



-
二氧化碳(CO2)和甲烷(CH4)是大气中最重要的两种温室气体,对全球总辐射强迫的贡献占所有长寿命温室气体的80%以上[1-2]。工业革命以来,受人为活动持续影响,大气中CO2和CH4含量迅速上升。世界气象组织全球大气观测网(World Meteorological Organization, Global Atmospheric Watch, WMO/GAW)的公报显示,2019年全球大气中CO2和CH4物质的量比年均值分别达410.50×10−6和1877.0×10−9,比工业革命前(1750年)分别增长了48%和160%[2]。大气中温室气体含量的持续上升导致全球变暖、海洋酸化和海平面上升等一系列气候与环境问题[3]。国内外众多学者对大气CO2和CH4等温室气体含量进行了大量观测研究[4-7],为探究温室气体源汇过程,评估和预测全球及区域尺度气候与环境变化提供了基础数据。
由于源汇和输送过程的复杂多样化,大气CO2和CH4呈明显的时空分布差异[5]。高精度的定点观测是掌握其特征的最有效、最直接的方式之一。全球最早的地面大气CO2观测始于1957年的美国夏威夷莫纳罗亚(Mauna Loa)站,迄今已持续60多年[8-9]。目前WMO/GAW已基于全球100多个国家的400多个大气本底站构建了可代表不同区域特征的温室气体观测网,为解析全球温室气体时空分布特征和源汇格局提供了数据支持[6,10-12]。
1991年起,我国青海瓦里关、北京上甸子、浙江临安和黑龙江龙凤山本底站先后加入WMO/GAW观测网,并陆续开展温室气体含量的高精度观测[13]。随着技术发展,大气CO2和CH4观测技术已由传统的气相色谱法或非色散红外法升级为以腔增强技术为基础的光学探测方法[14],其中具有代表性的包括波长扫描光腔衰荡光谱分析技术(wave scan cavity ring-down spectroscopy, WS-CRDS)和离轴积分腔输出光谱技术(off-axis integrated cell output spectroscopy,OA-ICOS)。为满足 “碳达峰,碳中和”国家战略实施需求,除WMO/GAW本底站外,我国各地正陆续开展基于光学技术的温室气体高精度在线连续观测,但所用仪器几乎完全依赖从美国Picarro公司或Los Gatos Research公司进口[15-18]。
为突破我国温室气体观测卡脖子核心技术,国内相关团队已基于OA-ICOS技术研发出商业化的高精度温室气体分析仪(由北京唯思德科技有限公司推广,GGA-311 CO2/CH4/H2O分析仪)[19-20]。本研究利用该国产设备,与目前国内外广泛使用的美国Picarro公司G-2401型高精度CO2/CH4/CO/H2O分析仪[15]和Agilent公司7890B型气相色谱仪[21-22]进行比对测试,通过精密度和线性等关键性能的针对性分析及野外站点(青海瓦里关全球大气本底站)试运行比对观测,结合WMO/GAW对本底温室气体观测质控要求,评估该国产高精度温室气体分析仪的基本性能指标,为我国建立大范围温室气体监测网络提供支撑。
-
GGA-311 CO2/CH4/H2O分析仪基于OA-ICOS技术,其利用近红外激光实时测量分子的光谱特征,使激光在光学谐振腔内多次反射(有效光程可达13 km)[23-24]。如图1所示,GGA-311 CO2/CH4/H2O分析仪主要由光学系统和电学系统组成,其中光学系统包括光学谐振腔、激光器、高反镜和光电二极管等;电学系统包括温度-压力输出模块、温度-压力控制模块和采集模块等。GGA-311 CO2/CH4/H2O分析仪主要采用两路近红外激光器作为光源,并利用时分复用的方法对两路激光进行区分。激光器驱动板控制激光器的温度和电流使两束激光同时进入光学谐振腔。光束在光学谐振腔内多次反射,大幅增加了有效吸收光程后出射。出射光经过聚焦透镜聚焦后,由光电探测器进行光信号采集,并转换为电信号输入至数据采集模块进行物质的量比计算。
-
GGA-311型分析仪测量过程中,只需设定进样时间和气体流量两个参数。与传统的气相色谱法和非色散红外法相比,该仪器具有响应频率和灵敏度高、不需要使用助燃气与载气、受环境因素(温度、气压等)影响更小、操作简便且维护成本低等优点[25]。
为评估GGA-311型分析仪性能,将其与目前国内使用广泛的进口型Picarro分析仪(G-2401)和气相色谱仪进行比对。由于气相色谱仪与GGA-311型分析仪和Picarro G-2401型分析仪原理不同(后两者均是基于光学方法),因此首先将GGA-311型分析仪和Picarro G-2401型分析仪同时接入标准气体(标气)开展测试。测试过程中,气体样品由高精度质量流量控制器(D07,北京七星华创流量计有限公司)控制流量(300 mL·min−1),经三通接头,分别进入GGA-311型分析仪和Picarro G-2401型分析仪(图2),完成同步测定后,经后置气泵排出。为避免进气不稳定和响应频率差异的影响,每瓶标气各进样测试30 min。通过对测试结果的比对分析,评估2台分析仪测定CO2和CH4的精密度和线性等基本性能。
搭配氢火焰离子化检测器的气相色谱系统(GC-FID)是广泛用于大气CO2和CH4观测的传统方法,在我国也有大范围的使用[26]。其原理:经除水后的样气由色谱柱分离,CH4先到达火焰离子化(FID)检测器,而CO2经镍催化炉转化为CH4后,再进入FID检测器。本研究所用气相色谱系统基于原“双通道气相色谱系统”[14]改进升级,可同步测定CO2、CH4、CO、N2O和SF6 的5个组分(图3),比对分析时,气相色谱系统也采用同一套标气接入系统测试。
本研究测试所用标气均以干洁空气为底气,存储于0.029 m3铝合金瓶 (美国Scott-Marrin公司),经多轮标校,可溯源至WMO/GAW一级标准(Primary Standard),测试标气CO2和CH4物质的量比如表1所示。
-
为进一步评估GGA-311型分析仪的性能,将其置于青海瓦里关全球大气本底站(36.17°N,100.54°E,海拔3810 m)开展比对观测试验。该站是WMO/GAW 在全球的31个大气本底基准观测站之一,安装有满足WMO/GAW观测质控标准的基于光腔衰荡光谱分析技术的Picarro G-2401 CO2/CH4高精度分析仪[27-28]。比对观测期间,为保证2台分析仪所测气体样品的实时一致,2台分析仪利用同一套进气和样品预处理单元,且将待测气体分为并联气路,分别接入GGA-311型分析仪和Picarro G-2401型分析仪.
-
为比较Picarro G-2401型分析仪、改进升级的“双通道气相色谱系统”和GGA-311型分析仪的精密度,选择摩尔比覆盖本底浓度变化范围的标气③④⑤接入测试系统,以响应值标准偏差(Standard Deviation,1σ)表示系统精密度。
