


-
砷(As)是一种广泛分布在水体、土壤和岩石等中的有毒致癌物质. 高砷地下水是指As浓度大于10 µg·L−1的地下水(世界卫生组织(WHO)规定的饮用水标准)[1],长期饮用高砷地下水会导致慢性砷中毒,严重威胁人体健康. 目前,高砷地下水广泛分布在世界各地,超过1.5亿人的饮水安全受到了威胁[2],据Rodriguez-Lado等的统计风险预测模型,中国受高砷地下水影响的人口约1960万[3]. 高砷地下水多是原生自然成因造成的,不同区域的原生高砷水成因存在一定差异.
微生物参与铁氧化物的还原性溶解被认为是高砷地下水形成的关键过程[4-5],在此过程中,溶解性有机碳(DOC)作为微生物代谢活动的主要碳源和能量来源,在一定程度上影响着元素的氧化还原反应和迁移转化[6-10]. DOC的同位素δ13CDOC可指示地下水中有机碳的来源,并反应微生物代谢活动. 溶解性无机碳(DIC)是微生物作用下有机质降解的重要产物,其稳定同位素δ13CDIC可用于判断地下水DIC的来源,揭示地下水中微生物对有机质的降解过程. 碳同位素在高砷地下水中主要用于地下水有机质来源的判别和有机质的微生物代谢指示两方面[11]. 因此,利用地下水稳定碳同位素表征微生物作用下有机质降解过程及其对砷富集的影响具有一定指示意义. 例如,周殷竹对河套盆地[12]和Guo等[13]对松嫩盆地的高砷地下水中δ13CDIC和δ13CDOC特征研究表明,铁氧化物还原性溶解过程中均有微生物参与并伴随着DOC的氧化分解共同导致地下水中砷的迁移和富集.
新疆奎屯地区是中国西北干旱区典型原生高砷水分布区,高砷地下水多分布在深层承压水层,同其他地区高砷地下水埋深在潜水层、浅层承压水层不同. 前期研究表明[14-16],该地区地下水多为碱性和还原性环境,铁氧化物矿物的还原性溶解是高砷地下水形成的主要机制,但微生物在该成因机制中起到的作用还不清楚. 因此,本研究以奎屯地区深层承压地下水为研究对象、地表水为对照并结合地下水水化学特征和稳定碳同位素分析,旨在查明研究区高砷地下水中微生物作用下有机质降解过程对砷富集的影响,进一步加深对新疆奎屯原生高砷地下水成因机制的认识,为地下水的保护及有效利用提供理论指导.
-
奎屯地区位于新疆天山山坡中段和准噶尔盆地西南部的奎屯河流域,深处于亚欧大陆内部,属于温带大陆性干旱荒漠气候,多年平均气温(7.3 ℃)较低,年平均降水量为165 mm,而蒸发量高达2080 mm,蒸发量远大于降水量. 奎屯河流域的地质范围从南部山区的古生代岩层延伸至南盆地中更新统的冰水沉积层,再到第四系松散沉积层的平原区[17]. 平原区地下水沉积物厚度在800—1400 m之间,含水层结构由单一的卵砂砾石潜水层逐渐转变为以黄灰色粉质黏土夹薄层砂、青灰色的砂和砂砾石、浅黄的粉细砂夹粘性土为主的多层承压水结构[18]. 南部山区以卵砾石区为主,以侏罗系、第三系与平原第四系接触,颗粒粒径大,平均厚度约为26 m. 地下水径流方向自南向北由上游山区流向下游平原区[19]. 地下水由单一结构潜水层过渡到多层结构潜水—承压水层,且水位埋深呈逐渐增加的趋势. 研究区地下水集中在深层承压水层,砷浓度分布极不均匀.
-
根据前期研究,奎屯地区126团和128团地下水中As浓度含量较高[20-21],本研究于2019年8月对奎屯河流域地表水(2组)和地下水(15组)共17组水样进行采集分析(如图1). 地表水采自距奎屯河源头流出后约3 km的位置,地下水水样中6组采集于126团,其余9组采集于128团(表1). 采集水样前清洗井孔,用清澈地下水润洗采样瓶3次后再采集水样. 地表水和地下水采样封存后分别用K(1—2)和C(1—15)进行标记分类.
用于阳离子分析(常量元素和微量元素)和总As测定的水样用浓硝酸(优级纯)将其酸化至pH<2并避光保存;阴离子、DIC和DOC分析的水样过滤后直接分装保存;无机碳及有机碳稳定同位素测试的样品分别加入HgCl2饱和溶液灭菌处理和浓磷酸(优级纯)酸化至pH<2密封保存[22]. 所有采集水样的样品瓶中不留气泡,并在低温4 ℃条件下冷藏保存. 同时现场记录采样点经纬度和井深,现场使用多参数便携式仪器(HI 8424,HANNA)测定水温、pH和Eh.
-
Na+和K+用火焰光度法测定;Ca2+和Mg2+采用EDTA间接络合滴定法测定;HCO3−和CO32−用双指示剂-中和滴定法测定;SO42−采用BaCl2滴定法测定;Cl−用硝酸银滴定法测定;Fe用TAS-990原子吸收分光光度计测定;总As用PF3-原子荧光光度计(北京普析)测定(标准曲线R2=0.999),仪器参数为温度200 ℃,载气气压为0.25—0.3 MPa.
DIC和DOC采用总有机碳分析仪1030(美国)测定,碳稳定同位素使用稳定同位素质谱仪(MAT 253)进行测定,测定工作由上海复昕化工技术服务有限公司完成. DIC碳稳定同位素测定方法采用准确度较高的气体逸出法. 首先,加入0.5 mL 85%H3PO4到进样瓶,用高纯氩气吹扫1 h去除CO2,再用1 mL注射器取0.8 mL样品注入进样瓶,然后进行60 ℃水浴加热1 h充分反应生成CO2,测定顶部CO2气体的无机13C. DOC稳定同位素测定采用湿法氧化法,首先将进样瓶使用马弗炉500 ℃燃烧5 h,冷却后加入0.5 mL水样,取50 µL 0.1 mol·L−1 AgNO3(催化剂)和0.5 mL 85%H3PO4加入进样瓶盖紧,用高纯氦气吹扫进样瓶1 h. 同时60 ℃水浴加热1 h,将吹扫气排除,以去除瓶中的残留气体,之后加入1 mL氧化剂,并在100 ℃水浴加热1 h,使DOC充分反应,测定进样瓶顶部CO2气体的有机13C.
DIC和DOC同位素测试精度可达0.04‰. DOC稳定同位素样品的标准样品采用美国国家标准技术研究所(NIST)认证的同位素标准物质USGS40(左旋谷氨酸,真值有机13C=−26.39‰±0.09‰)和USGS41(左旋谷氨酸,真值有机13C=+37.63‰±0.1‰)对未知样品进行较正,测试采用标准标准物质线性校正方法对测量值进行较正,每3至5个样品后分别加入所选的两种标准物质,选取有机碳13C值相差较大的两个标准物质,在相同参考条件下,将所选标准物质和待测样品采用相同的预处理流程处理,对其同位素比值进行测定,最终将标准物质的测量值与真值进行对比校正,根据线性回归,对样品的测量值进行校正.
-
采用ArcMap 10.7绘制研究区及采样点分布图;SPSS 25 进行数据统计和分析;Origin 2021 绘制散点图和箱线图.
-
由水样主要水化学指标统计表(表2)可知,研究区地表水pH值呈中性和氧化性环境;地下水的pH范围介于7.59—9.42之间,均值为8.63,整体呈弱碱性-碱性;Eh范围为−96—−7.5 mV,均值为−55.08 mV,地下水均处于还原性环境.
地下水中优势阳离子为Ca2+,其次为Mg2+和Na+,K+含量较低,阳离子的变异系数均大于1,说明地下水中阳离子浓度变化范围较大;阴离子Cl−含量占比最大,其次为SO42−和HCO3−,CO32−浓度最低,其中Cl−变异系数大于1,属于强变异. 地下水HCO3−质量浓度为49.44—125.52 mg·L−1,平均值为72.06 mg·L−1,地下水的HCO3−质量浓度小于地表水. 地下水中HCO3−和砷酸根离子均会吸附在铁氧化物矿物表面,矿物表面有限的吸附位置会使两种离子发生竞争吸附作用,当HCO3−形成碳酸盐复合物吸附在矿物表面时,As就会被释放到地下水中,导致地下水中As含量增加,HCO3−浓度也会降低[23],其次河流水体与大气CO2的交换作用也会使地表水HCO3−质量浓度高于地下水.
地表水As浓度均低于10 µg·L−1,为低砷水. 地下水As含量范围在2.45—460.38 µg·L−1之间,平均值为60.60 µg·L−1,73%的地下水As浓度大于10 µg·L−1,为高砷水. 126团地下水采样点As浓度范围在3.89—460.38 µg·L−1之间,平均值为120.81 µg·L−1,6组地下水样品中4组为高砷水;128团地下水采样点As浓度范围在2.45—48.77 µg·L−1之间,平均值为20.47 µg·L−1,9组地下水样品中7组为高砷水,2个团场高砷地下水检出率均较高. 整体看来,126团地下水样点As含量高于128团. 126团位于整个奎屯河流域地下水排泄区,径流条件更弱,更有利于地下水As的富集. 地下水中As的变异系数为1.83,大于1,属于强变异,表明该区域地下水中As的浓度变化较大. 奎屯高砷地下水主要分布在深层承压含水层中[14],本研究中约82%的高砷地下水分布在100 m以上的深层承压水中,与大同盆地(19—100 m)、河套平原(15—80 m)和江汉平原(15—40 m)的3个地区高砷地下水埋深分布在潜水、浅层承压水[10,24-25]有明显差异,不同采样点同一井深的地下水砷浓度可能相差很大. 这一原因主要是研究区地下水位埋深由南部山区到北部平原区呈现逐渐增加的趋势,由单一结构潜水层过渡到多层结构潜水—承压水层,即该地区奎屯河上游、中游和下游同一井深的地下水,赋存的含水层可能不同,不同含水层的地下水砷浓度变化较大.
-
地表水和地下水中DIC和DOC含量如图2所示,地表水和地下水DIC浓度均大于DOC浓度,地下水DIC和DOC浓度大多高于地表水. 地表水DIC的浓度范围为12.16—15.99 mg·L−1,平均值为14.08 mg·L−1,DOC的浓度范围在1.26—2.56 mg·L−1之间,平均值为1.91 mg·L−1;地下水的DIC浓度范围为10.84—45.28 mg·L−1,平均浓度为23.39 mg·L−1,DOC的浓度相比DIC较低,范围介于0.73—6.25 mg·L−1之间,均值为2.55 mg·L−1.
地表水DIC主要来源包括土壤中CO2、岩石风化溶解作用、水体与大气CO2的交换作用、大气降水的直接输入以及水中动植物的呼吸作用等[26];地下水DIC主要来源于微生物作用下有机质的代谢分解活动、空气中CO2的补偿以及碳酸盐矿物的溶解[27-30]. 前人研究表明,当[HCO3−]/[Ca2++Mg2+]≥2时,认为水体中的HCO3−主要来自碳酸盐岩的风化作用,当[HCO3−]/[Ca2++Mg2+]<2时,则水体的HCO3−存在多个潜在来源[31]. 在本研究中,根据所测得的离子数据计算出地表水[HCO3−]/[Ca2++Mg2+]的值均大于2,说明地表水中DIC主要来自碳酸盐岩风化作用的影响;地下水[HCO3−]/[Ca2++Mg2+]的值约60%小于2,表明地下水DIC受多种来源的共同影响. 地表水DOC值可反应水体的污染程度与人类活动对流域地表水体的影响[32],全球天然水体DOC平均含量为5 mg·L−1左右[33],若因人类生活污水和废水汇入,地表水DOC浓度会大于5 mg·L−1. 本研究地表水DOC的浓度范围在1.26—2.56 mg·L−1之间(图2),说明地表水并未受人为活动污染的影响.
