





-
As和Sb是自然界中天然存在的两种元素,因其为元素周期表同一主族上的相邻元素,两者也表现出了相似的化学性质和环境行为[1-2]。其中,As及其化合物因显而易见的毒害性一直受到关注[3]。近年来,随着Sb元素在环境中的累积及污染事件时有发生,锑化合物也受到科研工作者的关注[4-7]。在天然锑矿中,两种元素常常共存,也意味锑矿开采造成的环境问题中两种元素也是相伴存在[8-9]。因而,近年来科研工作者有积极探索同时处理两种元素污染的工程技术[10]。
在As和Sb的污染处理技术中,因处理目标和出水水质要求的不同而选用相应的处理技术[6]。其中,硫酸盐还原菌介导的硫化物沉淀处理技术因其成本低廉、用途广泛、多目标同时实现而占有一席之地[11]。近年来,有不少研究涉及硫酸盐还原菌用于水体As或Sb污染的治理[10-13],探讨硫酸盐还原菌把As或Sb从水相转移到固相中的潜力。这类研究表明,硫酸盐还原菌对水体中Sb的去除效率很高[10,13-14],但是除As效果却没有明确结论:有些研究表明硫酸盐还原菌能够有效去除水体中As[13,15],而另一些研究却发现硫酸盐还原菌对As的去除能力很有限[16-17]。而随着对Sb元素研究的深入,研究者也发现与As处理相似的问题,即硫酸盐还原菌会使固相中的Sb重新释放入水相中[18]。研究者认为,硫酸盐还原菌处理体系的pH值及其硫化物含量,可能是制约As或Sb去除效果的关键因素[11,18]。
不过,在有关硫酸盐还原菌去除水体中As或Sb的研究中,一个富有启示的结论是,当水体中有其他金属离子时,特别是Fe的存在能够显著降低水体As残留量[10,16-17,19],对Sb的去除也有促进作用[10]。在自然界中,矿石中Fe的存在并不罕见,在锑矿尾矿中也会发现大量的含铁矿物[20],探讨Fe元素在砷或锑迁移、转化中的作用具有现实意义[21]。在之前的研究中,研究者为提高硫酸盐还原菌的生理活性及去除砷的效率,也曾采用添加还原Fe粉或Fe(Ⅱ)的方式[10,16-17,22-25],但这些研究大多采用一个恒定的Fe加入量,对于不同的Fe加入量可能导致的pH变化、硫化物含量变化、铁硫化物生成量的不同及最终对砷和锑去除效率的影响还有待深入探讨。本实验将通过设置一系列的Fe(Ⅱ)浓度梯度,来揭示Fe(Ⅱ)对硫酸盐还原菌处理体系中砷和锑的去除机理和效率的影响,以期为将来的工程处理提供理论依据。
-
试验所用的SRB复合菌群从广西锑矿区的尾矿砂中筛选、富集而来,该菌群用于所有试验处理中。菌种的筛选、富集过程以及菌种鉴定在另一研究成果中已详述[10],本研究所用的培养基成分如下(g·L−1):KH2PO4 (0.5)、NH4Cl (1)、Na2SO4 (1)、MgSO4·7H2O (2)、乳酸钠(3.65)、抗坏血酸(0.1)、CaCl2 (0.1)和酵母膏(1)。
培养基主要成分所用药品为分析纯或更优级试剂,实验用水均为Millipore去离子水(18.2 MΩ·cm)。试验中用于菌种培养的所有血清瓶在使用前均在10%硝酸中浸泡24 h以上,防止残余的重金属或有机物污染,之后依次用清水、去离子水冲洗,并在60 ℃烘箱中烘干。
-
实验研究采用批处理模式(表1),所有试验处理均在22个50 mL血清瓶中进行:在超净台上,把灭菌后的培养液趁热分装在灭菌后的血清瓶中,每个血清瓶装入45 mL培养液,通入氩气,封口保存。其中,Fe/As比为2(T2)、4(T4)、10(T10)、20(T20)、40(T40)、100(T100)的处理各设3个平行,Fe/As比为0(T0)及不接种SRB的处理(CK)作为对照,各设2个平行。待冷却后,CK加入5 mL冷却培养液,其他处理接种对数期的菌液5 mL,所有血清瓶放于30 ℃恒温箱中静置培养约48 h,取样测定pH值和硫化物浓度(以此作为处理起点,即初始0点)。之后,CK组和T0组加入2.5 mL pH 4.01的苯二甲酸氢钾缓冲溶液,T2-T100处理组分别加入2.5 mL浓度为200、400、1000、2000、4000、10000 mg·L−1的Fe(Ⅱ)储备液。所有浓度的Fe(Ⅱ)储备液均由pH 4.01的苯二甲酸氢钾缓冲溶液在氩气保护下溶解和逐步稀释硫酸亚铁铵((NH4)2Fe(SO4)2·6H2O)制成。
加Fe(Ⅱ)后振荡3 h,按比例取样;取样后所有处理按比例加入As(Ⅲ)储备液(250 mg·L−1),使得初始As浓度均为5 mg·L−1,振荡9 h后按比例取样;取样后所有处理按比例加入Sb(Ⅲ)储备液(250 mg·L−1),使得初始Sb浓度均为5 mg·L−1,振荡12 h后按比例取样。所有处理放在30 ℃恒温箱培养,分别于第8 天、第15 天取样。样品溶液取出后,根据相应的处理节点(0、3 h(0.125 d)、12 h(0.5 d)、1 d、8 d、15 d),测定pH、Fe(Ⅱ)、硫化物、
SO2−4 、As(Ⅲ)及Sb(Ⅲ)等。As(Ⅲ)储备液和Sb(Ⅲ)储备液在使用当天分别由固态亚砷酸钠(NaAsO2,纯度99%,Fluka公司)和固态酒石酸锑钾(KSbC4H4O7,99%纯度,Acros Organics公司)在氩气保护下溶于除氧超纯水配制而成。除氧超纯水由煮沸的超纯水充入氩气冷却制成。 -
取样时一部分样品溶液立即用pHS-3C型数显酸度计(上海,雷磁)测定pH;另一部分样品经0.45 µm滤膜过滤后,测定硫化物、Fe(Ⅱ)、As(Ⅲ)、Sb(Ⅲ)及
SO2−4 等。硫化物总量采用亚甲蓝分光光度法于665 nm测定(GB/T 16489),Fe(Ⅱ)采用邻二氮杂菲分光光度法于510 nm测定(GB/T 11064.7—2013),SO2−4 的测定采用铬酸钡分光光度法于420 nm测定(HJ/T 342─2007),所用仪器均为756MC紫外/可见分光光度计(上海,菁华)。As(Ⅲ)和Sb(Ⅲ)测定均采用氢化物发生原子荧光光谱法(HG-AFS,海光AFS-2202E,北京)以测定总砷和总锑的形式进行,以液态砷标(100 μg·mL−1,2% HNO3,Accu Standard公司)和液态锑标(1000 μg·mL−1,中国计量科学研究院)作基准物质,以硫脲和抗坏血酸作为还原剂[20]。样品分析所用试剂均为分析纯及以上,所有测验用水均为Millipore去离子水。
-
数据处理及分析主要使用Office软件Excel 2010及统计分析软件SPSS 22.0,对原始数据进行运算、单因素方差分析及相关分析。
-
在加入Fe(Ⅱ)、As(Ⅲ)和Sb(Ⅲ)之前,接种培养2 d的硫酸盐还原菌pH为7.17,硫化物浓度为217.37 mg·L−1(图1)。加入Fe(Ⅱ)3 h后,Fe/As比不同的处理组pH都有不同程度的下降且Fe/As比越高,pH越低,T0—T100处理组pH值分别降为6.96、6.62、6.57、6.44、6.23、5.72和5.14;As(Ⅲ)和Sb(Ⅲ)的加入并没有造成pH的进一步降低,部分处理组pH甚至稍微提高;静置培养至第8天时pH值均有所升高,第15天时pH则又下降(图1a)。总体来看,不同处理组pH随时间的变化趋势较为一致,而在相同的时间点上pH则随着Fe/As比的升高而降低。
硫化物浓度的变化趋势和pH较为一致(图1b):Fe(Ⅱ)加入3 h后,不同处理组的硫化物浓度从初始217.37 mg·L−1都有下降,且Fe(Ⅱ)加入量越多,硫化物浓度减少也越多,T0—T100处理组硫化物浓度分别降为194.65、183.10、178.48、155.11、111.35、48.71、0.19 mg·L−1;As(Ⅲ)和Sb(Ⅲ)的加入并没有造成硫化物浓度的明显降低,部分处理组的硫化物含量很快就恢复到As(Ⅲ)和Sb(Ⅲ)加入前的浓度水平;第8天时各组硫化物浓度都有所升高,继续培养7 d后(即第15天)各组硫化物浓度则又有下降。除了T100组,其他处理组的硫化物浓度都维持在一个较高的水平,T0—T20组都超过100 mg·L−1,T40也一直在50 mg·L−1波动。
由于加入的Fe(Ⅱ)来自(NH4)2Fe(SO4)2·6H2O,所以不同浓度Fe(Ⅱ)的加入也引进了新的
SO2−4 ,因而,SO2−4 的残余量(mr )等于实测量(md )减去由Fe引入量(mFe 为各组的初始Fe(Ⅱ)含量),计算式如下:对数据进行修正后,不同处理组
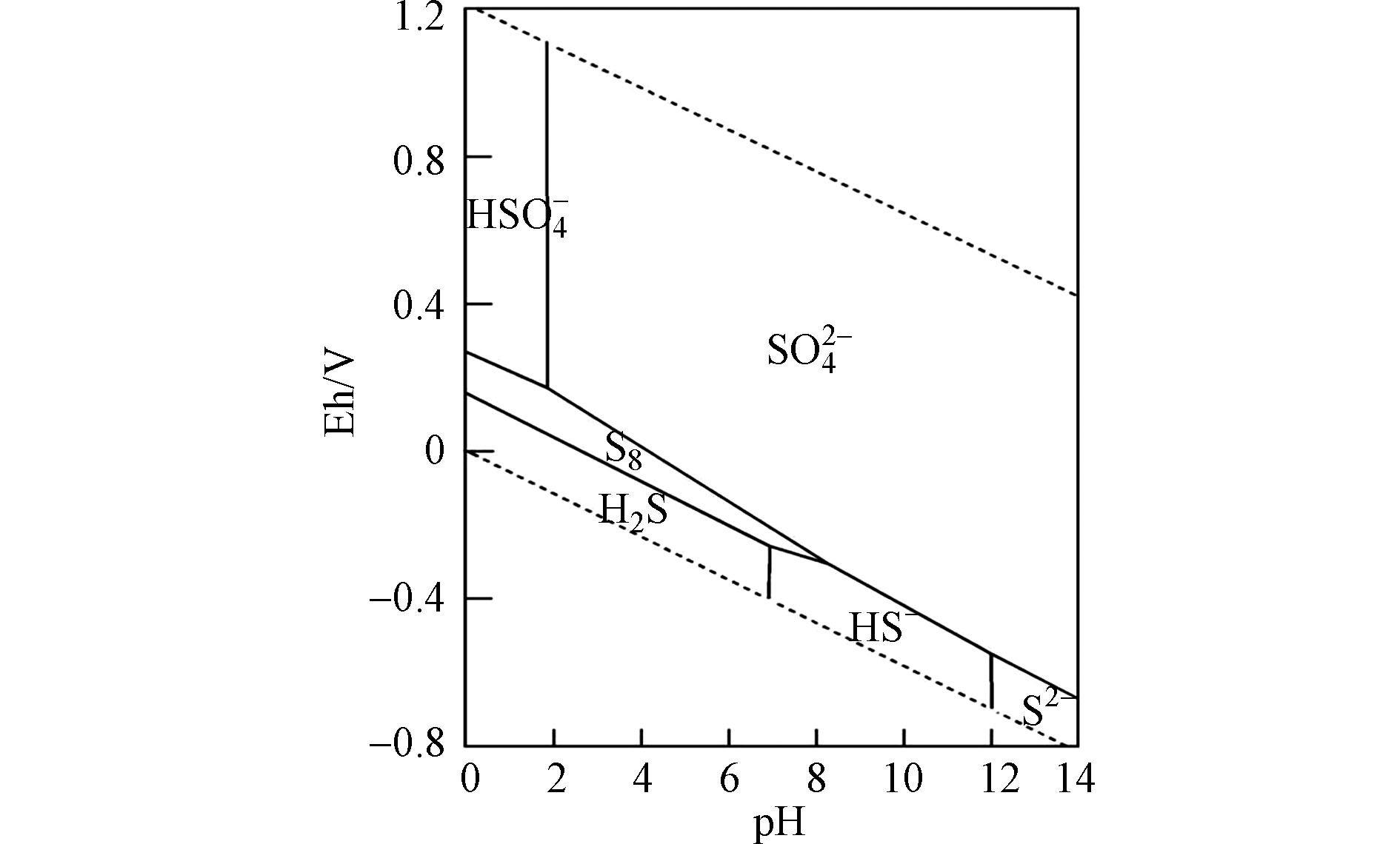
SO2−4 浓度的残余量表明(图2),与CK相比,硫酸盐还原菌的存在使得各组SO2−4 浓度均有不同程度的明显下降,其中,T0—T10组SO2−4 浓度差异不显著,T20—T100组SO2−4 浓度则随着Fe(Ⅱ)加入量的增加而明显下降。反过来说,较高浓度的Fe(Ⅱ)促进了基质中SO2−4 的转化与去除。从基质pH值、硫化物浓度随时间的变化状况及硫酸根的残余含量发现,Fe(Ⅱ)的不同加入量对初始条件一致的硫酸盐还原菌处理体系产生了显著影响。根据水体中硫元素的平衡相图(图3)及硫化氢的一级解离常数Ka1=1.3×10−7,加入Fe(Ⅱ)前基质pH约为7.17,表明此时硫化物主要以H2S及HS−两种形态共存,与Fe(Ⅱ)反应如下:
显然,这是一个消耗硫化物及产酸的过程,这也能直观解释加Fe(Ⅱ) 3 h后pH和硫化物浓度在T0—T100组的逐次递减。这里,一个隐含的pH影响因素是碳源(乳酸钠)的氧化消耗所产生的
HCO−3 [27-28],因
HCO−3 是两性离子,可在一定程度上调节体系的pH,避免基质过酸或过碱。所以,基质的pH值可认为是H2S、HS−、HCO−3 及CO2−3 解离平衡的结果[10]。通常,pH值低于5.5将对硫酸盐还原菌的生理活性产生影响[29],不过,在这个批量试验中,由于Fe(Ⅱ)及类金属加入前,48 h的微生物培养使得细菌生长进入了对数期且在此期间积累了足量的碱度和硫化物,这些目标元素的加入并未对微生物的生理活性造成较大冲击,这从第8天时pH和硫化物含量的升高能够得以证实。硫化物是
SO2−4 的还原产物,二者应是此消彼长的关系,但T0—T100处理组却呈现了硫化物浓度和SO2−4 残余含量的一致性。造成这种现象的原因,一方面是因为SO2−4 的消耗和硫化物的产生主要是在加入Fe(Ⅱ)前的48 h内,此时各处理组中二者的含量并无显著差异;另一方面,Fe(Ⅱ)的加入改变了两者的关系,随着初始Fe(Ⅱ)浓度的升高,对基质中硫化物的消耗增多,这既降低了硫化物对硫酸盐还原菌本身的毒害性[30]而利于硫化物的再产生又避免了硫化物转变为SO2−4 的可能性[31]。值得注意的是,尽管T0—T100组的SO2−4 残余浓度达到了显著性差异,但相差量最高也只有480 mg·L−1(占初始SO2−4 的23.6%),这可能是受碳源浓度制约,因为处理后期已没有足够的碳源来维持硫酸盐还原菌快速生长来消耗SO2−4 。 -
加入3 h后各组Fe(Ⅱ)的残余浓度表明(图4a),除了T40和T100组,其他各组Fe(Ⅱ)浓度均与T0组没有显著差异(P > 0.05),此时T0—T20组的Fe(Ⅱ)去除率达100%,T40组Fe(Ⅱ)去除率也达98%以上,T100去除率约为86%。在Fe(Ⅱ)残余浓度较高的T100组,As(Ⅲ)的加入造成了Fe(Ⅱ)的显著下降,而Sb(Ⅲ)的加入又显著提高了Fe(Ⅱ)的残余浓度(P < 0.05)。由于T0—T40组Fe(Ⅱ)的残余浓度较低,As(Ⅲ)和Sb(Ⅲ)的加入虽然使得各组Fe(Ⅱ)呈现与T100组相似的变化趋势,但各组Fe(Ⅱ)的浓度变化并不明显。第8天和第15天时,各组Fe(Ⅱ)的残余浓度有所起伏,不过,处理终点时除T100还有约109 mg·L−1的Fe(Ⅱ)外,其他各组的浓度均低于10 mg·L−1。
各组中As(Ⅲ)的残余浓度变化显示了硫酸盐还原菌去除As(Ⅲ)的效率和极限(图4b):与CK相比,各组As(Ⅲ)浓度均有所下降,且随加入处理时间的延长而进一步降低,但第15天时又有轻微上升;各组对比来看,T0—T100组的As(Ⅲ)浓度随Fe/As比的提高而降低。其中,T0—T4组的As(Ⅲ)浓度差异不明显,与对照相比,约降低了30%;T10组最终去除率达55.6%,T20—T100的As(Ⅲ)去除率在第8天时达到最高(均超过95%),第15天时残余浓度又有所升高,但去除率也均超过89%,基本达到0.5 mg·L−1的排放标准。
各组Sb(Ⅲ)残余浓度随时间的变化显示出,硫酸盐还原菌作用下各组Sb(Ⅲ)浓度均有明显下降(图4c):与As(Ⅲ)变化趋势不同的是,各组Sb(Ⅲ)的残余浓度在Sb(Ⅲ)加入12 h后(即第1天)达到最低,第8天时各组Sb(Ⅲ)浓度升高,第15天时又达到新的平衡,但也都高于第1天的残余量(T0组除外);各组对比来看,T0—T100组的Sb(Ⅲ)浓度随Fe/As比的提高而降低。其中,T0—T4组的Sb(Ⅲ)浓度差异不明显,与对照相比,约降低了82%;T10—T100组的最终去除率均达96%以上,对应的Sb(Ⅲ)残余浓度已经低于0.5 mg·L−1的排放标准。对处理终点时As(Ⅲ)和Sb(Ⅲ)的去除率(均以5 mg·L−1初始浓度计算)进行方差分析及多重比较(图5),可发现每一组的Sb(Ⅲ)去除率均高于As(Ⅲ),除T40和T100外,这种差异均达到显著性水平(P < 0.05)。总体来看,达到一定浓度(≥50 mg·L−1)的Fe(Ⅱ)显著提升了硫酸盐还原菌对两种元素的去除率,并且这种提升作用随Fe(Ⅱ)浓度的增加而强化,因而,500 mg·L−1Fe(Ⅱ)对硫酸盐还原菌去除As(Ⅲ)和Sb(Ⅲ)的促进作用最为明显,使得As(Ⅲ)和Sb(Ⅲ)的去除率从T0组的30.2%、83.8%分别提高到98.2%和100%。
与前人的研究结果相似的是,Fe(Ⅱ)的确能够提高As(Ⅲ)和Sb(Ⅲ)的去除率[10,16-17],并对As(Ⅲ)和Sb(Ⅲ)的去除效果有稳定作用。从图4可以看出,初始Fe(Ⅱ)越高,As(Ⅲ)和Sb(Ⅲ)的残余浓度越低,但两种元素达到最低浓度的时间有所差异。之前有研究者发现,硫酸盐还原菌对Sb的去除效果通常较好,在基质硫化物浓度不太高的情况下,基本可以达到污水排放标准(0.5 mg·L−1)[13-14]。不过,欧阳小雪等[32]在探讨硫酸盐还原菌去除Sb(Ⅲ)的实验研究中发现,处理后期出现了已沉淀Sb盐的复溶现象。最近也有研究者发现,硫酸盐还原菌会使已沉淀吸附在固相中的Sb化合物重新释放入水相中[18]。不过,初始Fe(Ⅱ)浓度对Sb的吸附作用有重要影响,Fe/As比小于10的几组处理中,Sb(Ⅲ)的去除率基本没有差别,这可能说明基质Fe浓度较低时,是不足以提供足够强度的吸附位点固定Sb(Ⅲ)的;在Fe/As比超过10的几组处理中,尽管基质中仍有较高浓度的硫化物存在(T100除外),但Sb(Ⅲ)的复溶现象并不明显。实验表明,加入适当的Fe(Ⅱ)的确有利于硫酸盐还原菌对Sb的去除,并稳定这种去除效果。
与Sb(Ⅲ)相比,Fe(Ⅱ)对硫酸盐还原菌除As的促进作用更为显著。另外,As的去除没有Sb的去除快,在第8天时达到最大去除量。不过,因除As的效率不如Sb,因而,处理后期As的复溶现象也不像Sb那么明显。从As的排放标准来看(0.5 mg·L−1),需要Fe/As比超过10才能达到出水标准。据此,可知As(Ⅲ)和Sb(Ⅲ)的去除机理可能略有不同。结合以往的研究[10],在基质中硫化物含量充足的情况下,Fe(Ⅱ)的迅速沉淀往往形成非晶形沉淀物,因而,在砷和锑的去除过程中,主要是无定形铁硫化物的吸附作用在起作用[33]。但是,Fe/As比低的几组中As和Sb的去除率,与T0组并无差别,这表明,FeS的吸附作用还会受到其他因素的制约。这种制约作用,很可能是pH和硫化物所致[11],而T0—T100组中均有pH较高及硫化物过量的问题,这显然对硫酸盐还原菌除As很不利。已有研究证实,控制基质pH值以避免硫代砷酸盐的生成,将极大地提高除As效率[29]。从这个意义上来说,较高浓度Fe(Ⅱ)的加入除了提供较多的吸附位点外,也降低了基质pH值,因而对除As效果有明显的促进。
-
对初始Fe(Ⅱ)加入量与其他指标(第15 天取样所测)进行Pearson相关性分析发现,初始Fe(Ⅱ)与基质pH、硫化物浓度、残余Fe(Ⅱ)浓度、残余As(Ⅲ)浓度、残余Sb(Ⅲ)浓度及残余
SO2−4 浓度等均呈极显著相关关系(P < 0.01):与理化指标pH、硫化物及SO2−4 等均呈现较强的负相关关系,与残余 Fe(Ⅱ)呈现正相关关系,与类金属的残余浓度呈现负相关关系(表2)。由于As(Ⅲ)和Sb(Ⅲ)在分析时采用残余浓度,初始Fe(Ⅱ)与As(Ⅲ)和Sb(Ⅲ)的负相关关系表明了初始Fe(Ⅱ)加入量的升高对As或Sb的去除有促进作用。此外,其他指标间也呈现一定的相关性,如残余的As(Ⅲ)和Sb(Ⅲ)与pH、硫化物均呈现较强的正相关性,表明pH、硫化物浓度的升高对基质中As(Ⅲ)和Sb(Ⅲ)的去除有不利影响;As(Ⅲ)和Sb(Ⅲ)的正相关系数为0.892(P < 0.01),表明它们的去除或共存受到相同的基质条件制约,反过来说,它们之间不存在竞争关系。同时,就As(Ⅲ)和Sb(Ⅲ)比较来看,As(Ⅲ)与其他指标的相关系数均高于Sb(Ⅲ),表明As(Ⅲ)的残余量对基质条件变化的响应更为敏感。对于硫酸盐还原菌处理砷污染废水来讲,较为普遍的结论是,由于中性或偏碱性条件下As(Ⅲ)易与过量的硫化物形成硫代亚砷酸盐而限制其去除[29],而Sb(Ⅲ)在硫化物过量时则相对惰性[3]。同时,As(Ⅲ)和Sb(Ⅲ)的正相关性说明,由于相似的化学性质,两者在相同的机制条件下呈现了较为一致的去除趋势,而铁硫化物也呈现出对As(Ⅲ)和Sb(Ⅲ)较为优越的去除潜力[3]。
-
不同浓度Fe(Ⅱ)的加入消耗了硫酸盐还原菌产生的碱度和硫化物,使得基质pH和硫化物含量均有所下降,不过低浓度Fe(Ⅱ)加入产生的黑色沉淀并不能明显促进硫酸盐还原菌对As(Ⅲ)和Sb(Ⅲ)的去除,达到一定浓度的Fe(Ⅱ)才能显著提高As(Ⅲ)和Sb(Ⅲ)的去除率。在相同的处理条件下,As(Ⅲ)和Sb(Ⅲ)的沉淀行为也有所不同,Sb(Ⅲ)沉淀速度更快、去除效率更高。总之,硫酸盐还原菌处理体系中As(Ⅲ)和Sb(Ⅲ)的去除效率受基质pH、硫化物、共存离子等因素制约,也受到自身化学性质的影响,适量Fe(Ⅱ)的加入提高了As(Ⅲ)和Sb(Ⅲ)的去除效率,并降低了固相中As(Ⅲ)和Sb(Ⅲ)复溶的可能性。
