

-
塑料制品在我们的生活中无处不在。2017年,全球塑料产量上升至3.35亿吨[1]。最常见的塑料制品包括聚乙烯(PE)、聚丙烯(PP)、聚氯乙烯(PVC)、聚对苯二甲酸乙二酯(PET)、聚苯乙烯(PS)等。关于海洋中塑料的研究最早发表于20世纪70年代 [2]。在2004年,Thompson等[3]首次提出微塑料的概念,并引起了广泛关注。目前的研究表明微塑料存在于海洋环境[4-5]、淡水环境[6-9]、沉积物[10-11]、土壤[12-13]以及生物体内[14-15]。微塑料在被生物摄食后可造成挤压、占位等,从而导致生物摄食效率降低、生长缓慢、受伤或死亡等 [16-17]。微塑料本身也会释放塑化剂、阻燃剂、抗氧化剂等有毒物质 [18-22] 。此外,微塑料表面还能吸附环境中的疏水性有机污染物,在被水生生物摄食后,会对生物体产生毒性效应 [23-25] 。
准确、高效的分析方法是研究微塑料的环境行为及生态毒理学效应的关键前提。欧盟海洋战略框架指令(MSFD)[26]以及美国国家海洋和大气管理局[27](NOAA)分别发布了监测海洋环境中微塑料的实验方法。然而,目前微塑料的提取和分离方法尚未标准化。
本文综述了已有研究报道的水样、土壤和沉积物、水生生物样品中微塑料的前处理方法,并针对现有方法的优缺点进行比较(表1),对进一步的研究方向进行了讨论。
-
筛分过滤法利用孔径较小的不锈钢或铜制滤网、筛网来截留微塑料,是水样中分离微塑料最常用的方法,也可用于样品密度分离上清液中微塑料的提取 [4, 28-30] 。在土壤或沉积物的预处理中,可通过较大的筛网进行预处理,减少样品体积,再进行密度分离,并通过过滤器或筛网过滤上清液,从而分离微塑料。过滤和筛分法采用的筛网孔径大小决定了分离微塑料的尺寸,文献报道的孔径范围一般在0.038 mm到4.75 mm之间[4, 28-30]。对于孔径较小的滤膜容易堵塞,一般在负压条件下进行,再通过异丙醇溶液(50%,体积分数)将滤膜上的微塑料洗脱,从而提高微塑料的分离效率 [67]。目前并没有标准化的孔径尺寸,导致不同研究结果之间难以进行比较。
-
密度分离法的原理是利用样品中微塑料与矿物质等杂质的密度差异来实现提取分离。微塑料的密度范围从0.80 g·cm−3(如硅胶)到1.60 g·cm−3(如PVC、PET)不等,而沉积物的密度通常为2.65 g·cm−3 [4]。首先向样品中加入高密度的饱和盐溶液,充分振荡、搅拌混合均匀,随后静置沉淀直至轻组分微塑料与重组分杂质分层,最后收集上层溶液中的微塑料。目前,密度分离法广泛应用于水样、土壤和沉积物中微塑料的提取。不同的盐溶液密度不同,导致提取效率各不相同。
-
NaCl作为密度分离中使用最多的盐类之一,具有价廉易得、无害等优点 [31] ,是MSFD[26]和NOAA[27]推荐使用的前处理方法。PP(密度0.8 g·cm−3)、聚酰胺(PA,密度1.13 g·cm−3)等密度较低的聚合物可通过NaCl达到分离的效果 [32]。然而,由于NaCl溶液密度(1.2 g·cm−3)的局限性,导致高密度的微塑料包括PET(密度1.37—1.45 g·cm−3)和PVC(密度1.16—1.58 g·cm−3)的提取效率较低。而PET和PVC的产量几乎占世界塑料产量的17% [1],通过NaCl溶液进行样品前处理,可能会导致环境中PET和PVC等高密度微塑料的浓度被低估。
-
碘化钠(NaI,密度1.8 g·cm−3)是一种用于分离微塑料的高密度溶液。NaI价格昂贵[33-34],研究人员通过减少样品量、回收NaI等方式来降低前处理的成本。Nuelle等[31]对样品通过NaCl分离结合空气溢流(AIO)进行预处理,使初始样品的质量降低80%,再用NaI进行密度分离。通过这两个步骤,既可以有效提取PVC、PET等高密度的微塑料,还能够减少NaI的使用量。Claessens等[34]将样品首先通过洗脱柱中向上的水流和曝气,从而减少样品量,再通过NaI进行密度分离,对PVC的提取效率大幅增加。为了研究NaI的可回收利用性,Kedzierski等[35]在10个循环使用过程后,测定了NaI的溶液密度和损失,发现NaI溶液的密度没有变化,损失为35.9%,证明通过回收NaI的方法可以大大降低前处理成本。Quinn等[36]对比了几种溶液(NaCl、NaBr、ZnBr2、NaI)对沉积物中微塑料的密度分离提取效率,发现NaCl和NaBr的回收率较低(<90%),而NaI和ZnBr2能够有效分离高密度的聚合物,可重现性高。此外,使用NaI和ZnBr2分离只需要对沉淀物进行一次洗涤,而NaCl需要3次洗涤 [36] 。
-
氯化锌(ZnCl2,密度1.6—1.7 g·cm−3)也可用于微塑料的提取和分离,通常与密度分离装置相结合使用 [32, 37-39] ,微塑料的回收率很高,而且使用成本不高。Coppock等[40]比较了NaCl、NaI和ZnCl2溶液进行样品前处理的成本和提取效率,发现ZnCl2是最有效、最便宜的方法。但是,该物质具有很大的危害性和腐蚀性。因此,在使用ZnCl2进行样品前处理时,需谨慎处置并回收利用。
-
饱和甲酸钾(K(HCOO))溶液的密度为1.6 g·cm−3,具有稳定性高、成本相对较低、粘度低、可通过过滤重复使用等特点,也被用于密度分离中[41-42]。二水钨酸钠(Na2WO4 ·2H2O)和聚钨酸钠(3 Na2WO4 ·9 WO3 2 H2O)在溶液中的密度都能达到1.4 g·cm−3,因此也可用于微塑料的密度分离[43-44]。但是,聚钨酸钠的价格相对昂贵,相比之下,一些研究者更推荐使用二水钨酸钠。
-
Crichton等[45]利用微塑料的亲脂性,建立了一种简单的油提取方法,从固体样品中提取微塑料。干燥的样品与水和菜籽油充分混匀,静置至油、水、矿物质完全分离,微塑料与油结合进入油层,经过转移过滤后提取微塑料,再用乙醇去除表面油脂。在不同环境样品(沉积物和海水)中,使用该油提取微塑料(纤维和碎片)的回收率达到92%—97%。近期,Mani等[46]的研究测试了蓖麻油对4种复杂环境基质中微塑料的分离效率,包括河流和海洋悬浮表面固体、海洋沙滩沉积物和农业土壤。加标回收试验中,该方法对几种微塑料的平均回收率为99%。Karlsson等[47]在盐饱和溶液中加入一滴橄榄油,促进收集上清液中的塑料颗粒,回收率从64%提高到82%。目前关于油提取的研究较少,在微塑料分离后还需洗涤剂清洗,似乎具有一定的局限性,但可以通过油与饱和溶液相结合,来提高微塑料回收率。油提取方法简单、安全、廉价、耗时短,是一种很有前景的方法,亟需进一步验证和优化。
-
基于密度分离的浮选装置通常与密度分离液(如ZnCl2)结合使用,主要是通过气体或液体作为流动相,产生上升流带动样品上浮,在上浮的过程中使微塑料从沉积物中分离出来。Imhof等[32]研发了塑料沉积物分离器(MPSS,图1),配有过滤器支架的可移动样品室可将微塑料颗粒直接转移到过滤器上,从而将样品与ZnCl2密度浮选液分离,提取沉积物中的微塑料。然而,Zobkov和Esiukova[48]对MPSS装置进行了评估,发现原始塑料的回收率与Imhof等报道相似,但老化塑料的回收率却低得多,仅为13%—39%。 此外,ZnCl2具有危险性和腐蚀性,pH值低,可能与沉积物中的成分(尤其是碳酸盐)反应,从而导致起泡,严重阻碍分离过程,该MPSS装置还需进一步的测试及优化。Coppock等[40]设计了便携式的沉积物中微塑料分离装置(图2),由PVC管、PVC球阀以及磁力搅拌棒组成,与MPSS原理相似,以ZnCl2作为密度浮选液在浮选过程中分离微塑料,回收率高达92%—98%。然而该方法中PVC管的磨损可能会污染样品,从而影响环境中PVC微塑料的测定。
-
样品中的有机质可能会对微塑料的测定产生干扰,因此需要在前处理过程中尽可能去除有机质,同时不影响微塑料聚合物的结构及形貌 [65,68] 。目前的研究中通常采用酸消解、碱消解、氧化消解以及酶消解等方法对样品进行预处理。
-
酸消解可以去除样品中的有机质,常用的酸包括 HCl[49]、HNO3 [49]、及混合酸[69]。文献报道HCl不能破坏所有的有机质,因此消解效率不高[50-52]。HNO3被广泛用于酸性消解。然而,HNO3可能会留下油性残留物或组织碎片,导致聚合物的损失或变色[34,53-54]。此外,一些聚合物(如尼龙、PET)容易在高温和高浓度下被酸腐蚀,因此需要选择合适的浓度和温度,从而在合理的反应时间内有效去除样品中的有机质。Naidoo等[55]研究发现HNO3(55%)加热至80 ℃可使鱼组织的消化速度提高26倍。然而,当消解液加热至60℃以上时,可能会造成微塑料的损失,需格外小心[56]。总的来说,酸消解法可能会破坏样品中的聚合物,导致环境样品中的微塑料含量被低估,因此需要首先优化实验中酸的浓度及温度,并谨慎使用。
-
利用NaOH或KOH等进行碱消解,可以水解化学键,使蛋白质变性从而消解水生生物组织[57]。使用KOH或NaOH[52]在60 ℃过夜[51]或60 ℃消解24 h[54],是有效的消解处理方法之一。KOH对有机质的去除和塑料的回收具有良好的效果[53,56]。Foekema等[58]研究了KOH溶液对北海鱼样品的消解,发现在2—3周后,有机质完全被破坏。但也有一些研究表明,碱消解会破坏或使塑料变色[54,56,59,60],留下油性残留物和骨质碎片[51,54],或在塑料表面重新沉积残留物,对样品的光谱信号产生干扰 [61]。
-
过氧化氢(H2O2,30%—35%)作为氧化剂,可有效消解有机质,并且对聚合物几乎没有降解作用[31,50,59]。消解温度是H2O2消解效率的关键因素。例如Cole等[52]报道,在室温下用H2O2 (35%)消解7 d,仅降解25%的有机质;而Avio等[62]报道用H2O2(15%)在50 ℃过夜,可有效去除有机质。除了通过H2O2进行氧化消解外,NOAA推荐采用H2O2(30%)与0.05 mol·L-1的硫酸亚铁溶液( Fenton试剂)在75 ℃下加热消解样品。Hurley等[63]研究了不同消解方法对富含有机质的污泥和土壤样品中8种常见微塑料的提取效率差异,包括H2O2、Fenton试剂氧化消解法,以及NaOH、KOH碱消解法。结果表明,H2O2(80.2%—108%)和Fenton试剂(86.9%—106%)对土壤及污泥中有机质的去除效率均优于NaOH(60.9%—68.6%)和KOH(34.5%—56.8%)。结合提取效率、对微塑料性质的影响以及对光谱信号的影响等多个因素的比较,最终发现Fenton试剂(40 ℃以下,pH值接近3)既能有效去除土壤和污泥中的有机组分,又不会破坏微塑料中的聚合物,具备高效、成本低以及消解快速等优点。
