



-
石油炼制和基础化学原料制造过程会产生大量的废催化剂。近年来,石化产品的需求量逐年增加、原料油劣质化和油品质量升级促使炼油催化剂生命周期缩短,卸出量增大。早在2016年,环境保护部联合国家发展和改革委员会、公安部发布《国家危险废物名录(2016年版)》,增加了HW50废催化剂类废物。最新发布的《国家危险废物名录(2021年版)》中,废催化剂主要包括加氢精制、采用钝镍剂的催化裂化、加氢裂化、催化重整等工艺过程产生的废催化剂[1]。
浸出毒性通常指固体废物遇水浸沥,浸出的有害物质迁移转化,对人和生态环境造成的污染。挥发性有机化合物(VOCs)具有较低的沸点,极易挥发和迁移。VOCs具有亲脂性、高毒性和刺激性,在生物体内产生致癌、致畸、致突变以及生理毒性,危害人类健康,属于浸出毒性重点关注的有害物质[2-3]。VOCs是废催化剂危险废物鉴别中重点关注的指标之一。GB5085.3—2007《危险废物鉴别标准浸出毒性鉴别》中涉及多项固体废物浸出液中VOCs指标,超过浓度限值的固体废物是具有浸出毒性特征的危险废物。
由于固体废物浸出液中VOCs种类复杂,含量极低,因此有必要对浸出液进行样品前处理。VOCs的前处理方法主要有吹扫捕集、顶空、溶剂萃取、固相微萃取等。溶剂萃取法成本低,但操作繁琐,需使用大量有机溶剂,提取过程中会损失部分VOCs。顶空法可避免VOCs的损失,测定范围广,操作简便,但不能浓缩VOCs,灵敏度低,无法满足痕量分析的要求。固相微萃取法选择性不高,萃取头种类较少,易受污染而导致萃取效率降低。吹扫捕集是将惰性气体通过吹扫管连续吹扫样品,将样品中的VOCs不断吹入气相,在捕集管中被吸附剂或冷阱捕集,然后迅速加热捕集管,VOCs被解吸,最后进行测定。吹扫捕集灵敏度高、线性范围宽,广泛应用于固体废物VOCs的痕量分析。固体废物浸出液中VOCs的测定方法主要有GB5085.3—2007中推荐的附录O、附录P和附录Q,以及7项固体废物中VOCs的分析标准。前处理技术采用顶空法和吹扫捕集,检测方法有GC-FID[4-7]、GC-MS[4,8-11]、GC-PID[4]、GC-HECD[4]。
目前,国内针对废催化剂浸出液中VOCs的测定鲜有报道。本文选取了3种不同炼油工艺产生的废催化剂,建立了吹扫捕集/气相色谱-质谱法测定废催化剂浸出液中VOCs的分析方法。
-
仪器和设备:气相色谱-质谱仪Aglient 7890B-5977B;吹扫捕集样品浓缩仪OI Analytical Eclipse 4760,样品瓶体积为40 mL;振荡设备:转速为(30±2)r·min−1的翻转式振荡装置;零顶空提取器(Zero-Headspace Extraction Vessel,ZHE):500—600 mL。
试剂:VOCs混合标准溶液(2000 mg·L−1)(购自伊诺凯),甲醇(色谱纯),内标标准溶液(氟苯、氯苯-D5、1,4-二氯苯-D4的混合溶液,2500 mg·L−1),替代物标准溶液(二溴氟甲烷、甲苯-D8、4-溴氟苯的混合溶液,2000 mg·L−1)。实验用水为超纯水,电阻率≥18 MΩ·cm。废催化剂分别来自于催化裂化(FCC)、吸附脱硫(S Zorb)和渣油加氢工艺产生的废催化剂。
-
吹扫捕集条件:吹扫流量40 mL·min−1;吹扫温度40 ℃;吹扫时间11 min;脱附温度190 ℃;脱附时间2 min;烘烤温度200 ℃;烘烤时间8 min。
气相色谱条件:色谱柱:DB-624UI,60 m×0.25 mm×1.4 μm,6%腈丙苯基、94%二甲基聚硅氧烷固定液;柱流量(恒流模式):1 mL·min−1;阻尼柱1:1.8 m×150 μm,弹性石英柱管,流量1.53 mL·min−1;阻尼柱2:0.7 m×150 μm,弹性石英柱管;进样口温度:250 ℃;载气:氦气;分流比:20:1;柱流量(恒流模式):1 mL·min−1;升温程序:初始温度38 ℃,保持4 min,以5 ℃·min−1速率升温至240 ℃。保持20 min。
质谱条件:扫描方式:全扫描(SCAN)及选择离子扫描(SIM)模式;扫描范围35—270 amu;离子化能量70 eV;接口温度280 ℃。
-
配制质量浓度分别为5、20、50、100、200 μg·L−1的VOCs混合标准溶液和替代物标准溶液,分别加入内标溶液,使每点的内标质量浓度均为50.0 μg·L−1,立即密封待测。
-
执行HJ/T 299—2007的方法制备废催化剂浸出液试样,浸提剂为超纯水。称取40 g废催化剂样品,快速转入ZHE,缓慢加压以排除顶空。根据样品的含水率,按液固比为10∶1(L·kg−1)计算所需浸提剂的体积。将ZHE固定在翻转式振荡装置上,23 ℃下振荡18 h,收集浸出液。取5 ml浸出液试样,再加入内标溶液和替代物溶液,使内标和替代物质量浓度均为50.0 μg·L−1,快速注入吹扫管中,内标法定量。
-
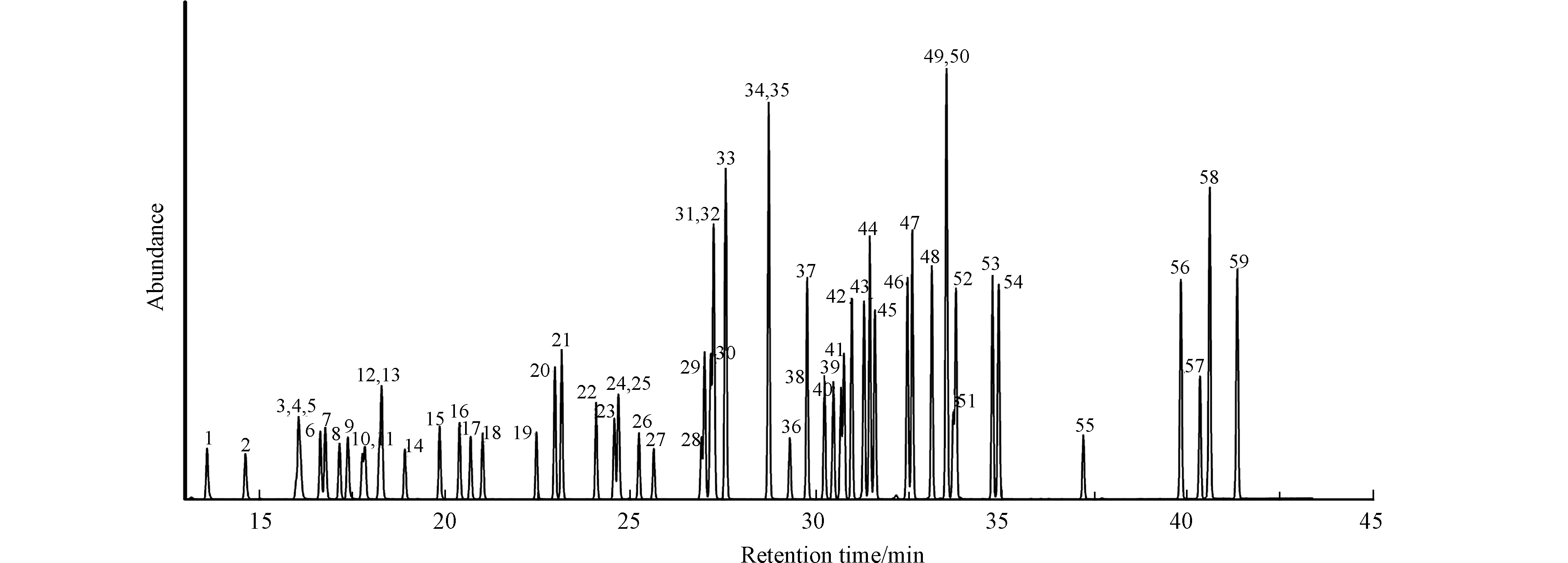
吹扫捕集/气相色谱-质谱法测定53种VOCs、3种内标和3种替代物的质谱的总离子流图如图1所示。吹扫捕集/气相色谱-质谱法的线性和检出限见表1,标准曲线的线性范围为5—200 μg·L−1,线性相关系数R2>0.99,方法检出限为0.1—2.0 μg·L−1,方法的线性程度和检出限均较为满意。
-
按照HJ/T 299—2007的方法制备废催化剂浸出液试样,浸提剂为超纯水。废催化剂分别来自于FCC、S Zorb和渣油加氢3种工艺产生的废催化剂。催化裂化是重质石油烃类在催化剂的作用下反应产生液化气、汽油和柴油等轻质油品的过程。FCC工艺所使用的催化剂属于Y型分子筛。S Zorb工艺是采用独特的吸附剂对汽油脱硫的过程。S Zorb催化剂的主要成分是含过渡金属的双金属氧化物。渣油加氢工艺是以渣油为原料加氢处理制得低硫燃料油,或加氢裂化制得轻质油的过程。渣油加氢催化剂主要是以大孔氧化铝、分子筛和复合金属氧化物为载体,以Mo、Mo-Ni、Mo-Co和Mo-W为活性组分与助剂一并合成的[12]。3种催化剂在使用过程中会负载有机物,严重时会造成积炭,失去活性成为废催化剂。负载上的一些有机物属于有毒物质,因此3种废催化剂属于《国家危险废物名录(2021年版)》中关注的固体废物。
图2分别为3种废催化剂浸出液的总离子流色谱图。3种废催化剂浸出液的测定结果及精密度见表2,其中氯仿、四氯化碳、苯、三氯乙烯、甲苯、四氯乙烯、氯苯、乙苯、间,对二甲苯、邻二甲苯、1,2-二氯苯、1,4-二氯苯属于GB5085.3—2007《危险废物鉴别标准浸出毒性鉴别》中涉及的11项VOCs指标。3种废催化剂浸出液中的这11项VOCs含量均低于GB5085.3—2007中浸出毒性鉴别限值。FCC、S Zorb和渣油加氢三种废催化剂浸出液中VOCs的相对标准偏差(RSD,n=3)分别为0.2%—0.6%、0.7%—3.9%和0.9%—4.3%,精密度较为满意。
-
为了考察吹扫捕集/气相色谱-质谱法的回收率,向3种废催化剂浸出液加入标准溶液和替代物,使VOCs加标量为50.0 μg·L−1。3种废催化剂浸出液加标50.0 μg·L−1的回收率试验结果见表3,回收率分别为78.7%—161.0%、64.9%—145.6%和74.0%—167.6%,替代物二溴氟甲烷、甲苯-D8、4-溴氟苯回收率满足70%—130%之间,大部分目标VOCs的回收率在70%—130%。以上结果表明,所建立的吹扫捕集/气相色谱-质谱法可以用于废催化剂浸出液中VOCs的监测。
少数目标VOCs的回收率偏高,主要原因是内标响应值的波动造成的。个别组分如正丙苯、叔丁基苯、仲丁基苯、4-异丙基甲苯、正丁基苯、六氯丁二烯等,加标回收率不够理想,该结果与陈德阳等[13]的文献保持一致。这可能是由于高碳数组分沸点较高,吹扫捕集的前处理方法不容易将高碳数组分从浸出液中提取出来,导致这些组分的回收率不高。
-
图2(a)可以看出,FCC废催化剂浸出液中大部分VOCs含量低于检出限。图2(b)和(c)表明,除了GB5085.3—2007所列的VOCs指标,S Zorb和渣油加氢废催化剂浸出液中含有大量以芳烃为主的VOCs,这些芳烃会对环境造成污染。如果芳烃的含量以正丙苯计算,S Zorb和渣油加氢废催化剂浸出液中芳烃总量分别为275 μg·L−1和6645 μg·L−1。S Zorb和渣油加氢废催化剂浸出液芳烃的组成和分布结果见图3。S Zorb废催化剂浸出液检出的化合物以C9—C11芳烃为主,其中C9芳烃、C10芳烃、C11芳烃的含量占比分别为17.7%、41.0%、31.7%。渣油加氢废催化剂检出的化合物以C10—C12芳烃为主,其中C10芳烃、C11芳烃、C12芳烃的含量占比分别为16.2%、49.1%、27.1%。结果表明,S Zorb和渣油加氢废催化剂浸出液中VOCs种类主要为C9+芳烃。
-
采用吹扫捕集/气相色谱-质谱法,建立了石油炼制工艺废催化剂浸出液中VOCs的检测方法。对线性范围、检出限、回收率及精密度进行了考察。结果表明该方法稳定性好、灵敏度高,可以满足废催化剂浸出液中VOCs的检测要求。不同石油炼制工艺产生的废催化剂,其负载VOCs的组成和分布可能差异较大,这对于废催化剂的认识、检测和治理提供了参考和支持。废催化剂浸出液的VOCs组成较为复杂,该方法针对的目标物数量有限,尚需开发新的方法进行分析测定。
吹扫捕集/气相色谱-质谱法测定石油炼制工艺废催化剂浸出液中挥发性有机物
Determination of volatile organic compounds in waste catalyst leaching liquor in petroleum refining process using P&T-GC-MS method
-
摘要: 采用吹扫捕集/气相色谱-质谱法,建立了石油炼制工艺废催化剂浸出液中挥发性有机物(VOCs)的检测方法。该方法标准曲线线性范围为5—200 μg L−1,线性相关系数R2>0.99,方法检出限为0.1—2.0 μg L−1。催化裂化(FCC)废催化剂浸出液、吸附脱硫(S Zorb)废催化剂浸出液和渣油加氢废催化剂浸出液的测定结果显示各组分的相对标准偏差(RSD)分别为0.2%—0.6%、0.7%—3.9%和0.9%—4.3%,基体加标回收率分别为78.7%—161.0%、64.9%—145.6%和74.0%—167.6%。所建立的方法可以满足废催化剂浸出液中VOCs的检测要求。FCC废催化剂浸出液中大部分VOCs的含量低于检出限。S Zorb废催化剂浸出液、渣油加氢废催化剂浸出液检出的化合物分别以C9—C11芳烃和C10—C12芳烃为主。所选取的3种废催化剂浸出液中目标VOCs含量均低于GB5085.3—2007中浸出毒性鉴别限值。Abstract: A method was established for the determination of volatile organic compounds (VOCs) in waste catalyst leaching liquor in petroleum refining process by gas chromatography-mass spectrometry (GC-MS) with purge and trap (P&T) pretreatment. The linear ranges were 5—200 μg L−1 and linear correlation coefficients were more than 0.99. The detection limits were 0.1—2.0 μg L−1. 3 kinds of waste catalysts from the semi synthetic fluid catalytic cracking (FCC), S Zorb and residue hydrotreating refinery process were investigated for leaching experiment. The recoveries of leaching liquors were 78.7%—161.0%, 64.9%—145.6% and 74.0%—167.6% with RSD of 0.2%—0.6%, 0.7%—3.9% and 0.9%—4.3% respectively. This method was suitable for the analysis of VOCs in waste catalyst leaching liquor.The concentration of most VOCs were below the detection limits in waste catalyst leaching liquor from FCC. C9—C11 aromatic hydrocarbons and C10—C12 aromatic hydrocarbons were the main compounds detected in waste catalyst leaching liquors from S Zorb and residue hydrotreating respectively. The contents of target VOCs in the leaching solution of the three waste catalysts selected were all lower than the limiting values for the identification of leaching toxicity in GB5085.3—2007.
