

 DownLoad:
DownLoad:



-
过氧乙酰硝酸酯(peroxyacetyl nitrate, PAN)是对流层大气中重要的活性有机含氮化合物,由挥发性有机物(VOCs)和氮氧化物(NOx)光化学反应产生[1],在大气化学过程中发挥着重要作用. PAN不存在天然源,比臭氧更适合作为光化学污染的指示剂[2]. PAN不溶于水、不易光解,但具有热不稳定性质[3-4]. PAN易热解释放氮氧化物(PAN的大气寿命在25 ℃时仅为30 min),是对流层中氮氧化物的重要储库;在低温条件下(PAN的大气寿命在−10 ℃时长达10 d)可远距离传输至偏远背景地区,成为当地大气中氮氧化物的重要来源[5-6],进而影响区域大气氧化性. 此外,PAN还具有显著的环境毒性,可毒害植物、刺激人眼、诱发突变等[7-8]. 鉴于PAN的重要大气化学影响和显著环境健康效应,其相关研究已经成为当前大气化学研究的热点之一.
对流层大气中PAN的体积分数通常处于10−9及以下水平,加之其极易热解,PAN的准确测量已成为当前研究中的难点. 大气中PAN的分析方法[9-14]已有较多报道,其中气相色谱-电子捕获检测法(GC-ECD)具有灵敏度高、抗干扰性强、设备成本低以及运输维修简便等特点,已成为当前国际上使用最广泛的分析方法. 目前基于GC-ECD的PAN分析方法必须通过标准物质的校准才能够进行准确定量. PAN极易热解的性质使其不易长期稳定保存,因此目前国内外仍无法提供稳定的商业化PAN标准物质. 当前PAN的标准物质主要通过湿法制备法[15]和光化学制备法[16-19]合成,而标准气体只能通过现场制备方式获得. 前者首先利用硝酸与过氧乙酸在液态烃中合成纯净PAN,随后通过气化及稀释制备出标准气体. 此种方法操作复杂繁琐、试剂消耗量大、花费时间较长,不适用于PAN的分析仪器外场标定. 后者主要利用丙酮光解与NOx反应合成PAN,操作简单且重复性好,容易在现场稳定制备PAN的标准气体[20],已在PAN分析仪标定中得到广泛应用. 该方法通常采用高浓度NOx与高浓度丙酮进行反应,随后对合成的高浓度PAN进行定量稀释,从而获得不同浓度梯度的PAN标准气体. 该方法必须提供过量[1, 19]丙酮才能够保障NOx向PAN的高效转换,并认为丙酮及其光解产物不会对GC-ECD检测造成影响[18]. 然而,研究表明高浓度NOx与高浓度丙酮光化学反应通常需要15 min至20 min[19]才能实现NOx向PAN的高效转化,所以气体在反应器停留时间内可能会对PAN的合成造成一定影响. 此外,丙酮光解的副产物实际上会在GC-ECD分析仪上有响应,且可能会影响PAN的准确标定.
为了更全面评估光化学制备法对PAN标准气体合成以及PAN分析仪标定的影响,本研究基于丙酮光解与NOx反应合成PAN原理搭建了一套同步稀释光化学合成系统,实现了低浓度NOx与高浓度丙酮快速混合的光化学合成PAN的方法,探讨了不同紫外光源及光照强度、产物残留等因素对PAN光化学合成的影响,获得了光化学稳定合成PAN的制备条件,并将其应用到GC-ECD-PAN分析仪的外场观测标定.
