

















-
岩溶地下水是泉城济南工农业生产和社会发展不可缺少的重要资源[1]. 上世纪80年代以来,随着城市化进程的不断加快,济南岩溶地下水开采量不断增大,工农业生产、旅游活动、生活污水等对岩溶地下水造成了一定程度的污染,水质逐年变差[2]. 特别是在泉域补给径流区,城郊和农村点源与面源污染交错,使得硝酸盐污染问题日益突出[3]. 饮用水中NO3-N含量过高容易造成儿童患高铁血红蛋白症,成人患胃癌、食管癌等,威胁人类健康[4-5]. 同时,岩溶区具有特殊的“地表—地下”双层结构,其含水层结构的特殊性决定了NO3−污染状况更为复杂[6-7].
近年来,许多学者针对岩溶水系统硝酸盐污染问题开展了大量研究,主要集中在西南亚热带岩溶区[8-10],而对于北方半湿润半干旱岩溶区的相关研究较少. 前人对于济南泉域主要开展了水文地质条件、岩溶含水层结构、水循环特征及水环境等方面的研究[11-13],特别是在岩溶地下水的水化学特征及水环境演化方面,部分学者对济南泉域岩溶地下水中主要元素、微量元素及稳定环境同位素进行分析,研究了济南泉域地下水补给来源、水文地球化学特征等问题[14-15]. 还有学者对济南泉域岩溶地下水水质变化规律及其影响因素进行了分析[16-17]. 但是在泉域尺度上采用水化学和NO3−氮氧双同位素分析岩溶地下水系统NO3−污染特征及污染来源的研究尚不多见.
本文以济南泉域为例,基于水化学和NO3−氮氧双同位素示踪技术,从水文地球化学角度,系统分析泉域岩溶水水化学特征,探究NO3−污染来源、分布及影响因素,并定量计算NO3−的各类贡献源比例,剖析NO3−的生物地球化学过程,以期为济南泉域岩溶水资源开发利用及生态环境保护提供科学依据.
-
济南位于暖温带大陆性季风气候区,多年平均气温14.3 ℃,多年平均降水量667.1 mm,降水主要集中在6—9月份,降水量在空间上自东南向西北递减. 流经济南的河流主要有黄河、玉符河、北大沙河、小清河. 黄河从济南市北部流过,玉符河和北大沙河自南部山区汇入黄河,小清河自西向东横穿市区,汇集了济南泉水后向东流入渤海[3].
济南泉域是微向北倾斜的单斜构造地质体,是我国典型的北方岩溶发育区,总面积约1500 km2(图1). 古生界寒武系、奥陶系碳酸盐岩地层成单斜状覆盖于变质岩系之上,与地形倾向基本一致,至北部隐伏于山前第四系地层之下[18]. 泉域北部及东、西郊有燕山期侵入的辉长岩体大面积分布;西部玉符河以西沿黄河地带,奥陶系埋藏于石炭、二叠系之下,呈北西—东南向分布. 特定的地质构造条件,是济南泉域含水层空间分布、地下水循环运动以及富水性的主要控制因素[19].
泉域碳酸盐岩裂隙岩溶含水系统由寒武系中统张夏组、上统凤山组及奥陶系含水层组成,岩性为灰岩、泥质灰岩、白云质灰岩、灰质白云岩和白云岩. 岩溶裂隙发育,连通性好,有利于地下水的补给、径流和汇集排泄[20]. 泉域由南向北依次为间接补给区、直接补给径流区和汇集排泄区[16]. 区内岩溶水接受大气降水补给后由南向北运动,遇辉长岩体受阻,上升出露成泉. 区域地表水与地下水水力联系密切,岩溶含水层极易受地表水的混合污染[21-22].
泉域内人类活动主要为农业生产活动,没有大型的污染工厂. 居民点呈点状分散式分布,家禽养殖普遍且化粪池广泛分布. 南部补给区和径流区耕地和林地面积占区域总面积的80%以上,区内施肥期主要集中在4月和8月,多用复合肥、尿素和碳酸氢铵等化学肥料.
-
2020年9月共采集研究区地表水和地下水样品30个,其中地表水4个、地下水26个(含泉水3个),采样位置分布见图1. 用液体采样器采集水样,地表水均采自水面以下0.5 m深处的水体样品. 地下水样品为深层地下水,包括民用井和专业监测井,采样井井深全部在150 m以上,水位埋深在60 m以上. 对于封闭式水井,抽取5 min后再取样,保证所取地下水新鲜;对于敞口式水井,用液体采样器采集水面以下0.5 m深处的水体样品. 现场使用便携式多参数水质参数仪(哈希HQ40D,America)测定各采样点水样的温度(T)、电导率(EC)、溶解氧(DO)、pH值,精度分别为0.1 °C、1 µS·cm−1、0.01 mg·L−1、0.01个pH单位. 水化学分析水样用洁净的600 mL聚乙烯瓶采集,并确保瓶内无气泡,用于室内阴、阳离子浓度检测. K+、Na+、Ca2+、Mg2+、采用ICP-OES分析;Cl−、NO3−、SO42−采用阴离子色谱仪测试,精度0.01 mg·L−1;NH4+采用纳氏试剂比色法测定,精度0.01 mg·L−1;NO2−采用α-萘铵比色法测定,精度0.01 mg·L−1;HCO3−、总硬度采用滴定法测定,精度0.01 mg·L−1. 以上水化学测试工作在山东省鲁南地质工程勘察院实验测试中心完成.
同位素分析水样用事先洗净的40 mL聚乙烯瓶采集,现场经0.22 μm混合纤维素滤膜过滤,冷藏后送至实验室进行硝酸盐δ15N、δ18O同位素分析. 其中,硝酸盐δ15N、δ18O同位素检测样品时利用特异性的反硝化细菌将硝态氮转化为N2O,再采用ISOPRIME100-Tracegas痕量气体-同位素质谱联用仪(英国Isoprime公司)完成N2O的N、O同位素测定,检测精度为0.01‰,δ15N测定结果标准偏差<0.4‰,δ18O测定结果标准偏差<0.22‰. 以上同位素测试工作在中国农业科学院农业环境稳定同位素实验室完成.
-
使用Aq·QA v.1.1软件绘制Piper三线图;采用MATLAB 9.3软件计算地下水水化学成分相关系数;其它图件均采用Origin 8.5软件进行绘制.
本研究基于15N、18O同位素值识别NO3−来源,使用稳定同位素混合模型MixSIAR计算NO3−各种来源的贡献率. MixSIAR模型是用于NO3−来源计算的可靠工具[23-24],通过定义k个来源n个混合物的j个同位素,考虑同位素分馏作用的影响,可表达如下[25]:
式中, Xij表示第i个样品中第j种同位素值(i=1,2,3,…,N;j=1,2,3,…,J);Sjk是第k种源中第j种同位素的值(k=1,2,3,…,K),μjk. 为平均值,
{\omega }_{jk}^{2} 为正态分布的方差;Cjk是第j种同位素在第k个源上的分馏系数,λjk为平均值,{T}_{jk}^{2} 为正态分布的方差;pk为第k个源的贡献率,由模型计算得到;qjk是同位素j在第k种源中的浓度;ɛij为残差,表示各混合物间未能确定的变量,其均值为0,{\sigma }_{j}^{2} 为正态分布的方差. -
对研究区内地下水和地表水的水化学参数进行分析,结果表明(表1),研究区地下水和地表水的pH值均大于7,最大值为8.93,呈弱碱性. 除G14采样点TDS异常低外,沿地下水流向岩溶地下水TDS同样呈现上升的趋势,且TDS分布在185.79—892.74 mg·L−1之间.Ca2+、Mg2+、SO42−、NO3−浓度在G4和G5采样点均较高,表明G4和G5采样点附近岩溶地下水环境污染较严重. 阳离子中Ca2+和Mg2+占绝对优势,阴离子中HCO3−和SO42−占绝对优势,其二者之和在地下水和地表水中均占阳离子和阴离子总量的80%以上.
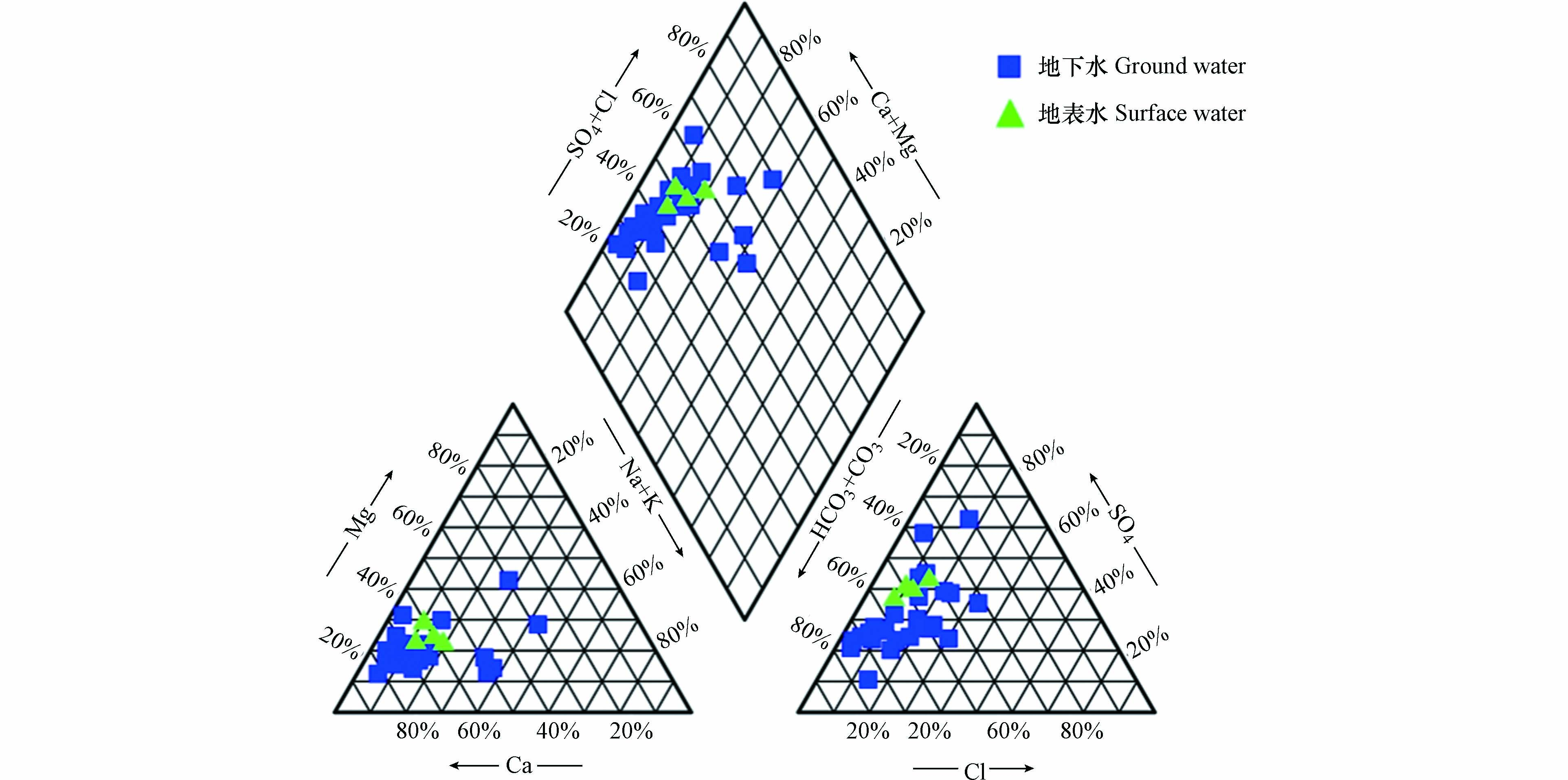
从研究区水化学Piper三线图可以看出(图2),采样点均落在菱形的左上区域,碱土金属(Ca、Mg)超过碱金属(K、Na),表明地下水化学性质以碱土金属和弱酸为主. 结合统计数据和Piper三线图可知,研究区地下水和地表水主要的阴、阳离子分别为HCO3−、Ca2+,根据舒卡列夫分类,研究区水化学类型以HCO3·SO4-Ca型为主,占所有样品的60%,主要分布在径流区和排泄区. 其次,HCO3-Ca型占所有样品的17%. 另外,还有个别水样为HCO3-Ca·Mg型、HCO3·SO4-Ca·Mg型、HCO3·SO4·Cl-Ca·Na型、SO4-Ca·Mg及SO4·Cl-Ca,表现出复杂的水化学类型体系.
-
济南泉域地下水NO3−浓度平均值(37.93 mg·L−1)高于地表水(10.15 mg·L−1). 对比我国地下水质量标准,在26个地下水样品中,ρ(NO3−N)≤2.0 mg·L−1的占8%,ρ(NO3−N)为2.0—5.0 mg·L−1的占23%,ρ(NO3−N)为5.0—20 mg·L−1的占65%,其中有4%的样品ρ(NO3−N)>20 mg·L−1,水质低于GB/T 14848—2017的Ⅲ类水标准;若按世界卫生组织标准[ρ(NO3−N)为10 mg·L−1]来衡量,则有27%的样品ρ(NO3−N)超标. 而4个地表水样品的ρ(NO3−N)均未超过3 mg·L−1,远低于地表水环境质量标准中集中式生活饮用水地表水源地ρ(NO3−N)≤10 mg·L−1的限值. 总体来说,研究区水质总体良好,仅少数采样点ρ(NO3−N)超标,存在点源污染的风险.