-
Key words:
- waste catalyst /
- leaching liquor /
- volatile organic compounds /
- purge and trap
-
近年来,抗生素的广泛使用导致其以不同的方式大量流入环境[1-2],其中包括盐酸土霉素。盐酸土霉素属于一种广谱抗菌素,在养殖业和农业中作为杀虫剂普遍使用,但是其很难被生物体完全吸收,盐酸土霉素的部分代谢产物及大量的残留会进入环境中,危害环境并给人类带来危害风险[3]。目前,针对抗生素的废水处理方法有水解、生物降解、高级氧化法等,其中,水解和生物降解均有耗时长、耗资大等缺点。高级氧化法是目前研究最为广泛一种废水处理方法,包括有芬顿氧化法[4-5]、光催化氧化法[6]和电化学氧化法[7-8]等。
高级氧化法主要利用具有强氧化性功能的羟基自由基(·OH)或硫酸根自由基(
SO⋅−4 )与废水中的目标污染物发生反应,氧化降解污染物并使其矿化。近年来,运用SO⋅−4 [9]的高级氧化技术逐渐成为热门,跟·OH[10-11]比较发现,SO⋅−4 具有较低的氧化还原电势(2.60 V),而且SO⋅−4 的使用寿命比·OH要更长久,能在降解污染物的同时保持节能和高效[12-14]。常规的高级氧化催化剂的催化活性较低,其中,一种新颖的金属-有机骨架材料(metal-organic frameworks, MOFs)引起人们广泛的关注。MOFs因其具有高比表面积、高稳定性和高孔隙率的特点被频繁运用。如PENG等[15]和ZHANG等[16]研究基于MOFs的新型材料MIL-53(Al)和H-UiO-66s对各类废水有机污染物的捕获时发现,MIL-53(Al)和H-UiO-66s均对有机污染物产生高效的吸附和去除,这表明MOFs具备去除污染物的性能。另外,MON等[17]和胡龙兴等[18]研究MOFs对水体污染的修复效果的结果表明,MOFs能有效的降解有机污染物。本研究采用水热法制备Co-MOF催化剂,并对其物化性质进行了表征。同时,考察了Co-MOF活化PS降解盐酸土霉素的性能及重复利用效果,并进一步对该降解反应的机理进行分析。1. 材料与方法
1.1 试剂和仪器
实验试剂:均苯三甲酸(GR,上海化学)、三乙胺(GR,国药控股)、六水合硝酸钴(AR,国药控股)、叔丁醇(CP,国药)、抗坏血酸(AR,国药)、盐酸土霉素(国药);其他试剂均为国药分析纯试剂,实验用水为去离子水。
实验仪器:KH-100型Teflon-lined反应釜(北京瑞昌伟业)、pHS-3C型pH计(上海仪电科学)、UV1900型紫外可见分光光度计(上海佑科)、SHA-C型水浴恒温振荡箱(常州澳华)、ZA3300型火焰原子吸收分光光度计(北京日立)。
1.2 实验方法
1) Co-MOF的制备。量取25 mL去离子水于烧杯中,加入1.45 g Co(NO3)2•6H2O、1.05 g均苯三甲酸和2 mL三乙胺,再将烧杯放置在磁力搅拌器上搅拌15 min,然后将溶液转移到Teflon-lined反应釜中,密封放置在190 ℃的电热鼓风干燥箱中24 h。将反应后所得离心(6 000 r·min−1)2 min获取沉淀物,用乙醇和去离子水各反复冲洗3次,再60 ℃真空烘干备用。
2) Co-MOF降解盐酸土霉素的实验。量取20 mg·L的盐酸土霉素于反应器中,加入一定量的Co-MOF和PS,在恒温震荡箱中反应5 min,特定时间取样,并用紫外分光光度计在353 nm波长处测定吸光度,根据标准曲线计算盐酸土霉素反应后的浓度。实验结束后收集定量的上清液,加入适量硝酸酸化至pH≤2,然后用火焰原子吸收分光光度计测定其中的钴含量。盐酸土霉素的降解率按照式(1)进行计算。
η=(C0−Ce)C0×100% (1) 式中:
η 为盐酸土霉素的降解率;C0和Ce分别为盐酸土霉素溶液初始浓度和平衡时的浓度,mg·L−1。2. 结果与讨论
2.1 Co-MOF表征
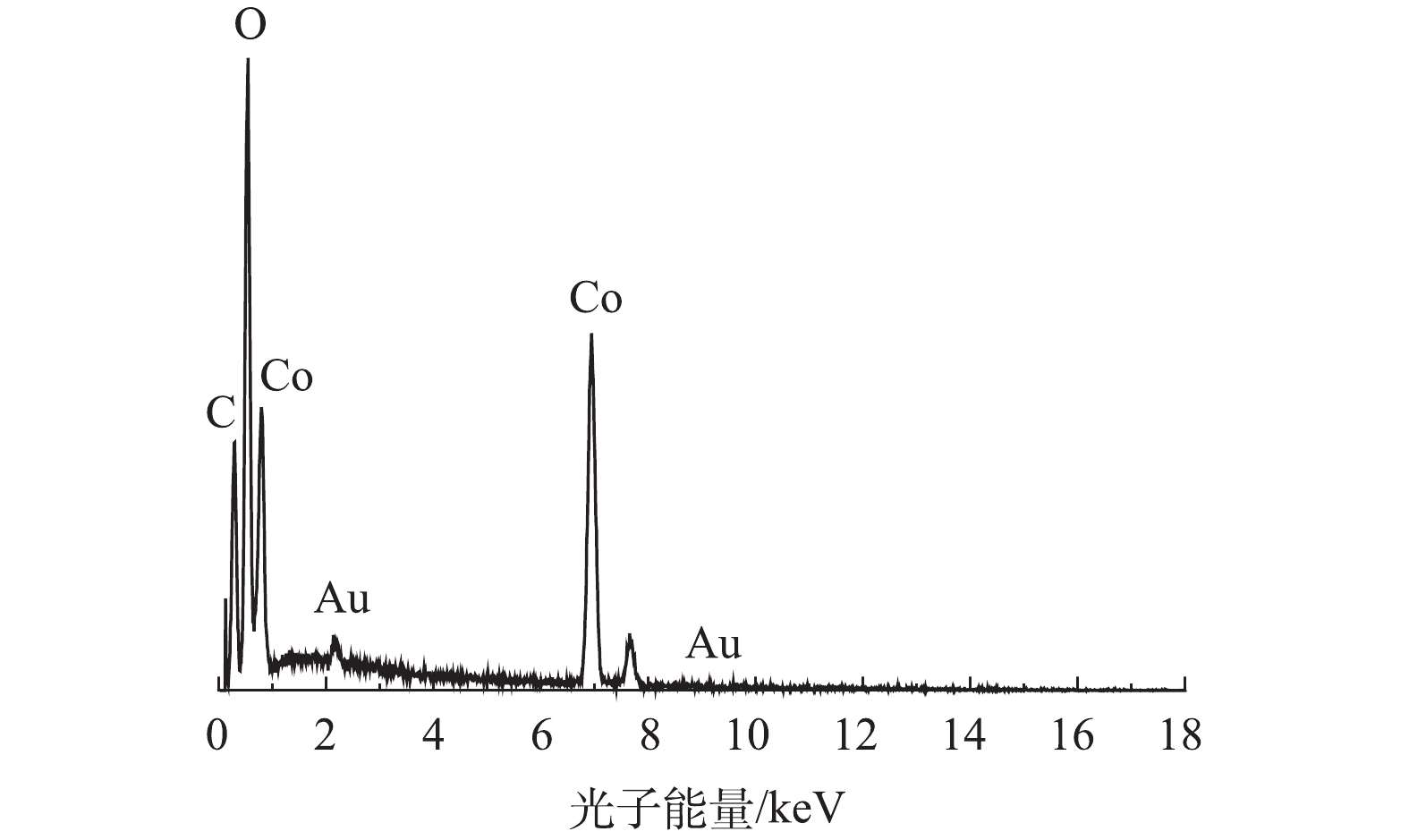
1)扫描电镜(SEM)和透射电镜(TEM)。通过SEM、TEM和EDS对制备的Co-MOF的形貌特征、粒径以及组成成分进行分析。如图1(a)和图1(b)的SEM表征所示,Co-MOF是由棒状晶体组成,每个棒状晶体的长度大约几百微米,宽约为30~100 μm,这与YAGHI等[19]的报道结果相似。如图2所示,Co-MOF含有C、O和Co元素,其中,Co元素的相对峰面积占比较大,这表明Co在Co-MOF结构中的重要性。如图1(c)和图1(d)的TEM表征所示,可见Co-MOF是由棒状晶体组成,并且每个棒状晶体表面平滑、多孔隙,这与PATTERSON等[20]的研究结果类似。
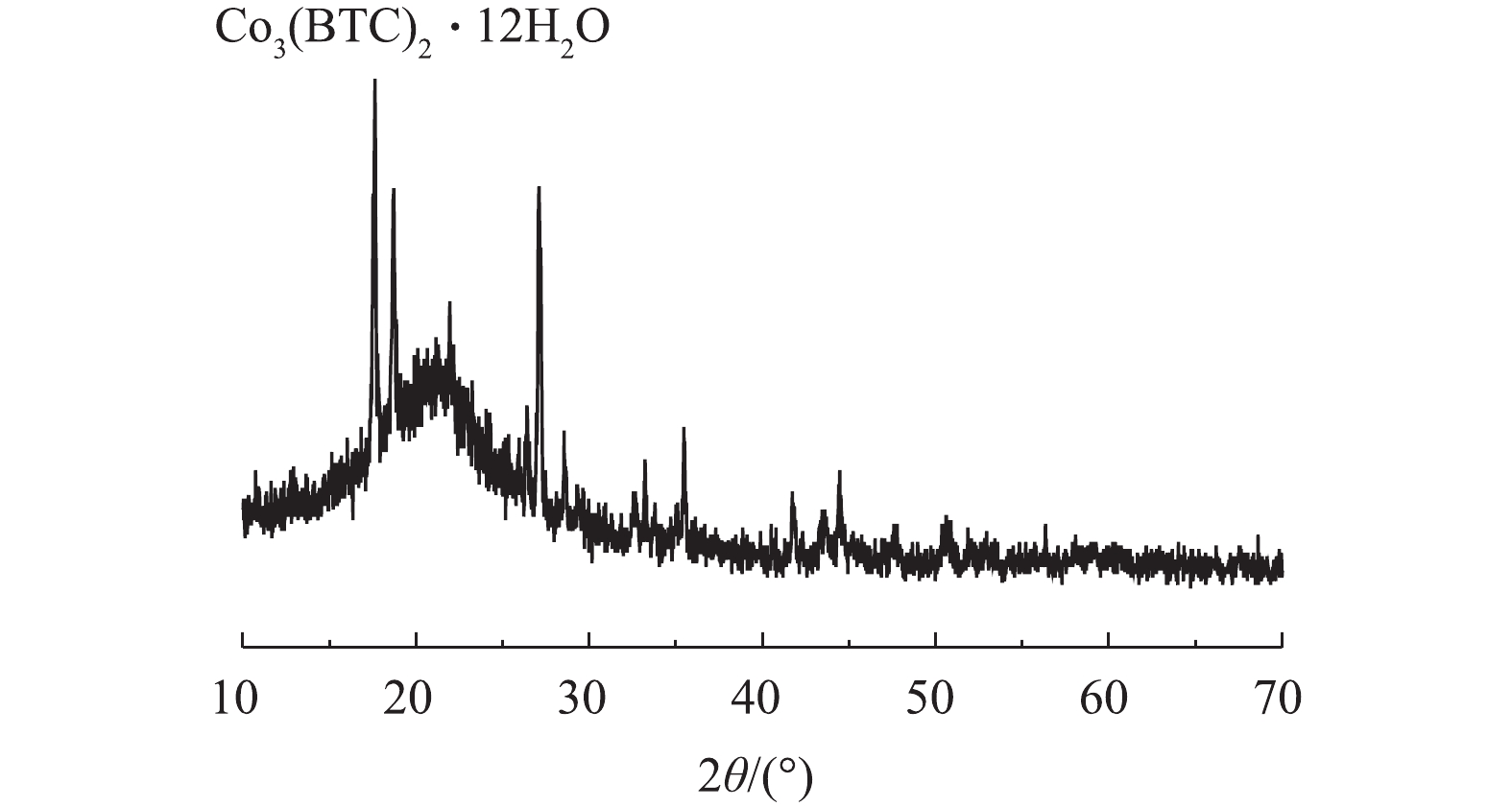
