越多,但是单个研究中,微塑料及涉及的重金属数量往往较少,考虑的影响因素也多局限在单一因素,较少有文章将多种微塑料/重金属、多种因素联系在一起进行综合比较. 此外,关于微塑料与重金属吸附作用的研究多集中于室内实验,也缺乏与真实水环境的对比.
本文以Web of Science核心数据库及Science Direct数据库作为数据源,对不同环境条件下不同微塑料对于不同重金属的吸附及其影响因素进行综合对比分析,并且还对室内实验结果与实际环境的差异进行研究,旨在探究微塑料吸附重金属的主要影响因素以及室内实验结果能否用来评估实际水环境中的微塑料对重金属的吸附水平.
1. 材料与方法(Materials and methods)
1.1 文献收集
本文搜集了2016—2022年期间发表的相关论文,数据库包括Web of Science(
https://www.webofscience.com/wos/alldb/basic-search ), Science Direct (https://www.sciencedirect.com/ ). 搜索关键词为“microplastics AND heavy metal AND adsorption” [31]. 参考《海水中微塑料的测定 傅里叶变换显微红外光谱法》(DB21/T 2751-2017),微塑料的搜索种类包括聚乙烯(polyethylene,PE)、聚丙烯(polypropylene, PP)、聚氯乙烯(polyvinyl chloride, PVC)、聚苯乙烯(polystyrene, PS)、聚氨酯(polyurethane, PU)、聚酰胺(nylon)、聚对苯二甲酸乙二脂(polyethylene terephthalate, PET);参考《海水水质标准》(GB3097-1997)和《地表水环境质量标准》(GB 3838-2002),重金属的搜索种类包括铜(Cu)、铅(Pb)、锌(Zn)、镉(Cd)、铬(Cr). 从文献中直接提取出吸附动力学及吸附等温线模型、微塑料粒径及老化程度. 不同微塑料及重金属的最大吸附量(Qmax 1.2 统计分析
当样本量大于3时,纳入统计分析. 使用SPSS 26.0对组间数据进行单因素方差分析(ANOVA),若P<0.05,则表示差异显著. 本文采用Origin 2022b进行数据描述性分析以及箱线图、弦图的制作.
2. 结果与讨论 (Results and discussion)
2.1 文献分析
共搜集到文献200篇,文献筛选流程见图1. 文献数量逐年递增(图2),说明该研究领域的研究热度也在逐年递增. 搜集到的文献中,室内实验文献共33篇,微塑料种类涉及PE、PET、PP、PVC、PS,重金属涉及Cu、Pb、Zn、Cd、Cr. 关于PS的论文有17篇, Pb的论文有19篇,说明PS与Pb是被研究的最多的微塑料和重金属,涉及Zn的研究最少,仅检索出4篇论文. 微塑料老化及粒径是关于微塑料对重金属吸附考虑最多的两个的因素,其中搜集到提及微塑料老化的文献有13篇,及微塑料粒径的文献共有33篇,粒径范围主要集中在0—2 mm. 关于真实水环境中微塑料吸附重金属的文献仅检索出6篇,远少于室内实验研究.
 图 2 关于微塑料与重金属吸附作用的已发表文献时间分布Figure 2. Temporal distribution of published papers on the adsorption of heavy metals with MPs
图 2 关于微塑料与重金属吸附作用的已发表文献时间分布Figure 2. Temporal distribution of published papers on the adsorption of heavy metals with MPs2.2 微塑料对重金属的吸附特征
由图3(a)可以看出,对于Cu、Pb、Cr等3种重金属,PS对它们的亲和力最强,其次是PVC、PE、PP与PET. PS特有的苯环结构可能是导致其对重金属吸附能力强的主要原因[32]. PVC和PP中都含有钛,但是PVC的吸附性能高于PP,这说明微塑料对重金属的吸附量不仅仅受单一的特性影响,而是所有这些特性的组合决定了微塑料与重金属的吸附作用[14]. PET在生产过程中会添加保护其免受老化和生物定殖的添加剂,这会导致其对于Pb的吸附量最弱 [14]. 而不同微塑料对于Cd的吸附量由大到小的顺序为,PVC>PP>PS>PE. 与其它微塑料相比,PVC可以为Cd提供更多的吸附位点[32]. Cd在PE上的吸附机制归因于范德华力,而PS则为π-π相互作用. 吸附机制的不同可能是导致其吸附量差异的主要原因[33].
 图 3 (a) 不同微塑料对同种重金属吸附量; (b) 同种微塑料对不同重金属吸附量;(c) 不同微塑料对不同重金属吸附量对比图Figure 3. (a)The adsorption capacity for the same heavy metal with different MPs ; (b)The adsorption capacity for different heavy metals with the same MPs; (c) Comparison of adsorption capacity of different heavy metals with different MPs.注: 箭头粗细表示吸附量的高低,箭头越粗表示吸附量越高,箭头越细表示吸附量越低.Note:The thickness of the arrow indicates the strength of the adsorption capacity. The thicker arrow indicates the higher adsorption capacity.