-
酶消解法包括使用纤维素酶、脂肪酶、甲壳素酶和蛋白酶等去除有机质和减少部分生物组织[52,64]。与化学消解不同,酶消解的危害性较小,并且不易对微塑料造成损害[51]。对于0.2 g的少量样品,Cole等[52]应用蛋白酶进行酶消解,97%的有机质被降解。然而,这种酶的成本较高,更适用于少量样品的消解[63]。酶消解的另一个缺点是处理样品耗时长,并且每种酶都需要最佳温度和pH值[70]。此外,根据样品的基质不同,有些有机质不能完全消化,需要后续处理去除未消解的碎片。如Karlsson等[47]使用了酶消解法结合H2O2进行再处理,才能够有效破坏所有有机质。
-
Felsing等[65]利用塑料颗粒的静电行为达到样品中微塑料提取分离的目的。将样品加入静电金属-塑料分离器,在去除99%的原始样品量的同时,对几种常见微塑料的回收率高达近100%。近期的研究报道了一种磁性提取方法,利用微塑料与Fe纳米颗粒疏水性结合,进而达到磁性提取的目的[66]。该方法对于海水、淡水和沉积物中几种常见微塑料的回收率为78%—93%,可用于密度分离或消解处理后样品中微塑料的进一步提取或饮用水等基质简单的样品前处理。然而对于土壤或沉积物中存在的亲脂性物质可能会导致非特异性结合,从而降低该方法的有效性。此外,Fe纳米颗粒可能会干扰微塑料的后续分析,尽管通过超声处理可以从微塑料表面去除Fe纳米颗粒,但可能会同时破坏微塑料,还需进一步深入研究。
-
环境样品和水生生物样品中微塑料的提取和分离方法并不统一,如何能够在去除样品杂质、不破坏微塑料性质的同时,保证微塑料回收率,是前处理的关键。几种提取分析方法并非独立,实验中应根据不同基质的样品,来选取最佳的前处理方法。针对水样、土壤和沉积物等样品,可使用Fenton试剂消解结合密度分离法,来提取分离微塑料。而处理生物样品时,则可使用KOH进行消解并结合密度分离法,去除杂质。未来的研究应从以下几个方面着手:
(1)结合每种方法的优势,选择更适合的方法组合,来达到最佳的提取和分离效果。比如首先通过静电分离或密度浮选装置等来大大降低样品量,再使用碘化钠等价格昂贵的密度分离浮选液,对微塑料进行进一步的提取和分离。
(2)对于文献最新报道的如油提取、磁提取、密度浮选装置等分离方法进行进一步的验证及优化。
(3)对多种提取和分离方法的一致性和准确性进行比较研究。通过开展不同介质中前处理方法比较研究和效果评价,从而筛选出最佳的预处理方法。进而分别建立水样、土壤和沉积物、生物样品等不同介质中微塑料的预处理标准方法,为深入研究微塑料的环境行为及生态毒理学效应奠定基础。
微塑料的提取分离方法研究进展
Research progress on the extraction and separation methods of microplastics
-
摘要: 微塑料作为海洋环境和陆生生态系统中的新型污染物,引起了广泛关注。然而目前微塑料的分析方法尚未标准化,不同研究结果间可比性较低。如何准确、高效地提取分离样品中的微塑料,是探究微塑料的环境行为及生态毒理学效应的关键前提。本文系统地综述了环境样品和水生生物样品中微塑料的前处理分析方法,包括筛分过滤法、密度分离法、消解法以及文献报道的其他方法,并对不同方法的优缺点及研究趋势进行了讨论和分析。结合不同前处理方法的优势,开展多种方法组合、比较等研究有利于微塑料分析方法的标准化。Abstract: Microplastic as an emerging pollutant in the marine environment and terrestrial ecosystems has attracted widespread attention. However, the analysis methods of microplastics have not been standardized at present, which hampered the comparability between different research results. How to accurately and efficiently extract microplastics in samples is a crucial prerequisite for exploring the environmental behavior and ecotoxicological effects of microplastics. The pretreatment analysis methods of microplastics in environmental samples and aquatic biological samples were systematically reviewed in this paper, including sieve filtration method, density separation method, digestion method and other methods reported in the literature. Besides, the advantages and disadvantages of different methods and research trends were discussed. Combining the advantages of different pretreatment methods, carrying out studies with various method combinations and comparisons is conducive to the standardization of microplastic analysis methods.
-
Key words:
- microplastics /
- extraction and separation /
- flotation /
- density separation /
- digestion
-
气溶胶颗粒物作为大气的重要组分,对气候演化和环境变化等方面有着重要影响. 大气颗粒物的散射效应使到达地球表面的太阳辐射减少,其降温效应可以部分抵消人类排放的温室气体所导致的全球变暖[1]. 气溶胶颗粒物可以成为云凝结核,进而影响到云和降水的物理过程[2]. 研究表明,三极地区对大气气溶胶的变化更为敏感[3]. 大气颗粒物沉降在青藏高原冰雪表面,促进冰雪的消融[4-5]. 青藏高原上黑碳(BC)和沙尘导致反射率降低约38%,造成的总辐射强迫约为18—32 W·m-2[6]. 高原南部扎当冰川上,BC和沙尘对老雪融化的贡献约为9%[7]. 藏东南冰川变化的研究表明,吸光性颗粒物对冰川融化的贡献为15%[8]. 数值模拟实验表明季风前期青藏高原南坡堆积的吸收性气溶胶加热大气形成热泵效应,导致雨季的提前和南亚夏季风的加剧[9]. Cong等发现珠峰北坡气溶胶中二羧酸与生物质燃烧指示物左旋葡聚糖和K+存在明显正相关关系,表明南亚的碳质气溶胶可以通过大尺度环流和局地山谷环流传输到青藏高原[10],大气观测、冰雪冰芯样品和模式模拟显示BC、重金属如汞、有机污染物POPs和微塑料能够跨境传输到青藏高原内部并对环境产生影响[11-14]. 南亚大气棕色云使低层大气变暖的程度和大气气溶胶相同,两者可以使气温在10年内增加0.25 K[15].
青藏高原北邻塔克拉玛干沙漠,西南部是塔尔沙漠,这些地区是青藏高原粉尘的潜在源区. 青藏高原东邻我国人口集中的地区,南邻南亚发展中国家,人为气溶胶也对青藏高原造成了影响. 目前对青藏高原大气颗粒物的研究主要有以下几种手段:1)对冰川区的冰雪及冰芯进行采样,分析微粒物化特征[16-18];2)利用卫星遥感等手段对大气气溶胶光学特征进行监测[19-20];3)直接采集大气气溶胶样品和定点监测[21-22]. 不同的研究手段有不同的利弊,冰芯记录侧重于过去大气粉尘的研究,通常为长期变化趋势;卫星监测和数据模拟可以在长时间和大尺度获取大气气溶胶浓度、光学性质等的信息,成为区域研究的重要工具[23]. 由于青藏高原复杂的地形和地表非均质反射,使得模拟结果和实地监测结果或多或少有偏差[24-25]. 研究表明MODIS AOD数据在拉萨不具有适用性,在高原东部的应用表明数据精度相对较低[26-27]. Sharma等对喜马拉雅地区气溶胶的模拟数值普遍低于实测值[5].
因此,急需地面观测气溶胶颗粒物数据对卫星监测和数值模拟数据来进行验证和校正. 高原西部大气颗粒物的监测数据较少,阿里地区地面实测数据的增加,给高原大气气溶胶的卫星监测和数据模拟研究提供了支撑和验证数据. 本文在大气颗粒物在线观测数据的基础上,获得阿里站大气颗粒物的浓度水平,进而分析其季节变化的基本特征,并综合运用HYSPLIT模型的聚类分析、潜在源贡献因子分析(PSCF)和浓度权重轨迹分析(CWT)方法,分析各个季节阿里站大气气溶胶的输送路径和可能来源,为青藏高原尤其是其西部地区的大气颗粒物研究提供数据支撑.
1. 材料与方法(Materials and methods)
1.1 监测地点
监测地点位于西藏自治区阿里地区日土县的中国科学院阿里荒漠环境综合观测研究站,地理位置为东经79.70°,北纬33.39°,海拔高度4270 m. 位于班公湖南部盆地. 整体自然环境为高寒荒漠. 研究期间,阿里站的平均气温为1.38 ℃,相对湿度为32%. 年降水量为161.24 mm,83%集中在7、8月份. 植被稀疏,归一化植被指数(NDVI)小于0.2.