如表2所示,Picarro G-2401型分析仪、GGA-311型分析仪和改进升级的“双通道气相色谱系统”对3瓶标气CO2测定结果的标准偏差范围分别为0.02×10−6、(0.10—0.18)×10−6和(0.30—0.64)×10−6,平均值分别为0.02×10−6、0.15×10−6和0.43×10−6。根据WMO/GAW实验室间比对分析质控标准的扩展目标(CO2: ±0.20×10−6)[2]可得,GGA-311型分析仪对标气③④⑤的CO2响应结果的精密度符合质控标准的扩展目标且优于改进升级的“双通道气相色谱系统”,但与Picarro G-2401型分析仪测定结果的精密度尚存在一定差距,主要由于GGA-311型分析仪采用两面高反射率镜片的增强腔,且因国产工艺等原因,其光腔内镜片反射率略低于Picarro G-2401型分析仪(99.999%以上)[29],其有效光程(约13 km)明显低于Picarro G-2401型分析仪的有效光程(16—20 km),导致其精密度稍逊于Picarro G-2401型分析仪。
如表3所示,Picarro G-2401型分析仪、GGA-311型分析仪和改进升级的“双通道气相色谱系统”对标气③④⑤的CH4测定结果的标准偏差范围分别为0.1×10−9、(1.2—1.3)×10−9和(3.0—3.4)×10−9,平均值分别为0.1×10−9、1.2×10−9和3.2×10−9。根据WMO/GAW实验室间比对分析质控标准(CH4: ±2.0×10−9)及其扩展目标(CH4: ±4.0×10−9)[30]可得,虽GGA-311型分析仪对3瓶标气的CH4响应结果与Picarro G-2401型分析仪测定结果的精密度仍存在一定差距,但满足WMO/GAW实验室间比对分析质控标准及其扩展目标,且优于改进升级的“双通道气相色谱系统”。总体而言,Picarro G-2401型分析仪和GGA-311型分析仪对标气CO2和CH4测试结果的精密度均满足WMO/GAW实验室间比对分析质控要求。
-
为明确几种仪器对CO2/CH4的线性响应性能[30],将标气①—⑤依次接入Picarro G-2401型分析仪和GGA-311型分析仪,另将标气②—⑤依次接入改进升级的“双通道气相色谱系统”(标气①因故未能参加测试),开展进样测试。为充分冲洗气路并获取足够用于统计分析的响应数据[31],Picarro G-2401型分析仪和GGA-311型分析仪对每瓶标气连续测试时间不少于30 min;改进升级的“双通道气相色谱系统”每瓶标气进样次数为35次以上。
3种分析仪对以上标气序列的响应结果如图4所示,结果显示,Picarro G-2401型分析仪、GGA-311型分析仪和改进升级的“双通道气相色谱系统”对CO2的一次线性拟合[14]相关系数(R2)分别为0.9999996、0.99998和0.9997,其中Picarro G-2401型分析仪、GGA-311型分析仪对CO2的二次线性拟合相关系数(R2)分别为0.9999996和0.999993;而对CH4的一次线性拟合[14]相关系数(R2)分别为0.999999、0.9993和0.99994,Picarro G-2401型分析仪、GGA-311型分析仪对CH4的二次线性拟合相关系数(R2)分别为0.9999993和0.99996。此外,Picarro G-2401型分析仪对CO2和CH4的一次线性拟合结果和二次线性拟合结果相近,且与文献报道结果保持一致[32]。相较于一次线性拟合的结果,GGA-311型分析仪对CO2和CH4的二次线性拟合结果明显较好,这主要由于基于激光原理的系统针对温室气体吸收一般是非线性,但Picarro G-2401型分析仪已基于长期测试结果在系统内部添加二次响应校正因子,而GGA-311型分析仪尚未加入校正因子。
进一步分析3台分析仪对标气响应的拟合残差[33](Picarro G-2401型分析仪和改进升级的“双通道气相色谱系统”对CO2和CH4进行一次线性拟合残差;GGA-311型分析仪对CO2和CH4进行二次线性拟合残差)。如图5所示,Picarro G-2401型分析仪对CO2物质的量比范围为(405.63—507.11)×10−6标气的残差测定范围为(−0.04—0.03)×10−6之内,对CH4物质的量比为(1988.7—2421.1)×10−9标气的残差测定范围为(−0.1—0.2)×10−9之内,CO2和CH4均在WMO/GAW规定的质控范围之内,表明Picarro G-2401型分析仪对5瓶标气的CO2和CH4的测试结果有良好的线性响应[34]。
GGA-311型分析仪对CO2物质的量比范围为(405.63—507.11)×10−6标气的残差测定范围为(−0.21—0.15)×10−6之内,对CH4物质的量比为(1988.7—2421.1)×10−9标气的残差测定范围为(−1.7—1.3)×10−9之内,CO2残差基本在WMO/GAW实验室间比对分析质控标准的扩展目标之内,而CH4残差均在WMO/GAW实验室间比对分析质控标准及其扩展目标之内,表明GGA-311型分析仪对5瓶标气的CO2和CH4的测试结果也有良好的线性响应。
改进升级的“双通道气相色谱系统”测试标气② — ⑤,其中CO2和CH4的测试结果残差范围分别为(-0.76—1.08)×10−6和(-1.5—1.9)×10−9,CH4均在WMO/GAW规定的范围之内,表明改进升级的“双通道气相色谱系统”对4瓶标气的CH4的测试结果有良好的线性响应,但针对CO2而言,因气相色谱系统通过镍转化炉将CO2转化为CH4,且大气CO2浓度比CH4高约200倍,因而在同一个FID检测器上,难以获得较高的精度。
比较而言,GGA-311型分析仪测试标气①—⑤的CO2测试结果与Picarro G-2401型分析仪的测试结果的残差相近,而改进升级的“双通道气相色谱系统”对标气CO2测试结果残差与另2台分析仪(Picarro G-2401型分析仪和GGA-311型分析仪)的结果差异较大。GGA-311型分析仪测试标气①—⑤CH4的测试结果与Picarro G-2401型分析仪的测试结果的残差存在一定差异,而改进升级的“双通道气相色谱系统”对标气CH4测试结果残差差于另2台分析仪。总体而言,GGA-311型分析仪可满足大气CO2和CH4的高精度连续在线观测[23]。
-
为测试和评估Picarro G-2401型分析仪和GGA-311型分析仪的实际观测性能,本研究利用2台分析仪分别在实验室和青海瓦里关大气本底站两种环境条件下开展比对观测研究。实验室比对测试期间,利用2台分析仪依次测定不少于30 min的标气⑥和不少于10 h的环境空气。根据响应值的校正结果,对比评估2台分析仪的模拟观测性能。如图6a所示,由于Picarro G-2401型分析仪和GGA-311型分析仪响应时间和光腔体积差异等原因,2台分析仪对标气⑥的CO2和CH4测定结果存在系统偏差,分别为(0.03±0.1)×10−6和(−3.8±0.6)×10−9。