全文HTML
-
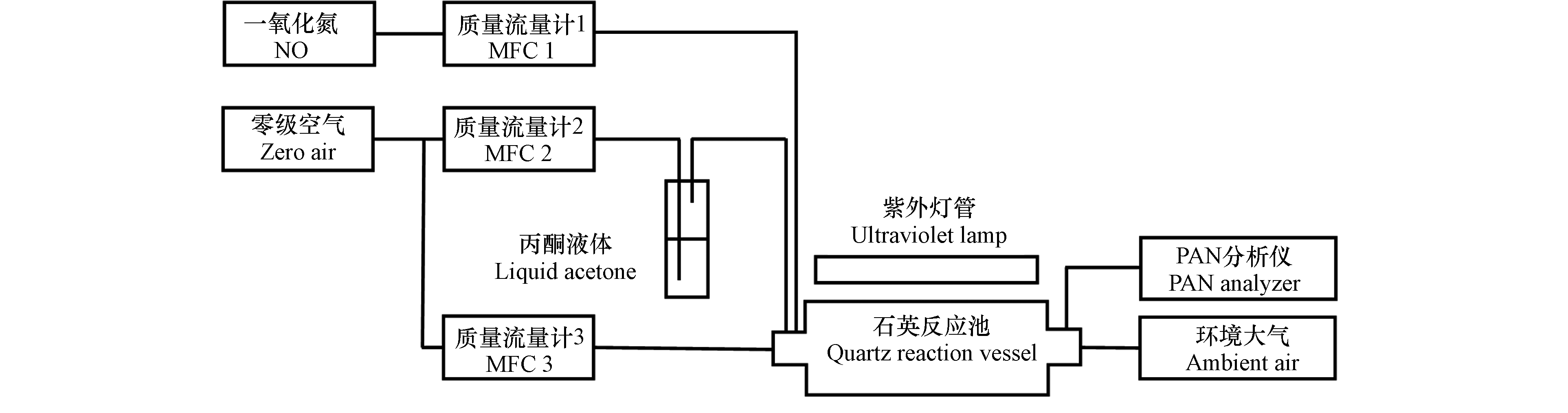
同步稀释光化学合成系统由气源、流量控制和合成反应等部分组成(如图1). 气源部分主要包括一氧化氮标准气体、利用鼓泡法产生的丙酮-空气混合气体以及稀释用零级空气. 流量控制部分主要采用电子质量流量计(MFC)分别控制一氧化氮标准气体、鼓泡用零级空气以及稀释用零级空气流量;合成反应部分为混合气源进入紫外灯照射的石英反应池进行同步稀释和光化学合成反应,其合成原理如下[21-23]:
丙酮在紫外灯照射下发生光解,产生乙酰基自由基
CH3C(O)⋅ ,其随后迅速与氧气结合生成过氧乙酰自由基CH3C(O)OO⋅ ,NO被氧化成NO2后快速与CH3C(O)OO⋅ 结合生成PAN.Meyrahn等指出丙酮和NOx体积分数分别在0.1%以上和10 × 10−6以下,可保证NOx向PAN的高效转化(大于90%)[18]. 为保障丙酮充足且混合气体在反应池中光化学反应充分,结合丙酮饱和蒸汽压和本研究光化学合成系统特点,各路气体流量设置如下:丙酮鼓泡用零级空气与稀释用零级空气流速分别为15 mL·min−1和500 mL·min−1,一氧化氮标准气体流速为2—96 mL·min−1范围内的梯度流量,实现PAN不同体积分数生成. PAN体积分数基于同步稀释的NO体积分数计算得出. 本合成系统中丙酮气体和PAN的体积分数计算如下式:
式中,[ac]为丙酮气体体积分数,VPac为丙酮(10 ℃)饱和蒸汽压(数值为1.541 × 104 Pa,根据安托因方程计算),Pair为标准大气压(数值为1.013 × 105 Pa),[PAN]表示合成的PAN体积分数,[NO]表示NO标准气体的体积分数,α表示NO向PAN的转化效率(取93% ± 3%[24]),vNO、vac和vtot分别表示实际校准的一氧化氮标准气体、鼓泡用零级空气和总混合气体的气体流速. 计算得出反应池中丙酮气体和NO体积分数分别约为0.4%和0.4 × 10−9—16 × 10−9,丙酮体积分数为NO的105倍以上. 合成的PAN体积分数范围约为0.31 × 10−9—13.37 × 10−9. 根据气体流量以及反应池容积(约500 mL),估算得出气体在反应池中停留时间在1 min左右. 在每个NO体积分数梯度下分别开展连续合成实验,合成达到稳定后连续测量10次,并开展长时间合成的连续稳定性实验. 实验开始前向系统内通入一段时间零级空气,随后测定系统内PAN峰色谱响应背景值以避免系统背景干扰.