-
由地表水和地下水中δ13CDIC和δ13CDOC关系图(图3)可知,研究区地表水δ13CDIC值范围为−2.88‰—−1.99‰(平均值为−2.44‰);地下水δ13CDIC值范围介于−9.13‰—0.58‰之间,平均值为−4.74‰. 与地表水相比,地下水中12个样点(80%)的δ13CDIC值偏负于−2.88‰,范围介于−9.13‰—−3.28‰之间,1个样点的δ13CDIC值大于−1.99‰,为0.58‰,接近0,另外2个样点的δ13CDIC值位于地表水δ13CDIC值范围内. 不同来源的DIC,其碳同位素具有不同的特征范围. 来源于碳酸盐岩溶解的DIC具有较大的δ13C值,而含水层中有机质的微生物降解过程,优先趋向利用较轻的12C,从而使产物中富集较轻的12C并导致13C发生分馏,其反应物中富集较大的13C,因此微生物对有机质的降解作用相对碳酸盐岩溶解的δ13CDIC值更偏负. 相关研究已证实有机质的生物降解过程会使δ13CDIC值负向移动[34],与地表水主要来源于碳酸盐岩溶解的DIC同位素δ13CDIC值范围相比,地下水有80%的δ13CDIC值更贫化,说明地下水DIC来源除受碳酸盐岩溶解的影响外,还存在微生物对有机质的降解作用. 微生物分解有机质释放的DIC,其δ13C值范围为−25‰—−18‰[35],而研究区地下水δ13CDIC值范围同微生物对有机质降解产生的δ13CDIC值范围相比偏正. 一方面,该区域地下水HCO3−受到含水层碳酸盐岩溶解的影响[36],会导致δ13CDIC值偏正;另外,已有研究表明在地下水缺氧的强还原环境中,产甲烷过程会使有机碳降解产生的CO2中富集13C,继而产生更多的DIC,所以产甲烷过程导致的碳同位素分馏所生成的DIC的δ13CDIC值会更偏正[37],研究区地下水氧化还原点位均小于0,深层承压水的还原缺氧环境,为产甲烷过程提供了有利条件,因此,研究区地下水中微生物作用下有机质降解产生的无机碳δ13C值不仅受到碳酸盐岩溶解的影响,还可能存在产甲烷的过程使δ13CDIC值偏正.
研究区地表水δ13CDOC值范围为−24.24‰—−18.81‰(平均值为−21.53‰),地下水δ13CDOC值范围介于−21.62‰—−13.79‰之间,平均值为−17.59‰,地表水的δ13CDOC值与地下水相比更贫化(图3). 地质来源碳和微生物活动产生的内源碳是地下水中DOC的主要碳源,此外,还存在从地表水通过地下径流过程进入到地下水的外源有机碳[25]. 不同碳源的DOC,其碳同位素具有不同的特征. C3植物(如树木、小麦、棉花等)的δ13CDOC值范围在−35‰—−20‰之间,C4植物(如玉米、高粱和甘蔗等)的δ13CDOC值介于−19‰—−8‰之间[34,38],景天酸代谢(CAM)植物的δ13CDOC值范围为−22‰—−10‰[39],CAM植物的同位素组成通常为中间值分布在C3和C4植物δ13CDOC值范围内. 土壤腐殖质中的δ13C值与该区域的植被类型有关,浅层地下水的DOC会受到人为活动和地表径流携带土壤有机质的影响. 奎屯地区经历多次地质运动中,垂直升降过程使得地表植被形成变迁,并在第四纪时期一直处于沉积地带的中心区域,形成了以泥质、黏土质和腐殖质为主的深厚沉积层地质条件[14,40],长期以微生物为媒介得生物地球化学过程演化成如今地下水中的沉积物. 地下水中δ13CDOC值分布于C3植物、C4植物和CAM植物的δ13CDOC值范围之间,埋深在浅层的地下水会受到地表入渗携带土壤有机质的影响,奎屯地区高砷地下水主要分布在100—200 m左右的深层承压水,相对封闭的水文地质条件使承压水受外源有机碳的影响可能性很小,因此,影响地下水δ13CDOC值大小的因素主要取决于地质条件造成地下水中沉积物富含的内源有机质,和微生物利用沉积物中有机质产生的内源碳以及通过地下径流汇入到浅层地下水的外源有机碳.
-
由地下水δ13CDIC值与ρ(HCO3−)呈显著负相关性(r=−0.541,P=0.037)的关系可知,δ13CDIC值越偏负,微生物活动越强,ρ(HCO3−)的浓度越高,说明微生物作用下有机质降解产生的HCO3−是地下水中DIC的重要来源之一. 由地下水δ13CDIC值与HCO3−关系图(图4)可以看出,高砷水主要分布在左侧区域,整体看来,砷浓度越高,δ13CDIC值越贫化,表示微生物作用下的有机质降解对地下水中As的富集具有重要意义.
研究区高砷地下水中δ13CDIC-δ13CDOC差值与δ13CDIC具有极显著正相关关系(r=0.729,P=0.013)(图5a),表明在δ13CDIC不断贫化过程中,DOC的氧化分解起一定作用. 当δ13CDIC-δ13CDOC值较大时,说明碳酸盐岩的溶解是DIC的主要来源;相反如果δ13CDIC-δ13CDOC值越小,则表明无机碳多来源于有机物的氧化分解越多,微生物活动越强烈. 由地下水中As含量可知,高砷水井深主要集中在100—200 m范围内,因此选取地下水(井深≥100 m)分析稳定碳同位素对砷富集的影响. 地下水(井深≥100 m)中As浓度与δ13CDIC-δ13CDOC的差值呈显著负相关关系(r=−0.726,P=0.017),随着砷浓度逐渐升高,δ13CDIC-δ13CDOC差值呈降低趋势(图5b),即砷浓度越高,δ13CDIC值越贫化,微生物活动越强烈,说明微生物对有机质的降解促进了地下水中As的富集.
研究区地下水所处的环境为还原性-弱碱性,地下水(井深≥100 m)中As与Fe具有极显著正相关关系(r=0.858,P=0.001)(图5c),随着地下水中Fe的浓度升高,As浓度也逐渐增加,说明在强还原环境下,铁氧化物表面高价态迁移性较差的Fe(Ⅲ)还原为溶解态和迁移性较强的Fe(Ⅱ),进而吸附在Fe(Ⅲ)上的砷被释放,使地下水中砷含量增加,这和前人的研究[14,21,23,41]一致. 地下水(井深≥100 m)中δ13CDIC-δ13CDOC的差值与Fe的浓度呈极显著负相关关系(r=−0.887,P=0.001)(图5d),结合图5(b、c),随着δ13CDIC-δ13CDOC差值的减小,Fe浓度越高且As的浓度也逐渐升高,说明微生物活动参与了含水层中铁氧化物的还原及砷的释放这一过程. 在这一生物地球化学过程中,溶解性有机碳和沉积物中有机质为微生物的代谢活动提供主要碳源和能量来源,有机碳在微生物的作用下被分解为无机碳形成碳的转化和分馏. 当微生物可利用的碳源增加时,可促进异养微生物的代谢,并消耗氧气,形成更有利于地下水As富集的还原环境.
我国北方典型高砷地下水分布区和南方典型高砷地下水分布区的地下水DIC与DOC同位素δ13C值、As和Fe质量浓度范围及埋深见表3. 研究区地下水中δ13CDIC值范围介于−9.13‰—0.58‰之间,整体变化范围同河套平原δ13CDIC值较接近,而比大同盆地和江汉平原更富集13C,最大值(0.58‰)和其他3个地区相比较正. 已有研究表明,在含水层中有丰富的有机质来源,微生物活动强烈,当O2、NO3−、Fe3+、SO42−等电子受体消耗殆尽时可能会进入到产甲烷阶段,该过程中发生明显的碳同位素分馏[42],地下水中的各项反应共同影响着体系内的无机碳和δ13C值的组成,因此,可能会导致砷、铁质量浓度较高的地下水中碳同位素δ13CDIC值更大. 铁氧化物矿物的还原性溶解均是这4个典型高砷水分布区砷释放的重要成因,微生物在此过程中发挥着重要作用. 其他3个地区地下水中均存在微生物对有机质的降解作用使δ13CDIC值偏负,新疆奎屯地区除了微生物对有机质降解作用和碳酸盐岩的溶解会影响外,还可能存在含水层相对封闭的水文地质条件下的产甲烷过程对δ13CDIC值的影响. 研究区地下水中δ13CDOC值范围为−21.62‰—−13.79‰,同河套平原(−22.9‰—−19.20‰)和江汉平原(−28.5‰—−19.60‰)相比偏正. 在强还原环境地下水中,微生物对有机质作用较强时,微生物优先利用更多的12C转化到降解产物DIC中,使得有机碳中更富集13C,推测新疆奎屯地区地下水中微生物活动较强.
-
(1)地下水中As浓度分布不均匀,平均值为60.60 µg·L−1,73%的地下水为高砷水,处于还原-弱碱性环境. 地下水中优势阴阳离子分别为Cl−和Ca2+. 地表水As浓度均为低于10 μg·L−1的低砷水,处于中性和氧化环境.
(2)地下水DIC浓度范围为10.84—45.28 mg·L−1,DOC浓度范围介于0.73—6.25 mg·L−1之间. 地下水DIC受多种来源的共同影响,地表水DIC主要来自碳酸盐岩风化作用的影响.
(3)地下水δ13CDIC值的范围在−9.13‰—0.58‰之间,80%的δ13CDIC值比地表水更贫化,δ13CDOC值的范围为−21.62‰—−13.79‰. 地下水中除了微生物作用下有机质降解,还存在碳酸盐岩溶解影响δ13CDIC值.
(4)地下水δ13CDIC-δ13CDOC差值与δ13CDIC呈正相关关系,有机碳的氧化分解对无机碳稳定同位素贫化过程起一定作用. 地下水中δ13CDIC-δ13CDOC差值与ρ(As)、ρ(Fe)均呈显著负相关关系,微生物参与了铁氧化物的还原性溶解并促进了As的富集.
新疆奎屯地区高砷地下水稳定碳同位素特征及其指示意义
Stable carbon isotope signatures of high arsenic groundwater and their indicative significancein in Kuitun area of Xinjiang
-
摘要: 地下水稳定碳同位素可以指示微生物作用下有机质降解过程对砷富集的影响. 新疆奎屯地区是中国西北干旱区典型原生高砷水分布区,本研究以地下水为研究对象,奎屯河地表水为对照,通过野外采样,并借助原子荧光、稳定同位素分析等技术手段,对地下水的水化学指标、溶解性无机碳(DIC)、溶解性有机碳(DOC)和碳稳定同位素进行测定与分析. 结果表明,研究区73%的地下水为高砷水,平均值为60.60 μg·L−1,呈还原-弱碱性环境,地表水均为低砷水,呈中性和氧化环境. 地下水中优势阴阳离子分别为Cl−和Ca2+. 地下水DIC受多种来源的共同影响,地表水DIC主要来自碳酸盐岩风化作用的影响. 地下水δ13CDIC值的范围为−9.13‰—0.58‰,δ13CDOC值的范围在−21.62‰ — −13.79‰之间. 地下水中除了微生物作用下有机质降解和碳酸盐岩溶解的影响外,还可能存在产甲烷过程对δ13CDIC值的影响. 地下水中δ13CDIC-δ13CDOC差值与ρ(As)、ρ(Fe)均呈显著负相关关系,表明微生物参与了铁氧化物的还原性溶解并促进了As的富集.Abstract: The stable carbon isotope of groundwater can indicate the influence of organic matter degradation process under the action of microorganisms on arsenic enrichment. Kuitun area of Xinjiang is a typical primary high arsenic water distribution area in the arid region of northwest China. In this study, the groundwater was taken as the research object, and the surface water of Kuitun River was taken as the control. The hydrochemical indicators, dissolved inorganic carbon (DIC), dissolved organic carbon (DOC) and carbon stable isotope of the groundwater were determined and analyzed. The results show that 73% of groundwater in the study area was high arsenic water, with an average value of 60.60 μg·L−1, presenting a reduction-weak alkaline environment, while the surface water was low arsenic water, presenting a neutral and oxidizing environment. The dominant anions in the groundwater were Cl− and Ca2+, respectively. The DIC of the groundwater was commonly influenced by various sources, and the DIC of the surface water mainly came from the influence of carbonate weathering.The value of δ13CDIC ranged from −9.13‰ to 0.58‰ and the values of δ13CDOC ranged from −21.62‰ to −13.79‰ in the groundwater. In addition to the degradation of organic matter and the dissolution of carbonate karst under the action of microorganisms, methanogenesis may also affect the δ13CDIC value in the groundwater. There was a negative correlation between δ13CDIC—δ13CDOC and As concentration also between δ13CDIC—δ13CDOC and Fe concentration in the groundwater, indicating that microorganisms participated in the reductive dissolution of iron oxides and promoted the enrichment of As.