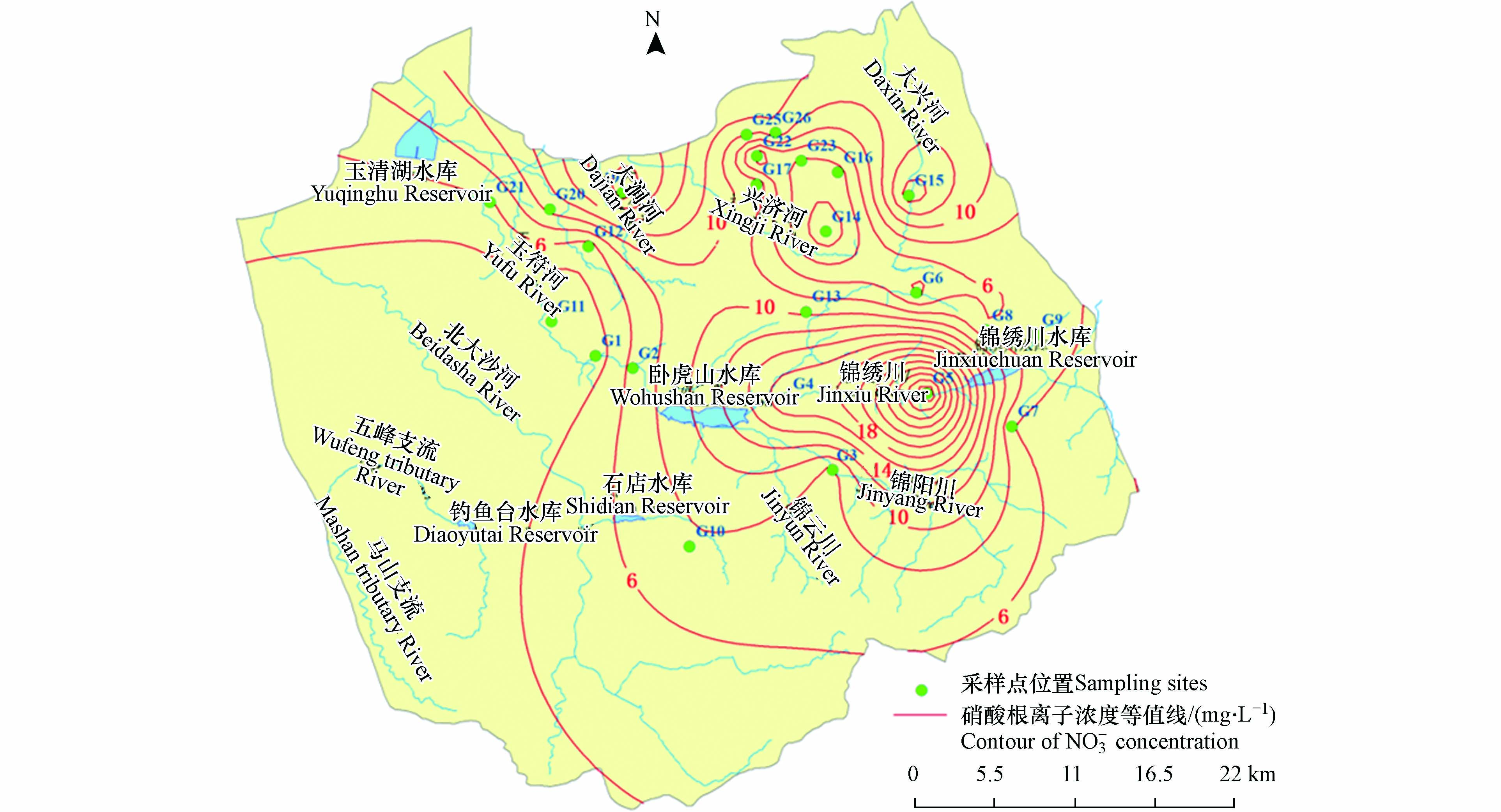
利用ArcGIS10.3软件反距离权重法进行插值,分析研究区地下水NO3−浓度的空间分布变化,结果如图3所示. 由图3和表2可知,上游补给区及下游汇集排泄区采样点地下水NO3−浓度较高,而径流区采样点地下水NO3−浓度相对偏低.NO3−浓度高值点出现在G4、 G5、G15、G19、 G20、G25、G26,浓度值均高于10 mg·L−1,其中G5采样点NO3−-N浓度高达32.90 mg·L−1. 结合采样点类型及周边环境,对其进行分析,G5采样点位于村内,地下水埋深约为7 m,该井为民井,井口保护设施不完善,生活污水及人畜粪便随降雨入渗后进入含水层,污染较严重. 其他NO3−浓度较高的采样点也均位于农村或城郊居民生活区,周边为耕地和工厂,NO3−污染原因可能是受到工业污染以及化肥施用的影响.
在空间上,从研究区南部上游至北部下游地下水NO3−浓度呈低—高—低—高的变化特征. 前面所述NO3−浓度超标的采样点,基本形成了3个NO3−高值区,均分布在上游补给区和下游排泄区.
土地利用类型对地下水和地表水中NO3−浓度有着直接影响[26-27]. 研究区内土地利用类型复杂多变,补给区和径流区以农业生产为主,耕地和林地面积占区域总面积的80%以上;汇集排泄区为城区,土地利用类型以建设用地为主. 研究区南部补给区和中部径流区地处泰山余脉,山地地形占地较广,村镇和人口相对较少,土地利用类型以次生林地、灌木丛和耕地为主,沿地下水流路径方向向北直至城郊,村镇和人口逐渐增多,适宜居住和开展农业活动,土地类型多样,以农田、村镇和次生林地为主. 由各采样点的土地利用类型可知,30个采样点中有22个位于农田或村镇附近,占比73.3%. 农田和村镇是人类农业活动密集的区域,其产生的污水中含有高浓度的NO3−,在岩溶地区,由于土壤层较薄,在降水过程中NO3−易通过雨水的携带进入土壤,并经岩溶裂隙和管道入渗影响地下水,同时通过地面冲刷和地表径流汇入河流.
-
通过研究区地表水和地下水HCO3−/Na+和Ca2+/Na+、Mg2+/Na+和Ca2+/Na+的关系,结合碳酸盐岩、硅酸盐岩和蒸发岩3个端元的范围值[28],来判断水岩作用对水化学成分的影响. 如图4所示,地表水和地下水均分布在靠近碳酸盐岩一侧,且地下水分布相对地表水更偏右上,说明两种水体的离子组成主要受到了碳酸盐岩溶解的影响,其中地下水受到的影响更为显著.
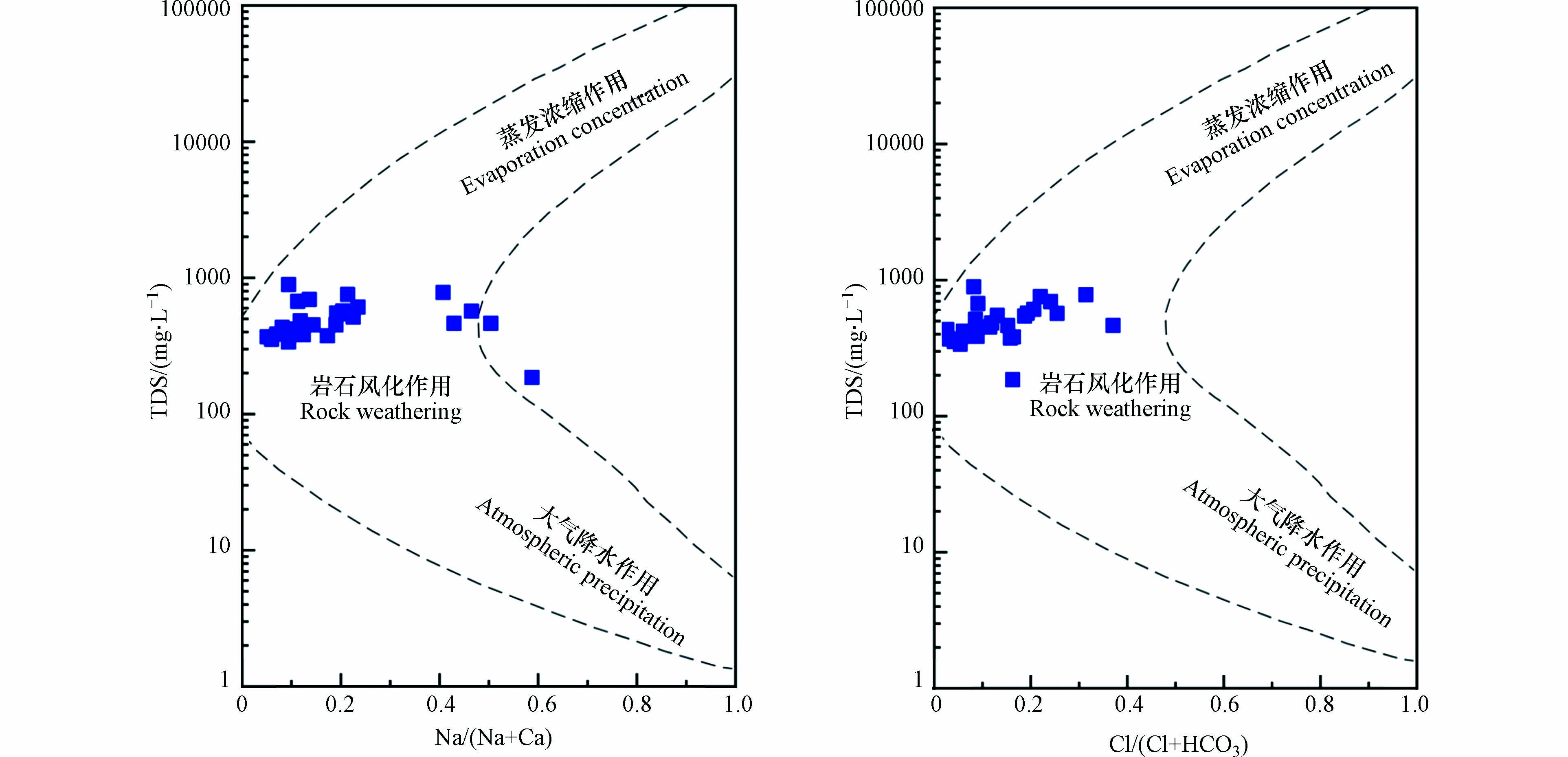
采用Gibbs图表征地下水主要离子的化学组成与变化规律,判断地下水是否受蒸发浓缩、岩石风化或者降水的作用,是分析水化学成分演化过程中主控作用的一种重要手段[29],可深入认识研究区地下水化学组分的成因及演化规律[30-31]. 从研究区地下水水化学Gibbs图可以看出,地下水样品大部分分布在模型的中部,TDS处于100—1000 mg·L−1之间,大部分样品的γ(Na+)/[γ(Na+)+γ(Ca2+)]和γ(Cl−)/[γ(Cl−)+γ(HCO3−)]值在0—0.5之间(图5),表明地下水主要阴、阳离子分别以HCO3−、Ca2+为主,且水化学组分特征主要受岩石风化作用的自然过程控制.
相关关系可以分析地下水水化学组分间的相似性,揭示不同离子是否具有同一来源. 采用统计学软件SPSS对研究区地下水水化学成分进行Pearson相关系数计算,从研究区地下水各化学组分之间的相关关系矩阵可以看出(表3),TDS与Ca2+、Mg2+、Cl−、SO42−、NO3−存在明显的线性相关关系,其中,TDS与Ca2+和SO42−的相关关系最为显著,相关关系分别为0.83和0.8,说明同样是这两种化学组分是TDS的重要来源. 地下水中的NO3−一般来源于大气降水及土壤淋滤,但是随着人类活动的增加,动物粪便、生活污水、化肥、农药等排放或使用导致地下水NO3−含量不断升高,从表1的水化学统计数据和表3相关系数矩阵可以看出,NO3−和SO42−均已成为地下水中主要的离子成分之一. Na+与Cl−显著相关,相关系数为0.7,表明研究区内含水层可能存在石盐矿物,受到水岩作用或淋滤作用影响,也可能是外源污染造成.
-
正常情况下由于天然地下水中NO3−含量很低,不会对人体产生危害,但是受城市化发展和农业活动影响,大量含氮肥料的使用以及人类、动物粪便和生活污水的排放等使得大量NO3−入渗进入含水层中,特别是在具有地表地下二元结构的岩溶含水层,入渗能力更强[32]. 人类长期暴露于过量NO3−环境中会引起高铁血红蛋白症,增加消化系统癌症的发病率[12]. 通过水水化学组分统计数据可以看出,研究区地下水中NO3−含量普遍较高,一部分样品NO3−超过了生活饮用水卫生标准. 由于地下水中Cl−相对其他离子成分要稳定得多,并且不同来源的NO3−与Cl−之间存在一定的区别,可采用γ(NO3−)/γ(Cl−)值来定性判断NO3−的来源. 从γ(NO3−)/γ(Cl−)比值关系(图6)可以看出,研究区大部分水样落在γ(NO3−)/γ(Cl−)<2以下,而且γ(Cl−)分布范围较大,表现出低γ(NO3−)/γ(Cl−),γ(Cl−)不稳定的特点,因此,判断研究区NO3−主要来自于动物粪便和生活污水.