2)X射线衍射(XRD)。通过XRD对制备的Co-MOF结构进行分析。如图3所示,在17.5°附近有明显的衍射峰,此峰对应Co3(BTC)2·12H2O的晶面(014),故这表明Co(NO3)2·6H2O和均苯三甲酸发生完美融合,这与以往报道[21-22]中的Co-MOF的XRD图谱相似。
2.2 不同体系对盐酸土霉素的降解效果
图4为不同催化剂反应体系下盐酸土霉素的降解效果对比。由图4可见,在第1组(未遮光)和第2组(遮光)中,单独的Co-MOF添加对盐酸土霉素的降解率分别为25.8%和3.9%,这是Co-MOF自身的光催化特性导致,如孙登荣等[23]研究MOFs材料的光催化性能时发现,MOFs具有光催化性能。在第3组实验中,单独的PS添加对盐酸土霉素的降解率为21.5%,这是由于PS的热活化反应导致。杨世迎等[24]开展的活化过硫酸盐高级氧化技术的研究结果表明,在热、光、过渡金属等外在条件的催化下,过硫酸盐均能通过吸收能量来使内部构造发生变化。在第4组实验中,Co-MOF/PS对盐酸土霉素的降解率为95.4%。这样的结果与预期一样,表明Co-MOF具有高效活化PS的性能。
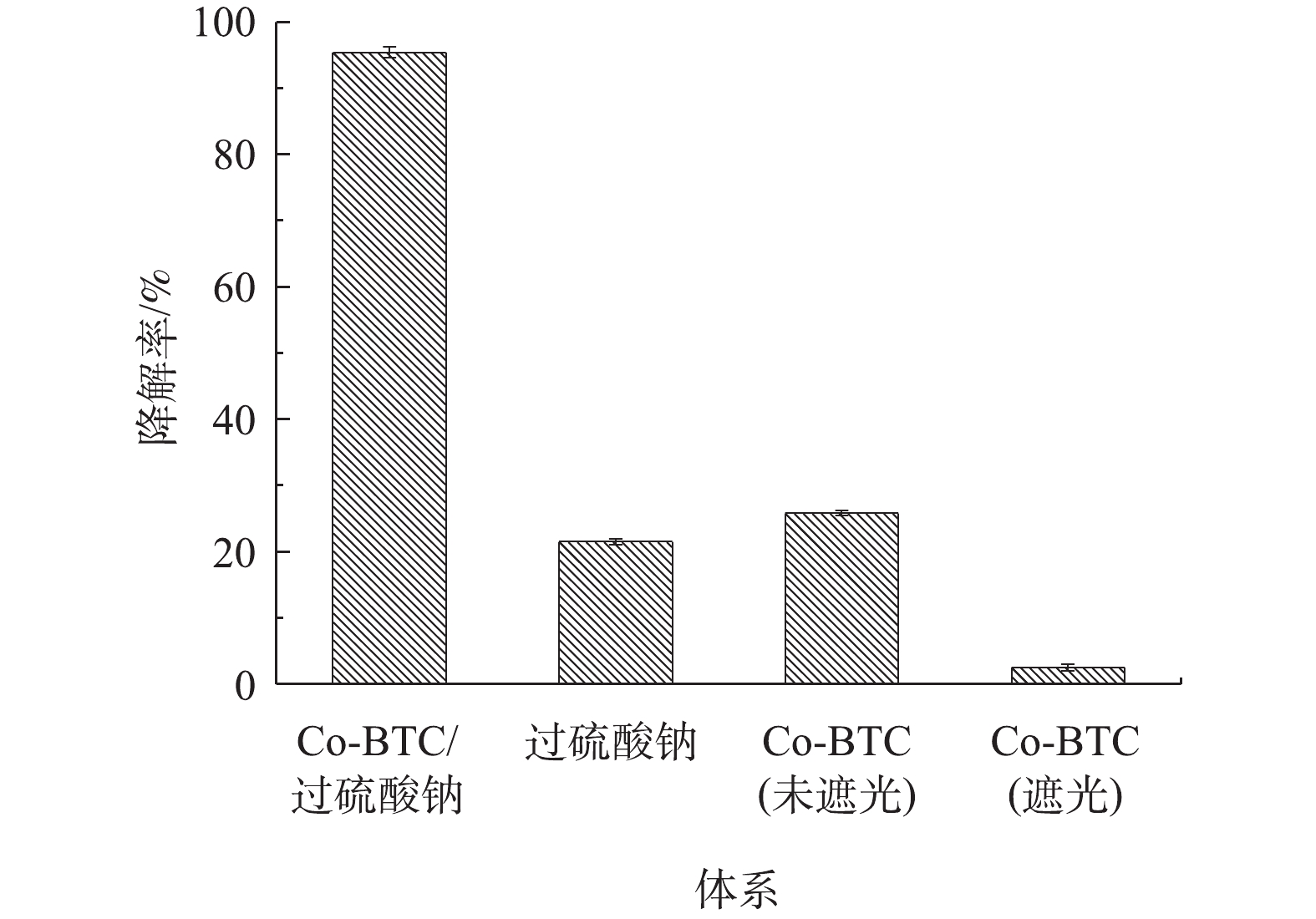 图 4 不同体系对降解盐酸土霉素的影响Figure 4. Effects of different systems on degradation of oxytetracycline hydrochloride
图 4 不同体系对降解盐酸土霉素的影响Figure 4. Effects of different systems on degradation of oxytetracycline hydrochloride2.3 影响Co-MOF降解盐酸土霉素的因素
1) pH对盐酸土霉素降解的影响。图5为不同pH对盐酸土霉素降解的影响。如图5所示,盐酸土霉素在pH=2.0~11.0时均有不同程度的降解,其中,当pH=5.0时,对盐酸土霉素的降解率最高为97.1%。当pH=2.0、3.0、9.0和11.0时,对盐酸土霉素的降解率分别为91.5%、93.1%、85.3%和79.8%,这与预估一致,一方面是由于
SO⋅−4 和·OH在pH≤3时与H+发生反应引起的(式(2)和式(3)),导致SO⋅−4 和·OH被清除;另一方面是由于SO⋅−4 与OH−和水发生反应引起的(式(4)和式(5)),导致SO⋅−4 被转化为·OH。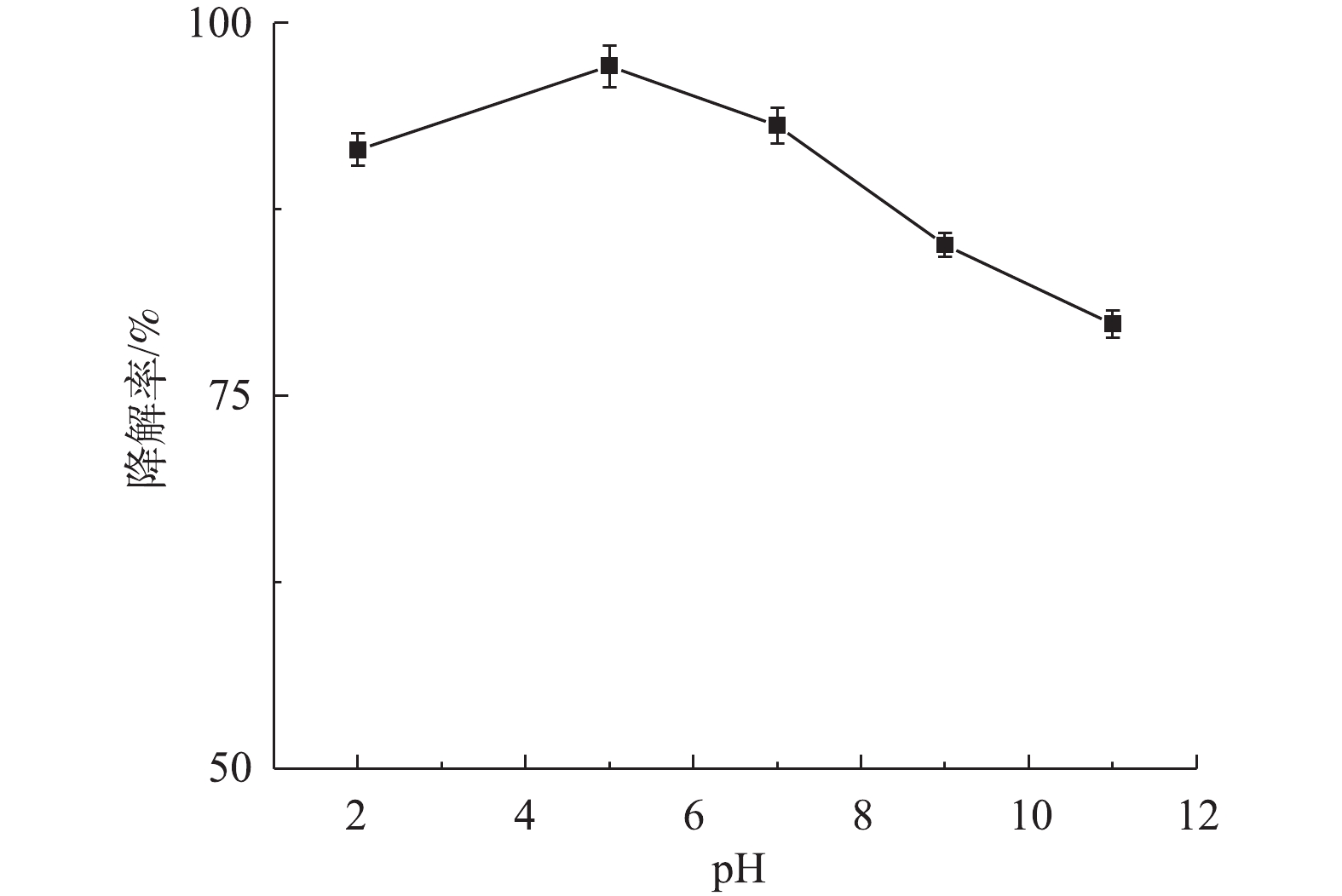 图 5 不同pH对降解盐酸土霉素的影响Figure 5. Effect of different pH on degradation of oxytetracycline hydrochloride
图 5 不同pH对降解盐酸土霉素的影响Figure 5. Effect of different pH on degradation of oxytetracycline hydrochloride⋅OH+H++e−→H2O (2) SO4⋅−+H++e−→HSO4− (3) SO4⋅−+OH−→SO22−+⋅OH (4) SO4⋅−+H2O→SO22−+OH+H+ (5) 2) PS投加量对盐酸土霉素降解的影响。由图6可知,当PS添加量为从50 mg增加到100 mg时,对盐酸土霉素的降解率从78.9%上升到95.8%,这表明盐酸土霉素的降解率随着PS投加量的增加而变大。而PS投加量达到150 mg时,对盐酸土霉素的降解率下降,原因是由于过多的PS会使
SO⋅−4 量超过临界值,使SO⋅−4 被PS捕获清除[25]。根据图6中拟合曲线结果,Co-MOF活化PS降解盐酸土霉素的动力学曲线符合准二级动力学模型,50 mg和100 mg PS添加量对应的的拟合曲线中R2分别为0.992 6和0.995 6,而150 mg PS添加量的拟合曲线中的R2最低为0.989 3,由于100 mg PS添加量的拟合曲线中R2高于50 mg添加量,所以确认本研究中最佳PS投加量为100 mg。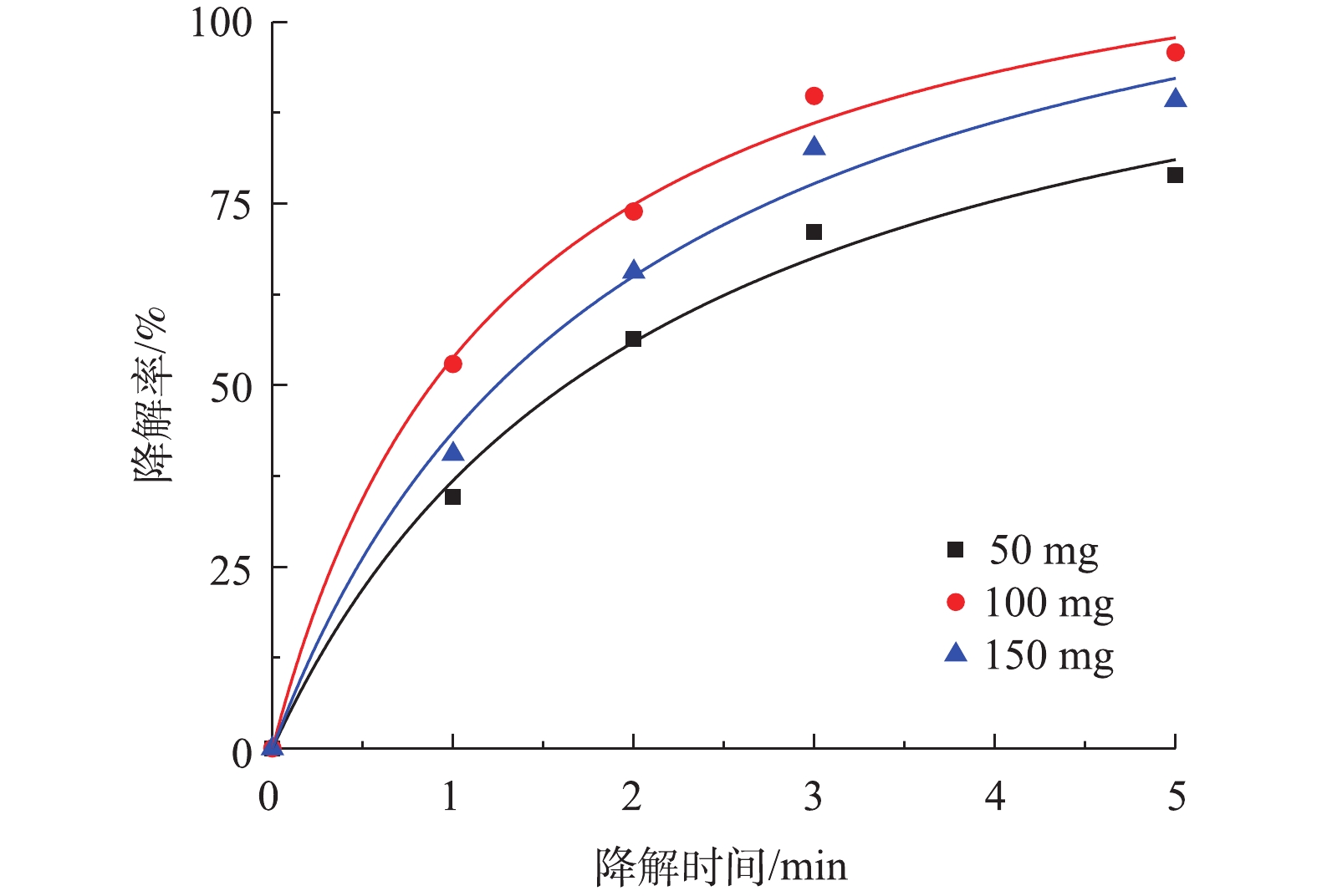 图 6 PS投加量对降解盐酸土霉素的影响及二级动力学曲线Figure 6. Effect of PS dosage on degradation of oxytetracycline hydrochloride and second-order kinetic curve
图 6 PS投加量对降解盐酸土霉素的影响及二级动力学曲线Figure 6. Effect of PS dosage on degradation of oxytetracycline hydrochloride and second-order kinetic curve3) Co-MOF投加量对盐酸土霉素降解的影响。由图7可知,5 mg Co-MOF添加量对盐酸土霉素的降解率为77.6%,10 mg Co-MOF添加量对盐酸土霉素的降解率为95.1%,而15 mg Co-MOF添加量对盐酸土霉素的降解率为87.2%,这表明随着Co-MOF投加量的增加,盐酸土霉素去除率呈现先增后减的趋势。造成这种情况的原因是:一方面是由于过多的