而对环境空气(图6b)的同步测定则显示,2台分析仪对CO2和CH4测定结果总体趋势相近,但系统偏差分别达(6.57±0.50)×10−6和(102.1±2.5)×10−9,明显大于对标气中CO2和CH4测定结果的系统偏差。结合2台分析仪对标气⑥和实验室空气的差异发现:测试过程中,实验室环境空气未进行除水处理,而标气(底气为干洁空气)中几乎不含水汽(<30×10−6)。据文献报道,水汽可通过稀释和光谱效应影响光增强技术分析仪的测定结果[35-38]。因此,2台分析仪对相同标气和实验室空气测定结果偏差幅度的不同,主要原因可能是实验室空气中水汽导致的,因为Picarro系统中已经内置水汽校正因子[36],而国产仪器尚未加入此参数。由此也说明,在利用近红外光学法对大气CO2和CH4进行分析时,必须对气体进行严格干燥,才能保证较好的分析精度。
2021年5—6月,利用GGA-311型分析仪和Picarro G-2401型分析仪在青海瓦里关站开展为期1周的比对测试。另外需要说明的是由于2台分析仪响应时间不同(GGA-311型分析仪:约6.25 s;Picarro G-2401型分析仪:2 s),为了方便比较,将每小时内的所有物质的量比做平均运算,统一为小时平均值。如图7所示,2台分析仪对瓦里关站大气CO2物质的量比波动的测定结果基本一致,而对CH4物质的量比波动的测定结果存在细微误差。统计显示,2套系统对CO2和CH4系统偏差范围在±0.1×10−6和±2.0×10−9[2]的观测结果占总数据量的27.98%和47.62%;范围在±0.2×10−6和±4.0×10−9的观测结果占总数据量的51.79%和79.76%。导致2台分析仪比对测定结果的细小差异,尤其是大气CO2和CH4物质的量比剧烈波动时[30],主要也是因为两台仪器响应时间差异所致。
此外,瓦里关站进气预处理系统配置超低温冷阱(-60 ℃),可尽量减少待测空气样品中水汽(<30×10−6)的影响。2台分析仪比对测试结果的系统偏差也远小于实验室环境空气比对测定结果的系统偏差,因此进一步证实,水汽对GGA-311型分析仪的CO2和CH4测定存在一定影响。必要而严格的样气除水流程,可大幅降低2台分析仪比对测定结果的系统偏差,提高观测结果的可比性。
-
(1)GGA-311型分析仪对CO2和CH4的精密度测定结果均值分别为0.15×10−6和1.2×10−9,满足WMO/GAW实验室间比对分析质控标准的扩展目标。
(2)线性测试结果显示,GGA-311型分析仪对5瓶不同物质的量比的CO2和CH4标气的响应值与标称值之间线性相关系数(R2)分别为0.999993和0.99996,且二次线性拟合残差均在WMO/GAW实验室间比对分析质控标准的扩展目标之内,表明GGA-311型分析仪具有良好的线性响应。
(3)比对测试结果表明,GGA-311型分析仪和Picarro G-2401分析仪对CO2和CH4测试结果的波动基本一致,但受2台分析仪系统响应等因素影响,导致不同程度的系统偏差,其中系统偏差处于±0.20×10−6和±4.0×10−9范围内的数据分别占总数据量的51.79%和79.76%。整体而言,GGA-311型分析仪满足大气CO2和CH4高精度连续观测需求。
致谢:感谢青海瓦里关大气本底站业务人员的辛勤工作。
国产高精度温室气体分析仪性能评估
Evaluation on the domestic invented high precision greenhouse gas analyzer
-
摘要: 在国家碳中和战略实施背景下,我国将大范围开展温室气体高精度监测,而目前国内温室气体高精度分析仪几乎完全依赖进口。本研究针对国产GGA-311型高精度温室气体分析仪开展综合性能评估研究。结果显示,该分析仪对CO2和CH4的分析精密度分别达0.15×10−6(物质的量比)和1.2×10−9,达到WMO/GAW实验室间比对分析质控标准的扩展目标(CO2: ±0.2×10−6; CH4: ±4.0×10−9);其线性拟合相关系数(R2)分别为0.999993和0.99996。实验室和青海瓦里关全球大气本底站比对测试结果显示,该分析仪与进口Picarro G-2401型分析仪均能较好地捕捉本底CO2和CH4变化特征,但受水汽等因素影响,两套系统CO2和CH4偏差分别处于±0.2×10−6和±4.0×10−9范围内的数据占总数据的51.79%和79.76%,因此国产高精度温室气体分析仪在应用过程中,必须针对样气进行严格干燥,方能保证观测结果的可靠性。
-
关键词:
- 二氧化碳 /
- 甲烷 /
- 离轴积分腔输出光谱 /
- 波长扫描光腔衰荡光谱 /
- 观测
Abstract: In the context of the implementation of the national carbon neutral strategy, China will build up high-density greenhouse gases monitoring network at a large scale, while the current domestic high-precision greenhouse gas analyzers were dependent on imports. In this study, a systematic test was carried out for the domestic invented GGA-311 greenhouse gas high-precision analyzer and its applicability was evaluated. The results showed that the precision of the analyzer for CO2 and CH4 reached 0.15×10−6 (molar ratio) and 1.2×10−9, respectively, meeting the extended goals of WMO/GAW laboratory comparability goals (CO2: ±0.2×10−6; CH4: ± 4.0×10−9); The linear correlation coefficients (R2) were 0.999993 and 0.99996, respectively. Parallel observations at the laboratory and Waliguan atmospheric background station showed that GGA-311 analyzer and imported Picarro G-2401 can both well capture the characteristics of background CO2 and CH4 changes. However, due to the influence of factors such as water vapor, the deviations of CO2 and CH4 in the range of ±0.2×10−6 and ±4.0×10−9 for the two systems accounted for 51.79% and 79.76% of the total data, respectively. Therefore, the sample must be strictly dried to ensure the measurement quality when using the domestic invented GGA-311 greenhouse gas high-precision analyzer. -