Cl−结合NO3−/Cl−比值的方法是水化学中常用来识别NO3−来源的手段之一[33]. 低浓度的Cl−和较高的NO3−/Cl−值表明NO3−主要来源于化学肥料,而高浓度的Cl−和较低的NO3−/Cl−值表明NO3−主要来源于人畜粪便和生活污水. 由图6可知,NO3−与Cl−之间无良好的线性关系,且相关性也较弱,说明NO3−和Cl−的来源并不完全相同. 样品点主要分布在低Cl−和高NO3−/Cl−比值区域,分布较分散,表明研究区地下水和地表水中NO3−主要来源于粪便和污水,并且不同区域受化肥与污水和有机肥的影响程度差异较大. 地下水与地表水的样品点分布相似,表明两种水体有相似的NO3−来源.
一般情况下,如果环境中没有大量NH4+积累,矿化和硝化作用产生的NO3−的δ15N值与初始反应物质的δ15N值一致,具有较小的同位素分馏[6]. 因为大量NH4+在环境中积累,反应不完全,一方面会毒害环境中的微生物,中止或减缓生化反应;另一方面由于同位素动力学分馏将使产物NO3−富集14N. 在本研究中,绝大多数地下水样品中的NH4+浓度极低,上述几个反应带来的同位素分馏可以认为是不显著的. 鉴于雨水中具有较低含量的NH4+,且是主要的氮素形态,而NO3−含量也非常低,雨水在稀释地下水的同时不可能成为主要的地下水NO3−来源. 研究区范围较大,补给区和径流区主要人类活动为种植业、养殖业及手工业,汇集排泄区位于城区,大量的农用化肥、工业污水和生活污水造成了较严重的点源污染,是NO3−含量分布差异较大的主要原因. 从研究区地下水和地表水中硝酸盐氮氧同位素的特征值(图7)可知,δ15N值范围在1.05‰—14.43‰,δ18O值范围在−7.92‰—22.94‰,大部分位于粪便和污水的范围内,少部分位于土壤氮及降雨和化肥中NH4+范围内,说明研究区粪便和污水是地下水和地表水NO3−污染的主要来源,其次为土壤氮,这一研究结论与水化学分析结果一致.
-
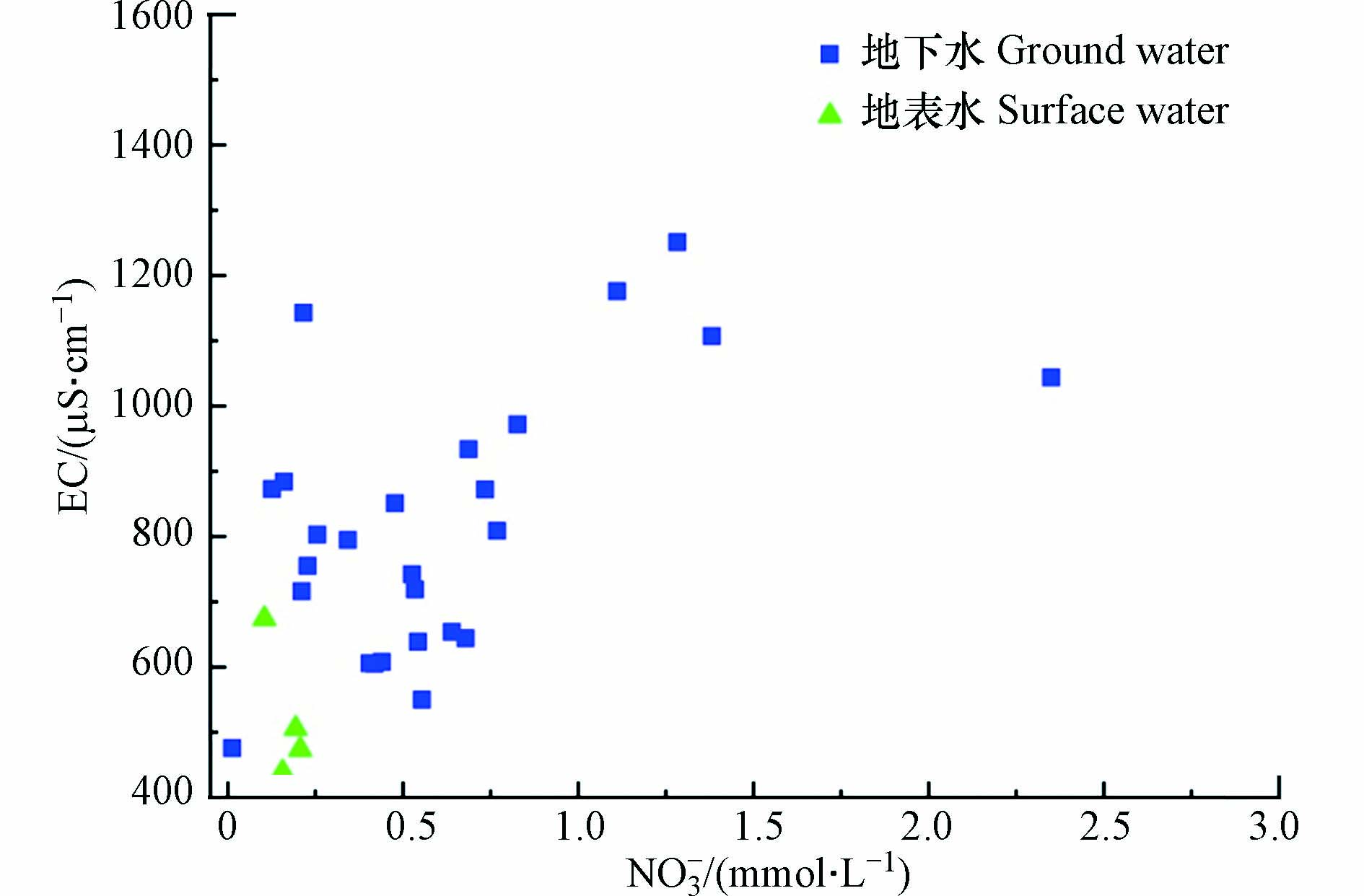
Cl−和NO3−的关系以及EC和NO3−的关系能初步判断水中NO3−浓度的变化是源于无N形态转化的混合过程还是由反硝化作用引起[20]. 若NO3−浓度的增加伴随Cl−浓度和EC的增加,则说明水中NO3−浓度可能主要受到无N形态转化的混合过程的影响,反之则可能有反硝化作用发生. 如图6和图8所示,整体上NO3−与Cl−以及NO3−与EC之间均没有显著的线性关系和良好的相关性,但仍然趋向于随NO3−浓度的升高,Cl−和EC均有所升高,表明研究区NO3−主要受到无N形态转化的混合过程的影响,而无明显的反硝化作用发生.
由图9可以看出,研究区地下水与地表水中NO3−的δ15N值和δ18O值均与NO3-N浓度的倒数呈正相关,表明NO3−N属于混合来源. 其中,26个地下水样品δ15N平均值为8.04‰,4个地表水样品δ15N平均值为11.16‰,较高的δ15N值域落在粪便和污水区域,再次说明粪便和污水是该区地下水和地表水中NO3−N的主要污染源,其他来源的N(包括土壤氮和化肥)的比例相对较少,可能有少部分的混合.
水中溶解氧(DO)含量的多少是衡量水体自净能力的重要指标,也是判断是否发生反硝化作用的前提. 有研究发现地下水中发生反硝化作用时DO浓度上限为2 mg·L−1,而反硝化作用引起的δ15N和δ18O之比接近2:1[34-35]. 本次采集的地下水中DO浓度在1.65—9.06 mg·L−1的范围内,平均值为6.34 mg·L−1;地表水中DO浓度在7.66—16.28 mg·L−1之间,平均值为10.38 mg·L−1,说明研究区地下水和地表水环境发生反硝化作用的可能性非常小.
硝化作用是指NH4+在氧化环境下经硝化细菌作用,形成NO3−的过程,属于耗氧反应,氧元素在反应过程中会有明显的变化[36]. 因此,可以利用
{\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} 的值判别是否发生硝化作用[37]. 根据研究区地下水中{\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} 特征值可知,{\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} 分布范围为−7.92‰—22.94‰,平均值1.83‰,大部分落在−10‰—10‰范围内,说明硝化作用是研究区地下水NO3−-N转换的主要过程. -
结合济南泉域地下水和地表水的
{\delta ^{15}}{{\rm{N}}_{{\rm{N}}{{\rm{O}}_3}}} 和{\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} ,使用MixSIAR模型计算得出粪便和污水、土壤氮、降雨和化肥中的NH4+、化肥、大气沉降5个潜在NO3−来源的贡献率,地下水中分别为39.9%、27.4%、22.9%、6.6%、3.3%,地表水中分别为51.5%、19.2%、15.7%、8.9%、4.7%(表4).从MixSIAR模型计算结果可以看出,地下水和地表水的主要NO3−贡献源均为粪便和污水,贡献率分别高达39.9%和51.5%,其次为土壤氮及降雨和化肥中的NH4+. 此外与人类活动有关的化肥输入分别占地下水和地表水NO3−总来源的6.6%和8.9%,贡献率相对较低,大气沉降的贡献率最低,说明粪便和污水中的NO3−是济南泉域NO3−污染的主要来源. 总体而言,人为因素的NO3−来源高于自然因素的NO3−来源,表明研究区具有较高的硝酸盐污染风险,这与已有相关研究[38-39]结果较为一致.
-
(1)研究区地下水和地表水的水化学条件在自然和人为因素影响下,水化学场变化较大. 地下水NO3−污染较为严重;地表水中NO3−浓度较低,不存在NO3−污染. 较高的ρ(Cl-)和EC水平说明局部区域存在点源污染.
(2)研究区地下水和地表水中NO3−主要来源于粪便和污水,其次为土壤氮.NO3−浓度变化主要受到混合过程影响,硝化作用是研究区地下水NO3−-N转换的主要过程,无明显反硝化作用发生.
(3)研究区正处于城市化进程的加速阶段,人类活动的加剧对地下水的影响也越来越明显. 因此,在农村居住区应加强牲畜粪便管理、加强排污管道建设、提高污水处理率、防止污水渗漏;在农业种植区应控制化肥施用量、提高氮肥利用率;在城镇区域应加强居民生活污水处理和排放管理,以防止该区地下水NO3−污染进一步恶化.
济南泉域岩溶水系统硝酸盐空间分布及溯源解析
Spatial distribution and traceability analysis of nitrate in karst water system in Jinan spring basin
-
摘要: 人为活动产生的硝酸盐(NO3−)污染是北方岩溶泉域面临的最普遍的环境问题之一. 以济南泉域为研究对象,于2020年9月采集地下水和地表水样品共30件,综合利用水化学分析方法耦合硝酸盐氮氧同位素(
δ15NNO3 和δ18ONO3 )示踪技术,分析研究区NO3−来源及空间分布. 结果表明,研究区主要阴、阳离子浓度从高到低依次为HCO3−>SO42−>Cl−->NO3−、Ca2+>Na+>Mg2+>K+,水化学类型以HCO3·SO4-Ca为主. 地下水NO3−浓度平均值(37.93 mg·L−1)高于地表水(10.15 mg·L−1). 有7个地下水采样点明显受到NO3−污染,超标率为27%,上游补给区及下游汇集排泄区采样点地下水NO3−浓度较高,而径流区采样点地下水NO3−浓度相对偏低. NO3−浓度主要受到无N形态转化的混合过程的影响,反硝化作用并不显著. δ15N和δ18O值范围分别为1.05‰—14.43‰和−7.92‰—22.94‰,MixSIAR模型计算结果显示,粪便和污水对地下水和地表水NO3−的贡献率最大,分别为39.9%和51.5%,其次为土壤氮及降雨和化肥中的NH4+,大气沉降的贡献率最低,说明粪便和污水是该区NO3−污染的主要来源,也可能有土壤有机氮和化肥的混合.Abstract: Nitrate pollution caused by human activities is one of the most common environmental problems in karst spring areas in north China. A total of 30 groundwater and surface water samples were collected in Jinan Spring Basin in September 2020. The source and spatial distribution of nitrate pollution in the study area were analyzed by using hydrochemical analysis method coupled with nitrate nitrogen and oxygen double isotope tracer technology. The results show that the sequences of major anions and cations in water were HCO3−>SO42−>Cl−>NO3− and Ca2+>Na+>Mg2+>K+, respectively. And the main water chemistry type was HCO3·SO4-Ca. The average NO3− concentration in groundwater (37.93 mg·L−1) was higher than that in surface water (10.15 mg·L−1). Waters of 7 sampling sites were obviously polluted by nitrate, exceeding standard rate was 27%, and the concentration of NO3−-N in groundwater at the sampling points of upstream recharge area and downstream drainage area was higher than from runoff area. The concentration of NO3− was mainly affected by the mixing process, while no obvious denitrification occurred. The values ofδ15NNO3 andδ18ONO3 ranged from 1.05‰ to 14.43‰ and −7.92‰ to 22.94‰, respectively. According to the calculation results of Bayesian isotope mixing model (MixSIAR), manure and sewage was mainly nitrate source of the groundwater and surface water, accounting for 39.3% and 52.3% of the total contribution, respectively. Soil nitrogen and NH4+ in rainfall and fertilizer were the second, and the contribution rate of atmospheric deposition was the lowest. The results indicated that manure and sewage was the main source of nitrate pollution in this area, and there may be a mixture of soil organic nitrogen and chemical fertilizer.-
Key words:
- karst water system /
- hydrochemical characteristics /
- nitrate /
- spatial distribution /
- pollution source.