-
塑料的商业生产始于20世纪50年代[1],现广泛应用于包装、医疗、农业等行业,仅2019年全球塑料产量就高达3.68亿吨[2]。塑料在光照辐射、机械磨损、风化侵蚀、动物和微生物的作用下,可逐渐分解成粒径更小的塑料颗粒[3]。微塑料(microplastics, MPs)的概念最早出现在2004年Science发表的一篇文章[4],定义为粒径小于5 mm的塑料颗粒[5],粒径小于100 nm的被称为“纳米塑料”(nanoplastics, NPs)[6]。MPs通过大气、洋流等作用在全球范围内长距离运输[7],并在环境中持续存在和积累。水体[8]、沉积物[9]、土壤[10]、大气[11]甚至深海和极地都能检测到MPs[7]。尽管多项研究回顾了MPs在水环境中的发生、分布、生态风险及水体MPs与其他污染物的环境地球化学行为[8, 12-13],但关于陆地MPs的综述论文却很少[14-15]。陆地MPs是海洋MPs的主要来源,其MPs污染程度可能是海洋的4—23倍[16]。土壤作为陆地系统中MPs的汇[17],对MPs的储存和转移起着至关重要的作用[18]。因此,充分认识MPs在土壤环境中的丰度、来源、迁移和生态毒性对于科学评估和源头控制土壤MPs污染十分关键。
在Web of Science核心数据库中以“microplastics”和“soil”为关键词进行了搜索(截至2021年8月21日),产生了608篇文献。通过共现网络分析(图1),发现土壤环境MPs的研究始于2016年,相关研究主要包括:1)土壤类型,全球学者普遍注重农田土壤MPs的研究;2)MPs的来源,包括未合理处置的塑料垃圾、污泥堆肥、有机肥料的施用、污水灌溉和地膜覆盖等;3)MPs的分析方法,包括采样、分离(筛分、密度分离、消解等)、鉴定(目检法、光谱法、热解质谱分析法等);4)土壤MPs的丰度、类型(如聚丙烯(PP)、聚乙烯(PE)、聚苯乙烯(PS))、形状(如纤维、薄膜、碎片、颗粒等);5)MPs的生物效应,包括对植物、动物和微生物的影响。由此可见,MPs的来源、种类、分布、检测方法及生态健康风险是当前土壤MPs污染研究的热点方向。已发表的文献中,Praveena等[19]、陈雅兰等[20]较为全面的综述了土壤中MPs的提取与鉴定方法,郝爱红等[14]、Zhao等[15]从土壤中MPs的来源、迁移、分析方法、污染特征和生态风险等方面入手,揭示了土壤MPs的归宿和生态风险,但有关土壤MPs与多种有害污染物共同暴露的生物毒性、土壤中老化或降解MPs的生态风险鲜有报道。有学者对全球土壤MPs污染做了简单的总结[17, 21],但所收集的数据不够全面。因此,本文在总结最新国内外研究进展的基础上,从土壤环境中MPs的来源、丰度、迁移及其生态健康风险方面进行了综述,并提出了相关领域未来的研究重点。相比先前的研究,本文更加全面的总结了土壤中MPs的丰度,通过绘制分布图以更加直观的形式展现了全球土壤MPs污染,并将土壤老化/降解MPs的生态风险以及MPs的复合污染毒性和潜在生态风险展开了系统地回顾和展望,填补该领域综述论文的空白。本文将为评估土壤MPs潜在的生态健康风险提供有价值的参考。
 图 1 已发表论文中以“微塑料”、“土壤”为关键词的共现网络分析图[22]。Figure 1. Co-occurrence network analysis of published research papers with “microplastics” and “soil” as keywords每个节点(关键词)大小与其出现频次成正比,连线颜色表示论文发表年份,数据截至2021年8月21日
图 1 已发表论文中以“微塑料”、“土壤”为关键词的共现网络分析图[22]。Figure 1. Co-occurrence network analysis of published research papers with “microplastics” and “soil” as keywords每个节点(关键词)大小与其出现频次成正比,连线颜色表示论文发表年份,数据截至2021年8月21日1. 土壤MPs的来源、丰度和迁移 (Source, abundance and migration of soil MPs)
1.1 土壤MPs的来源
土壤中MPs的来源十分广泛(图2),人们日常生活(如未合理处置的塑料垃圾)和农业活动(如污泥堆肥、有机肥施用、地膜覆盖及农田灌溉等)产生的MPs会直接进入土壤[23-26],或通过地表径流[27]和大气沉降[28]间接输送到土壤环境。
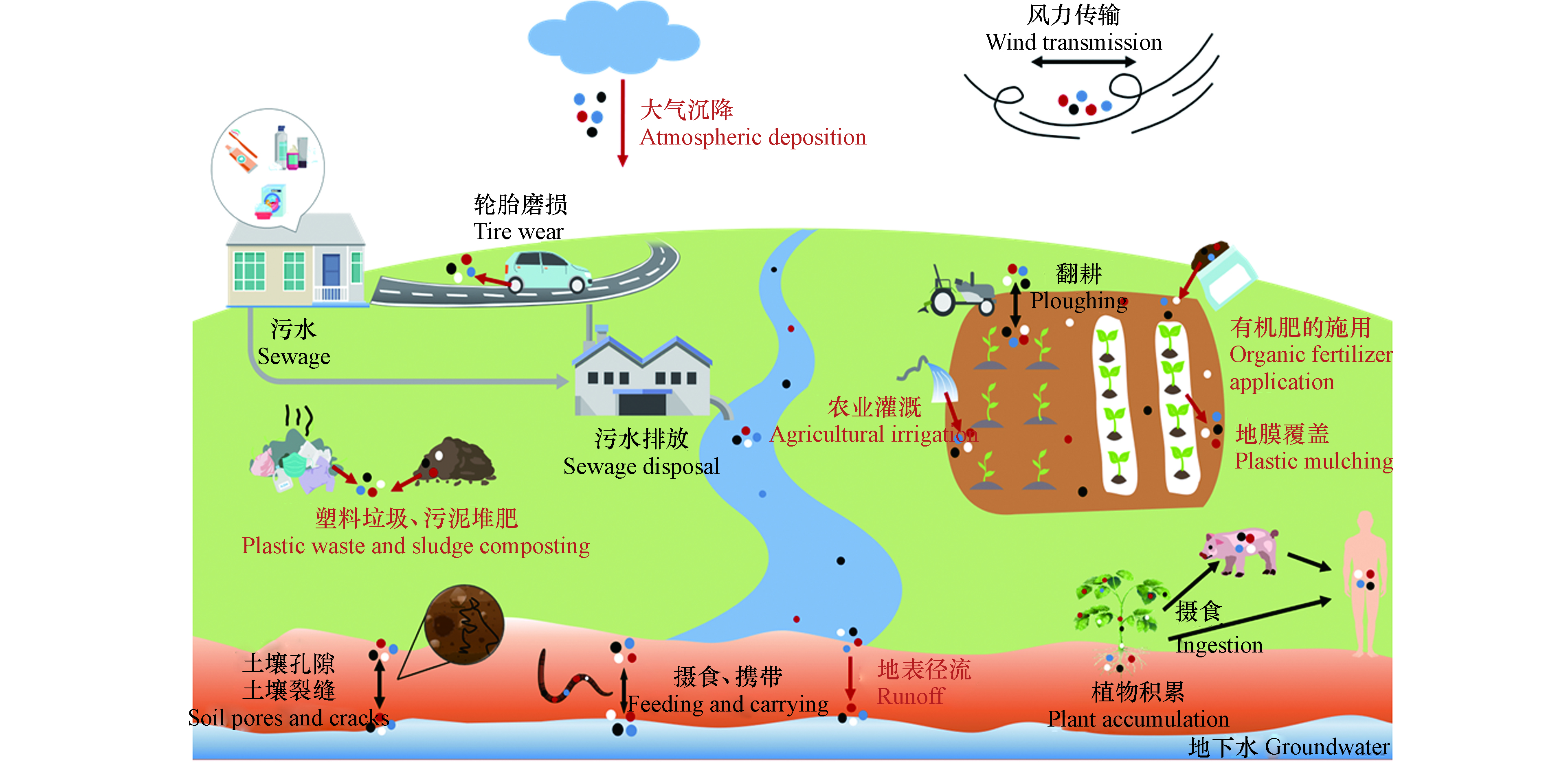 图 2 土壤中MPs的来源与迁移Figure 2. Origin and migration of MPs in soil(红色和黑色箭头/文字分别表示MPs的来源和迁移路径)
图 2 土壤中MPs的来源与迁移Figure 2. Origin and migration of MPs in soil(红色和黑色箭头/文字分别表示MPs的来源和迁移路径)1.1.1 未合理处置的塑料垃圾
土壤中存在着与水环境类似、种类繁多的MPs碎片[29],它们与塑料污染密不可分。根据目前的塑料废弃物管理趋势预测,2050年全球产生的塑料垃圾中将有120万吨进入垃圾填埋场或自然环境[30],必然会对生态环境造成影响。日常生活使用的一次性塑料袋/瓶、口罩/手套、衣服等均含有塑料,如使用后被随意丢弃在路边或非法倾倒地点[31],会造成附近土壤塑料污染。作为塑料垃圾的重要组成部分,塑料袋全球每年的消费量约为5000—10000亿个,其中900多亿个塑料袋不可回收[32],可在环境中老化降解生成MPs。自2020年新冠疫情爆发以来,大量一次性口罩排放到环境中。据估计,2020年全球生产的一次性口罩约520亿个[33]。每片新口罩中可释放(183.0±78.4)个MPs,而使用过的口罩因附着了空气中的MPs会释放更多的MPs(每片(1246.6±403.5) 个) [34]。由此,未合理处置的一次性口罩引起的土壤塑料和MPs污染不容忽视。
1.1.2 污泥堆肥的长期积累
污泥堆肥可能导致土壤MPs的增加[24]。生活废水经污水处理厂,可大大减少MPs(去除率约99%)向水环境直接排放[24],但未被处理的MPs通常积聚在污泥中[35],由于污泥含有丰富的N、P、K等营养元素[36],许多地区将污泥用作农田肥料[24],MPs便由此进入土壤。不同国家污泥中MPs的含量与经济发展水平、人口密度和废物处置等因素有关[37]。对于经济发达、人口密度高的国家,因使用药品、个人护理品(PPCPs)及洗衣产生的污水量大[38],污泥中MPs的含量相应较高。在欧洲和北美地区,每年通过污泥堆肥进入农田的MPs分别有约6.3×104—4.3×105和4.4×104—3.0×105吨[39]。土壤MPs的丰度随污泥施用量的增加而增加[24]。研究发现,在农田中仅施用一次污泥,15年后该区域土壤中仍可检测出塑料纤维[40],表明MPs在土壤中难以降解,会产生持久性污染。
1.1.3 有机肥料的施用
有机肥料的重复施用除了会引起重金属和抗生素等污染残留[41],还会导致土壤MPs污染,而后者常常被人们忽视[42]。研究发现,有机肥中普遍含有的MPs可能来自运输饲料的塑料管道、储存消毒剂或抗生素的塑料瓶[43]。江西鹰潭,猪粪中MPs的平均年丰度约为(1250±640)个·kg−1(干重),施用了猪粪的农田中MPs的年均累积量约为(1.25±0.61)个·kg−1[42];施用猪粪22年后的农田中MPs丰度((43.8±16.2)个·kg−1)明显高于未施用猪粪的农田((16.4±2.7)个·kg−1)[42]。德国是全球对肥料质量要求最严格的国家之一,但每年通过施用有机肥进入农田的MPs高达3.5×1010—2.2×1012个[26]。我国作为有机肥生产和使用大国,据估计,我国每年通过有机肥进入农田土壤中的MPs可达52.4—26400吨[3]。但该数据仅仅基于德国波恩、斯洛文尼亚等地区关于有机肥中塑料污染的报道[23, 26, 44],并结合我国有机肥每年实际施用量(2200万吨左右)来进行估算的,该估算忽略了粒径小于0.5 mm的MPs,且缺乏我国有机肥中关于MPs丰度的报道,因此,未来的研究中还应多关注我国有机肥中MPs的污染情况,以便全面评估我国通过有机肥进入土壤的MPs量。
1.1.4 农业灌溉和地表径流
农业灌溉是MPs进入土壤的又一重要途径。据统计,全球每年生活污水产生量超过356 km3,处理后的出水中有23.8 km3主要用于农业灌溉[45]。生活污水中含有大量源于PPCPs和衣物的MPs。虽然常规的处理工艺可有效去除污水中绝大部分MPs,但出水中仍有残留的MPs通过农业灌溉进入土壤环境[15]。在部分水资源匮乏的国家,未经处理的污水也会被用于灌溉农田[23]。据报道,全球约有3.6×105 km2的农田是使用未处理或者部分处理的生活污水进行灌溉的[46],必然会向土壤中输入更多的MPs。此外,天然水体中也存在MPs,例如:我国长江水中MPs高达6.6×103个·m−3[47],珠江水中MPs的丰度介于397—7924个·m−3之间[48],即使在偏远的内陆湖泊沿岸也有大量MPs存在,如青藏高原湖泊中MPs丰度可达(625±411)个·m−3[49]。这些水环境中的MPs也可通过灌溉或随地表径流进入土壤环境中。随着研究的深入,人们开始对生态环境敏感区(如青藏高原[49]、沙漠[50]、黄土高原[51])MPs污染进行研究,作为东南亚多条河流重要发源地的青藏高原,无处不在的MPs可能使其污染范围不断扩大到其他水系,或通过地表径流进入土壤环境,而该地区生态环境脆弱,存在调查难度大、恢复年限长等问题,未来的研究应该更加注重生态环境敏感区MPs污染及其健康风险评价。
1.1.5 地膜的广泛应用