目前,氰化法是最普遍的提金工艺[1],但过程中会产生大量的氰化废水,该类废水具有成分复杂、含盐量高、处理难度大等特点,因其含有大量的氰化物,直接排放会造成严重的污染问题[2-3]。在国内已经得到工业实践的方法有酸化法、碱性氯化法和SO2-空气法,但存在环境二次污染,处理成本高、过程难以控制、处理不达标等问题。其他的化学氧化法[4]、离子交换法[5]、化学沉淀法[6]等仍处于实验室阶段。因此,寻找一种短流程、高效低耗的处理方法,实现节能减排得到黄金冶炼企业的普遍关注。
电絮凝法常用处理城市污水与工业废水,此外,该方法还可用于重金属离子和无机离子的去除[7]。DAS等[8]采用集成臭氧辅助电絮凝法去除钢铁工业废水中的氰化物,在臭氧生成率1.33 mg·L−1、臭氧氧化时间40 min、电流密度100 A·m−2的和电解时间30 min的条件下,总氰质量浓度由150 mg·L−1降为0.1 mg·L−1,但电解过程中阳极易形成包裹极板表面的氧化膜,使极板与溶液接触面积减少,阻止阳极溶解,造成极板钝化[9],牺牲阳极在电解体系中不断被损耗,需要定期更换极板以确保反应正常进行[10]。电解氧化法是指利用直流电进行氧化还原反应,达到污染物分解的方法,常用于氰化废水[11-12]、电镀废水[13]的处理,研究表明,电解氧化法可以有效降解氰化废水中的金属氰络合物。LEI等[14]采用三维电化学体系处理氰化物废水,以煤基电极为主电极的三维电极体系,施加电压4 V,处理时间5 h,极板间距10 mm,活性炭颗粒用量2 g,CNT、Cu、Zn、CN−和SCN−的去除率分别为94.14%、94.53%、98.14%、98.55%和93.13%。曾鑫辉等[15]采用电解氧化法处理金矿废水,在电压6 V、pH=9、电解时间3 h、极板间距1.5 cm、NaCl添加量10 g·L−1的条件下,CNT、COD和铜氰络合物的去除率达到97.2%、96%和97.7%。Cl−的添加虽然有利于污染物的去除,但是处理后余氯浓度高,增加了环境风险。絮凝是指添加适当的絮凝剂使水或液体中悬浮微粒集聚变大,或形成絮团,从而加快粒子的聚沉,达到固-液分离的目的。常用于处理染料/纺织废水、农业废水、食品加工工业废水、纸浆和造纸废水、制革废水、垃圾渗滤液、废水中的重金属离子等[16]。聚硅酸铝铁是一种新型无机高分子絮凝剂,是在聚硅酸及传统铝盐、铁盐基础上发展起来的聚硅酸与金属盐的复合产物。该絮凝剂的大分子链上所带正电荷密度高,因絮凝剂溶解性、电中和及吸附架桥能力强、易制备、价格低廉等特点,成为无机高分子絮凝剂研究的热点[17]。许小洁等[18]研究了联合硅藻土与聚合氯化铝强化混凝对原水中重金属离子的去除,聚合氯化铝添加量为30 mg·L−1,硅藻土添加量为1.5 g·L−1时,重金属Cu、Pb和Cd的去除率分别达到57.5%、83.7%和22.2%。
针对电絮凝法存在阳极钝化,电化学氧化法存在电解质添加量大,处理后余氯浓度高,处理成本高等问题,制备绿色无氯絮凝剂,本研究采用絮凝-电解氧化联合工艺,研究了氰化废水处理过程中重金属离子和氰化物的去除规律及过程机理,以期为氰化提金废水的治理提供参考。
1. 材料与方法
1.1 实验原料
实验所用含氰废水来源于陕西某黄金冶炼厂,废水pH为9,其主要组成为 1 600.20 mg·L−1 CNT、− 439.74 mg·L−1 CN、257.35 mg·L−1 Cu、0.36 mg·L−1Fe、672.80 mg·L−1Zn。可以看出,此废水中CNT、CN−和Zn的含量均较高,其中CNT质量浓度大于800 mg·L−1,属于高浓度的氰化提金废水,Zn元素含量高达672.80 mg·L−1。所用其他化学试剂均为分析纯。
1.2 PSAF的制备
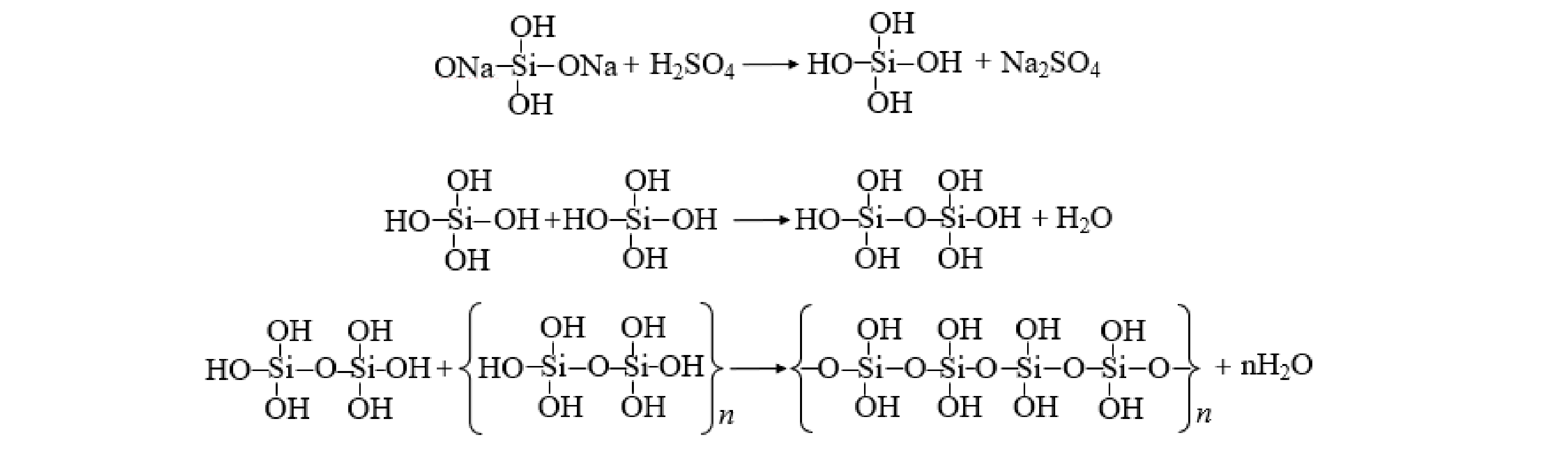
首先,向100 mL的0.5 mol·L−1的硅酸钠溶液中加入20%(体积比)硫酸溶液,磁力搅拌至溶液pH=2,所得溶液在室温下陈化30 min,得到聚硅酸 (PSA) ,反应过程如图1所示。其次,在聚硅酸中先加入1.297 g的Al2(SO4)3·18H2O粉末,然后连续搅拌10 min,再加入0.400 g的Fe2(SO4)3粉末。最后,将略显绿色透明溶液在30 ℃陈化24 h。
1.3 实验步骤
室温下,在150 mL烧杯中加入100 mL废水,使用硫酸或氢氧化钠调节pH,加入一定量的PSAF置于搅拌器,120 r·min−1快速搅拌30 s,再60 r·min−1慢速搅拌,静止一定时间,实验后固液分离,沉淀物采用去离子水洗至pH为7左右置于电热鼓风干燥箱中于60 ℃烘干。随后以钛板为阴极,石墨为阳极,采用一阴两阳三极板体系对过滤后的溶液进行电解氧化实验,在不同的电压、电解时间、极板间距下电解反应,实验后固液分离,对溶液取样分析,测定CNT、CN−、Cu、Zn离子的含量。
1.4 分析表征
采用硝酸银容量法(HJ484-2009)分析测定CN−、CNT的含量,采用原子吸收光谱仪分析测定Cu、Fe、Zn等金属离子的浓度,并根据式(1)计算各离子的去除率。采用傅立叶变换红外光谱(FTIR)分析絮凝样品,采用D/MAX2200型X射线衍射仪分析沉淀物,采用马尔文Zeta电位仪测定Zeta电位。
E=C0−CeC0×100% (1) 式中:E为各离子的去除率,%;C0为各离子的初始质量浓度,mg·L−1;Ce为絮凝或电解氧化处理后溶液中各离子的质量浓度,mg·L−1。
2. 结果与讨论
2.1 絮凝条件对去除率的影响
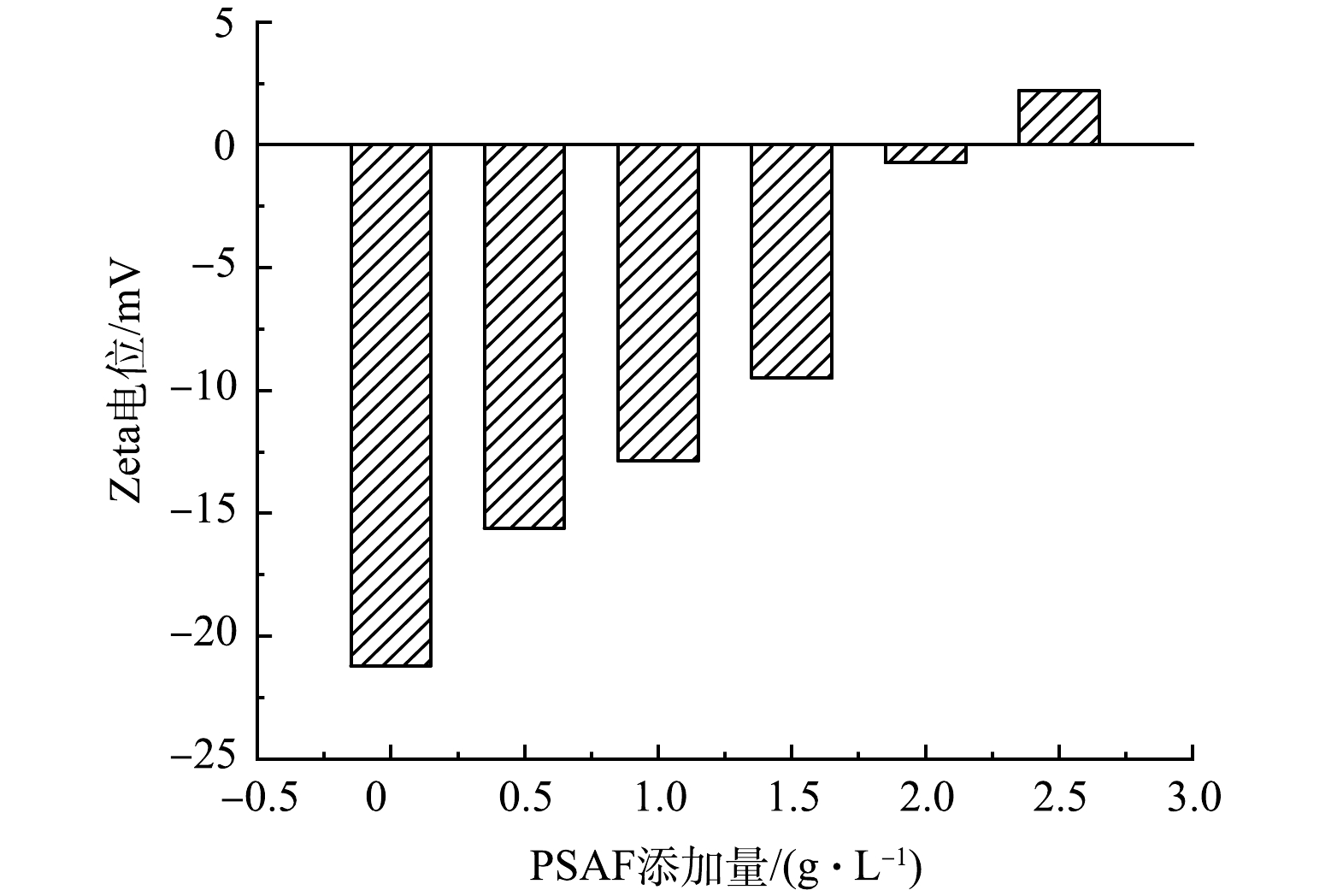
1) PSAF添加量的影响。常温条件下,取絮凝时间为30 min,pH为9,PSAF添加量分别为0.5、1、1.5、2、2.5 g·L−1进行絮凝实验,结果如图2所示。随着PSAF添加量的增加,废水中的CNT、CN−、Zn、Cu离子去除率先升高后趋于平缓,pH则先降低后趋于平缓,在PSAF添加量2 g·L−1时,CNT、CN−、Zn、Cu离子去除率均达到最大值,分别为43.9%、100%、85.14%、36.88%,pH为7。因为随着PSAF添加量的增加,PSAF与CN−、Zn(CN)42−、Cu(CN)32−发生的凝结点位增多,形成大量螯合絮体的网捕、扫卷作用增强,PSAF所带的正电荷中和了污染物所带的负电荷,使污染物失稳而达到去除效果[12],从而增加了CNT、CN−、Zn、Cu离子的去除率。
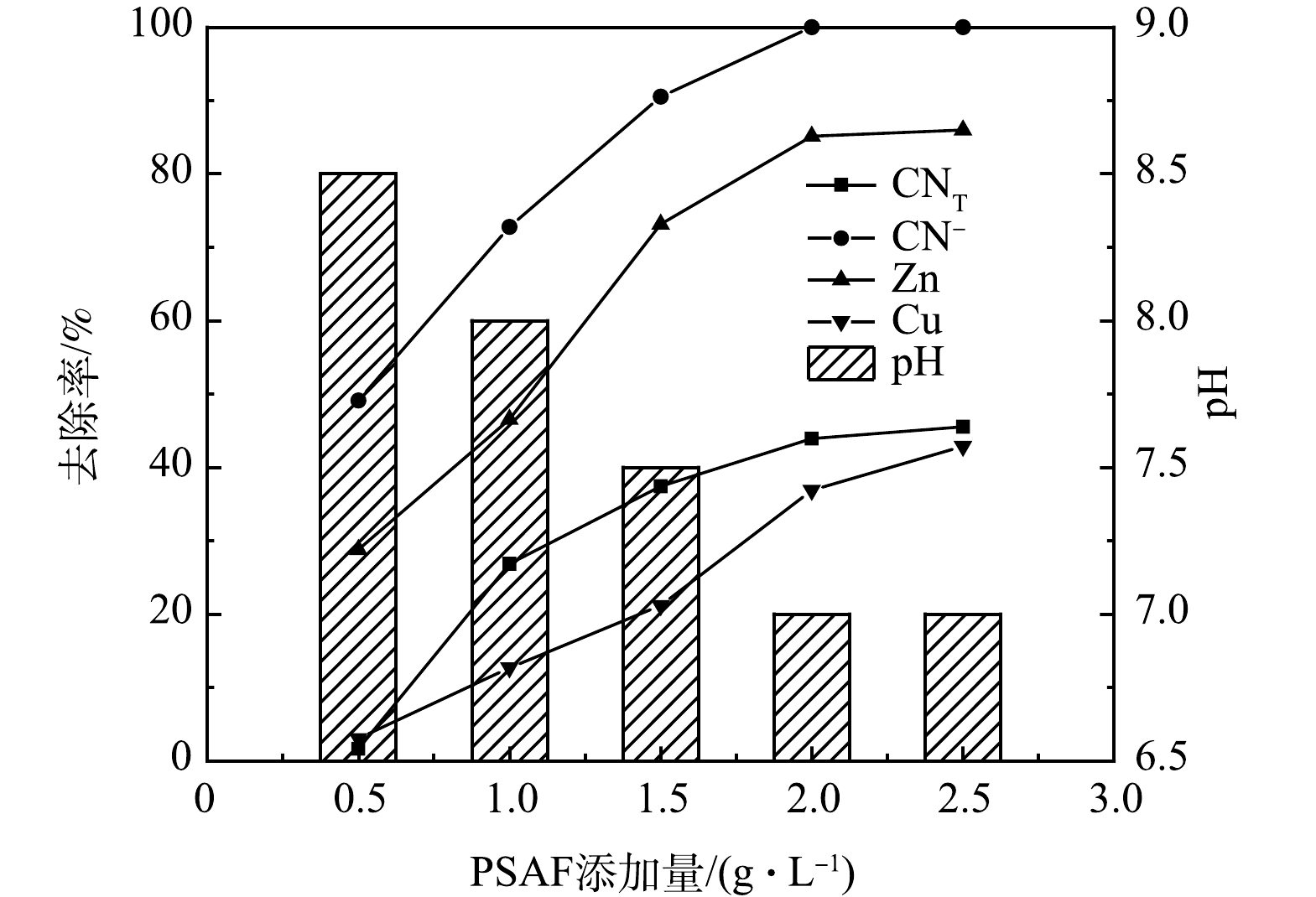 图 2 PSAF添加量对去除率的影响及pH的变化Figure 2. Effects of PSAF dosage on the removal rates and pH change