1.2 监测仪器
监测仪器为德国GRIMM公司生产的EDM365颗粒物在线监测仪,该仪器提供31个粒径通道的颗粒物数浓度,同时给出PM10、PM2.5、PM1(空气动力学粒径)的质量浓度. 样气通过Nafion管自动启动除湿功能. 原始数据的时间分辨率设定为5 min,在剔除异常值的基础上,将质量浓度数据依次合并成逐时算术平均值、逐日算术平均值和逐月算术平均值.
1.3 数据分析
由于阿里站气象数据缺失较多,再分析数据和气象数据的拟合较好,因此除了湿度和降水数据来自阿里站气象数据,其他气象数据来自ERA5再分析结果,下载地址为(https://cds.climate.copernicus.eu/cdsapp#!/search?type=dataset). 归一化植被指数(NDVI)数据来自MODIS的MOD13Q1影像(时间分辨率为16 d,空间分辨率为250 m). 气溶胶光学厚度(AOD)数据来自MODIS的MCD19A2影像(时间分辨率为1 d,空间分辨率为1 km). 对数据的处理和分析主要用到Excel、Spss、ArcGis和Matlab等软件,对数据的绘图用的是Origin软件. 后向轨迹分析、PSCF分析、CWT分析用的数据为美国国家环境预报中心(NCEP) 的全球资料同化系统(GDAS) 数据,使用的软件为TrajStat. 根据青藏高原地区的大气环流特征将研究时段划分为四个季节:冬季(2018-12-01至2019-02-28),季风前期(2019-03-01至2019-05-31),季风期(2019-06-01至2019-09-30),以及季风后期(2019-10-01至2019-11-30).
2. 结果与讨论(Results and discussion)
2.1 阿里站大气颗粒物质量浓度基本特征
仪器监测数据的时段为2018-12-01至2019-11-30. 在监测期间,阿里站PM10的日均质量浓度为(10.51±8.62) μg·m−3(0.47—78.91 μg·m−3),PM2.5的日均质量浓度为(4.05±2.36) μg·m−3(0.31—12.91 μg·m−3),PM1的日均质量浓度为(2.47±1.56) μg·m−3(0.13—10.64 μg·m−3),与高原城市地区如西宁[28]、拉萨[29]相比,浓度较低. PM2.5、PM10远低于国家规定的颗粒物年平均一级浓度限值(GB 3095—2012),说明阿里站颗粒物浓度可以作为高原大气颗粒物浓度的本底值. Chen等统计分析了2016年6月至2017年5月阿里地区的PM2.5和PM10浓度值,分别为33.1 μg·m−3和87.3 μg·m-3[30],PM10和PM2.5约是本研究的8倍. 这是因为Chen的数据来自阿里噶尔县狮泉河镇,本研究的数据来自阿里日土县野外站点,研究地点受人为排放的影响不同. 阿里地区属于高寒荒漠环境,对阿里站周边植被覆盖率的统计结果表明,NDVI<0.2,裸地沙土对颗粒物浓度的贡献大. 这里用PM2.5/PM10的比值来表征粗细颗粒的相对贡献. PM2.5/PM10平均比值为0.39,小于狮泉河镇的0.42[30],说明阿里地区粗颗粒物(空气动力学粒径>2.5 μm 的颗粒物)在PM10中占比较高,颗粒物浓度受人为影响较小.
将北京、拉萨、阿里、珠峰、Hyytiala、JGM站的大气颗粒物质量浓度进行比较(表1),意在对比中国大城市、高原城市、高原野外站点、极地区颗粒物的不同特征. PM2.5的比较为北京>拉萨>珠峰>Hyytiala>阿里>JGM站. PM2.5来源主要为燃烧源、大气二次转化形成的颗粒物,城市PM2.5显著高于背景站点. 2019年,北京市PM2.5为42 μg·m−3,比PM2.5年均值二级浓度限值(35 μg·m−3)高20%,但显著小于2013年PM2.5值,是其的1/2. 2013年国家发布了《大气污染防治行动计划》,为了达到计划目标,北京市先后实行了《北京市2013—2017年清洁空气行动计划》、《京津冀及周边地区2017—2018年秋冬季大气污染综合治理攻坚行动方案》,煤炭使用减少和能源结构优化使北京市大气污染状况好转,颗粒物浓度逐渐降低[31];拉萨污染物排放(工业污染、交通污染)相对较少,PM2.5略低于北京,比背景站高;阿里、珠峰、Hyytiala、JGM站为大气本底站,受到人为干扰少,PM2.5浓度均小于10 μg·m−3. 将不同站点PM2.5/PM10进行比较,发现Hyytiala>北京>JGM>珠峰>阿里>拉萨. Hyytiala站是芬兰南部靠近北极圈的背景站点,位于北方针叶林带,颗粒物来源主要为自然来源. 森林植被排放的挥发性有机物形成有机气溶胶是颗粒物的主要来源,PM2.5中有机物占比为73%. 颗粒物以细颗粒(空气动力学粒径<2.5 μm的颗粒物)为主,PM2.5/PM10高达0.85[32];北京市人为污染物形成的细颗粒物使颗粒物浓度比值PM2.5/PM10较高;JGM站位于南极最大的无冰区James Ross岛,裸露的沉积物和大风天气易产生沙尘天气,使JGM站粗颗粒物占比较高[33];珠峰站和阿里站处于高寒荒漠或草原环境,裸地为粗颗粒的供给提供了有利条件,PM2.5/PM10比值较低;对单颗粒化学元素的研究表明,拉萨市颗粒物的主要元素为硅、铝和钙,主要来自矿物粉尘(沙尘天气、建筑用材),拉萨市颗粒物以粗颗粒物为主,PM2.5/PM10为0.38[34].
表 1 不同站点颗粒物水平的比较(顺序为北京、拉萨、阿里站、珠峰站、Hyytiala站、JGM站)Table 1. Comparison of atmospheric particulate mass concentration at different stations地点 Site PM10/(μg·m−3) PM2.5/(μg·m−3) PM2.5/PM10 时间 Date 北京[31] 68 42 0.62 2019 拉萨[30] 87.3 33.1 0.38 2016-06—2017-05 阿里 10.51±8.62 4.05±2.36 0.39 2018-11—2019-12 珠峰[35] 11.49±13.81 4.75±3.87 0.41 2018—2020 Hyytiala(芬兰)[36] 5.83 4.55 0.78 2010—2017 JGM(南极)[33] 6.4±1.4 3.1±1 0.48 2018-01—2018-03 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.2 阿里站颗粒物质量浓度季节变化特征
图1为阿里站颗粒物质量浓度和气象因子的时间序列图,可以看出,颗粒物质量浓度有着明显的季节变化特征,冬季和季风前期明显大于季风期和季风后期. PM10的季节变化规律为:冬季(15.4 μg·m−3)>季风前期(14.06 μg·m−3)>季风后期(6.14 μg·m−3)≈季风期(5.92 μg·m−3);PM2.5的变化规律为:季风前期(5.4 μg·m−3)≈冬季(5.12 μg·m−3)>季风后期(2.91 μg·m−3)≈季风期(2.64 μg·m−3);PM1的变化规律为:冬季(3.16 μg·m−3)≈季风前期(3.15 μg·m−3)>季风后期(2.0 μg·m−3)≈季风期(1.59 μg·m−3). PM2.5和PM1在冬季的浓度值约等于季风前期的值,PM10和PM2.5在季风期的值与季风后期相当. 虽然PM10在冬季大于季风前期,但冬季的值仅比季风前期高9.5%. 因此,PM10、PM2.5、PM1具有一致的季节变化规律.
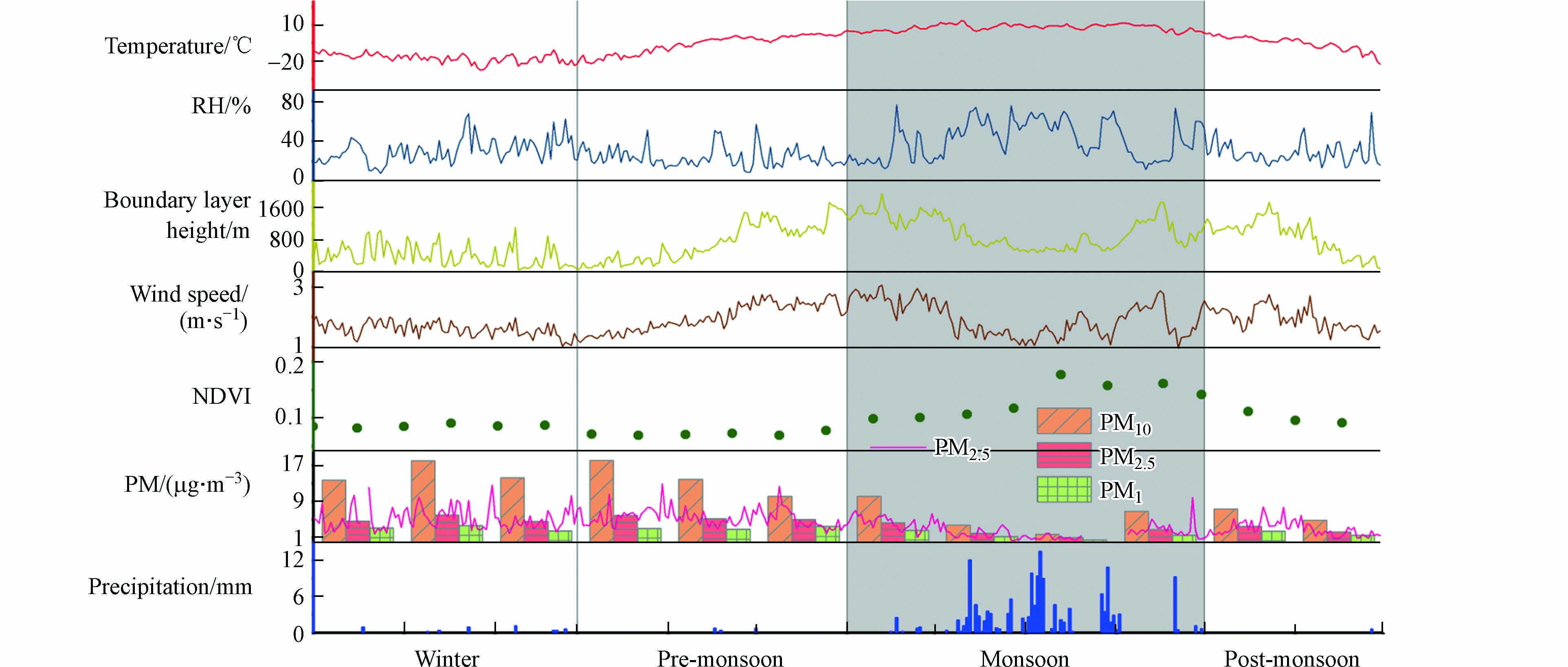 图 1 阿里站颗粒物质量浓度及气象因子的时间序列图Figure 1. Time series of meteorological parameters and PM at Ngari Station
图 1 阿里站颗粒物质量浓度及气象因子的时间序列图Figure 1. Time series of meteorological parameters and PM at Ngari Station由于阿里地区污染物排放源少,因此主要从气象方面分析颗粒物质量浓度的变化原因. 据图1分析,冬季,阿里地区处于干寒的状态,相对湿度低,植被覆盖率极低,归一化植被指数(NDVI)小于0.1,表土疏松,易产生浮尘现象. 冬季阿里站的平均边界层高度低于400 m,风速较小,加上逆温层的阻挡作用,大气的容积变小,扩散能力弱. 稳定的大气条件使颗粒物不断积累,浓度变高[37]. 季风前期,阿里站的NDVI和相对湿度依旧较低,地面沙尘易被扬起,颗粒物浓度水平较高. 边界层高度和风速不断变高,使大气扩散能力变强的同时受远程气团影响变大. 3—5月颗粒物浓度呈现变低的趋势,但季风前期PM平均值依旧较高季风期间尤其是7、8月份,阿里站受到西南季风的影响,气象因素发生显著变化. 湿度的变高使加热大气的感热变少,限制了对流混合的发展,边界层高度和风速相应变低[38]. NDVI的增加和风速的减小,使风对地面尘土的搬运能力变低. 颗粒物的湿沉降作用和降水的清除作用使颗粒物尤其是粗颗粒物显著减少,质量浓度在一年之内最低[39]. 季风后期,随着湿度和NDVI变低,颗粒物质量浓度开始回升. 由于季风后期温度和边界层高度比冬春高,扩散条件较之好,颗粒物浓度相对冬季和季风前期较低.