地膜是农田土壤MPs污染的重要来源[23, 25]。2016年全球农用塑料薄膜市场交易量为400万吨,预计到2030年将以每年5.6%的速度增长[25]。全球约有1.29×105 km2的农田覆盖有地膜[52],我国地膜使用量最大,占全世界地膜覆盖面积的90%[17]。从田地中去除地膜费时费力,大量被残留的地膜在阳光辐射等作用下逐步破碎裂解,形成MPs[29]。农田土壤中MPs的含量随覆盖时间的延长逐渐增加[17]。在我国石河子市,随着地膜连续覆盖时间从5年增加至30年,MPs丰度从10.10 mg·kg−1增加到了61.05 mg·kg−1[53]。目前,大力研制与推广的环保型可降解地膜是解决塑料污染最有效的途径,但研究表明,MPs对污染物(如抗生素、农药等)的吸附能力大小排序为:老化可降解MPs>可降解MPs>非可降解MPs,且老化程度越高对污染物的吸附量越大[54-55],在这种情况下可降解地膜的使用,特别是地膜在环境中不可避免的老化行为,可能会给环境带来更大的生态危害,在未来的农业发展中应该重视这一问题。
1.1.6 大气沉降输入
土壤MPs也有部分来自大气中悬浮的塑料颗粒。多项研究表明,大气中存在MPs,如南海西北部大气中MPs的丰度为(0.035±0.015)n·m−3[56]。大气中的MPs主要来源于建筑材料、纺织品磨损、灰尘、道路油漆、轮胎和制动器磨损[57]。轮胎磨损产生的MPs主要来自各种车辆,全球车辆轮胎磨损的MPs排放量为人均0.81 kg·a−1[58],飞机轮胎磨损释放的MPs相对较少,约占荷兰轮胎磨损MPs排放总量的2%[58]。空气中密度小的大塑料颗粒和MPs可通过大气沉降和风力传输沉积在城市或乡村陆地表面[59],还可传输到偏远、人烟稀少的地区[28]。据报道,我国烟台市大气MPs沉降通量达1.5×105个·(m2 a)−1[60];法国巴黎大气MPs沉降通量达2—355个·(m2 d)−1,且该地区每年有3—10吨的纤维被大气沉降物沉积[59]。由此可见,大气沉降是MPs沉积到陆地的重要途径。值得思考的是,粒径小于50 μm的MPs可以重新悬浮到大气中[61],增加人体吸入MPs的风险,而多数国家并没有将大气中的MPs作为空气污染的一部分进行监测,为了明晰MPs对人类健康构成的潜在风险,将MPs纳入空气污染的监测范围迫在眉睫,尤其是在MPs污染严重的大城市。
总体来看,国内外大量关于土壤中MPs的来源研究仅停留在对来源的简单陈述,只有少部分做了MPs的溯源追踪方法。目前,环境中MPs的溯源方法主要集中于水体和沉积物,通过非仪器分析法(目视分析法、密度分析法、灼烧分析法等)从MPs的颜色、形状、密度等特性初步判识MPs的外观及用途[62],或通过仪器检测(光谱分析法、显微分析法、色谱质谱分析法等)判识MPs的化学成分及结构[63],两者相结合可追溯环境中MPs的来源。从已有研究成果来看,土壤MPs的溯源依旧没有可靠且简单易行的检测方法。值得注意的是,进入到环境的塑料碎片和MPs,由于各种物理化学作用,最终会破碎形成NPs,更小的粒径以及颜色、形状等特性不够显著增加了对MPs来源追溯的难度,因此亟需建立适合更小粒径的NPs的检测方法和理化指标。
1.2 土壤MPs的迁移行为
MPs在土壤中可发生水平和垂直迁移[64],其迁移行为受土壤和MPs理化性质的影响[21, 65]。土壤的理化性质(包括孔隙度、土壤质地、矿物和腐殖质含量等)对MPs的迁移有重要影响。土壤的孔隙大小由其质地决定,可直接影响MPs的迁移[30],砂土表面的MPs在渗透作用下可垂直迁移至距地表1.5—7.5 cm的土壤中[66]。由于土壤裂缝,干燥气候可能会加速MPs向下移动[66]。土壤矿物和腐殖酸共存时会增加MPs的垂直传输距离(9—10 cm)[67]。Wu等[68]发现, PS微球的迁移能力随土壤矿物(Fe/Al氧化物)含量的增高而降低,这是由于带负电的MPs与带正电的Fe/Al氧化物发生静电吸引所致。此外,MPs的特性(包括粒径、形状、电荷和表面化学等)也会影响其在土壤中的迁移。当MPs的粒径小于土壤孔隙尺寸时,MPs能通过土壤孔隙和裂缝向下移动,粒径小的MPs也容易被土壤动物摄食而转移到更深层的土壤中[69-70]。由于MPs与土壤团聚体的相互作用不同,不同形状的MPs可能对土壤中MPs的迁移产生阻塞作用影响其迁移行为[65]。如:塑料微球和微粒比微纤维更易下移到土壤深层,因为微纤维与土壤颗粒缠结形成土块后无法迁移[71]。高密度的MPs(如PET(聚对苯二甲酸乙二醇酯))可能会因重力作用而促进其在土壤中的迁移[72]。表面含有羧基、磺酸基、低密度氨基官能团的PS微球,比含有高密度氨基官能团的PS微球更易在海沙中迁移,这是由于带正电的高密度氨基MPs与带负电的沙粒之间存在静电吸引,从而阻碍MPs的迁移行为[73]。
除了在土壤内部迁移外,土壤中的MPs也会在风力、气流、地表径流等作用下迁移到空气和水等环境介质中[64, 66]。土壤表面的MPs尤其是微纤维等轻质塑料颗粒,可以被风和气流抬升到空气中,最终长距离传播到其他陆地或地表水中[59]。此外,地表径流可促使MPs进入深层土壤甚至含水层。据报道,澳大利亚维多利亚州地下水中MPs的平均丰度为38个·L−1[74],向地下水迁移的MPs可能带来新的环境问题,但目前仍缺乏对地下水MPs污染的环境风险预测、评估和防控研究。
1.3 全球土壤环境中MPs的丰度
我们收集了全球不同地区土壤环境中检出的MPs的理化性质和丰度,绘制了图3。目前,虽然只有少量研究报道了土壤环境中MPs的丰度情况,但可看出MPs广泛存在于多种土壤中(如农业土壤、公园土壤、湿地土壤、沙漠土壤等),其丰度从几个·kg−1到数万个·kg−1不等,多数地区土壤MPs丰度在0—5×103个·kg−1之间,粒径大多小于1 mm[75-77];MPs形状有纤维、薄膜、碎片、颗粒等,PP、PE、PS是土壤中最主要的聚合物类型。土壤环境中MPs的丰度普遍高于水和沉积物中的[8],说明土壤环境是MPs重要的汇。在全球范围内,亚洲、欧洲、北美、大洋洲的土壤环境中都发现了MPs,且不同地区丰度差异较大。从图3中可看出,智利梅利皮利亚县田地因长期施用污泥导致土壤MPs丰度高达18000—41000个·kg−1,明显高于其他地区[24];西班牙东南部穆尔西亚蔬菜农田土壤和墨西哥坎佩切家庭花园土壤中也检测到了数量较高的MPs,丰度分别为(2116±1024)个·kg−1和(870±1900)个·kg−1[78-79];但德国石勒苏益格-荷尔斯泰因州农田表层土壤中MPs仅有(5.8±8)个·kg−1[80],且该国弗兰科尼亚中部农田中MPs的丰度最低,仅为(0.34±0.36)个·kg−1[81]。
 图 3 文献报道的全球部分地区土壤环境MPs污染情况Figure 3. MPs pollution of soil environment in some regions of the world reported in the literatures(数据更新于2021年10月,没有标记的区域不代表没有MPs污染)(Updated in October 2021, unmarked areas do not represent no MPs contamination)
图 3 文献报道的全球部分地区土壤环境MPs污染情况Figure 3. MPs pollution of soil environment in some regions of the world reported in the literatures(数据更新于2021年10月,没有标记的区域不代表没有MPs污染)(Updated in October 2021, unmarked areas do not represent no MPs contamination)作为最大的塑料生产国和消费国[82],我国土壤MPs污染引起了越来越多的关注。在我国大多数受人为活动影响较少的土壤中MPs含量较低,如山东东营黄河三角洲湿地无植物覆盖的土壤和长江沿岸休耕的土壤中MPs丰度仅为60个·kg−1[83]和(28.4±22.0)个·kg−1[84];但农业土壤中MPs的含量通常较高,如:云南滇池柴河流域土壤MPs丰度为7100—42960个·kg−1[85];湖北武汉、山东寿光的农田土壤中也含有较高丰度的MPs(4.3×104—6.2×105、275—4165个·kg−1)[76-77],这可能是塑料地膜老化降解、污泥施用和污水灌溉所致。而少数地区如黄土高原[51]、上海菜地[75]等农田土壤中MPs丰度较小。在工业活动频繁的地区,也可能会引入较高丰度的MPs,广东贵屿电子废物拆解区土壤中MPs的丰度达34100个·kg−1[86]。沿海地区可通过海水养殖、旅游和港口建设等活动引入大量MPs[87]。一些偏远地区也存在少量MPs,可能是通过游客活动、卡车轮胎磨损和农用地膜引入的[88],或与大气传输有关。
土壤中MPs的垂直分布没有明显的规律[76]。例如我国上海郊区[75]、山东寿光[76]和德国石勒苏益格-荷尔斯泰因州[80]农田中表层土壤MPs丰度高于深层土壤MPs丰度,黄土高原[51]、山东胶州湾菜地和果园土壤[89]、毛里求斯农业土壤[90]中深层土壤含有更多的MPs,而我国云南滇池柴河流域农田[85]和墨西哥家庭花园[79]的表层和深层土壤MPs含量无显著差异。不同地区土壤MPs垂直分布可能会受到土壤翻耕、地表径流等因素的影响[51],动物的摄食和排泄行为也可能影响MPs在表层和深层土壤之间的垂直转移[58, 64]。此外,少数研究还报道了土壤质地、植被覆盖、栽培时间、恢复年限等与MPs丰度的关系[50, 76, 85]。例如: 我国山东寿光的农业土壤和砂质壤土中MPs丰度显著高于粉质壤土[76],毛乌素沙漠土壤MPs丰度高于草地和林地[50];设施栽培时间>25与<10年的农田土壤中MPs丰度差异不显著,表明早期的设施栽培措施导致土壤中MPs的累积数量不高[85]。由此可见,土壤中MPs无处不在,不同地区土壤MPs污染水平之间的差异是人类农业活动、工业生产等因素共同作用的结果。值得注意的是,已有研究采用的分离、计数MPs的方法不一,在单位上也有区别,可能会低估或高估了土壤中MPs的真实污染水平。因此,未来的研究亟需建立土壤MPs分离和检测标准。在深层土壤中,MPs受阳光辐照的影响减小,且可降解塑料的微生物种群较少[91],这意味着土壤深处MPs的老化降解可能减慢,其持久性可能会更长。那么,除了表层土壤,检测深层土壤中MPs的含量才能全面评估土壤中MPs的污染状况。