图 3 (a) 不同微塑料对同种重金属吸附量; (b) 同种微塑料对不同重金属吸附量;(c) 不同微塑料对不同重金属吸附量对比图Figure 3. (a)The adsorption capacity for the same heavy metal with different MPs ; (b)The adsorption capacity for different heavy metals with the same MPs; (c) Comparison of adsorption capacity of different heavy metals with different MPs.注: 箭头粗细表示吸附量的高低,箭头越粗表示吸附量越高,箭头越细表示吸附量越低.Note:The thickness of the arrow indicates the strength of the adsorption capacity. The thicker arrow indicates the higher adsorption capacity.由图3(b)可以看出,针对不同重金属,PE、PS、PVC对Pb吸附量最大,PP对Pb的吸附量也相对较大,主要是因为在这些重金属中,Pb的水合离子半径最小[34],而对于等电荷金属的静电吸附,吸附亲和力与离子半径呈负相关. 尽管Cu的水合离子半径与Cd相似,但PS对Cu的吸附量比Cd高. 这一结果可以用Cu比Cd更强的络合能力来解释[34]. 而PVC的结晶度较小,更有利于其对Cd的吸附[32]. 对于PP,其对重金属的吸附量的大小顺序为Cd>Pb>Cu>Cr. 重金属在PP颗粒上的吸附主要集中在表面,此时PP对不同重金属的吸附机理可能是导致吸附量产生差异的主要原因[35].
微塑料对重金属吸附量差异主要与微塑料官能团、结构,重金属水合离子半径等因素有关. 由图3(c)可以看出,PS较其它微塑料更易吸附重金属,而Pb则更容易被微塑料所吸附.
2.3 微塑料对重金属吸附的影响因素
(1)微塑料老化 微塑料老化会改变微塑料表面性质而影响其对重金属的吸附行为,老化方式与老化程度的不同也对微塑料自身官能团、结构产生影响[36-38]. 老化微塑料对重金属的吸附量往往大于新鲜微塑料(图4). Hadiuzzaman等通过光降解的方式对PE进行人工老化,发现老化PE与新鲜PE相比,生成了新的含氧官能团,导致老化PE对Pb吸附量比新鲜PE高[36]. Li等发现老化后的PE比表面积增加,同时生成新的官能团,对Cr的吸附量比新鲜PE高[37]. 微塑料老化后,其表面存在更多的孔洞或裂缝,导致与重金属接触的表面积更大,吸收污染物的量提高[38]. Liu等通过对比PE与PP的老化过程,发现两者官能团生成顺序不同,这表明不同微塑料的老化机制存在差异[39]. 结合图3(a)发现,其实验结果中PE对于Cd的吸附数据偏高,该实验在PE老化时采用了长江水振荡老化18个月的方式,老化时间较长. 这表明老化微塑料对重金属的吸附量随着老化时间的延长而增加[38, 40]. Lin等通过FTIR观察到PP光化学老化过程中,羟基、羰基和乙烯基的形成,由此判断官能团的变化可以用来评价PP的老化程度[41]. Fan通过实验发现老化PP的zeta电位由老化前的-5.36mV下降至-9.52 mV,这也进一步促进了PP对Pb的吸附[42],但是在其实验结果中,老化PP对Pb吸附量达到1126 μg·g−1,远高于新鲜PP对Pb吸附量,通过实验条件对比发现,该实验中新鲜微塑料的吸附量在正常范围内,因此推断其老化方式是导致吸附量偏离正常值的主要原因[42]. Liu等也发现在老化过程中,与化学氧化作用相比,光老化起主要作用,这表明老化程度确实与老化方式有关[39]. PS塑料老化后吸附量中值略大于新鲜PS,但是总体吸附量与新鲜PS差异并不显著. Xi等实验发现,老化PS与原始PS相比,含氧官能团增多,对重金属吸附量增强[43]. 由此可以推测对于PS塑料来说,老化程度会影响其对重金属的吸附量,但是影响程度较小.
 图 4 新鲜微塑料与老化微塑料吸附量对比Figure 4. Comparison of adsorption capacity of fresh MPs and aged MPs
图 4 新鲜微塑料与老化微塑料吸附量对比Figure 4. Comparison of adsorption capacity of fresh MPs and aged MPs自然环境中的微塑料老化增大了微塑料对于重金属的吸附作用,进而强化了微塑料作为重金属污染物的载体作用,从而加大了微塑料和重金属对生态环境的联合风险[44- 45].
(2)微塑料粒径 不同粒径微塑料的比表面积不同,进而对重金属的吸附量也不同[29, 46]. 由图5可以看出,微塑料对重金属的吸附量总体趋势随粒径的增加而减小. Wang等通过对比3种不同粒径的PE对Cd的吸附量发现,粒径是决定Cd吸附到PE上的主要因素,较小的颗粒具有更大的比表面积和更多的吸附点位[46],从而导致粒径较小的微塑料对重金属吸附量更大. 而Zhou等研究中PE的粒径虽然大于Wang等采用的微塑料,但是由于其取自商业材料,表面较为粗糙,导致其吸附量更大[34]. Gao等比较了不同粒径的PP与重金属之间的亲和性,结果表明,随着PP粒径的增大,Pb、Cu、Cd在PP上的吸附量显著降低[29]. Shen等探究了PP对Pb吸附量,虽然粒径最小,但是其吸附量小于其它粒径微塑料,这可能是吸附实验中微塑料初始浓度太低所导致的[47]. 而当粒径小于0.5 mm时,PS对Pb的吸附量随粒径变化较为明显,通过条件对比发现,除了粒径,pH值与重金属初始浓度的不同也对两组微塑料对重金属的吸附量有一定的影响,进一步导致了吸附量的剧烈变化. 值得指出的是,在微塑料粒径较小时表现出的较大变动(图5b, 5c, 5f),本文由于样本量所限还无法确认并揭示其准确原因,但这一现象值得进一步探究. 粒径越小的微塑料越容易成为环境中重金属迁移的载体,被生物摄食并在体内积聚,进而表现出更高的毒性[48-50].