SO∙−4 会产生自我清除反应;另一方面由于过多的Co-MOF会增加溶液内的浊度。根据图7中拟合曲线结果,Co-MOF活化PS降解盐酸土霉素的动力学曲线符合准二级动力学模型,其中5 mg Co-MOF添加量的曲线拟合度R2最小为0.989 6,而10 mg和15 mg Co-MOF添加量的曲线拟合度R2分别为0.993 7和0.992 0,由于10 mg Co-MOF添加量的拟合曲线中的R2最高,所以确定本研究中最佳Co-MOF投加量为10 mg。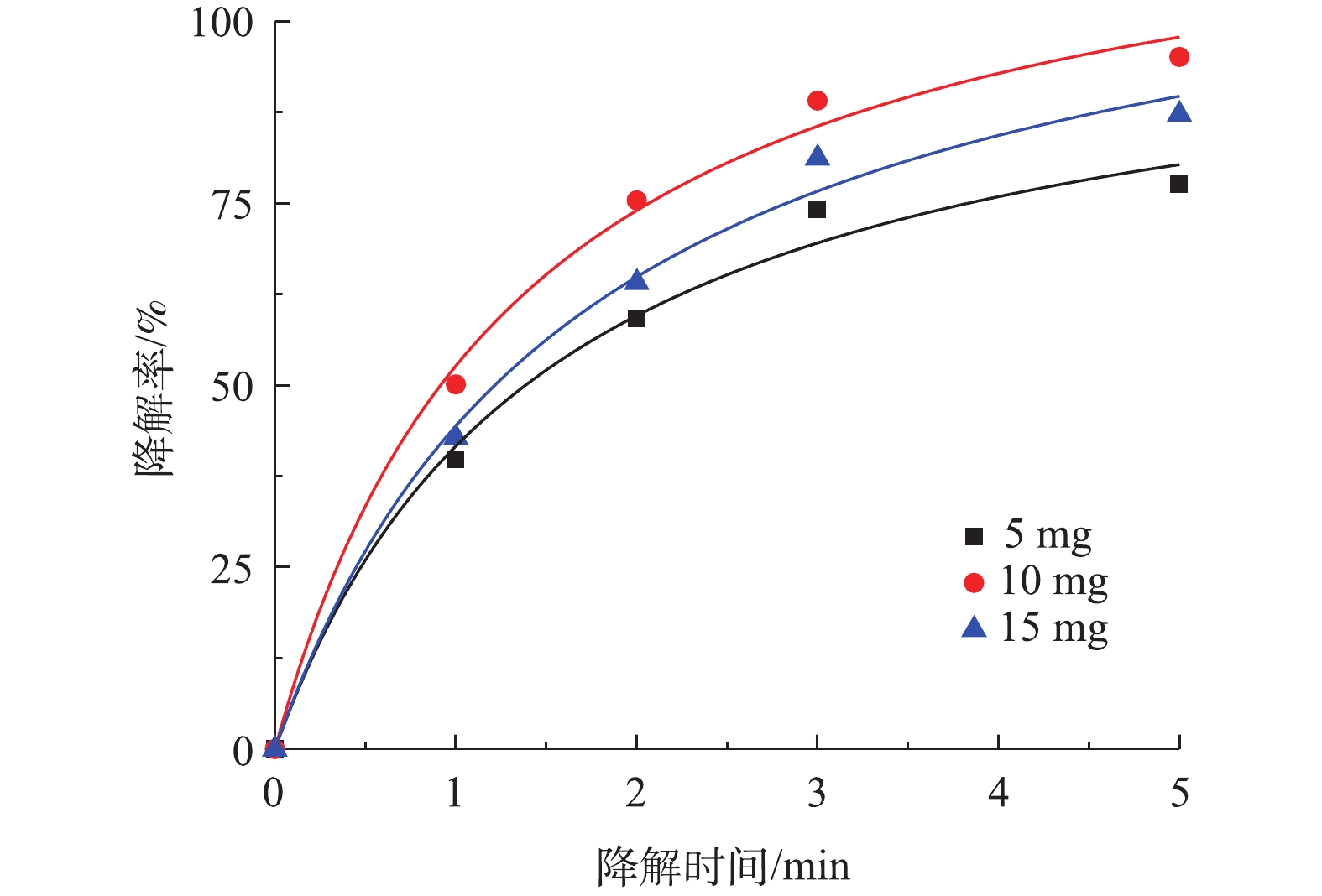 图 7 Co-MOF投加量对降解盐酸土霉素的影响及二级动力学曲线Figure 7. Effect of Co-MOF dosage on degradation of oxytetracycline hydrochloride and second-order kinetic curve
图 7 Co-MOF投加量对降解盐酸土霉素的影响及二级动力学曲线Figure 7. Effect of Co-MOF dosage on degradation of oxytetracycline hydrochloride and second-order kinetic curve4)温度对盐酸土霉素降解的影响。图8为不同反应温度对盐酸土霉素降解的影响。如图8所示,在25 ℃时,对盐酸土霉素的降解率有82.1%;在30 ℃时,对盐酸土霉素的降解率上升到92.3%;而当45 ℃时,对盐酸土霉素的降解率高达95.6%。这表明随着反应温度的升高,对盐酸土霉素的降解率也随之增大。在Co-MOF/PS体系中,
SO⋅−4 的生成不仅被Co-MOF的活化性能影响,还被温度影响。随着温度的增加,加快体系中PS的热激活行为,加速电子转移,使PS的O—O键更易于断裂,SO⋅−4 的生成更多更快,从而提高对盐酸土霉素的降解率。以往的报道[26-27]也证实这点,如刘红梅等[28]开展的热活化过硫酸盐降解土壤和地下水中污染物的研究发现,当温度过低时并不能对过硫酸盐进行有效的活化并生成SO∙−4 ,反而在升温过程中SO⋅−4 的生成速率会加快,进而促进盐酸土霉素的降解;但当温度过高时也会受到抑制影响,原因是由于自由基被高温清除。龙安华等[29]关于活化过硫酸盐降解水污染的研究结果也表明,在热活化过硫酸盐的过程中,随着温度的变化,pH、离子强度及有机质耗氧等也会产生相应的改变,所以温度会影响到污染物的降解率。根据图8的结果可见,Co-MOF活化PS降解盐酸土霉素的动力学曲线符合准二级动力学模型,其中30 ℃的曲线拟合度R2最高为0.991 0,故确定30 ℃为本次研究的最佳反应温度。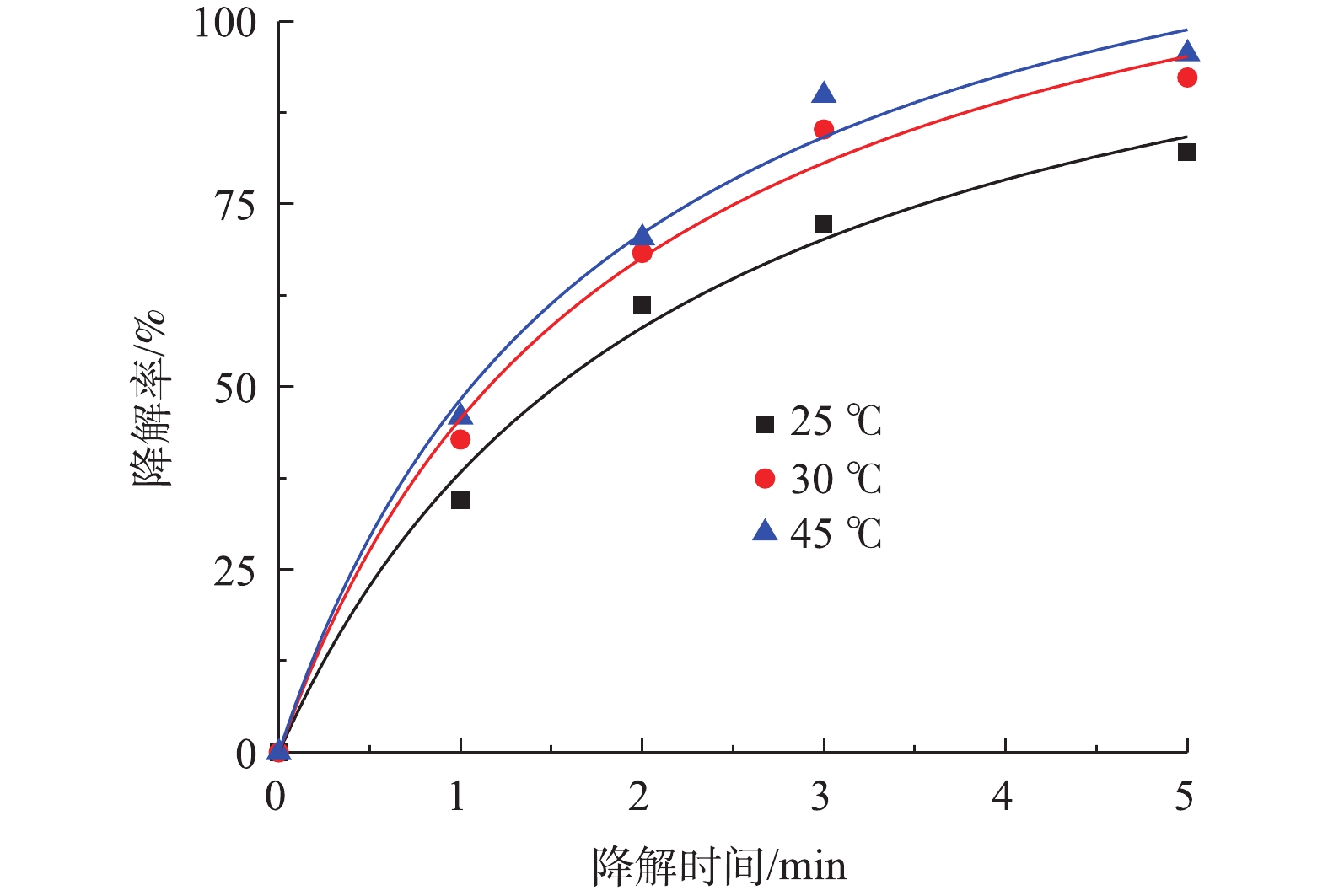 图 8 不同温度对盐酸土霉素的去除影响及二级动力学曲线Figure 8. Effects of different temperatures on the removal of oxytetracycline hydrochloride and second-order kinetic curves
图 8 不同温度对盐酸土霉素的去除影响及二级动力学曲线Figure 8. Effects of different temperatures on the removal of oxytetracycline hydrochloride and second-order kinetic curves2.4 Co-MOF重复利用对降解盐酸土霉素的影响
催化剂的稳定性是检验催化剂性能的重要指标,图9为Co-MOF重复利用对降解盐酸土霉素的影响。如图9所示,Co-MOF第4次运行后,对盐酸土霉素的降解率下降为82.1%,这表明Co-MOF具备较高的稳定性。其中,导致盐酸土霉素降解率降低的原因是:一方面由于Co-MOF活化PS降解盐酸土霉素时,长时间浸泡导致Co(Ⅱ)的活性位点损失;另一方面是由于Co2+随着多次循环出现微量损失(式(6))。第4次运行结束后收集定量的上清液,经检测显示钴含量为1×10−4 mg·L−1,而国标(GB 25467-2010)中的钴排放标准为总钴含量≤1 mg·L−1,故本次研究中Co-MOF的使用不会对环境造成二次污染。
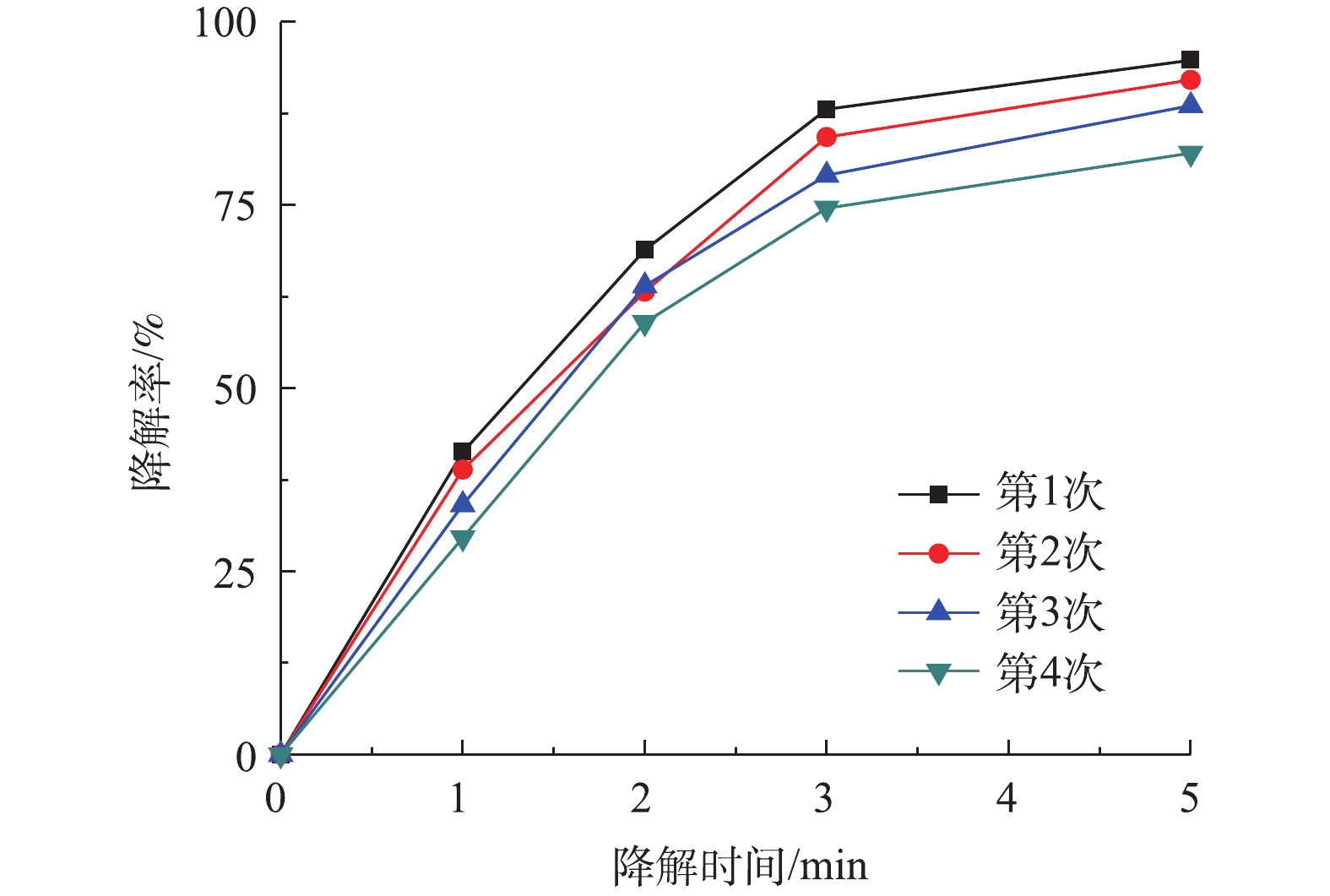 图 9 重复利用Co-MOF对降解盐酸土霉素的影响Figure 9. Effect of recycled Co-MOF on degradation of oxytetracycline hydrochloride