-
岩溶地下水是泉城济南工农业生产和社会发展不可缺少的重要资源[1]. 上世纪80年代以来,随着城市化进程的不断加快,济南岩溶地下水开采量不断增大,工农业生产、旅游活动、生活污水等对岩溶地下水造成了一定程度的污染,水质逐年变差[2]. 特别是在泉域补给径流区,城郊和农村点源与面源污染交错,使得硝酸盐污染问题日益突出[3]. 饮用水中NO3-N含量过高容易造成儿童患高铁血红蛋白症,成人患胃癌、食管癌等,威胁人类健康[4-5]. 同时,岩溶区具有特殊的“地表—地下”双层结构,其含水层结构的特殊性决定了NO3−污染状况更为复杂[6-7].
近年来,许多学者针对岩溶水系统硝酸盐污染问题开展了大量研究,主要集中在西南亚热带岩溶区[8-10],而对于北方半湿润半干旱岩溶区的相关研究较少. 前人对于济南泉域主要开展了水文地质条件、岩溶含水层结构、水循环特征及水环境等方面的研究[11-13],特别是在岩溶地下水的水化学特征及水环境演化方面,部分学者对济南泉域岩溶地下水中主要元素、微量元素及稳定环境同位素进行分析,研究了济南泉域地下水补给来源、水文地球化学特征等问题[14-15]. 还有学者对济南泉域岩溶地下水水质变化规律及其影响因素进行了分析[16-17]. 但是在泉域尺度上采用水化学和NO3−氮氧双同位素分析岩溶地下水系统NO3−污染特征及污染来源的研究尚不多见.
本文以济南泉域为例,基于水化学和NO3−氮氧双同位素示踪技术,从水文地球化学角度,系统分析泉域岩溶水水化学特征,探究NO3−污染来源、分布及影响因素,并定量计算NO3−的各类贡献源比例,剖析NO3−的生物地球化学过程,以期为济南泉域岩溶水资源开发利用及生态环境保护提供科学依据.
1. 材料与方法(Materials and methods)
1.1 研究区概况
济南位于暖温带大陆性季风气候区,多年平均气温14.3 ℃,多年平均降水量667.1 mm,降水主要集中在6—9月份,降水量在空间上自东南向西北递减. 流经济南的河流主要有黄河、玉符河、北大沙河、小清河. 黄河从济南市北部流过,玉符河和北大沙河自南部山区汇入黄河,小清河自西向东横穿市区,汇集了济南泉水后向东流入渤海[3].
济南泉域是微向北倾斜的单斜构造地质体,是我国典型的北方岩溶发育区,总面积约1500 km2(图1). 古生界寒武系、奥陶系碳酸盐岩地层成单斜状覆盖于变质岩系之上,与地形倾向基本一致,至北部隐伏于山前第四系地层之下[18]. 泉域北部及东、西郊有燕山期侵入的辉长岩体大面积分布;西部玉符河以西沿黄河地带,奥陶系埋藏于石炭、二叠系之下,呈北西—东南向分布. 特定的地质构造条件,是济南泉域含水层空间分布、地下水循环运动以及富水性的主要控制因素[19].
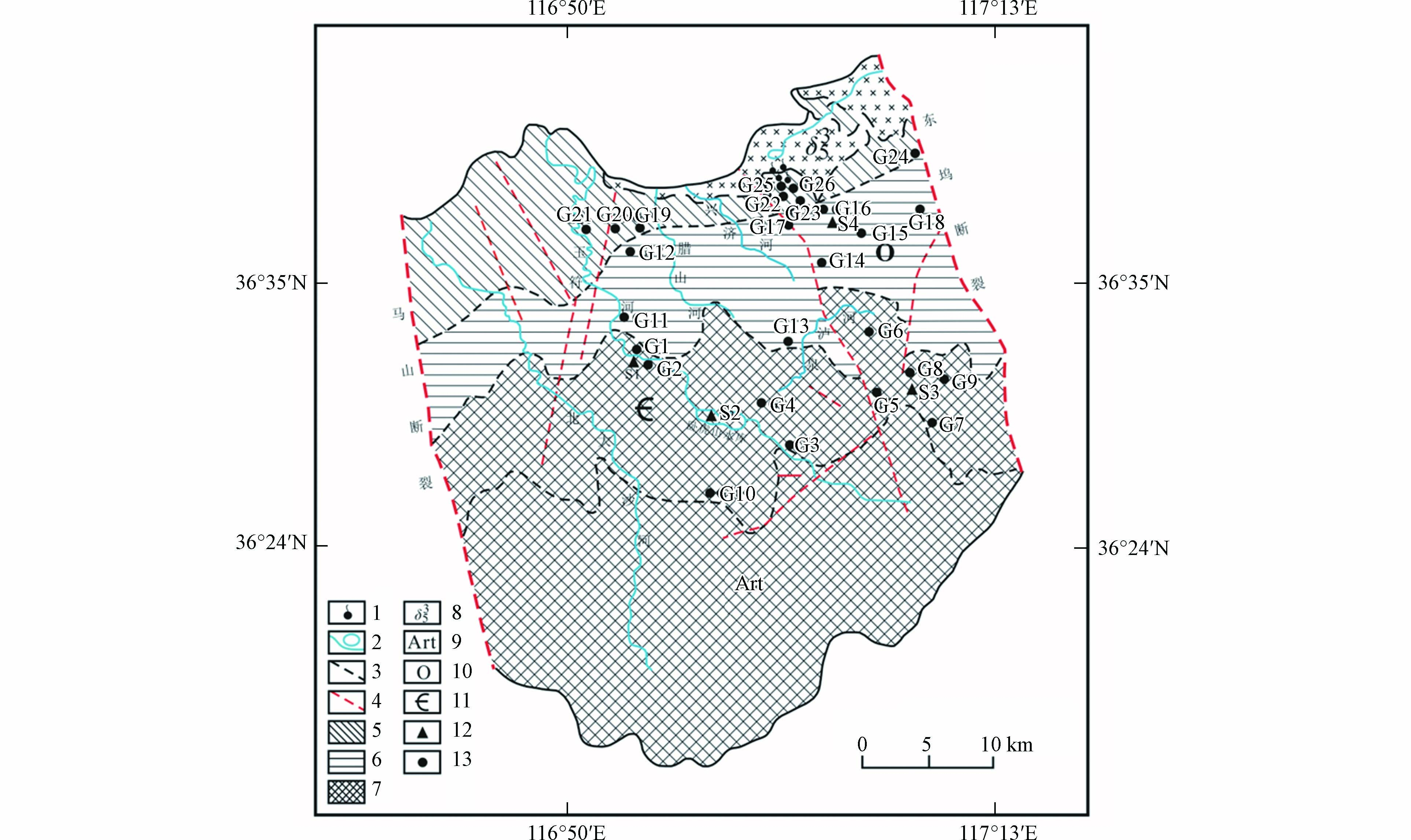 图 1 研究区水文地质简图及采样点分布Figure 1. Hydrogeological sketch of the study area and location of sampling sites1.泉群; 2.河流水库; 3.地层分界线; 4.断层; 5.排泄区; 6.直接补给区; 7.间接补给区; 8.岩浆岩; 9.太古代变质岩; 10.奥陶系碳酸盐岩; 11.寒武系碳酸盐岩; 12.地表水采样点; 13.地下水采样点1.Spring groups; 2.River and reservoir; 3.Stratigraphic boundary; 4.Fault; 5.Discharge area; 6.Direct recharge area; 7.Indirect recharge area; 8.Magmatite; 9.Metamorphite; 10.Ordovician Carbonatite; 11.Cambrian Carbonatite; 12. Surface water sampling sites; 13.Groundwater sampling sites
图 1 研究区水文地质简图及采样点分布Figure 1. Hydrogeological sketch of the study area and location of sampling sites1.泉群; 2.河流水库; 3.地层分界线; 4.断层; 5.排泄区; 6.直接补给区; 7.间接补给区; 8.岩浆岩; 9.太古代变质岩; 10.奥陶系碳酸盐岩; 11.寒武系碳酸盐岩; 12.地表水采样点; 13.地下水采样点1.Spring groups; 2.River and reservoir; 3.Stratigraphic boundary; 4.Fault; 5.Discharge area; 6.Direct recharge area; 7.Indirect recharge area; 8.Magmatite; 9.Metamorphite; 10.Ordovician Carbonatite; 11.Cambrian Carbonatite; 12. Surface water sampling sites; 13.Groundwater sampling sites泉域碳酸盐岩裂隙岩溶含水系统由寒武系中统张夏组、上统凤山组及奥陶系含水层组成,岩性为灰岩、泥质灰岩、白云质灰岩、灰质白云岩和白云岩. 岩溶裂隙发育,连通性好,有利于地下水的补给、径流和汇集排泄[20]. 泉域由南向北依次为间接补给区、直接补给径流区和汇集排泄区[16]. 区内岩溶水接受大气降水补给后由南向北运动,遇辉长岩体受阻,上升出露成泉. 区域地表水与地下水水力联系密切,岩溶含水层极易受地表水的混合污染[21-22].
泉域内人类活动主要为农业生产活动,没有大型的污染工厂. 居民点呈点状分散式分布,家禽养殖普遍且化粪池广泛分布. 南部补给区和径流区耕地和林地面积占区域总面积的80%以上,区内施肥期主要集中在4月和8月,多用复合肥、尿素和碳酸氢铵等化学肥料.
1.2 样品采集与测试
2020年9月共采集研究区地表水和地下水样品30个,其中地表水4个、地下水26个(含泉水3个),采样位置分布见图1. 用液体采样器采集水样,地表水均采自水面以下0.5 m深处的水体样品. 地下水样品为深层地下水,包括民用井和专业监测井,采样井井深全部在150 m以上,水位埋深在60 m以上. 对于封闭式水井,抽取5 min后再取样,保证所取地下水新鲜;对于敞口式水井,用液体采样器采集水面以下0.5 m深处的水体样品. 现场使用便携式多参数水质参数仪(哈希HQ40D,America)测定各采样点水样的温度(T)、电导率(EC)、溶解氧(DO)、pH值,精度分别为0.1 °C、1 µS·cm−1、0.01 mg·L−1、0.01个pH单位. 水化学分析水样用洁净的600 mL聚乙烯瓶采集,并确保瓶内无气泡,用于室内阴、阳离子浓度检测. K+、Na+、Ca2+、Mg2+、采用ICP-OES分析;Cl−、NO3−、SO42−采用阴离子色谱仪测试,精度0.01 mg·L−1;NH4+采用纳氏试剂比色法测定,精度0.01 mg·L−1;NO2−采用α-萘铵比色法测定,精度0.01 mg·L−1;HCO3−、总硬度采用滴定法测定,精度0.01 mg·L−1. 以上水化学测试工作在山东省鲁南地质工程勘察院实验测试中心完成.
同位素分析水样用事先洗净的40 mL聚乙烯瓶采集,现场经0.22 μm混合纤维素滤膜过滤,冷藏后送至实验室进行硝酸盐δ15N、δ18O同位素分析. 其中,硝酸盐δ15N、δ18O同位素检测样品时利用特异性的反硝化细菌将硝态氮转化为N2O,再采用ISOPRIME100-Tracegas痕量气体-同位素质谱联用仪(英国Isoprime公司)完成N2O的N、O同位素测定,检测精度为0.01‰,δ15N测定结果标准偏差<0.4‰,δ18O测定结果标准偏差<0.22‰. 以上同位素测试工作在中国农业科学院农业环境稳定同位素实验室完成.
1.3 数据处理与分析
使用Aq·QA v.1.1软件绘制Piper三线图;采用MATLAB 9.3软件计算地下水水化学成分相关系数;其它图件均采用Origin 8.5软件进行绘制.
本研究基于15N、18O同位素值识别NO3−来源,使用稳定同位素混合模型MixSIAR计算NO3−各种来源的贡献率. MixSIAR模型是用于NO3−来源计算的可靠工具[23-24],通过定义k个来源n个混合物的j个同位素,考虑同位素分馏作用的影响,可表达如下[25]:
Xij=∑Kk=1pkqjk(Sjk+Cjk)∑Kk=1pkqjk+εij \begin{array}{c} S_{jk}~N(μ_{jk,} {\omega }_{jk}^{2} )\\ C_{jk}~N(λ_{jk,} {T}_{jk}^{2} )\\ \varepsilon_{ij}~N(0_{,} {\sigma }_{j}^{2} ) \end{array}{c} 式中, Xij表示第i个样品中第j种同位素值(i=1,2,3,…,N;j=1,2,3,…,J);Sjk是第k种源中第j种同位素的值(k=1,2,3,…,K),μjk. 为平均值,
{\omega }_{jk}^{2} {T}_{jk}^{2} {\sigma }_{j}^{2} 2. 结果与讨论(Results and discussion)
2.1 水化学及硝酸盐污染特征
2.1.1 水化学特征
对研究区内地下水和地表水的水化学参数进行分析,结果表明(表1),研究区地下水和地表水的pH值均大于7,最大值为8.93,呈弱碱性. 除G14采样点TDS异常低外,沿地下水流向岩溶地下水TDS同样呈现上升的趋势,且TDS分布在185.79—892.74 mg·L−1之间.Ca2+、Mg2+、SO42−、NO3−浓度在G4和G5采样点均较高,表明G4和G5采样点附近岩溶地下水环境污染较严重. 阳离子中Ca2+和Mg2+占绝对优势,阴离子中HCO3−和SO42−占绝对优势,其二者之和在地下水和地表水中均占阳离子和阴离子总量的80%以上.
