


-
能源和环境问题是人类社会可持续发展所面临的最重要制约因素。我国碳交易市场于2017年正式启动,意味着能源发展必须走低碳清洁化道路。我国正处在统一碳交融交易市场关键时期,碳减排量与碳减排水平需要一个衡量标准及相应的函数关系,但目前缺乏相应的碳交易手段和政策制定依据。碳交易市场的启动会推动、倒逼发电企业进行发电结构调整,化解高碳落后产能,煤电低碳清洁转型升级、灵活性改造势在必行。把煤电和生物质能“撮合”在一起进行发电试点,将推动中国能源结构转型和煤电绿色发展,为煤电低碳清洁发展带来了新途径,燃煤耦合生物质发电的零碳排放优势将把握住电力产业低碳转型发展机遇[1-2]。
生物质能是一种清洁可再生能源,也是6种可再生能源中唯一可以转化为气、液、固体的零碳能源。近年来生物质发电逐渐兴起,利用生物质替代矿物燃料进行发电可以有效减少CO2和SO2排放。生物质与煤混合燃烧发电已被纳入国家产业规划,缺乏科学、公正的生物质应用量在线检测技术依旧是燃煤耦合生物质发电所面临的最大问题。目前较为先进的后端检测方法,主要包括SO2浓度检测法和14C含量检测法[3-6]。由于生物质含有一些碱金属,会与S发生反应,影响SO2浓度监测结果。生物质与大气中的14C活度相等,煤种14C活度几乎为零,因此14C含量检测法的准确性、可靠性仍然处于探索性阶段[7]。14C技术分析设备多为价格昂贵的核磁、加速器质谱(AMS)等精密仪器[8-14],影响了生物质应用量后端检测技术的发展。
目前,生物质应用量后端检测技术亟待解决实时检测、设备昂贵等问题。研究发现中红外激光同位素检测技术可实现快速实时检测稳定碳同位素比值δ13C (13CO2/12CO2)[15-23]。Castrillo等采用近红外激光设备测试CO2气体中的13C/12C同位素比值(δ),设备系统误差为0.5‰ [24]。传统光谱法相对质谱法测量灵敏度较低,但中红外量子级联激光器和腔衰荡光谱法(QCL-CRDS)相结合可以最大程度地提高气体光学检测的灵敏度,是目前最前沿的超低气体浓度检测技术,其具有体积小、操作简单、价格便宜、对环境敏感性低等优势,大大提高了现场布控和在线分析的能力,在生态、油气、环境监测等领域发挥着重要的角色。生物质与煤燃烧释放的CO2中均含有稳定碳同位素13C,生物质燃烧释放CO2气体中所含稳定碳同位素重,而煤燃烧过程中释放的碳同位素轻,可依据各自δ13C值的差异进行煤和生物质掺混比的判定[25-26]。现有碳同位素中红外激光检测设备检测误差仅为±0.025‰,可满足生物质应用量的精确评测需求,同时该检测仪造价较低,可实现对质谱等昂贵设备的替代,但与之相对应的评测方法国内外尚属空白[18, 27-28]。
本文研究建立基于稳定碳同位素中红外激光检测仪监测生物质掺烧比的实时在线检测分析方法十分重要,可为政府补贴政策的实施提供技术支撑,同时该分析方法可用于追踪“碳溯源”,推动碳排放监测和碳交易市场的快速发展,促进清洁能源的健康有序发展和碳交易市场建立建全定价机制。
-
实验选取山西煤、贵州煤和内蒙古煤3种煤样,以及玉米秸秆、棉花秸秆、木屑和稻壳4种生物质进行混燃实验,实验中实际测试煤样和生物质的工业分析、元素分析和热值测定结果见表1。实验煤样品的制备主要经过破碎、研磨和筛分3个过程。首先将样品破碎成小颗粒(粒径小于1 cm);然后使用粉碎机对其进行粉碎;最后用标准试验筛粉状样品进行筛分,取粒径60—80目的样品进行混燃实验。生物质粉末样品购买于惠丰秸秆农产品加工企业。
-
自主搭建小型混燃实验系统,该系统主要包括:刚瓶、流量控制计、燃烧炉、净化装置、碳同位素中红外激光检测设备,如图1。使用气体总流量为200 mL·min−1,气体组分包括33%O2和67%空气,富氧条件可保证煤和生物质的充分燃烧。实验过程主要包括:将装有混合燃料的石英舟(78 mm(长)×11 mm(宽)×15 mm(高))放置在燃烧炉中不锈钢管的恒温段,O2和空气经质量流量计后进行充分混合后通入燃烧炉,燃烧温度850 ℃;充分燃烧的混合烟气经过滤器后进入碳同位素中红外激光检测设备进行碳同位素比值δ13C(13CO2/12CO2)的检测。
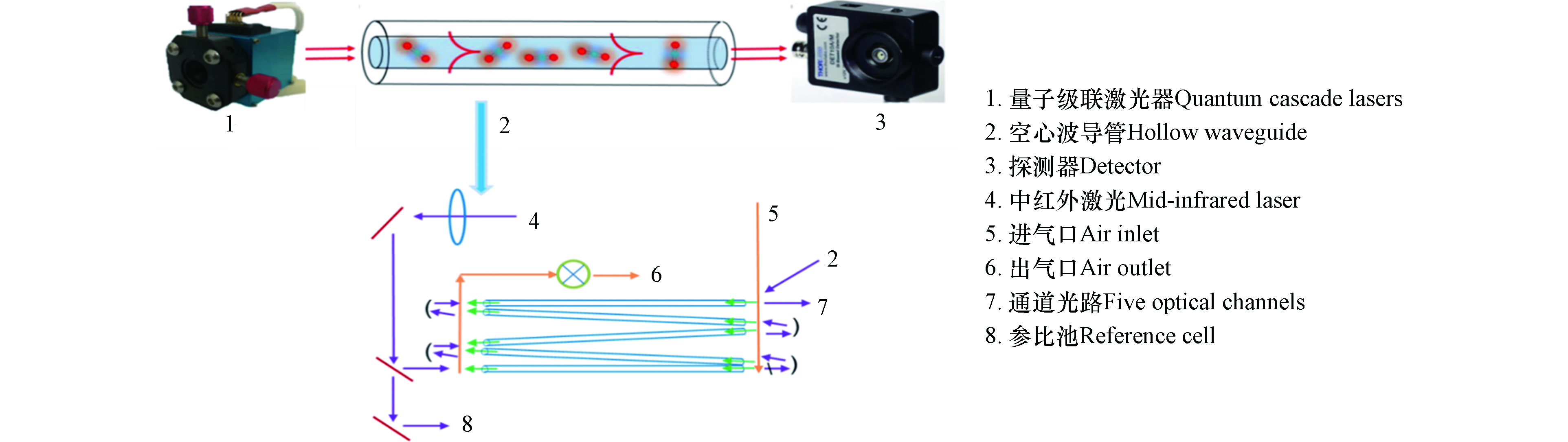
碳同位素中红外激光检测设备(团队自主研发)基于不断成熟发展量子级联激光器设计开发一套多通道光学检测核心器件,其结构如图2所示。该检测设备主要包括量子级联激光器、空心波导管和探测器,其中空心波导中包含一套五通道耦合光路系统。实验设计主要包括:一种煤样和一种或多种生物质进行混燃,一种生物质与一种或多种煤样进行混燃。生物质燃料的掺烧比主要为5%、10%、20%和30%;为了进一步建立准确的分析方法,将玉米与山西煤的掺烧比设计范围为0—100%,其中在0—30%之内增加2.5%、7.5%、15%和25%的掺烧比例。
本研究中为了验证稳定碳同位素激光检测设备的技术优势及建立分析方法的准确性,同时采用加速质谱仪(AMS,3MV TN-4130,High Voltage Engineering Europa B.V.,HVEE)对混燃烟气的14C浓度进行了离线测试,燃烧过程如上所述,所得烟气经集气袋收集后在AMS设备上进行14C浓度检测,得到14C/12C的值。另外,采用SO2浓度检测方对混燃烟气中SO2浓度进行了离线对比分析。
-
稳定碳同位素中红外激光检测设备所测试同位素比值δ13C(13C/12C)按照如下公式定义:
式(1)中,Rp是样品中碳元素重轻同位素丰度之比(13Cp/12Cp),Rs是国际通用标准物的重轻同位素丰度之比(13Cs/12Cs)。不同煤样与不同生物质的δ13C值之间具有显著差异,依据所述δ13C差异性,通过如下线性回归方程计算混燃物质中生物质的掺烧比例:
式(2)中,y为碳同位素激光检测设备显示的δ13C值,x为碳量基掺烧比,a为通过配比不同煤种与不同生物质混燃所得线性回归方程的斜率,b为煤样的δ13C值;式(3)中,r为质量基掺烧比,CC为煤样碳含量,CB为生物质碳含量。同时,可用发热量进行矫正后得到线性回归方程(4)、(5),其中x1为热量基掺烧比,a1为通过配比不同煤种与不同生物质混燃所得线性回归方程的斜率,式(5)中,r1为质量基掺烧比,QC为煤样的发热量,QB为生物质发热量。经碳含量和发热量矫正后可排除水分、灰分、挥发分等干扰因素的影响,有利于更准确的判断生物质掺烧比。
另外,生物质混燃比还可通过14C/12C的值R进行判定:
式(6)中,yR为AMS检测的14C/12C的值,aR为通过配比不同煤种与不同生物质混燃所得线性回归方程的斜率,xR为碳量基掺烧比,b为煤样的14C/12C值;式(7)中,r2为质量基掺烧比。经发热量进行矫正后得到线性回归方程(8)、(9),式(8)中xR1为热量基掺烧比,式(9)中,r3为质量基掺烧比。
-
判定生物质掺烧比时易受到含水量、灰分、挥发分的影响,导致线性方程拟合优度R2较差,需要对其进行碳含量和发热量矫正,以获取更准确的线性拟合方程,使其拟合优度R2均达到0.990以上。纯煤样和生物质样品充分燃烧后经碳同位素红外激光检测设备所得碳同位素比值(δ13C)见表2。
表2可知,纯煤样(山西、贵州、内蒙古)的δ13C值具有差异性,各生物质δ13C值也呈现一定的区别,尤其是玉米秸秆的δ13C值为−11.91,说明该生物质的碳同位素较重,则玉米秸秆中所含13C比重偏多,而棉花秸秆、木屑和稻壳之间的δ13C值属于相似范围−26—−27,说明三者所含13C的比重较近似。本文依据各燃料之间δ13C值的差异性建立函数关系进行生物质掺烧比的判断。
-
煤和生物质充分混燃烟气经碳同位素中红外激光检测设备测定其稳定碳同位素比值δ13C,未对设计生物质掺烧比进行矫正时,所得拟合线性方程见图3—5。
图3a为山西煤与不同比例(0—30%)的玉米秸秆混燃所得线性回归方程,其拟合优度R2=0.996,说明此方程的线性关系较好,当玉米秸秆的掺烧比为20%时,误差分析为6.1%,说明用此公式判断玉米秸秆的掺烧比存在很大的不准确性。若将玉米秸秆的掺烧比设计为0—100%(图3b),则所得方程的线性关系较差,拟合优度R2=0.986,用此方程判断玉米秸秆的掺烧比同样存在很大的误差。图3a和3b所得线性回归方程的斜率存在较大差异,说明未经矫正的线性方程由于受到含水量、灰分等因素的干扰,不适合用于判断生物质的掺烧比。图3c为山西煤与不同比例的木屑混燃的线性方程,可知方程的拟合优度R2=0.998,线性关系较好,但由于受含水量、灰分等的影响,对生物质掺烧比(20%)进行判断时仍存在较大误差;同样图3d和图3e分别为山西煤和不同比例的棉花秸秆、稻壳进行混燃所得线性方程,若使用目前所得函数关系判断生物质掺烧比亦存在较大误差。
图3f为山西煤同时与两种生物质混燃的线性方程,其生物质的总掺烧比为两种掺烧量相同生物质的加和。由拟合线性关系的斜率变化可知,同时掺混两种生物质时,所得δ13C值与生物质掺混比关系较敏感。由山西煤、玉米、木屑三者混燃所得拟合线性方程可知,生物质中仅玉米秸秆碳同位比值δ13C(δ13C=-11.91)较大,当玉米秸秆与其它生物质一起混燃时所得线性方程的斜率(0.02左右)与δ13C值的关系过于敏感,生物质掺烧比的变化易引起δ13C值较大波动,用所得线性方程判断生物质混燃比将带来较大的误差。图3(g、h)为山西煤和木屑、棉花秸秆、稻壳等3种生物质混燃的线性方程,由于添加多种生物质,且受到含水量、灰分等的影响,线性拟合关系并不理想,因此不能准确判断生物质的混燃比。图3h为山西煤和玉米秸秆、木屑、棉花秸秆、稻壳等4种生物质混燃的线性方程,同样由于玉米秸秆的δ13C值与其他生物质差别较大,混燃玉米秸秆后所得线性关系斜率绝对值较低,不利于进行生物质混燃比的判断,亦说明此检测分析方法用于玉米秸秆与其他生物质同时进行混燃时不利于生物质掺烧比的判断。
由于我国玉米秸秆产量较大,因此使用该检测分析方法针对玉米秸秆与多种煤样混燃进行了研究。图4为玉米秸秆与内蒙古煤、贵州煤、山西煤混燃线性方程。由图4结果可知,玉米秸秆与不同煤样混燃的线性关系均较好。图4a为玉米秸秆和内蒙古煤的混燃线性方程,其拟合优度R2=0.996,线性关系较好,但由于受含水量、灰分等的影响,当玉米秸秆的混燃比为20%,误差分析在2.9%。图4b为玉米秸秆和贵州煤的线性拟合方程,拟合优度R2=0.998,线性关系较好,由于含水量和灰分等的影响,判断玉米秸秆混燃比将出现误差(混燃比20%时,误差分析在10%)。图4(c、d)分别为玉米秸秆和山西、贵州煤以及玉米秸秆和内蒙古煤和贵州煤的混燃线性方程,由于未排除干扰因素(含水量、灰分等)的影响,用以上两个线性方程判断玉米秸秆的掺烧比均存在不准确性。