致谢 感谢2016级农业资源与环境专业杨晶同学在试验处理中提供帮助,感谢中国科学院地球化学研究所环境地球化学国家重点实验室提供平台和便利。
Fe(Ⅱ)浓度对硫酸盐还原菌去除水体中砷和锑的影响
Effect of different contents of Fe(Ⅱ) on removal of arsenic and antimony from water by sulfate reducing bacteria
-
摘要: 在硫酸盐还原菌处理体系中,加入浓度分别为10、20、50、100、200、500 mg·L−1的Fe(Ⅱ),探讨不同浓度的Fe(Ⅱ)对硫酸盐还原菌去除As(Ⅲ)和Sb(Ⅲ)(初始浓度均为5 mg·L−1)的影响。结果显示,不同浓度Fe(Ⅱ)的加入对体系pH、硫化物含量及
SO2−4 残余量均产生了显著影响;10 mg·L−1和20 mg·L−1的Fe(Ⅱ)对硫酸盐还原菌去除As(Ⅲ)和Sb(Ⅲ)的影响并不显著,随着Fe(Ⅱ)浓度的升高,体系中As(Ⅲ)和Sb(Ⅲ)的去除率均有明显提高;经过15 d的静置处理,500 mg·L−1 Fe(Ⅱ)对硫酸盐还原菌去除As(Ⅲ)和Sb(Ⅲ)的促进作用最为明显,使得As(Ⅲ)和Sb(Ⅲ)的去除率从不加Fe(Ⅱ)时的30.2%、83.8%分别提高到98.2%、100%;比较每个Fe(Ⅱ)浓度下As(Ⅲ)和Sb(Ⅲ)的去除率发现,Sb(Ⅲ)的去除率均高于As(Ⅲ)。研究表明,硫酸盐还原菌处理体系中As(Ⅲ)和Sb(Ⅲ)的去除效率将受基质pH、硫化物、共存离子等因素制约,也受到自身化学性质的影响,适量Fe(Ⅱ)的加入提高了As(Ⅲ)和Sb(Ⅲ)的去除效率,并降低了固相中As(Ⅲ)和Sb(Ⅲ)复溶的可能性。Abstract: In the treatment system of sulfate-reducing bacteria, Fe(Ⅱ) reagent at a specific concentration (10, 20, 50, 100, 200, and 500 mg·L−1) was added at each time to a treatment system of sulfate-reducing bacteria (SRB), to investigate the effect of concentration of Fe(Ⅱ) on the removal of As(Ⅲ) and Sb(Ⅲ)—initial concentrations of both were at 5 mg·L−1—by SRB. The results showed that all Fe(Ⅱ) reagents had a significant effect on the pH, sulfide content, and residualSO2−4 of the treatment system, 10 and 20 mg·L−1 of Fe(Ⅱ) had no significant effect on the removal of As(Ⅲ) and Sb(Ⅲ) by SRB. However, the removal rate of As(Ⅲ) and Sb(Ⅲ) in the system improved with the Fe(Ⅱ) concentration and thus, 500 mg·L−1 Fe(Ⅱ) had the largest effect on the removal of As(Ⅲ) and Sb(Ⅲ) by SRB (compared to the control when adding no Fe(Ⅱ), the removal rate of As(Ⅲ) and Sb(Ⅲ) increased from 30.2% and 83.8% to 98.2% and 100% in a 15-day static treatment, respectively). In each treatment, the removal rate of Sb(Ⅲ) was higher than that of As(Ⅲ). This study indicates that the removal efficiency of As(Ⅲ) and Sb(Ⅲ) in the SRB treatment system is influenced by substrate pH, sulfide, coexisting ions, etc., as well as their own chemical properties and the addition of appropriate amount of Fe(Ⅱ) can improve the removal efficiency and at the same time, reduce the resolution of As(Ⅲ) and Sb(Ⅲ) from the solid phase.-
Key words:
- sulfate-reducing bacteria /
- Fe(Ⅱ) /
- arsenic /
- antimony
-
羟胺作为重要的活性氮化合物,在地球氮循环中起着至关重要的作用[1-3]. 大量研究表明多种氨氧化微生物催化的氨氧化[4-5],氮固化[6]、硝化和反硝化[7]等过程中均有羟胺化合物的产生,为了深入了解羟胺在地球氮循环中的作用,研究羟胺在环境中的迁移转化是十分必要的.
NOM作为环境中普遍存在的复杂有机混合物,包含羟基、酮基、羧基等多种活性基团[8-9],对多种含氮化合物的环境迁移转化有着重要影响. 例如,环境中的苯胺化合物可以共价结合NOM分子[10];氨作为农业土壤氮肥中含量最丰富的一种形式,可与NOM结合而被称之为氨固化[11]; 3,4-二氯苯胺是敌草隆除草剂降解过程中产生的主要代谢物,它能够不可逆地与NOM结合,从而降低其毒性[12]. 在NOM与上述含氮化合物的相互作用过程中,NOM的醛、酮官能团被认为是主要的作用位点.
羟胺也可以与羰基反应生成肟类产物[13],已有多篇研究报道了羟胺与NOM的相互作用以探究NOM对羟胺的吸收作用[14]或测定含羰基的NOM组分[15]. Thorn等通过液体15N NMR光谱法对羟胺衍生的NOM产物进行了表征,证明肟类化合物是其主要衍生物[16]. 然而,由于NOM组成极其复杂,传统的方法很难分离羟胺衍生的NOM组分从而获得更全面的分子信息. 因此,尽管羟胺衍生的NOM的分子组成对环境化学家更好地理解有机质对全球氮循环中羟胺转化的影响具有重要意义,但到目前为止尚未见报道.
FTICR-MS,因其具有超高的质量检测准确度和分辨率,目前已成为表征NOM[17]及其消毒副产物[18]分子组成的重要分析工具. 通过对NOM中存在的小分子有机物的精确质量数测定,从而获得其分子式信息,在此基础上,通过各种可视化图分析,可进一步区分NOM分子所属化合物种类,如脂类、木质素类、鞣酸类、稠环芳烃类等,还可进一步分析化合物的疏水性、芳香度、不饱和度等性质[19-20]. 基于此,本研究采用FTICR-MS对羟胺衍生的NOM产物进行分子表征,采用15N同位素标记的羟胺作为起始反应物,以保证所得到的15N同位素标记羟胺衍生NOM产物能排除NOM中其他组分的干扰,从而被特异性地识别检测;同时考察了时间对羟胺与NOM反应的影响.
1. 材料与方法(Materials and Methods)
1.1 试剂与样品制备
15N标记盐酸羟胺 (H215NOH.HCl, >98%) 购置于 Sigma-Aldrich 公司(Steinheim, Germany),NOM标准品购置于美国腐殖酸学会,超纯水制备于Milli-Q system (Millipore, Milford, MA, USA),甲醇 (色谱纯) 购置于Fisher Scientific 公司(Fair Lawn, N.J., USA),其他试剂均为国产分析纯试剂.
称取2.0 mg NOM标准品溶解于100 mL超纯水中制备成20.0 mg·L−1 NOM储备液. 取10 mL NOM储备液4份,分别加入50倍质量过量的15N标记盐酸羟胺,在室温条件分别振荡反应2、4、8、10 h,然后,采用全自动固相萃取装置(睿科公司,厦门)和安捷伦Bond Elut PPL固相萃取柱((0.5 g/6 mL, Varian, Palo Alto, CA, U.S.A.)对上述样品进行固相萃取以去除残留的15N标记盐酸羟胺并富集浓缩样品[21],具体操作步骤如下:(1)用甲酸调节反应样品 pH至2.0;(2)在固相萃取装置上安装好PPL柱,先后用 12 mL 甲醇(色谱纯)和 12 mL 0.1%的甲酸水溶液淋洗活化柱子;(3)PPL柱缓慢加入10 mL样品,样品以1 mL·min−1 的流速通过固相萃取柱以富集目标化合物;(4)样品流完后加入 12 mL 0.1% 甲酸水溶液淋洗柱子,以除去不保留成分;(5)氮吹干燥柱子,然后用 12 mL 甲醇洗脱,收集洗脱液;(6)氮吹洗脱液至甲醇完全挥发,FTICR-MS测定前用 1 mL甲醇溶解.