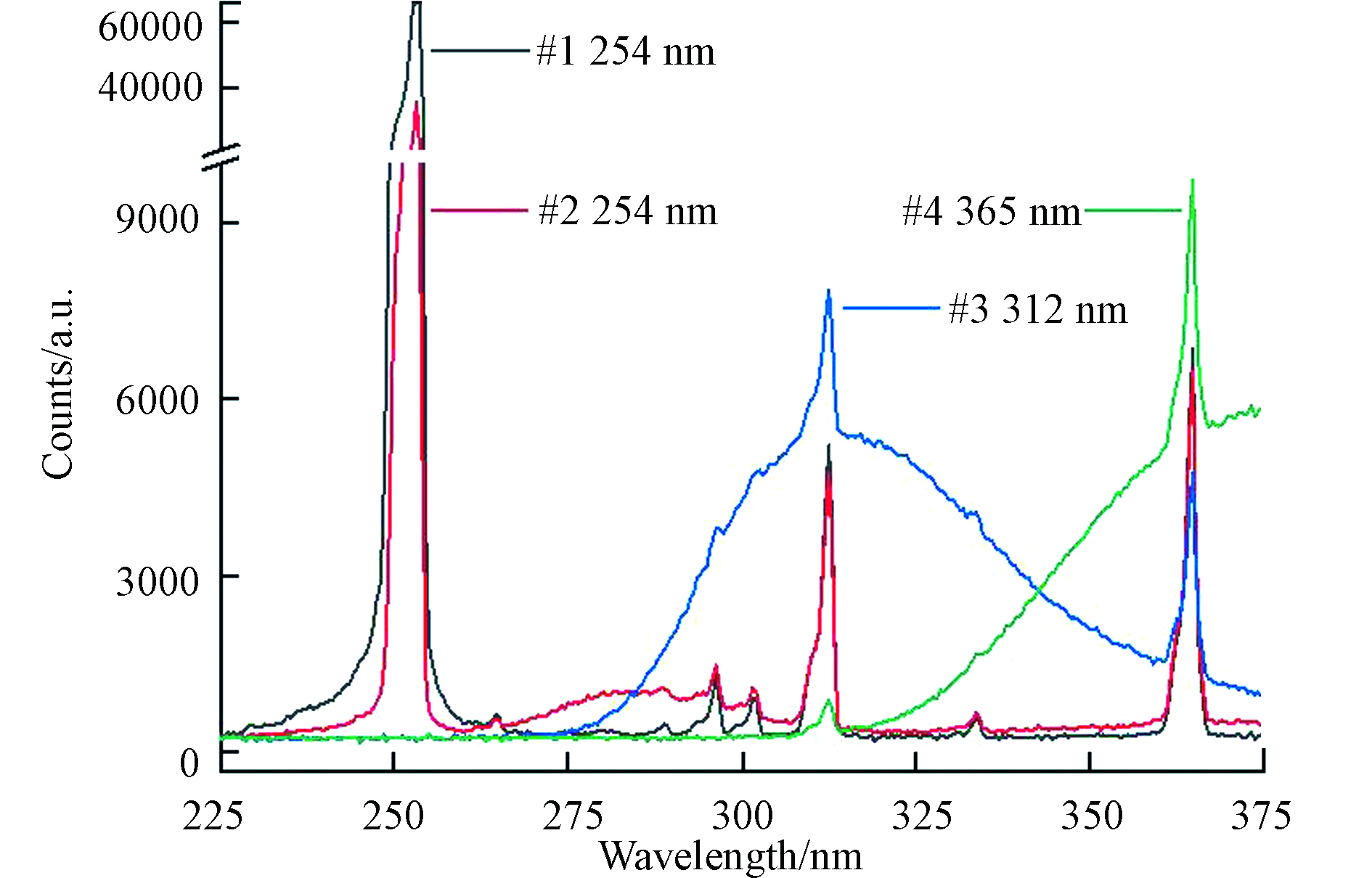
本研究使用的零级空气由AG-5000型号空气发生器(中科汇恒公司)产生的压缩空气经除水、脱烃和净化等处理后获得;一氧化氮标准气体为中国计量科学研究院研制的低浓度气体,体积分数为98 × 10−9,剩余充填气体为氮气;丙酮为分析纯丙酮液体(国药集团化学试剂公司). 为保证丙酮气化速率的稳定,利用半导体制冷装置控制丙酮液体温度为10 ℃. 电子质量流量计选用GT-130D型号流量计(天津吉思特仪器仪表有限公司),其中控制一氧化氮标准气体和鼓泡用零级空气的流量计量程范围为1—100 mL·min−1,控制稀释用零级空气的流量计量程范围为1—1000 mL·min−1. 质量流量计在实验温度下由皂膜流量计校准,精密度RSD < 2%. 本研究采用4种紫外灯(光谱图如图2,灯管规格:6 W),主峰波长分别为254 nm(#1和#2)、312 nm(#3)和365 nm(#4). 通过遮挡发光灯管长度改变光照强度(如全灯管、3/4灯管、半灯管及1/4灯管). 本研究使用的石英反应池为薄壁圆柱型腔体(外径60 mm,长180 mm).