图 9 重复利用Co-MOF对降解盐酸土霉素的影响Figure 9. Effect of recycled Co-MOF on degradation of oxytetracycline hydrochlorideCo2++S2O82−→SO4⋅−+SO42−+Co3+ (6) 2.5 反应机理
1) Co-MOF降解盐酸土霉素反应前后的XPS表征。通过XPS对Co-MOF降解盐酸土霉素反应前后的组分变化进行分析。图10为Co-MOF反应前后的XPS图谱。如图10(a)所示,Co-MOF材料的宽扫描光电子能谱呈现Co2p、O1s和C1s 3个光谱图。在Co2p核心级XPS光谱中(图10(b)),反应前的Co2p1/2和Co2p3/2的结合能对应781.4 eV和800.4 eV;在786.2 eV的峰值是由辅峰造成的,由Co的sp3杂化所形成。有研究[30]表明,在低结合能一侧的781.4 eV若是归属于Co(Ⅱ),就表明Co在材料中的主要存在状态是Co(Ⅱ)氧化态,其在pH=2时没明显变化,而在pH=11时Co2p1/2对应的强度明显增强,这表明Co-MOF在酸性条件下更稳定。由图10(a)可知,在pH=11时反应后的Co2p的峰值有所升高,这是由于碱性条件下Co3+被还原为Co2+所致(式(7))。图10(c)显示的是O1s的光谱,反应前和当体系的pH=2反应后的结合能峰均为532.4 eV,表明CO-MOF中的羧基没有受到损坏,结合C1s的XPS光谱中(图10(d))的数据,其结合能没有发生明显的变化。综上所述,Co-MOF具有良好的稳定性。
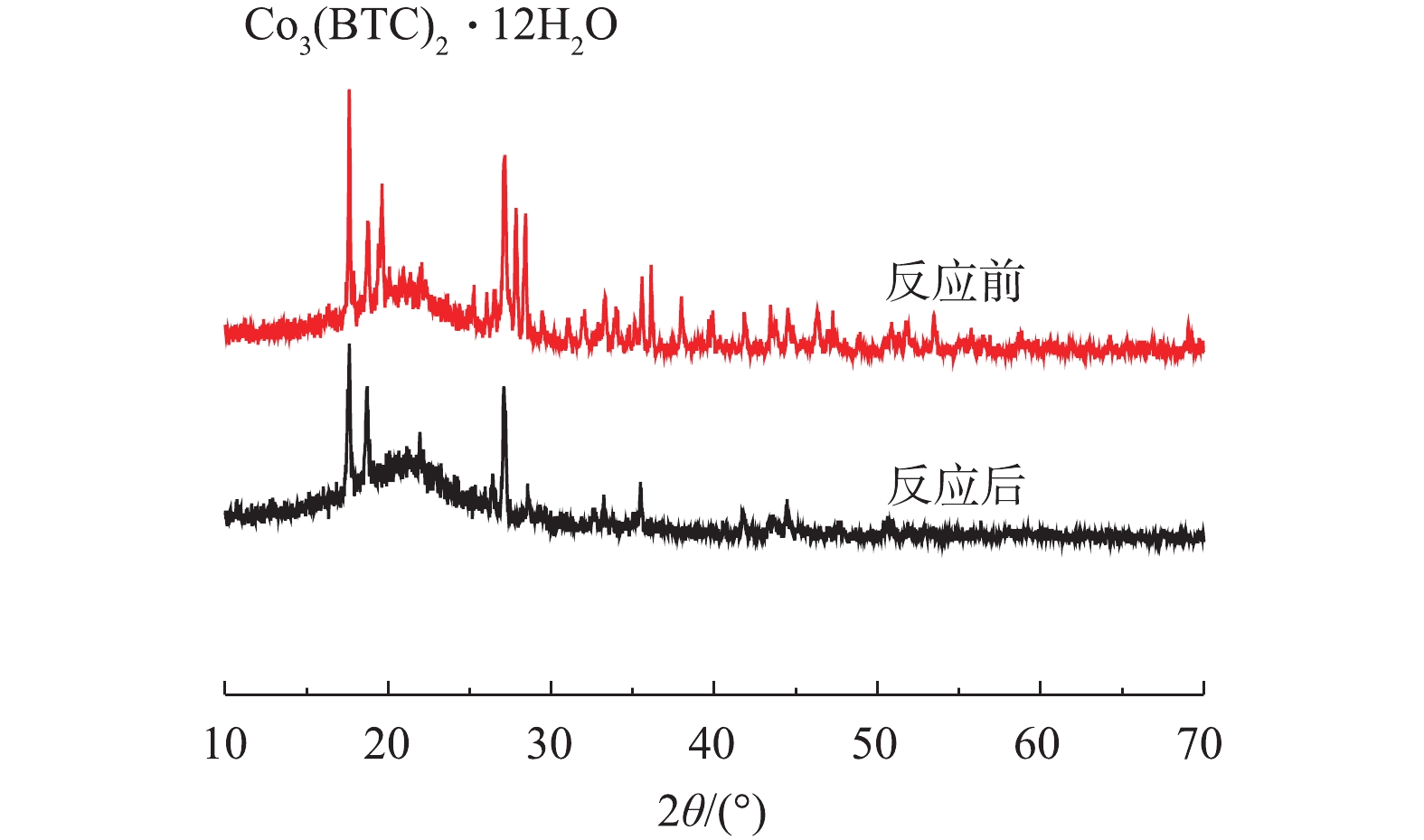
Co3++OH−→3/2Co2++H−+1/2O2 (7) 2) Co-MOF降解盐酸土霉素反应前后的XRD表征。如图11所示,在Co-MOF降解盐酸土霉素反应完成后,XRD图谱中Co3(BTC)2·12H2O对应的峰强度降低,这表明Co-MOF材料在降解盐酸土霉素的过程中有损失。
3)自由基淬灭剂对Co-MOF降解盐酸土霉素的影响。为了确定Co-MOF活化PS所产生的自由基种类,采用自由基捕获实验以验证活化体系中可能存在的自由基。众所周知,抗坏血酸和乙醇均为·OH和
SO⋅−4 的淬灭剂,而叔丁醇仅为·OH的淬灭剂。如图12所示,加入叔丁醇的实验组中,盐酸土霉素的降解率与无添加组相比有微弱下降,而加入抗坏血酸和乙醇的实验组中,盐酸土霉素的降解率无添加组相比大幅度下降,这表明在对盐酸土霉素降解的实验过程中同时涉及到SO⋅−4 和·OH,且SO⋅−4 是起主导作用的。在pH=2的条件下,加入抗坏血酸、乙醇和叔丁醇后,相比较空白条件,盐酸土霉素的降解率反而得到提升,加入叔丁醇的实验组对盐酸土霉素的降解率的提升效果微弱,这表明·OH在酸性条件下更易于生成,如陈震等[31]研究溶液pH对·OH的影响时发现,酸性环境更易于·OH的生成。当pH=11时,在加入淬灭剂后,发现对于盐酸土霉素降解效果的抑制明显高于空白条件下,原因是由于碱性环境有利于·OH的去除。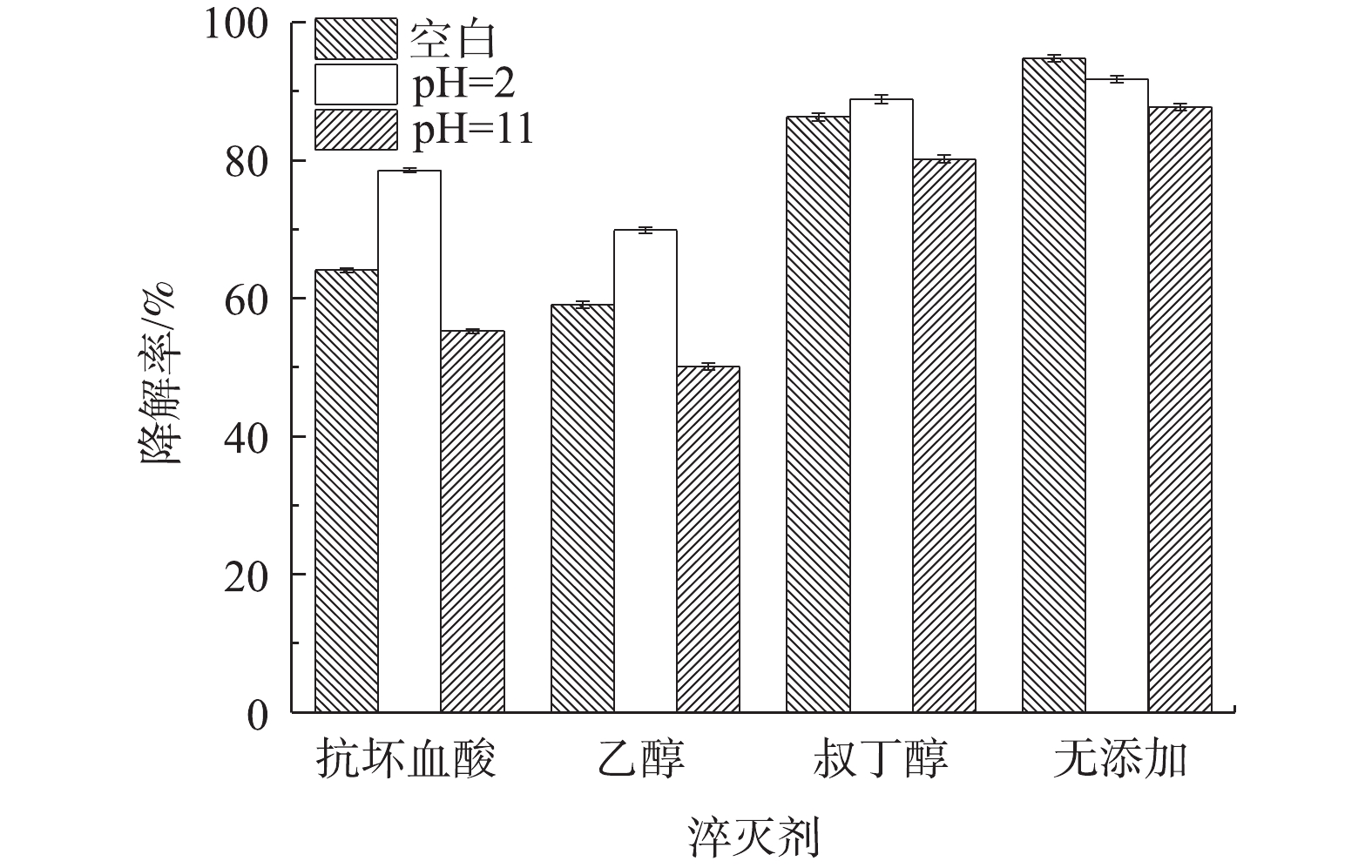 图 12 自由基淬灭剂在不同pH下对降解盐酸土霉素的影响Figure 12. Effect of free radical quencher on the degradation of oxytetracycline hydrochloride at different pHs
图 12 自由基淬灭剂在不同pH下对降解盐酸土霉素的影响Figure 12. Effect of free radical quencher on the degradation of oxytetracycline hydrochloride at different pHs3. 结论
1) SEM、TEM和XRD的表征结果表明,采用水热法成功制备出Co-MOF。该催化剂具有较好的降解性能,在Co-MOF浓度为200 mg·L−1、PS浓度为2 000 mg·L−1时,5 min后Co-MOF/PS对盐酸土霉素的降解率达到95.4%。
2) Co-MOF投加量、PS投加量和温度对Co-MOF降解盐酸土霉素有较大的影响,pH对降解盐酸土霉素的影响不大。其中,当pH=5、温度30 ℃、Co-MOF浓度为200 mg·L−1、PS浓度为2 000 mg·L−1时,Co-MOF/PS对盐酸土霉素的降解率可达到最高。
3)重复利用Co-MOF降解盐酸土霉素结果表明,Co-MOF具有可重复利用性能。Co-MOF反应前后的XRD和XPS数据表明,Co-MOF具有良好的稳定性。自由基捕获结果表明,在该降解反应过程中,