表 1 研究区水化学指标数据Table 1. Hydrochemical indexes in the study area采样点Sampling sites T/℃ pH EC/(µS·cm−1) ρ/(mg·L−1) DO K+ Na+ Ca2+ Mg2+ NH4+−N Cl− HCO3− SO42− NO2−−N NO3−−N 总硬度Total hardness TDS 间接补给区Indirect recharge area G1 19.5 7.79 716 7.05 3.08 57.99 77.06 16.92 0.01 39.54 220.77 133.03 <0.005 2.95 262.14 463.86 G2 24.5 7.62 851 7.74 0.92 30.66 129.50 15.65 0.02 35.25 234.71 174.23 <0.005 6.68 387.85 550.61 G3 17.6 7.28 742 3.99 2.14 18.30 109.26 16.71 <0.01 26.41 283.51 85.23 <0.005 7.36 341.67 451.70 G4 18.1 7.20 1251 6.70 3.41 21.05 203.13 36.36 0.05 27.10 304.43 346.05 0.006 17.96 657.02 892.74 G5 17.1 7.18 1044 6.56 4.35 18.60 146.35 28.55 0.01 30.71 309.07 118.82 <0.005 32.90 483.07 671.46 G6 22.2 7.47 755 5.45 1.91 11.02 124.82 15.17 0.03 9.15 339.28 79.34 0.180 3.19 374.17 433.32 G7 16.0 7.59 605 7.07 0.83 4.80 89.72 26.28 <0.01 9.31 288.16 59.99 <0.005 5.86 332.28 369.61 G8 21.3 7.75 608 8.77 0.92 6.10 95.85 15.46 0.01 10.70 246.33 65.73 <0.005 6.15 303.02 353.71 G9 21.8 7.56 719 7.87 2.96 12.27 111.80 15.40 <0.01 17.11 267.24 84.84 <0.005 7.48 342.62 420.78 G10 17.7 7.48 550 8.41 1.49 8.52 82.00 15.69 0.01 12.44 218.44 61.87 <0.005 7.75 269.40 337.55 直接补给区Direct recharge area G11 17.9 7.59 884 4.75 2.37 80.86 93.06 16.72 <0.01 75.30 220.77 168.19 <0.005 2.24 301.27 569.83 G12 20.5 7.28 795 8.13 0.48 24.70 105.21 17.54 <0.01 40.75 313.72 70.27 <0.005 4.79 334.97 451.47 G13 18.8 7.60 644 9.06 0.78 8.69 109.62 10.18 0.01 21.78 223.09 71.79 0.006 9.50 315.69 386.67 G14 17.7 8.93 476 1.92 8.38 24.30 17.07 10.29 1.45 14.87 76.69 68.23 0.094 0.17 84.99 185.79 G15 17.5 7.27 1176 7.40 2.03 43.97 161.77 34.61 0.15 81.75 290.48 205.02 0.043 15.55 546.50 754.93 G16 18.9 7.92 1143 2.45 28.54 84.67 123.25 18.80 0.02 69.11 151.05 352.93 <0.005 3.02 385.20 782.09 G17 19.2 7.14 934 6.44 0.54 34.81 135.93 20.41 0.02 68.86 288.16 108.07 0.006 9.62 423.51 571.36 G18 20.9 7.95 654 8.83 0.67 7.34 97.12 20.75 0.02 18.79 250.98 67.90 <0.005 8.95 327.99 386.36 排泄区Discharge area G19 19.7 7.01 1107 6.19 0.46 28.12 178.32 22.62 0.01 99.67 313.72 106.83 <0.005 19.34 538.48 694.81 G20 21.7 7.24 809 8.65 0.71 15.67 117.16 21.29 0.02 39.18 290.48 78.38 <0.005 10.76 380.26 483.21 G21 21.4 7.61 606 6.41 1.07 18.61 89.13 15.81 0.02 39.17 209.15 67.95 <0.005 5.67 287.70 375.12 G22 18.8 7.05 873 1.65 4.15 31.06 106.91 35.19 4.01 42.27 453.15 42.32 0.006 1.76 411.90 514.11 G23 21.9 7.92 803 3.72 3.46 49.98 49.18 42.94 0.05 90.18 153.37 131.52 0.006 3.57 299.64 464.22 G24 17.7 7.58 639 6.91 1.05 13.05 92.11 16.74 0.02 36.85 188.23 82.60 <0.005 7.60 298.96 382.54 G25 19.2 7.37 872 6.32 1.50 32.49 125.64 22.48 0.02 62.16 269.57 104.44 <0.005 10.28 406.31 543.44 G26 19.3 7.29 972 6.31 1.29 41.90 136.66 23.29 0.02 75.43 290.48 121.88 <0.005 11.57 437.17 610.62 地表水Surface water S1 24.3 8.56 676 16.28 4.57 27.83 80.16 18.44 0.04 35.58 164.99 135.83 <0.005 1.44 276.10 397.93 S2 26.3 8.36 508 8.74 3.02 17.40 63.77 15.61 0.28 22.29 146.40 95.30 0.088 2.69 223.52 310.53 S3 21.8 8.31 441 8.82 1.90 10.75 59.48 12.36 0.02 13.26 144.08 75.73 0.009 2.17 199.46 260.79 S4 22.3 8.11 476 7.66 1.47 11.12 59.75 17.37 0.10 16.02 137.11 88.27 0.009 2.87 220.76 279.50 | Show Table DownLoad:
CSV
DownLoad:
CSV
从研究区水化学Piper三线图可以看出(图2),采样点均落在菱形的左上区域,碱土金属(Ca、Mg)超过碱金属(K、Na),表明地下水化学性质以碱土金属和弱酸为主. 结合统计数据和Piper三线图可知,研究区地下水和地表水主要的阴、阳离子分别为HCO3−、Ca2+,根据舒卡列夫分类,研究区水化学类型以HCO3·SO4-Ca型为主,占所有样品的60%,主要分布在径流区和排泄区. 其次,HCO3-Ca型占所有样品的17%. 另外,还有个别水样为HCO3-Ca·Mg型、HCO3·SO4-Ca·Mg型、HCO3·SO4·Cl-Ca·Na型、SO4-Ca·Mg及SO4·Cl-Ca,表现出复杂的水化学类型体系.
2.1.2 硝酸盐污染特征
济南泉域地下水NO3−浓度平均值(37.93 mg·L−1)高于地表水(10.15 mg·L−1). 对比我国地下水质量标准,在26个地下水样品中,ρ(NO3−N)≤2.0 mg·L−1的占8%,ρ(NO3−N)为2.0—5.0 mg·L−1的占23%,ρ(NO3−N)为5.0—20 mg·L−1的占65%,其中有4%的样品ρ(NO3−N)>20 mg·L−1,水质低于GB/T 14848—2017的Ⅲ类水标准;若按世界卫生组织标准[ρ(NO3−N)为10 mg·L−1]来衡量,则有27%的样品ρ(NO3−N)超标. 而4个地表水样品的ρ(NO3−N)均未超过3 mg·L−1,远低于地表水环境质量标准中集中式生活饮用水地表水源地ρ(NO3−N)≤10 mg·L−1的限值. 总体来说,研究区水质总体良好,仅少数采样点ρ(NO3−N)超标,存在点源污染的风险.
利用ArcGIS10.3软件反距离权重法进行插值,分析研究区地下水NO3−浓度的空间分布变化,结果如图3所示. 由图3和表2可知,上游补给区及下游汇集排泄区采样点地下水NO3−浓度较高,而径流区采样点地下水NO3−浓度相对偏低.NO3−浓度高值点出现在G4、 G5、G15、G19、 G20、G25、G26,浓度值均高于10 mg·L−1,其中G5采样点NO3−-N浓度高达32.90 mg·L−1. 结合采样点类型及周边环境,对其进行分析,G5采样点位于村内,地下水埋深约为7 m,该井为民井,井口保护设施不完善,生活污水及人畜粪便随降雨入渗后进入含水层,污染较严重. 其他NO3−浓度较高的采样点也均位于农村或城郊居民生活区,周边为耕地和工厂,NO3−污染原因可能是受到工业污染以及化肥施用的影响.
表 2 地下水NO3-N浓度>10 mg·L−1采样点分布Table 2. Distribution of groundwater concentration of NO3-N>10 mg·L−1 sampling sites采样点Sampling sites NO3−N/(mg·L−1) 分区Partition G4 17.96 间接补给区Indirect recharge area G5 32.90 间接补给区Indirect recharge area G15 15.55 直接补给区Direct recharge area G19 19.34 排泄区Discharge area G20 10.76 排泄区Discharge area G25 10.28 排泄区Discharge area G26 11.57 排泄区Discharge area | Show TableDownLoad:
CSV
在空间上,从研究区南部上游至北部下游地下水NO3−浓度呈低—高—低—高的变化特征. 前面所述NO3−浓度超标的采样点,基本形成了3个NO3−高值区,均分布在上游补给区和下游排泄区.
土地利用类型对地下水和地表水中NO3−浓度有着直接影响[26-27]. 研究区内土地利用类型复杂多变,补给区和径流区以农业生产为主,耕地和林地面积占区域总面积的80%以上;汇集排泄区为城区,土地利用类型以建设用地为主. 研究区南部补给区和中部径流区地处泰山余脉,山地地形占地较广,村镇和人口相对较少,土地利用类型以次生林地、灌木丛和耕地为主,沿地下水流路径方向向北直至城郊,村镇和人口逐渐增多,适宜居住和开展农业活动,土地类型多样,以农田、村镇和次生林地为主. 由各采样点的土地利用类型可知,30个采样点中有22个位于农田或村镇附近,占比73.3%. 农田和村镇是人类农业活动密集的区域,其产生的污水中含有高浓度的NO3−,在岩溶地区,由于土壤层较薄,在降水过程中NO3−易通过雨水的携带进入土壤,并经岩溶裂隙和管道入渗影响地下水,同时通过地面冲刷和地表径流汇入河流.
2.2 水化学组分成因及演化
通过研究区地表水和地下水HCO3−/Na+和Ca2+/Na+、Mg2+/Na+和Ca2+/Na+的关系,结合碳酸盐岩、硅酸盐岩和蒸发岩3个端元的范围值[28],来判断水岩作用对水化学成分的影响. 如图4所示,地表水和地下水均分布在靠近碳酸盐岩一侧,且地下水分布相对地表水更偏右上,说明两种水体的离子组成主要受到了碳酸盐岩溶解的影响,其中地下水受到的影响更为显著.
 图 4 地下水与地表水中HCO3−/Na+、Mg2+/Na+与Ca2+/Na+的关系Figure 4. Relationships of HCO3-/Na+ and Mg2+/Na+ with Ca2+/Na+ in groundwater and surface water
图 4 地下水与地表水中HCO3−/Na+、Mg2+/Na+与Ca2+/Na+的关系Figure 4. Relationships of HCO3-/Na+ and Mg2+/Na+ with Ca2+/Na+ in groundwater and surface water采用Gibbs图表征地下水主要离子的化学组成与变化规律,判断地下水是否受蒸发浓缩、岩石风化或者降水的作用,是分析水化学成分演化过程中主控作用的一种重要手段[29],可深入认识研究区地下水化学组分的成因及演化规律[30-31]. 从研究区地下水水化学Gibbs图可以看出,地下水样品大部分分布在模型的中部,TDS处于100—1000 mg·L−1之间,大部分样品的γ(Na+)/[γ(Na+)+γ(Ca2+)]和γ(Cl−)/[γ(Cl−)+γ(HCO3−)]值在0—0.5之间(图5),表明地下水主要阴、阳离子分别以HCO3−、Ca2+为主,且水化学组分特征主要受岩石风化作用的自然过程控制.
相关关系可以分析地下水水化学组分间的相似性,揭示不同离子是否具有同一来源. 采用统计学软件SPSS对研究区地下水水化学成分进行Pearson相关系数计算,从研究区地下水各化学组分之间的相关关系矩阵可以看出(表3),TDS与Ca2+、Mg2+、Cl−、SO42−、NO3−存在明显的线性相关关系,其中,TDS与Ca2+和SO42−的相关关系最为显著,相关关系分别为0.83和0.8,说明同样是这两种化学组分是TDS的重要来源. 地下水中的NO3−一般来源于大气降水及土壤淋滤,但是随着人类活动的增加,动物粪便、生活污水、化肥、农药等排放或使用导致地下水NO3−含量不断升高,从表1的水化学统计数据和表3相关系数矩阵可以看出,NO3−和SO42−均已成为地下水中主要的离子成分之一. Na+与Cl−显著相关,相关系数为0.7,表明研究区内含水层可能存在石盐矿物,受到水岩作用或淋滤作用影响,也可能是外源污染造成.