由于阿里站颗粒物质量浓度存在着明显的季节变化,为了探究不同季节影响阿里地区颗粒物浓度的气团来源,综合运用轨迹聚类分析、PSCF分析和CWT分析进行研究. 图2为阿里站不同季节的气团轨迹聚类分析,可以看出阿里站主要受偏西气团影响,轨迹主要来自中亚和南亚地区.
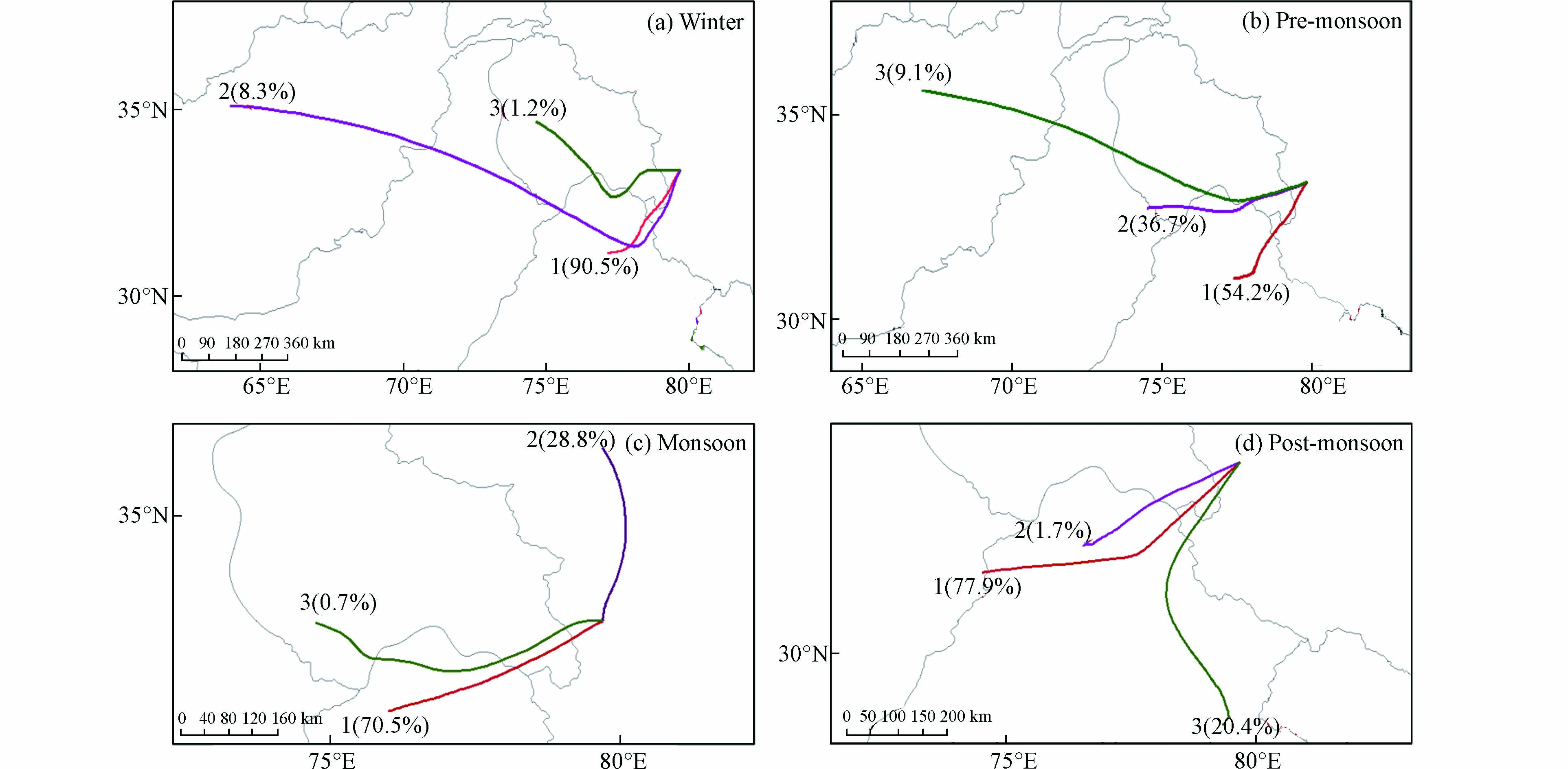 图 2 阿里站冬季、季风前期、季风期和季风后期的气团后向轨迹聚类分析Figure 2. The seasonal distribution of airflow back-trajectories clusters in the Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon
图 2 阿里站冬季、季风前期、季风期和季风后期的气团后向轨迹聚类分析Figure 2. The seasonal distribution of airflow back-trajectories clusters in the Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon冬季和季风前期受西风急流增强的影响,部分气团来自阿富汗等远源地区(~10%),而季风期和季风后期西风急流减弱,远源地区的贡献消失,主要表现为印度和巴基斯坦北部的近源贡献. 与其他季节不同,季风期有28.8%的气团来自新疆南部,这与Duo等在拉萨的研究一致[40]. 将阿里站气团后向轨迹经过的地区划成0.25°×0.25°的网格,每个网格污染轨迹端点的个数与经过网格的所有轨迹端点的比值即为该网格PSCF值,高值区代表着可能的潜在源区[41]. 污染轨迹通常由标准限值、平均值界定. 阿里地区为生态脆弱区,属于一类区,于是使用环境空气质量标准(GB 3095—2012)颗粒物浓度年均值一级浓度限值(PM10=40 μg·m−3;PM2.5=15 μg·m−3)区分污染轨迹. 由于阿里地区PM2.5浓度过小,污染轨迹过少,PSCF分析结果不理想,于是在这里不予讨论PM2.5的PSCF分析结果. PSCF分析表明(图3),PM10的主要潜在源区分布在印度北部、克什米尔地区、巴基斯坦北部和阿里西部地区. 季风前期和季风后期PM10的潜在源区范围较广. 冬季,由于边界层低,受到远程传输气团的影响较小,PM10潜在源区范围较小. 季风期,暖湿气团使颗粒物产生沉降,受远程传输的影响不明显. 在后向轨迹的基础上,CWT分析可以求出潜在源区贡献给阿里站颗粒物浓度的权重浓度,得出对阿里地区颗粒物浓度影响较大的潜在源区[42]. 由于不用界定污染轨迹,CWT分析得出的潜在源区范围明显大于PSCF分析(图4). 如图4、图5和图6所示,PM10、PM2.5和PM1的潜在源区范围和颗粒物主要源区相似. 对阿里地区颗粒物质量浓度贡献最大的地区是印度西北部和克什米尔,其次巴基斯坦北部,我国和田地区和阿里地区也有一定程度的贡献.