SO⋅−4 和·OH对盐酸土霉素的降解均有贡献,其中,SO⋅−4 的贡献最大。 -
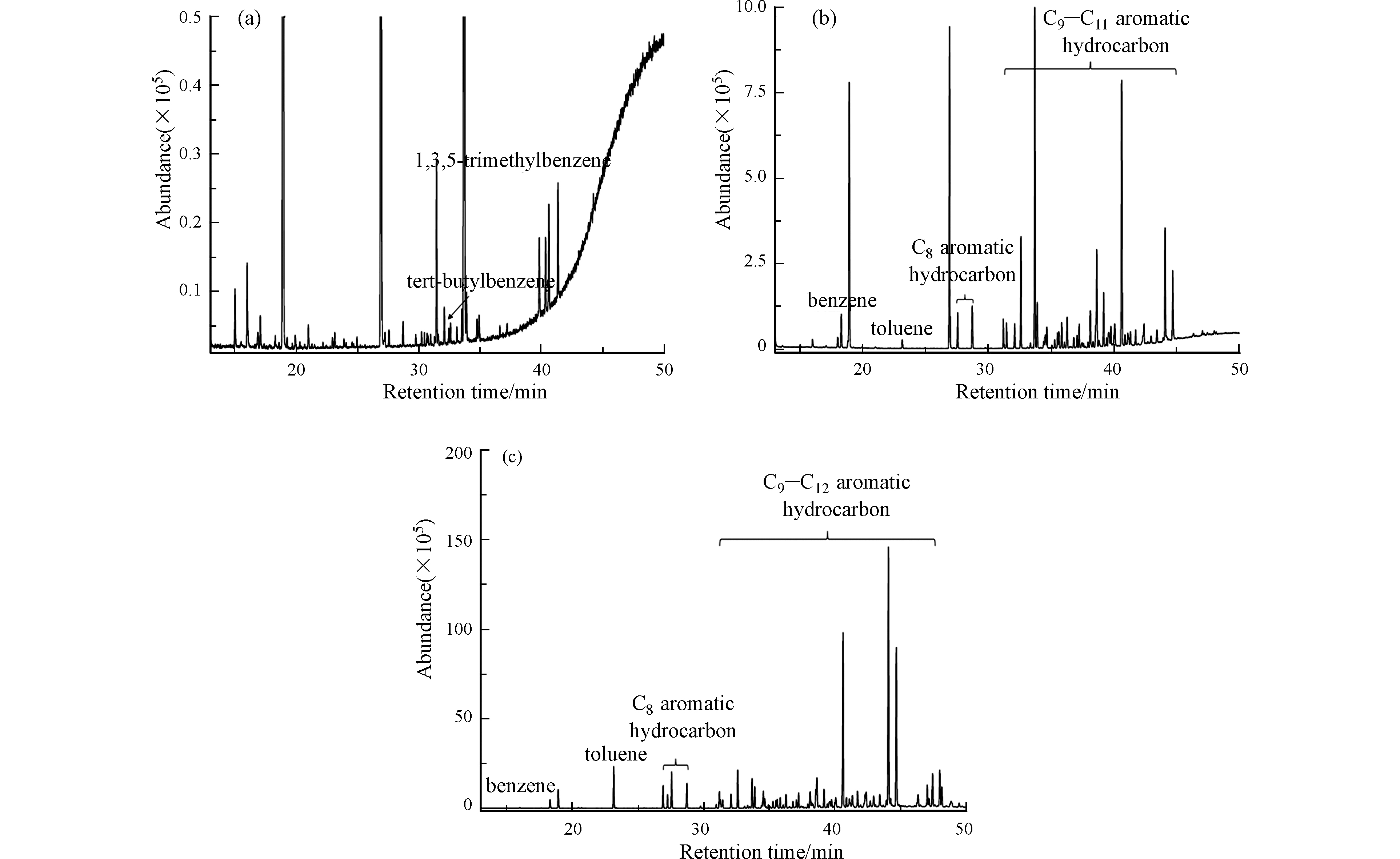
图 2 FCC(a)、S Zorb(b)和渣油加氢(c)废催化剂浸出液的总离子流色谱图
Figure 2. Total ion flow chromatogram of waste catalyst leaching liquor from FCC(a), S Zorb(b)and residue hydrotreating(c)
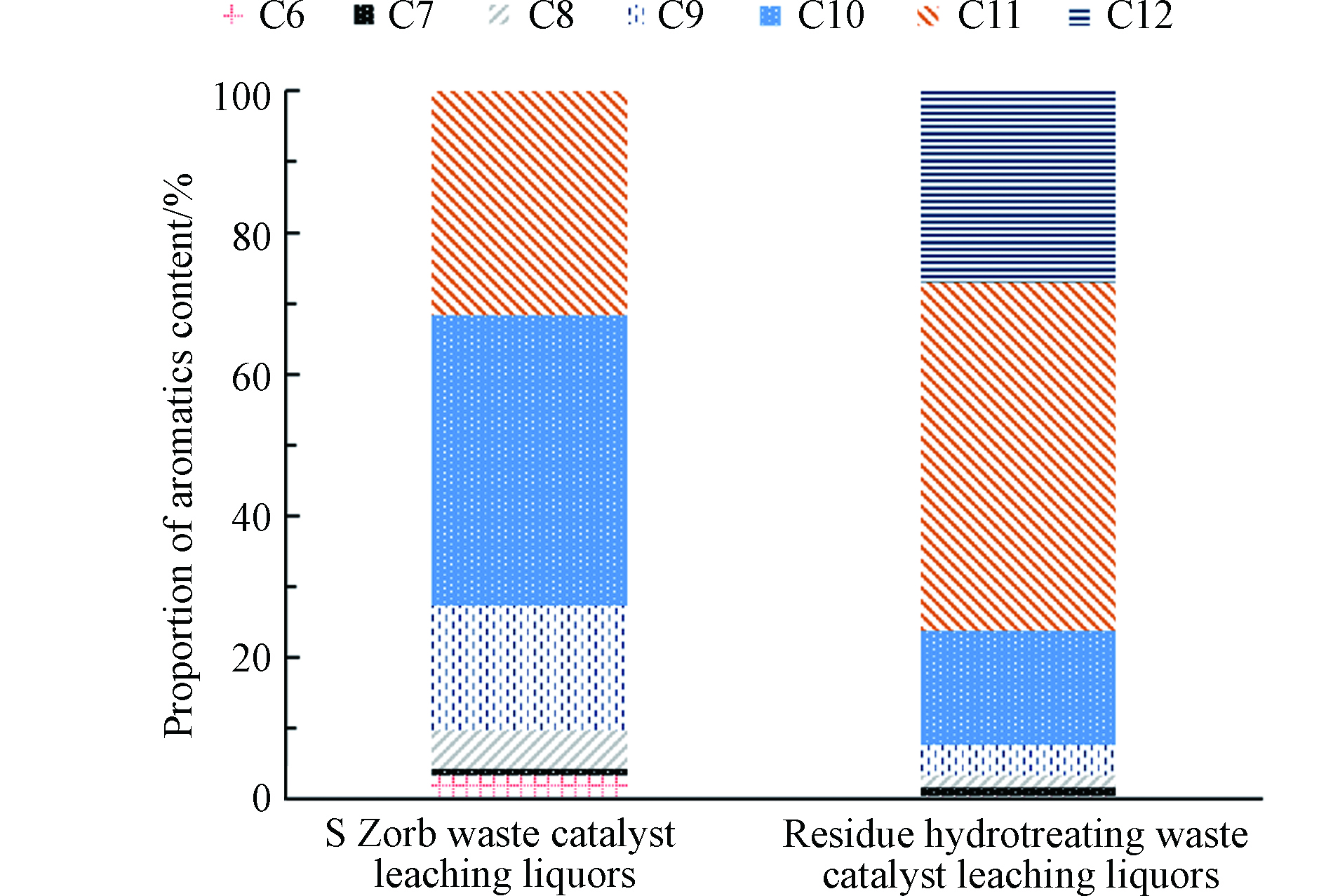
图 3 S Zorb和渣油加氢废催化剂浸出液VOCs的组成和分布
Figure 3. Composition and distribution of VOCs in waste catalyst leaching liquors from S Zorb and residue hydrotreating
表 1 吹扫捕集/气相色谱-质谱法的校准曲线和检出限
Table 1. Calibration curves and detection limits of P&T-GC-MS
VOC种类 Type of VOCs 校准曲线 Calibration curves R2 检出限/(μg·L−1) Detection limits 反-1,2-二氯乙烯 y = 0.120x+0.003 0.9999 0.8 1,1-二氯乙烷 y = 0.246x+0.009 0.9998 0.6 顺-1,2-二氯乙烯 y = 0.196x+0.005 0.9999 0.7 2-丁酮 y = 0.035x+0.008 1.0000 2.0 2,2-二氯丙烷 y = 0.142x 0.9999 1.5 溴氯甲烷 y = 0.102x+0.003 0.9999 0.7 氯仿 y = 0.323x+0.011 0.9998 0.6 二溴氟甲烷 y = 0.176x 1.0000 0.4 1,1,1-三氯乙烷 y = 0.242x−0.002 0.9998 1.2 四氯化碳 y = 0.175x−0.002 0.9997 1.2 1,1-二氯丙烯 y = 0.126x−0.003 0.9999 0.3 苯 y = 0.545x+0.010 0.9999 0.3 1,2-二氯乙烷 y = 0.226x+0.007 0.9998 0.2 三氯乙烯 y = 0.136x+0.002 0.9999 0.3 1,2-二氯丙烷 y = 0.168x 1.0000 0.2 二溴甲烷 y = 0.122x+0.006 0.9991 0.4 一溴二氯甲烷 y = 0.238x−0.008 0.9998 0.2 4-甲基-2-戊酮 y = 0.045x−0.005 0.9987 0.4 甲苯-D8 y = 0.587x+0.010 0.9997 0.3 甲苯 y = 0.449x−0.001 1.0000 0.3 1,1,2-三氯乙烷 y = 0.180x+0.004 0.9997 0.3 四氯乙烯 y = 0.107x+0.003 0.9998 0.4 1,3-二氯丙烷 y = 0.357x+0.005 0.9997 0.2 2-己酮 y = 0.166x−0.012 0.9997 0.3 二溴氯甲烷 y = 0.226x−0.016 0.9995 0.2 1,2-二溴乙烷 y = 0.229x−0.005 1.0000 0.1 氯苯 y = 0.580x+0.010 0.9999 0.2 1,1,1,2-四氯乙烷 y = 0.277x−0.012 0.9997 0.5 乙苯 y = 0.289x−0.007 0.9999 0.3 1,1,2-三氯丙烷 y = 0.483x+0.001 0.9999 0.4 间,对二甲苯 y = 0.733x−0.022 0.9998 0.4 邻二甲苯 y = 0.448x−0.022 0.9996 0.5 苯乙烯 y = 0.660x−0.061 0.9991 0.6 溴仿 y = 0.171x−0.022 0.9972 0.4 异丙苯 y = 1.530x−0.025 0.9997 0.3 4-溴氟苯 y = 0.464x+0.009 0.9995 0.2 1,1,2,2-四氯乙烷 y = 0.567x−0.008 0.9999 1.0 溴苯 y = 0.426x+0.018 0.9990 0.2 1,2,3-三氯丙烷 y = 0.335x+0.007 0.9956 1.6 正丙苯 y = 1.270x+0.022 0.9962 0.6 2-氯甲苯 y = 1.251x+0.011 0.9997 0.3 1,3,5-三甲基苯 y = 1.526x−0.061 1.0000 0.5 4-氯甲苯 y = 