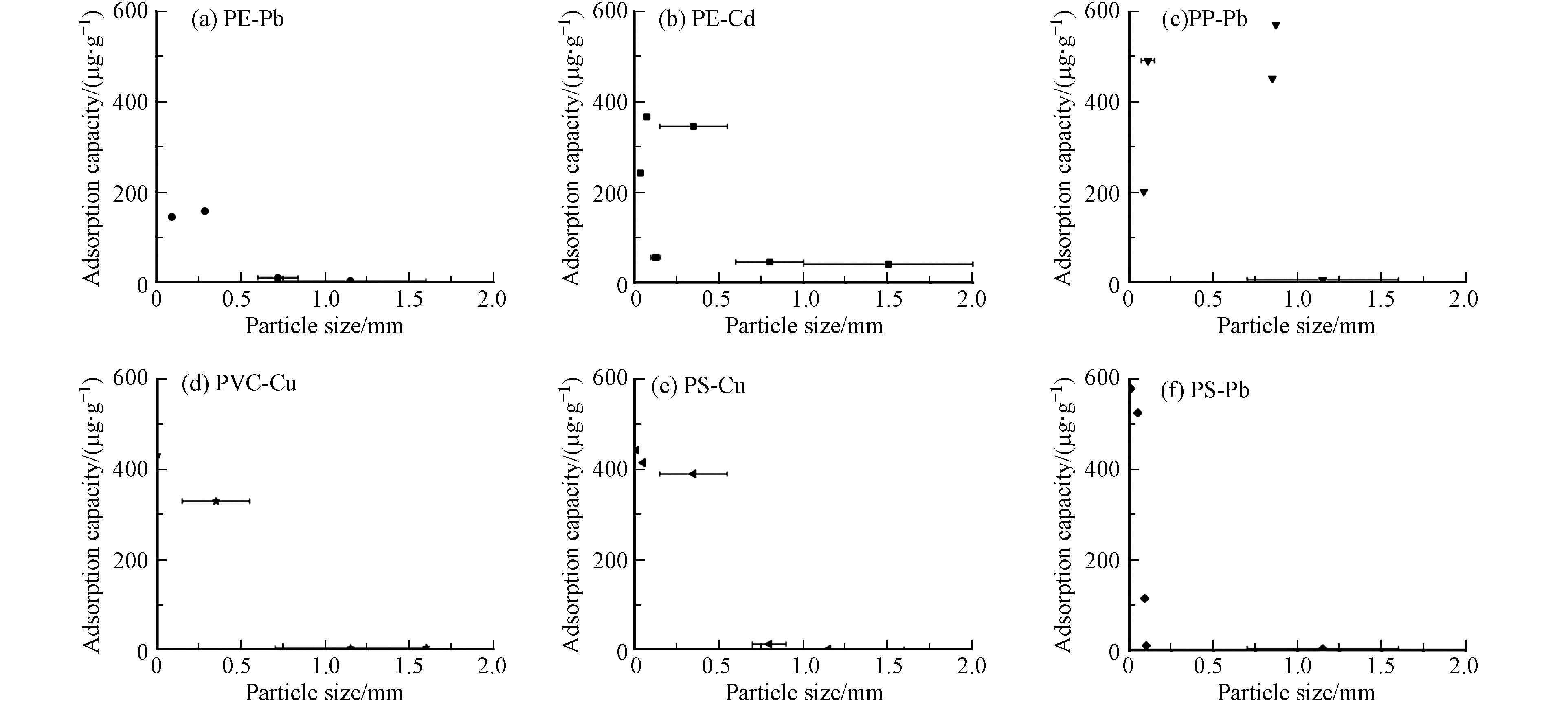 图 5 不同粒径微塑料对不同重金属吸附量对比Figure 5. Comparison of adsorption capacity of heavy metals with MPs with different particle sizes注:误差棒表示粒径范围Note: Error bars indicate particle size ranges
图 5 不同粒径微塑料对不同重金属吸附量对比Figure 5. Comparison of adsorption capacity of heavy metals with MPs with different particle sizes注:误差棒表示粒径范围Note: Error bars indicate particle size ranges(3)盐度 盐度对微塑料对重金属吸附的影响较为显著. 当盐度从5增加到35时,PS和PET颗粒对Cu的吸附量呈小幅下降趋势. 盐度的增加会破坏吸附剂的电荷平衡,中和吸附剂的表面电荷,从而抑制静电作用[51]. Barus等研究了PS在不同盐度下对Pb、Zn和Cu等3种金属的吸附量,其吸附量均随盐度的增加而降低[22]. 当盐度增加时,微塑料不仅会与重金属相互作用,还会与其它离子相互作用. 海水中Na、Ca、Mg等离子的浓度增加,导致电解质离子的竞争效应[15, 40]. 随着离子强度增加到一定水平,特别是较高浓度的Na离子可能会激烈竞争微塑料表面的阳离子交换位点,进一步抑制重金属和微塑料相互作用,导致吸附量下降. 此外,Ho等发现,在一定盐度条件下,Ca对Pb的抑制作用远大于Na,可能的原因是Ca和Pb具有相同的价态,因此阻止了Pb与微塑料有限活性位点的结合[52].
(4)pH值 微塑料对重金属的吸附会受pH影响. Holmes等研究发现,PE对Cd、Co、Ni和Pb的吸附量随着pH值的增加而增加,但对Cr的吸附量降低[30]. 随着pH从2.0增加到6.0,Pb在不同微塑料上的吸附均增加. 在Pb吸附到微塑料上的过程中,溶液 pH 影响溶液中 Pb物种的分布和微塑料的表面性质. 该现象可归因于带负电的微塑料与带正电的Pb之间的静电吸引力. pH值的增加会加速重金属在微塑料中的吸附过程[15, 24]. 值得注意的是由于气候变化的影响,海水pH值总体呈现酸化趋势 [53-54],海水酸化总体上不利于微塑料对于重金属的吸附,一定程度上削弱了微塑料对于重金属的携带和运输作用.