2. 土壤MPs污染的生态健康风险(Ecological health risks of soil MPs pollution)
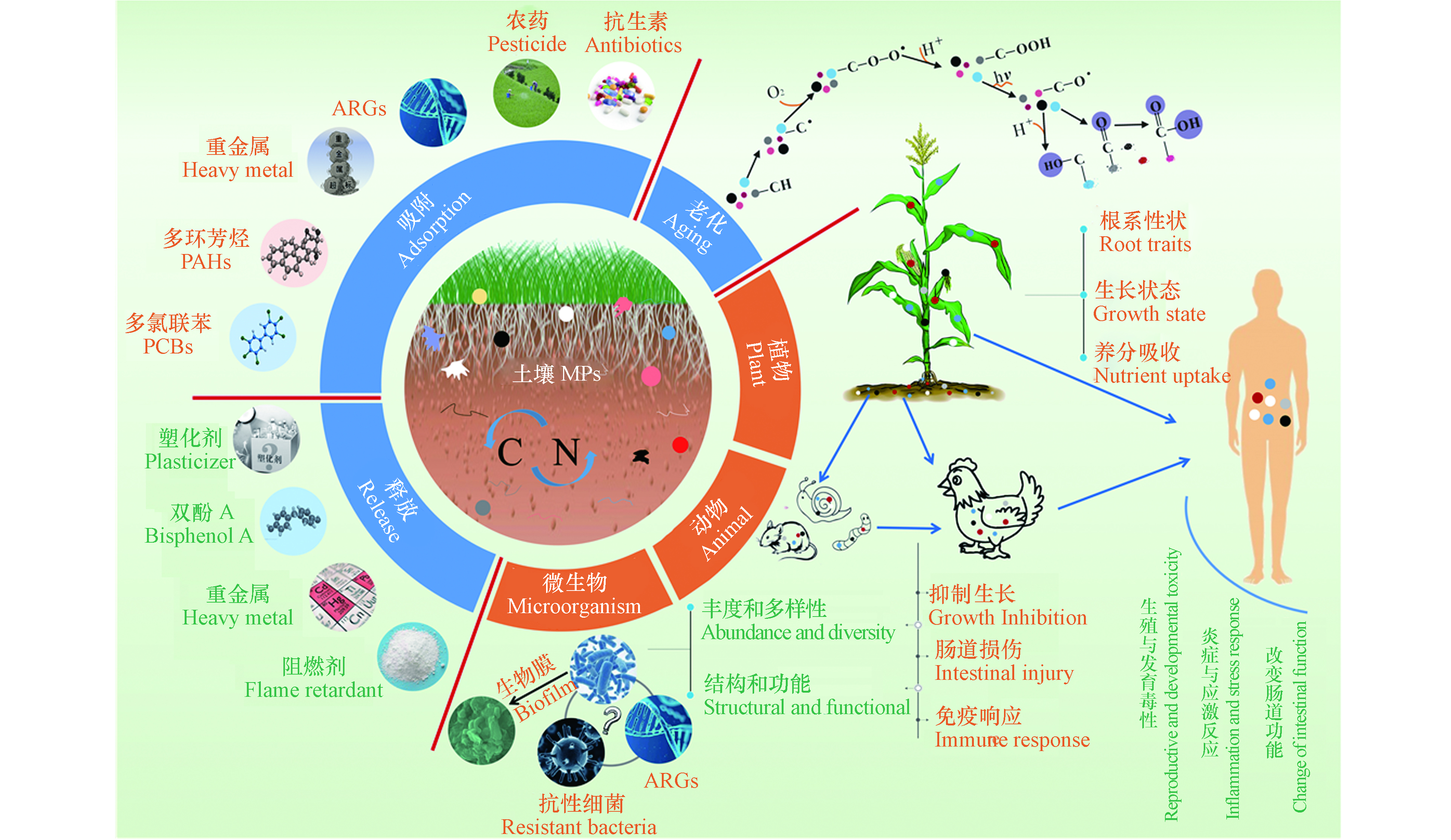
土壤MPs可通过多种途径对生态系统构成潜在威胁(图4)。MPs的存在可直接影响土壤动植物、微生物的生长[92-94],后经食物链的积累和传递可能对人体健康构成潜在威胁[79]。土壤MPs在土壤环境中能够吸附多种污染物质(如重金属、抗生素、农药等)[58, 95],或与自身释放的添加剂(如增塑剂、抗氧化剂、阻燃剂等)形成复合污染[96],这会给土壤动植物的生长带来极大的危害,而土壤环境中的MPs大多处于老化/降解状态,较原生MPs对污染物表现为更高的吸附能力[97],可能会对土壤生态系统构成更大的威胁。
2.1 MPs对陆生植物的影响
MPs进入农业土壤会对植物产生暴露,阻塞种子孔隙、限制根吸收水和养分[92],影响植物的芽高、生物量和发芽率等[98-100]。Bosker 等[101]发现,绿色荧光塑料颗粒(50、500、4800 nm, 107个·mL−1)因堵塞种子的荚膜孔道会限制水芹种子发芽。而含PP、高密度聚乙烯(HDPE)、低密度聚乙烯(LDPE)和PET的土壤MPs能促进番茄植株的生长,但会延迟结果和降低果实产量[102]。MPs还可通过改变土壤结构、容重、持水能力和营养成分[103-104],间接影响植物根系性状、生长状态和养分吸收[99, 105]。de Souza Machado等[100]发现,MPs污染使得土壤容重降低,通气增加,有助于植物根系渗透到土壤中。然而,MPs(如微纤维)也会缠住幼根,阻碍幼苗的生长[92]。
MPs对植物生长的影响与其类型、暴露浓度、粒径等因素有关。de Souza Machado等[105]发现,PA、PE、HDPE、PP(均为2.0%)均会改变大葱的生物量、元素组成和根系性状,其影响程度因聚合物类型而异。Boots等[98]对比研究了生物降解的聚乳酸(PLA, 65.6 μm, 0.1% W/W)和难降解合成纤维((丙烯酸(AA)和尼龙混合物), 0.001% W/W)对黑麦草发芽的影响,发现两种MPs均会降低发芽率,PLA还会降低芽高。Qi等[99]也报道了类似的结果,即1%的淀粉基生物降解塑料和PE均抑制了小麦生长,且前者比后者的抑制作用更强。由此,生物降解材料来源的MPs对植物可能产生更强的毒性效应,值得进一步研究。一些研究表明粒径大小不同的MPs对植物的影响也不同,与5 μm PS(10、50、100 mg·L−1)相比,100 nm PS对蚕豆的生长抑制作用、遗传毒性和氧化损伤更强[106]。但目前,对于MPs在植物中的积累和转运以及对植物的毒性作用和机制等的认识仍不清楚。
2.2 MPs对陆生动物的影响
MPs被动物摄入后会影响其摄食行为、生长和繁殖[107]。与水生动物相比,MPs对陆生动物影响的生态毒理学研究非常有限,且主要集中在无脊椎动物(如蚯蚓)[93]。已有研究证实MPs暴露对蚯蚓的毒性作用主要包括抑制生长、体重减轻、肠道损伤、免疫响应、肠道微生物群落的改变,以及死亡率增加[70, 108-109]。少数研究报道了土壤MPs也会影响蜗牛[110]、土壤线虫[111]、小鼠[112]等的健康。MPs对动物的影响存在剂量-效应关系。Huerta Lwanga等[107]发现,0.2%的PE(<150 μm)对蚯蚓(Lumbricidae)的生长和存活没有影响,但较高的添加量(1.2%)有抑制作用。Cao等[108]同样发现,低剂量(≤0.5%)的PS(58 μm)对蚯蚓生长的影响不明显,但高剂量(1%、2%)的MPs显著抑制了蚯蚓的生长,死亡率达40%。PS(0.05—0.1 μm)在高暴露量(10%)下可观察到蚯蚓肠道微生物群的明显变化[113]。虽然低浓度MPs暴露不会明显影响动物的生长和引起动物死亡,但会诱使动物组织病理损伤和免疫响应[70]。在评估MPs对动物健康的影响时,粒径是除暴露剂量之外的重要影响因素,Lei等[111]研究了不同粒径的PS(0.1、0.5、1.0、2.0、5.0 μm)对土壤线虫(Caenorhabditis elegans)的影响,发现相同质量浓度(1 mg·L−1)下1.0 μm PS暴露后土壤线虫的存活率最低。然而,对于MPs对陆生动物的潜在影响,如MPs在动物组织中的积累和运输、MPs对动物的毒性作用和机制等方面的认识仍存在空白。
2.3 MPs对土壤微生物的影响
MPs内含或吸附的有机物可为微生物提供碳源[21],微生物在MPs表面定殖后形成生物膜[114],继而构成具有特殊微生物群落组成和功能的“塑料圈”[115]。研究发现,电子拆解厂区域的MPs(如PP、聚碳酸酯(PC)和ABS)及其周围环境的细菌群落存在显著差异,这可能是因为MPs为微生物提供了新的生态位[116],或通过改变土壤理化性质(如破坏土壤结构、降低土壤密度、改变土壤持水能力等)影响了微生物的群落结构和功能[65, 117]。添加MPs后土壤微生物群落多样性的影响研究还处于起步阶段,Huang、Judy等[118-119]认为,HDPE(<2 mm, 0.1%、0.25%、0.5%、1% W/W)、PVC(<2 mm, 0.01%、0.1%、0.25%、0.5%、1% W/W)、PET(<2 mm, 0.1%、0.25%、0.5%、1% W/W)和LDPE(2 mm×2 mm, 0.076 g·kg−1)的存在并没有显著改变土壤微生物群落的丰度和多样性。但也有研究发现土壤中添加低或高浓度(1%、5%)的LDPE(678 μm)和高浓度(5%)的PVC(18 μm)均显著增加了β变形杆菌目(Betaproteobacteriales)和假单胞菌目(Pseudomonadales)的相对丰度,而高浓度的PVC(18 μm, 5%)显著降低了鞘脂单胞菌科(Sphingomonadaceae)的丰度[120]。这些研究结果之间的差异可能与MPs的类型、浓度、以及土壤的理化性质有关。不同类型的MPs对微生物活性影响不同,PP颗粒(<180 μm, 7%、28%)对土壤微生物活性有积极影响[103],然而,Lozano等[94]发现PP碎片(<5 mm, 20%)会降低土壤微生物活性,PS颗粒(32.6 nm±11.9 nm, 1000 ng·g−1)、LDPE(643 μm, 17%)也对土壤微生物活性显示出负面影响[65, 121],de Souza Machado等[105]的研究也报道了类似的结果,但在这些研究中,MPs粒径、形状、大小和浓度各不相同,因此很难得出MPs对微生物毒性的一般性结论。
此外,MPs作为致病菌和耐药菌的载体[122],可能影响土壤中ARGs的分布和迁移。MPs与ARGs在环境中广泛共存,由于ARGs对人类健康的潜在不利影响,其传播越来越受到关注。水生环境中,多项研究表明MPs(如PVC、聚乙烯醇(PVA))可影响ARGs的分布和传播[123]。在土壤中,PS(0.08—0.10 mm, 0.1%)的存在已被证实会增加抗生素和ARGs的保留时间[124],Lu等[125]也得出了类似的结果,MPs可促进土壤中ARGs丰度和数量,但还需要更多的证据来证实MPs污染是否促进ARGs在土壤环境中传播的结论。此外,Zhu等[126]发现土壤温度和湿度的升高均显著提高了MPs上ARGs的丰度,因此,在全球气候变化的情况下,土壤MPs对ARGs影响需引起更多的关注。
2.4 MPs对人类健康的潜在影响
MPs可通过改变土壤理化性质、降低土壤肥力,影响土壤的生态功能和粮食生产[127],对人类的生存和发展产生潜在影响。MPs也可经陆生食物链传递进入人体。MPs及其吸附的污染物可在动植物体内积累[79],食用植物可以从土壤中吸收和积累微型(0.2 μm)荧光PS珠[128],100 nm PS可以在蚕豆、生菜根中积累,然后运输到茎叶[106]。一些重要的家禽(如鸡)也可食用MPs[79],而当人们食用被污染的家禽或蔬菜时,MPs可能在人体内大量积累。据估计,在墨西哥每人每年通过食用鸡肉就可摄入840个塑料颗粒[79],MPs一旦进入人体,可能引起炎症与应激反应、产生生殖与发育毒性,或改变肠道微生物的组成和功能[129]。MPs(<150 μm)可能会从肠腔转移到淋巴和循环系统,进而导致全身暴露[129]。Schirinzi等[130]证明了MPs(PS, 10 μm)和NPs(PS, 40、250 nm)可诱导人体细胞发生氧化应激,并在细胞水平上引起细胞毒性。MPs和NPs与免疫系统作用还可能会导致免疫毒性,进而引发不良反应(即免疫抑制、免疫激活和异常炎症反应)[131]。Prata[132]还发现,由于摄入MPs引起的慢性炎症和刺激可能会因DNA损伤而导致癌症。此外,常见的塑料添加剂,如邻苯二甲酸盐、阻燃剂、双酚A等,与生殖和发育障碍有关,可能引发乳腺癌、血液感染、青春期过早和生殖器缺陷[133]。目前开展的土壤MPs由食物链传递被吸食进入人体的研究还比较少,但已经在人类食物[129]和粪便[134]中检测到了MPs,甚至在人类胎盘、婴儿粪便、婴儿内脏中也发现了MPs的存在[135],虽然没有证据表明这些MPs是来源于土壤环境,但该结果应该足以引起人们对土壤MPs的重视。此外,大气MPs或许能通过反射阳光辐射对气候有冷却效果[136],而土壤中的MPs通过扬尘进入大气环境是否也有同样的效应,进而引起一系列的生态健康问题,如气候变化、水文调节及粮食安全等[137]。