1.225x+0.028 0.9995 0.3 叔丁基苯 y = 1.196x−0.082 0.9998 0.3 1,2,4-三甲基苯 y = 1.614x−0.046 1.0000 0.5 仲丁基苯 y = 1.729x−0.074 1.0000 0.5 1,3-二氯苯 y = 0.895x+0.026 0.9997 0.3 4-异丙基甲苯 y = 1.523x−0.061 1.0000 0.6 1,4-二氯苯 y = 0.936x+0.021 0.9999 0.2 正丁基苯 y = 1.356x−0.061 1.0000 0.6 1,2-二氯苯 y = 0.966x+0.022 0.9999 0.4 1,2-二溴-3-氯丙烷 y = 0.180x−0.017 0.9989 0.4 1,2,4-三氯苯 y = 0.720x−0.010 1.0000 0.7 六氯丁二烯 y = 0.234x 1.0000 2.0 萘 y = 2.596x−0.097 0.9984 0.8 1,2,3-三氯苯 y = 0.760x+0.023 0.9992 0.8 注:表中x为目标VOCs含量与相对应内标物含量的比值,y为目标VOCs峰面积与相对应内标物峰面积的比值. Note: in the table, x is the ratio of target VOCs content to corresponding internal standard content, and y is the ratio of target VOCs peak area to corresponding internal standard peak area.  下载: 导出CSV
下载: 导出CSV
表 2 废催化剂浸出液中VOCs测定结果及精密度
Table 2. Determination results and precisions of VOCs in waste catalyst leaching liquors
VOC种类Type of VOCs FCC废催化剂浸出液Waste catalyst leaching liquor from FCC S Zorb废催化剂浸出液Waste catalyst leaching liquor from S Zorb 渣油加氢废催化剂浸出液Waste catalyst leaching liquor from residue hydrotreating S/(μg·L−1) 平均值/(μg·L−1) RSD/% S/(μg·L−1) 平均值/(μg·L−1) RSD/% S/(μg·L−1) 平均值/(μg·L−1) RSD/% 氯仿 — — — — — — — — — 四氯化碳 — — — — — — — — — 苯 — — — 0.089 9.3 1.0 0.279 24.1 1.2 三氯乙烯 — — — — — — — — — 甲苯 — — — 0.028 2.40 1.2 1.217 87.3 1.4 四氯乙烯 — — — — — — — — — 氯苯 — — — — — — — — — 乙苯 — — — — — — 0.233 25.0 0.9 间,对二甲苯 — — — 0.064 5.10 1.2 0.405 36.5 1.1 邻二甲苯 — — — 0.163 9.72 1.7 0.470 44.1 1.1 苯乙烯 — — — — — — — — — 异丙苯 — — — — — — 0.083 4.86 1.7 正丙苯 — — — — — — 0.157 4.80 3.3 1,3,5-三甲基苯 0.024 3.89 0.6 0.070 6.47 1.1 0.617 16.9 3.7 叔丁基苯 0.005 3.44 0.2 0.139 3.55 3.9 — — — 1,2,4-三甲基苯 — — — 0.137 18.9 0.7 2.767 67.0 4.1 仲丁基苯 — — — — — — 0.283 7.79 3.6 4-异丙基甲苯 — — — — — — 0.246 6.96 3.5 1,4-二氯苯 — — — — — — — — — 正丁基苯 — — — — — — 0.362 8.45 4.3 1,2-二氯苯 — — — — — — — — — 萘 — — — 1.387 39.2 3.5 13.9 364 3.8 注:S为标准偏差(μg·L−1);表中未列出的VOCs表示其质量浓度低于方法检出限. Note: S is the standard deviation(μg·L−1);VOCs not listed in the table indicated that their mass concentrations were lower than the detection limits of the method.