2.4 微塑料对重金属的吸附过程模拟
微塑料对重金属的吸附动力学模拟模型主要有Pseudo-first order model(PFO)模型、Pseudo-second order(PSO)模型,而PSO模型往往有更好的模拟结果[33, 48, 55](表1),这表明吸附过程不仅包括污染物吸附到微塑料上的不同位点,还涉及多重吸附,传质和离子扩散等不同阶段[59]. 通过绘制
qt t1/2 表 1 重金属在微塑料上的吸附等温线和动力学模型Table 1. Adsorption isotherms and kinetic models of heavy metals with MPs微塑料类型Type 重金属类型Type 吸附动力学模型Adsorption kinetics 吸附等温线模型Adsorption isotherm 参考文献Reference PS/PP/PEPVC/PET Cd/Co/Cr/CuNi/Pb/Zn NM Langmuir [14] PE Cd/Co/CrCu/Ni/Pb/ NM Freundlich/Langmuir [15] PS/PET Cu PSO Freundlich [21] PE Pb(Ⅱ) PSO Freundlich [26] PVC/PE/PS Pb PSO PVC-Pb(Ⅱ): BETPS/PE-Pb(Ⅱ): Freundlich [33] PS Pb/Cu/Cd/Ni/Zn NM Freundlich [38] PP/PE/PET Cu/Cr/Pb PSO NM [48] PE Cr PFO Langmuir [56] PS/PVC Cu/Zn PFO NM [57] PE Cr/Cu/Ag/CdHg/Ni/Co/Pb/Zn PFO Non-linear Langmuir, Freundlich [58] 注: NM:未提及 Note: NM: Not Mentioned | Show Table DownLoad:
CSV
DownLoad:
CSV
Freundlich模型、Langmuir模型与Brunauer-Emmett-Teller(BET)模型,都能较好地拟合微塑料与重金属吸附等温线. Freundlich模型表明吸附过程发生在微塑料的异质表面,涉及单层和多层吸附[21, 38],往往为物理吸附,重金属通过弱键吸附在微塑料上,并在微塑料表面形成一层薄膜[60]. 该模型适用于较多微塑料与重金属吸附场景,可以很好的拟合PVC对Pb的吸附[33],同时也适用于PE对Cr的吸附[56]. Langmuir等温线模型通常用于描述单层吸附. 该模型假设吸附剂表面是均匀的,并且表面上的所有吸附位点具有相同的吸附物亲和力[61]. Godoy同时使用Freundlich与Langmuir模型对5种不同的微塑料与重金属进行拟合,结果Langmuir模型具有更好的相关系数. BET等温线模型是从Langmuir等温线模型扩展得出的多层吸附系统的一种特殊形式[61]. 该模型多用于液固平衡系统,能较好的拟合PS与PE对Pb的吸附过程[33].
2.5 真实水环境与室内实验吸附量对比
由图6可知,真实水环境中和室内实验条件下微塑料均呈现出对重金属的富集作用,但真实水环境中微塑料对重金属的吸附量小于室内实验中微塑料对重金属的吸附. 造成这一结果的原因可能有以下几点:
 图 6 不同环境下微塑料与重金属吸附量对比Figure 6. Comparison of adsorption capacity of heavy metals with MPs in different environments
图 6 不同环境下微塑料与重金属吸附量对比Figure 6. Comparison of adsorption capacity of heavy metals with MPs in different environments第一,真实水环境中微塑料及重金属浓度与室内实验条件有较大差异. Wang等研究发现,随着微塑料浓度的增加,微塑料对重金属的吸附量显著增加,当微塑料浓度从1.0 g·L−1增加到5.0 g·L−1时,微塑料对Cu的吸附量从40.3 µg·g−1增加到了55.1 µg·g-1[25]. 但是随着微塑料浓度的持续增加,微塑料表面的空白吸附点数量增加,导致微塑料的单位吸附容量下降[55]. 室内实验中,微塑料与重金属浓度通常维持在在10—100 g·L−1、1—20 mg·L−1之间,而真实水环境中微塑料与重金属浓度要远低于该区间. 室内实验中微塑料与重金属的浓度范围跨度较大,这也导致了微塑料对重金属的吸附量数据离散程度大于真实水环境. 同时,真实水环境中微塑料对重金属的吸附行为并不明显,这也可能是真实水环境中重金属环境浓度较低导致.
第二,环境中存在悬浮颗粒物. 除了微塑料,重金属还会与水中的其它颗粒物共存,并且它们对重金属有较高的亲和力[62]. Qi等比较了悬浮颗粒物、沉积物以及微塑料对Pb的吸附作用,结果表明,在中性条件下,这三者之间对Pb都存在竞争性吸附[63]. 悬浮颗粒物与沉积物对重金属的吸附量大于微塑料[64—65],这说明微塑料在水环境中作为重金属载体的功能与悬浮颗粒物、沉积物相比较弱,很难对环境产生重大影响[66].
第三,真实水环境中有多种重金属离子共存. Gao发现,Cu、Pb、Cd共存的混合溶液的吸附容量低于单一金属溶液的吸附容量. 由于3种金属离子的存在,金属离子在微塑料表面的吸附是竞争的[29]. 微塑料可以从Cu和Cd共存的溶液中选择性吸附Cu和Cd,但重金属离子会在微塑料表面竞争活性吸附位点. Shen通过共存离子实验也发现,Cu的存在会干扰Pb的吸附[47]. 同时,当混合重金属溶液浓度较低时,共存体系的存在对吸附行为影响较小. 随着重金属溶液浓度的增加,微塑料对重金属的吸附才开始受到抑制[35].