1.2 设备
FTICR-MS(Solarix 15T, Infinity cell,Bruker Daltonik, GmbH, Bremen, Germany)配备电喷雾/基质辅助激光解吸双离子源(ESI/MALDI). 本研究均采用ESI 源进行样品离子化,进样方式采用直接连续进样,进样流速为120 μL·h−1;离子源喷针电压设置为 4.0 kV,样品采用负离子宽谱扫描模式进行分析,离子累积时间设置为0.06 s,四百万字节时域采集瞬变模式,每个样品采集300扫描并叠加(每个样品重复测定3次); 质量扫描范围(m/z)100—1600. 仪器测定样品前,采用10 mmol·L−1 甲酸钠(50%异丙醇水溶液)校准液进行外标校准,样品测定后获得数据采用自制的NOM中常见已知CHO分子式列表作为内标校准文件进行进一步的内标校准,校准后的质量误差均小于百万分之0.5. 质谱数据采用Data Analysis软件进行分析 (Bruker Daltonics, version 5.3).
1.3 分子式匹配方法
采用Data Analysis (Bruker Daltonics, version 5.3)软件对所得数据进行质谱峰确认和可能分子式匹配,分子式匹配条件如下:,确认质谱峰信噪比(S/N)大于等于4,质量检测误差值小于等于百万分之0.5,仅考虑C、H、O、N、S元素,元素个数范围设置如下:12C (0—∞), 1H (0—∞), 16O (0—∞), 14N (0—3 ), 15N (0—2 ), and 32S (0—1);进一步采用如下规则[22] 对得到的可能分子式进行进一步筛选:氢碳比值(H/C)范围: 0.3<H/C<2.2,氧碳比值范围(O/C):O/C<1.2,不饱和度(DBE)值需为整数且小于40,分子式组成满足偶氮规则,同时H、C、N元素个数满足:H<=2C+2+N,如果一个质核比(m/z)离子有多个可能分子式时,采用杂元素之和最小规则[23]筛选出正确分子式(即选择N、S元素个数之和最小的分子式是正确的),保留3次重复测定结果中共同鉴定到的分子式结果并转换成Excel表数据进行作图分析.
根据已有文献报道,计算每个分子式的不饱和度减氧元素个数值(DBE-O)[24],修正芳香指数(AI-mod) [25];根据AI-mod 和 H/C 值范围将所得分子式分子划分为4类[26]: 第一类(Group 1)属于燃烧源稠环多环芳烃类化合物 (AI_mod > 0.66), 第二类(Group 2)属于维管植物源多酚类化合物 (0.66 ≥AI_mod > 0.5), 第三类(Group 3)属于高不饱和酚类化合物(AI_mod ≤ 0.50 and H/C <1.5), 第四类(Group 4)属于脂肪族化合物 (2.0 ≥ H/C ≥ 1.5).
2. 结果与讨论(Results and Discussion)
2.1 FTICR-MS检测15N同位素标记羟胺衍生化NOM产物
羟胺作为活性氮化合物可直接与NOM中的含羰基分子反应形成多种羟胺衍生化NOM产物[16],然而,由于NOM中本身含有的含氮化合物同分异构体的存在,导致羟胺衍生化NOM产物很难被FTICR-MS特异性识别检测. 本研究采用15N同位素标记羟胺作为反应物,当与NOM发生反应后形成15N同位素标记羟胺衍生化NOM产物,而NOM本身含氮分子的15N同位素峰通常掩盖在FTICR-MS的噪音信号以下,不会对15N同位素标记羟胺衍生化NOM产物的质谱信号造成干扰,从而能在FTICR-MS检测时特异性识别此类产物. 实验结果如图1 所示. 以质谱图标称m/z 389和m/z 460为例,可以看到,当NOM与15N同位素标记羟胺在室温条件下反应10 h后,与原NOM样品的质谱图相比(图1 A、C),明显有15N同位素标记羟胺衍生化NOM产物峰出现(图1 B、D),且这些质谱峰不受NOM中其他离子峰干扰,产物分子式种类主要包括CHO15N1, CHO15N2, CHOS115N1 和 CHON115N1等4类. 以上实验结果表明,当NOM与15N同位素标记羟胺反应10 h后,会明显产生15N同位素标记羟胺衍生化NOM产物,且这些产物峰可以被FTICR-MS特异性检测识别.
 图 1 NOM样品FTICR-MS质谱图Figure 1. Mass spectra of NOM at nominal m/z 389 (A) and m/z 460 (C); Mass spectra of NOM reacting with -labelled hydroxylamine in 10 h, at nominal m/z 389 (B), at nominal m/z 460(D). Molecular formulas of the detected 15N- labelled-hydroxylamine derivatized-NOM components were listed under the spectra(A)标称m/z 389, (C) 标称m/z 460;NOM与15N标记羟胺反应10 h后的样品FTICR-MS质谱图(B)标称m/z 389, (D) 标称m/z 460. 15N标记羟胺衍生NOM产物分子式标记在谱图下方
图 1 NOM样品FTICR-MS质谱图Figure 1. Mass spectra of NOM at nominal m/z 389 (A) and m/z 460 (C); Mass spectra of NOM reacting with -labelled hydroxylamine in 10 h, at nominal m/z 389 (B), at nominal m/z 460(D). Molecular formulas of the detected 15N- labelled-hydroxylamine derivatized-NOM components were listed under the spectra(A)标称m/z 389, (C) 标称m/z 460;NOM与15N标记羟胺反应10 h后的样品FTICR-MS质谱图(B)标称m/z 389, (D) 标称m/z 460. 15N标记羟胺衍生NOM产物分子式标记在谱图下方2.2 15N同位素标记羟胺衍生化NOM产物的分子组成分析
为了进一步分析羟胺衍生化NOM产物的分子组成,对整个质谱数据依据1.3节部分描述进行分子式匹配和筛选,得到所有检测到的15N同位素标记羟胺衍生化NOM产物的分子式结果. 分析结果如图2所示,共鉴定到2137个15N同位素标记羟胺衍生化NOM产物分子式(图2B),其中,包括CHO15N1分子式1346个(约占鉴定总数的63%),CHO15N2分子式194个(约占鉴定总数的9%),CHON115N1分子式221个(约占鉴定总数的10%),CHOS115N1分子式376个(约占鉴定总数的18%). 而对于反应前的NOM样品,共鉴定到CHO分子式2902个(约占鉴定总数的51%),CHON1分子式929个(约占鉴定总数的17%),CHOS1分子式1831个(约占鉴定总数的32%). 通过比较分析,可以看到,约40%以上的NOM中的CHO分子组分可以与15N同位素标记羟胺发生反应形成CHO15N1羟胺衍生NOM产物,而仅有低于7%的NOM中的CHO分子组分可以形成CHO15N2羟胺衍生NOM产物;对于NOM中的CHON1和CHOS1分子组分,仅有约23.7%的CHON1和约20.7%的CHOS1分子组分可与羟胺反应形成相应的CHON115N1 和CHOS115N1产物. 据此,可以推测,这些与羟胺反应的NOM分子中很可能至少含有一个羰基基团. Baluha等的研究表明,苏万尼河富里酸中约39%的CHO分子组分含有一到两个醛基或者酮基基团[27],此研究结果为本研究的推测提供了支持证据. 而形成CHO15N2羟胺衍生物的数目较少的原因可能是由于空间位阻、氢键作用或者电荷效应等导致NOM的CHO分子很难与两个羟胺分子连续发生反应.
 图 2 NOM与15N标记羟胺反应前(A)后(B)鉴定到的分子式数目和占比图;NOM样品(C)和NOM与15N标记羟胺反应10 h后(D)鉴定分子式的Van Krevelen图Figure 2. Number and percentage of molecular formulas identified in NOM (A) and NOM reacting with 15N-labelled hydroxylamine in 10 h (B); Van Krevelen diagrams of the identified formulas in NOM (C) and NOM reacting with 15N- labelled hydroxylamine in 10 h (D)
图 2 NOM与15N标记羟胺反应前(A)后(B)鉴定到的分子式数目和占比图;NOM样品(C)和NOM与15N标记羟胺反应10 h后(D)鉴定分子式的Van Krevelen图Figure 2. Number and percentage of molecular formulas identified in NOM (A) and NOM reacting with 15N-labelled hydroxylamine in 10 h (B); Van Krevelen diagrams of the identified formulas in NOM (C) and NOM reacting with 15N- labelled hydroxylamine in 10 h (D)通过Van Krevelen (VK)图分析,可以进一步推测化合物的理化属性和分子种类,通过AI_mod 和 H/C值的范围可以将化合物划分为4类[26] (如图2C、D所示). 可以看到,15N同位素标记羟胺衍生化NOM产物绝大部分属于维管植物源多酚类(Group 2)和高不饱和酚类化合物(Group 3)(图2D);与原始NOM样品的VK图(图2C)比较发现,NOM组分在VK图上的点分布范围几乎完全覆盖羟胺衍生化NOM产物组分的点分布,表明羟胺衍生化NOM产物组分的H/C 和O/C值与原NOM组分相比没有发生较大变化,对这一结果的可能解释是羟胺衍生化NOM产物很可能是通过羟胺与NOM组分所含的羰基发生肟化反应形成[16],因为发生肟化反应时,羟胺衍生化NOM产物分子式与原NOM分子相比H元素个数仅增加了1,O元素个数无变化,同时增加了1个15N元素. 为了进一步证明此推测,基于肟化反应,根据羟胺衍生化NOM产物分子式还原出相应的NOM反应物分子式,如果被还原出的分子式在原NOM样品中被鉴定到,则被定义为母-子分子对,同时认为此产物应该是通过肟化反应产生的. 通过数据分析发现(结果如图3A所示),对于CHO15N1羟胺衍生化NOM产物,可以找到1160个母-子分子对(约占总数的86.1%),对于CHO15N2羟胺衍生化NOM产物,共鉴定到172个母-子分子对 (约占总数的86.4%),对于CHON115N1羟胺衍生化NOM产物,共鉴定到198个母-子分子对 (约占总数的89.6%),对于CHOS115N1羟胺衍生化NOM产物,共鉴定到357个母-子分子对 (约占总数的94.9%). 此结果表明,86%以上的羟胺衍生化NOM产物很可能是通过肟化反应形成.