-
PAN分析仪为中国科学院生态环境研究中心自主研制TGC-PAN分析仪. 分析仪基于配有电子捕获检测器的气相色谱仪(GC-ECD)设计而成,采用气泵抽气和定量环(1 mL)定量方式直接进样,利用电驱动两位十通阀进行气路切换. 气体样品经低温(5 ℃)毛细管色谱柱进行分离,在ECD检测器(温度为38 ℃[25])进行检测. 载气和尾吹气采用高纯氮气(99.999%),流速分别为25 mL·min−1和15 mL·min−1. 分析仪的每个样品分析周期为10 min.
-
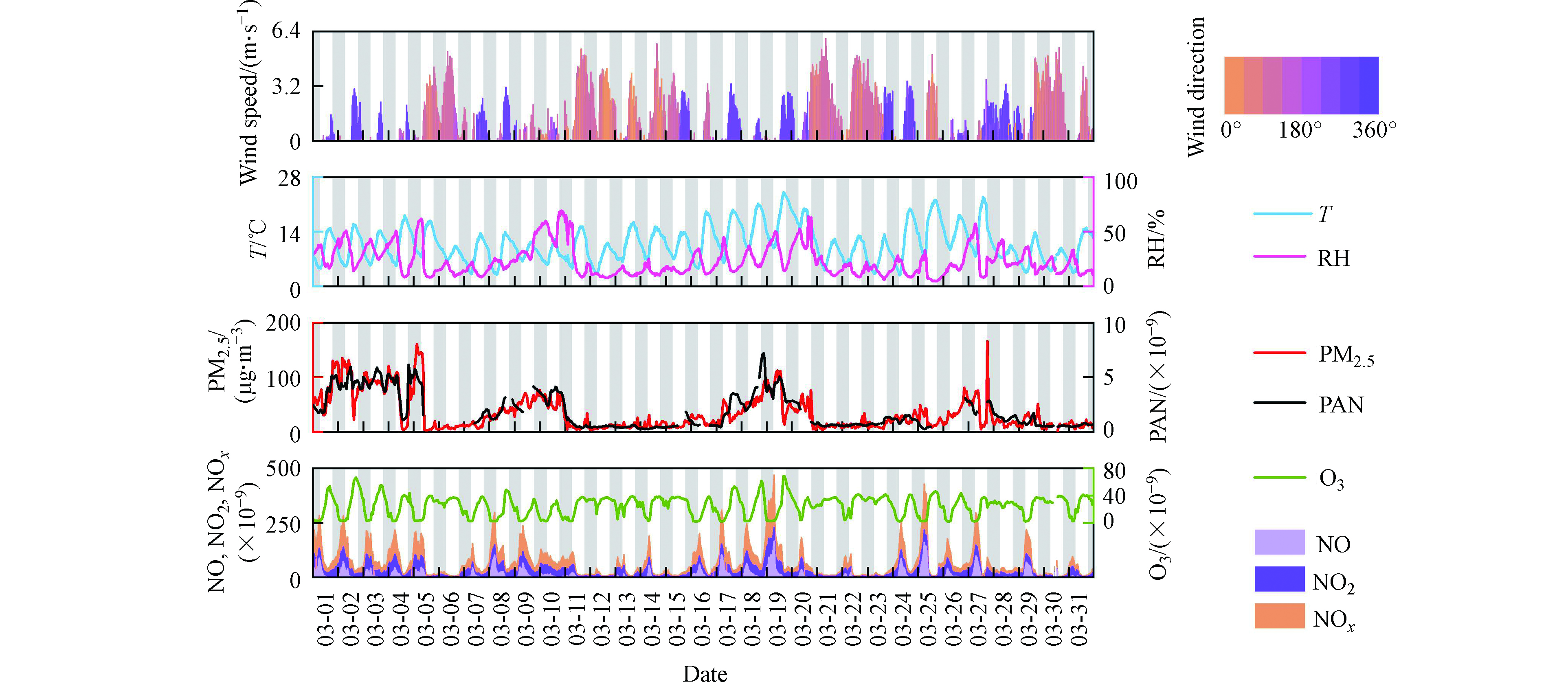
本研究于2019年3月1日至3月31日期间利用TGC-PAN分析仪对北京大气中PAN进行观测,观测频率为每10 min 1次,观测期间采用本研究优化的PAN光化学合成方法每周对分析仪进行一次校准. 观测站点位于中国科学院生态环境研究中心环境技术楼8层(40°0′N,116°20′E),距地高度约为20 m. 观测点位于北京市北四环与北五环之间,周围主要为教学楼、居民区以及公园,绿化覆盖率较高. 观测点西侧150 m处有1条东北-西南走向双向四车道公路,南侧200 m处有1条东西走向双向两车道公路,工作日早晚高峰车辆较多,平时交通流量不大. 观测期内的气象参数(风速风向、气温、相对湿度)及气态污染物(PM2.5、NO、NO2、NOx、O3)浓度数据来自中国科学院生态环境研究中心监测台站.