第四,真实水环境中溶液介质与室内实验环境有差异. 不同环境水体也会对吸附产生影响,在废水和灌溉水中,有机物的存在增加了塑料对Pb和Cr的吸附. Fu等发现以雨水为溶液介质,微塑料对重金属的吸附量高于纯水,这主要是由于雨水中存在有机物,会影响吸附过程,提高吸附效率[67]. 而海水中盐度和pH值的共同作用有时会增加微塑料对重金属的吸附,有时会减少微塑料对重金属的吸附,这可能是由于阳离子之间对微塑料吸附位点的竞争所致[14].
3. 结论(Conclusion)
(1)不同微塑料对不同重金属的吸附量不同,微塑料官能团、结构,重金属水合离子半径等是影响其吸附量的主要因素. 其中,PS较其它微塑料更易吸附重金属,而Pb则更容易被微塑料吸附. 另外,微塑料老化程度、粒径大小以及盐度、pH值等因素也都会影响微塑料对重金属吸附.
(2)在一般情况下,重金属与微塑料的吸附动力学模型可以用PSO模型进行很好的解释,而吸附等温线模型需要由微塑料与重金属类型共同决定.
(3)真实水环境中微塑料颗粒对重金属的吸附量小于室内实验结果,其主要原因为:微塑料与悬浮颗粒物以及各种重金属之间均存在竞争吸附,不同溶液介质pH值、盐度、有机物均存在差异,室内实验中微塑料与重金属的浓度设置远高于实际环境.
-
图 2 关于微塑料与重金属吸附作用的已发表文献时间分布
Figure 2. Temporal distribution of published papers on the adsorption of heavy metals with MPs
图 3 (a) 不同微塑料对同种重金属吸附量; (b) 同种微塑料对不同重金属吸附量;(c) 不同微塑料对不同重金属吸附量对比图
Figure 3. (a)The adsorption capacity for the same heavy metal with different MPs ; (b)The adsorption capacity for different heavy metals with the same MPs; (c) Comparison of adsorption capacity of different heavy metals with different MPs.
图 4 新鲜微塑料与老化微塑料吸附量对比
Figure 4. Comparison of adsorption capacity of fresh MPs and aged MPs
图 5 不同粒径微塑料对不同重金属吸附量对比
Figure 5. Comparison of adsorption capacity of heavy metals with MPs with different particle sizes
图 6 不同环境下微塑料与重金属吸附量对比
Figure 6. Comparison of adsorption capacity of heavy metals with MPs in different environments
表 1 重金属在微塑料上的吸附等温线和动力学模型
Table 1. Adsorption isotherms and kinetic models of heavy metals with MPs
微塑料类型Type 重金属类型Type 吸附动力学模型Adsorption kinetics 吸附等温线模型Adsorption isotherm 参考文献Reference PS/PP/PEPVC/PET Cd/Co/Cr/CuNi/Pb/Zn NM Langmuir [14] PE Cd/Co/CrCu/Ni/Pb/ NM Freundlich/Langmuir [15] PS/PET Cu PSO Freundlich [21] PE Pb(Ⅱ) PSO Freundlich [26] PVC/PE/PS Pb PSO PVC-Pb(Ⅱ): BETPS/PE-Pb(Ⅱ): Freundlich [33] PS Pb/Cu/Cd/Ni/Zn NM Freundlich [38] PP/PE/PET Cu/Cr/Pb PSO NM [48] PE Cr PFO Langmuir [56] PS/PVC Cu/Zn PFO NM [57] PE Cr/Cu/Ag/CdHg/Ni/Co/Pb/Zn PFO Non-linear Langmuir, Freundlich [58] 注: NM:未提及 Note: NM: Not Mentioned
下载: 导出CSV
-
[1] ANDRADY A L. Microplastics in the marine environment [J]. Marine Pollution Bulletin, 2011, 62(8): 1596-1605. doi: 10.1016/j.marpolbul.2011.05.030 [2] GEYER R, JAMBECK J R, LAW K L. Production, use, and fate of all plastics ever made [J]. Science Advances, 2017, 3(7): e1700782. doi: 10.1126/sciadv.1700782 [3] COLE M, LINDEQUE P, HALSBAND C, et al. Microplastics as contaminants in the marine environment: A review [J]. Marine Pollution Bulletin, 2011, 62(12): 2588-2597. doi: 10.1016/j.marpolbul.2011.09.025 [4] NAPPER I E, THOMPSON R C. Plastic debris in the marine environment: History and future challenges [J]. Global Challenges, 2020, 4(6): 1900081. doi: 10.1002/gch2.201900081 [5] BHATTACHARYA P, LIN S J, TURNER J, et al. Physical adsorption of charged plastic nanoparticles affects algal photosynthesis [J]. Journal of Physical Chemistry C, 2010, 114: 16556-16561. doi: 10.1021/jp1054759 [6] YANG Y Y, LIU G H, SONG W J, et al. Plastics in the marine environment are reservoirs for antibiotic and metal resistance genes [J]. Environment International, 2019, 123: 79-86. doi: 10.1016/j.envint.2018.11.061 [7] AVIO C G, GORBI S, REGOLI F. Plastics and microplastics in the oceans: From emerging pollutants to emerged threat [J]. Marine Environmental Research, 2017, 128: 2-11. doi: 10.1016/j.marenvres.2016.05.012 [8] HAN M, NIU X R, TANG M, et al. Distribution of microplastics in surface water of the lower Yellow River near estuary [J]. The Science of the Total Environment, 2020, 707: 135601. doi: 10.1016/j.scitotenv.2019.135601 [9] NABIZADEH R, SAJADI M, RASTKARI N, et al. Microplastic pollution on the Persian Gulf shoreline: A case study of Bandar Abbas city, Hormozgan Province, Iran [J]. Marine Pollution Bulletin, 2019, 145: 536-546. doi: 10.1016/j.marpolbul.2019.06.048 [10] NEL H A, FRONEMAN P W. A quantitative analysis of microplastic pollution along the south-eastern coastline of South Africa [J]. Marine Pollution Bulletin, 2015, 101(1): 274-279. doi: 10.1016/j.marpolbul.2015.09.043 [11] XU P, PENG G Y, SU L, et al. Microplastic risk assessment in surface waters: A case study in the Changjiang Estuary, China [J]. Marine Pollution Bulletin, 2018, 133: 647-654. doi: 10.1016/j.marpolbul.2018.06.020 [12] ZHANG J X, ZHANG C L, DENG Y X, et al. Microplastics in the surface water of small-scale estuaries in Shanghai [J]. Marine Pollution Bulletin, 2019, 