 图 3 (A)基于肟化反应的母子-分子对个数和百分比图;(B)NOM中CHO分子以及与羟胺反应10 h形成CHO15N1 和 CHO15N2产物的CHO分子的VK图,(C)NOM中CHON1分子以及与羟胺反应10 h形成CHON115N1产物的CHON1分子的VK图,(D)NOM中CHOS1分子以及与羟胺反应10 h形成CHOS115N1产物的CHOS1分子的VK图Figure 3. (A) Number and percentage of the precursor-product molecule pairs based on oximation reaction; (B) Van Krevelen diagrams of CHO formulas in NOM, precursors of CHO15N1 and CHO15N2 formulas obtained in NOM reacting with 15N-labelled hydroxylamine in 10 h; (C) Van Krevelen diagrams of CHON1 formulas in NOM and precursors of CHON115N1 formulas obtained in NOM reacting with 15N- labelled hydroxylamine in 10 h; (D) Van Krevelen diagrams of CHOS1 formulas in NOM and precursors of CHOS115N1 formulas obtained in NOM reacting with 15N- labelled hydroxylamine in 10 h
图 3 (A)基于肟化反应的母子-分子对个数和百分比图;(B)NOM中CHO分子以及与羟胺反应10 h形成CHO15N1 和 CHO15N2产物的CHO分子的VK图,(C)NOM中CHON1分子以及与羟胺反应10 h形成CHON115N1产物的CHON1分子的VK图,(D)NOM中CHOS1分子以及与羟胺反应10 h形成CHOS115N1产物的CHOS1分子的VK图Figure 3. (A) Number and percentage of the precursor-product molecule pairs based on oximation reaction; (B) Van Krevelen diagrams of CHO formulas in NOM, precursors of CHO15N1 and CHO15N2 formulas obtained in NOM reacting with 15N-labelled hydroxylamine in 10 h; (C) Van Krevelen diagrams of CHON1 formulas in NOM and precursors of CHON115N1 formulas obtained in NOM reacting with 15N- labelled hydroxylamine in 10 h; (D) Van Krevelen diagrams of CHOS1 formulas in NOM and precursors of CHOS115N1 formulas obtained in NOM reacting with 15N- labelled hydroxylamine in 10 h通过对上述鉴定到的羟胺衍生化NOM产物的母分子的VK图分析发现(图3B、C、D),H/C值范围在0.4—1.4同时O/C值范围在0.3—0.9的NOM CHO分子,H/C值范围在0.6—1.1同时O/C值范围在0.5—0.75的NOM CHON1分子,以及H/C值范围在0.7—1.5同时O/C值范围在0.42—0.9的NOM CHOS1分子更容易与15N同位素标记羟胺反应形成单肟产物,且这些分子大部分属于维管植物源多酚和高不饱和酚类化合物;而H/C 值范围在0.6—1.2同时O/C值范围在0.3—0.7的NOM CHO分子可以与羟胺形成双肟产物. 其他被检测到的NOM分子,如H/C值大于1.4或者O/C值大于0.9的NOM CHO分子,H/C值大于1.1或者O/C值大于0.75的NOM CHON1分子,以及H/C 值大于1.5或者O/C值大于0.9的NOM CHOS1分子,在此实验条件下不与羟胺发生反应形成肟化产物. 此实验结果表明,低H/C值和O/C值NOM组分可能具有较高的羟胺反应活性. 低H/C值和O/C值意味着分子具有较高的不饱和度和较低的氧化度,据此可以推测,这些分子可能含有更多的羰基基团和较少的羧基基团,从而导致其对羟胺的反应活性较高.
2.3 反应时间对15N同位素标记羟胺衍生化NOM产物的影响
为了进一步研究NOM组分对羟胺的反应活性,实验考察了反应时间对15N同位素标记羟胺衍生化NOM产物的影响,过量的羟胺与NOM在室温条件下分别反应0、2、4、8、10 h后,分别过固相萃取柱进行浓缩萃取并进行FTICR-MS检测. 本研究主要关注CHO15N1类羟胺衍生NOM产物,由于此类产物的占比较高. 所得质谱结果的部分质谱图如图4所示,可以看到,有一些15N同位素标记羟胺衍生化NOM产物的峰强度会随反应时间的增加而逐渐增大(图4A),对应其NOM反应物的峰强度随时间延长而降低(图4D),这一结果较容易解释,即随着反应时间的延长,反应物的量会随着产物量的增加而降低;有一些产物峰强度随反应时间延长没有发生明显变化(图4B),而对应NOM反应物的峰强度却随反应时间延长而降低(图4E),这一结果较为异常,正常情况下,反应物峰强度应该不会发生明显变化,造成此异常结果的原因可能是此类NOM反应物除了生成CHO15N1类产物,还有可能发生了其他化学反应,具体原因有待进一步研究验证;有一些产物峰强度随反应时间增加而降低(图4C),对于其NOM反应物的峰强度在反应2 h后几乎均降低到质谱噪音峰强度水平(图4F),这一结果表明此类NOM反应物在反应2 h后已经反应完全,对应产物峰强度在2 h后进一步降低,推测此类产物有进一步转化为其他产物的可能性. 总之,对于15N同位素标记羟胺衍生化NOM产物而言,在反应10 h后,产物峰强度几乎不再变化. 此结果表明,15N同位素标记羟胺与NOM的反应10 h后可能已达到平衡. CHO15N1类羟胺衍生化NOM产物的形成具有高度的时间依赖性,很可能是由于NOM组分对羟胺的反应活性不同所造成的.
 图 4 15N同位素标记羟胺与NOM反应0, 2, 4, 8 和 10 h后的FTICR-MS质谱图(A)m/z 411.142检测的质谱峰,(B)m/z 499.064检测的质谱峰, (C)m/z 469.127检测的质谱峰, (D) m/z 395.134检测的质谱峰, (E) m/z 483.056检测的质谱峰, (F) m/z 453.119检测的质谱峰Figure 4. Mass spectra of NOM reacting with 15N-labelled hydroxylamine in 0, 2, 4, 8 and 10 h (A) the detected peak at m/z 411.142, (B) at m/z 499.064, (C) at m/z 469.127, (D) at m/z 395.134, (E) at m/z 483.056, (F) at m/z 453.119
图 4 15N同位素标记羟胺与NOM反应0, 2, 4, 8 和 10 h后的FTICR-MS质谱图(A)m/z 411.142检测的质谱峰,(B)m/z 499.064检测的质谱峰, (C)m/z 469.127检测的质谱峰, (D) m/z 395.134检测的质谱峰, (E) m/z 483.056检测的质谱峰, (F) m/z 453.119检测的质谱峰Figure 4. Mass spectra of NOM reacting with 15N-labelled hydroxylamine in 0, 2, 4, 8 and 10 h (A) the detected peak at m/z 411.142, (B) at m/z 499.064, (C) at m/z 469.127, (D) at m/z 395.134, (E) at m/z 483.056, (F) at m/z 453.119本研究进一步比较了反应2 h和10 h的CHO15N1产物的分子组成,结果如图5所示. 可以看到,反应2 h单独鉴定到的CHO15N1类分子式数目为84个,而10 h单独鉴定到314个,两个时间点共同鉴定到的分子式数目为1032个(图5B);VK图结果(图5A)显示,大部分2 h单独鉴定到的分子的O/C值较低,而大部分10 h单独鉴定到的分子的O/C值较高;DBE-O对分子式数目图[24](图5C)显示,10 h单独鉴定到的分子中,DBE-O值偏负的分子数目占比明显高于2 h单独鉴定到的分子,DBE-O值偏负表明分子中氧个数大于双键和环的个数[28]. 此结果表明,一些高氧化且饱和的NOM的CHO化合物对羟胺可能具有较低的反应活性,只有当延长反应时间时才可能形成羟胺衍生化NOM产物.
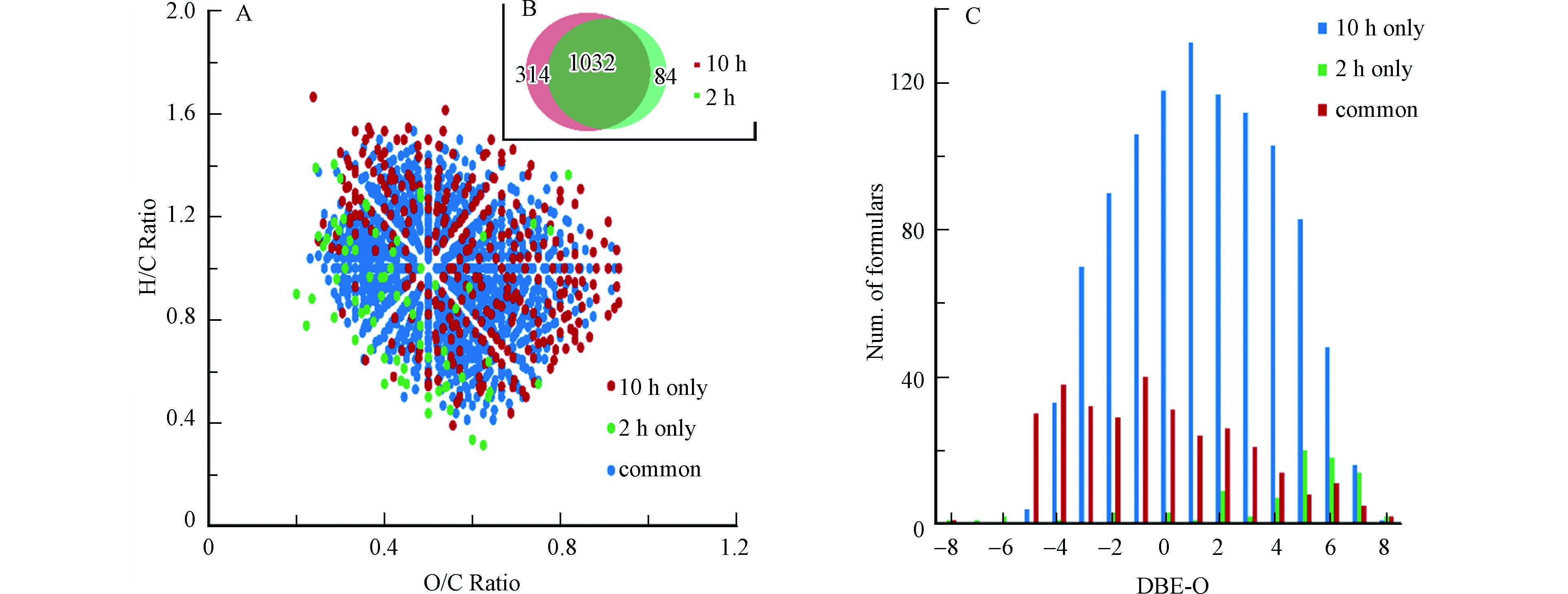 图 5 15N同位素标记羟胺与NOM反应2 h和10 h的CHO15N1产物的分子组成比较Figure 5. Comparison of the identified CHO15N1 formulas in NOM reacting with 15N-labelled hydroxylamine in 2 h and 10 h(A)VK图,(B)分子式数目比较图,(C)DBE-O对分子式数目图(A) Van Krevelen diagrams, (B) the number of the obtained formulas and (C) the plot of DBE-O vs. number of formulas
图 5 15N同位素标记羟胺与NOM反应2 h和10 h的CHO15N1产物的分子组成比较Figure 5. Comparison of the identified CHO15N1 formulas in NOM reacting with 15N-labelled hydroxylamine in 2 h and 10 h(A)VK图,(B)分子式数目比较图,(C)DBE-O对分子式数目图(A) Van Krevelen diagrams, (B) the number of the obtained formulas and (C) the plot of DBE-O vs. number of formulas对于反应2 h和10 h共同鉴定到的CHO15N1产物的分子,为了考察反应时间对其影响,同一产物离子在10 h的质谱峰强度除以其在2 h的质谱峰强度被定义为该离子的相对峰强度(RI),为了减小测定误差的影响,RI值小于等于0.8被定义为该离子的峰强度在反应10 h时明显小于反应2 h的峰强度,代表该类产物在反应10 h的产量低于其在2 h 的产量;RI值大于0.8且小于1.2被定义为该离子的峰强度在反应10 h时与其反应2 h的峰强度无明显差异,代表该类产物在反应10 h与在2 h 的产量相当;RI值大于等于1.2被定义为该离子的峰强度在反应10 h时明显大于其反应2 h的峰强度,代表该类产物在反应10 h的产量大于其反应2 h的产量. 通过分析发现,对于1032个共同鉴定到的CHO15N1产物的分子(结果如图6B所示),RI值大于等于1.2的分子式有613个(约占总数的60%),RI值在0.8—1.2之间的分子式有364个(约占总数的35%),而RI值小于等于0.8的分子式仅有55个(约占总数的5%). 此结果表明,有60%共同鉴定分子的产量随反应时间的延长而增大,说明产生此类产物的NOM反应物可能由于反应活性较弱,并不能在较短反应时间内与羟胺反应完全,而延长反应时间会增加羟胺衍生NOM产物的产量;有35%共同鉴定分子的产量随反应时间的延长无明显变化,说明产生此类产物的NOM反应物可能由于反应活性较强,在较短反应时间内便能与羟胺反应完全形成单肟化产物或随时间延长进一步形成其他产物;而仅有5%共同鉴定分子的产量随反应时间的延长而降低,说明产生此类产物的NOM反应物可能含有多个羰基基团,随反应时间延长可能进一步生成多肟化产物或其他不稳定产物,从而导致这些单肟化产物随反应时间延长而产量下降.