表 3 地下水水化学成分相关系数矩阵Table 3. Correlation coefficient matrix of groundwater hydrochemical compositionpH EC TDS K+ Na+ Ca2+ Mg2+ HCO3− Cl− NO3− SO42− pH 1 −0.54 −0.54 0.36 0.1 −0.75 −0.35 −0.82 −0.28 −0.52 −0.03 EC 1 0.98 0.29 0.49 0.82 0.57 0.37 0.63 0.55 0.74 TDS 1 0.31 0.49 0.83 0.54 0.33 0.57 0.54 0.80 K+ 1 0.57 −0.06 −0.01 −0.37 0.15 −0.18 0.61 Na+ 1 0.01 0.19 −0.25 0.70 −0.22 0.60 Ca2+ 1 0.32 0.59 0.28 0.68 0.49 Mg2+ 1 0.36 0.44 0.33 0.32 HCO3− 1 −0.01 0.32 −0.17 Cl− 1 0.15 0.36 NO3− 1 0.19 SO42− 1 | Show TableDownLoad:
CSV
2.3 泉域硝酸盐溯源分析
2.3.1 硝酸盐来源定性辨识
正常情况下由于天然地下水中NO3−含量很低,不会对人体产生危害,但是受城市化发展和农业活动影响,大量含氮肥料的使用以及人类、动物粪便和生活污水的排放等使得大量NO3−入渗进入含水层中,特别是在具有地表地下二元结构的岩溶含水层,入渗能力更强[32]. 人类长期暴露于过量NO3−环境中会引起高铁血红蛋白症,增加消化系统癌症的发病率[12]. 通过水水化学组分统计数据可以看出,研究区地下水中NO3−含量普遍较高,一部分样品NO3−超过了生活饮用水卫生标准. 由于地下水中Cl−相对其他离子成分要稳定得多,并且不同来源的NO3−与Cl−之间存在一定的区别,可采用γ(NO3−)/γ(Cl−)值来定性判断NO3−的来源. 从γ(NO3−)/γ(Cl−)比值关系(图6)可以看出,研究区大部分水样落在γ(NO3−)/γ(Cl−)<2以下,而且γ(Cl−)分布范围较大,表现出低γ(NO3−)/γ(Cl−),γ(Cl−)不稳定的特点,因此,判断研究区NO3−主要来自于动物粪便和生活污水.
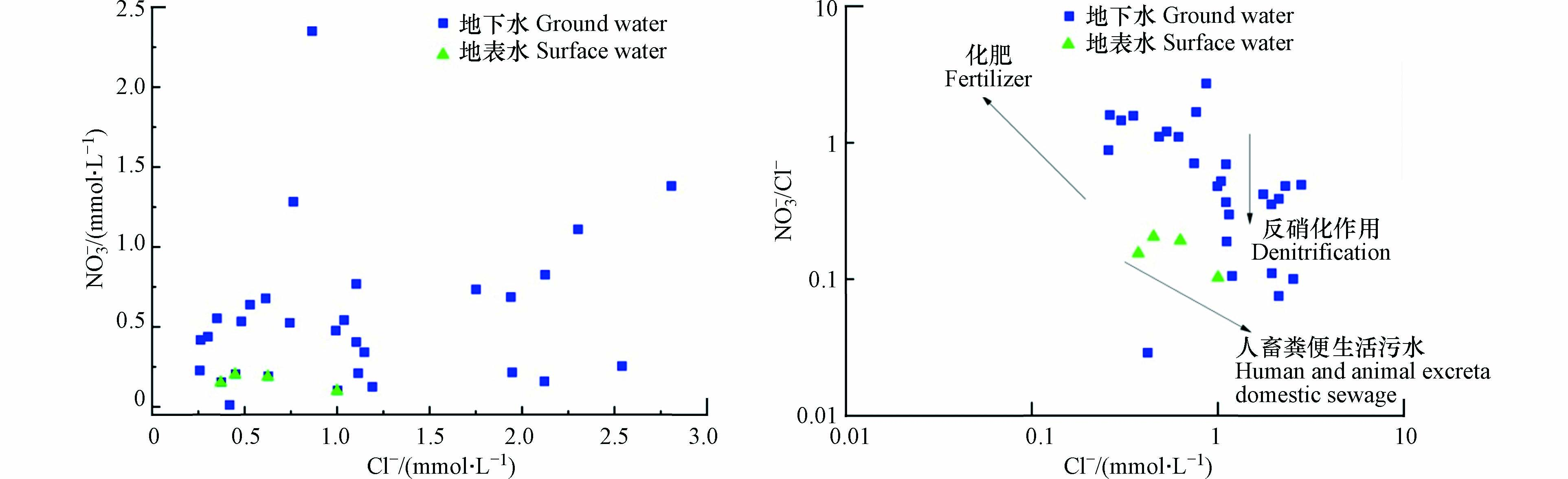 图 6 地下水与地表水中NO3−与Cl−及Cl−−NO3−/Cl−相关关系Figure 6. Relationship of NO3−−Cl− and Cl−−NO3−/Cl− in groundwater and surface water
图 6 地下水与地表水中NO3−与Cl−及Cl−−NO3−/Cl−相关关系Figure 6. Relationship of NO3−−Cl− and Cl−−NO3−/Cl− in groundwater and surface waterCl−结合NO3−/Cl−比值的方法是水化学中常用来识别NO3−来源的手段之一[33]. 低浓度的Cl−和较高的NO3−/Cl−值表明NO3−主要来源于化学肥料,而高浓度的Cl−和较低的NO3−/Cl−值表明NO3−主要来源于人畜粪便和生活污水. 由图6可知,NO3−与Cl−之间无良好的线性关系,且相关性也较弱,说明NO3−和Cl−的来源并不完全相同. 样品点主要分布在低Cl−和高NO3−/Cl−比值区域,分布较分散,表明研究区地下水和地表水中NO3−主要来源于粪便和污水,并且不同区域受化肥与污水和有机肥的影响程度差异较大. 地下水与地表水的样品点分布相似,表明两种水体有相似的NO3−来源.
一般情况下,如果环境中没有大量NH4+积累,矿化和硝化作用产生的NO3−的δ15N值与初始反应物质的δ15N值一致,具有较小的同位素分馏[6]. 因为大量NH4+在环境中积累,反应不完全,一方面会毒害环境中的微生物,中止或减缓生化反应;另一方面由于同位素动力学分馏将使产物NO3−富集14N. 在本研究中,绝大多数地下水样品中的NH4+浓度极低,上述几个反应带来的同位素分馏可以认为是不显著的. 鉴于雨水中具有较低含量的NH4+,且是主要的氮素形态,而NO3−含量也非常低,雨水在稀释地下水的同时不可能成为主要的地下水NO3−来源. 研究区范围较大,补给区和径流区主要人类活动为种植业、养殖业及手工业,汇集排泄区位于城区,大量的农用化肥、工业污水和生活污水造成了较严重的点源污染,是NO3−含量分布差异较大的主要原因. 从研究区地下水和地表水中硝酸盐氮氧同位素的特征值(图7)可知,δ15N值范围在1.05‰—14.43‰,δ18O值范围在−7.92‰—22.94‰,大部分位于粪便和污水的范围内,少部分位于土壤氮及降雨和化肥中NH4+范围内,说明研究区粪便和污水是地下水和地表水NO3−污染的主要来源,其次为土壤氮,这一研究结论与水化学分析结果一致.
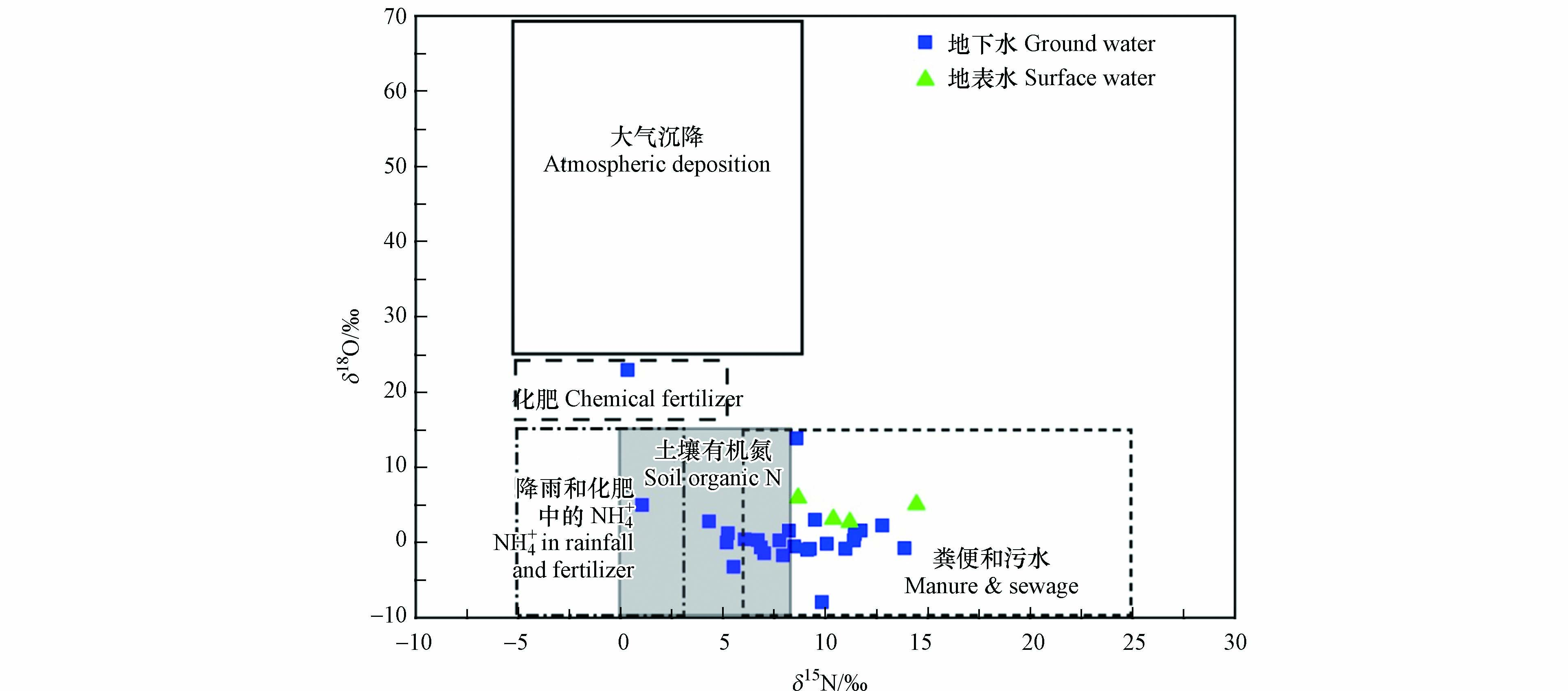 图 7 硝酸盐氮氧同位素特征值分布图Figure 7. Eigenvalue distribution of nitrate nitrogen and oxygen isotope
图 7 硝酸盐氮氧同位素特征值分布图Figure 7. Eigenvalue distribution of nitrate nitrogen and oxygen isotope2.3.2 反硝化作用的判别
Cl−和NO3−的关系以及EC和NO3−的关系能初步判断水中NO3−浓度的变化是源于无N形态转化的混合过程还是由反硝化作用引起[20]. 若NO3−浓度的增加伴随Cl−浓度和EC的增加,则说明水中NO3−浓度可能主要受到无N形态转化的混合过程的影响,反之则可能有反硝化作用发生. 如图6和图8所示,整体上NO3−与Cl−以及NO3−与EC之间均没有显著的线性关系和良好的相关性,但仍然趋向于随NO3−浓度的升高,Cl−和EC均有所升高,表明研究区NO3−主要受到无N形态转化的混合过程的影响,而无明显的反硝化作用发生.
由图9可以看出,研究区地下水与地表水中NO3−的δ15N值和δ18O值均与NO3-N浓度的倒数呈正相关,表明NO3−N属于混合来源. 其中,26个地下水样品δ15N平均值为8.04‰,4个地表水样品δ15N平均值为11.16‰,较高的δ15N值域落在粪便和污水区域,再次说明粪便和污水是该区地下水和地表水中NO3−N的主要污染源,其他来源的N(包括土壤氮和化肥)的比例相对较少,可能有少部分的混合.
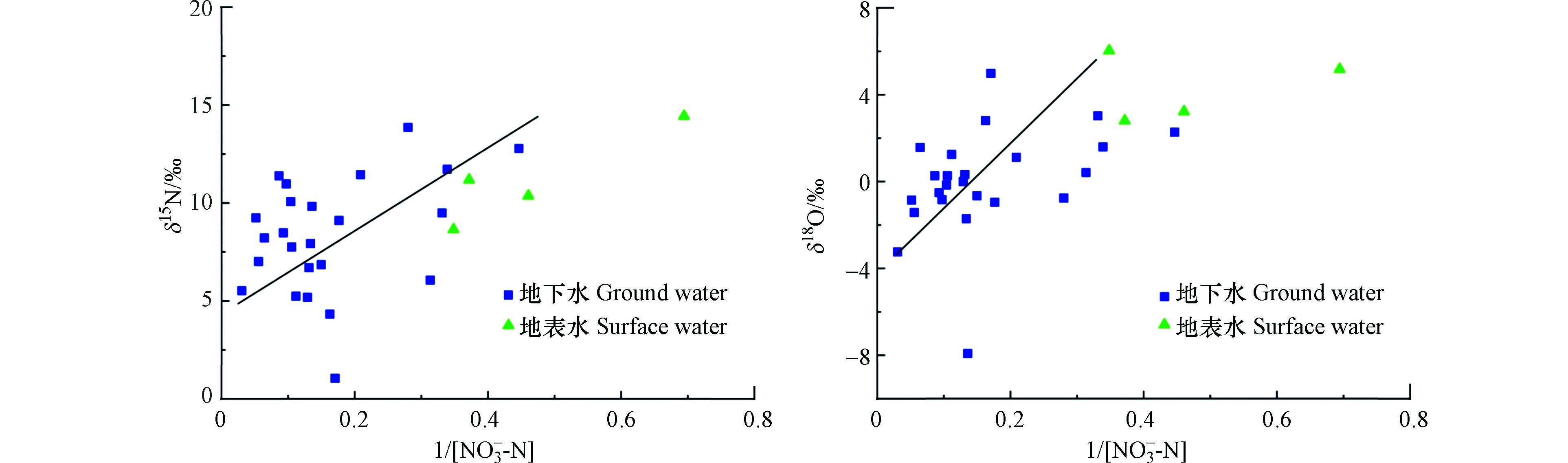 图 9 地下水与地表水中δ15N-1/[NO3−N]和δ18O-1/[NO3−N]关系Figure 9. Relationships of δ15N, δ18O and 1/[NO3−N] in groundwater and surface water
图 9 地下水与地表水中δ15N-1/[NO3−N]和δ18O-1/[NO3−N]关系Figure 9. Relationships of δ15N, δ18O and 1/[NO3−N] in groundwater and surface water水中溶解氧(DO)含量的多少是衡量水体自净能力的重要指标,也是判断是否发生反硝化作用的前提. 有研究发现地下水中发生反硝化作用时DO浓度上限为2 mg·L−1,而反硝化作用引起的δ15N和δ18O之比接近2:1[34-35]. 本次采集的地下水中DO浓度在1.65—9.06 mg·L−1的范围内,平均值为6.34 mg·L−1;地表水中DO浓度在7.66—16.28 mg·L−1之间,平均值为10.38 mg·L−1,说明研究区地下水和地表水环境发生反硝化作用的可能性非常小.