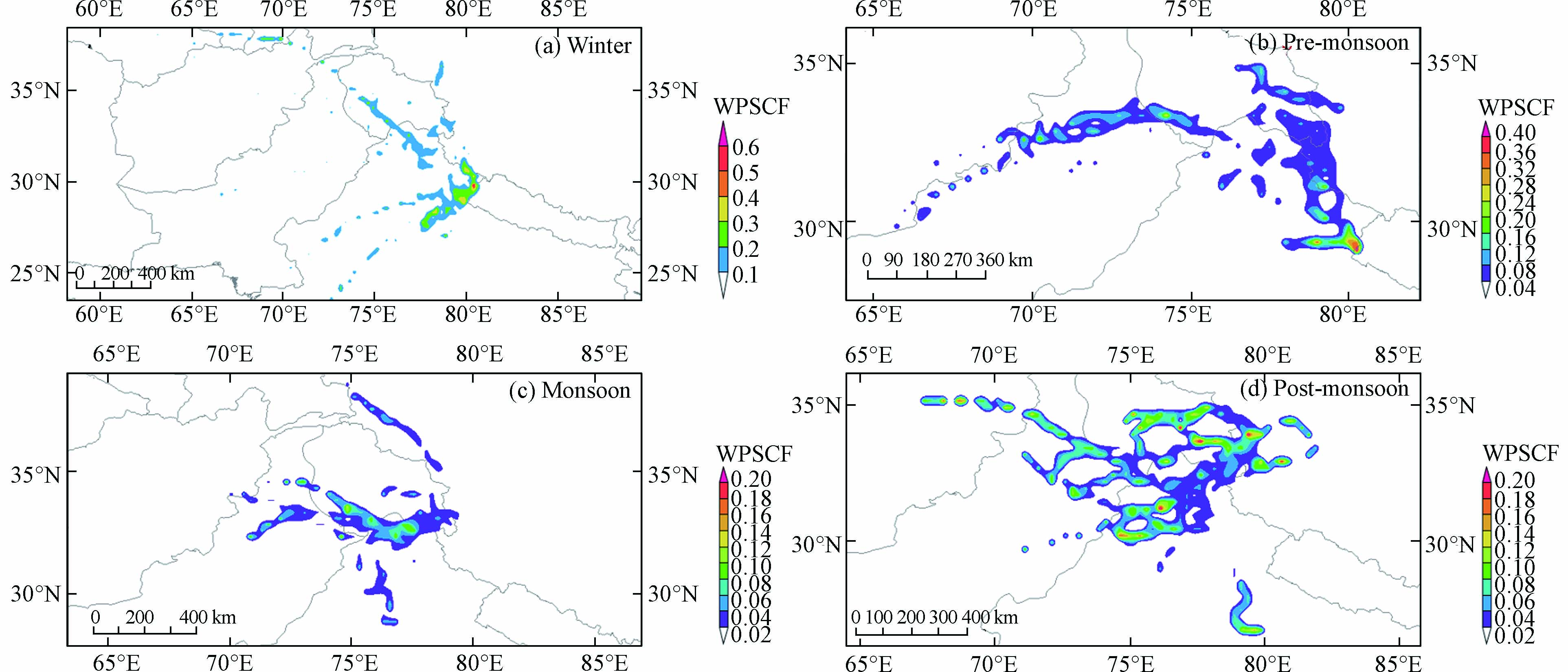 图 3 阿里站PM10的PSCF分析结果Figure 3. Weighted potential source contribution function for PM10 in Ngar(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon
图 3 阿里站PM10的PSCF分析结果Figure 3. Weighted potential source contribution function for PM10 in Ngar(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon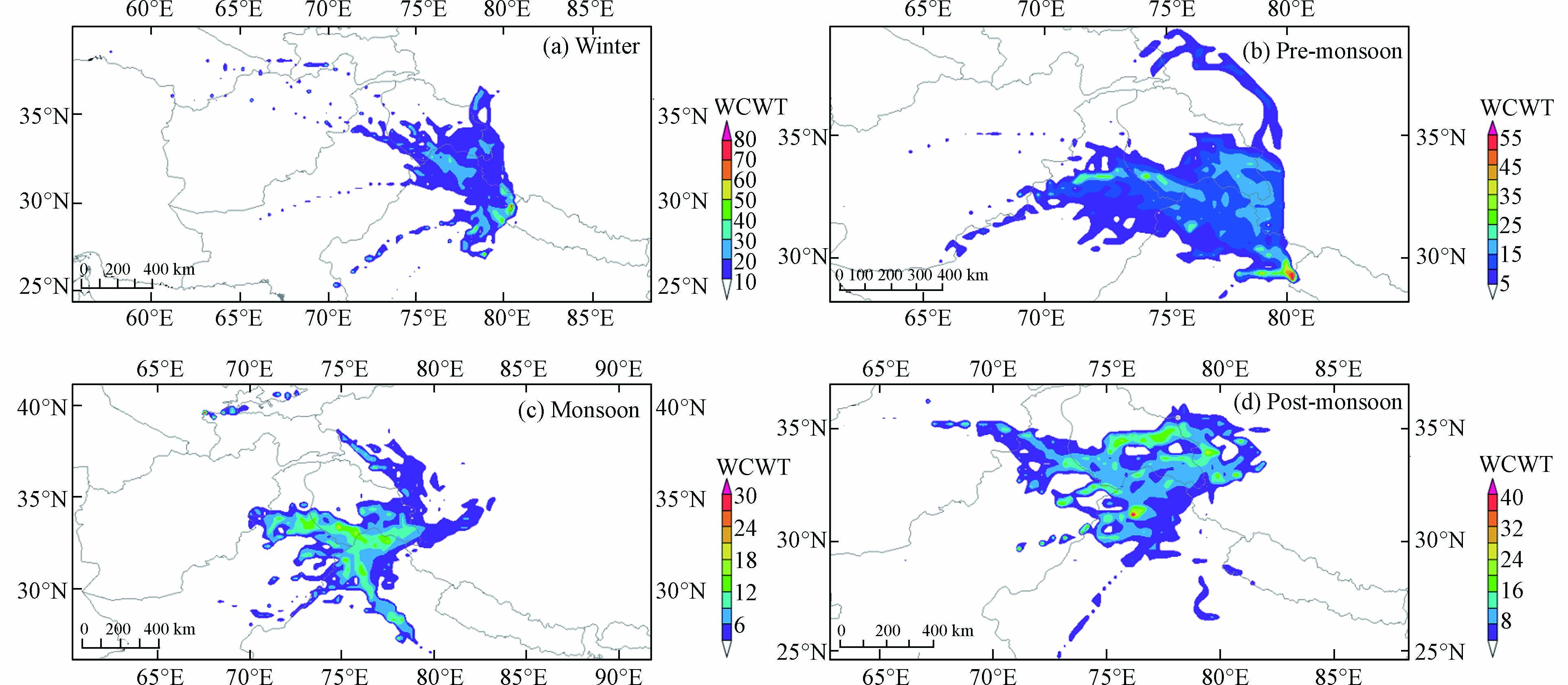 图 4 阿里站PM10的CWT分析结果Figure 4. Weighted concentration-weighted trajectory for PM10 in Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter,(b) pre-monsoon,(c) monsoon and (d) post-monsoon
图 4 阿里站PM10的CWT分析结果Figure 4. Weighted concentration-weighted trajectory for PM10 in Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter,(b) pre-monsoon,(c) monsoon and (d) post-monsoon2.3 MODIS AOD 与实测颗粒物质量浓度的比较
当前遥感获取的主要气溶胶参数为光学厚度(AOD),表征大气颗粒物对入射的辐射产生的消光性质. 将AOD数据和地面监测数据联系起来,可增进人们对气溶胶辐射效应的机制研究. 图7为大气颗粒物质量浓度与AOD的月变化图. 图7可以看出,12月、7月和8月AOD的变化与颗粒物浓度变化相差较大. 除去12月、7月和8月,颗粒物浓度与AOD呈现相同的月变化特征,颗粒物质量浓度高的时期(1—6月),所对应的AOD较高,颗粒物质量浓度低的时期(9—11月),AOD较低. 7、8月份,西南季风带来了暖湿气团,使阿里地区湿度变高,相对湿度超过50%. 研究表明每增加3—6 mm,水汽对太阳短波辐射的吸收增加1%[43]. 太阳短波辐射的减少也就是水汽的增加使AOD变高. 颗粒物监测仪器GRIMM EDM 365自带除湿系统,数据是相对湿度较低状态下的质量浓度,而卫星影像是在环境湿度下的监测,两者的湿度条件不同[44]. 加之AOD和颗粒物质量浓度监测高度不同等原因,使7、8月份AOD值高而颗粒物质量浓度低的不匹配现象. MCD19A2 AOD数据使用的MAIAC算法改进了亮表面的反演,但仍存在一定偏差. 12月份,阿里站NDVI小于0.1,地表覆盖类型为裸地,较高的反照率影响卫星对颗粒物监测的敏感性[45]. AOD指介质的消光系数在垂直方向上的积分,而颗粒物浓度数据是对近地面站点的监测. 冬季,较低的边界层和逆温层阻止了颗粒物的扩散,使近地面浓度变高. 因此,监测原理的不同使AOD、颗粒物浓度的变化趋势出现不一致现象. 需要对高原颗粒物浓度数据和AOD数据进行混合层高度和相对湿度等影响因素的订正,以提高数据的相关系数[46].