由以上分析可知,无论是单一煤样和单一或多种生物质混燃,或者单一生物质和单一或多种煤样混燃,由于受到含水量、灰分等其他干扰因素的影响,用本文的碳同位素激光检测-分析方法判定生物质的掺烧比均存在一定的误差,需要排除以上因素的干扰,也从侧面证明此检测-分析方法可用于判定掺烧生物是否含水量过多、或掺进其他非生物质的物质,为准确判断生物质的掺烧比提供了多重保障。为了准确获得生物质掺烧比,对多种煤样和生物质混燃数据进行了碳含量和发热量矫正,用燃料最本征的数值来判定生物质的掺烧比。
-
燃料的碳(C)含量与燃料充分燃烧后所释放的CO2量直接相关,也最能真实的反应出碳同位素比值(δ13C=13CO2/12CO2)变化情况,以下所得线性方程均采用燃料的含碳量(详细数据见表1)进行矫正。图5均为经碳含量矫正后所得拟合线性方程,由图5可知,经碳含量矫正后的数据排除了水分、灰分等多种因素的干扰后,拟合优度R2>0.995,线性关系得到进一步改善。图5(a、b)为山西煤和不同掺混比玉米秸秆混燃所得线性方程,两条线性方程的斜率相似(0.096≈0.1),说明碳基线性方程的斜率与玉米秸秆的掺混比关系不大,为山西煤和玉米秸秆混燃的真实线性函数关系,当玉米秸秆的掺烧比为20%时,误差分析在2.0%,准确性得到了很大的提高。同样图5(c、d)分别为山西煤与稻壳、棉花秸秆以及木屑混燃的碳基线性拟合方程,由于3种生物质的δ13C值相近,因此与山西煤混燃时所得线性方程的斜率均相似(−0.077—−0.079),说明碳基线性关系均为煤与生物质混燃的真实线性函数关系。当生物质掺烧比为20%时,误差分析可控制在2.0%以内。
图6为玉米秸秆与一种或两种煤样混燃的碳基线性拟合方程,其中图6a为玉米秸秆和内蒙古煤混燃的线性拟合方程,拟合优度R2=0.999,当玉米秸秆掺烧比为20%时,误差分析为0.5%,说明线性关系较好,此公式用于判断玉米秸秆的掺烧比较准确。图6b为玉米秸秆与贵州煤混燃的碳基线性方程,其拟合优度R2=0.998,当玉米秸秆掺烧比为20%时,误差分析为0.5%,说明用此拟合线性方程判断玉米秸秆的掺烧比较准确。图6c为玉米秸秆与内蒙古煤、贵州煤同时进行混燃(煤样掺烧比平均分配),所得线性方程y=0.093x−21.83的拟合优度R2=0.993,当玉米秸秆掺烧比为20%时,误差分析为0.3%,用此公式判断玉米秸秆与内蒙古、贵州煤混燃的准确性较高。由以上分析数据可知,生物质与煤混燃数据经碳含量矫正后能得到其真实的线性关系,且误差分析较低,说明采用该检测-分析方法所得碳基线性方程可用于准确判断生物质掺烧比,同样该检测-分析方法可为碳交易市场和碳排放监测提供可靠数据支撑,促进我国碳交易市场的快速发展。
同时,本研究中使用加速器质谱(AMS)和二氧化硫(SO2)浓度检测对山西煤和玉米秸秆混燃烟气的14C/12C值及SO2浓度进行了离线分析,如图7所示。图7a为山西煤和不同掺混比玉米秸秆混燃烟气经AMS测试所得14C/12C值与碳基掺混比拟合线性方程,拟合优度R2=0.989,当碳基掺烧比为20%时,误差分析为11%,采用此线性关系判定生物质掺烧比存在较大误差。导致以上结果的原因可能由于燃烧过程中通入空气中含有现代碳,以及制样过程中引入了现代碳,或者煤本身含有现代碳,生物质中14C丰度仅为10−12,受到以上干扰因素的影响造成分析结果不准确。图7b为山西煤与棉花秸秆混燃烟气经气相色谱(GC)测试SO2浓度所得线性关系,由图可知检测混合烟气中SO2浓度仅能进行定性测量,不能判断生物质掺烧比。分析原因可能是生物质中含硫元素较少,且生物质中的碱金属与硫发生反应,影响SO2气体的排放,因此造成SO2检测浓度出现跳点现象,不能用于定量判断生物质掺烧比。
通过对比采用碳同位素红外激光设备、AMS及GC色谱测试煤与生物质混燃烟气的δ13C值、14C/12C值及SO2浓度来判定生物质掺烧比,发现碳同位素激光检测设备所得线性拟合度较好,用于判断生物质掺烧比误差分析较低,同时可进行在线监测生物质掺烧比。
-
为排除煤和生物质混燃烟气中水分、灰分等干扰因素的影响,除了使用碳含量矫正外,还可使用燃料的发热量进行矫正,此处所使用的发热量为空干基最低发热量。燃料的发热量与电厂的发电量直接相关,用发热量进行矫正可直接为电厂提供生物质掺烧比与发电量之间获得参考数据,如图8所示。图8为山西煤和不同生物质混燃经发热量矫正后所得拟合线性方程,其中图8(a、b)为山西煤和不同掺烧比例玉米秸秆的混燃线性关系,两者斜率较相似,其拟合优度R2>0.997,说明检测-分析方法经发热量矫正后得到了山西煤与玉米秸秆的真实线性关系,当玉米秸秆掺烧比为20%时,误差分析为1.8%。图8 (c—e)为山西煤分别和棉花秸秆、木屑、稻壳混燃经发热量矫正后所得拟合线性方程,斜率分布在−0.0762—−0.0793之间。
当生物质掺烧比为20%时,3组线性方程的误差分析均在2.0%之内,说明所得线性方程经燃料发热量矫正后均能反应煤与生物质混燃的真实线性关系。图8f为山西煤与棉花秸秆、木屑混燃经发热量矫正所得的拟合线性方程,拟合优度R2=0.999,排除干扰因素影响后斜率绝对值由0.039提高到0.074,且所得斜率与山西煤单独与棉花秸秆、木屑混燃的斜率相近似,说明经发热量矫正可获得煤与多种生物质混燃的真实函数关系,当生物质混燃比为20%时,误差分析为1.5%,且生物质总掺烧比不受两种生物质混合比例变化的影响(图8g,横坐标为两种生物质掺混比例:0%木屑+20%棉花秸秆、5%木屑+15%棉花秸秆、10%木屑+10%棉花秸秆、15%木屑+5%棉花秸秆、20%木屑+0%棉花秸秆)。山西煤与棉花秸秆、木屑和稻壳3种生物质同时混燃经发热量矫正后所得线性方程见图8h,拟合优度R2=0.998,斜率为−0.073,与图8g两种生物与山西煤混燃所得线性方程斜率相似,进一步说明该检测-分析方法用于判别生物质掺烧比受生物质种类及混合比例的影响较小。
图9a为玉米秸秆与内蒙古煤混燃经发热量矫正后所得线性拟合方程,拟合优度R2=0.999,当玉米秸秆掺混量为20%时,误差分析为1.0%。图9b为玉米秸秆和贵州煤混燃经发热量矫正后所得线性方程,拟合优度R2=0.998,当玉米秸秆的混燃比为20%时,误差分析为1.8%。图9c为玉米秸秆同时与内蒙古煤、贵州煤进行混燃,其数据经发热量矫正后所得斜率为0.094,处于0.092—0.095之间,说明该检测-分析方法可用于检测玉米秸秆同时与内蒙古煤、贵州煤进行混燃,其拟合优度R2=0.993,当玉米秸秆的混燃比为20%时误差分析为0.35‰,用于准确判断玉米秸秆与多种煤样混燃的掺烧比。图9d为山西煤和不同掺混比玉米秸秆混燃烟气经AMS测试所得14C/12C值与热值基掺混比拟合线性方程,拟合优度R2=0.989,当热值基生物质掺烧比为20%时,误差分析为8.7%,因此采用该线性关系判定生物质掺烧比存在较大的不准确性。
由经发热量矫正后数据可知,本文中采用碳同位素中红外激光检测设备测试碳同位素比值用于判断生物质的掺烧比是准确可靠的,发热量矫正后所得线性方程可直接为电厂核定生物质掺烧比与发电量之间的关系提供重要参考数据。
-
为了进一步证明该碳同位素中红外激光检测设备不受水分含量的影响,设计了一系列关于不同含水量的实验,见表3。
表3为实验中向燃料中添加不同量水分后进行充分燃烧,所得烟气经碳同位素激光检测设备在线分析烟气的δ13C值,对比燃料添加不同量水分及表2中各燃料的δ13C值,发现添加水分对δ13C值几乎没有影响,说明该碳同位素中红外激光检测设备不受含水量的影响,生物值含水量不会对碳同位素值的检测造成干扰。若加水量过多会导致未经矫正的原始数据拟合线性关系式较差,同样也可以从线性拟合关系式中反映出所混燃生物质是否含有大量水分或掺混其它非生物质的物质,可为电厂准确判定生物质掺烧比及真实性提供重要的数据支撑。
-
不同燃料的燃烧过程均具有差异性,碳同位素中红外激光检测设备可在线监测燃料燃烧过程中碳同位素比值的变化情况,从而推测燃料的燃烧进程及燃烧完全所使用的时间,图10为碳同位素中红外激光检测设备在线监测山西煤及不同生物质燃烧过程的δ13C值变化情况。
图10a为山西煤燃烧过程的δ13C值变化过程,由图中曲线可知开始前2 min内,山西煤燃烧过程比较迅速,释放较多的CO2气体,δ13C值低于-21.61;燃烧5 min后,所得烟气的δ13C值变化缓慢,说明山西煤已基本燃烧完毕,所需时间大概为13 min。图10b为玉米秸秆的燃烧过程,由于玉米秸秆属于生物质类,其燃烧特性优于煤样,燃烧2 min所测得δ13C值接近-10.60,燃烧5 min后,δ13C值接近-11.61;继续进行燃烧时,其δ13C值出现明显下降,说明玉米秸秆在5 min内几乎燃烧完全;图10c为山西煤(80%)和玉米秸秆(20%)混燃过程中δ13C值随燃烧时间的变化过程,由图中曲线可知,前2 min内所测得δ13C值为-17.50,此δ13C值为玉米秸秆和山西煤混燃释放CO2所含稳定碳同位素13C,由于生物质具有优于煤样燃烧的性质,推测前2 min内玉米秸秆优先燃烧,山西煤部分燃烧。随后,δ13C值在5 min内出现明显降低为-19.50,主要为剩余山西煤燃烧释放CO2所含稳定碳同位素13C;后续燃烧过程中δ13C值变化不明显。以上变化过程说明山西煤和玉米秸秆混燃时,由于生物质碳含量较煤样低,可快速将所含碳燃尽释放CO2,因此可推测玉米秸秆燃烧速率优于山西煤。图10d为山西煤同时与玉米秸秆、棉花秸秆混燃过程的δ13C值变化情况,由图中曲线可知开始2 min内所得δ13C值为-19.81,依据实验所测得玉米秸秆、棉花秸秆的δ13C值推测此过程可能为玉米秸秆优先燃烧,而棉花秸秆和山西煤部分燃烧;燃烧5 min时δ13C值出现明显下降为-21.0,依据棉花秸秆的δ13C值为-26.10推测此过程可能主要为所剩棉花秸秆的燃烧;随后燃烧10 min时,δ13C值上升为-20.61,此值接近山西煤的δ13C值,推测此过程可能主要为所剩山西煤的燃烧。此曲线变化情况反映了多种生物质混燃时由于其性质的不同,出现先后燃烧的情况。由于玉米秸秆和棉花秸秆的δ13C值有较大区别,因此混燃过程中所得δ13C值会出现上下跳跃的情况,不易得到准确的线性关系,解释了玉米秸秆不能与其他生物质混燃的直接原因。
通过在线监测煤样和生物质的混燃过程可知生物质的燃烧特性优于煤样燃烧特性,主要由于生物质的着火点比煤低很多,挥发分含量多,能够在短时间内迅速燃烧释放出CO2。在电厂实际发电过程中,燃料需要不断输入到锅炉内进行燃烧,采用碳同位素激光检测设备在线监测烟气中所含CO2的δ13C值可能是上下稍微浮动的数据点,因此需要多次采集数据点,能更准确的判断生物质的真实掺烧比.