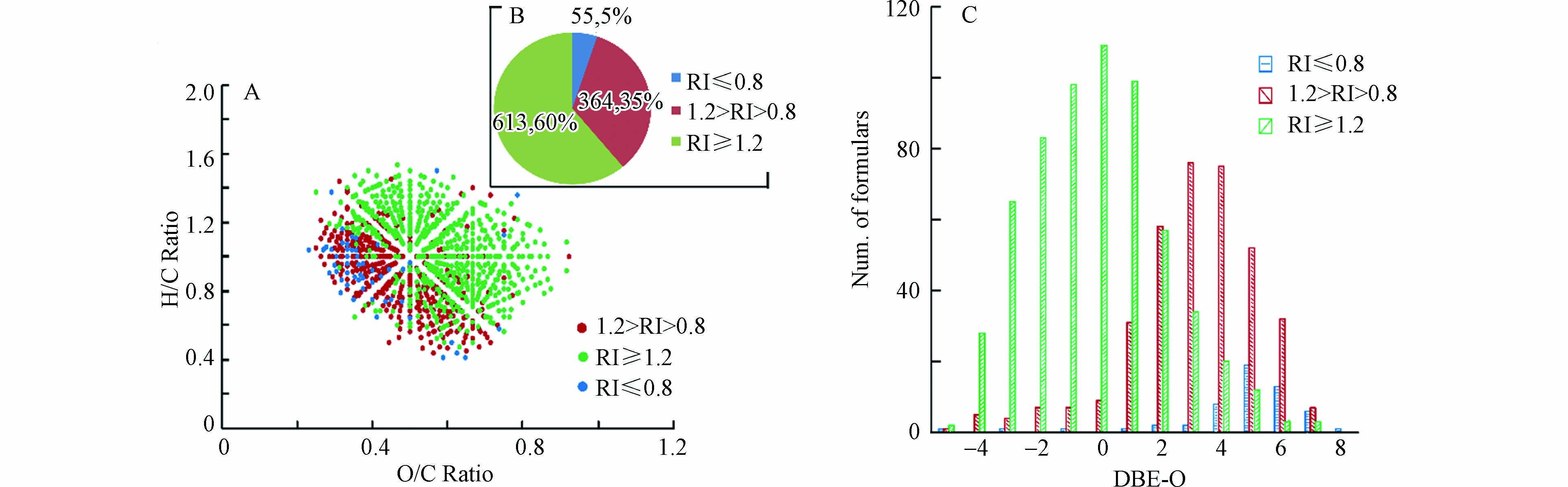 图 6 15N同位素标记羟胺与NOM反应2 h和10 h共同鉴定的不同RI值的CHO15N1产物的分子组成比较Figure 6. Comparison of the commonly identified CHO15N1 formulas with different RI values in NOM reacting with 15N-labelled hydroxylamine in 2 h and 10 h(A)VK图,(B)分子式数目比较图,(C)DBE-O对分子式数目图.(A) Van Krevelen diagrams, (B) the number and percentage of the obtained formulas and (C) the plot of DBE-O vs. number of formulas
图 6 15N同位素标记羟胺与NOM反应2 h和10 h共同鉴定的不同RI值的CHO15N1产物的分子组成比较Figure 6. Comparison of the commonly identified CHO15N1 formulas with different RI values in NOM reacting with 15N-labelled hydroxylamine in 2 h and 10 h(A)VK图,(B)分子式数目比较图,(C)DBE-O对分子式数目图.(A) Van Krevelen diagrams, (B) the number and percentage of the obtained formulas and (C) the plot of DBE-O vs. number of formulasVK图和DBE-O对分子式数目图结果(图6A、C)显示,RI值小于1.2的共同鉴定分子大部分具有较低的O/C值且DBE-O值偏正的分子式数目较多,而RI值大于等于1.2的共同鉴定分子大部分具有较高的O/C值且DBE-O值偏负的分子式数目较多,此结果说明高氧化度高饱和的NOM CHO分子对羟胺的反应活性相对较弱,需要较长的反应时间才能反应完全,而低氧化度高不饱和的NOM CHO分子对羟胺的反应活性相对较强,其反应完全时间较短甚至有些可以生成其他衍生产物.
事实上,RI值小于等于0.8的共同鉴定分子的进一步肟化产物确实在此研究实验中被检测到,图7A是检测质谱结果的一个举例;约有29%的此类分子可以进一步与羟胺反应形成双肟化产物,而其他71%的分子可能并不稳定或转化成其他产物(图7B).
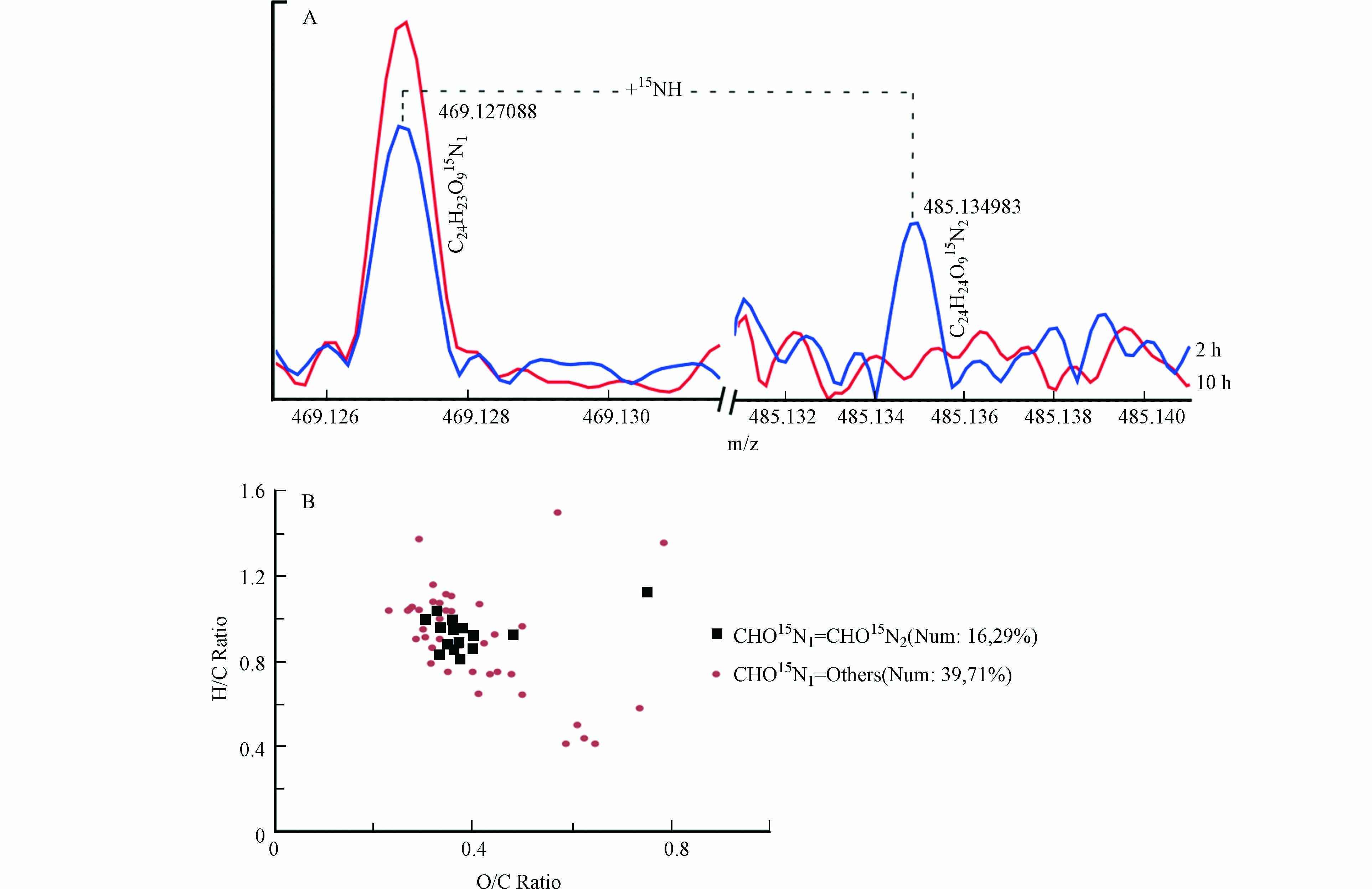 图 7 (A)15N同位素标记羟胺与NOM反应2、10 h后的FTICR-MS质谱图m/z 469.127和m/z 485.134;(B)形成双肟化产物或者其他产物的RI值小于等于0.8的共同鉴定CHO15N1产物的VK图Figure 7. (A) Mass spectra of NOM reacting with 15N-labelled hydroxylamine in 2 and 10 h at m/z 469.127 and m/z 485.134; (B) Van Krevelen diagrams of formulas with RI lower than 0.8 formed dioxime products and other products
图 7 (A)15N同位素标记羟胺与NOM反应2、10 h后的FTICR-MS质谱图m/z 469.127和m/z 485.134;(B)形成双肟化产物或者其他产物的RI值小于等于0.8的共同鉴定CHO15N1产物的VK图Figure 7. (A) Mass spectra of NOM reacting with 15N-labelled hydroxylamine in 2 and 10 h at m/z 469.127 and m/z 485.134; (B) Van Krevelen diagrams of formulas with RI lower than 0.8 formed dioxime products and other products3. 结论 (Conclusion)
羟胺作为地球氮循环过程中重要的活性氮化合物,可以与环境中普遍存在的NOM发生反应,从而转化成其他含氮产物. 本研究采用FTICR-MS超高分辨质谱结合15N同位素标记技术全面表征了羟胺与NOM在自然条件下发生反应的产物分子信息,共鉴定到2000多个羟胺衍生化NOM产物,主要包括CHO15N1、CHO15N2、CHOS115N1和CHON115N1 等4类分子产物,其中大部分产物可能属于维管植物源多分和高不饱和酚类化合物. 86%以上的产物可能是通过肟化反应形成,数据可视化分析表明,高氧化高饱和的NOM CHO化合物可能与羟胺的反应活性较弱,而低氧化高不饱和度的CHO化合物与羟胺的反应活性较强,其中部分可形成双肟化产物或其他产物. 本研究揭示了NOM不同组分对羟胺的反应活性以及相应产物的分子信息,可为深入探究羟胺在地球氮循环过程中的迁移转化和相应转化机制研究提供更全面的基础信息.