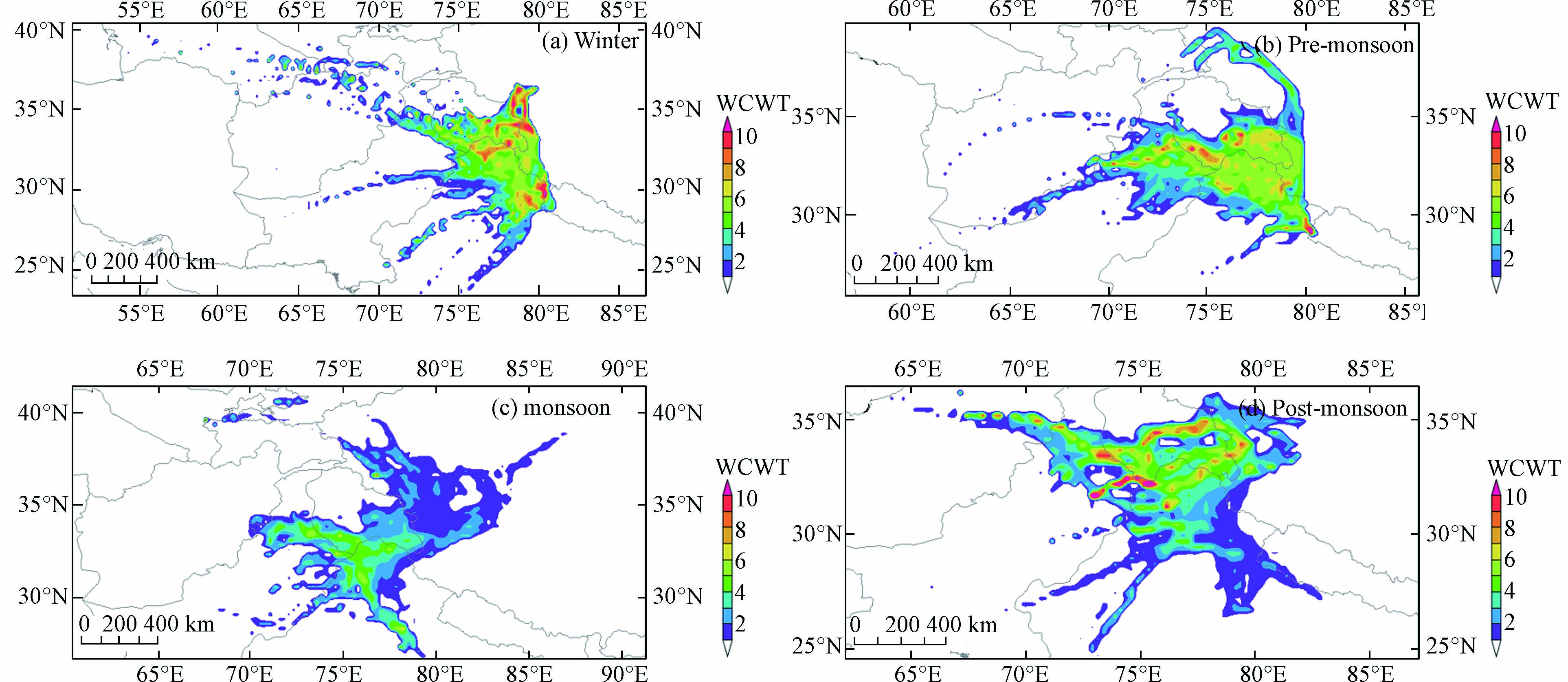 图 5 阿里站PM2.5的CWT分析结果Figure 5. Weighted concentration-weighted trajectory for PM2.5 in Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon
图 5 阿里站PM2.5的CWT分析结果Figure 5. Weighted concentration-weighted trajectory for PM2.5 in Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon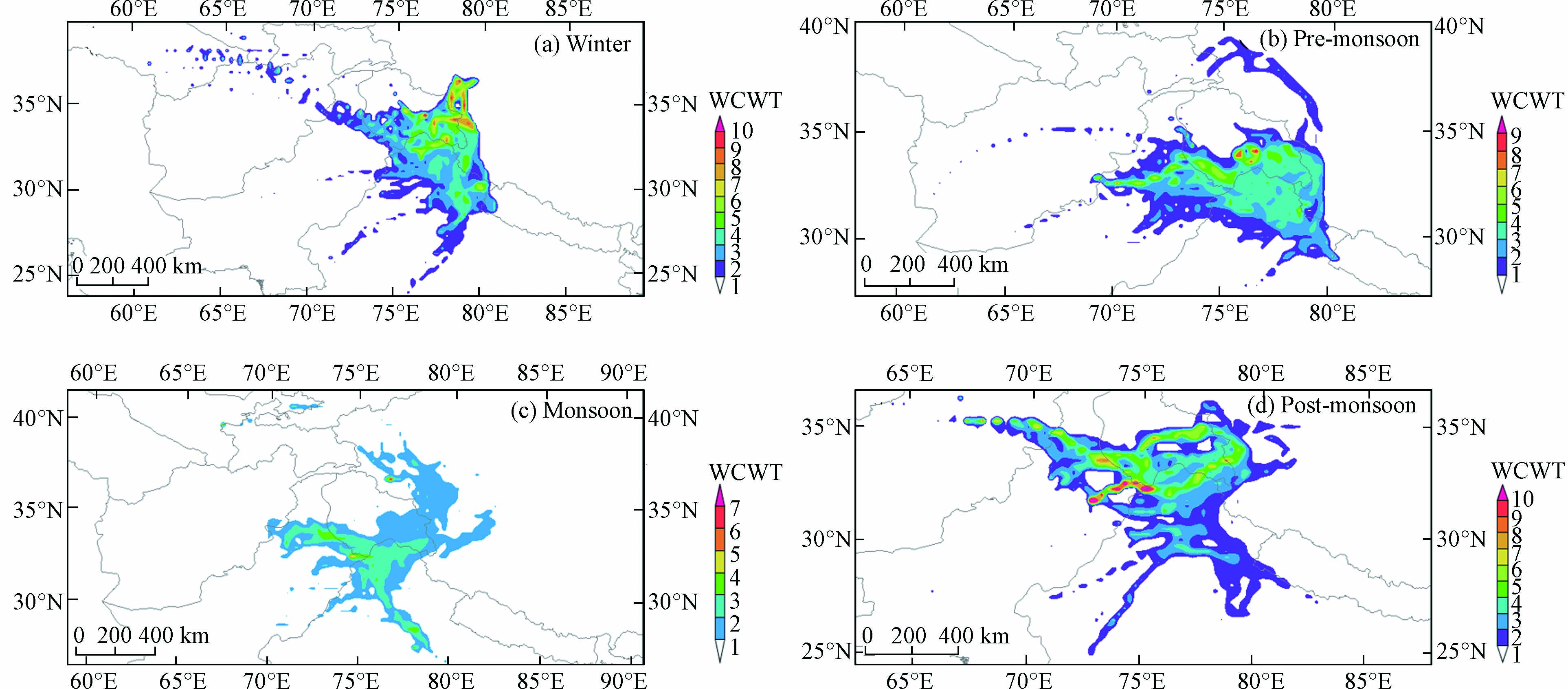 图 6 阿里站PM1的CWT分析结果Figure 6. Weighted concentration-weighted trajectory for PM1 in Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon
图 6 阿里站PM1的CWT分析结果Figure 6. Weighted concentration-weighted trajectory for PM1 in Ngari(a)冬季,(b)季风前期,(c)季风期,(d)季风后期(a) winter, (b) pre-monsoon, (c) monsoon and (d) post-monsoon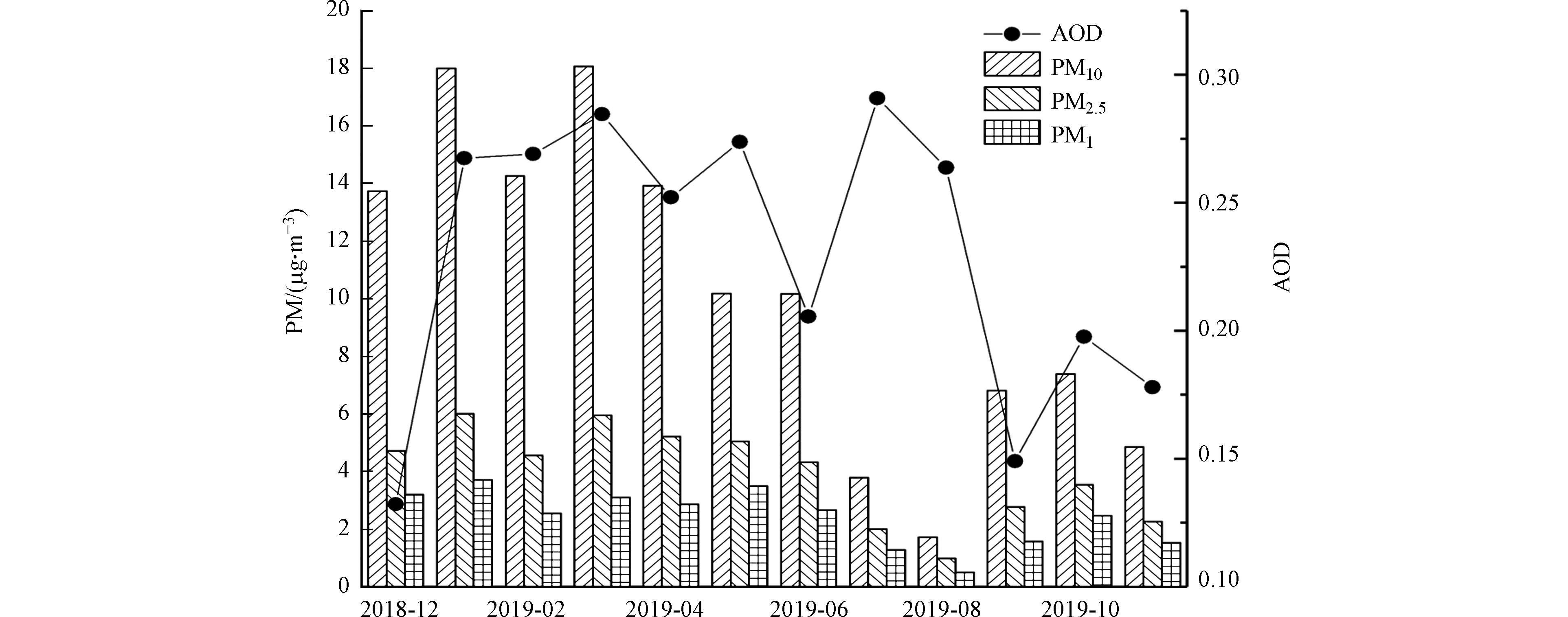 图 7 阿里站PM和AOD月变化图Figure 7. The monthly variations of PM and MODIS AOD at Ngari station
图 7 阿里站PM和AOD月变化图Figure 7. The monthly variations of PM and MODIS AOD at Ngari station3. 结论(Conclusion)
通过对阿里站大气颗粒物浓度的实测和传输气团轨迹的聚类分析,得出以下主要结论:
(1) 阿里站PM10、PM2.5和PM1浓度均较低,分别为(10.51±8.62) μg·m−3、(4.05±2.36) μg·m−3和(2.47±1.56) μg·m−3,PM10、PM2.5远小于PM10(40 μg·m−3)、PM2.5(15 μg·m−3)年平均颗粒物浓度一级标准,表明了阿里站洁净的大气本底特征.
(2)阿里站大气颗粒物浓度存在着明显的季节变化,冬季和季风前期显著大于季风期和季风后期. 冬春季节温度低、大气边界层低,污染物容积能力小. 且冬春季植被覆盖率极低,沙尘频繁,颗粒物浓度高于其他季节. 季风期风速小,颗粒物携带能力弱,降水较多、湿度高、植被覆盖率高,大气颗粒物浓度最低. 聚类分析、PSCF和CWT分析结果表明,印度北部是阿里站颗粒物浓度的主要潜在源区,贡献值较高.