硝化作用是指NH4+在氧化环境下经硝化细菌作用,形成NO3−的过程,属于耗氧反应,氧元素在反应过程中会有明显的变化[36]. 因此,可以利用
{\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} {\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} {\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} 2.3.3 硝酸盐来源定量解析
结合济南泉域地下水和地表水的
{\delta ^{15}}{{\rm{N}}_{{\rm{N}}{{\rm{O}}_3}}} {\delta ^{18}}{{\rm{O}}_{{\rm{N}}{{\rm{O}}_3}}} 表 4 地下水和地表水NO3−不同来源的贡献率(%)Table 4. Contribution rate of potential source to NO3− in groundwater and surface water来源Source 地下水Ground water 地表水Surface water 最小值Min 最大值Max 平均值±标准差Mean±SD 最小值Min 最大值Max 平均值±标准差Mean±SD 粪便和污水Manure & nitrogen 21.3 61.0 39.9±13.1 41.2 62.7 51.5±9.0 土壤有机氮Soil organic nitrogen 15.5 35.3 27.4±6.4 10.8 24.2 19.2±5.9 降雨和化肥中的NH4+NH4+ in rainfall and fertilizer 12.2 34.2 22.9±6.9 13.3 18.2 15.7±2.1 化肥Chemical fertilizer 1.3 25.3 6.6±5.1 7.8 10.5 8.9±1.2 大气沉降Atmospheric deposition 0.5 21.0 3.3±4.6 3.6 5.9 4.7±1.1 | Show TableDownLoad:
CSV
从MixSIAR模型计算结果可以看出,地下水和地表水的主要NO3−贡献源均为粪便和污水,贡献率分别高达39.9%和51.5%,其次为土壤氮及降雨和化肥中的NH4+. 此外与人类活动有关的化肥输入分别占地下水和地表水NO3−总来源的6.6%和8.9%,贡献率相对较低,大气沉降的贡献率最低,说明粪便和污水中的NO3−是济南泉域NO3−污染的主要来源. 总体而言,人为因素的NO3−来源高于自然因素的NO3−来源,表明研究区具有较高的硝酸盐污染风险,这与已有相关研究[38-39]结果较为一致.
3. 结论(Conclusion)
(1)研究区地下水和地表水的水化学条件在自然和人为因素影响下,水化学场变化较大. 地下水NO3−污染较为严重;地表水中NO3−浓度较低,不存在NO3−污染. 较高的ρ(Cl-)和EC水平说明局部区域存在点源污染.
(2)研究区地下水和地表水中NO3−主要来源于粪便和污水,其次为土壤氮.NO3−浓度变化主要受到混合过程影响,硝化作用是研究区地下水NO3−-N转换的主要过程,无明显反硝化作用发生.
(3)研究区正处于城市化进程的加速阶段,人类活动的加剧对地下水的影响也越来越明显. 因此,在农村居住区应加强牲畜粪便管理、加强排污管道建设、提高污水处理率、防止污水渗漏;在农业种植区应控制化肥施用量、提高氮肥利用率;在城镇区域应加强居民生活污水处理和排放管理,以防止该区地下水NO3−污染进一步恶化.
-
图 1 研究区水文地质简图及采样点分布
Figure 1. Hydrogeological sketch of the study area and location of sampling sites
图 4 地下水与地表水中HCO3−/Na+、Mg2+/Na+与Ca2+/Na+的关系
Figure 4. Relationships of HCO3-/Na+ and Mg2+/Na+ with Ca2+/Na+ in groundwater and surface water
图 6 地下水与地表水中NO3−与Cl−及Cl−−NO3−/Cl−相关关系
Figure 6. Relationship of NO3−−Cl− and Cl−−NO3−/Cl− in groundwater and surface water
图 7 硝酸盐氮氧同位素特征值分布图
Figure 7. Eigenvalue distribution of nitrate nitrogen and oxygen isotope
图 9 地下水与地表水中δ15N-1/[NO3−N]和δ18O-1/[NO3−N]关系
Figure 9. Relationships of δ15N, δ18O and 1/[NO3−N] in groundwater and surface water
表 1 研究区水化学指标数据
Table 1. Hydrochemical indexes in the study area
采样点Sampling sites T/℃ pH EC/(µS·cm−1) ρ/(mg·L−1) DO K+ Na+ Ca2+ Mg2+ NH4+−N Cl− HCO3− SO42− NO2−−N NO3−−N 总硬度Total hardness TDS 间接补给区Indirect recharge area G1 19.5 7.79 716 7.05 3.08 57.99 77.06 16.92 0.01 39.54 220.77 133.03 <0.005 2.95 262.14 463.86 G2 24.5 7.62 851 7.74 0.92 30.66 129.50 15.65 0.02 35.25 234.71 174.23 <0.005 6.68 387.85 550.61 G3 17.6 7.28 742 3.99 2.14 18.30 109.26 16.71 <0.01 26.41 283.51 85.23 <0.005 7.36 341.67 451.70 G4 18.1 7.20 1251 6.70 3.41 21.05 203.13 36.36 0.05 27.10 304.43 346.05 0.006 17.96 657.02 892.74 G5 17.1 7.18 1044 6.56 4.35 18.60 146.35 28.55 0.01 30.71 309.07 118.82 <0.005 32.90 483.07 671.46 G6 22.2 7.47 755 5.45 1.91 11.02 124.82 15.17 0.03 9.15 339.28 79.34 0.180 3.19 374.17 433.32 G7 16.0 7.59 605 7.07 0.83 4.80 89.72 26.28 <0.01 9.31 288.16 59.99 <0.005 5.86 332.28 369.61 G8 21.3 7.75 608 8.77 0.92 6.10 95.85 15.46 0.01 10.70 246.33 65.73 <0.005 6.15 303.02 353.71 G9 21.8 7.56 719 7.87 2.96 12.27 111.80 15.40 <0.01 17.11 267.24 84.84 <0.005 7.48 342.62 420.78 G10 17.7 7.48 550 8.41 1.49 8.52 82.00 15.69 0.01 12.44 218.44 61.87 <0.005 7.75 269.40 337.55 直接补给区Direct recharge area G11 17.9 7.59 884 4.75 2.37 80.86 93.06 16.72 <0.01 75.30 220.77 168.19 <0.005 2.24 301.27 569.83 G12 20.5 7.28 795 8.13 0.48 24.70 105.21 17.54 <0.01 40.75 313.72 70.27 <0.005 4.79 334.97 451.47 G13 18.8 7.60 644 9.06 0.78 8.69 109.62 10.18 0.01 21.78 223.09 71.79 0.006 9.50 315.69 386.67 G14 17.7 8.93 476 1.92 8.38 24.30 17.07 10.29 1.45 14.87 76.69 68.23 0.094 0.17 84.99 185.79 G15 17.5 7.27 1176 7.40 2.03 43.97 161.77 34.61 0.15 81.75 290.48 205.02 0.043 15.55 546.50 754.93 G16 18.9 7.92 1143 2.45 28.54 84.67 123.25 18.80 0.02 69.11 151.05 352.93 <0.005 3.02 385.20 782.09 G17 19.2 7.14 934 6.44 0.54 34.81 135.93 20.41 0.02 68.86 288.16 108.07 0.006 9.62 423.51 571.36 G18 20.9 7.95 654 8.83 0.67 7.34 97.12 20.75 0.02 18.79 250.98 67.90 <0.005 8.95 327.99 386.36 排泄区Discharge area G19 19.7 7.01 1107 6.19 0.46 28.12 178.32 22.62 0.01 99.67 313.72 106.83 <0.005 19.34 538.48 694.81 G20 21.7 7.24 809 8.65 0.71 15.67 117.16 21.29 0.02 39.18 290.48 78.38 <0.005 10.76 380.26 483.21 G21 21.4 7.61 606 6.41 1.07 18.61 89.13 15.81 0.02 39.17 209.15 67.95 <0.005 5.67 287.70 375.12 G22 18.8 7.05 873 1.65 4.15 31.06 106.91 35.19 4.01 42.27 453.15 42.32 0.006 1.76 411.90 514.11 G23 21.9 7.92 803 3.72 3.46 49.98 49.18 42.94 0.05 90.18 153.37 131.52 0.006 3.57 299.64 464.22 G24 17.7 7.58 639 6.91 1.05 13.05 92.11 16.74 0.02 36.85 188.23 82.60 <0.005 7.60 298.96 382.54 G25 19.2 7.37 872 6.32 1.50 32.49 125.64 22.48 0.02 62.16 269.57 104.44 <0.005 10.28 406.31 543.44 G26 19.3 7.29 972 6.31 1.29 41.90 136.66 23.29 0.02 75.43 290.48 121.88 <0.005 11.57 437.17 610.62 地表水Surface water S1 24.3 8.56 676 16.28 4.57 27.83 80.16 18.44 0.04 35.58 164.99 135.83 <0.005 1.44 276.10 397.93 S2 26.3 8.36 508 8.74 3.02 17.40 63.77 15.61 0.28 22.29 146.40 95.30 0.088 2.69 223.52 310.53 S3 21.8 8.31 441 8.82 1.90 10.75 59.48 12.36 0.02 13.26 144.08 75.73 0.009 2.17 199.46 260.79 S4 22.3 8.11 476 7.66 1.47 11.12 59.75 17.37 0.10 16.02 137.11 88.27 0.009 2.87 220.76 279.50
下载: 导出CSV
表 2 地下水NO3-N浓度>10 mg·L−1采样点分布
Table 2. Distribution of groundwater concentration of NO3-N>10 mg·L−1 sampling sites
采样点Sampling sites NO3−N/(mg·L−1) 分区Partition G4 17.96 间接补给区Indirect recharge area G5 32.90 间接补给区Indirect recharge area G15 15.55 直接补给区Direct recharge area G19 19.34 排泄区Discharge area G20 10.76 排泄区Discharge area G25 10.28 排泄区Discharge area G26 11.57 排泄区Discharge area
下载: 导出CSV
表 3 地下水水化学成分相关系数矩阵
Table 3. Correlation coefficient matrix of groundwater hydrochemical composition
pH EC TDS K+ Na+ Ca2+ Mg2+ HCO3− Cl− NO3− SO42− pH 1 −0.54 −0.54 0.36 0.1 −0.75 −0.35 −0.82 −0.28 −0.52 −0.03 EC 1 0.98 0.29 0.49 0.82 0.57 0.37 0.63 0.55 0.74 TDS 1 0.31 0.49 0.83 0.54 0.33 0.57 0.54 0.80 K+ 1 0.57 −0.06 −0.01 −0.37 0.15 −0.18 0.61 Na+ 1 0.01 0.19 −0.25 0.70 −0.22 0.60 Ca2+ 1 0.32 0.59 0.28 0.68 0.49 Mg2+ 1 0.36 0.44 0.33 0.32 HCO3− 1 −0.01 0.32 −0.17 Cl− 1 0.15 0.36 NO3− 1 0.19 SO42− 1
下载: 导出CSV
表 4 地下水和地表水NO3−不同来源的贡献率(%)
Table 4. Contribution rate of potential source to NO3− in groundwater and surface water
来源Source 地下水Ground water 地表水Surface water 最小值Min 最大值Max 平均值±标准差Mean±SD 最小值Min 最大值Max 平均值±标准差Mean±SD 粪便和污水Manure & nitrogen 21.3 61.0 39.9±13.1 41.2 62.7 51.5±9.0 土壤有机氮Soil organic nitrogen 15.5 35.3 27.4±6.4 10.8 24.2 19.2±5.9 降雨和化肥中的NH4+NH4+ in rainfall and fertilizer 12.2 34.2 22.9±6.9 13.3 18.2 15.7±2.1 化肥Chemical fertilizer 1.3 25.3 6.6±5.1 7.8 10.5 8.9±1.2 大气沉降Atmospheric deposition 0.5 21.0 3.3±4.6 3.6 5.9 4.7±1.1
下载: 导出CSV
-
[1] KANG F X, JIN M G, QIN P R. Sustainable yield of a Karst aquifer system: A case study of Jinan springs in Northern China [J]. Hydrogeology Journal, 2011, 19(4): 851-863. doi: 10.1007/s10040-011-0725-2 [2] GUO Y, QIN D J, LI L, et al. A complicated Karst spring system: Identified by Karst springs using water level, hydrogeochemical, and isotopic data in Jinan, China [J]. Water, 2019, 11(5): 947. doi: 10.3390/w11050947 [3] 杨丽芝, 刘春华, 祁晓凡. 济南泉水水化学特征变异研究 [J]. 水资源与水工程学报, 2016, 27(1): 59-64. doi: 10.11705/j.issn.1672-643X.2016.01.10 YANG L Z, LIU C H, QI X F. Study on characteristic variation of hydro-chemistry of Jinan spring [J]. Journal of Water Resources and Water Engineering, 2016, 27(1): 59-64(in Chinese). doi: 10.11705/j.issn.1672-643X.2016.01.10
[4] KENDALL C, MCDONNELL J. Isotope tracers in catchment hydrology[M]. Amsterdam: Elsevier, 1998, 517-576. [5] 梁永平, 王维泰, 赵春红, 等. 中国北方岩溶水变化特征及其环境问题 [J]. 中国岩溶, 2013, 32(1): 34-42. doi: 10.3969/j.issn.1001-4810.2013.01.006 LIANG Y P, WANG W T, ZHAO C H, et al. Variations of Karst water and environmental problems in North China [J]. Carsologica Sinica, 2013, 32(1): 34-42(in Chinese). doi: 10.3969/j.issn.1001-4810.2013.01.006
[6] 王开然, 郭芳, 姜光辉, 等. 15N和18O在桂林岩溶水氮污染源示踪中的应用 [J]. 中国环境科学, 2014, 34(9): 2223-2230. WANG K R, GUO F, JIANG G H, et al. Application of 15N and 18O to nitrogen pollution source in Karst water in Eastern Guilin [J]. China Environmental Science, 2014, 34(9): 2223-2230(in Chinese).