下载: 导出CSV
表 3 废催化剂浸出液基体加标回收率测定结果
Table 3. Determination of recovery rates of waste catalyst leaching liquors by matrix standard addition
VOC种类Type of VOCs FCC废催化剂浸出液Waste catalyst leaching liquor from FCC S Zorb废催化剂浸出液Waste catalyst leaching liquor from S Zorb 渣油加氢废催化剂浸出液Waste catalyst leaching liquor from residue hydrotreating 实际样品含量/(μg·L−) Content in practical sample 加标后浓度/(μg·L−1)Content after standard addition 回收率/%Recovery rate 实际样品含量/(μg·L−1)Content in practical sample 加标后浓度/(μg·L−1)Content after standard addition 回收率/%Recovery rate 实际样品含量/(μg·L−1)Content in practical sample 加标后浓度/(μg·L−1)Content after standard addition 回收率/%Recovery rate 反-1,2-二氯乙烯 — 80.5 161.0 — 72.8 145.6 — 67.8 135.6 1,1-二氯乙烷 — 68.5 137.0 — 61.1 122.2 — 83.8 167.6 顺-1,2-二氯乙烯 — 63.8 127.6 — 55.9 111.9 — 78.2 156.4 2-丁酮 — 60.7 121.4 — 55.0 109.9 — 82.8 165.6 2,2-二氯丙烷 — 71.4 142.9 — 52.2 104.4 — 77.7 155.5 溴氯甲烷 — 65.9 131.8 — 57.1 114.1 — 80.3 160.5 氯仿 — 54.8 109.7 — 48.1 96.2 — 65.8 131.7 二溴氟甲烷 — 52.3 104.6 — 45.0 90.0 — 62.8 125.5 1,1,1-三氯乙烷 — 54.8 109.7 — 45.1 90.2 — 53.4 106.9 四氯化碳 — 47.9 95.9 — 38.6 77.3 — 43.4 86.8 1,1-二氯丙烯 — 50.1 100.1 — 43.8 87.5 — 55.1 110.2 苯 — 61.2 122.5 9.3 58.3 97.9 24.1 92.0 135.7 1,2-二氯乙烷 — 60.2 120.5 — 52.9 105.9 — 71.3 142.6 三氯乙烯 — 50.5 101.1 — 44.0 88.0 — 57.3 114.7 1,2-二氯丙烷 — 58.3 116.7 — 50.7 101.4 — 66.5 133.1 二溴甲烷 — 59.6 119.3 — 51.7 103.4 — 69.9 139.8 一溴二氯甲烷 — 51.6 103.2 — 44.0 88.0 — 62.6 125.3 4-甲基-2-戊酮 — 62.1 124.2 — 52.6 105.2 — 75.3 150.6 甲苯-D8 — 48.4 96.9 — 42.6 85.3 — 54.9 109.7 甲苯 — 49.6 99.1 2.40 45.1 85.3 87.3 136.2 97.8 1,1,2-三氯乙烷 — 55.4 110.8 — 49.2 98.4 — 65.6 131.1 四氯乙烯 — 41.7 83.4 — 36.0 72.1 — 43.4 86.9 1,3-二氯丙烷 — 56.1 112.1 — 50.3 100.6 — 67.2 134.4 2-己酮 — 52.4 104.9 — 46.4 92.7 — 70.4 140.8 二溴氯甲烷 — 51.6 103.1 — 45.4 90.7 — 61.9 123.8 1,2-二溴乙烷 — 55.4 110.9 — 49.1 98.1 — 67.6 135.1 氯苯 — 52.0 104.1 — 45.2 90.3 — 58.3 116.6 1,1,1,2-四氯乙烷 — 51.8 103.6 — 43.4 86.8 — 56.9 113.8 乙苯 — 46.5 93.0 — 40.3 80.6 25.0 72.9 95.9 1,1,2-三氯丙烷 — 53.6 107.3 — 45.6 91.1 — 60.3 120.6 间,对二甲苯 — 47.0 93.9 5.10 42.5 74.7 36.5 84.6 96.2 邻二甲苯 — 50.9 101.7 9.72 47.7 75.9 44.1 95.9 103.6 苯乙烯 — 48.1 96.2 — 41.3 82.6 — 56.9 113.8 溴仿 — 50.4 100.7 — 43.1 86.3 — 60.8 121.6 异丙苯 — 47.5 95.0 — 41.1 82.2 4.86 52.1 94.5 4-溴氟苯 — 57.9 115.8 — 50.4 100.8 — 64.4 128.7 1,1,2,2-四氯乙烷 — 59.4 118.9 — 49.1 98.3 — 70.4 140.8 溴苯 — 54.9 109.8 — 48.1 96.1 — 58.8 117.6 1,2,3-三氯丙烷 — 45.0 90.1 — 42.6 85.2 — 53.6 107.3 正丙苯 — 42.1 84.3 — 37.5 75.0 4.80 50.7 91.9 2-氯甲苯 — 53.2 106.4 — 46.4 92.7 — 56.2 112.3 1,3,5-三甲基苯 3.89 51.5 95.3 6.47 45.4 77.8 16.9 67.5 101.3 4-氯甲苯 — 51.6 103.3 — 45.3 90.5 — 53.8 107.6 叔丁基苯 3.44 45.3 83.7 3.55 39.7 72.2 — 49.5 99.0 1,2,4-三甲基苯 — 53.0 106.0 18.9 56.5 75.2 67.0 128.1 122.2 仲丁基苯 — 42.5 85.0 — 36.5 72.9 7.79 47.2 78.9 1,3-二氯苯 — 53.9 107.7 — 46.5 92.9 — 54.4 108.8 4-异丙基甲苯 — 45.7 91.4 — 38.9 77.9 6.96 48.4 82.9 1,4-二氯苯 — 53.8 107.7 — 47.2 94.4 — 56.1 112.2 正丁基苯 — 42.3 84.7 — 37.1 74.2 8.45 47.7 78.6 1,2-二氯苯 — 58.5 116.9 — 49.8 99.5 — 59.6 119.2 1,2-二溴-3-氯丙烷 — 59.3 118.6 — 50.6 101.1 — 74.7 149.5 1,2,4-三氯苯 — 60.8 121.6 — 51.6 103.2 — 49.2 98.4 六氯丁二烯 — 39.3 78.7 — 32.5 64.9 — 37.0 74.0 萘 — 64.8 129.5 39.2 85.2 92.1 364 430.2 131.7 1,2,3-三氯苯 — 62.0 124.0 — 52.4 104.9 — 47.1 94.3 注:VOCs的加标量均为50.0 μg·L−1. Note: adding standard amounts of VOCs were 50.0 μg·L−1.
下载: 导出CSV
-
[1] 生态环境部, 国家发展和改革委员会, 公安部, 等. 国家危险废物名录(2021年版) [R/OL]. 2020: [000014672/2020-01495]. http://www.mee.gov.cn/xxgk2018/xxgk/xxgk02/202011/t20201127_810202.html. Ministry of Ecology and Environment, National Development and Reform Commission, Ministry of Public Security, et al. National hazardous waste list (2021 Edition) [R/OL]. 2020: [000014672/2020-01495]. http://www.mee.gov.cn/xxgk2018/xxgk/xxgk02/202011/t20201127_ 810202. html.
[2] 胡桐搏, 张长平, 刘浩, 等. VOCs废气危害及处理技术进展 [J]. 化工管理, 2018(10): 125-127. doi: 10.3969/j.issn.1008-4800.2018.10.088 HU T B, ZHANG C P, LIU H, et al. The damage and development of treatment technique of VOCs [J]. Chemical Enterprise Management, 2018(10): 125-127(in Chinese). doi: 10.3969/j.issn.1008-4800.2018.10.088
[3] 齐安安, 周小平, 雷春妮, 等. 兰州市功能区环境空气中挥发性有机物关键活性组分与来源解析 [J]. 环境化学, 2020, 39(11): 3083-3093. doi: 10.7524/j.issn.0254-6108.2019080402 QI A A, ZHOU X P, LEI C N, et al. Key active components and sources of volatile organic compounds in ambient air of Lanzhou City [J]. Environmental Chemistry, 2020, 39(11): 3083-3093(in Chinese). doi: 10.7524/j.issn.0254-6108.2019080402
[4] 国家环境保护总局, 国家质量监督检验检疫总局. 中华人民共和国国家标准: GB5085.3—2007 [S]. 北京: 中国环境科学出版社, 2007. State Environmental Protection Administration of China, General Administration of quality supervision, inspection and Quarantine of the people's Republic of China. National standards of China: GB5085.3—2007 [S]. Beijing: China Environmental Science Press, 2007.
[5] 中华人民共和国环境保护部. 中华人民共和国环保行业标准: 固体废物 挥发性有机物的测定 顶空-气相色谱法 HJ 760—2015[S]. 北京: 中国环境科学出版社, 2015. Ministry of Environmental Protection of the People's Republic of China. Environmental Protection Standard of the People's Republic of China: Solid waste—Determination of volatile organic compounds—Headspace-gas chromatography method. HJ 760—2015[S]. Beijing: China Environment Science Press, 2015(in Chinese).
[6] 环境保护部. 中华人民共和国国家环境保护标准: HJ 874—2017 [S]. 北京: 中国环境科学出版社, 2017. Ministry of Environmental Protection. National standards of China: HJ 874—2017 [S]. Beijing: China Environmental Science Press, 2017.
[7] 生态环境部. 中华人民共和国国家环境保护标准: HJ 975—2018 [S]. 北京: 中国环境科学出版社, 2018. Ministry of Ecology and Environment. National standards of China: HJ 975—2018 [S]. Beijing: China Environmental Science Press, 2018.
[8] 环境保护部. 中华人民共和国国家环境保护标准: HJ 643—2013 [S]. 北京: 中国环境科学出版社, 2013. Ministry of Environmental Protection. National standards of China: HJ 643—2013 [S]. Beijing: China Environmental Science Press, 2013.
[9] 环境保护部. 中华人民共和国国家环境保护标准: HJ 713—2014 [S]. 北京: 中国环境科学出版社, 2014. Ministry of Environmental Protection. National standards of China: HJ 713—2014 [S]. Beijing: China Environmental Science Press, 2014.
[10] 环境保护部. 中华人民共和国国家环境保护标准: HJ 714—2014 [S]. 北京: 中国环境科学出版社, 2014. Ministry of Environmental Protection. National standards of China: HJ 714—2014 [S]. Beijing: China Environmental Science Press, 2014.
[11] 生态环境部. 中华人民共和国国家环境保护标准: HJ 976—2018 [S]. 北京: 中国环境科学出版社, 2018. Ministry of Ecology and Environment. National standards of China: HJ 976—2018 [S]. Beijing: China Environmental Science Press, 2018.
[12] 王爽, 丁巍, 赵德智, 等. 渣油加氢催化剂酸性、孔结构及分散度对催化活性的影响 [J]. 化工进展, 2015, 34(9): 3317-3322. doi: 10.16085/j.issn.1000-6613.2015.09.017 WANG S, DING W, ZHAO D Z, et al. Influence on catalytic activity in residue hydrogenation by acidity, pore structure and dispersion [J]. Chemical Industry and Engineering Progress, 2015, 34(9): 3317-3322(in Chinese). doi: 10.16085/j.issn.1000-6613.2015.09.017
[13] 陈德阳, 江思雨. 吹扫捕集-气质联用法测定固体废物浸出液中64种挥发性有机物[J]. 环境科学导刊, 2018, 37(增刊1): 142-148. CHEN D Y, JIANG S Y. Determination of sixty-four kinds of volatile organic compounds in solid waste leaching liquor by purging and trapping-gas chromatography and mass spectrometry[J]. Environmental Science Survey, 2018, 37(Sup 1): 142-148(in Chinese).
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 1995
- HTML全文浏览数: 1995
- PDF下载数: 65
- 施引文献: 0