-
本文采用碳同位素中红外激光检测设备在线监测煤耦合生物质发电的混燃比,该检测精度较高,所得数据线性拟合关系较好,判定生物质掺烧比误差较小,且该设备与市场现有AMS设备相比成本低,具有经济竞争力。所得δ13C值经碳基量矫正后可得到CO2释放量与生物质掺烧比的真实函数关系,因此该分析方法不仅可为发电厂判断生物质掺烧比提供可靠的技术支撑,还可为国家目前急需发展的碳交易市场及碳排放监测提供衡量标准,从而推动我国碳交易市场的健康发展。同时经发热量矫正的拟合曲线可为电厂掺烧生物质所提供发电量提供重要数据,可协助电厂辨别生物质中是否添加过多水分或添加其他非生物质的物质。该分析方法正处于由实验室规模向中试示范装置转换的阶段,以进一步验证该分析方法的可行性及准确性。未来工作中需结合电厂实际监测环境对该分析方法进行集成开发,并进一步增加分析设备的连续分析时间和寿命,降低使用成本,以便后续在电厂中进行该技术的推广和应用。
稳定碳同位素激光检测仪监测生物质燃料掺烧比的研究
Monitoring biomass-coal fuel blending ratio by stable carbon-isotope laser detector
-
摘要: 稳定碳同位素激光技术可用于检测煤和生物质的碳同位素比值δ13C,以确定燃煤耦合生物质的掺烧比,且不受含水量影响。本文通过对煤(山西煤、内蒙古煤、贵州煤)和生物质(玉米秸秆、棉花秸秆、木屑、稻壳)混燃烟气进行稳定碳同位素比值测定,在一定的生物质掺烧比范围(碳量基、热量基)内建立在线分析方法得到拟合线性方程y=ax+b,拟合优度R2>0.99。所得分析方法可用于在线监测生物质掺烧比变化情况,当生物质掺烧比为20%时,误差分析控制在0.5%—2.0%以内。此外,该检测-分析方法能够实时、准确地监测生物质掺烧比,可为国家针对生物质使用量补贴政策的制定和实施提供技术支撑;同时可用于追踪“碳溯源”,推动碳排放监测和碳交易市场的快速发展。Abstract: The stable carbon isotope laser technology can be used to detect the carbon isotope ratio of coal and biomass (δ13C) to determine the blending ratio of coal-fired coupled biomass, and is not affected by water content. In this paper, the stable carbon isotope ratio was measured in the flue gas of coal (Shanxi coal, Inner Mongolia coal or Guizhou coal) and biomass (cornstalks, cotton stalks, wood chips or rice husks). The online analysis method was established considering a certain range of biomass blend ratio (carbon-based and calorific value-based) to get the fitted linear equation y = ax + b, and the goodness-of-fit (R2) exceeded 0.99. The obtained analysis method can be used to on-line monitor the change of the biomass content in the blend. When the biomass blending ratio is 20%, the analysis error is controlled within 0.5%—2.0%. In addition, this detection-analysis method can real-time monitor the biomass blending ratio accurately, which provides technical support for the development and implementation of the national subsidy policy for biomass usage and promotes the rapid development of carbon emission monitoring and the carbon trading market.
-
能源和环境问题是人类社会可持续发展所面临的最重要制约因素。我国碳交易市场于2017年正式启动,意味着能源发展必须走低碳清洁化道路。我国正处在统一碳交融交易市场关键时期,碳减排量与碳减排水平需要一个衡量标准及相应的函数关系,但目前缺乏相应的碳交易手段和政策制定依据。碳交易市场的启动会推动、倒逼发电企业进行发电结构调整,化解高碳落后产能,煤电低碳清洁转型升级、灵活性改造势在必行。把煤电和生物质能“撮合”在一起进行发电试点,将推动中国能源结构转型和煤电绿色发展,为煤电低碳清洁发展带来了新途径,燃煤耦合生物质发电的零碳排放优势将把握住电力产业低碳转型发展机遇[1-2]。
生物质能是一种清洁可再生能源,也是6种可再生能源中唯一可以转化为气、液、固体的零碳能源。近年来生物质发电逐渐兴起,利用生物质替代矿物燃料进行发电可以有效减少CO2和SO2排放。生物质与煤混合燃烧发电已被纳入国家产业规划,缺乏科学、公正的生物质应用量在线检测技术依旧是燃煤耦合生物质发电所面临的最大问题。目前较为先进的后端检测方法,主要包括SO2浓度检测法和14C含量检测法[3-6]。由于生物质含有一些碱金属,会与S发生反应,影响SO2浓度监测结果。生物质与大气中的14C活度相等,煤种14C活度几乎为零,因此14C含量检测法的准确性、可靠性仍然处于探索性阶段[7]。14C技术分析设备多为价格昂贵的核磁、加速器质谱(AMS)等精密仪器[8-14],影响了生物质应用量后端检测技术的发展。
目前,生物质应用量后端检测技术亟待解决实时检测、设备昂贵等问题。研究发现中红外激光同位素检测技术可实现快速实时检测稳定碳同位素比值δ13C (13CO2/12CO2)[15-23]。Castrillo等采用近红外激光设备测试CO2气体中的13C/12C同位素比值(δ),设备系统误差为0.5‰ [24]。传统光谱法相对质谱法测量灵敏度较低,但中红外量子级联激光器和腔衰荡光谱法(QCL-CRDS)相结合可以最大程度地提高气体光学检测的灵敏度,是目前最前沿的超低气体浓度检测技术,其具有体积小、操作简单、价格便宜、对环境敏感性低等优势,大大提高了现场布控和在线分析的能力,在生态、油气、环境监测等领域发挥着重要的角色。生物质与煤燃烧释放的CO2中均含有稳定碳同位素13C,生物质燃烧释放CO2气体中所含稳定碳同位素重,而煤燃烧过程中释放的碳同位素轻,可依据各自δ13C值的差异进行煤和生物质掺混比的判定[25-26]。现有碳同位素中红外激光检测设备检测误差仅为±0.025‰,可满足生物质应用量的精确评测需求,同时该检测仪造价较低,可实现对质谱等昂贵设备的替代,但与之相对应的评测方法国内外尚属空白[18, 27-28]。
本文研究建立基于稳定碳同位素中红外激光检测仪监测生物质掺烧比的实时在线检测分析方法十分重要,可为政府补贴政策的实施提供技术支撑,同时该分析方法可用于追踪“碳溯源”,推动碳排放监测和碳交易市场的快速发展,促进清洁能源的健康有序发展和碳交易市场建立建全定价机制。
1. 材料与方法(Materials and methods)
1.1 实验原料
实验选取山西煤、贵州煤和内蒙古煤3种煤样,以及玉米秸秆、棉花秸秆、木屑和稻壳4种生物质进行混燃实验,实验中实际测试煤样和生物质的工业分析、元素分析和热值测定结果见表1。实验煤样品的制备主要经过破碎、研磨和筛分3个过程。首先将样品破碎成小颗粒(粒径小于1 cm);然后使用粉碎机对其进行粉碎;最后用标准试验筛粉状样品进行筛分,取粒径60—80目的样品进行混燃实验。生物质粉末样品购买于惠丰秸秆农产品加工企业。