149: 110569. doi: 10.1016/j.marpolbul.2019.110569 [13] LUSHER A L, TIRELLI V, O’CONNOR I, et al. Microplastics in Arctic polar waters: The first reported values of particles in surface and sub-surface samples [J]. Scientific Reports, 2015, 5: 14947. doi: 10.1038/srep14947 [14] GODOY V, BLÁZQUEZ G, CALERO M, et al. The potential of microplastics as carriers of metals[J]. Environmental Pollution(Barking, Essex: 1987), 2019, 255(Pt 3): 113363. [15] HOLMES L A, TURNER A, THOMPSON R C. Interactions between trace metals and plastic production pellets under estuarine conditions [J]. Marine Chemistry, 2014, 167: 25-32. doi: 10.1016/j.marchem.2014.06.001 [16] TOURINHO P S, KOČí V, LOUREIRO S, et al. Partitioning of chemical contaminants to microplastics: Sorption mechanisms, environmental distribution and effects on toxicity and bioaccumulation[J]. Environmental Pollution(Barking, Essex: 1987), 2019, (Pt B): 252: 1246-1256. [17] WANG F, SHIH K M, LI X Y. The partition behavior of perfluorooctanesulfonate (PFOS) and perfluorooctanesulfonamide (FOSA) on microplastics [J]. Chemosphere, 2015, 119: 841-847. doi: 10.1016/j.chemosphere.2014.08.047 [18] SANTANA M F M, MOREIRA F T, TURRA A. Trophic transference of microplastics under a low exposure scenario: Insights on the likelihood of particle cascading along marine food-webs [J]. Marine Pollution Bulletin, 2017, 121(1/2): 154-159. [19] CARBERY M, O'CONNOR W, PALANISAMI T. Trophic transfer of microplastics and mixed contaminants in the marine food web and implications for human health [J]. Environment International, 2018, 115: 400-409. doi: 10.1016/j.envint.2018.03.007 [20] ZHANG G Z, PAN Z K, HOU X W, et al. Distribution and bioaccumulation of heavy metals in food web of Nansi Lake, China [J]. Environmental Earth Sciences, 2015, 73(5): 2429-2439. doi: 10.1007/s12665-014-3592-z [21] WANG X X, ZHANG R X, LI Z Y, et al. Adsorption properties and influencing factors of Cu(II) on polystyrene and polyethylene terephthalate microplastics in seawater [J]. Science of the Total Environment, 2022, 812: 152573. doi: 10.1016/j.scitotenv.2021.152573 [22] BARUS B S, CHEN K, CAI M G, et al. Heavy metal adsorption and release on polystyrene particles at various salinities [J]. Frontiers in Marine Science, 2021, 8: 671802. doi: 10.3389/fmars.2021.671802 [23] GUO X, WANG J L. Projecting the sorption capacity of heavy metal ions onto microplastics in global aquatic environments using artificial neural networks [J]. Journal of Hazardous Materials, 2021, 402: 123709. doi: 10.1016/j.jhazmat.2020.123709 [24] PURWIYANTO A I S, SUTEJA Y, Trisno, et al. Concentration and adsorption of Pb and Cu in microplastics: Case study in aquatic environment [J]. Marine Pollution Bulletin, 2020, 158: 111380. doi: 10.1016/j.marpolbul.2020.111380 [25] WANG Q, ZHANG Y, WANGJIN X X, et al. The adsorption behavior of metals in aqueous solution by microplastics effected by UV radiation [J]. Journal of Environmental Sciences (China), 2020, 87: 272-280. doi: 10.1016/j.jes.2019.07.006 [26] LIU S, HUANG J H, ZHANG W, et al. Investigation of the adsorption behavior of Pb(II) onto natural-aged microplastics as affected by salt ions [J]. Journal of Hazardous Materials, 2022, 431: 128643. doi: 10.1016/j.jhazmat.2022.128643 [27] NAQASH N, PRAKASH S, KAPOOR D, et al. Interaction of freshwater microplastics with biota and heavy metals: A review [J]. Environmental Chemistry Letters, 2020, 18(6): 1813-1824. doi: 10.1007/s10311-020-01044-3 [28] CECHINEL M A P, ULSON DE SOUZA S M A G, ULSON DE SOUZA A A. Study of lead (II) adsorption onto activated carbon originating from cow bone [J]. Journal of Cleaner Production, 2014, 65: 342-349. doi: 10.1016/j.jclepro.2013.08.020 [29] GAO F L, LI J X, SUN C J, et al. Study on the capability and characteristics of heavy metals enriched on microplastics in marine environment [J]. Marine Pollution Bulletin, 2019, 144: 61-67. doi: 10.1016/j.marpolbul.2019.04.039 [30] HOLMES L A, TURNER A, THOMPSON R C. Adsorption of trace metals to plastic resin pellets in the marine environment [J]. Environmental Pollution, 2012, 160: 42-48. doi: 10.1016/j.envpol.2011.08.052 [31] LIU S T, SHI J F, WANG J, et al. Interactions between microplastics and heavy metals in aquatic environments: A review [J]. Frontiers in Microbiology, 2021, 12: 652520. doi: 