2.5 MPs与其他污染物的复合污染毒性及生态风险
MPs因疏水性强、比表面积大[138],可以吸附多种有机和无机污染物,如多环芳烃(PAHs)、多氯联苯(PCBs)、重金属等[58, 95],或与自身释放的添加剂(如增塑剂、抗氧化剂、阻燃剂等)形成复合污染[96],从而影响土壤动植物的生长。对植物来说,Gao等[96]发现当加入邻苯二甲酸二丁酯(DBP)时,PS (100—1000 nm、>10000 nm)加重了DBP诱导的植物毒性,增强了对生菜(Lactuca sativa L. var . ramosa Hort)的负面影响,且小粒径PS(100—1000 nm)对生菜的不利影响略大。Liu等[139]发现土壤中PE(200—250 μm,0.5%、1%、2%、5%、8% W/W)和菲(100 mg·kg−1)共同污染比单一处理对小麦幼苗(Triticum aestivum L. cv. NAU 9918)的毒性更强,PE的单一污染破坏了小麦叶片的光合系统,而PE和菲复合污染则加剧了这种破坏。MPs与土壤中重金属等无机污染物的复合污染也引起了人们的关注。Dong等[140]研究发现,在As(Ⅲ)存在下,大尺寸的PS(5 µm)可以迁移到胡萝卜的叶和根部,这是由于As(Ⅲ)增加了PS表面的负电荷,同时As(Ⅲ)也会导致细胞壁扭曲和变形,并导致更多的MPs进入胡萝卜,降低其质量。另一项研究表明,PET(<2 mm)还可以作为载体将重金属运输到小麦根际区域[141]。而Zong等[142]的研究表明,与单一重金属处理相比,PS(0.5 µm, 100 mg·L−1)与Cu2+、Cd2+的结合增加了小麦中叶绿素含量,增强了光合作用,减少了活性氧(ROS)的积累,表明PS(0.5 µm, 100 mg·L−1)对Cu2+、Cd2+的生物利用度和毒性具有缓解作用。对动物来说,Zhou等[143]发现PP(<150 μm, 0.03%、0.3%、0.6%、0.9%)与重金属(Cd, 8 mg·kg−1)二者联合暴露会对蚯蚓(Eisenia foetida)产生更强的负面影响,降低蚯蚓的生长速度并增加其死亡率。而另一项研究却发现,PVC可能通过吸附/结合As(Ⅴ),降低As(Ⅴ)的生物利用度来缓解As(Ⅴ)对肠道菌群的影响,从而防止As(Ⅴ)的减少和总砷在肠道中的积累,降低对蚯蚓(Metaphire californica)的毒性[144]。然而,Sun等[145]发现,MPs(40—50 μm, 10 mg·kg−1、300 mg·kg−1)可显著增加毒氟磷杀虫剂在蚯蚓体(Eisenia fetida)内的生物蓄积性,加重对蚯蚓的氧化损伤和干扰代谢。Boughattas等[146]将MPs(100 µg·kg−1)和除草剂2,4-二氯苯氧乙酸(2-4-D)(7 mg·kg−1)共同暴露于土壤中,结果表明,MPs增加了蚯蚓中的2,4-D生物积累,破坏了溶酶体膜的稳定性和氧化状态,并增加了抗氧化基因的表达。
目前,不管是对MPs的单一毒性研究还是与其他污染物的复合毒性研究,都存在受试动植物类别有限、土壤类型单一、研究周期短等问题,且MPs的种类、大小和浓度与实际土壤环境有一定的差异,如实验室研究中所用MPs浓度往往会高于实际土壤环境中MPs的最大浓度(6.7%)[147],未来的研究应在环境相关浓度条件下评估生态效应。更重要的是,没有充分考虑自然环境因素,真实土壤环境中MPs更多是处于老化或被生物膜定殖的状态,这无疑增加了MPs上的吸附位点,可能使得MPs上吸附的污染物更多,对陆地生态系统构成更严重的威胁。此外,粒径较小的MPs,特别是NPs,可能对陆地生态系统的健康风险更大[21],应作为重点评估的对象。
2.6 MPs的老化/降解及其潜在生态风险
MPs在土壤中的长期积累可以进一步老化或降解[21]。除光照辐射、机械磨损、风化侵蚀外,土壤环境中动物群和微生物(如细菌和真菌)也可以降解MPs[21, 107, 148]。从土壤中分离得到的假单胞菌属细菌AKS2对LDPE的降解率在45 d内达到4%—6%[149],在地膜中分离得到的红球菌C208对PE塑料薄膜的降解率在30 d内达8%[150]。但目前从土壤中分离出能降解MPs的菌株种类较少,因此探究用于降解土壤MPs的微生物可能是进一步研究的方向之一。而生物体可以通过咬、咀嚼或消化碎片来物理降解MPs[151-152]。蜡螟(Waxworms)、印度谷螟(Indian Mealmoths)已被证实能吞食PE并在其肠道微生物的帮助下降解塑料聚合物[153]。此外,大麦虫(Zophobas Morio)、黄粉虫(Tenebrio molitor)、蚯蚓等均具有降解MPs的能力[154-156]。老化/降解会改变MPs的表面结构、疏水性、结晶度和比表面积,并增加MPs表面C—O、C=O、—OH等含氧官能团的数量[8, 97],导致老化或降解MPs具有更高的吸附能力,使其可以吸附其他污染物质,对土壤生态系统构成更大的威胁。
目前,关于土壤MPs的老化或降解对陆地生态系统的危害研究并不多,主要是以下几个方面。首先,长期风化会使MPs分解成为NPs,许多研究已证明粒径较小的NPs可能较MPs具有更大的环境流动性和毒性[111]。Muhammad等[157]发现家蚕(Bombyx mori)暴露于PS MPs(5—5.9 μm, 10 μg·mL−1)的个体在感染后存活得更好,而暴露于PS NPs(50—100 nm, 10 μg·mL−1)的个体则表现出更高的死亡率。Liu等[158]也得出了类似的结果,相较于100 nm PS NPs,20 nm PS NPs(0.1—100 μg·L−1)对线虫(Caenorhabditis elegans)表现出更强的毒性。其次,老化MPs对污染物表现为更强的吸附能力,且老化的可降解MPs更强[51, 159]。Zhang等[159]研究发现,搁浅的PS泡沫对土霉素的吸附能力高于原始PS泡沫的吸附能力,Fan等[55]的研究也发现通过紫外线的老化过程,PLA、PVC对四环素、环丙沙星的吸附能力增加,且可降解PLA表现出更好的吸附能力,这些研究表明更多的有机污染物可以吸附并浓缩到老化的MPs上,形成的复合污染可能对生物体造成更严重的危害。最后,一些研究还探究了在超纯水和模拟肠液中,抗生素在原生/老化MPs上的解吸行为,发现与原生MPs相比,抗生素在老化MPs上解吸量更大,且模拟肠液中的抗生素解吸量比超纯水中大,这可能会对生物体造成更严重的危害[55]。除了老化MPs对生物体的危害外,也可能会带来其他的环境问题,如老化后形成的NPs由于粒径太小,如何从土壤环境中检测丰度及去除也是一大难题。综上,老化MPs的生态毒性问题及其带来的环境污染问题值得高度关注。
3. 结论与展望(Conclusion and perspective)
(1)土壤MPs的来源途径很多,包括未合理处置的塑料垃圾、污泥堆肥、有机肥的施用、农业灌溉、地膜覆盖等,但当前的研究仅停留在对土壤MPs来源的描述上,很少聚焦MPs的溯源研究,现有的技术条件无法将MPs从环境中根除,因此从源头管控就显得尤为重要。但如今土壤MPs溯源几乎处于空白状态,建议加强这方面的研究,为土壤中MPs的源头控制提供关键支撑。
(2)MPs污染在全球土壤环境中普遍存在,应加大力度调查土壤MPs丰度。不同地点、土地类型、不同深度土壤中MPs污染水平和特征存在较大差异,频繁的农业活动导致农田土壤MPs污染较为严重,PE、PP、PS是土壤中最常见的MPs类型。通过大气传输、植物积累、动物摄食、翻耕等多种途径,MPs最终可迁移到深层土壤甚至含水层,因此检测深层土壤中MPs的含量才能全面评估土壤MPs的污染状况。迁移到地下水中的MPs可能带来新的环境问题,但相关的环境风险预测、评估和防控仍缺乏。
(3)土壤MPs的存在会对动植物的生长产生不同影响,关于这方面的研究存在暴露时间短、受试动植物类别有限、土壤类型单一以及MPs种类、粒径大小和浓度与实际土壤环境有一定差异等问题,未来应结合实际土壤环境状况加强这方面的研究。土壤MPs经陆生食物链的传递和积累,可能对人类健康构成严重威胁,但关于环境相关浓度土壤MPs对不同类型动植物的阈值毒性水平及其在食物链中转移的研究还不足,这些问题在后续研究中需重点考虑,以全面揭示陆地生态系统中MPs带来的生态风险。
(4)MPs因疏水性强、比表面积大,可以吸附多种有机和无机污染物,从而影响土壤生物的生长,MPs还可与自身释放的添加剂等形成复合污染,使得MPs的环境行为更加复杂。但目前关于土壤MPs与其携带的污染物结合和释放的机理尚不清楚,与多种有害污染物共同暴露对陆生生物的毒性效应和人体健康的风险亟待研究。未来的研究重点应关注MPs进入到土壤中如何参与其他元素(如重金属)和污染物的环境地球化学行为及生物效应。
(5)土壤MPs的存在可改变微生物的群落结构和功能,反过来,在微生物、土壤动物、光照辐射等作用下MPs可进一步老化或降解,可能对土壤生态系统构成更大的威胁。但MPs影响土壤微生物的机制和途径暂不明晰,未来探究MPs对微生物群落结构、微生物活性的影响,MPs对全球生态系统和生物地球化学循环及对ARGs的影响是研究的重点方向之一。此外,还应寻找绿色、高效且环保的控制措施,以减少生物体对MPs的吸收,并降低其在土壤生态系统中的迁移。
-

图 2 研究区地表水和地下水中溶解性无机碳和有机碳的含量
Figure 2. Contents of dissolved inorganic carbon and organic carbon in surface water and groundwater in the study area
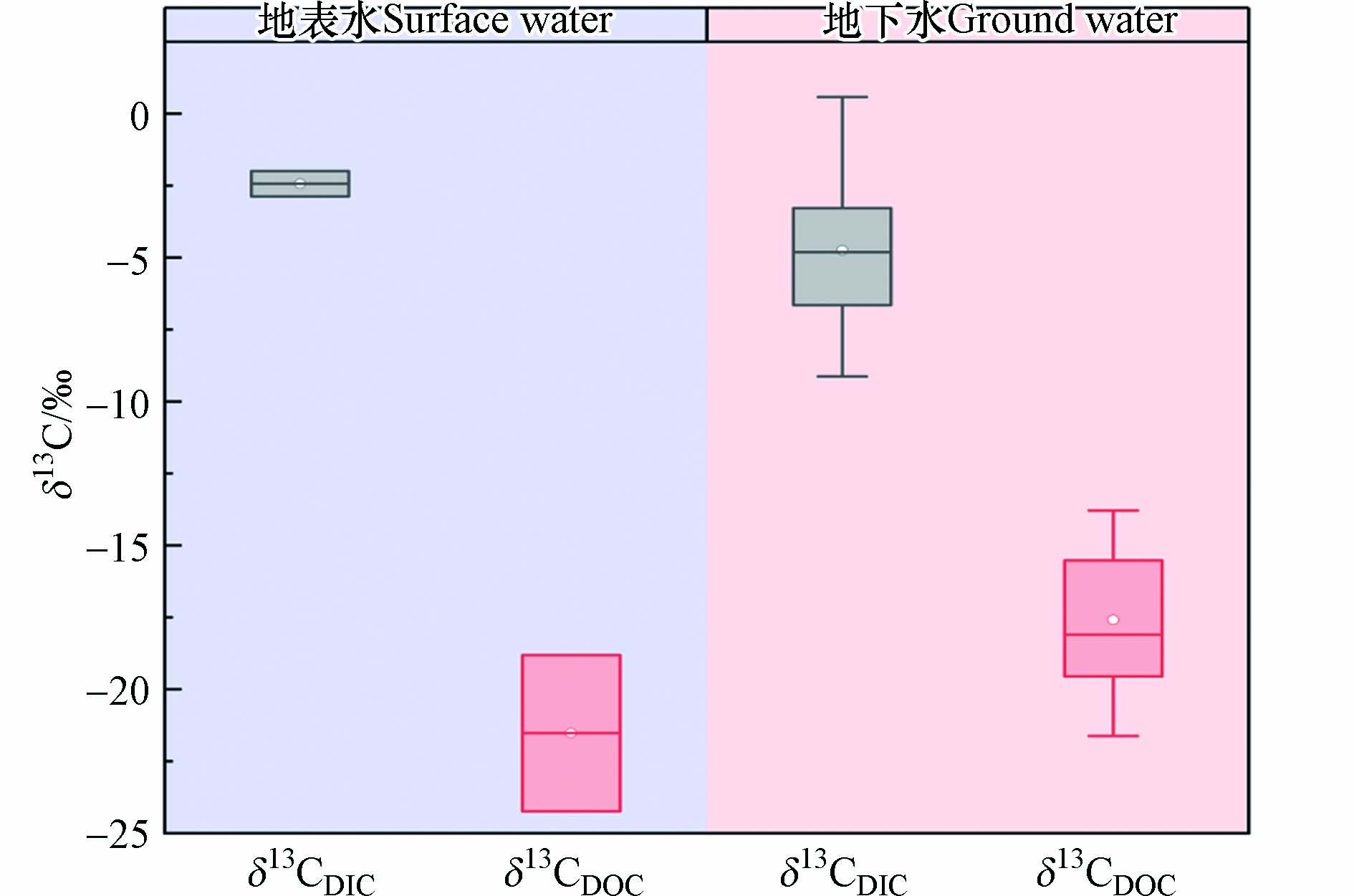
图 3 研究区地表水和地下水中δ13CDIC和δ13CDOC关系图