表 1 煤和生物质工业分析、元素分析和热值测定Table 1. Industrial analysis, element analysis, and calorific value determination of coal and biomass样品/组分Sample/component 内蒙古煤Inner Mongolia coal 贵州煤Guizhou coal 山西煤Shanxi coal 玉米秸秆Cornstalks 稻壳Rice husks 棉花秸秆Cotton stalks 木屑Wood chips 水分Mad/% 6.83 4.77 5.58 7.59 6.54 7.77 7.01 灰分Aad/% 2.07 11.81 3.76 7.76 12.74 2.50 1.9 挥发分Vad/% 30.00 30.52 28.90 67.53 62.90 70.86 76.33 固定碳FCad/% 61.10 52.90 61.76 17.12 17.83 18.88 14.76 碳Cad/% 73.56 66.23 73.85 41.24 40.39 44.77 45.47 氢Ht,ad% 4.91 4.16 4.60 6.13 5.61 6.30 6.43 氮Nad/% 0.98 0.91 1.08 1.49 0.49 1.18 0.40 全硫St,ad/% 0.59 0.21 0.20 0.21 0.11 0.14 0.08 氧Oad/% 11.82 12.44 11.55 36.43 34.86 38.21 39.49 空干基低位发热量Qnet,ad/(J·g−1) 27060 24030 26400 15030 14660 16140 16580 空干基高位发热量Qgr,ad/(J·g−1) 28080 24890 27350 16290 15810 17440 17900 干基高位发热量Qgr,d/(J·g−1) 30130 26130 28960 17630 16920 18910 19250 注:本文发热量矫正使用的为空干基低位发热量. Note: Low calorific value at air-dry basis was used in the calorific value correction in this study. | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 实验装置
自主搭建小型混燃实验系统,该系统主要包括:刚瓶、流量控制计、燃烧炉、净化装置、碳同位素中红外激光检测设备,如图1。使用气体总流量为200 mL·min−1,气体组分包括33%O2和67%空气,富氧条件可保证煤和生物质的充分燃烧。实验过程主要包括:将装有混合燃料的石英舟(78 mm(长)×11 mm(宽)×15 mm(高))放置在燃烧炉中不锈钢管的恒温段,O2和空气经质量流量计后进行充分混合后通入燃烧炉,燃烧温度850 ℃;充分燃烧的混合烟气经过滤器后进入碳同位素中红外激光检测设备进行碳同位素比值δ13C(13CO2/12CO2)的检测。
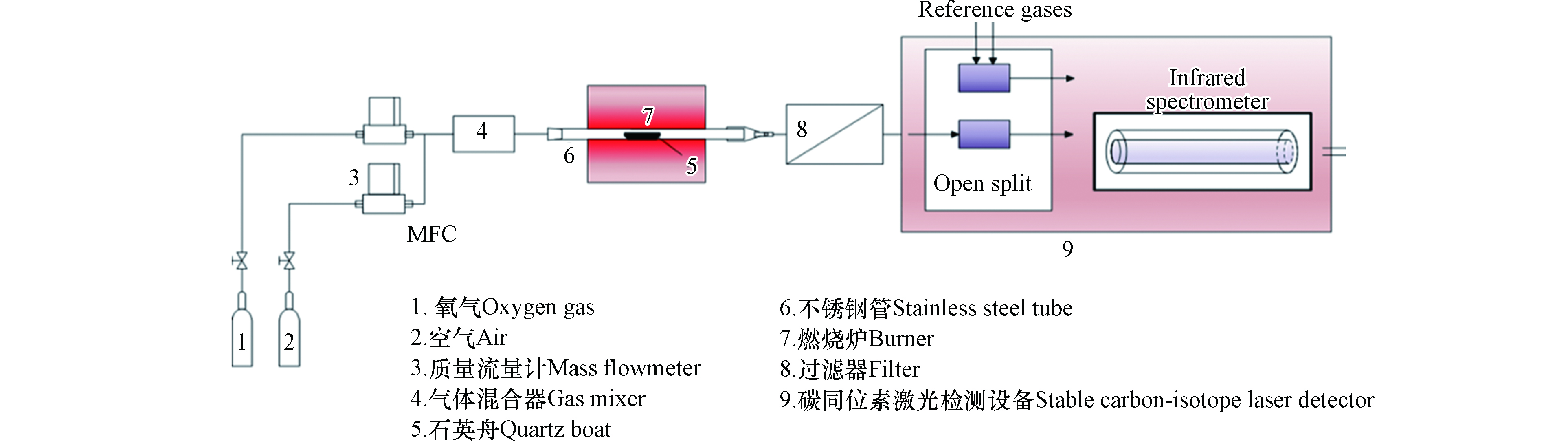 图 1 小型煤耦合生物质混燃在线检测系统Figure 1. On-line detection system for small-scale co-combustion of coal and biomass
图 1 小型煤耦合生物质混燃在线检测系统Figure 1. On-line detection system for small-scale co-combustion of coal and biomass碳同位素中红外激光检测设备(团队自主研发)基于不断成熟发展量子级联激光器设计开发一套多通道光学检测核心器件,其结构如图2所示。该检测设备主要包括量子级联激光器、空心波导管和探测器,其中空心波导中包含一套五通道耦合光路系统。实验设计主要包括:一种煤样和一种或多种生物质进行混燃,一种生物质与一种或多种煤样进行混燃。生物质燃料的掺烧比主要为5%、10%、20%和30%;为了进一步建立准确的分析方法,将玉米与山西煤的掺烧比设计范围为0—100%,其中在0—30%之内增加2.5%、7.5%、15%和25%的掺烧比例。
本研究中为了验证稳定碳同位素激光检测设备的技术优势及建立分析方法的准确性,同时采用加速质谱仪(AMS,3MV TN-4130,High Voltage Engineering Europa B.V.,HVEE)对混燃烟气的14C浓度进行了离线测试,燃烧过程如上所述,所得烟气经集气袋收集后在AMS设备上进行14C浓度检测,得到14C/12C的值。另外,采用SO2浓度检测方对混燃烟气中SO2浓度进行了离线对比分析。
1.3 分析方法
稳定碳同位素中红外激光检测设备所测试同位素比值δ13C(13C/12C)按照如下公式定义:
δ13C=[(Rp/Rs−1)]×1000 (1) 式(1)中,Rp是样品中碳元素重轻同位素丰度之比(13Cp/12Cp),Rs是国际通用标准物的重轻同位素丰度之比(13Cs/12Cs)。不同煤样与不同生物质的δ13C值之间具有显著差异,依据所述δ13C差异性,通过如下线性回归方程计算混燃物质中生物质的掺烧比例:
y=ax+b (2) r=CC×x(CC−CB)×x+CB (3) y=a1x1+b (4) r1=QC×x1(QC−QB)×x1+QB (5) 式(2)中,y为碳同位素激光检测设备显示的δ13C值,x为碳量基掺烧比,a为通过配比不同煤种与不同生物质混燃所得线性回归方程的斜率,b为煤样的δ13C值;式(3)中,r为质量基掺烧比,CC为煤样碳含量,CB为生物质碳含量。同时,可用发热量进行矫正后得到线性回归方程(4)、(5),其中x1为热量基掺烧比,a1为通过配比不同煤种与不同生物质混燃所得线性回归方程的斜率,式(5)中,r1为质量基掺烧比,QC为煤样的发热量,QB为生物质发热量。经碳含量和发热量矫正后可排除水分、灰分、挥发分等干扰因素的影响,有利于更准确的判断生物质掺烧比。
另外,生物质混燃比还可通过14C/12C的值R进行判定:
yR=aRxR+bR (6) r2=CC×xR(CC−CB)×xR+CB (7) yR=aR1xR1+bR (8) r3=QC×xR1(QC−QB)×xR1+QB (9) 式(6)中,yR为AMS检测的14C/12C的值,aR为通过配比不同煤种与不同生物质混燃所得线性回归方程的斜率,xR为碳量基掺烧比,b为煤样的14C/12C值;式(7)中,r2为质量基掺烧比。经发热量进行矫正后得到线性回归方程(8)、(9),式(8)中xR1为热量基掺烧比,式(9)中,r3为质量基掺烧比。
2. 结果与讨论(Results and discussion)
判定生物质掺烧比时易受到含水量、灰分、挥发分的影响,导致线性方程拟合优度R2较差,需要对其进行碳含量和发热量矫正,以获取更准确的线性拟合方程,使其拟合优度R2均达到0.990以上。纯煤样和生物质样品充分燃烧后经碳同位素红外激光检测设备所得碳同位素比值(δ13C)见表2。
表 2 纯燃料样品的碳同位素比值(δ13C)Table 2. The 13C proportion (δ13C) in pure coal and biomass samples样品Sample 山西煤Shanxi coal 贵州煤Guizhou coal 内蒙古煤Inner Mongolia coal 玉米秸秆Cornstalks 棉花秸秆Cotton stalks 木屑Wood chips 稻壳Rice husks δ13C −20.61 −21.01 −21.91 −11.91 −26.09 −26.99 −27.02 | Show TableDownLoad:
CSV
表2可知,纯煤样(山西、贵州、内蒙古)的δ13C值具有差异性,各生物质δ13C值也呈现一定的区别,尤其是玉米秸秆的δ13C值为−11.91,说明该生物质的碳同位素较重,则玉米秸秆中所含13C比重偏多,而棉花秸秆、木屑和稻壳之间的δ13C值属于相似范围−26—−27,说明三者所含13C的比重较近似。本文依据各燃料之间δ13C值的差异性建立函数关系进行生物质掺烧比的判断。
2.1 初始数据拟合线性方程
煤和生物质充分混燃烟气经碳同位素中红外激光检测设备测定其稳定碳同位素比值δ13C,未对设计生物质掺烧比进行矫正时,所得拟合线性方程见图3—5。
 图 3 山西煤与单一或两种生物质混燃所得线性回归方程Figure 3. The linear regression equations of co-combustion Shanxi coal and various biomass samples(a、b)玉米秸秆,(c)木屑,(d)棉花秸秆,(e)稻壳,(f)玉米秸秆+木屑,(g)木屑+棉花秸秆+稻壳,(h)玉米+木屑+棉花秸秆+稻壳(a,b) cornstalks, (c) wood, (d) cotton stalk, (e) rice hucks, (f) corn stalks+wood, (g) wood chips+cotton stalks+rice hucks, (h) cornstalks+wood chips+cotton stalks+rice hucks