图 2 PSAF添加量对去除率的影响及pH的变化Figure 2. Effects of PSAF dosage on the removal rates and pH change为了研究不同PSAF添加量反应机制,进行了Zeta电位分析。有研究[19]表明,Zeta电位绝对值越小,即越趋近于0时,粒子之间斥力越小,越容易团聚。由图3可见,随着PSAF添加量的增加,Zeta电位不断增加,未经处理废水的Zeta电位为−21.2 mV,添加量增加到2.0 g·L−1时,Zeta电位为−0.72 mV。这是因为水解后产生的聚羟基络合物带正电荷,随着添加量的增加,正电荷数量不断增加,絮凝过程中双电层吸附及电中和能力增强,使得电位降低,趋近于零点时,颗粒凝聚且脱稳沉降[20]。在中性体系中,带负电荷的络合物在絮凝过程中能够吸附在氢氧化铁絮体表面。
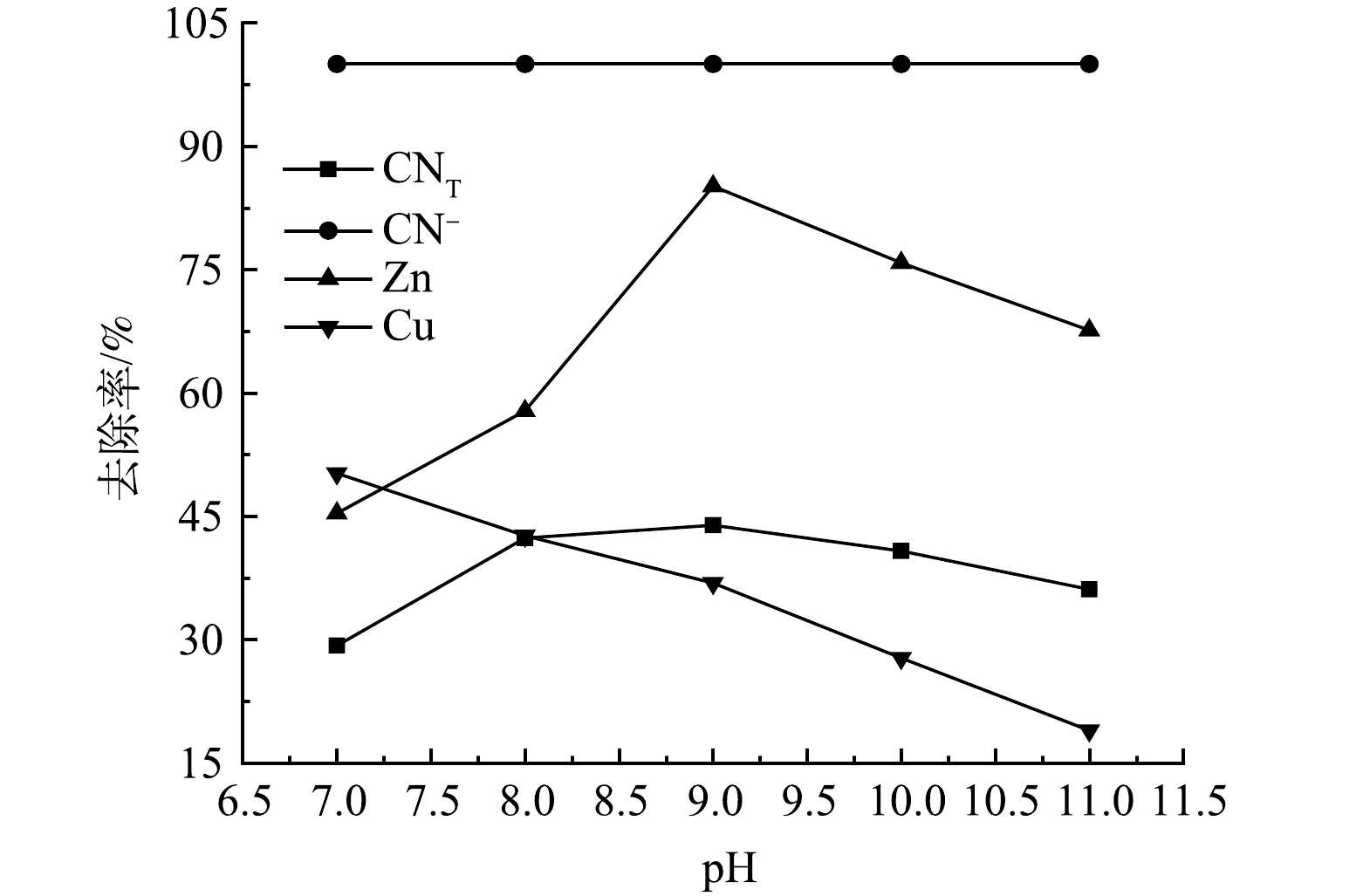
2)废水pH的影响。水中的H+和OH−均参与絮凝剂的水解反应,因此,pH可影响絮凝剂的水解速度、水解产物的存在形态和性能。在PSAF添加量为2 g·L−1,废水pH分别为7、8、9、10、11的条件下进行絮凝实验,结果如图4所示。可见,随着废水pH的增加,废水中的CN−去除率很快就达到了最大值,随后不再发生明显变化,而CNT、Zn离子去除率呈现先升高后降低的变化趋势,Cu离子去除率随着pH的增大而升高,当pH为9时,CNT、CN−、Zn、Cu离子去除率分别为43.9%、100%、85.14%、36.88%。这是因为随着pH的减小,H+会与PSAF水解的羟基络合阳离子Me(OH)2+、Me(OH)2+、Me2(OH)24+竞争,从而削弱带正电的PSAF和带负电的CN−、Cu(CN)32−、Zn(CN)42−之间的静电吸引力,因此,pH较低时不利于污染物的去除。但在强碱条件下,PSAF的大分子结构易被破坏,各物质的去除率会下降[15]。
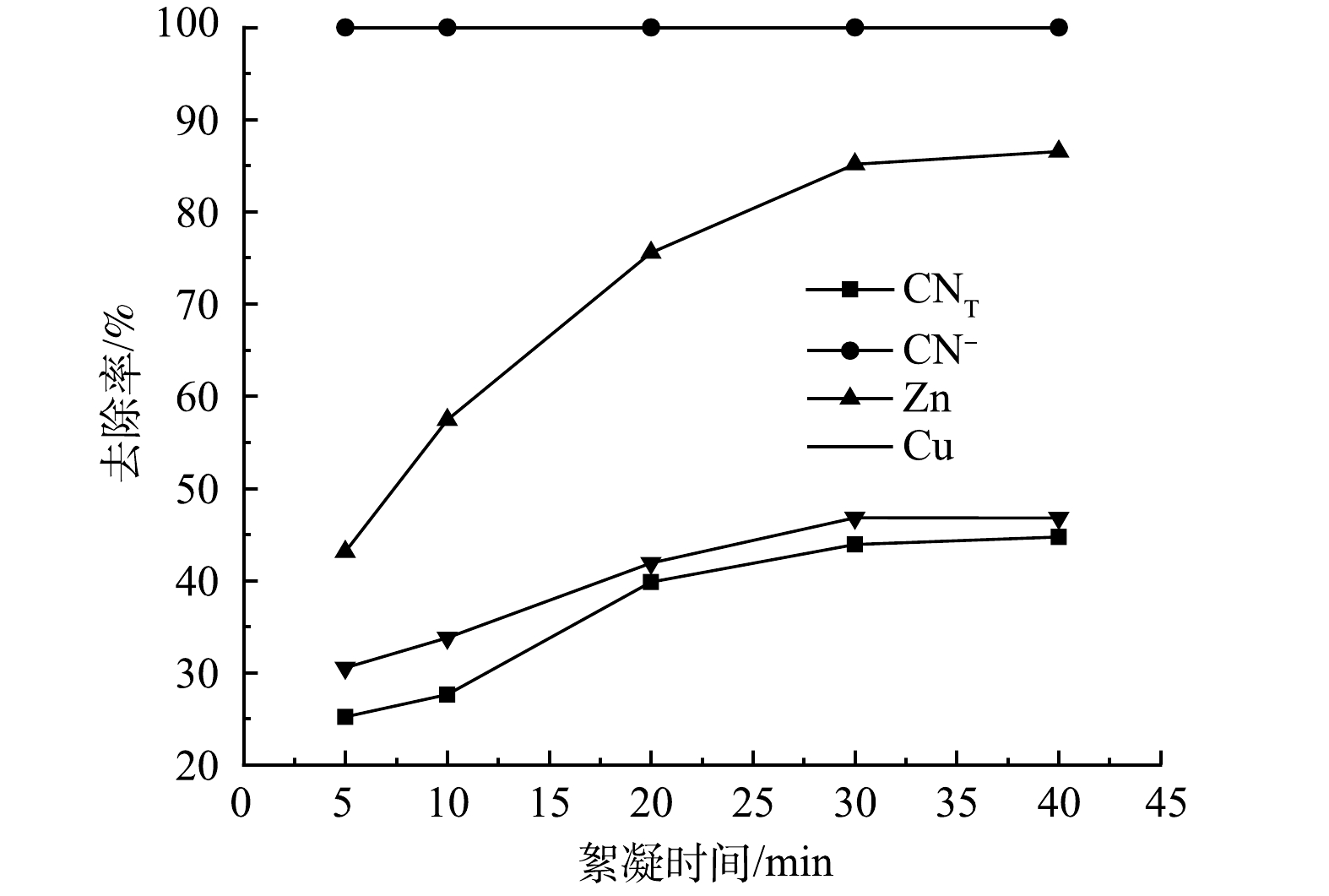
3) 絮凝时间的影响。在pH为9,絮凝时间分别为5、10、20、30、40 min的条件下进行絮凝实验,结果如图5所示。可见,随着絮凝时间的增加,CNT、Zn、Cu离子去除率不断增加,当絮凝时间增加到30 min时,CNT、CN−、Zn、Cu离子去除率均达到最高值,分别为43.9%、100%、85.14%、46.80%。考虑到时间成本,絮凝时间选择30 min最为合适。此时,废水的pH由原来的9降至7。
4) FTIR和XRD的表征分析。对制备的PSAF和PSAF絮凝沉淀物进行FTIR光谱分析,结果如图6(a)所示。制备的PSAF在3 309 cm−1和1 634 cm−1处的特征峰可归于羟基的振动[21-23],PSAF絮凝沉淀物对应的特征峰发生了偏移,分别为3 450 cm−1和1 637 cm−1,且峰面积变小,说明羟基发生了交换。在2 136 cm−1处出现了C
≡ 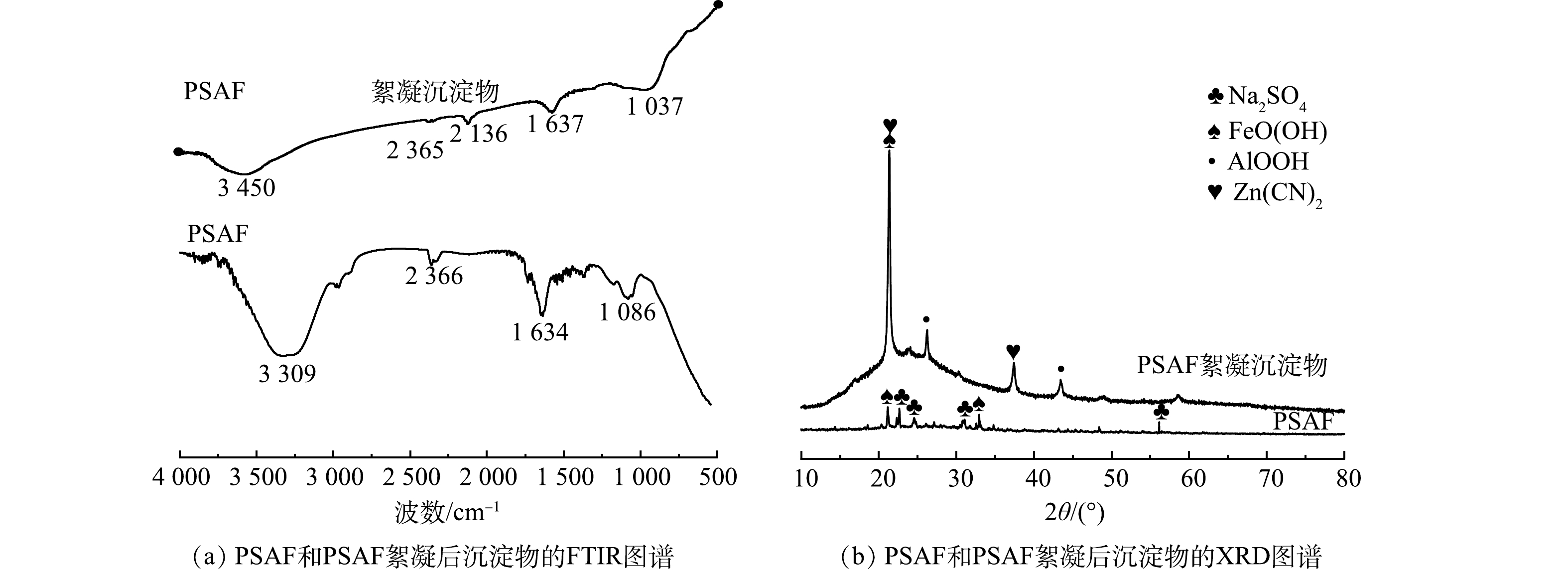 图 6 PSAF和PSAF絮凝后沉淀物的FTIR图谱和XRD图谱Figure 6. FTIR spectra and XRD patterns of PSAF and PSAF flocculated precipitates
图 6 PSAF和PSAF絮凝后沉淀物的FTIR图谱和XRD图谱Figure 6. FTIR spectra and XRD patterns of PSAF and PSAF flocculated precipitatesR-OH+CN−=RCN+OH− (2) 2R-OH+Zn(CN)2−4=R2Zn(CN)4+2OH− (3) 2R-OH+Cu(CN)2−3=R2Cu(CN)3+2OH− (4) 根据Zeta电位、FTIR分析可知,絮凝过程存在电荷中和吸附与化学吸附,Zn(CN)42−、Cu(CN)32−、CN−的吸附量分别为567.88、89.76、439.74 mg·L−1。离子的水合作用与配合离子中的电荷密度(电荷/元素数)有关,该比值越高,稳定溶液中离子所需的水分子越多,水合的种类也越多,就越不易被吸附,Zn(CN)42−电荷密度为2/9,Cu(CN)32−电荷密度为2/7,CN−电荷密度为1/2,因此,羟基对提金废水中阴离子的化学吸附能力为Zn(CN)42−>Cu(CN)32−>CN−[25],而Cu(CN)32−的总吸附量仅达到89.76 mg·L−1,远远小于Zn(CN)42−、CN−的吸附量,随着异号离子的加入,异号离子之间有强烈的吸附作用,发生电中和吸附。化学吸附需要活化能,因此,化学吸附在絮凝过程中并非起主要作用。
分别对制备的PSAF、PSAF絮凝沉淀物进行XRD分析表征,结果如图6(b)所示。由分析结果可以看出,制备的PSAF主要是Na2SO4和FeOOH的吸收峰,絮凝后沉淀物出现了AlOOH、Zn(CN)2的吸收峰。游离Na+和SO42−在合成PSAF絮凝剂时形成Na2SO4,干燥过程形成Na2SO4晶体[19],絮凝后Na2SO4峰消失,是因为絮凝时将PSAF加入废水中,Na2SO4溶解;在PSAF中无法观察到Fe2(SO4)3、Fe2O3、Fe3O4、Al2(SO4)3和SiO2等衍射晶体的光谱,表明Fe、Al和Si聚合成一些新的化合物,而不是保持原料的简单混合物[21];同时,絮凝时羟基络合离子Fe(OH)2+、Al(OH)2+发生了脱水反应,产生的FeO+、AlO+消耗水中的OH−,导致FeOOH、AlOOH的出现(式(5)~式(8))。由于PSAF属于酸性絮凝剂,导致体系pH降低,Zn(CN)42−分解得到Zn(CN)2白色沉淀(式(9))。
Fe(OH)+2→FeO++H2O (5) Al(OH)+2→AlO++H2O (6) FeO++OH−→FeO(OH) (7) AlO++OH−→AlO(OH) (8) Zn(CN)2−4+2H+=Zn(CN)2+HCN (9) 2.2 电解条件对去除率的影响
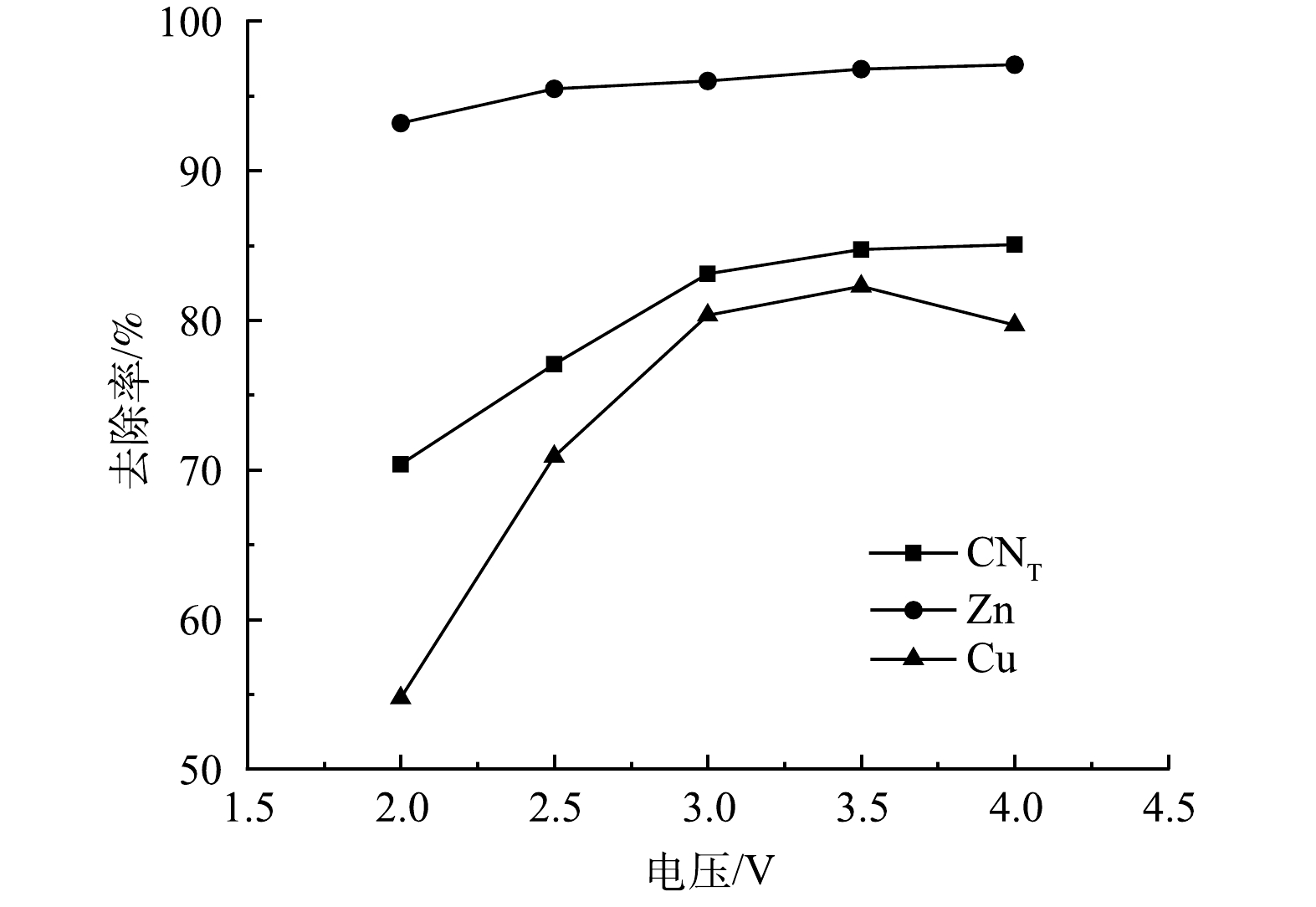
1) 电解电压的影响。取电解时间为1 h、极板间距为10 mm,对絮凝后液分别在2、2.5、3、3.5、4 V下进行电解氧化实验,结果如图7所示。随着电压的增加,废水中各离子去除率逐渐升高,随后不再发生明显变化。电压为3 V时,CNT、CN−、Zn、Cu去除率均达到最大值,分别为83.11%、100%、96.00%、80.34%。是因为电压较低时,阴离子在电场作用下通过定向迁移向阳极表面移动,并在表面吸附,系统主要依靠吸附和电吸附过程,去除率相对较低。而随着电压的升高,阳极电压高于析氧电位(0.401 V),溶液中的氰化物因析氧而被氧化(式(10)~式(13)),达到·OH的电位时(2.8 V),利用·OH的强氧化性,攻击含氰基的化合物,将C和N氧化成CO2和NO2(式(14)~式(17))[27]。释放出来的金属离子大部分在阴极板电沉积而被去除(式(18)~式(20)),使得各离子去除率大大增加[28]。