(3)由于盛行风的改变和边界层高度变化等原因,12月、7月和8月AOD与颗粒物浓度的差异较大,遥感数据和地面监测数据需要进行订正,为高原遥感反演颗粒物浓度提供数据支撑.
-


表 1 样品前处理方法汇总
Table 1. Summary of methods for sample pretreatment.
前处理方法Pretreatment methods 样品基质Sample matrix 优点Advantages 缺点Disadvantage 参考文献Reference 筛分过滤法 过滤筛分 水、固体样品浮选上清液 可快速分离;通过不同孔径滤网,可对微塑料按照粒径分类 没有标准化的孔径尺寸,不同研究可比性低 [4, 28-30] 密度分离法 NaCl 水、土、沉积物、生物 无毒、无害、成本低 对高密度微塑料提取效率低 [26, 27, 31- 32] NaI 水、土、沉积物、生物 密度高、安全、可重复使用、提取效率高 价格昂贵 [31, 33-36] ZnCl2 水、土、沉积物、生物 密度高、提取效率高、成本低 腐蚀性、危害性 [32, 37-40] 甲酸钾 水、土、沉积物、生物 稳定性好、成本低 目前应用研究较少 [41-42] 聚钨酸钠 水、土、沉积物、生物 密度高、成本较低 吸湿性强 [43-44] 油 土、沉积物 成本低、易操作 需要对微塑料进行进一步清洗;目前应用研究较少 [45-47] 密度分离浮选装置 土、沉积物 直接分离,能够有效减小样品量 需要与密度浮选液结合,还需进一步验证及优化 [32, 40, 48] 消解法 酸消解 (HCl) 水、土、沉积物、生物 − 不能破坏所有有机质 [49-52] 酸消解 (HNO3) 水、土、沉积物、生物 能够去除大部分有机质 可能会造成PET等聚合物溶解 [34, 49, 53-56] 碱消解 (NaOH/KOH) 水、土、沉积物、生物 能去除大部分有机质;对大部分聚合物没有破坏性 可能使塑料变色;沉积残留物对光谱信号产生干扰 [51-54, 56-61] 氧化消解 (H2O2) 水、土、沉积物、生物 能去除大部分有机质; 对部分聚合物有破坏性 [31, 50, 52, 59, 62] 氧化消解 (Fenton试剂) 水、土、沉积物、生物 能去除有机质、提取效率高、对光谱信号无影响 − [27, 63] 酶消解 水、土、沉积物、生物 危害小、不会对聚合物造成损害 成本高、耗时长 [51-52, 63-64] 其他方法 静电分离装置 沉积物 能够将样品量减小99% 不适用于少量样品 [65] 磁提取法 水、沉积物 对大部分聚合物提取效率高 对于复杂样品需与其他方法结合,更适用于饮用水等基质简单的样品;需进一步优化 [66]
下载: 导出CSV
-
[1] PlasticsEurope, Plastics - the Facts 2017: An analysis of European plastics production, demand and waste data[R], 2017. Available from: https://www.plasticseurope.org/application/files/5715/1717/4180/Plastics_the_facts_2017_FINAL_for_website_one_page.pdf [2] CARPENTER E, SMITH K L. Plastics on the Sargasso Sea surface [J]. Science, 1972, 175(4027): 1240-1241. doi: 10.1126/science.175.4027.1240 [3] THOMPSON R C, OLSEN Y, MITCHELL R P, et al. Lost at sea: Where is all the plastic? [J]. Science, 2004, 304(5672): 838-838. doi: 10.1126/science.1094559 [4] HIDALGO-RUZ V, GUTOW L, THOMPSON R C, et al. Microplastics in the marine environment: A review of the methods used for identification and quantification [J]. Environmental Science & Technology, 2012, 46(6): 3060-3075. [5] ZHANG W W, ZHANG S F, ZHANG Z Y, et al. Microplastic pollution in the surface waters of the Bohai Sea, China [J]. Environmental Pollution, 2017, 231: 541-548. doi: 10.1016/j.envpol.2017.08.058 [6] EERKES-MEDRANO D, THOMPSON R C, ALDRIDGE D C. Microplastics in freshwater systems: A review of the emerging threats, identification of knowledge gaps and prioritisation of research needs [J]. Water Research, 2015, 75: 63-82. doi: 10.1016/j.watres.2015.02.012 [7] BLETTLER M C M, ABRIAL E, KHAN F R, et al. Freshwater plastic pollution: Recognizing research biases and identifying knowledge gaps [J]. Water Research, 2018, 143: 416-424. doi: 10.1016/j.watres.2018.06.015 [8] LI J Y, LIU H H, CHEN J P. Microplastics in freshwater systems: A review on occurrence, environmental effects, and methods for microplastics detection [J]. Water Research, 2018, 137: 362-374. doi: 10.1016/j.watres.2017.12.056 [9] KOELMANS A A, MOHAMED N H, HERMSEN E, et al. Microplastics in freshwaters and drinking water: Critical review and assessment of data quality [J]. Water Research, 2019, 155: 410-422. doi: 10.1016/j.watres.2019.02.054 [10] PRATA J C, DA C J P, DUARTE A C, et al. Methods for sampling and detection of microplastics in water and sediment: A critical review [J]. Trends in Analytical Chemistry, 2019, 110: 150-159. doi: 10.1016/j.trac.2018.10.029 [11] DING L, MAO R F, GUO X T, et al. Microplastics in surface waters and sediments of the Wei River, in the northwest of China [J]. Science of the Total Environment, 2019, 667: 427-434. doi: 10.1016/j.scitotenv.2019.02.332 [12] HE D F, LUO Y M, LU S B, et al. Microplastics in soils: Analytical methods, pollution characteristics and ecological risks [J]. Trends in Analytical Chemistry, 2018, 109: 163-172. doi: 10.1016/j.trac.2018.10.006 [13] MÖLLER J N, LÖDER M G J, LAFORSCH C. Finding microplastics in soils: A review of analytical methods [J]. Environmental Science & Technology, 2020, 54(4): 2078-2090. [14] RIBEIRO F, O'BRIEN J W, GALLOWAY T, et al. Accumulation and fate of nano- and micro-plastics and associated contaminants in organisms [J]. Trends in Analytical Chemistry, 2018, 111: 139-147. [15] SHAHABALDIN R, JUNBOUM P, MOHD F M D, et al. Microplastics pollution in different aquatic environments and biota: A review of recent studies [J]. Marine Pollution Bulletin, 2018, 133: 191-208. doi: 10.1016/j.marpolbul.2018.05.022 [16] BROWNE M A, DISSANAYAKE A, GALLOWAY T, et al. Ingested microscopic plastic translocates to the circulatory system of the Mussel, Mytilus edulis (L. ) [J]. Environmental Science & Technology, 2008, 42(13): 5026-5031. [17] DERRAIK J G B. The Pollution of the marine environment by plastic debris: A review [J]. Marine Pollution Bulletin, 2002, 44(9): 842-852. doi: 10.1016/S0025-326X(02)00220-5 [18] TEUTEN E L, SAQUING J M, KNAPPE D R, et al. Transport and release of chemicals from plastics to the environment and to wildlife [J]. Philosophical transactions - Royal Society. Biological Sciences, 2009, 364(1526): 2027-2045. doi: 10.1098/rstb.2008.0284 [19] SUHRHOFF T J, SCHOLZ-BOTTCHER B M. Qualitative impact of salinity, UV radiation and turbulence on leaching of organic plastic additives from four common plastics - A lab experiment [J]. Marine Pollution Bulletin, 2016, 102(1): 84-94. doi: 10.1016/j.marpolbul.2015.11.054 [20] KOELMANS A A, BESSELING E, FOEKEMA E M. Leaching of plastic additives to marine organisms [J]. Environmental Pollution, 2014, 187: 49-54. doi: 10.1016/j.envpol.2013.12.013 [21] ROCHMAN C M, HOH E, HENTSCHEL B T, et al. Long-term field measurement of sorption of organic contaminants to five types of plastic pellets: implications for plastic marine debris [J]. Environmental Science & Technology, 2013, 47(3): 1646-1654. [22] LIU X M, SHI H H, XIE B, et al. Microplastics as both a sink and a source of Bisphenol A in the marine environment [J]. Environmental Science & Technology, 2019, 53(17): 10188-10196. [23] ROCHMAN C M, MANZANO C, HENTSCHEL B T, et al. Polystyrene plastic: A source and sink for polycyclic aromatic hydrocarbons in the marine environment [J]. Environmental Science & Technology, 2013, 47(24): 13976-13984. [24] WARDROP P, SHIMETA J, NUGEGODA D, et al. Chemical pollutants sorbed to ingested microbeads from personal care products accumulate in fish [J]. Environmental Science & Technology, 2016, 50(7): 4037-4044. [25] SONG X W, WU X F, SONG X P, et al. Sorption and desorption of petroleum hydrocarbons on biodegradable and nondegradable microplastics [J]. Chemosphere, 2020,273: 128553. [26] MSFD Technical Subgroup on Marine Litter, Guidance on monitoring of marine litter in european seas. A guidance document within the common implementation strategy for the marine strategy framework directive[M]. European Commission, 2013. [27] MASURA J, BAKER J, FOSTER G, et al. Laboratory methods for the analysis of microplastics in the marine environment: Recommendations for quantifying synthetic particles in waters and sediments[R], NOAA Technical Memorandum, 2015. Available from: https://marinedebris.noaa.gov/sites/ default/files/publications-files/noaa_microplastics_methods_manual.pdf. [28] CLAESSENS M, MEESTER S D, LANDUYT L V, et al. Occurrence and distribution of microplastics in marine sediments along the Belgian coast [J]. Marine Pollution Bulletin, 2011, 62(10): 2199-2204. doi: 10.1016/j.marpolbul.2011.06.030 [29] KUSUI T, NODA M. International survey on the distribution of stranded and buried litter on beaches along the Sea of Japan [J]. Marine Pollution Bulletin, 2003, 47(1/6): 175-179. [30] SUL J A I D, SPENGLER A, COSTA M F. Here, there and everywhere. Small plastic fragments and pellets on beaches of Fernando de Noronha (Equatorial Western Atlantic) [J]. Marine Pollution Bulletin, 2009, 58(8): 1236-1238. doi: 10.1016/j.marpolbul.2009.05.004 [31] NUELLE M T, DEKIFF J H, REMY D, et al. A new analytical approach for monitoring microplastics in marine sediments [J]. Environmental Pollution, 2014, 184: 161-169. doi: 10.1016/j.envpol.2013.07.027 [32] IMHOF H K, SCHMID J, NIESSNER R, et al. A novel, highly efficient method for the separation and quantification of plastic particles in sediments of aquatic environments [J]. Limnology & Oceanography