图 3 山西煤与单一或两种生物质混燃所得线性回归方程Figure 3. The linear regression equations of co-combustion Shanxi coal and various biomass samples(a、b)玉米秸秆,(c)木屑,(d)棉花秸秆,(e)稻壳,(f)玉米秸秆+木屑,(g)木屑+棉花秸秆+稻壳,(h)玉米+木屑+棉花秸秆+稻壳(a,b) cornstalks, (c) wood, (d) cotton stalk, (e) rice hucks, (f) corn stalks+wood, (g) wood chips+cotton stalks+rice hucks, (h) cornstalks+wood chips+cotton stalks+rice hucks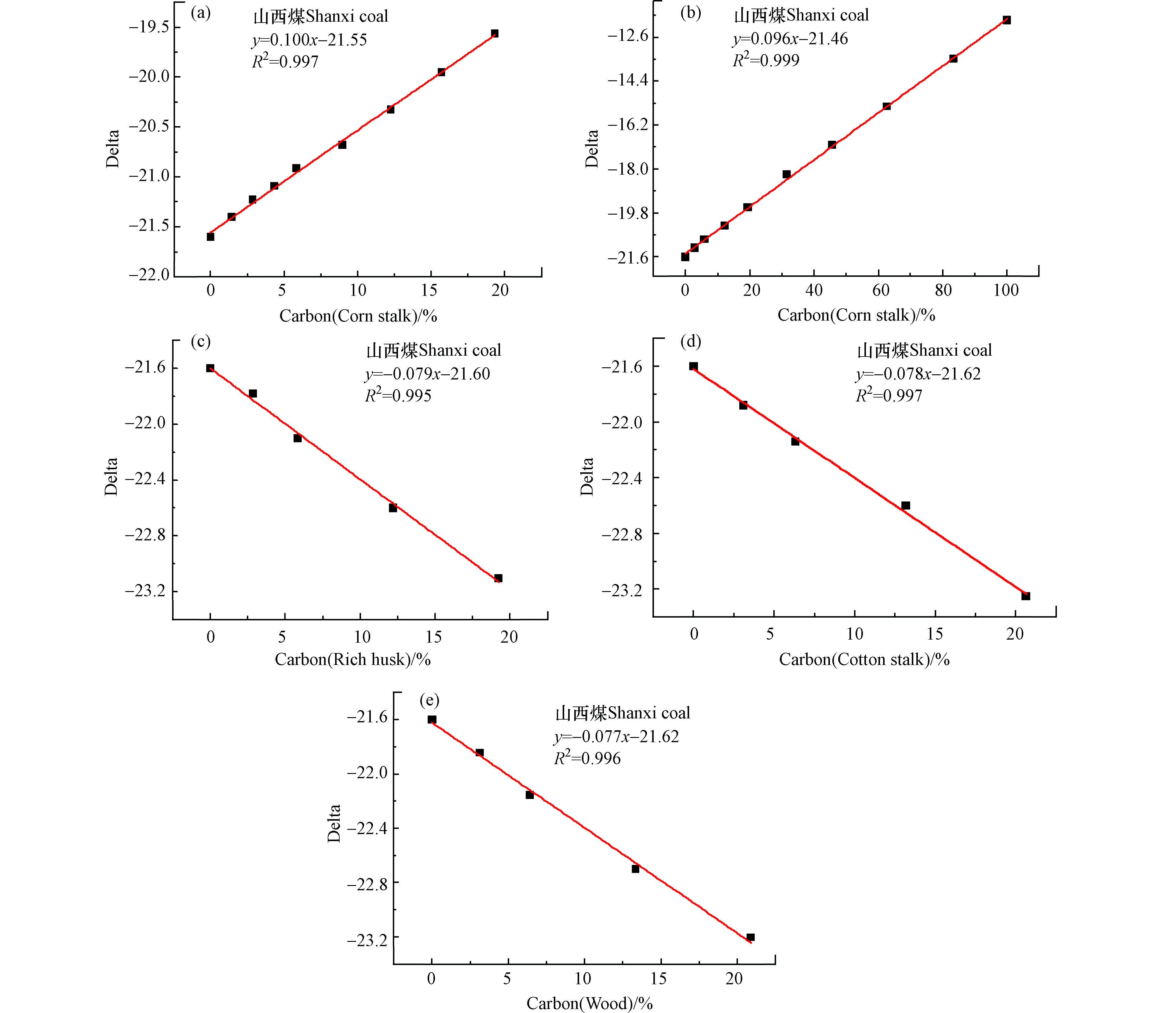 图 5 山西煤与单一或多种生物质混燃的碳基掺烧比线性方程Figure 5. The linear regression equations of co-combustion Shanxi coal and various biomass samples based on carbon content (a,b) cornstalks, (c) rich hucks, (d) cotton stalks, (e) wood(a、b)玉米秸秆,(c) 稻壳,(d)棉花秸秆,(e) 木屑
图 5 山西煤与单一或多种生物质混燃的碳基掺烧比线性方程Figure 5. The linear regression equations of co-combustion Shanxi coal and various biomass samples based on carbon content (a,b) cornstalks, (c) rich hucks, (d) cotton stalks, (e) wood(a、b)玉米秸秆,(c) 稻壳,(d)棉花秸秆,(e) 木屑图3a为山西煤与不同比例(0—30%)的玉米秸秆混燃所得线性回归方程,其拟合优度R2=0.996,说明此方程的线性关系较好,当玉米秸秆的掺烧比为20%时,误差分析为6.1%,说明用此公式判断玉米秸秆的掺烧比存在很大的不准确性。若将玉米秸秆的掺烧比设计为0—100%(图3b),则所得方程的线性关系较差,拟合优度R2=0.986,用此方程判断玉米秸秆的掺烧比同样存在很大的误差。图3a和3b所得线性回归方程的斜率存在较大差异,说明未经矫正的线性方程由于受到含水量、灰分等因素的干扰,不适合用于判断生物质的掺烧比。图3c为山西煤与不同比例的木屑混燃的线性方程,可知方程的拟合优度R2=0.998,线性关系较好,但由于受含水量、灰分等的影响,对生物质掺烧比(20%)进行判断时仍存在较大误差;同样图3d和图3e分别为山西煤和不同比例的棉花秸秆、稻壳进行混燃所得线性方程,若使用目前所得函数关系判断生物质掺烧比亦存在较大误差。
图3f为山西煤同时与两种生物质混燃的线性方程,其生物质的总掺烧比为两种掺烧量相同生物质的加和。由拟合线性关系的斜率变化可知,同时掺混两种生物质时,所得δ13C值与生物质掺混比关系较敏感。由山西煤、玉米、木屑三者混燃所得拟合线性方程可知,生物质中仅玉米秸秆碳同位比值δ13C(δ13C=-11.91)较大,当玉米秸秆与其它生物质一起混燃时所得线性方程的斜率(0.02左右)与δ13C值的关系过于敏感,生物质掺烧比的变化易引起δ13C值较大波动,用所得线性方程判断生物质混燃比将带来较大的误差。图3(g、h)为山西煤和木屑、棉花秸秆、稻壳等3种生物质混燃的线性方程,由于添加多种生物质,且受到含水量、灰分等的影响,线性拟合关系并不理想,因此不能准确判断生物质的混燃比。图3h为山西煤和玉米秸秆、木屑、棉花秸秆、稻壳等4种生物质混燃的线性方程,同样由于玉米秸秆的δ13C值与其他生物质差别较大,混燃玉米秸秆后所得线性关系斜率绝对值较低,不利于进行生物质混燃比的判断,亦说明此检测分析方法用于玉米秸秆与其他生物质同时进行混燃时不利于生物质掺烧比的判断。
由于我国玉米秸秆产量较大,因此使用该检测分析方法针对玉米秸秆与多种煤样混燃进行了研究。图4为玉米秸秆与内蒙古煤、贵州煤、山西煤混燃线性方程。由图4结果可知,玉米秸秆与不同煤样混燃的线性关系均较好。图4a为玉米秸秆和内蒙古煤的混燃线性方程,其拟合优度R2=0.996,线性关系较好,但由于受含水量、灰分等的影响,当玉米秸秆的混燃比为20%,误差分析在2.9%。图4b为玉米秸秆和贵州煤的线性拟合方程,拟合优度R2=0.998,线性关系较好,由于含水量和灰分等的影响,判断玉米秸秆混燃比将出现误差(混燃比20%时,误差分析在10%)。图4(c、d)分别为玉米秸秆和山西、贵州煤以及玉米秸秆和内蒙古煤和贵州煤的混燃线性方程,由于未排除干扰因素(含水量、灰分等)的影响,用以上两个线性方程判断玉米秸秆的掺烧比均存在不准确性。
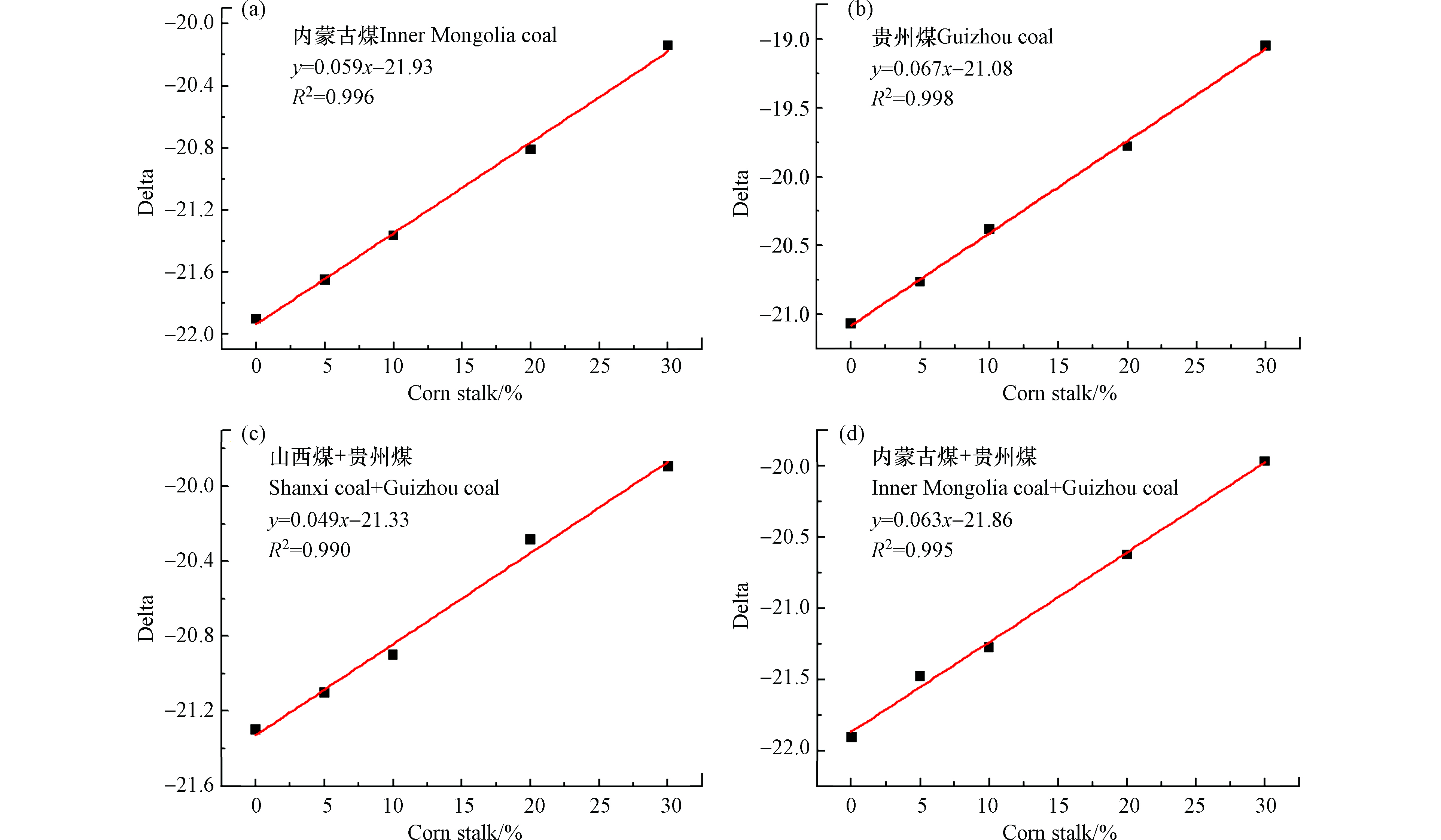 图 4 玉米秸秆与一种或两种煤混燃所得线性方程Figure 4. The linear regression equations of co-combustion cornstalks and various coal samples corresponding to mass content (a) Inner Mongolia coal, (b) Guizhou coal, (c) Shanxi coal+ Guizhou coal, (d) Inner Mongolia coal+Guizhou coal(a)内蒙古煤,(b)贵州煤,(c)山西煤+贵州煤,(d)内蒙古煤+贵州煤
图 4 玉米秸秆与一种或两种煤混燃所得线性方程Figure 4. The linear regression equations of co-combustion cornstalks and various coal samples corresponding to mass content (a) Inner Mongolia coal, (b) Guizhou coal, (c) Shanxi coal+ Guizhou coal, (d) Inner Mongolia coal+Guizhou coal(a)内蒙古煤,(b)贵州煤,(c)山西煤+贵州煤,(d)内蒙古煤+贵州煤由以上分析可知,无论是单一煤样和单一或多种生物质混燃,或者单一生物质和单一或多种煤样混燃,由于受到含水量、灰分等其他干扰因素的影响,用本文的碳同位素激光检测-分析方法判定生物质的掺烧比均存在一定的误差,需要排除以上因素的干扰,也从侧面证明此检测-分析方法可用于判定掺烧生物是否含水量过多、或掺进其他非生物质的物质,为准确判断生物质的掺烧比提供了多重保障。为了准确获得生物质掺烧比,对多种煤样和生物质混燃数据进行了碳含量和发热量矫正,用燃料最本征的数值来判定生物质的掺烧比。
2.2 碳含量矫正的线性方程
燃料的碳(C)含量与燃料充分燃烧后所释放的CO2量直接相关,也最能真实的反应出碳同位素比值(δ13C=13CO2/12CO2)变化情况,以下所得线性方程均采用燃料的含碳量(详细数据见表1)进行矫正。图5均为经碳含量矫正后所得拟合线性方程,由图5可知,经碳含量矫正后的数据排除了水分、灰分等多种因素的干扰后,拟合优度R2>0.995,线性关系得到进一步改善。图5(a、b)为山西煤和不同掺混比玉米秸秆混燃所得线性方程,两条线性方程的斜率相似(0.096≈0.1),说明碳基线性方程的斜率与玉米秸秆的掺混比关系不大,为山西煤和玉米秸秆混燃的真实线性函数关系,当玉米秸秆的掺烧比为20%时,误差分析在2.0%,准确性得到了很大的提高。同样图5(c、d)分别为山西煤与稻壳、棉花秸秆以及木屑混燃的碳基线性拟合方程,由于3种生物质的δ13C值相近,因此与山西煤混燃时所得线性方程的斜率均相似(−0.077—−0.079),说明碳基线性关系均为煤与生物质混燃的真实线性函数关系。当生物质掺烧比为20%时,误差分析可控制在2.0%以内。