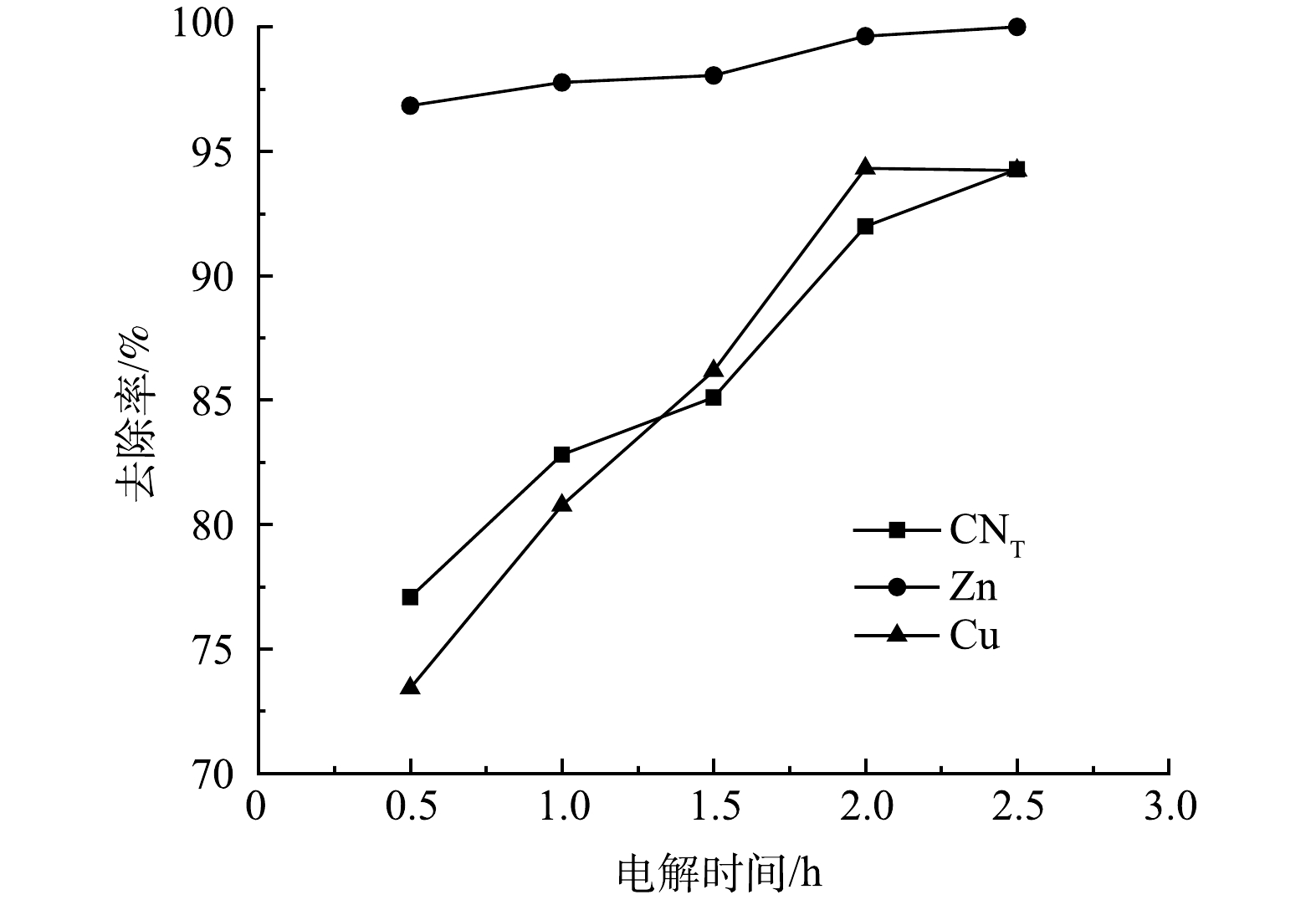
4OH−−4e−→2H2O+O2 (10) Zn(CN)2−4+2O2→Zn2++4CNO− (11) 2Cu(CN)2−3+3O2→2Cu++6CNO− (12) 2CNO−+O2-2e−→2CO2+N2 (13) 2H2O−2e−→2HO⋅+2H+ (14) OH−−e−→HO⋅ (15) Zn(CN)2−4+20⋅OH→Zn2++4CO2+2N2+4OH−+8H2O (16) 2Cu(CN)2−3+32⋅OH→2Cu2++6CO2+3N2+8OH−+12H2O (17) Zn2++2e−→Zn (18) Cu++e−→Cu (19) Cu2++2e−→Cu (20) 2)电解时间的影响。取电解电压为3 V,电解时间分别为0.5、1、1.5、2、2.5 h进行电解氧化实验,结果如图8所示。随着电解时间的增加,废水中的CN−去除率很快就达到了最大值,随后不再发生明显变化,而CNT、Zn、Cu离子去除率呈现逐渐上升的趋势。反应初期,废水中离子浓度较高,离子的迁移速率相对较快,阴阳极发生剧烈化学反应,Zn(CN)42−、Cu(CN)32−在阳极发生氧化反应,金属离子在阴极发生电沉积,2 h后,溶液中参与反应的离子浓度逐渐降低,迁移速率减慢,被还原的金属离子附着在极板表面。电解时间为2 h时,CNT、CN−、Zn、Cu离子去除率均达到最大值,分别为91.98%、100%、99.63%、94.31%。电解后存在少量沉淀,经XRD检测只存在C峰,是因为阳极的石墨板脱落少量的碳,而Zn(CN)42−、Cu(CN)32−中的氰根被氧化为CO2、N2,释放出来的Zn、Cu离子在阴极以电沉积的形式被去除。
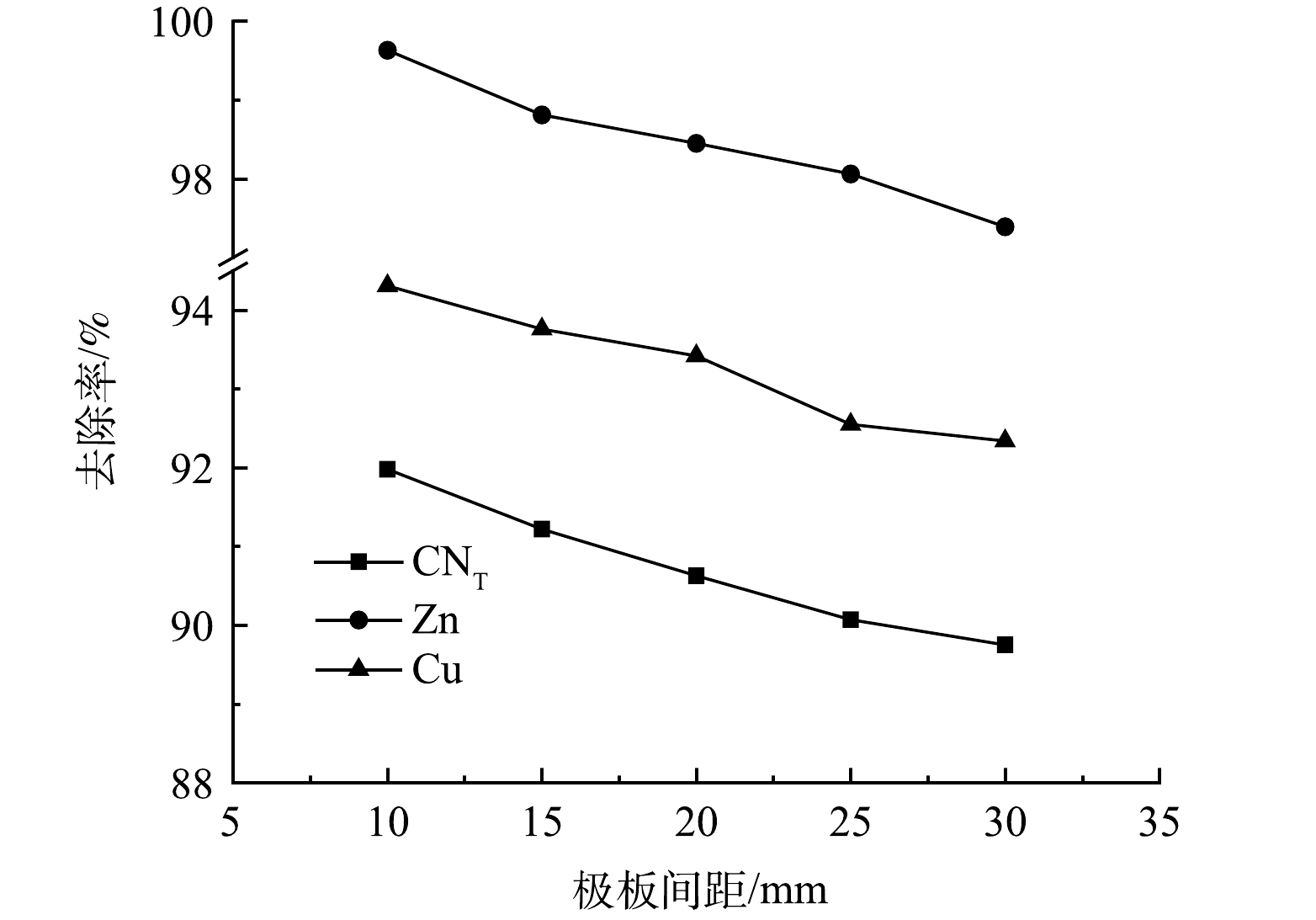
3)极板间距的影响。取电解时间为2 h,极板间距分别为10、15、20、25、30 mm进行电解氧化实验,结果如图9所示。随着极板间距的增大,CNT、Zn、Cu离子的去除率均呈现下降趋势,当极板间距为10 mm时,去除率分别为91.98%、99.63%、94.31%。随着极板间距增加,电子传递距离增加,电能损耗增加,极板间的电场强度减小,极板间带电离子Cu(CN)32−、Zn(CN)42−的迁移速率减小,且电解过程中产生的O2的扩散距离增长,从而降低各离子的去除效率[11]。因此,选择最佳极板间距为10 mm。
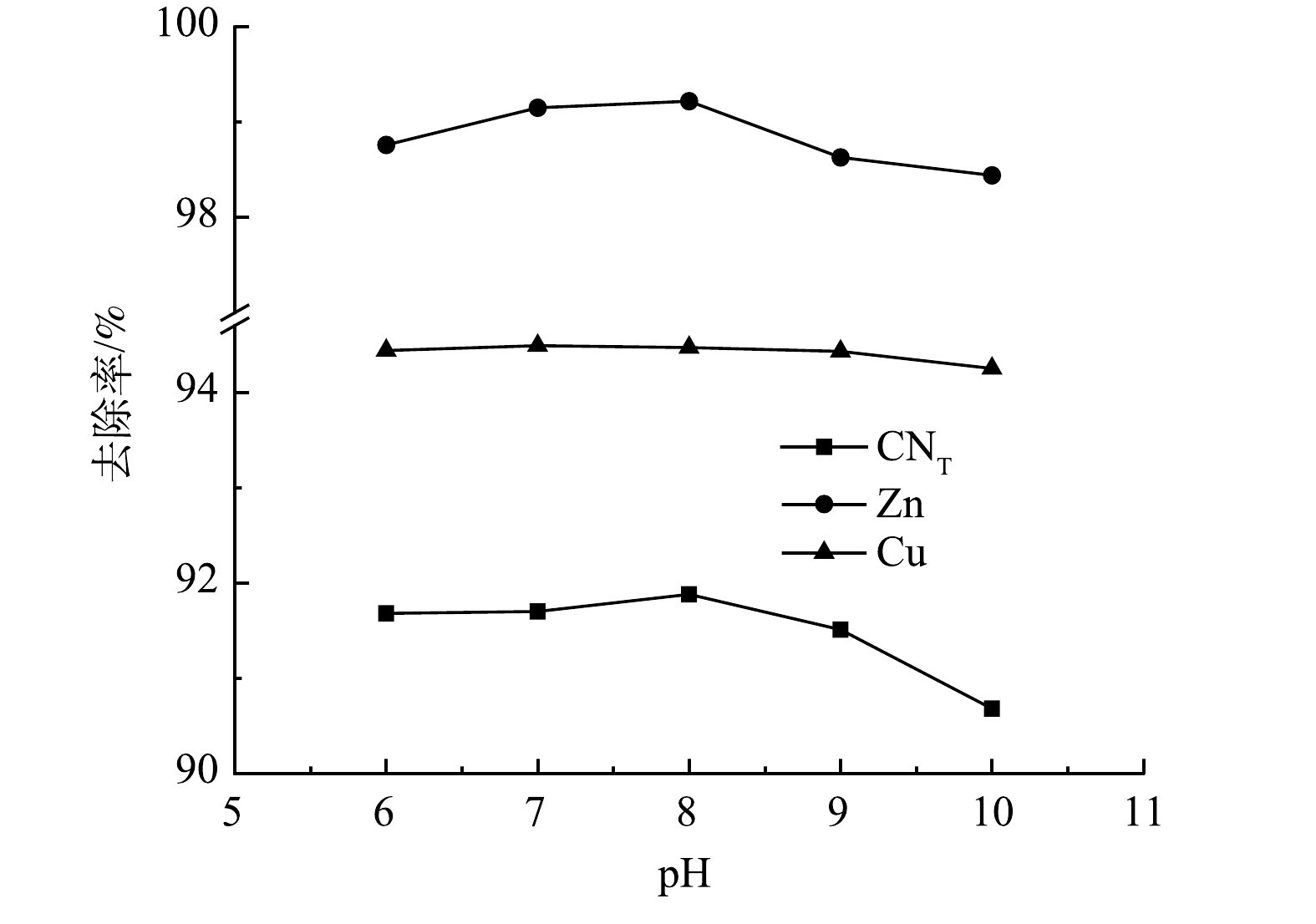
4) pH的影响。取极板间距为10 mm,絮凝以后调节pH分别为6、7、8、9、10进行电解氧化实验,结果如图10所示。随着pH的增加,溶液中的CNT、Zn离子的去除率略微增大后减小,Cu离子去除率没有明显变化,当pH为7时,去除率分别为91.70%、99.15%、94.49%。阳极氧化主要是阳极产生的氧气起作用,而氧气的产生是由电解水而得到,因此絮凝后废水的pH对电氧化效果没有明显影响。考虑到药剂成本问题,选择不改变废水pH。
2.3 平行实验
在PSAF添加量为2 g·L−1、絮凝时间为30 min、pH为9、电压为3 V、电解时间为2 h、极板间距为10 mm的条件下,进行氰化提金废水的絮凝-电解氧化验证实验,结果如表1所示。从3组平行实验所测得的结果来看,CNT、CN−、Zn、Cu的去除率相对稳定,平均去除率分别为91.70%、100%、99.15%、94.49%,说明采用絮凝-电解氧化处理氰化提金废水的技术是有效可行的。
表 1 平行实验结果Table 1. Parallel experiment results组分 序号 沉淀后液/(mg·L−1) 电解后液/(mg·L−1) 去除率/% 平均去除率/% CNT A 921.56 138.10 91.37 91.70 B 909.71 134.10 91.62 C 906.35 126.26 92.11 CN- A 0 0 100 100 B 0 0 100 C 0 0 100 Zn A 121.75 7.44 98.89 99.15 B 109.55 5.1 99.24 C 83.47 4.63 99.31 Cu A 173.02 17.87 93.06 94.49 B 166.15 13.41 94.79 C 163.60 11.3 95.61 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.4 机理分析
综上所述,絮凝-电解氧化联合处理氰化提金废水可分为PSAF絮凝和电解氧化两个阶段,反应机理示意图如图11所示。首先,加入的PSAF发生水解,生成羟基络合阳离子及羟基络合物,羟基络合阳离子与废水中的Zn(CN)42−、Cu(CN)32−、CN−通过正负电荷中和吸附去除,同时伴有羟基络合物化学吸附过程,化学吸附时,羟基络合物的羟基起主要作用,其与废水中Zn(CN)42−、Cu(CN)32−、CN−发生离子交换,产生RCN、R2Zn(CN)4、R2Cu(CN)3。其次,絮凝后液采用电解氧化处理,废水中剩余的Zn(CN)42−、Cu(CN)32−在电场作用下迁移至阳极,在阳极发生氧化反应,生成的O2、·OH将迁移至阳极的Zn(CN)42−、Cu(CN)32−氧化为N2和CO2,同时释放出Zn2+、Cu+定向迁移至阴极还原析出金属单质。
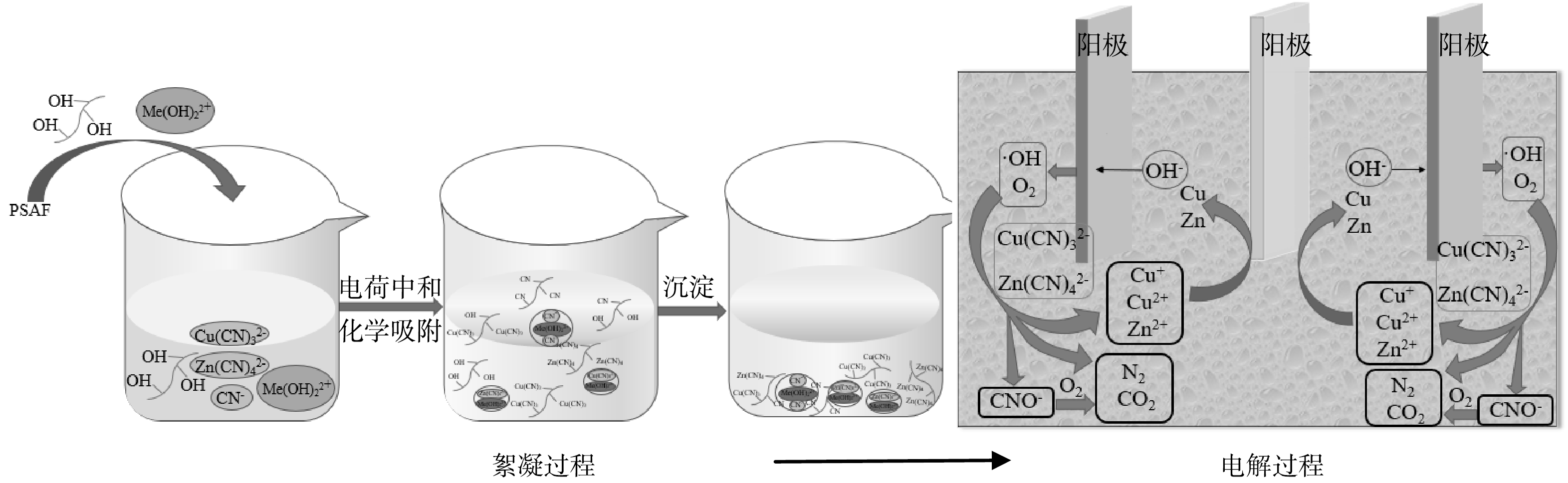 图 11 絮凝-电解氧化机理图Figure 11. Schematic diagram of flocculation-electrolytic oxidation mechanism
图 11 絮凝-电解氧化机理图Figure 11. Schematic diagram of flocculation-electrolytic oxidation mechanism3. 结论
1)采用絮凝-电解氧化联合技术处理氰化废水的思路是可行的。在室温条件下,当聚合硅酸铝铁添加量为2 g·L−1、絮凝时间30 min、pH为9时,废水中CNT、CN−、Cu、Zn离子的去除率分别为42.97%、100%、84.40%、34.88%。以电中和吸附的方式为主,伴随着化学吸附将污染物去除,同时生成了Zn(CN)2沉淀。
2)当电解时间为2 h,外加电压为3 V,极板间距为10 mm时,废水中CNT、CN−、Cu、Zn离子的总去除率分别为91.70%、100%、99.15%、94.49%。
3)无氯絮凝剂PSAF对氰化废水的处理是有效的。絮凝-电解氧化联合技术处理氰化废水,减少了氯对水环境的风险,且具有成本小,处理效果好等优点,对黄金冶炼行业的环境友好发展具有重要意义。
-
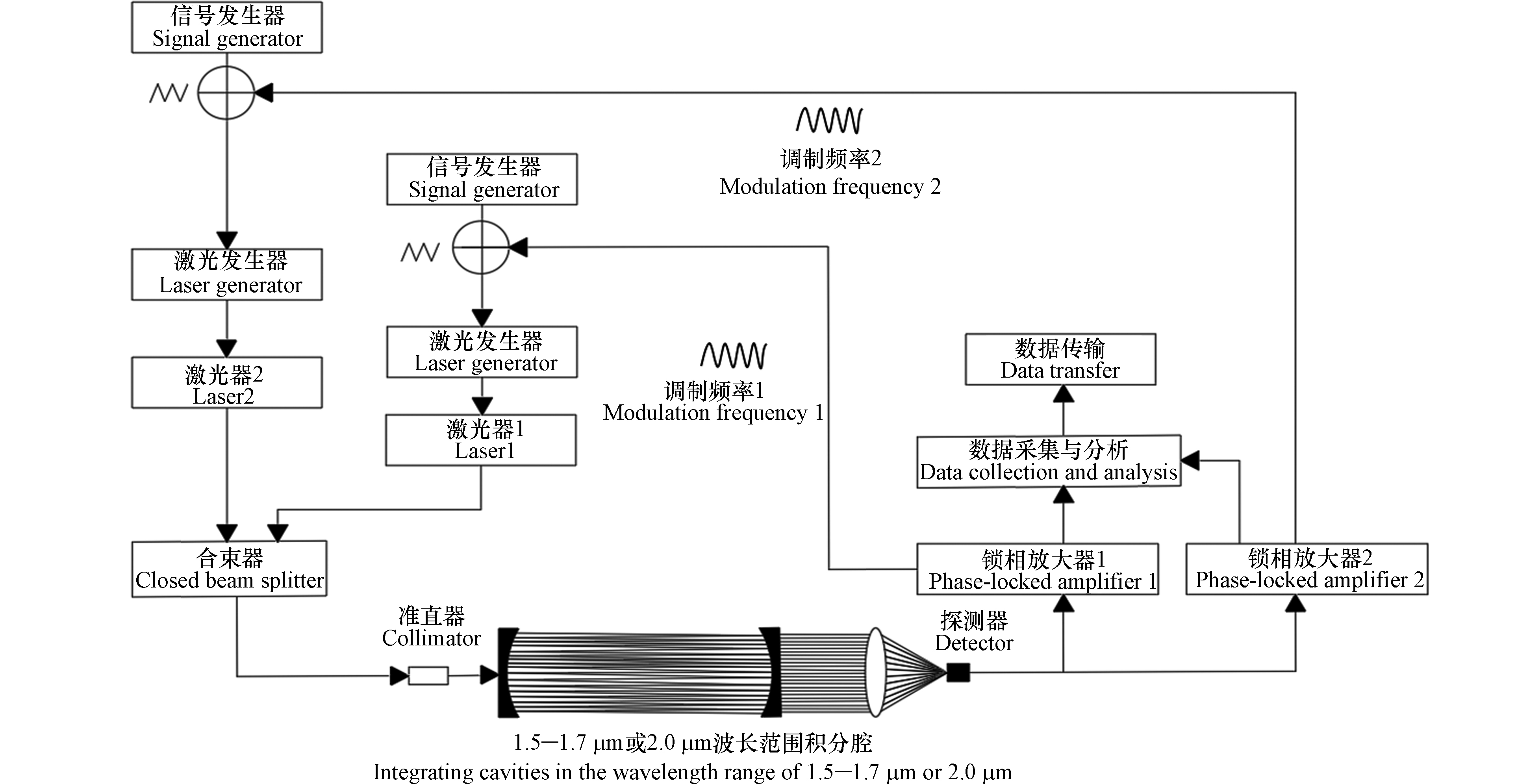
图 1 GGA-311 CO2/CH4/H2O分析仪原理图
Figure 1. Schematic diagram of GGA-311 CO2/CH4/H2O analyzer
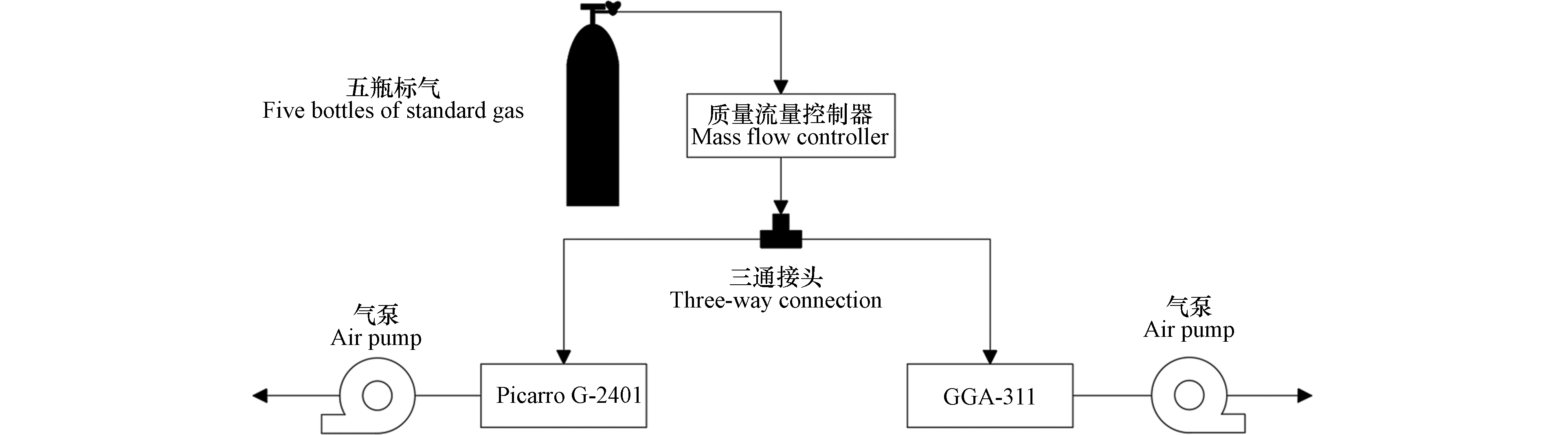