Figure 3. Relationship between δ13CDIC and δ13CDOC in surface water and groundwater in the study area
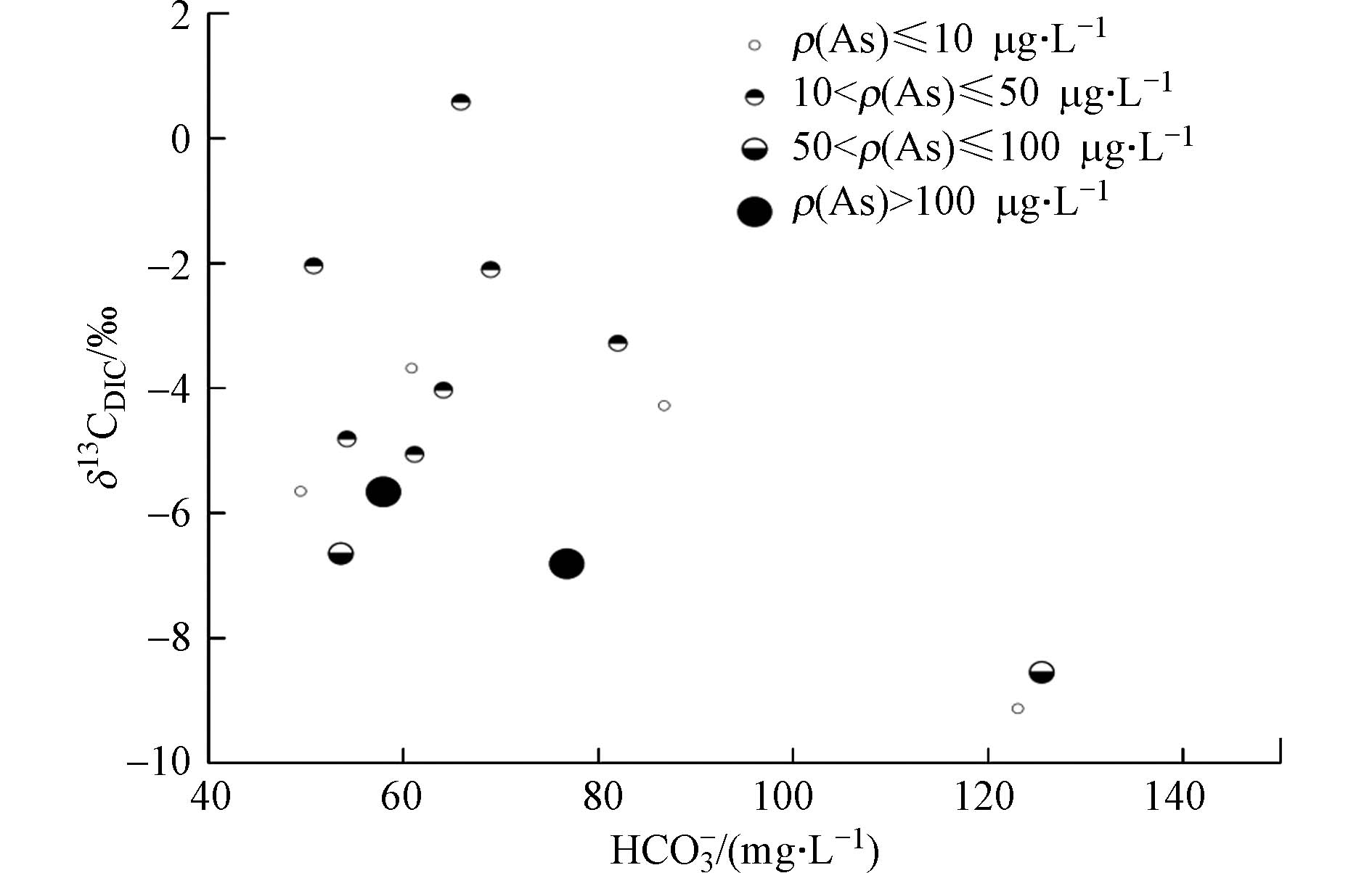
图 4 地下水中δ13CDIC与HCO3-关系图
Figure 4. Relationship between δ13CDIC and HCO3- in groundwater
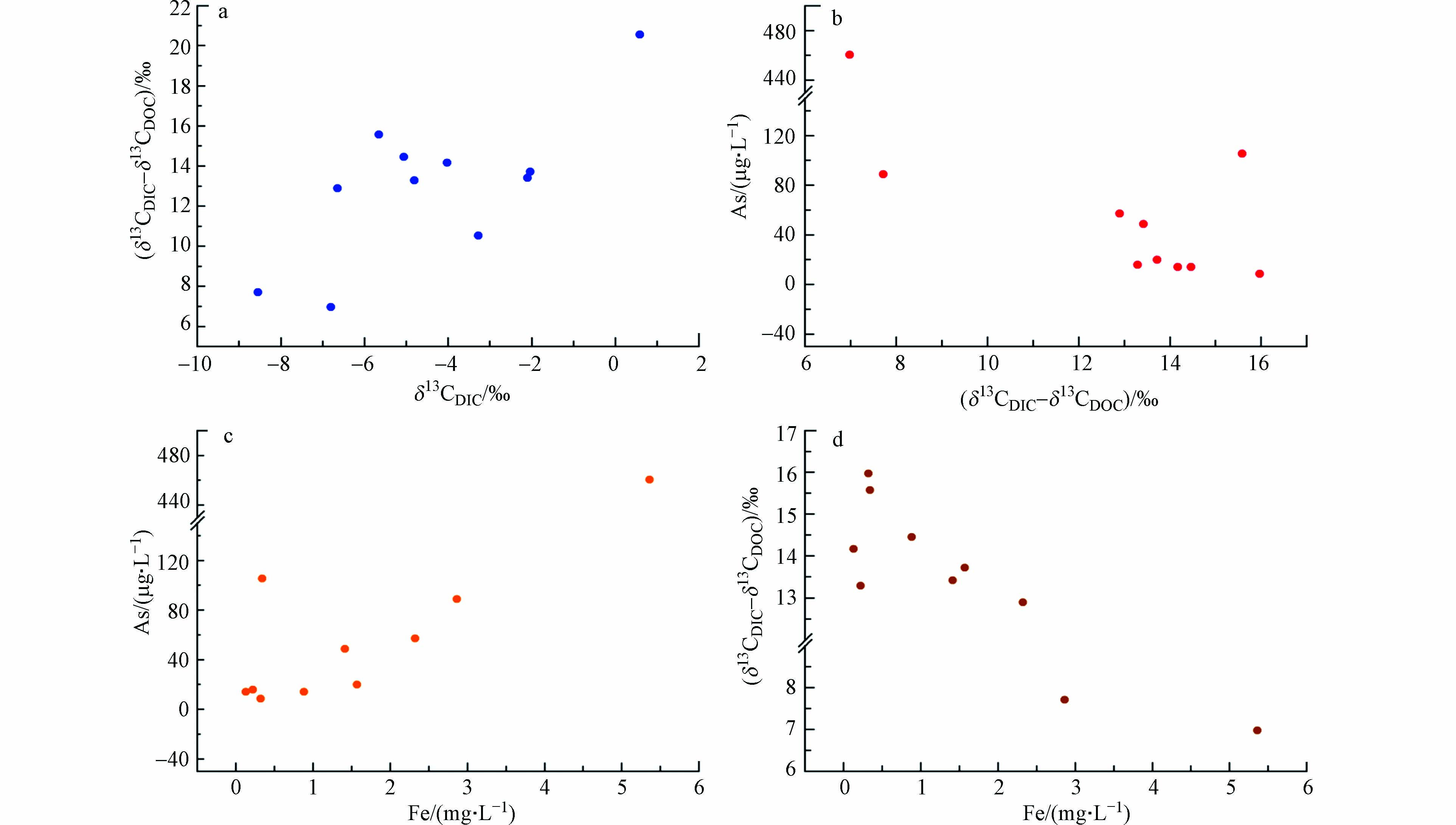
图 5 δ13CDIC-δ13CDOC与δ13CDIC(a)和As浓度与δ13CDIC-δ13CDOC(b)、Fe浓度(c)以及δ13CDIC-δ13CDOC与Fe浓度(d)关系图
Figure 5. Relationship between δ13CDIC-δ13CDOC and δ13CDIC (a) and As concentration and δ13CDIC-δ13CDOC (b), Fe concentration (c) and relationship between δ13CDIC-δ13CDOC and Fe concentrations (d)
表 1 研究区采样点信息
Table 1. Information on sampling sites in the study area
采样点编号Sampling number 经度Longitude 纬度Latitude 井深/m Well depth 采样地点Location of sampling K1 84.7277 44.1247 — 奎屯河 K2 84.7519 44.1605 — 奎屯河 C1 84.0812 45.0314 200 126团 C2 84.0641 45.0257 180 126团 C3 84.1032 45.0210 200 126团 C4 84.0825 45.0215 180 126团 C5 84.0808 45.0222 160 126团 C6 84.1322 45.0230 70 126团 C7 84.3551 45.0217 130 128团 C8 84.3553 45.0221 80 128团 C9 84.3747 44.5852 60 128团 C10 84.3739 44.5848 60 128团 C11 84.3659 44.5905 200 128团 C12 84.4051 44.5836 100 128团 C13 84.4038 44.5558 80 128团 C14 84.4004 44.5723 120 128团 C15 84.3508 45.0513 100 128团  下载: 导出CSV
下载: 导出CSV
表 2 水样主要水化学指标统计表
Table 2. Statistical table of main water chemical indicators of water samples
指标 Index pH K+/(mg·L−1) Na+/(mg·L−1) Ca2+/(mg·L−1) Mg2+/(mg·L−1) 地表水 6.66—7.06(6.86) 1.18 273.50—280.41(276.96) 47.33 10.67—32.80(21.73) 地下水 7.59—9.42(8.63) 0.02—1.25(0.46) 4.58—503.07(107.62) 5.01—544.42(151.57) 1.37—467.36(110.11) 指标 Index Eh/mV CO32−/(mg·L−1) HCO3−/(mg·L−1) Cl−/(mg·L−1) SO42−/(mg·L−1) As/(μg·L−1) 地表水 21.3—28.5(24.90) — 157—179(168) 24.69—50.07(37.38) 536—640(588) 6.27—6.76(6.52) 地下水 −96— −7.5(−55.08) 2.45—9.81(5.19) 49.44—125.52(72.06) 18.88—1829.43(527.35) 0—1415.70(506.56) 2.45—460.38(60.60) 注:括号内为平均值. Note: Average values in brackets
下载: 导出CSV
表 3 不同地区地下水中δ13CDIC与δ13CDOC值、As和Fe质量浓度及井深范围
Table 3. δ13CDIC and δ13CDOC values, As and Fe mass concentrations and well depth ranges in groundwater from different regions
下载: 导出CSV
-
[1] 贾永锋, 郭华明. 高砷地下水研究的热点及发展趋势 [J]. 地球科学进展, 2013, 28(1): 51-61. doi: 10.11867/j.issn.1001-8166.2013.01.0051 JIA Y F, GUO H M. Hot topics and trends in the study of high arsenic groundwater [J]. Advances in Earth Science, 2013, 28(1): 51-61(in Chinese). doi: 10.11867/j.issn.1001-8166.2013.01.0051
[2] GILLISPIE E C, ANDUJAR E, POLIZZOTTO M L. Chemical controls on abiotic and biotic release of geogenic arsenic from Pleistocene aquifer sediments to groundwater [J]. Environmental Science. Processes & Impacts, 2016, 18(8): 1090-1103. [3] RODRÍGUEZ-LADO L, SUN G F, BERG M, et al. Groundwater arsenic contamination throughout China [J]. Science, 2013, 341(6148): 866-868. doi: 10.1126/science.1237484 [4] AHMED K M, BHATTACHARYA P, HASAN M A, et al. Arsenic enrichment in groundwater of the alluvial aquifers in Bangladesh: An overview [J]. Applied Geochemistry, 2004, 19(2): 181-200. doi: 10.1016/j.apgeochem.2003.09.006 [5] SMEDLEY P L, KINNIBURGH D G. A review of the source, behaviour and distribution of arsenic in natural waters [J]. Applied Geochemistry, 2002, 17(5): 517-568. doi: 10.1016/S0883-2927(02)00018-5 [6] 于凯. 高砷地下水系统中有机质来源及其对砷动态变化的影响研究: 以江汉平原为例[D]. 武汉: 中国地质大学, 2016. YU K. The sources and influences of dissolved organic matter on temporal variations of groundwater arsenic concentrations— a case study in Jianghan plain[D]. Wuhan: China University of Geosciences, 2016 (in Chinese).
[7] 周殷竹. 基于碳、铁稳定同位素的高砷地下水生物地球化学研究[D]. 北京: 中国地质大学(北京), 2018. ZHOU Y Z. Biogeochemical processes in high arsenic groundwater in the northwestern Hetao Basin, Inner Mongolia: Evidences from hydrogeochemistry and stable isotopes[D]. Beijing: China University of Geosciences, 2018 (in Chinese).
[8] YU K, GAN Y Q, ZHOU A G, et al. Organic carbon sources and controlling processes on aquifer arsenic cycling in the Jianghan Plain, central China [J]. Chemosphere, 2018, 208: 773-781. doi: 10.1016/j.chemosphere.2018.05.188 [9] 韩莉, 甘义群, 于凯. 江汉平原高砷地下水中溶解性有机质来源的稳定碳同位素示踪研究[J]. 地质学报, 2015, 89(S1): 266-268. HAN L, GAN Y Q, YU K. Stable carbon isotope tracing study on the source of dissolved organic matter in high arsenic groundwater in Jianghan plain[J]. Acta Geologica Sinica, 2015, 89(Sup 1): 266-268 (in Chinese).