图6为玉米秸秆与一种或两种煤样混燃的碳基线性拟合方程,其中图6a为玉米秸秆和内蒙古煤混燃的线性拟合方程,拟合优度R2=0.999,当玉米秸秆掺烧比为20%时,误差分析为0.5%,说明线性关系较好,此公式用于判断玉米秸秆的掺烧比较准确。图6b为玉米秸秆与贵州煤混燃的碳基线性方程,其拟合优度R2=0.998,当玉米秸秆掺烧比为20%时,误差分析为0.5%,说明用此拟合线性方程判断玉米秸秆的掺烧比较准确。图6c为玉米秸秆与内蒙古煤、贵州煤同时进行混燃(煤样掺烧比平均分配),所得线性方程y=0.093x−21.83的拟合优度R2=0.993,当玉米秸秆掺烧比为20%时,误差分析为0.3%,用此公式判断玉米秸秆与内蒙古、贵州煤混燃的准确性较高。由以上分析数据可知,生物质与煤混燃数据经碳含量矫正后能得到其真实的线性关系,且误差分析较低,说明采用该检测-分析方法所得碳基线性方程可用于准确判断生物质掺烧比,同样该检测-分析方法可为碳交易市场和碳排放监测提供可靠数据支撑,促进我国碳交易市场的快速发展。
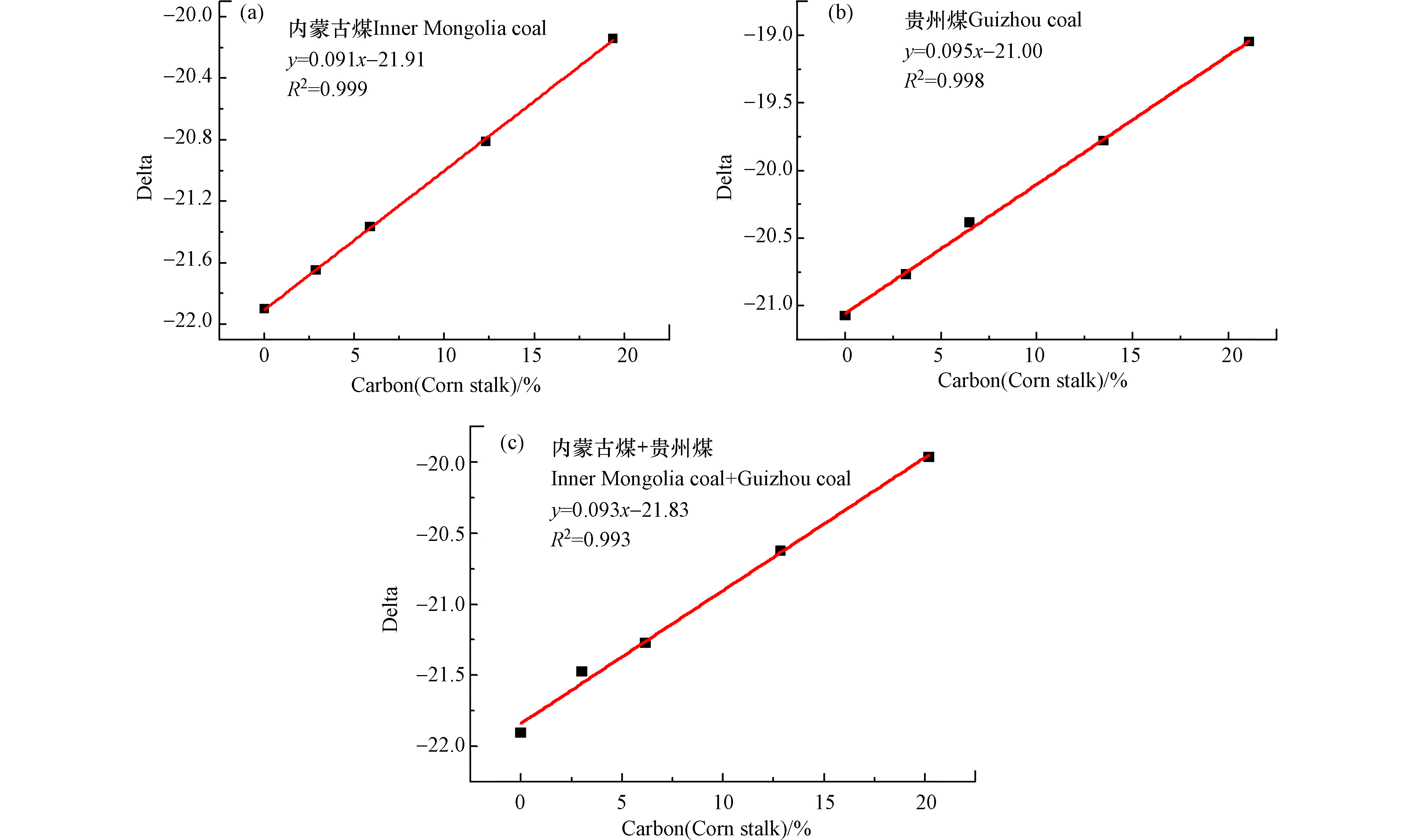 图 6 玉米秸秆与一种或两种煤混燃的碳基掺烧比线性方程Figure 6. The linear regression equations of co-combustion cornstalks and various coal samples based on carbon content(a)内蒙古煤,(b)贵州煤,(c)内蒙古煤+贵州煤(a) Inner Mongolia coal, (b) Guizhou coal, (c) Inner Mongolia coal+Guizhou coal
图 6 玉米秸秆与一种或两种煤混燃的碳基掺烧比线性方程Figure 6. The linear regression equations of co-combustion cornstalks and various coal samples based on carbon content(a)内蒙古煤,(b)贵州煤,(c)内蒙古煤+贵州煤(a) Inner Mongolia coal, (b) Guizhou coal, (c) Inner Mongolia coal+Guizhou coal同时,本研究中使用加速器质谱(AMS)和二氧化硫(SO2)浓度检测对山西煤和玉米秸秆混燃烟气的14C/12C值及SO2浓度进行了离线分析,如图7所示。图7a为山西煤和不同掺混比玉米秸秆混燃烟气经AMS测试所得14C/12C值与碳基掺混比拟合线性方程,拟合优度R2=0.989,当碳基掺烧比为20%时,误差分析为11%,采用此线性关系判定生物质掺烧比存在较大误差。导致以上结果的原因可能由于燃烧过程中通入空气中含有现代碳,以及制样过程中引入了现代碳,或者煤本身含有现代碳,生物质中14C丰度仅为10−12,受到以上干扰因素的影响造成分析结果不准确。图7b为山西煤与棉花秸秆混燃烟气经气相色谱(GC)测试SO2浓度所得线性关系,由图可知检测混合烟气中SO2浓度仅能进行定性测量,不能判断生物质掺烧比。分析原因可能是生物质中含硫元素较少,且生物质中的碱金属与硫发生反应,影响SO2气体的排放,因此造成SO2检测浓度出现跳点现象,不能用于定量判断生物质掺烧比。
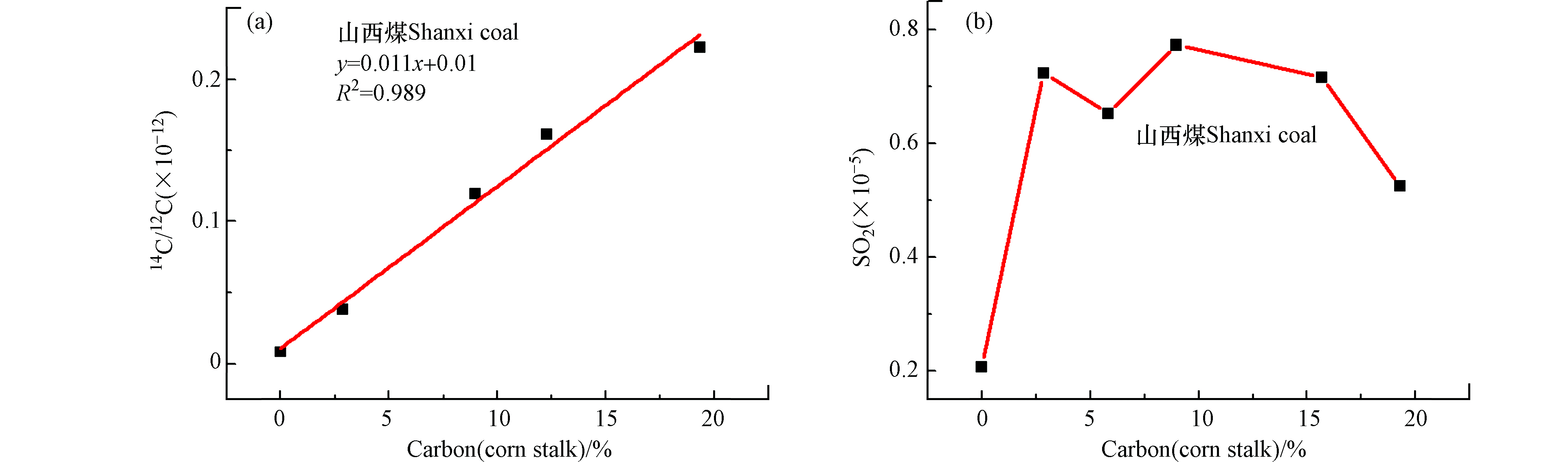 图 7 山西煤与玉米秸秆混燃 (a)加速器质谱测14C/12C值判定生物质混燃比线性方程,(b)SO2浓度测试Figure 7. The linear regression equations of co-combustion Shanxi coal and cornstalks based on carbon content (a) 14C detection, (b) SO2 concentration
图 7 山西煤与玉米秸秆混燃 (a)加速器质谱测14C/12C值判定生物质混燃比线性方程,(b)SO2浓度测试Figure 7. The linear regression equations of co-combustion Shanxi coal and cornstalks based on carbon content (a) 14C detection, (b) SO2 concentration通过对比采用碳同位素红外激光设备、AMS及GC色谱测试煤与生物质混燃烟气的δ13C值、14C/12C值及SO2浓度来判定生物质掺烧比,发现碳同位素激光检测设备所得线性拟合度较好,用于判断生物质掺烧比误差分析较低,同时可进行在线监测生物质掺烧比。
2.3 热量矫正的线性方程
为排除煤和生物质混燃烟气中水分、灰分等干扰因素的影响,除了使用碳含量矫正外,还可使用燃料的发热量进行矫正,此处所使用的发热量为空干基最低发热量。燃料的发热量与电厂的发电量直接相关,用发热量进行矫正可直接为电厂提供生物质掺烧比与发电量之间获得参考数据,如图8所示。图8为山西煤和不同生物质混燃经发热量矫正后所得拟合线性方程,其中图8(a、b)为山西煤和不同掺烧比例玉米秸秆的混燃线性关系,两者斜率较相似,其拟合优度R2>0.997,说明检测-分析方法经发热量矫正后得到了山西煤与玉米秸秆的真实线性关系,当玉米秸秆掺烧比为20%时,误差分析为1.8%。图8 (c—e)为山西煤分别和棉花秸秆、木屑、稻壳混燃经发热量矫正后所得拟合线性方程,斜率分布在−0.0762—−0.0793之间。
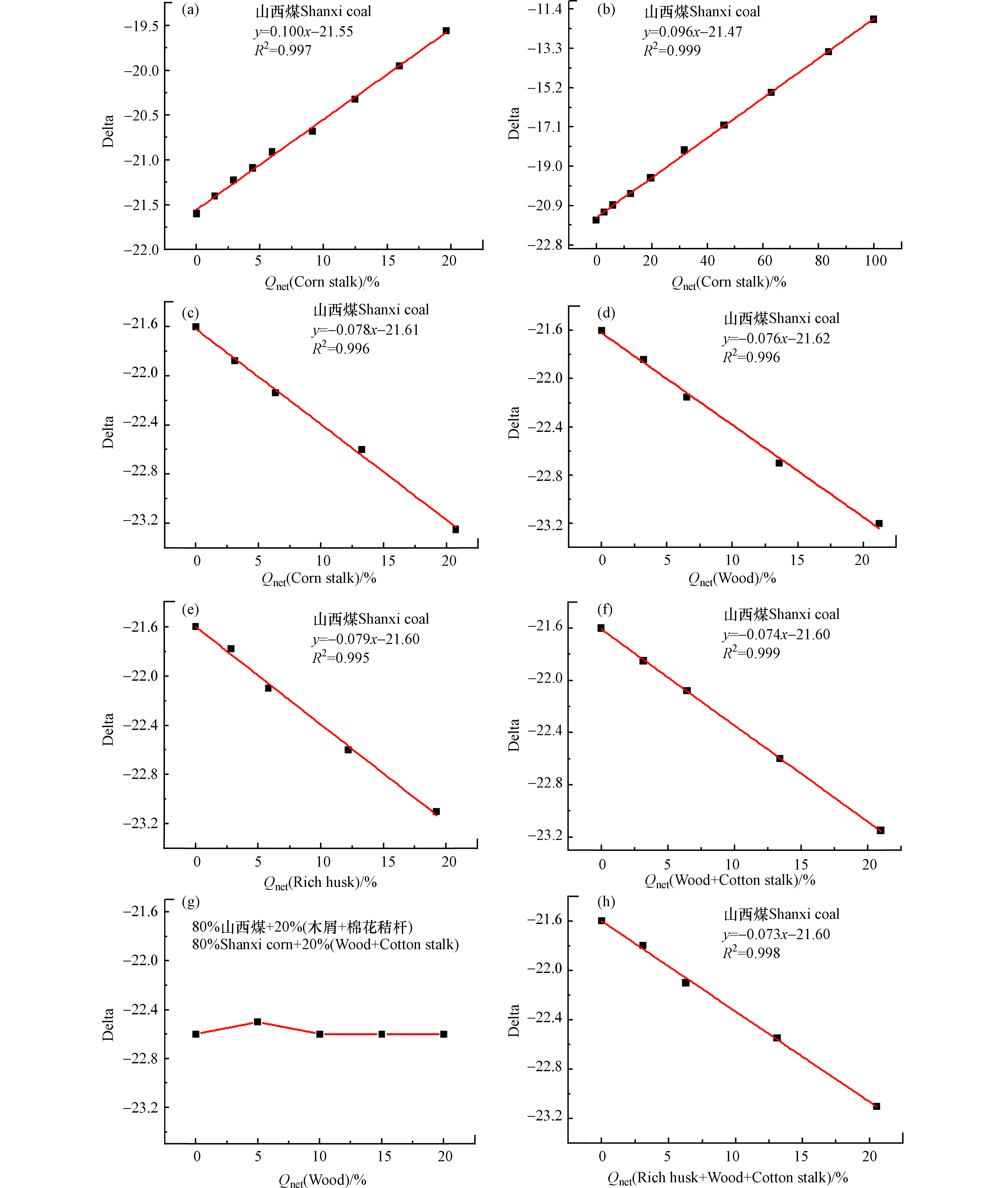 图 8 山西煤与一种或多种生物质混燃经发热量矫正所得线性方程Figure 8. The linear regression equations of co-combustion Shanxi coal and various biomass samples based on Qnet(a、b)玉米秸秆,(c)棉花秸秆,(d)木屑,(e)稻壳,(f、g)木屑+棉花秸秆,(h)棉花秸秆+木屑+稻壳(a,b) cornstalks, (c) cotton stalks, (d) wood, (e) rich hucks, (f,g) wood + cotton stalks, (h) rice huck + wood + cotton stalk
图 8 山西煤与一种或多种生物质混燃经发热量矫正所得线性方程Figure 8. The linear regression equations of co-combustion Shanxi coal and various biomass samples based on Qnet(a、b)玉米秸秆,(c)棉花秸秆,(d)木屑,(e)稻壳,(f、g)木屑+棉花秸秆,(h)棉花秸秆+木屑+稻壳(a,b) cornstalks, (c) cotton stalks, (d) wood, (e) rich hucks, (f,g) wood + cotton stalks, (h) rice huck + wood + cotton stalk当生物质掺烧比为20%时,3组线性方程的误差分析均在2.0%之内,说明所得线性方程经燃料发热量矫正后均能反应煤与生物质混燃的真实线性关系。图8f为山西煤与棉花秸秆、木屑混燃经发热量矫正所得的拟合线性方程,拟合优度R2=0.999,排除干扰因素影响后斜率绝对值由0.039提高到0.074,且所得斜率与山西煤单独与棉花秸秆、木屑混燃的斜率相近似,说明经发热量矫正可获得煤与多种生物质混燃的真实函数关系,当生物质混燃比为20%时,误差分析为1.5%,且生物质总掺烧比不受两种生物质混合比例变化的影响(图8g,横坐标为两种生物质掺混比例:0%木屑+20%棉花秸秆、5%木屑+15%棉花秸秆、10%木屑+10%棉花秸秆、15%木屑+5%棉花秸秆、20%木屑+0%棉花秸秆)。山西煤与棉花秸秆、木屑和稻壳3种生物质同时混燃经发热量矫正后所得线性方程见图8h,拟合优度R2=0.998,斜率为−0.073,与图8g两种生物与山西煤混燃所得线性方程斜率相似,进一步说明该检测-分析方法用于判别生物质掺烧比受生物质种类及混合比例的影响较小。
图9a为玉米秸秆与内蒙古煤混燃经发热量矫正后所得线性拟合方程,拟合优度R2=0.999,当玉米秸秆掺混量为20%时,误差分析为1.0%。图9b为玉米秸秆和贵州煤混燃经发热量矫正后所得线性方程,拟合优度R2=0.998,当玉米秸秆的混燃比为20%时,误差分析为1.8%。图9c为玉米秸秆同时与内蒙古煤、贵州煤进行混燃,其数据经发热量矫正后所得斜率为0.094,处于0.092—0.095之间,说明该检测-分析方法可用于检测玉米秸秆同时与内蒙古煤、贵州煤进行混燃,其拟合优度R2=0.993,当玉米秸秆的混燃比为20%时误差分析为0.35‰,用于准确判断玉米秸秆与多种煤样混燃的掺烧比。图9d为山西煤和不同掺混比玉米秸秆混燃烟气经AMS测试所得14C/12C值与热值基掺混比拟合线性方程,拟合优度R2=0.989,当热值基生物质掺烧比为20%时,误差分析为8.7%,因此采用该线性关系判定生物质掺烧比存在较大的不准确性。