图 2 Picarro G2401和GGA-311型分析仪测试系统示意图
Figure 2. Schematic diagram of test system for Picarro G-2401 & GGA-311 analyzer
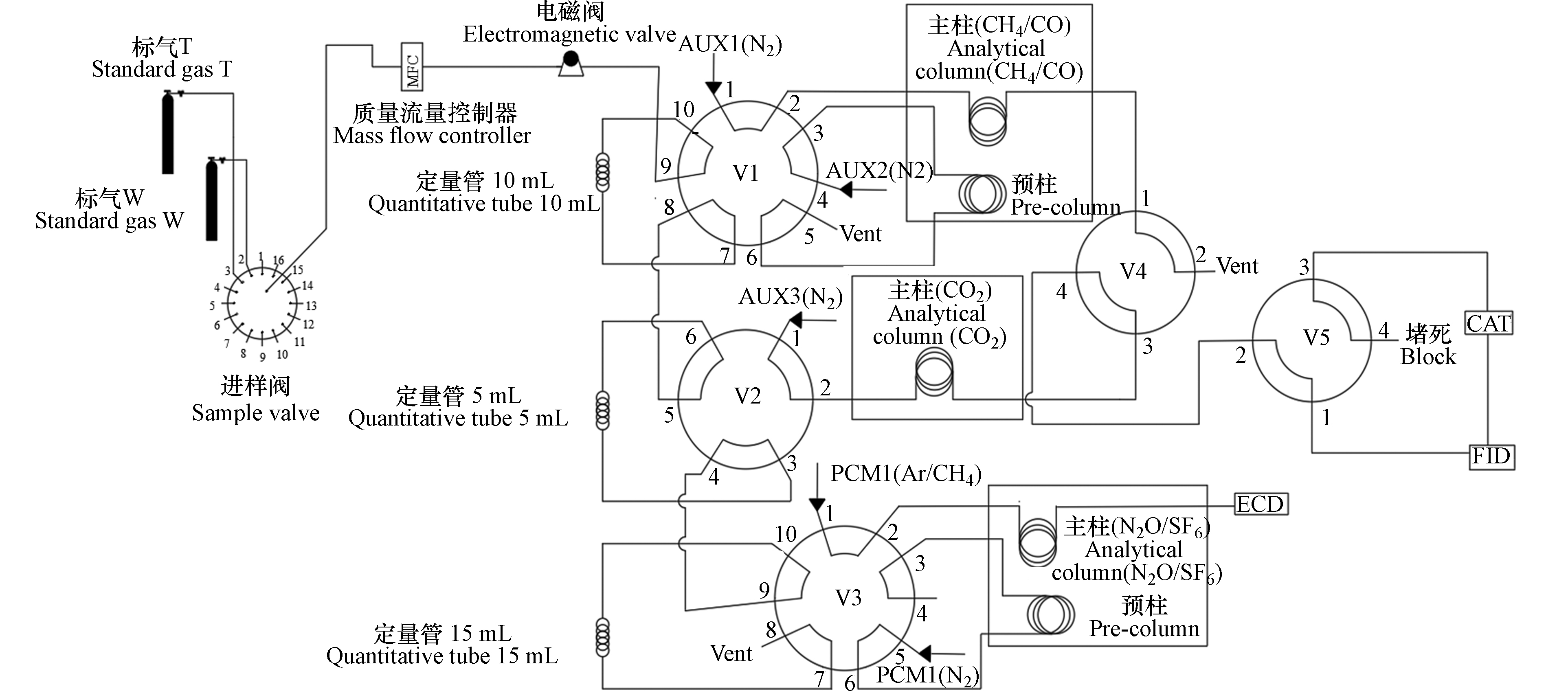
图 3 升级双通道气相色谱原理图
Figure 3. Schematic diagram of upgraded dual-channel gas chromatography
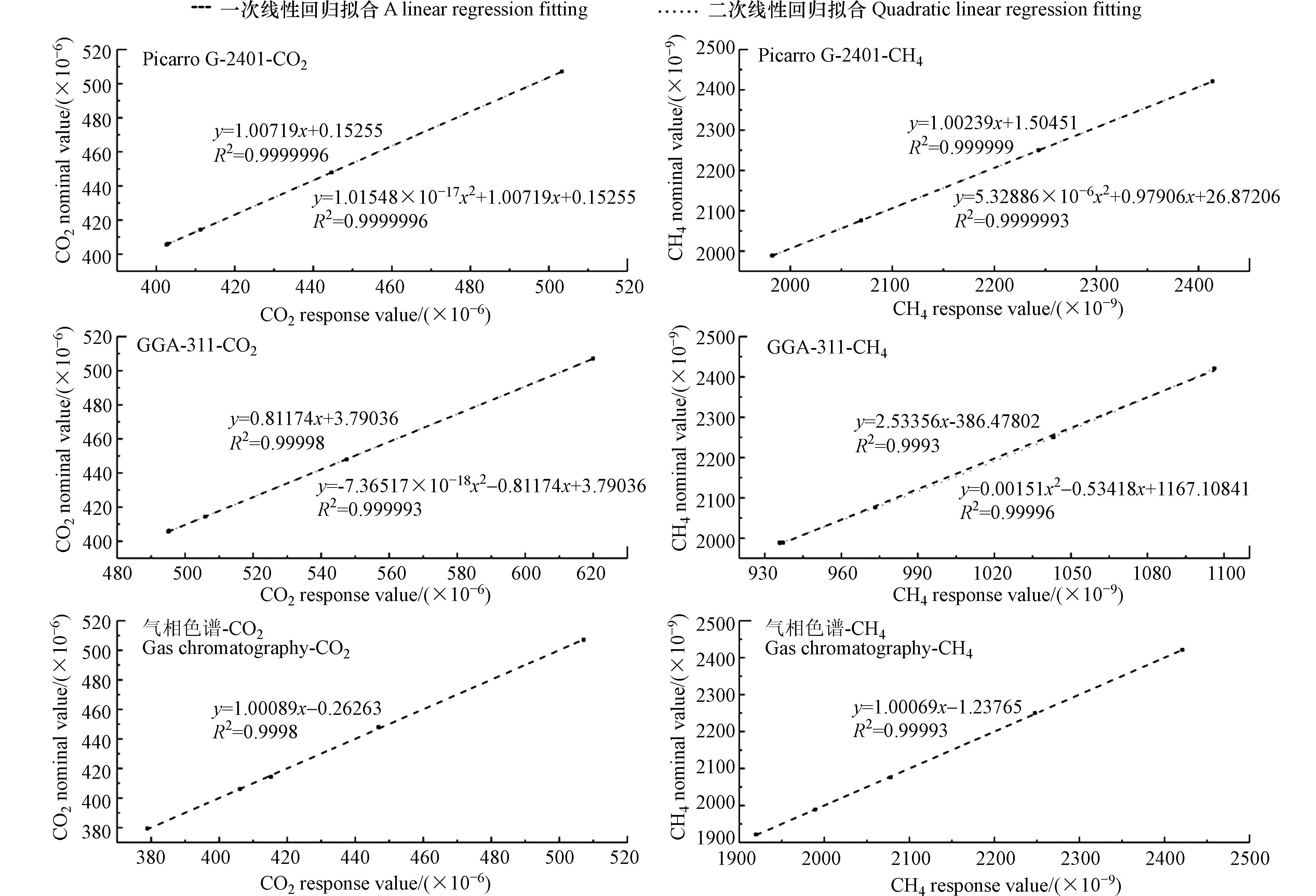
图 4 三台分析仪对标气响应值的拟合结果
Figure 4. The fitting regressions of the three analyzers’ responses to the standard gases
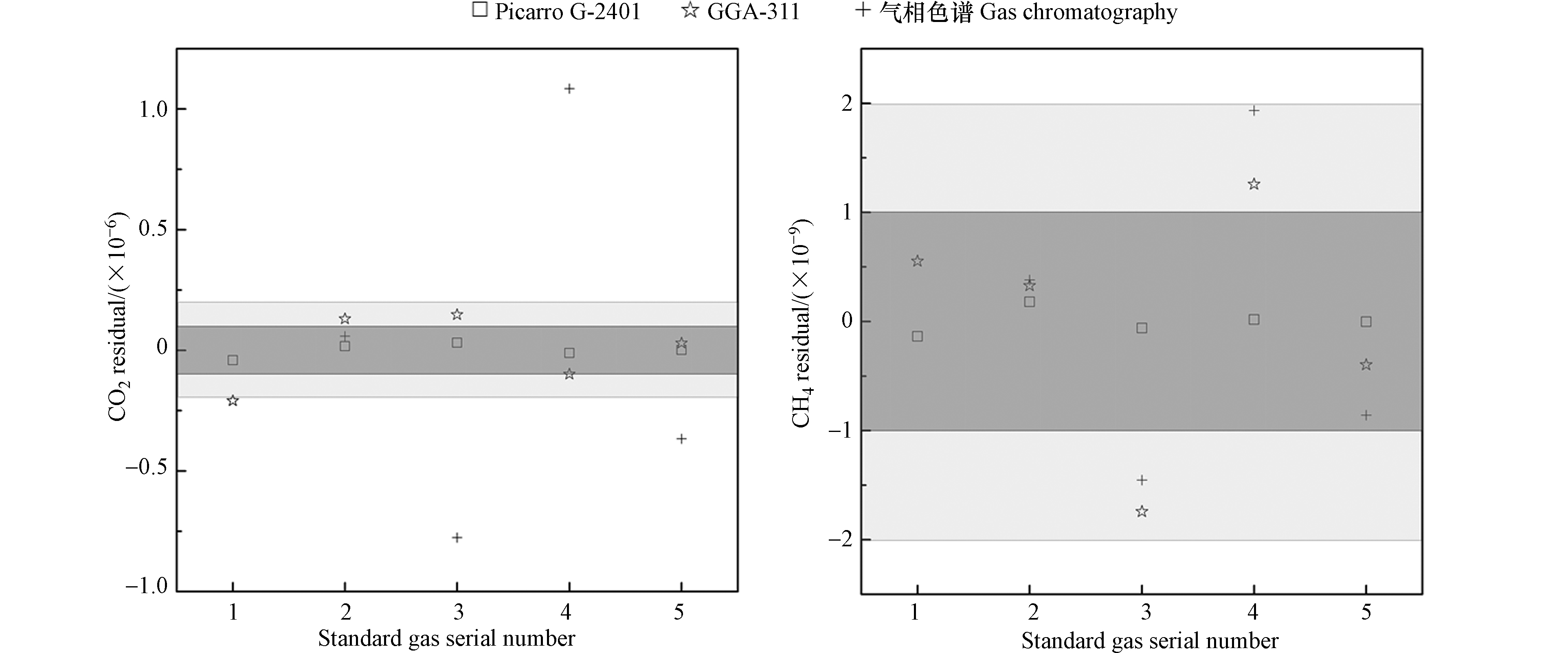
图 5 标气拟合残差情况
Figure 5. Residuals of standard gases fitting. Light grey band indicates the comparability goals of WMO/GAW and grey band denotes the extended comparability goals of WMO/GAW.
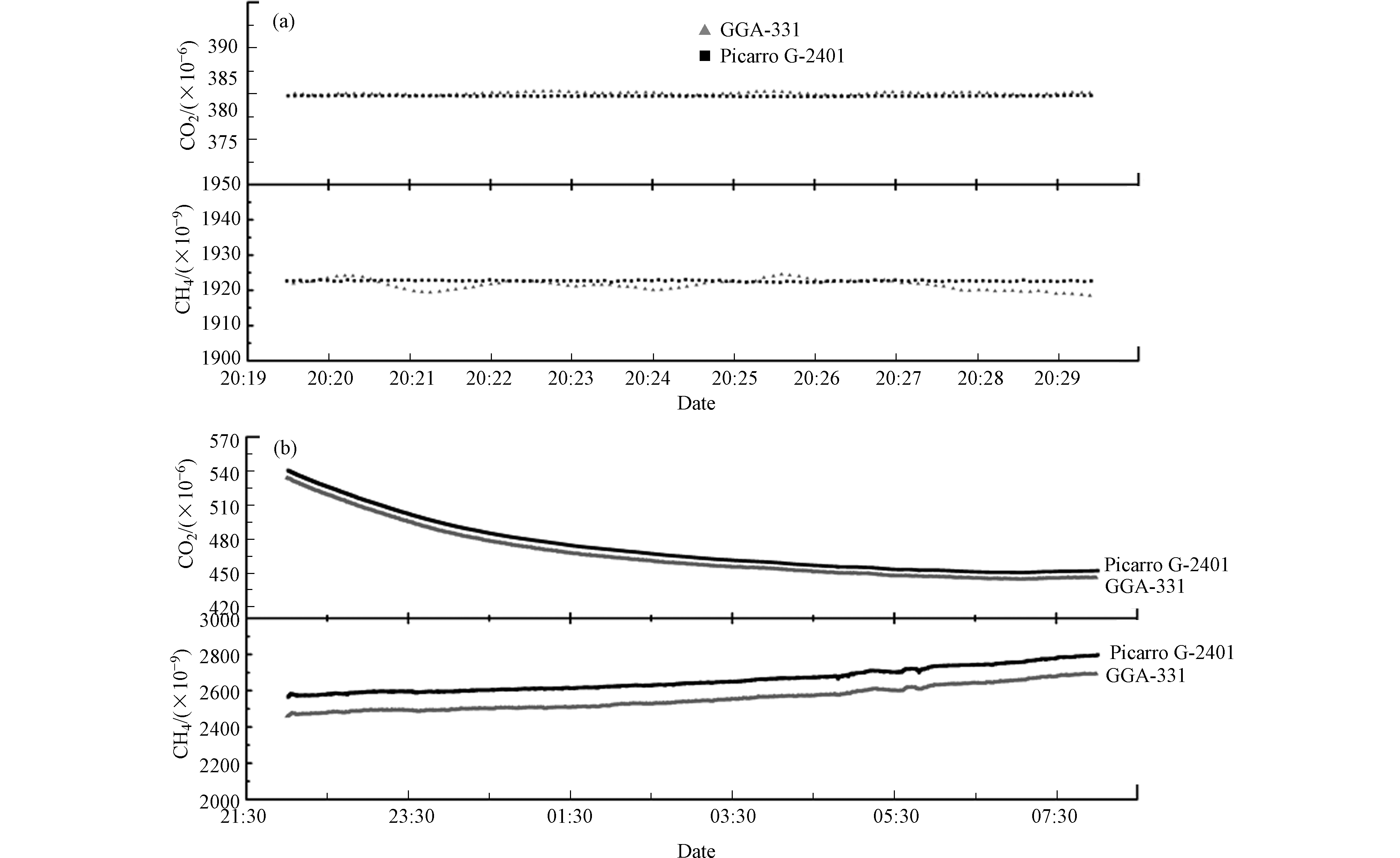
图 6 标气⑥(a)和实验室空气(b)的比对测试结果
Figure 6. Comparation test results of standard gas⑥(a) and laboratory air (b)
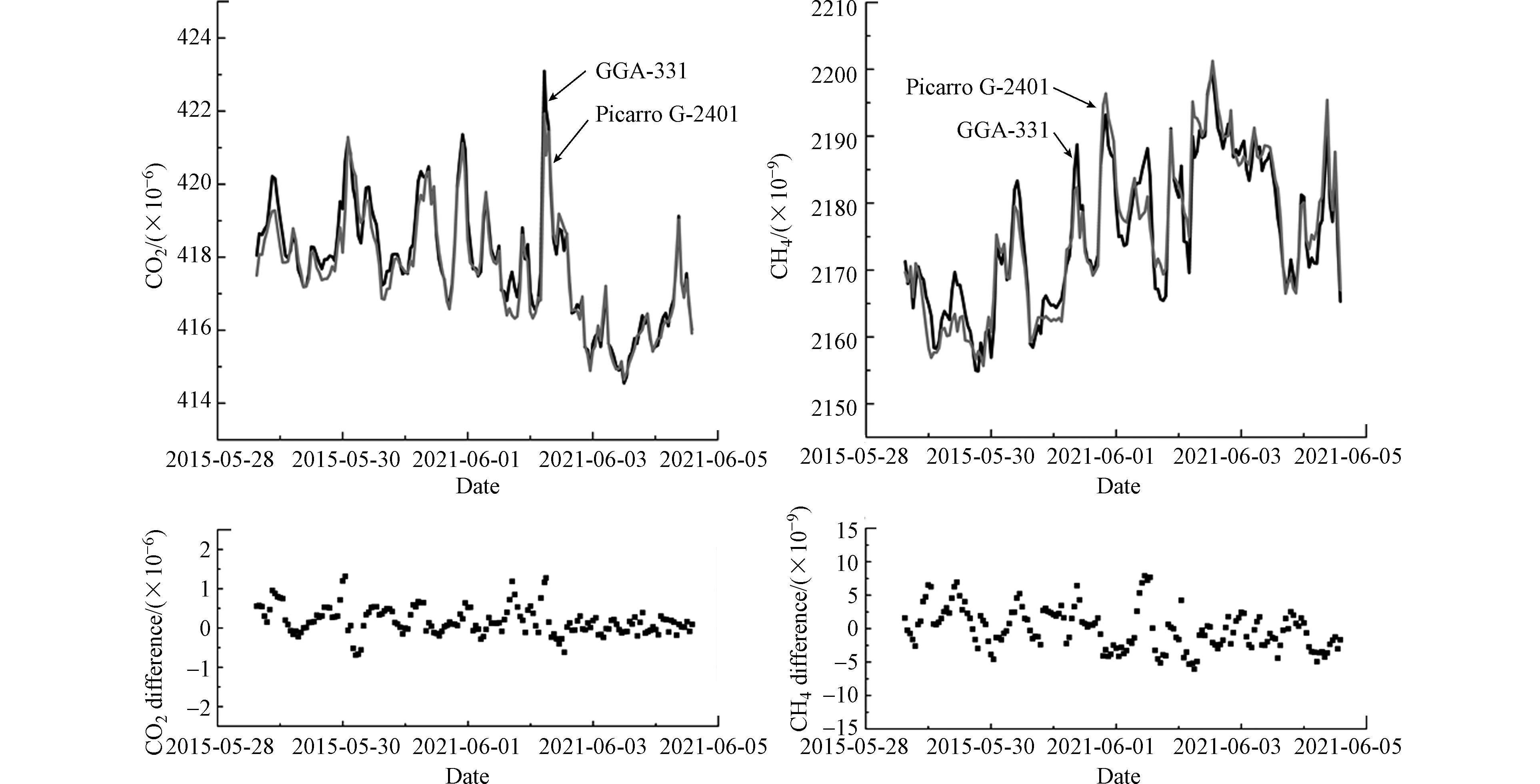
图 7 在青海瓦里关站比对结果
Figure 7. Parallel observation results by the two instruments at Qinghai Waliguan station
表 1 测试标气及标称值
Table 1. Molar ratio of standard gases for the system test
序号Serial number 标气编号Standard gas number CO2(×10−6) CH4(×10−9) ① CC738035 405.63 1988.9 ② CC738026 406.01 1988.7 ③ CC738076 414.38 2075.8 ④ CC738025 448.00 2249.9 ⑤ CC738082 507.11 2421.1 ⑥ CC738039 379.43 1921.4
下载: 导出CSV
表 2 标气CO2精密度测试结果
Table 2. CO2 precision test results of standard gases
序号Serial number CO2标称值(×10−6)CO2 nominal value 标准偏差(1σ)(×10−6)Standard deviation Picarro G-2401 GGA-311 气相色谱Gas chromatography ③ 414.38 0.02 0.18 0.30 ④ 448.00 0.02 0.16 0.64 ⑤ 507.11 0.02 0.10 0.34
下载: 导出CSV
表 3 标气CH4精密度测试结果
Table 3. CH4 precision test results of standard gases
序号Serial number CH4标称值(×10−9)CH4 nominal value 标准偏差(1σ)(×10−9)Standard deviation Picarro G-2401 GGA-311 气相色谱Gas chromatography ③ 2075.8 0.1 1.2 3.3 ④ 2249.9 0.1 1.3 3.0 ⑤ 2421.1 0.1 1.2 3.4
下载: 导出CSV
-
[1] HOFMANN D, BUTLER J, CONWAY T, et al. The NOAA Annual Greenhouse Gas Index (AGGI)[R]. Agu general assembly, 2014. [2] WMO. Greenhouse Gas Bulletin: The state of greenhouse gases in the atmosphere based on global observations through 2019[R]. 2020, [3] DANIEL J, SOLOMON S. On the climate forcing of carbon monoxide [J]. Journal of Glaciology and Geocrylogy, 1998, 103(D11): 13249-13260. [4] SATAR E, BERHANU T A, BRUNNER D, et al. Continuous CO2/CH4/CO measurements (2012—2014) at Beromünster tall tower station in Switzerland [J]. Biogeosciences, 2016, 13(9): 2623-2635. doi: 10.5194/bg-13-2623-2016 [5] TIEMOKO T D, RAMONET M, YOROBA F, et al. Analysis of the temporal variability of CO2, CH4 and CO concentrations at Lamto, West Africa [J]. Tellus B:Chemical and Physical Meteorology, 2021, 73(1): 1-24. [6] YAZIDI A E, RAMONET M, CIAIS P, et al. Identification of spikes associated with local sources in continuous time series of atmospheric CO, CO2 and CH4 [J]. Atmospheric Measurement Techniques, 2018, 11(3): 1599-1614. doi: 10.5194/amt-11-1599-2018 [7] 唐孝炎, 张远航, 邵敏. 大气环境化学 (第 2 版)[M]. 北京: 高等教育出版社, 2006: 55-90. TANG J Y, ZHANG Y H, SHAO M. Atmospheric Environmental Chemistry (Version 2)[M]. Beijing: Higher Education Press, 2006: 55-90 (in Chinese).