[10] XIE X J, WANG Y X, ELLIS A, et al. Multiple isotope (O, S and C) approach elucidates the enrichment of arsenic in the groundwater from the Datong Basin, Northern China [J]. Journal of Hydrology, 2013, 498: 103-112. doi: 10.1016/j.jhydrol.2013.06.024 [11] 张亚男. 江汉平原含水层铁还原砷释放过程中有机质转化机制研究[D]. 武汉: 中国地质大学, 2021. ZHANG Y N. Mechanisms of organic matter transformation during dissimilated iron reduction and arsenic mobilization in aquifer of Jianghan plain[D]. Wuhan: China University of Geosciences, 2021 (in Chinese).
[12] 周殷竹. 内蒙古河套盆地地下水碳同位素特征及生物地球化学意义[D]. 北京: 中国地质大学(北京), 2014. ZHOU Y Z. Characteristics of C isotopes and its biogeochemistry significance in high as groundwater system in the Hetao Basin, Inner Mongolia[D]. Beijing: China University of Geosciences, 2014 (in Chinese).
[13] GUO H M, ZHANG D, WEN D G, et al. Arsenic mobilization in aquifers of the southwest Songnen Basin, P. R. China: Evidences from chemical and isotopic characteristics [J]. Science of the Total Environment, 2014, 490: 590-602. doi: 10.1016/j.scitotenv.2014.05.050 [14] 罗艳丽, 李晶, 蒋平安, 等. 新疆奎屯原生高砷地下水的分布、类型及成因分析 [J]. 环境科学学报, 2017, 37(8): 2897-2903. LUO Y L, LI J, JIANG P A, et al. Distribution, classification and cause analysis of geogenic high-arsenic groundwater in Kuitun, Xinjiang [J]. Acta Scientiae Circumstantiae, 2017, 37(8): 2897-2903(in Chinese).
[15] 罗艳丽, 李晶, 蒋平安, 等. 新疆高砷地区地下水水化学特征及其成因分析 [J]. 干旱区资源与环境, 2017, 31(8): 116-121. doi: 10.13448/j.cnki.jalre.2017.256 LUO Y L, LI J, JIANG P A, et al. Hydro-chemical characteristics and the formations for groundwater in Kuitun, Xinjiang [J]. Journal of Arid Land Resources and Environment, 2017, 31(8): 116-121(in Chinese). doi: 10.13448/j.cnki.jalre.2017.256
[16] 邓雯文, 罗艳丽, 王翔, 等. 新疆奎屯地区地下水中砷和盐的分布特征及成因分析 [J]. 环境污染与防治, 2021, 43(11): 1404-1409. DENG W W, LUO Y L, WANG X, et al. Distribution characteristics and cause analysis of arsenic and salinity in groundwater of Kuitun area, Xinjiang [J]. Environmental Pollution & Control, 2021, 43(11): 1404-1409(in Chinese).
[17] 张慧. 基于RS与GIS的天山奎屯河流域冰川变化研究[D]. 兰州: 西北师范大学, 2015. ZHANG H. Research of glacier change based on RS and GIS in the Kuytun River Basin, Tianshan mountains[D]. Lanzhou: Northwest Normal University, 2015 (in Chinese).
[18] 南峰, 李有利, 邱祝礼. 新疆奎屯河流域山前河流地貌特征及演化 [J]. 水土保持研究, 2005, 12(4): 10-13. doi: 10.3969/j.issn.1005-3409.2005.04.004 NAN F, LI Y L, QIU Z L. Characteristics and evolutions of fluvial geomorphology in foreland of the Kuitun River valley [J]. Research of Soil and Water Conservation, 2005, 12(4): 10-13(in Chinese). doi: 10.3969/j.issn.1005-3409.2005.04.004
[19] 王丽娟, 王文科, 王哲, 等. 奎屯河流域包气带盐分分布特征与影响因素 [J]. 地球科学与环境学报, 2008, 30(4): 429-433. WANG L J, WANG W K, WANG Z, et al. Distribution regularity and the formative factor soil salt of the Kuitun River Basin [J]. Journal of Earth Sciences and Environment, 2008, 30(4): 429-433(in Chinese).
[20] 王翔, 罗艳丽, 邓雯文, 等. 新疆奎屯地区高砷地下水DOM三维荧光特征 [J]. 中国环境科学, 2020, 40(11): 4974-4981. WANG X, LUO Y L, DENG W W, et al. The 3D-EEM characteristics of DOM in high arsenic groundwater of Kuitun, XinjiangFull text replacement [J]. China Environmental Science, 2020, 40(11): 4974-4981(in Chinese).
[21] 王翔. 奎屯河下游区域地下水中砷的释放过程研究[D]. 乌鲁木齐: 新疆农业大学, 2021. WANG X. Mobilization processes of arsenic in groundwater of Kuitun River downsteam[D]. Urumqi: Xinjiang Agricultural University, 2021 (in Chinese).
[22] 周殷竹, 郭华明, 逯海. 高砷地下水中溶解性有机碳和无机碳稳定同位素特征 [J]. 现代地质, 2015, 29(2): 252-259. ZHOU Y Z, GUO H M, LU H. Stable isotope characteristics of dissolved organic carbon and inorganic carbon in high arsenic groundwater [J]. Geoscience, 2015, 29(2): 252-259(in Chinese).
[23] 袁翰卿, 李巧, 陶洪飞, 等. 新疆奎屯河流域地下水砷富集因素 [J]. 环境化学, 2020, 39(2): 524-530. doi: 10.7524/j.issn.0254-6108.2019051403 YUAN H Q, LI Q, TAO H F, et al. Groundwater arsenic enrichment factors of Kuitun River Basin, Xinjiang [J]. Environmental Chemistry, 2020, 39(2): 524-530(in Chinese). doi: 10.7524/j.issn.0254-6108.2019051403
[24] FONYUY E W, ATEKWANA E A. Dissolved inorganic carbon evolution and stable carbon isotope fractionation in acid mine drainage contaminated streams: Insights from a laboratory study [J]. Applied Geochemistry, 2008, 23(9): 2634-2648. doi: 10.1016/j.apgeochem.2008.05.012 [25] 袁晓芳, 邓娅敏, 杜尧, 等. 江汉平原高砷地下水稳定碳同位素特征及其指示意义 [J]. 地质科技通报, 2020, 39(5): 156-163. YUAN X F, DENG Y M, DU Y, et al. Characteristics of stable carbon isotopes and its implications on arsenic enrichment in shallow groundwater of the Jianghan Plain [J]. Bulletin of Geological Science and Technology, 2020, 39(5): 156-163(in Chinese).
[26] 史婷婷. 岩溶流域水循环过程碳汇效应研究: 以湖北香溪河流域为例[D]. 武汉: 中国地质大学, 2012. SHI T T. The carbon sequestration effect of water cycle in a Karst Basin— a case study of Xiangxi River Basin, Hubei[D]. Wuhan: China University of Geosciences, 2012 (in Chinese).
[27] YANG C, TELMER K, VEIZER J. Chemical dynamics of the “St. Lawrence” riverine system: δDH2O, δ18OH2O, δ13CDIC, δ34Ssulfate, and dissolved 87Sr/86Sr [J]. Geochimica et Cosmochimica Acta, 1996, 60(5): 851-866. doi: 10.1016/0016-7037(95)00445-9 [28] BARTH J A C, CRONIN A A, DUNLOP J, et al. Influence of carbonates on the riverine carbon cycle in an anthropogenically dominated catchment basin: Evidence from major elements and stable carbon isotopes in the Lagan River (N. Ireland) [J]. Chemical Geology, 2003, 200(3/4): 203-216. [29] 邹君宇, 韩贵琳. 河流中碳、硫稳定同位素的研究进展 [J]. 地球与环境, 2015, 43(1): 111-122. doi: 10.14050/j.cnki.1672-9250.2015.01.016 ZOU J Y, HAN G L. Research review on carbon and sulfur stable isotopes in rivers [J]. Earth and Environment, 2015, 43(1): 111-122(in Chinese). doi: 10.14050/j.cnki.1672-9250.2015.01.016
[30] ZHOU Y Z, GUO H M, ZHANG Z, et al. Characteristics and implication of stable carbon isotope in high arsenic groundwater systems in the northwest Hetao Basin, Inner Mongolia, China [J]. Journal of Asian Earth Sciences, 2018, 163: 70-79. doi: 10.1016/j.jseaes.2018.05.018 [31] 姚冠荣, 高全洲. 河流碳循环对全球变化的响应与反馈 [J]. 地理科学进展, 2005, 24(5): 50-60. doi: 10.11820/dlkxjz.2005.05.006 YAO G R, GAO Q Z. The feedback and response of the riverine carbon cycle to global changes [J]. Progress in Geography, 2005, 24(5): 50-60(in Chinese). doi: 10.11820/dlkxjz.2005.05.006
[32] 丁薇. 河流水体有机碳研究综述 [J]. 安徽农学通报, 2017, 23(9): 28-29,71. doi: 10.3969/j.issn.1007-7731.2017.09.010 DING W. The review of the riverine organic carbon [J]. Anhui Agricultural Science Bulletin, 2017, 23(9): 28-29,71(in Chinese). doi: 10.3969/j.issn.1007-7731.2017.09.010
[33] 魏秀国. 河流有机质生物地球化学研究进展 [J]. 生态环境, 2007, 16(3): 1063-1067. WEI X G. Progress in the study of biogeochemistry of riverine organic matter [J]. Ecology and Environment, 2007, 16(3): 1063-1067(in Chinese).
[34] CLARK I D, FRITZ P. Environmental Isotopes in Hydrogeology[M].New York: Lewis Publishing House, 1997. [35] TRUESDELL A H, HULSTON J R. Isotopic evidence on environments of geothermal systems[M]//The Terrestrial Environment, A. Amsterdam: Elsevier, 1980: 179-226. [36] 江军, 鲜虎胜, 李巧, 等. 奎屯河流域地下水地球化学特征及其对砷运移的影响 [J]. 环境化学, 2021, 40(6): 1775-1786. doi: 10.7524/j.issn.0254-6108.2020062702 JIANG J, XIAN H S, LI Q, et al. Groundwater geochemistry and its implications for arsenic mobilization in Kuitun River Basin, Xinjiang [J]. Environmental Chemistry, 2021, 40(6): 1775-1786(in Chinese). doi: 10.7524/j.issn.0254-6108.2020062702
[37] 吕航. 地下水中石油烃生物降解的同位素地球化学研究[D]. 长春: 吉林大学, 2011. (LÜ/LV/LU/LYU) H. Isotope geochemistry of biodegradation processes in a petroleum hydrocarbon-contaminated aquifer[D]. Changchun: Jilin University, 2011 (in Chinese).
[38] 郑永飞, 陈江峰. 稳定同位素地球化学[M]. 北京: 科学出版社, 2000. ZHENG Y F, CHEN J F. Isotopic geochemistry[M]. Beijing: Science Press, 2000(in Chinese).
[39] 陈世苹, 白永飞, 韩兴国. 稳定性碳同位素技术在生态学研究中的应用 [J]. 植物生态学报, 2002, 26(5): 549-560. doi: 10.3321/j.issn:1005-264X.2002.05.007 CHEN S P, BAI Y F, HAN X G. Applications of stable carbon isotope techniques to ecological research [J]. Acta Phytoecologica Sinica, 2002, 26(5): 549-560(in Chinese). doi: 10.3321/j.issn:1005-264X.2002.05.007
[40] 洪里. 新疆奎屯北部车排子地区高氟、高砷水的病害与形成环境的初步研究 [J]. 新疆环境保护, 1983, 5(1): 22-28. HONG L. Preliminary study on diseases and formation environment of high fluorine and high arsenic water in Chepaizi area of northern Kuitun, Xinjiang [J]. Environmental Protection of Xinjiang, 1983, 5(1): 22-28(in Chinese).
[41] 宿彦鹏, 李巧, 陶洪飞, 等. 新疆奎屯河流域地下水砷超标原因分析 [J]. 长江科学院院报, 2022, 39(3): 54-59. doi: 10.11988/ckyyb.20201188 SU Y P, LI Q, TAO H F, et al. Causes of excessive arsenic in groundwater of Kuitun River Basin in Xinjiang [J]. Journal of Yangtze River Scientific Research Institute, 2022, 39(3): 54-59(in Chinese). doi: 10.11988/ckyyb.20201188
[42] HUANG S B, WANG Y X, MA T, et al. Lin1 dissolved organic matter to sedimentary organic matter from a fluvio-lacustrine aquifer at Jianghan Plain, China by EEM-PARAFAC and hydrochemical analyses [J]. Science of the Total Environment, 2015, 529: 131-139. doi: 10.1016/j.scitotenv.2015.05.051 期刊类型引用(1)
1. 贺凯凯,朱峰,王彦堂,郝春明. 湖南锡矿山高锑地下水稳定碳同位素特征及其富锑意义. 科学技术与工程. 2024(22): 9672-9680 .  百度学术
百度学术
其他类型引用(0)
-

 点击查看大图
点击查看大图

计量
- 文章访问数: 1650
- HTML全文浏览数: 1650
- PDF下载数: 76
- 施引文献: 1