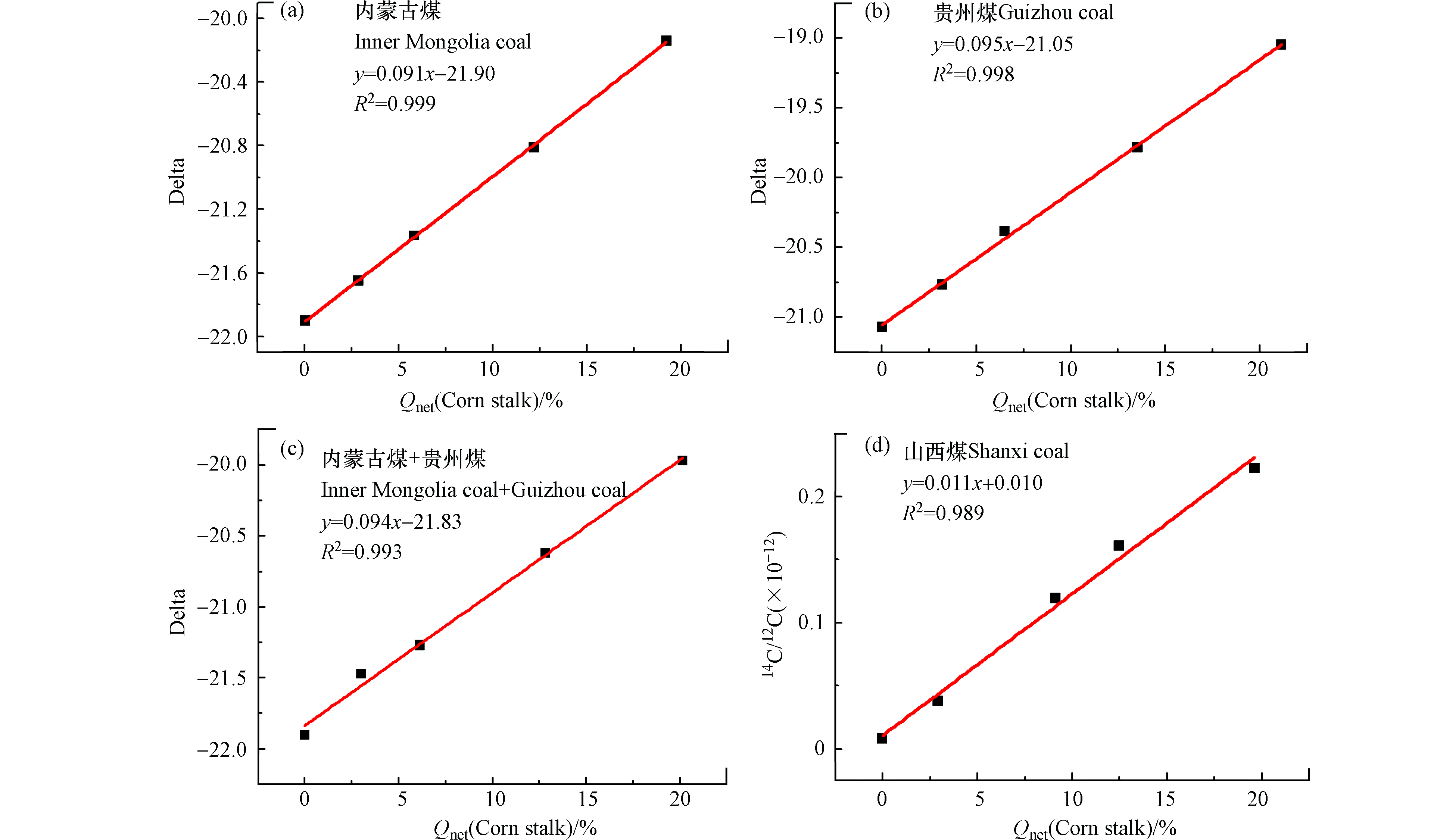 图 9 玉米秸秆与单一煤样或两种煤样混燃经发热量矫正后所得线性方程Figure 9. The linear regression equations of co-combustion cornstalks The linear regression equations of co-combustion cornstalks and various coal samples based on Qnet(a)内蒙古煤,(b)贵州煤,(c)内蒙古煤+贵州煤;(d)山西煤与玉米秸秆混燃烟气经AMS测定14C/12C值判定掺烧比的热值基线性方程(a) Inner Mongolia coal, (b) Guizhou coal, (c) Inner Mongolia coal + Guizhou coal. The linear regression equations of 14C detection for co-combustion Shanxi coal and cornstalks based on Qnet (d)
图 9 玉米秸秆与单一煤样或两种煤样混燃经发热量矫正后所得线性方程Figure 9. The linear regression equations of co-combustion cornstalks The linear regression equations of co-combustion cornstalks and various coal samples based on Qnet(a)内蒙古煤,(b)贵州煤,(c)内蒙古煤+贵州煤;(d)山西煤与玉米秸秆混燃烟气经AMS测定14C/12C值判定掺烧比的热值基线性方程(a) Inner Mongolia coal, (b) Guizhou coal, (c) Inner Mongolia coal + Guizhou coal. The linear regression equations of 14C detection for co-combustion Shanxi coal and cornstalks based on Qnet (d)由经发热量矫正后数据可知,本文中采用碳同位素中红外激光检测设备测试碳同位素比值用于判断生物质的掺烧比是准确可靠的,发热量矫正后所得线性方程可直接为电厂核定生物质掺烧比与发电量之间的关系提供重要参考数据。
2.4 含水量对碳同位素比值(δ13C)的影响
为了进一步证明该碳同位素中红外激光检测设备不受水分含量的影响,设计了一系列关于不同含水量的实验,见表3。
表 3 燃料样品不同含水量的δ13C值Table 3. The δ13C values of fuel samples with different water contents样品 Sample 煤量/g Coal 生物质量/g Biomass 加水量/% Water δ13C值 δ13C value 山西煤 0.50 0 20 -20.60 山西煤 0.50 0 50 -20.61 贵州煤 0.50 0 20 -21.02 贵州煤 0.50 0 50 -21.01 内蒙古煤 0.50 0 20 -21.91 内蒙古煤 0.50 0 50 -21.92 玉米秸秆 0 0.50 20 -11.90 玉米秸秆 0 0.50 50 -11.92 木屑 0 0.50 20 -26.98 木屑 0 0.50 50 -26.97 棉花秸秆 0 0.50 20 -26.08 棉花秸秆 0 0.50 50 -26.09 稻壳 0 0.50 20 -27.01 稻壳 0 0.50 50 -27.02 | Show TableDownLoad:
CSV
表3为实验中向燃料中添加不同量水分后进行充分燃烧,所得烟气经碳同位素激光检测设备在线分析烟气的δ13C值,对比燃料添加不同量水分及表2中各燃料的δ13C值,发现添加水分对δ13C值几乎没有影响,说明该碳同位素中红外激光检测设备不受含水量的影响,生物值含水量不会对碳同位素值的检测造成干扰。若加水量过多会导致未经矫正的原始数据拟合线性关系式较差,同样也可以从线性拟合关系式中反映出所混燃生物质是否含有大量水分或掺混其它非生物质的物质,可为电厂准确判定生物质掺烧比及真实性提供重要的数据支撑。
2.5 燃料碳同位素比值(δ13C)与燃烧过程的关系
不同燃料的燃烧过程均具有差异性,碳同位素中红外激光检测设备可在线监测燃料燃烧过程中碳同位素比值的变化情况,从而推测燃料的燃烧进程及燃烧完全所使用的时间,图10为碳同位素中红外激光检测设备在线监测山西煤及不同生物质燃烧过程的δ13C值变化情况。
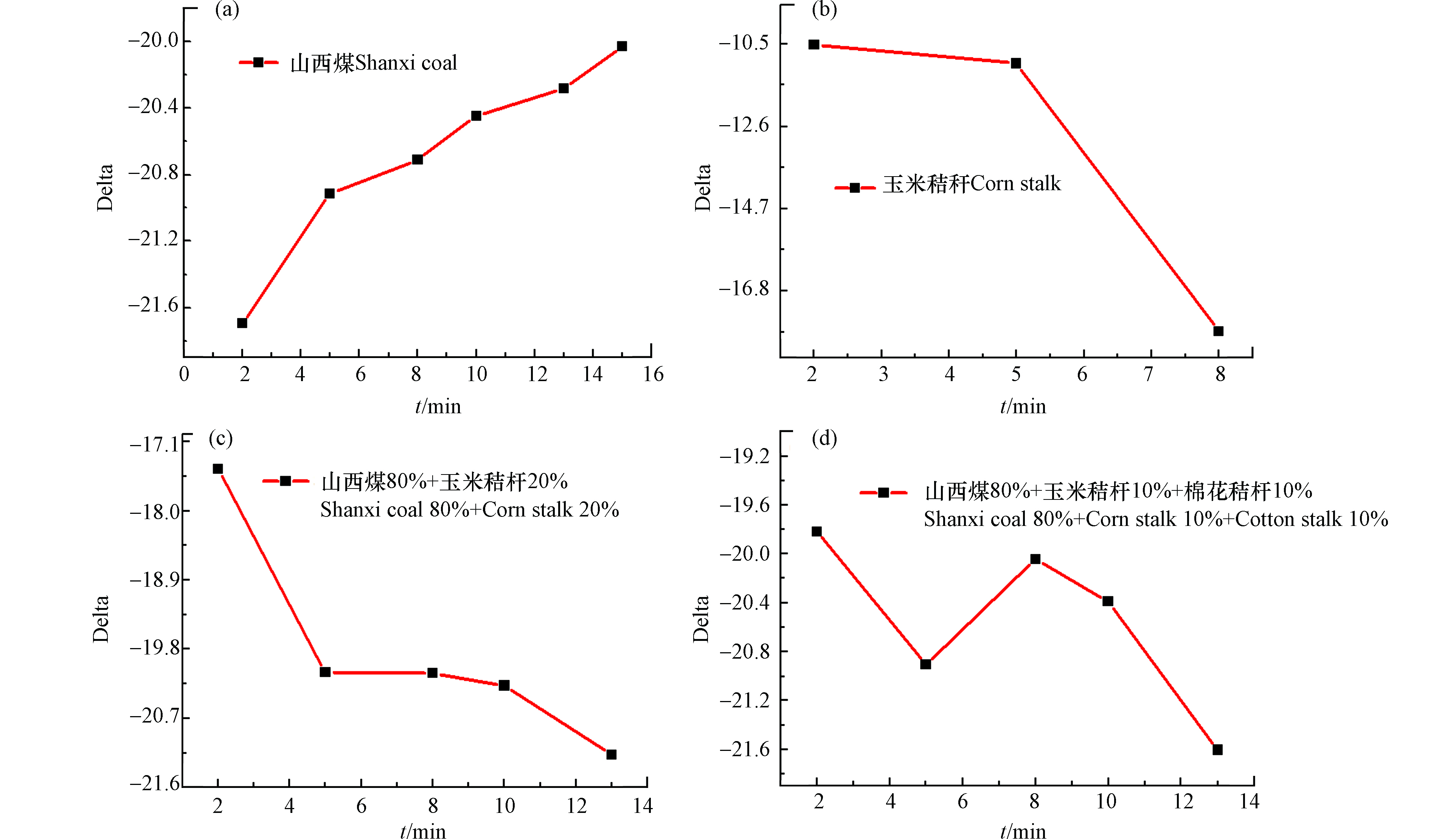 图 10 燃料的碳同位素比值δ13C与燃烧时间的关系Figure 10. The changes in the delta value in the combustion processes of Shanxi coal and different biomass samples(a)山西煤,(b)玉米秸秆,(c)山西煤+玉米秸秆,(d)山西煤+玉米秸秆+棉花秸秆(A) Shanxi coal, (B) Cornstalks, (C) Shanxi coal+Cornstalks, (D) Shanxi coal+cornstalks+cotton stalks
图 10 燃料的碳同位素比值δ13C与燃烧时间的关系Figure 10. The changes in the delta value in the combustion processes of Shanxi coal and different biomass samples(a)山西煤,(b)玉米秸秆,(c)山西煤+玉米秸秆,(d)山西煤+玉米秸秆+棉花秸秆(A) Shanxi coal, (B) Cornstalks, (C) Shanxi coal+Cornstalks, (D) Shanxi coal+cornstalks+cotton stalks图10a为山西煤燃烧过程的δ13C值变化过程,由图中曲线可知开始前2 min内,山西煤燃烧过程比较迅速,释放较多的CO2气体,δ13C值低于-21.61;燃烧5 min后,所得烟气的δ13C值变化缓慢,说明山西煤已基本燃烧完毕,所需时间大概为13 min。图10b为玉米秸秆的燃烧过程,由于玉米秸秆属于生物质类,其燃烧特性优于煤样,燃烧2 min所测得δ13C值接近-10.60,燃烧5 min后,δ13C值接近-11.61;继续进行燃烧时,其δ13C值出现明显下降,说明玉米秸秆在5 min内几乎燃烧完全;图10c为山西煤(80%)和玉米秸秆(20%)混燃过程中δ13C值随燃烧时间的变化过程,由图中曲线可知,前2 min内所测得δ13C值为-17.50,此δ13C值为玉米秸秆和山西煤混燃释放CO2所含稳定碳同位素13C,由于生物质具有优于煤样燃烧的性质,推测前2 min内玉米秸秆优先燃烧,山西煤部分燃烧。随后,δ13C值在5 min内出现明显降低为-19.50,主要为剩余山西煤燃烧释放CO2所含稳定碳同位素13C;后续燃烧过程中δ13C值变化不明显。以上变化过程说明山西煤和玉米秸秆混燃时,由于生物质碳含量较煤样低,可快速将所含碳燃尽释放CO2,因此可推测玉米秸秆燃烧速率优于山西煤。图10d为山西煤同时与玉米秸秆、棉花秸秆混燃过程的δ13C值变化情况,由图中曲线可知开始2 min内所得δ13C值为-19.81,依据实验所测得玉米秸秆、棉花秸秆的δ13C值推测此过程可能为玉米秸秆优先燃烧,而棉花秸秆和山西煤部分燃烧;燃烧5 min时δ13C值出现明显下降为-21.0,依据棉花秸秆的δ13C值为-26.10推测此过程可能主要为所剩棉花秸秆的燃烧;随后燃烧10 min时,δ13C值上升为-20.61,此值接近山西煤的δ13C值,推测此过程可能主要为所剩山西煤的燃烧。此曲线变化情况反映了多种生物质混燃时由于其性质的不同,出现先后燃烧的情况。由于玉米秸秆和棉花秸秆的δ13C值有较大区别,因此混燃过程中所得δ13C值会出现上下跳跃的情况,不易得到准确的线性关系,解释了玉米秸秆不能与其他生物质混燃的直接原因。
通过在线监测煤样和生物质的混燃过程可知生物质的燃烧特性优于煤样燃烧特性,主要由于生物质的着火点比煤低很多,挥发分含量多,能够在短时间内迅速燃烧释放出CO2。在电厂实际发电过程中,燃料需要不断输入到锅炉内进行燃烧,采用碳同位素激光检测设备在线监测烟气中所含CO2的δ13C值可能是上下稍微浮动的数据点,因此需要多次采集数据点,能更准确的判断生物质的真实掺烧比.