-

图 1 处理期间各组pH(a)和硫化物含量(b)随时间的变化
Figure 1. Variation of pH (a) and total sulfide contents (b) over time in every treatment group during processing
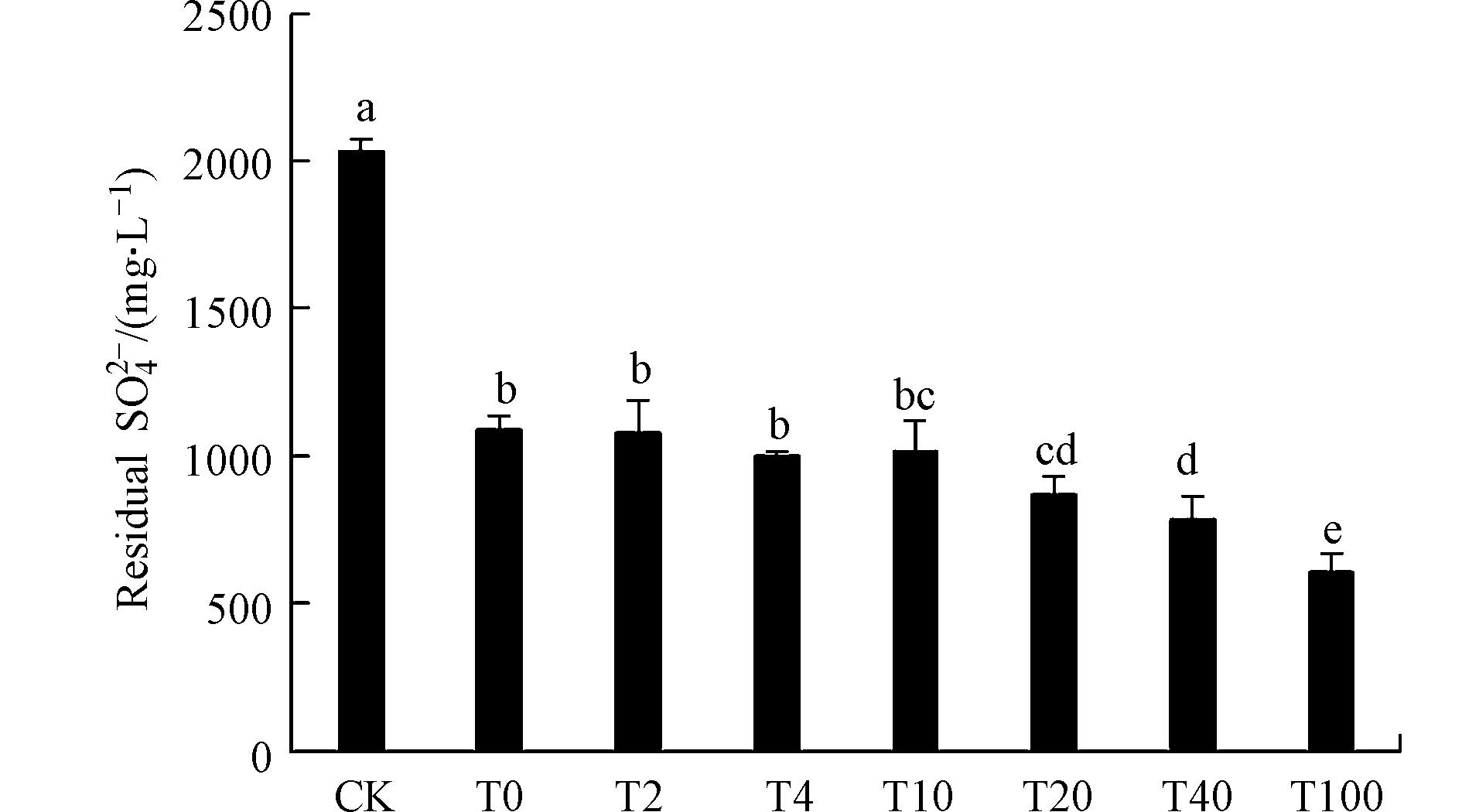
图 2 处理终点时各组SO42-的残余浓度
Figure 2. Residual concentration of SO42- in every treatment group in the end of processing
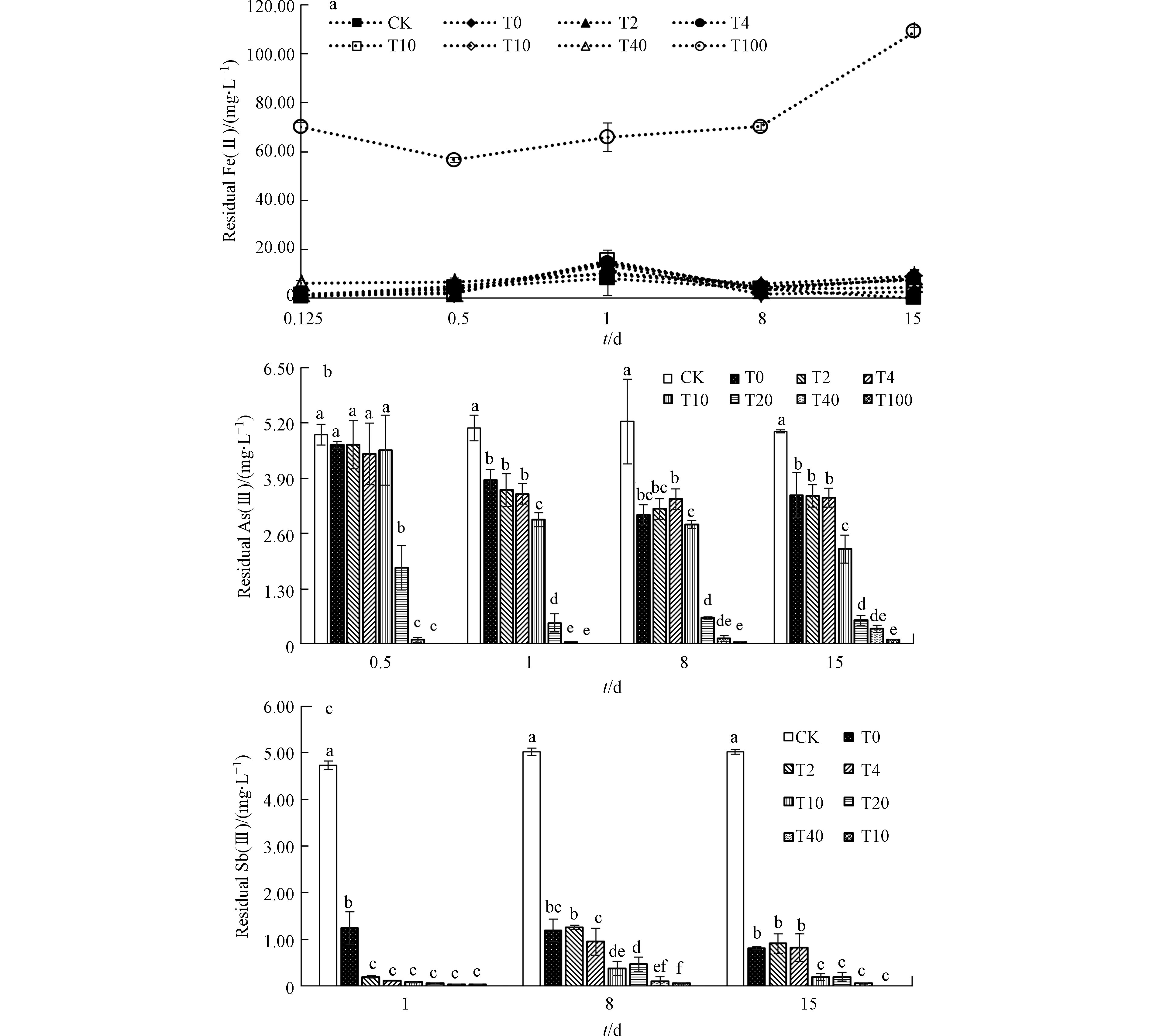
图 4 处理期间各组Fe(Ⅱ)(a)、As(Ⅲ)(b)和Sb(Ⅲ)(c)残余浓度随时间的变化
Figure 4. Variation of residual Fe(Ⅱ) (a), As(Ⅲ) (b), and Sb(Ⅲ) (c) concentration over time in every treatment group during processing
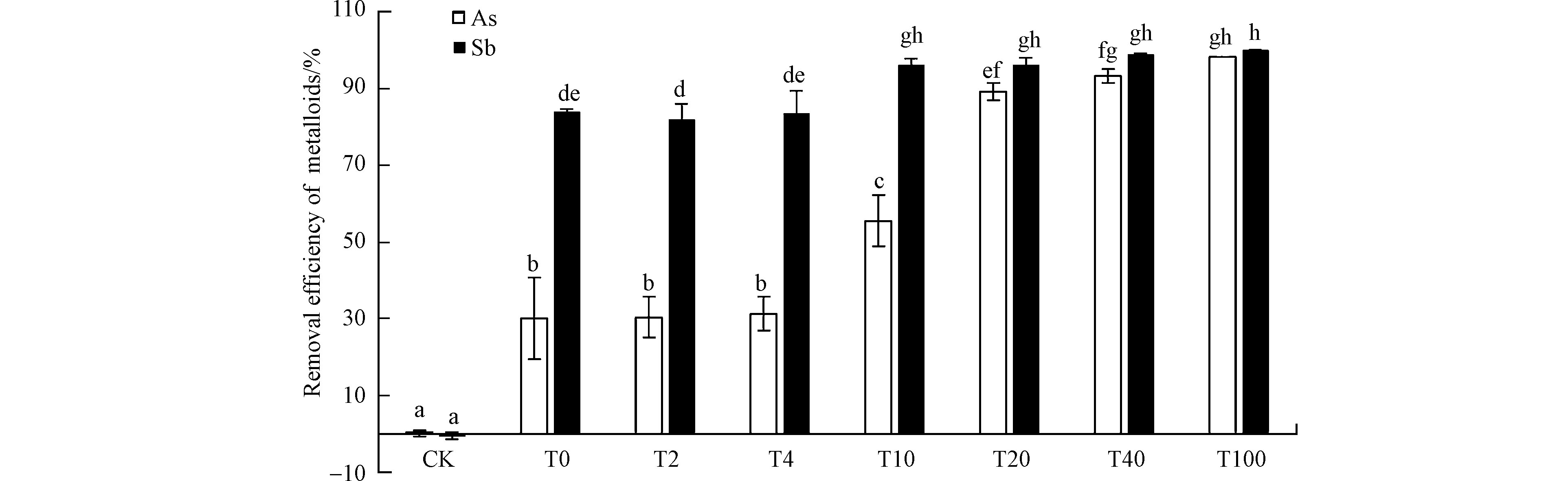
图 5 处理终点时类金属的去除率比较(均值±标准差,所有数据进行统一排序和多重比较)
Figure 5. Comparison of removal efficiency of metalloids in every treatment type in the end of processing (means ± standard, all data conform to uniform sorting and multiple comparisons)
表 1 批量处理试验设计
Table 1. Batch mode test design
处理组编号Treatment number SRB Fe(Ⅱ)/(mg·L−1) As(Ⅲ) (5 mg·L−1) Sb(Ⅲ) (5 mg·L−1) CK - - + + T0 + - + + T2 + 10 + + T4 + 20 + + T10 + 50 + + T20 + 100 + + T40 + 200 + + T100 + 500 + +  下载: 导出CSV
下载: 导出CSV
表 2 15 d的处理后基质各指标间的相关关系
Table 2. Correlation between the indicators in the matrix after 15 days’ treatment
初始Fe(Ⅱ)Initial Fe(Ⅱ) pH 硫化物Total sulfide Fe(Ⅱ) As(Ⅲ) Sb(Ⅲ) pH −0.977** 硫化物Total sulfide −0.938** 0.974** Fe(Ⅱ) 0.927** −0.854** −0.745** As(Ⅲ) −0.767** 0.837** 0.903** −0.519* Sb(Ⅲ) −0.673** 0.736** 0.806** −0.435 0.892** SO42- −0.889** 0.908** 0.905** −0.742** 0.810** 0.662** 注:**,P < 0.01; *,P < 0.05.