[8] KEELING C D. The concentration and isotopic abundances of carbon dioxide in the atmosphere [J]. Tellus, 1960, 12(2): 200-203. doi: 10.3402/tellusa.v12i2.9366 [9] KEELING C D, BACASTOW R B, BAINBRIDGE A E, et al. Atmospheric carbon dioxide variations at Mauna Loa Observatory, Hawaii [J]. Tellus, 1976, 28(6): 538-551. doi: 10.3402/tellusa.v28i6.11322 [10] CONIL S, HELLE J, LANGRENE L, et al. Continuous atmospheric CO2, CH4 and CO measurements at the Observatoire Pérenne de l'Environnement (OPE) station in France from 2011 to 2018 [J]. Atmospheric Measurement Techniques, 2019, 12(6): 6361-6383. [11] CUNNOLD M D, STEELE P L, FRASER J P, et al. In situ measurements of atmospheric methane at GAGE/AGAGE sites during 1985-2000 and resulting source inferences [J]. Journal of Geophysical Research:Atmospheres, 2002, 107(D14): 1-20. [12] DERWENT R, RYALL D, MANNING A, et al. Continuous observations of carbon dioxide at Mace Head, Ireland from 1995 to 1999 and its net European ecosystem exchange [J]. Atmospheric environment, 2002, 36(17): 2799-2807. doi: 10.1016/S1352-2310(02)00203-0 [13] ZHOU L X, LIU L X, ZHANG X C, et al. Preliminary results on network observation of greenhouse gases at China GAW station [J]. Journal of Applied Meteorolgical Science, 2008, 19(6): 641-645. [14] 方双喜, 周凌晞, 张芳, 等. 双通道气相色谱法观测本底大气中的CH4、CO、N2O和SF6 [J]. 环境科学学报, 2010, 30(1): 363-371. FANG S X, ZHOU L X, ZHANG F, et al. Dual channel GC system for measuring background atmospheric CH4, CO, N2O and SF6 [J]. Acta Scientiae Circumstantiae, 2010, 30(1): 363-371(in Chinese).
[15] CROSSON E R. A cavity ring-down analyzer for measuring atmospheric levels of methane, carbon dioxide, and water vapor [J]. Applied Physics B, 2008, 92: 403-408. doi: 10.1007/s00340-008-3135-y [16] WELP L R, KEELING R E, WEISS R F, et al. Design and performance of a Nafion dryer for continuous operation at CO2 and CH4 air monitoring sites [J]. Atmospheric Measurement Techniques, 2013, 6(5): 1217-1226. doi: 10.5194/amt-6-1217-2013 [17] FANG S X, TANS P P, BO Y, et al. Study of atmospheric CO2 and CH4 at Longfengshan WMO/GAW regional station: the variations, trends, influence of local sources/sinks, and transport [J]. Science China Earth Sciences, 2017, 60: 1886-1895. doi: 10.1007/s11430-016-9066-3 [18] MAHESH P, SREENIVAS G, RAO P, et al. High-precision surface-level CO2 and CH4 using off-axis integrated cavity output spectroscopy (OA-ICOS) over Shadnagar, India [J]. International Journal of Remote Sensing, 2015, 36(22): 5754-5765. doi: 10.1080/01431161.2015.1104744 [19] WANG J J, TIAN X, DONG Y, et al. High-sensitivity off-axis integrated cavity output spectroscopy implementing wavelength modulation and white noise perturbation [J]. Optics Letters, 2019, 44(22): 3298-3301. [20] 田兴, 曹渊, 王静静, 等. 基于离轴腔增强吸收光谱双组分CH4/H2O高灵敏度探测研究 [J]. 光谱学与光谱分析, 2019, 39(10): 3078-3083. TIAN X, CAO Y, WANG J J, et al. High sensitivity detection of two-component CH4/H2O based on off-axis cavity enhanced absorption spectroscopy [J]. Spectroscopy and Spectral Analysis, 2019, 39(10): 3078-3083(in Chinese).
[21] CHRISTIANSEN J R, OUTHWAITE J, SMUKLER S M. Comparison of CO2, CH4 and N2O soil-atmosphere exchange measured in static chambers with cavity ring-down spectroscopy and gas chromatography [J]. Agricultural and forest meteorology, 2015, 211-212: 48-57. doi: 10.1016/j.agrformet.2015.06.004 [22] 顾帅, 周凌晞, 刘立新, 等. 静态箱-气相色谱法CO2和CH4通量观测的质控方法研究 [J]. 气象, 2010, 36(8): 87-101. doi: 10.7519/j.issn.1000-0526.2010.08.012 GU S, ZHOU L X, LIU L X, et al. Research of quality control measures in greenhouse gases flux observation using static closed chamber-GC technique [J]. Meteorological Monthly, 2010, 36(8): 87-101(in Chinese). doi: 10.7519/j.issn.1000-0526.2010.08.012
[23] ZHENG K Y, ZHENG C T, HE Q X, et al. Near-infrared acetylene sensor system using off-axis integrated-cavity output spectroscopy and two measurement schemes [J]. Optics Express, 2018, 26(20): 26205-26216. doi: 10.1364/OE.26.026205 [24] WANG K Y, SHAO L G, CHEN J J, et al. A dual-laser sensor based on off-axis integrated cavity output spectroscopy and time-division multiplexing method [J]. Sensors, 2020, 20(21): 6192. doi: 10.3390/s20216192 [25] 臧昆鹏, 周凌晞, 方双喜, 等. 新型CO2和CH4混合标气标校流程及方法 [J]. 环境化学, 2011, 30(2): 511-516. ZANG K P, ZHOU L X, FANG S X, et al. A new system for calibration and propagation of mixed CO2 and CH4 standards [J]. Environmental Chemistry, 2011, 30(2): 511-516(in Chinese).
[26] 周凌晞, 汤洁, 张晓春, 等. 气相色谱法观测本底大气中的甲烷和二氧化碳 [J]. 环境科学学报, 1998, 18(2): 356-361. doi: 10.13671/j.hjkxxb.1998.04.004 ZHOU L X, TANG J, ZHANG X C, et al. In-situ gas chromatographic measurement of atmospheric methane and carbon dioxide [J]. Acta Scientiae Circumstantiae, 1998, 18(2): 356-361(in Chinese). doi: 10.13671/j.hjkxxb.1998.04.004
[27] LIU S, FANG S X, LIU P, et al. Measurement report: Changing characteristics of atmospheric CH4 in the Tibetan Plateau: records from 1994 to 2019 at the Mount Waliguan station [J]. Atmospheric Chemistry and Physics, 2021, 21(1): 393-413. doi: 10.5194/acp-21-393-2021 [28] ZHANG F, ZHOU L X, NOVELLI P C, et al. Evaluation of in situ measurements of atmospheric carbon monoxide at Mount Waliguan, China [J]. Atmospheric Chemistry and Physics, 2011, 11(11): 5195-5206. doi: 10.5194/acp-11-5195-2011 [29] 王剑琼, 薛丽梅, 张国庆, 等. 不同方法测量大气二氧化碳浓度的特征分析 [J]. 青海环境, 2015, 25(2): 75-78. doi: 10.3969/j.issn.1007-2454.2015.02.007 WANG J Q, XUE L M, ZHANG G Q, et al. Analysis of characteristics of atmospheric carbon dioxide concentration measured by different methods [J]. Journal of Qinghai Environment, 2015, 25(2): 75-78(in Chinese). doi: 10.3969/j.issn.1007-2454.2015.02.007
[30] KWOK Y C, LAURENT O, GUEMRI A, et al. Comprehensive laboratory and field testing of cavity ring-down spectroscopy analyzers measuring H2O, CO2, CH4 and CO [J]. Atmospheric Measurement Techniques, 2015, 8(9): 3867-3892. doi: 10.5194/amt-8-3867-2015 [31] ANNETTE F, CHRISTOPH G, HUILIN C, et al. The IAGOS-core greenhouse gas package: a measurement system for continuous airborne observations of CO2, CH4, H2O and CO [J]. Tellus B:Chemical and Physical Meteorology, 2015, 67(1): 1-19. [32] FLOWERS B A, POWERS H H, DUBEY M K, et al. Inter-comparison of two high-accuracy fast-response spectroscopic sensors of carbon dioxide: A case study [J]. Atmospheric Measurement Techniques, 2012, 5(5): 991-997. doi: 10.5194/amt-5-991-2012 [33] 崔俊富, 陈金伟, 崔伟. 残差在线性回归分析中的作用研究 [J]. 牡丹江大学学报, 2020, 29(10): 84-88. doi: 10.3969/j.issn.1008-8717.2020.10.018 CUI J F, CHEN J W, CUI W. Research about residuals in linear regression analysis [J]. Journal of Mudanjiang University, 2020, 29(10): 84-88(in Chinese). doi: 10.3969/j.issn.1008-8717.2020.10.018
[34] MAHATA K, PANDAY A, SINGH A, et al. Seasonal and diurnal variations in methane and carbon dioxide in the Kathmandu Valley in the foothills of the central Himalayas [J]. Atmospheric Measurement Techniques, 2017, 17(20): 12573-12596. [35] FANG S X, ZHOU L X, MASARIE K A, et al. Study of atmospheric CH4 mole fractions at three WMO/GAW stations in China [J]. Journal of Geophysical Research:Atmospheres, 2013, 118(10): 4874-4886. doi: 10.1002/jgrd.50284 [36] CHEN H J, WINDERLICH J, GERBIG C, et al. High-accuracy continuous airborne measurements of greenhouse gases (CO2 and CH4) using the cavity ring-down spectroscopy (CRDS) technique [J]. Atmospheric Measurement Techniques, 2010, 3(2): 375-386. doi: 10.5194/amt-3-375-2010 [37] ZELLWEGER C, EMMENEGGER L, FIRDAUS M, et al. Assessment of recent advances in measurement techniques for atmospheric carbon dioxide and methane observations [J]. Atmospheric Measurement Techniques, 2016, 9(9): 4737-4757. doi: 10.5194/amt-9-4737-2016 [38] MORGAN E J, LAVRIČ J V, SEIFERT T, et al. Continuous measurements of greenhouse gases and atmospheric oxygen at the Namib Desert Atmospheric Observatory [J]. Atmospheric Measurement Techniques, 2015, 8(6): 2233-2250. doi: 10.5194/amt-8-2233-2015 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5986
- HTML全文浏览数: 5986
- PDF下载数: 150
- 施引文献: 0