3. 结论(Conclusion)
本文采用碳同位素中红外激光检测设备在线监测煤耦合生物质发电的混燃比,该检测精度较高,所得数据线性拟合关系较好,判定生物质掺烧比误差较小,且该设备与市场现有AMS设备相比成本低,具有经济竞争力。所得δ13C值经碳基量矫正后可得到CO2释放量与生物质掺烧比的真实函数关系,因此该分析方法不仅可为发电厂判断生物质掺烧比提供可靠的技术支撑,还可为国家目前急需发展的碳交易市场及碳排放监测提供衡量标准,从而推动我国碳交易市场的健康发展。同时经发热量矫正的拟合曲线可为电厂掺烧生物质所提供发电量提供重要数据,可协助电厂辨别生物质中是否添加过多水分或添加其他非生物质的物质。该分析方法正处于由实验室规模向中试示范装置转换的阶段,以进一步验证该分析方法的可行性及准确性。未来工作中需结合电厂实际监测环境对该分析方法进行集成开发,并进一步增加分析设备的连续分析时间和寿命,降低使用成本,以便后续在电厂中进行该技术的推广和应用。
-
图 1 小型煤耦合生物质混燃在线检测系统
Figure 1. On-line detection system for small-scale co-combustion of coal and biomass
图 3 山西煤与单一或两种生物质混燃所得线性回归方程
Figure 3. The linear regression equations of co-combustion Shanxi coal and various biomass samples
图 5 山西煤与单一或多种生物质混燃的碳基掺烧比线性方程
Figure 5. The linear regression equations of co-combustion Shanxi coal and various biomass samples based on carbon content (a,b) cornstalks, (c) rich hucks, (d) cotton stalks, (e) wood
图 4 玉米秸秆与一种或两种煤混燃所得线性方程
Figure 4. The linear regression equations of co-combustion cornstalks and various coal samples corresponding to mass content (a) Inner Mongolia coal, (b) Guizhou coal, (c) Shanxi coal+ Guizhou coal, (d) Inner Mongolia coal+Guizhou coal
图 6 玉米秸秆与一种或两种煤混燃的碳基掺烧比线性方程
Figure 6. The linear regression equations of co-combustion cornstalks and various coal samples based on carbon content
图 7 山西煤与玉米秸秆混燃 (a)加速器质谱测14C/12C值判定生物质混燃比线性方程,(b)SO2浓度测试
Figure 7. The linear regression equations of co-combustion Shanxi coal and cornstalks based on carbon content (a) 14C detection, (b) SO2 concentration
图 8 山西煤与一种或多种生物质混燃经发热量矫正所得线性方程
Figure 8. The linear regression equations of co-combustion Shanxi coal and various biomass samples based on Qnet
图 9 玉米秸秆与单一煤样或两种煤样混燃经发热量矫正后所得线性方程
Figure 9. The linear regression equations of co-combustion cornstalks The linear regression equations of co-combustion cornstalks and various coal samples based on Qnet
图 10 燃料的碳同位素比值δ13C与燃烧时间的关系
Figure 10. The changes in the delta value in the combustion processes of Shanxi coal and different biomass samples
表 1 煤和生物质工业分析、元素分析和热值测定
Table 1. Industrial analysis, element analysis, and calorific value determination of coal and biomass
样品/组分Sample/component 内蒙古煤Inner Mongolia coal 贵州煤Guizhou coal 山西煤Shanxi coal 玉米秸秆Cornstalks 稻壳Rice husks 棉花秸秆Cotton stalks 木屑Wood chips 水分Mad/% 6.83 4.77 5.58 7.59 6.54 7.77 7.01 灰分Aad/% 2.07 11.81 3.76 7.76 12.74 2.50 1.9 挥发分Vad/% 30.00 30.52 28.90 67.53 62.90 70.86 76.33 固定碳FCad/% 61.10 52.90 61.76 17.12 17.83 18.88 14.76 碳Cad/% 73.56 66.23 73.85 41.24 40.39 44.77 45.47 氢Ht,ad% 4.91 4.16 4.60 6.13 5.61 6.30 6.43 氮Nad/% 0.98 0.91 1.08 1.49 0.49 1.18 0.40 全硫St,ad/% 0.59 0.21 0.20 0.21 0.11 0.14 0.08 氧Oad/% 11.82 12.44 11.55 36.43 34.86 38.21 39.49 空干基低位发热量Qnet,ad/(J·g−1) 27060 24030 26400 15030 14660 16140 16580 空干基高位发热量Qgr,ad/(J·g−1) 28080 24890 27350 16290 15810 17440 17900 干基高位发热量Qgr,d/(J·g−1) 30130 26130 28960 17630 16920 18910 19250 注:本文发热量矫正使用的为空干基低位发热量. Note: Low calorific value at air-dry basis was used in the calorific value correction in this study.
下载: 导出CSV
表 2 纯燃料样品的碳同位素比值(δ13C)
Table 2. The 13C proportion (δ13C) in pure coal and biomass samples
样品Sample 山西煤Shanxi coal 贵州煤Guizhou coal 内蒙古煤Inner Mongolia coal 玉米秸秆Cornstalks 棉花秸秆Cotton stalks 木屑Wood chips 稻壳Rice husks δ13C −20.61 −21.01 −21.91 −11.91 −26.09 −26.99 −27.02
下载: 导出CSV
表 3 燃料样品不同含水量的δ13C值
Table 3. The δ13C values of fuel samples with different water contents
样品 Sample 煤量/g Coal 生物质量/g Biomass 加水量/% Water δ13C值 δ13C value 山西煤 0.50 0 20 -20.60 山西煤 0.50 0 50 -20.61 贵州煤 0.50 0 20 -21.02 贵州煤 0.50 0 50 -21.01 内蒙古煤 0.50 0 20 -21.91 内蒙古煤 0.50 0 50 -21.92 玉米秸秆 0 0.50 20 -11.90 玉米秸秆 0 0.50 50 -11.92 木屑 0 0.50 20 -26.98 木屑 0 0.50 50 -26.97 棉花秸秆 0 0.50 20 -26.08 棉花秸秆 0 0.50 50 -26.09 稻壳 0 0.50 20 -27.01 稻壳 0 0.50 50 -27.02
下载: 导出CSV
-
[1] TANG Y X, LUO Z Y, YU C J, et al. Determination of biomass-coal blending ratio by 14C measurement in co-firing flue gas [J]. Journal of Zhejiang University-SCIENCE A, 2019, 20(7): 475-486. doi: 10.1631/jzus.A1900006 [2] AGBOR E, ZHANG X L, KUMAR A. A review of biomass co-firing in North America [J]. Renewable and Sustainable Energy Reviews, 2014, 40: 930-943. doi: 10.1016/j.rser.2014.07.195 [3] RODRÍGUEZ L S, BERMEJO MUÑOZ J M, ZAMBON A, et al. Determination of the biomass content of end-of-life tyres[M]//Biomass Volume Estimation and Valorization for Energy. InTech, 2017 . [4] MOHN J, SZIDAT S, FELLNER J, et al. Determination of biogenic and fossil CO2 emitted by waste incineration based on 14CO2 and mass balances [J]. Bioresource Technology, 2008, 99(14): 6471-6479. doi: 10.1016/j.biortech.2007.11.042 [5] CONTRERAS M L, GANESH N, RODILLA I, et al. Assess of biomass co-firing under oxy-fuel conditions on Hg speciation and ash deposit formation [J]. Fuel, 2018, 215: 395-405. doi: 10.1016/j.fuel.2017.11.081 [6] MOHN J, SZIDAT S, ZEYER K, et al. Fossil and biogenic CO2 from waste incineration based on a yearlong radiocarbon study [J]. Waste Management, 2012, 32(8): 1516-1520. doi: 10.1016/j.wasman.2012.04.002 [7] STABER W, FLAMME S, FELLNER J. Methods for determining the biomass content of waste[J]. Waste Management & Research: the Journal for a Sustainable Circular Economy, 2008, 26(1): 78-87. [8] NORTON G A, DEVLIN S L. Determining the modern carbon content of biobased products using radiocarbon analysis [J]. Bioresource Technology, 2006, 97(16): 2084-2090. doi: 10.1016/j.biortech.2005.08.017 [9] CALCAGNILE L, QUARTA G, D’ELIA M, et al. Radiocarbon AMS determination of the biogenic component in CO2 emitted from waste incineration [J]. Nuclear Instruments and Methods in Physics Research Section B:Beam Interactions With Materials and Atoms, 2011, 269(24): 3158-3162. doi: 10.1016/j.nimb.2011.04.020 [10] QUARTA G, CICERI G, MARTINOTTI V, et al. Bringing AMS radiocarbon into the Anthropocene: Potential and drawbacks in the determination of the bio-fraction in industrial emissions and in carbon-based products [J]. Nuclear Instruments and Methods in Physics Research Section B:Beam Interactions With Materials and Atoms, 2015, 361: 521-525. doi: 10.1016/j.nimb.2015.01.058 [11] LU J Y, FU L L, LI X M, et al. Capture efficiency of coal/biomass co-combustion ash in an electrostatic field [J]. Particuology, 2018, 40: 80-87. doi: 10.1016/j.partic.2017.10.006 [12] QUARTA G, CALCAGNILE L, CIPRIANO D, et al. AMS-14C determination of the biogenic-fossil fractions in flue gases [J]. Radiocarbon, 2018, 60(5): 1327-1333. doi: 10.1017/RDC.2018.88 [13] VOGEL J S, NELSON D E, SOUTHON J R. 14C background levels in an accelerator mass spectrometry system [J]. Radiocarbon, 1987, 29(3): 323-333. doi: 10.1017/S0033822200043733 [14] LOWE D C. Problems associated with the use of coal as a source of 14C-free background material [J]. Radiocarbon, 1989, 31(2): 117-120. doi: 10.1017/S0033822200044775 [15] CRAIG H. Carbon 13 in plants and the relationships between carbon 13 and carbon 14 variations in nature [J]. The Journal of Geology, 1954, 62(2): 115-149. doi: 10.1086/626141 [16] COUSIN J, CHEN W D, FOURMENTIN M, et al. Laser spectroscopic monitoring of gas emission and measurements of the 13C/12C isotope ratio in CO2 from a wood-based combustion [J]. Journal of Quantitative Spectroscopy and Radiative Transfer, 2008, 109(1): 151-167. doi: 10.1016/j.jqsrt.2007.05.010 [17] FRIEDRICHS G, BOCK J, TEMPS F, et al. Toward continuous monitoring of seawater 13CO2/12CO2isotope ratio andpCO2: Performance of cavity ringdown spectroscopy and gas matrix effects [J]. Limnology and Oceanography:Methods, 2010, 8(10): 539-551. doi: 10.4319/lom.2010.8.539 [18] WEIDMANN D, WYSOCKI G, OPPENHEIMER C, et al. Development of a compact quantum cascade laser spectrometer for field measurements of CO2 isotopes [J]. Applied Physics B, 2005, 80(2): 255-260. doi: 10.1007/s00340-004-1639-7 [19] GHOSH P, BRAND W A. Stable isotope ratio mass spectrometry in global climate change research [J]. International Journal of Mass Spectrometry, 2003, 228(1): 1-33. doi: 10.1016/S1387-3806(03)00289-6 [20] BECKER J F, SAUKE T B, LOEWENSTEIN M. Stable isotope analysis using tunable diode laser spectroscopy [J]. Applied Optics, 1992, 31(12): 1921. doi: 10.1364/AO.31.001921 [21] TANS P P, BERRY J A, KEELING R F. Oceanic 13C/12C observations: A new window on ocean CO2 uptake [J]. Global Biogeochemical Cycles, 1993, 7(2): 353-368. doi: 10.1029/93GB00053 [22] GAGLIARDI G, CASTRILLO A, IANNONE R Q, et al. High-precision determination of the 13CO2/ 12CO2 isotope ratio using a portable 2.008-μm diode-laser spectrometer [J]. Applied Physics B, 2003, 77(1): 119-124. doi: 10.1007/s00340-003-1240-5 [23] SCHULZE B, WIRTH C, LINKE P, et al. Laser ablation-combustion-GC-IRMS—a new method for online analysis of intra-annual variation of δ13C in tree rings [J]. Tree Physiology, 2004, 24(11): 1193-1201. doi: 10.1093/treephys/24.11.1193 [24] CASTRILLO A, CASA G, KERSTEL E, et al. Diode laser absorption spectrometry for 13CO2/ 12CO2 isotope ratio analysis: Investigation on precision and accuracy levels [J]. Applied Physics B, 2005, 81(6): 863-869. doi: 10.1007/s00340-005-1949-4 [25] MOOK W G, van der PLICHT J. Reporting 14C activities and concentrations [J]. Radiocarbon, 1999, 41(3): 227-239. doi: 10.1017/S0033822200057106 [26] MURNICK D E, PEER B J. Laser-based analysis of carbon isotope ratios [J]. Science, 1994, 263(5149): 945-947. doi: 10.1126/science.8310291 [27] MCMANUS J B, ZAHNISER M S, NELSON D D, et al. Infrared laser spectrometer with balanced absorption for measurement of isotopic ratios of carbon gases [J]. Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy, 2002, 58(11): 2465-2479. doi: 10.1016/S1386-1425(02)00064-1 [28] ALLARD P, MAIORANI A, TEDESCO D, et al. Isotopic study of the origin of sulfur and carbon in Solfatara fumaroles, Campi Flegrei caldera[J]. Journal of Volcanology and Geothermal Research, 1991, 48(1/2): 139-159. 期刊类型引用(2)
1. 张世鑫,赵云增,许燕飞,郭涛,江建忠,曹林涛,邓磊. 流化床锅炉多源废弃物耦合利用技术现状. 节能技术. 2024(02): 186-192 .  百度学术
百度学术
2. 王睿思,孙堂磊,杨延涛,刘鹏,雷廷宙. 基于AMS的~(14)C法对生物质与煤混燃掺烧比测定的研究进展. 林产工业. 2023(10): 33-39 . 百度学术
其他类型引用(1)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2665
- HTML全文浏览数: 2665
- PDF下载数: 100
- 施引文献: 3