Methods, 2012, 10: 524-537. [33] CRAWFORD C B, QUINN B. 9-Microplastic separation techniques[M]. Microplastic Pollutants. Amsterdam: Elsevier Science, 2017: 203-218. [34] CLAESSENS M, VAN C L, VANDEGEHUCHTE M B, et al. New techniques for the detection of microplastics in sediments and field collected organisms [J]. Marine Pollution Bulletin, 2013, 70(1/2): 227-233. [35] KEDZIERSKI M, LE T V, C G, et al. Efficient microplastics extraction from sand. A cost-effective methodology based on sodium iodide recycling [J]. Marine Pollution Bulletin, 2017, 115(1/2): 120-129. [36] QUINN B, MURPHY F, EWINS C. Validation of density separation for the rapid recovery of microplastics from sediment [J]. Analytical Methods, 2016, 9(9): 1491-1498. [37] DRIS R, IMHOF H, SANCHEZ W, et al. Beyond the ocean: contamination of freshwater ecosystems with (micro-)plastic particles [J]. Environmental Chemistry, 2015, 12(5): 539-550. doi: 10.1071/EN14172 [38] IMHOF H K, WIESHEU A C, ANGER P M, et al. Variation in plastic abundance at different lake beach zones-A case study [J]. Science of the Total Environment, 2017, 613/614: 530-537. [39] HORTON A A, SVENDSEN C, WILLIAMS R J, et al. Large microplastic particles in sediments of tributaries of the River Thames, UK-Abundance, sources and methods for effective quantification [J]. Marine Pollution Bulletin, 2016, 114(1): 218-226. [40] COPPOCK R L, COLE M, LINDEQUE P K, et al. A small-scale, portable method for extracting microplastics from marine sediments [J]. Environmental Pollution, 2017, 230: 829-837. doi: 10.1016/j.envpol.2017.07.017 [41] ZHANG K, SU J, XIONG X, et al. Microplastic pollution of lakeshore sediments from remote lakes in Tibet plateau, China [J]. Environmental Pollution, 2016, 219: 450-455. doi: 10.1016/j.envpol.2016.05.048 [42] XIONG X, ZHANG K, CHEN X C, et al. Sources and distribution of microplastics in China's largest inland lake - Qinghai Lake [J]. Environmental Pollution, 2018, 235: 899-906. doi: 10.1016/j.envpol.2017.12.081 [43] CORCORAN P L, BIESINGER M C, GRIFI M. Plastics and Beaches: A Degrading Relationship [J]. Marine Pollution Bulletin, 2009, 58(1): 80-84. doi: 10.1016/j.marpolbul.2008.08.022 [44] PAGTER E, FRIAS J, NASH R. Microplastics in Galway Bay: A comparison of sampling and separation methods [J]. Marine Pollution Bulletin, 2018, 135: 932-940. doi: 10.1016/j.marpolbul.2018.08.013 [45] CRICHTON E M, NOL M, GIES E A, et al. A novel, density-independent and FTIR-compatible approach for the rapid extraction of microplastics from aquatic sediments [J]. Analytical Methods, 2017, 9(9): 1419-1428. doi: 10.1039/C6AY02733D [46] MANI T, FREHLAND S, KALBERER A, et al. Using castor oil to separate microplastics from four different environmental matrices [J]. Analytical Methods, 2019, 11(13): 1788-1794. doi: 10.1039/C8AY02559B [47] KARLSSON T M, VETHAAK A D, ALMROTH B C, et al. Screening for microplastics in sediment, water, marine invertebrates and fish: Method development and microplastic accumulation [J]. Marine Pollution Bulletin, 2017, 122(1/2): 403-408. [48] ZOBKOV M B, ESIUKOVA E E. Evaluation of the munich plastic sediment separator efficiency in extraction of microplastics from natural marine bottom sediments [J]. Limnology & Oceanography Methods, 2017, 15(11): 967-978. [49] DESFORGES J P W, GALBRAITH M, ROSS P S. Ingestion of microplastics by Zooplankton in the Northeast Pacific Ocean [J]. Archives of Environmental Contamination & Toxicology, 2015, 69(3): 320-330. [50] ZHAO S Y, DANLEY M, WARD J E, et al. An approach for extraction, characterization and quantitation of microplastic in natural marine snow using Raman microscopy [J]. Analytical Methods, 2016, 9(9): 1470-1478. [51] MAES T, JESSOP R, WELLNER N, et al. A rapid-screening approach to detect and quantify microplastics based on fluorescent tagging with Nile Red [J]. Scientific Reports, 2017, 7: 44501. doi: 10.1038/srep44501 [52] COLE M, LINDEQUE P, HALSBAND C, et al. Microplastics as contaminants in the marine environment: A review [J]. Marine Pollution Bulletin, 2011, 62(12): 2588-2597. doi: 10.1016/j.marpolbul.2011.09.025 [53] CATARINO A I, THOMPSON R, SANDERSON W, et al. Development and optimization of a standard method for extraction of microplastics in mussels by enzyme digestion of soft tissues [J]. Environmental Toxicology and Chemistry, 2017, 36(4): 947-951. doi: 10.1002/etc.3608 [54] DEHAUT A, CASSONE A L, FRERE L, et al. Microplastics in seafood: Benchmark protocol for their extraction and characterization [J]. Environmental Pollution, 2016, 215: 223-233. doi: 10.1016/j.envpol.2016.05.018 [55] NAIDOO T, GOORDIYAL K, GLASSOM D, Are nitric acid (HNO3) digestions efficient in isolating microplastics from Juvenile Fish? [J]. Water Air & Soil Pollution, 2017, 228(12): 470. [56] MUNNO K, HELM P A, JACKSON D A, et al. Impacts of temperature and selected chemical digestion methods on microplastic particles [J]. Environmental Toxicology & Chemistry, 2017, 37(1): 91-98. [57] 李陵云, 朱静敏, 李佳娜, 等. 水生生物样品中微塑料的提取和分离方法综述 [J]. 海洋环境科学, 2019, 38(2): 187-191. doi: 10.12111/j.mes20190204 LI L L, ZHU J M, LI J N, et al. Review on methods for extraction and isolation of microplastics in aquatic organisms [J]. Marine Environmental Science, 2019, 38(2): 187-191(in Chinese). doi: 10.12111/j.mes20190204
[58] FOEKEMA E M, GRUIJTER C D, MERGIA M T, et al. Plastic in North Sea Fish [J]. Environmental Science & Technology, 2013, 47(15): 8818-8824. [59] QIU Q X, TAN Z, WANG J D, et al. Extraction, enumeration and identification methods for monitoring microplastics in the environment [J]. Estuarine Coastal & Shelf Science, 2016, 176: 102-109. [60] KUHN S, WERVEN V B, OYEN V A, et al. The use of potassium hydroxide (KOH) solution as a suitable approach to isolate plastics ingested by marine organisms [J]. Marine Pollution Bulletin, 2017, 115(1/2): 86-90. [61] WAGNER J, WANG Z M, GHOSAL S, et al. Novel method for the extraction and identification of microplastics in Ocean Trawl and Fish Gut Matrices [J]. Analytical Methods, 2016, 9(9): 1479-1490. [62] AVIO C G, GORBI S, REGOLI F. Experimental development of a new protocol for extraction and characterization of microplastics in fish tissues: First observations in commercial species from Adriatic Sea [J]. Marine Environmental Research, 2015, 111: 18-26. doi: 10.1016/j.marenvres.2015.06.014 [63] HURLEY R R, LUSHER A L, OLSEN M, et al. Validation of a method for extracting microplastics from complex, organic-rich, environmental matrices [J]. Environmental Science & Technology, 2018, 52(13): 7409-7417. [64] LÖDER M G J, GERDTS G. Methodology Used for the detection and identification of microplastics—A critical appraisal[B]. Springer, Cham, 2015: 201-227. https://doi.org/10.1007/978-3-319-16510-3_8 [65] FELSING S, KOCHLEUS C, BUCHINGER S, et al. A new approach in separating microplastics from environmental samples based on their electrostatic behavior [J]. Environmental Pollution, 2018, 234: 20-28. doi: 10.1016/j.envpol.2017.11.013 [66] GRBIC J, NGUYEN B, GUO E, et al. Magnetic extraction of microplastics from environmental samples [J]. Environmental Science & Technology Letters, 2019, 6(2): 68-72. [67] 王昆, 林坤德, 袁东星. 环境样品中微塑料的分析方法研究进展 [J]. 环境化学, 2017, 36(1): 27-36. doi: 10.7524/j.issn.0254-6108.2017.01.2016051704 WANG K, LIN K D, YUAN D X. Research progress on the analysis of microplastics in the environment [J]. Environmental Chemistry, 2017, 36(1): 27-36(in Chinese). doi: 10.7524/j.issn.0254-6108.2017.01.2016051704
[68] MILLER M E, KROON F J, MOTTI C A. Recovering microplastics from marine samples: A review of current practices [J]. Marine Pollution Bulletin, 2017, 123(1/2): 6-18. [69] DEVRIESE L I, VAN D M, MYRA D, et al. Microplastic contamination in brown shrimp (Crangon crangon, Linnaeus 1758) from coastal waters of the Southern North Sea and Channel area [J]. Marine Pollution Bulletin, 2015, 98(1/2): 179-187. [70] LUSHER A, WELDEN N, SOBRAL P, et al. Sampling, isolating and identifying microplastics ingested by fish and invertebrates [J]. Analytical Methods, 2016, 9(9): 1346-1360. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 12828
- HTML全文浏览数: 12828
- PDF下载数: 729
- 施引文献: 0