10.3389/fmicb.2021.652520 [32] GUO X T, HU G L, FAN X Y, et al. Sorption properties of cadmium on microplastics: The common practice experiment and A two-dimensional correlation spectroscopic study [J]. Ecotoxicology and Environmental Safety, 2020, 190: 110118. doi: 10.1016/j.ecoenv.2019.110118 [33] LIN Z K L, HU Y W, YUAN Y J, et al. Comparative analysis of kinetics and mechanisms for Pb(II) sorption onto three kinds of microplastics [J]. Ecotoxicology and Environmental Safety, 2021, 208: 111451. doi: 10.1016/j.ecoenv.2020.111451 [34] ZOU J Y, LIU X P, ZHANG D M, et al. Adsorption of three bivalent metals by four chemical distinct microplastics [J]. Chemosphere, 2020, 248: 126064. doi: 10.1016/j.chemosphere.2020.126064 [35] FAN T Y, ZHAO J, CHEN Y X, et al. Coexistence and adsorption properties of heavy metals by polypropylene microplastics [J]. Adsorption Science & #X0026; Technology, 2021, 2021: 4938749. [36] HADIUZZAMAN M, SALEHI M, FUJIWARA T. Plastic litter fate and contaminant transport within the urban environment, photodegradation, fragmentation, and heavy metal uptake from storm runoff [J]. Environmental Research, 2022, 212: 113183. doi: 10.1016/j.envres.2022.113183 [37] LI Y H, ZHANG Y, SU F, et al. Adsorption behaviour of microplastics on the heavy metal Cr(VI) before and after ageing [J]. Chemosphere, 2022, 302: 134865. doi: 10.1016/j.chemosphere.2022.134865 [38] MAO R F, LANG M F, YU X Q, et al. Aging mechanism of microplastics with UV irradiation and its effects on the adsorption of heavy metals [J]. Journal of Hazardous Materials, 2020, 393: 122515. doi: 10.1016/j.jhazmat.2020.122515 [39] LIU P, WU X W, HUANG H X Y, et al. Simulation of natural aging property of microplastics in Yangtze River water samples via a rooftop exposure protocol [J]. Science of the Total Environment, 2021, 785: 147265. doi: 10.1016/j.scitotenv.2021.147265 [40] GUO X T, PANG J W, CHEN S Y, et al. Sorption properties of tylosin on four different microplastics [J]. Chemosphere, 2018, 209: 240-245. doi: 10.1016/j.chemosphere.2018.06.100 [41] LIN W H, KUO J, LO S L. Effect of light irradiation on heavy metal adsorption onto microplastics [J]. Chemosphere, 2021, 285: 131457. doi: 10.1016/j.chemosphere.2021.131457 [42] FAN X L, MA Z X, ZOU Y F, et al. Investigation on the adsorption and desorption behaviors of heavy metals by tire wear particles with or without UV ageing processes [J]. Environmental Research, 2021, 195: 110858. doi: 10.1016/j.envres.2021.110858 [43] XI X L, DING D J, ZHOU H L, et al. Interactions of pristine and aged nanoplastics with heavy metals: Enhanced adsorption and transport in saturated porous media [J]. Journal of Hazardous Materials, 2022, 437: 129311. doi: 10.1016/j.jhazmat.2022.129311 [44] VROOM R J E, KOELMANS A A, BESSELING E, et al. Aging of microplastics promotes their ingestion by marine zooplankton [J]. Environmental Pollution, 2017, 231: 987-996. doi: 10.1016/j.envpol.2017.08.088 [45] HAN X X, VOGT R D, ZHOU J Y, et al. Increased Cu(II) adsorption onto UV-aged polyethylene, polypropylene, and polyethylene terephthalate microplastic particles in seawater [J]. Frontiers in Marine Science, 2021, 8: 770606. doi: 10.3389/fmars.2021.770606 [46] WANG F Y, YANG W W, CHENG P, et al. Adsorption characteristics of cadmium onto microplastics from aqueous solutions [J]. Chemosphere, 2019, 235: 1073-1080. doi: 10.1016/j.chemosphere.2019.06.196 [47] SHEN M C, SONG B, ZENG G M, et al. Surfactant changes lead adsorption behaviors and mechanisms on microplastics [J]. Chemical Engineering Journal, 2021, 405: 126989. doi: 10.1016/j.cej.2020.126989 [48] HAN X X, WANG S Y, YU X, et al. Kinetics and size effects on adsorption of Cu(II), Cr(III), and Pb(II) onto polyethylene, polypropylene, and polyethylene terephthalate microplastic particles [J]. Frontiers in Marine Science, 2021, 8: 785146. doi: 10.3389/fmars.2021.785146 [49] REHSE S, KLOAS W, ZARFL C. Short-term exposure with high concentrations of pristine microplastic particles leads to immobilisation of Daphnia magna [J]. Chemosphere, 2016, 153: 91-99. doi: 10.1016/j.chemosphere.2016.02.133 [50] YUAN W K, ZHOU Y F, CHEN Y L, et al. Toxicological effects of microplastics and heavy metals on the Daphnia magna [J]. Science of the Total Environment, 2020, 746: 141254. doi: 10.1016/j.scitotenv.2020.141254 [51] LIU S, HUANG J H, ZHANG W, et al. Microplastics as a vehicle of heavy metals in aquatic environments: A review of adsorption factors, mechanisms, and biological effects [J]. Journal of Environmental Management, 2022, 302: 113995. doi: 10.1016/j.jenvman.2021.113995 [52] HO W K, LEUNG K S-Y. The crucial role of heavy metals on the interaction of engineered nanoparticles with polystyrene microplastics [J]. Water Research, 2021, 201: 117317. doi: 10.1016/j.watres.2021.117317 [53] ORR J C, FABRY V J, AUMONT O, et al. Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms [J]. Nature, 2005, 437(7059): 681-686. doi: 10.1038/nature04095 [54] 石莉, 桂静, 吴克勤. 海洋酸化及国际研究动态 [J]. 海洋科学进展, 2011, 29(1): 122-128. doi: 10.3969/j.issn.1671-6647.2011.01.015 SHI L, GUI J, WU K Q. Developments in international studies on ocean acidification [J]. Advances in Marine Science, 2011, 29(1): 122-128(in Chinese). doi: 10.3969/j.issn.1671-6647.2011.01.015
[55] WU P F, CAI Z W, JIN H B, et al. Adsorption mechanisms of five bisphenol analogues on PVC microplastics[J]. The Science of the Total Environment, 2019, 650(Pt 1): 671-678. [56] ZON N F, ISKENDAR A, AZMAN S, et al. Sorptive behaviour of chromium on polyethylene microbeads in artificial seawater [J]. MATEC Web of Conferences, 2018, 250: 06001. doi: 10.1051/matecconf/201825006001 [57] BRENNECKE D, DUARTE B, PAIVA F, et al. Microplastics as vector for heavy metal contamination from the marine environment [J]. Estuarine, Coastal and Shelf Science, 2016, 178: 189-195. doi: 10.1016/j.ecss.2015.12.003 [58] TURNER A, HOLMES L A. Adsorption of trace metals by microplastic pellets in fresh water [J]. Environmental Chemistry, 2015, 12(5): 600. doi: 10.1071/EN14143 [59] YU F, YANG C F, HUANG G Q, et al. Interfacial interaction between diverse microplastics and tetracycline by adsorption in an aqueous solution [J]. The Science of the Total Environment, 2020, 721: 137729. doi: 10.1016/j.scitotenv.2020.137729 [60] SANDHYARANI N. Surface modification methods for electrochemical biosensors[M]//Electrochemical Biosensors. Amsterdam: Elsevier, 2019: 45-75. [61] SAADI R, SAADI Z, FAZAELI R, et al. Monolayer and multilayer adsorption isotherm models for sorption from aqueous media [J]. Korean Journal of Chemical Engineering, 2015, 32(5): 787-799. doi: 10.1007/s11814-015-0053-7 [62] CIFFROY P, MONNIN L, GARNIER J M, et al. Modelling geochemical and kinetic processes involved in lead (Pb) remobilization during resuspension events of contaminated sediments [J]. Science of the Total Environment, 2019, 679: 159-171. doi: 10.1016/j.scitotenv.2019.04.192 [63] QI K, LU N, ZHANG S Q, et al. Uptake of Pb(II) onto microplastic-associated biofilms in freshwater: Adsorption and combined toxicity in comparison to natural solid substrates [J]. Journal of Hazardous Materials, 2021, 411: 125115. doi: 10.1016/j.jhazmat.2021.125115 [64] XU X D, CAO Z M, ZHANG Z X, et al. Spatial distribution and pollution assessment of heavy metals in the surface sediments of the Bohai and Yellow Seas [J]. Marine Pollution Bulletin, 2016, 110(1): 596-602. doi: 10.1016/j.marpolbul.2016.05.079 [65] RüGNER H, SCHWIENTEK M, MILAČIČ R, et al. Particle bound pollutants in rivers: Results from suspended sediment sampling in Globaqua River Basins [J]. Science of the Total Environment, 2019, 647: 645-652. doi: 10.1016/j.scitotenv.2018.08.027 [66] CHEN C C, ZHU X S, XU H, et al. Copper adsorption to microplastics and natural particles in seawater: A comparison of kinetics, isotherms, and bioavailability [J]. Environmental Science & Technology, 2021, 55(20): 13923-13931. [67] FU Q M, TAN X F, YE S J, et al. Mechanism analysis of heavy metal lead captured by natural-aged microplastics [J]. Chemosphere, 2021, 270: 128624. doi: 10.1016/j.chemosphere.2020.128624 期刊类型引用(5)
1. 朱晓艳,王琪琛,姜懿真,武忠,柳钟惠,陈吉孝,王钰琳,袁宇翔. 微塑料对稻田土壤-水界面重金属分布及迁移的影响. 水生态学杂志. 2024(03): 10-20 .  百度学术
百度学术
2. 刘文娟,郭玉峰,邓文博. 光老化对不同粒径聚苯乙烯微塑料吸附Cu(Ⅱ)的影响. 环境科学. 2024(06): 3708-3715 . 百度学术
3. 张欣宇,唐玉朝,王坤,黄显怀,尹翠琴,高和气,伍昌年,黄健,李卫华. 原位生成纳米MnO_2胶体对Mn~(2+)的吸附机理实验研究. 环境科学与技术. 2024(04): 16-25 . 百度学术
4. 杨蓉,赵凡,桂向阳,曹峰赫,李博文,陈明. 老化作用对微塑料与镉在运河沿岸土壤中共迁移影响. 中国环境科学. 2024(11): 6260-6270 . 百度学术
5. 龚慧山,徐友宁,陈华清,柯海玲. 某地陈家沟河水中重金属元素时空变化及影响因素研究. 西北地质. 2023(04): 169-184 . 百度学术
其他类型引用(4)
-

 点击查看大图
点击查看大图

计量
- 文章访问数: 7754
- HTML全文浏览数: 7754
- PDF下载数: 188
- 施引文献: 9