[7] LIANG Y P, GAO X B, ZHAO C H, et al. Review: Characterization, evolution, and environmental issues of Karst water systems in Northern China [J]. Hydrogeology Journal, 2018, 26(5): 1371-1385. doi: 10.1007/s10040-018-1792-4 [8] 段世辉, 蒋勇军, 张远瞩, 等. 岩溶槽谷区地下河硝酸盐来源及其环境效应: 以重庆龙凤槽谷地下河系统为例 [J]. 环境科学, 2019, 40(4): 1715-1725. DUAN S H, JIANG Y J, ZHANG Y Z, et al. Sources of nitrate in groundwater and its environmental effects in Karst trough valleys: A case study of an underground river system in the Longfeng trough valley, Chongqing [J]. Environmental Science, 2019, 40(4): 1715-1725(in Chinese).
[9] 赵然, 韩志伟, 申春华, 等. 典型岩溶地下河流域水体中硝酸盐源解析 [J]. 环境科学, 2020, 41(6): 2664-2670. ZHAO R, HAN Z W, SHEN C H, et al. Identifying nitrate sources in a typical Karst underground river basin [J]. Environmental Science, 2020, 41(6): 2664-2670(in Chinese).
[10] 李学先, 吴攀, 查学芳, 等. 基于水化学及稳定同位素的岩溶山区城镇水体硝酸盐来源示踪 [J]. 环境科学学报, 2021, 41(4): 1428-1439. LI X X, WU P, ZHA X F, et al. Tracing nitrate sources in urban waters of Karst mountainous area using hydrochemistry and stable isotope [J]. Acta Scientiae Circumstantiae, 2021, 41(4): 1428-1439(in Chinese).
[11] ZHANG Z X, WANG W P, QU S S, et al. A new perspective to explore the hydraulic connectivity of Karst aquifer system in Jinan spring catchment, China [J]. Water, 2018, 10(10): 1368. doi: 10.3390/w10101368 [12] GAO S, LI C S, JIA C, et al. Health risk assessment of groundwater nitrate contamination: A case study of a typical Karst hydrogeological unit in East China [J]. Environmental Science and Pollution Research International, 2020, 27(9): 9274-9287. doi: 10.1007/s11356-019-07075-w [13] 李严, 王家乐, 靳孟贵, 等. 运用水文时间序列分析识别济南泉域岩溶发育特征 [J]. 地球科学, 2021, 46(7): 2583-2593. LI Y, WANG J L, JIN M G, et al. Hydrodynamic characteristics of Jinan Karst spring system identified by hydrologic time-series data [J]. Earth Science, 2021, 46(7): 2583-2593(in Chinese).
[14] 高宗军, 徐军祥, 王世臣, 等. 济南岩溶水微量元素分布特征及其水文地质意义 [J]. 地学前缘, 2014, 21(4): 135-146. GAO Z J, XU J X, WANG S C, et al. The distribution characteristics and hydrogeological significance of trace elements in Karst water, Jinan, China [J]. Earth Science Frontiers, 2014, 21(4): 135-146(in Chinese).
[15] ZHOU J, XING L T, ZHANG F J, et al. Chemical characteristics research on Karst water in Jinan spring area [J]. Advanced Materials Research, 2015, 1092/1093: 593-596. doi: 10.4028/www.scientific.net/AMR.1092-1093.593 [16] 孙斌, 邢立亭, 李常锁. 趵突泉泉域岩溶水典型污染组分变化特征及污染途径 [J]. 中国岩溶, 2018, 37(6): 810-818. SUN B, XING L T, LI C S. Variation of typical pollution components and pollution way of Karst water in Baotu Spring region [J]. Carsologica Sinica, 2018, 37(6): 810-818(in Chinese).
[17] 管清花, 李福林, 王爱芹, 等. 济南市岩溶泉域地下水化学特征与水环境演化 [J]. 中国岩溶, 2019, 38(5): 653-662. doi: 10.11932/karst20190501 GUAN Q H, LI F L, WANG A Q, et al. Hydrochemistry characteristics and evolution of Karst spring groundwater system in Jinan [J]. Carsologica Sinica, 2019, 38(5): 653-662(in Chinese). doi: 10.11932/karst20190501
[18] 邢立亭, 周娟, 宋广增, 等. 济南四大泉群泉水补给来源混合比探讨 [J]. 地学前缘, 2018, 25(3): 260-272. XING L T, ZHOU J, SONG G Z, et al. Mixing ratios of recharging water sources for the four largest spring groups in Jinan [J]. Earth Science Frontiers, 2018, 25(3): 260-272(in Chinese).
[19] 殷秀兰, 王庆兵, 凤蔚. 济南岩溶泉域泉群区水化学与环境同位素研究 [J]. 地质学报, 2017, 91(7): 1651-1660. doi: 10.3969/j.issn.0001-5717.2017.07.016 YIN X L, WANG Q B, FENG W. Hydro-chemical and isotopic study of the Karst spring catchment in Jinan [J]. Acta Geologica Sinica, 2017, 91(7): 1651-1660(in Chinese). doi: 10.3969/j.issn.0001-5717.2017.07.016
[20] 高帅, 李常锁, 贾超, 等. 济南趵突泉泉域岩溶水化学特征时空差异性研究[J]. 地质学报, 2019, 93(S1): 61-70. GAO S, LI C S, JIA C, et al. Spatiotemporal difference study of Karst hydrochemical characteristics in the Baotu Spring area of Jinan[J]. Acta Geologica Sinica, 2019, 93(Sup 1): 61-70(in Chinese).
[21] 徐慧珍, 李文鹏, 殷秀兰, 等. 济南泉域浅层地下水水化学同位素研究 [J]. 水文地质工程地质, 2008, 35(3): 65-69,98. doi: 10.3969/j.issn.1000-3665.2008.03.017 XU H Z, LI W P, YIN X L, et al. Hydrochemistry and isotopes of shallow groundwater in the Jinan spring catchment [J]. Hydrogeology & Engineering Geology, 2008, 35(3): 65-69,98(in Chinese). doi: 10.3969/j.issn.1000-3665.2008.03.017
[22] 王珺瑜, 王家乐, 靳孟贵. 济南泉域岩溶水水化学特征及其成因 [J]. 地球科学, 2017, 42(5): 821-831. WANG J Y, WANG J L, JIN M G. Hydrochemical characteristics and formation causes of Karst water in Jinan spring catchment [J]. Earth Science, 2017, 42(5): 821-831(in Chinese).
[23] MATIATOS I. Nitrate source identification in groundwater of multiple land-use areas by combining isotopes and multivariate statistical analysis: A case study of Asopos Basin (Central Greece) [J]. Science of the Total Environment, 2016, 541: 802-814. doi: 10.1016/j.scitotenv.2015.09.134 [24] YU L, ZHENG T Y, ZHENG X L, et al. Nitrate source apportionment in groundwater using Bayesian isotope mixing model based on nitrogen isotope fractionation [J]. Science of the Total Environment, 2020, 718: 137242. doi: 10.1016/j.scitotenv.2020.137242 [25] PARNELL A C, INGER R, BEARHOP S, et al. Source partitioning using stable isotopes: Coping with too much variation [J]. PLoS One, 2010, 5(3): e9672. doi: 10.1371/journal.pone.0009672 [26] 金赞芳, 胡晶, 吴爱静, 等. 基于多同位素的不同土地利用区域水体硝酸盐源解析 [J]. 环境科学, 2021, 42(4): 1696-1705. JIN Z F, HU J, WU A J, et al. Identify the nitrate sources in different land use areas based on multiple isotopes [J]. Environmental Science, 2021, 42(4): 1696-1705(in Chinese).
[27] 王雨旸, 杨平恒. 张洁茹. 重庆市老龙洞地下河流域硝酸盐来源和生物地球化学过程的识别[J]. 环境科学, 2022, 43(10): 4470-4479. WANG Y Y, YANG P H, ZHANG J R. Sources and biogeochemical processes of nitrate in the Laolongdong karst underground river basin, Chongqing [J]. Environmental Science, 2022, 43(10): 4470-4479 (in Chinese).
[28] XU Z F, LIU C Q. Water geochemistry of the Xijiang Basin rivers, South China: Chemical weathering and CO2 consumption [J]. Applied Geochemistry, 2010, 25: 1603-1614. doi: 10.1016/j.apgeochem.2010.08.012 [29] MARANDI A, SHAND P. Groundwater chemistry and the Gibbs diagram [J]. Applied Geochemistry, 2018, 97: 209-212. doi: 10.1016/j.apgeochem.2018.07.009 [30] 王瑞, 李潇瀚. 百泉泉域岩溶地下水水化学演化特征及成因 [J]. 中国岩溶, 2021, 40(3): 398-408. WANG R, LI X H. Hydrochemical characteristics and genesis of Karst groundwater in the Baiquan spring catchment [J]. Carsologica Sinica, 2021, 40(3): 398-408(in Chinese).
[31] 任孝宗, 杨小平. 鄂尔多斯沙区天然水体水化学组成及其成因 [J]. 地理学报, 2021, 76(9): 2224-2239. doi: 10.11821/dlxb202109013 REN X Z, YANG X P. Hydrochemical compositions of natural waters in Ordos Deserts and their influencing factors [J]. Acta Geographica Sinica, 2021, 76(9): 2224-2239(in Chinese). doi: 10.11821/dlxb202109013
[32] ZHANG P, YUE F J, WANG X D, et al. Antecedent rainfall and land use controlling the fate of nitrogen in Karst urban rivers, elucidated by an isotopic approach [J]. Journal of Hydrology, 2021, 592: 125803. doi: 10.1016/j.jhydrol.2020.125803 [33] WANG K R, CHEN H W, LI F L, et al. Spatial distribution characteristics of nitrogen pollution in a typical Karst groundwater system [J]. Arabian Journal of Geosciences, 2020, 13(10): 1-9. [34] 盛婷, 杨平恒, 谢国文, 等. 基于δ15N和δ18O的农业区地下河硝酸盐污染来源 [J]. 环境科学, 2018, 39(10): 4547-4555. SHENG T, YANG P H, XIE G W, et al. Nitrate-nitrogen pollution sources of an underground river in Karst agricultural area using 15N and 18O isotope technique [J]. Environmental Science, 2018, 39(10): 4547-4555(in Chinese).
[35] WELLS N S, CLOUGH T J, JOHNSON-BEEBOUT S E, et al. Effects of denitrification and transport on the isotopic composition of nitrate (δ18O, δ15N) in freshwater systems [J]. Science of the Total Environment, 2019, 651: 2228-2234. doi: 10.1016/j.scitotenv.2018.10.065 [36] YANG P H, WANG Y Y, WU X Y, et al. Nitrate sources and biogeochemical processes in Karst underground rivers impacted by different anthropogenic input characteristics [J]. Environmental Pollution, 2020, 265: 114835. doi: 10.1016/j.envpol.2020.114835 [37] 路路, 戴尔阜, 程千钉, 等. 基于水环境化学及稳定同位素联合示踪的土地利用类型对地下水体氮素归趋影响 [J]. 地理学报, 2019, 74(9): 1878-1889. doi: 10.11821/dlxb201909013 LU L, DAI E F, CHENG Q D, et al. The sources and fate of nitrogen in groundwater under different land use types: Stable isotope combined with a hydrochemical approach [J]. Acta Geographica Sinica, 2019, 74(9): 1878-1889(in Chinese). doi: 10.11821/dlxb201909013
[38] MING X X, GROVES C, WU X Y, et al. Nitrate migration and transformations in groundwater quantified by dual nitrate isotopes and hydrochemistry in a Karst World Heritage site [J]. Science of the Total Environment, 2020, 735: 138907. doi: 10.1016/j.scitotenv.2020.138907 [39] GIBRILLA A, FIANKO J R, GANYAGLO S, et al. Nitrate contamination and source apportionment in surface and groundwater in Ghana using dual isotopes (15N and 18O-NO3) and a Bayesian isotope mixing model [J]. Journal of Contaminant Hydrology, 2020, 233: 103658. doi: 10.1016/j.jconhyd.2020.103658 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1626
- HTML全文浏览数: 1626
- PDF下载数: 98
- 施引文献: 0