下载: 导出CSV
-
[1] LIU J L, YAO J, DURAN R, et al. Bacterial shifts during in-situ mineralization bio-treatment to non-ferrous metal (loid) tailings [J]. Environmental Pollution, 2019, 255: 113165. doi: 10.1016/j.envpol.2019.113165 [2] KULP T R, MILLER L G, BRAIOTTA F, et al. Microbiological reduction of Sb(Ⅴ)in anoxic freshwater sediments [J]. Environmental Science & Technology, 2014, 48(1): 218-226. [3] HAN Y, SEONG H J, CHON C, et al. Interaction of Sb(Ⅲ) with iron sulfide under anoxic conditions: Similarities and differences compared to As(Ⅲ) interactions [J]. Chemosphere, 2018, 195: 762-770. doi: 10.1016/j.chemosphere.2017.12.133 [4] REN M, DING S, FU Z, et al. Seasonal antimony pollution caused by high mobility of antimony in sediments: In situ evidence and mechanical interpretation [J]. Journal of Hazardous Materials, 2019, 367: 427-436. doi: 10.1016/j.jhazmat.2018.12.101 [5] BURTON E D, HOCKMANN K, KARIMIAN N, et al. Antimony mobility in reducing environments: The effect of microbial iron (Ⅲ)-reduction and associated secondary mineralization [J]. Geochimica et Cosmochimica Acta, 2019, 245: 278-289. doi: 10.1016/j.gca.2018.11.005 [6] UNGUREANU G, SANTOS S, RUI B, et al. Arsenic and antimony in water and wastewater: Overview of removal techniques with special reference to latest advances in adsorption [J]. Journal of Environmental Management, 2015, 151: 326-342. [7] HE M, WANG X, WU F, et al. Antimony pollution in China [J]. Science of the Total Environment, 2012, 421-422(3): 41-50. [8] SUN X, LI B, HAN F, et al. Impacts of arsenic and antimony co-contamination on sedimentary microbial communities in rivers with different pollution gradients [J]. Microbial Ecology, 2019, 78(3): 589-602. doi: 10.1007/s00248-019-01327-5 [9] ARSIC M, TEASDALE P R, WELSH D T, et al. Diffusive gradients in thin films reveals differences in antimony and arsenic mobility in a contaminated wetland sediment during an oxic-anoxic transition [J]. Environmental Science & Technology, 2018, 52(3): 1118-1127. [10] LIU F, ZHANG G, LIU S, et al. Bioremoval of arsenic and antimony from wastewater by a mixed culture of sulfate-reducing bacteria using lactate and ethanol as carbon sources [J]. International Biodeterioration & Biodegradation, 2018, 126: 152-159. [11] ALAM R, MCPHEDRAN K. Applications of biological sulfate reduction for remediation of arsenic–A review [J]. Chemosphere, 2019, 222: 932-944. doi: 10.1016/j.chemosphere.2019.01.194 [12] DE MATOS L P, COSTA P F, MOREIRA M, et al. Simultaneous removal of sulfate and arsenic using immobilized non-traditional SRB mixed culture and alternative low-cost carbon sources [J]. Chemical Engineering Journal, 2018, 334: 1630-1641. doi: 10.1016/j.cej.2017.11.035 [13] WANG H, CHEN F, MU S, et al. Removal of antimony (Sb(Ⅴ)) from Sb mine drainage: Biological sulfate reduction and sulfide oxidation–precipitation [J]. Bioresource Technology, 2013, 146(10): 799-802. [14] ZHANG G, OUYANG X, LI H, et al. Bioremoval of antimony from contaminated waters by a mixed batch culture of sulfate-reducing bacteria [J]. International Biodeterioration & Biodegradation, 2016, 115: 148-155. [15] TECLU D, TIVCHEV G, LAING M, et al. Bioremoval of arsenic species from contaminated waters by sulphate-reducing bacteria [J]. Water Research, 2008, 42(19): 4885-4893. doi: 10.1016/j.watres.2008.09.010 [16] SAHINKAYA E, YURTSEVER A, TOKER Y, et al. Biotreatment of As-containing simulated acid mine drainage using laboratory scale sulfate reducing upflow anaerobic sludge blanket reactor [J]. Minerals Engineering, 2015, 75: 133-139. doi: 10.1016/j.mineng.2014.08.012 [17] ALTUN M, SAHINKAYA E, DURUKAN I, et al. Arsenic removal in a sulfidogenic fixed-bed column bioreactor [J]. Journal of Hazardous Materials, 2014, 269(1): 31-37. [18] YE L, CHEN H, JING C. Sulfate-Reducing bacteria mobilize adsorbed antimonate by thioantimonate formation [J]. Environmental Science & Technology Letters, 2019, 6(7): 418-422. [19] LEE M K, SAUNDERS J A, WILSON T, et al. Field-scale bioremediation of arsenic-contaminated groundwater using sulfate-reducing bacteria and biogenic pyrite [J]. Bioremediation Journal, 2019, 23(1): 1-21. doi: 10.1080/10889868.2018.1516617 [20] FU Z, ZHANG G, LI H, et al. Influence of reducing conditions on the release of antimony and arsenic from a tailings sediment [J]. Journal of Soils and Sediments, 2016, 16(10): 2471-2481. doi: 10.1007/s11368-016-1484-4 [21] KARIMIAN N, JOHNSTON S G, BURTON E D. Iron and sulfur cycling in acid sulfate soil wetlands under dynamic redox conditions: A review [J]. Chemosphere, 2018, 197: 803-816. doi: 10.1016/j.chemosphere.2018.01.096 [22] ZACARÍAS-ESTRADA O L, BALLINAS-CASARRUBIAS L, MONTERO-CABRERA M E, et al. Arsenic removal and activity of a sulfate reducing bacteria-enriched anaerobic sludge using zero valent iron as electron donor [J]. Journal of Hazardous Materials, 2020, 384: 121392. doi: 10.1016/j.jhazmat.2019.121392 [23] BAI H, KANG Y, QUAN H, et al. Treatment of acid mine drainage by sulfate reducing bacteria with iron in bench scale runs [J]. Bioresource Technology, 2013, 128: 818-822. doi: 10.1016/j.biortech.2012.10.070 [24] 王悦, 周孜迈, 邓文娜, 等. 两种体系去除水体中的砷 [J]. 环境化学, 2018, 37(12): 2613-2620. doi: 10.7524/j.issn.0254-6108.2018033001 WANG Y, ZHOU Z M, DENG W N, et al. A study on the removal of arsenic from water by two systems [J]. Environmental Chemistry, 2018, 37(12): 2613-2620(in Chinese). doi: 10.7524/j.issn.0254-6108.2018033001
[25] 胡一帆, 王文兵, 仵彦卿. 弱磁场强化零价铁去除水中砷的效果 [J]. 环境化学, 2019, 38(5): 1074-1081. doi: 10.1002/etc.4383 HU Y F, WANG W B, WU Y Q. The role of weak magnetic field in accelerating the removal of arsenic by zero valent iron [J]. Environmental Chemistry, 2019, 38(5): 1074-1081(in Chinese). doi: 10.1002/etc.4383
[26] LIANG H C, BILLIN S J, WILLIS W B, et al. Designing a wastewater treatment plant to remove sulfate at an iron mine [J]. Proceedings of the Water Environment Federation, 2010, 2010(11): 5664-5674. doi: 10.2175/193864710798193671 [27] NEVATALO L M, MAKINEN A E, KAKSONEN A H, et al. Biological hydrogen sulfide production in an ethanol–lactate fed fluidized-bed bioreactor [J]. Bioresource Technology, 2010, 101(1): 276-284. doi: 10.1016/j.biortech.2009.07.042 [28] THAUER R K, JUNGERMANN K, DECKER K, et al. Energy conservation in chemotrophic anaerobic bacteria [J]. Bacteriological Reviews, 1977, 41(1): 100-180. doi: 10.1128/br.41.1.100-180.1977 [29] SUN J, HONG Y, GUO J, et al. Arsenite removal without thioarsenite formation in a sulfidogenic system driven by sulfur reducing bacteria under acidic conditions [J]. Water Research, 2019: 362-370. [30] 姚琪, 黄建洪, 杨磊, 等. 硫酸盐生物还原过程中涉硫组分代谢特性 [J]. 环境工程学报, 2018, 12(10): 2783-2790. doi: 10.12030/j.cjee.201802082 YAO Q, HUANG J H, YANG L, et al. Characteristic of metabolism for sulfur-containing components during sulfate bioreduction process [J]. Chinese Journal of Environmental Engineering, 2018, 12(10): 2783-2790(in Chinese). doi: 10.12030/j.cjee.201802082
[31] SAALFIELD S L, BOSTICK B C. Changes in iron, sulfur, and arsenic speciation associated with bacterial sulfate reduction in ferrihydrite-rich systems [J]. Environmental Science & Technology, 2009, 43(23): 8787-8793. [32] 欧阳小雪, 张国平, 李海霞, 等. 用硫酸盐还原菌去除废水中锑的实验研究 [J]. 地球与环境, 2014, 42(5): 663-668. OUYANG X X, ZHANG G P, LI H X, et al. Removal of antimony in synthetic wastewater by sulfate-reducing bacteria [J]. Earth and Environment, 2014, 42(5): 663-668(in Chinese).
[33] WATSON J H P, ELLWOOD D C, DENG Q, et al. Heavy metal adsorption on bacterially produced FeS [J]. Minerals Engineering, 1995, 8(10): 1097-1108. doi: 10.1016/0892-6875(95)00075-2 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2895
- HTML全文浏览数: 2895
- PDF下载数: 37
- 施引文献: 0


