1.1. 同步稀释光化学合成系统



1.2. PAN分析仪
1.3. 观测时间及站点介绍
-
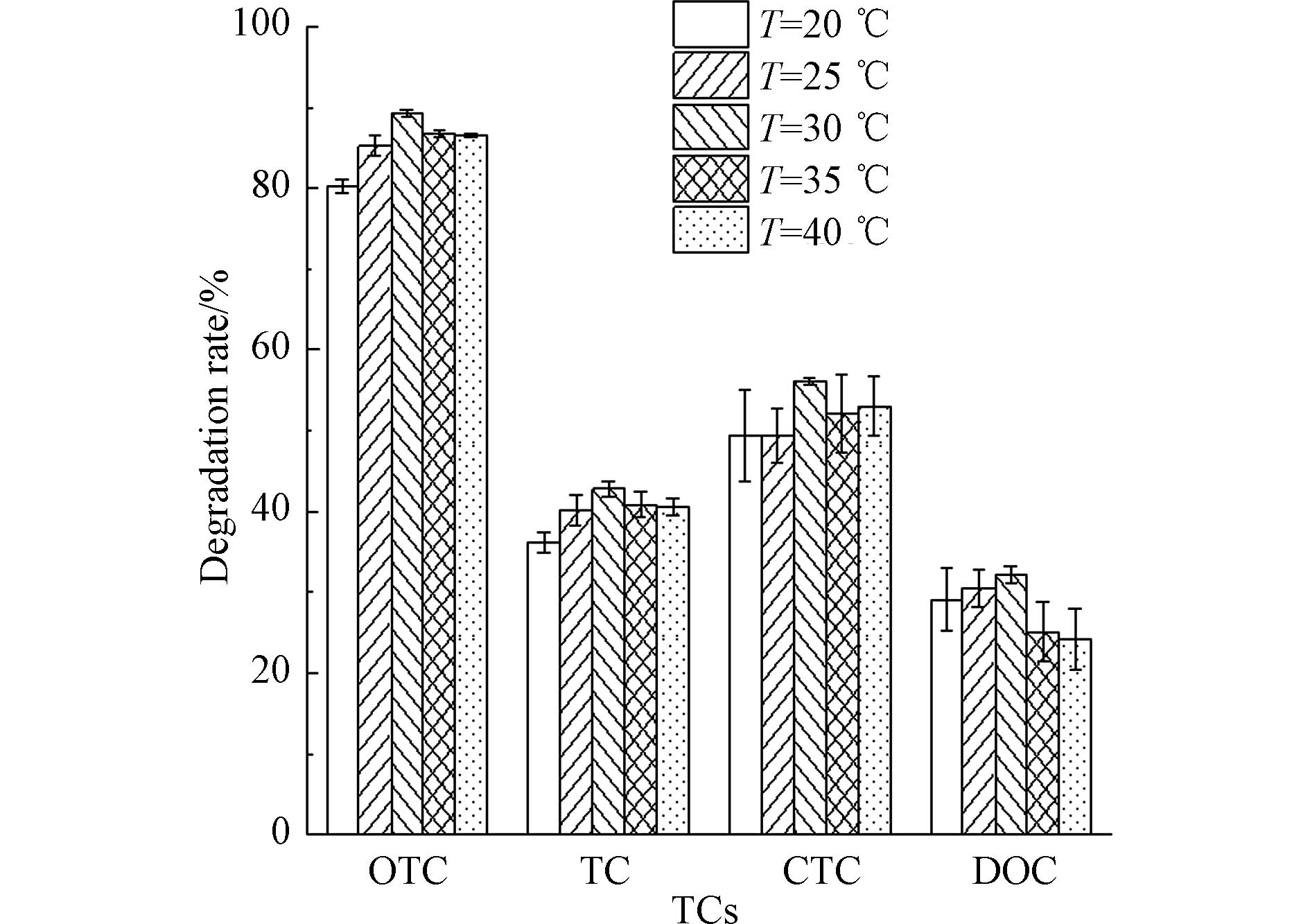
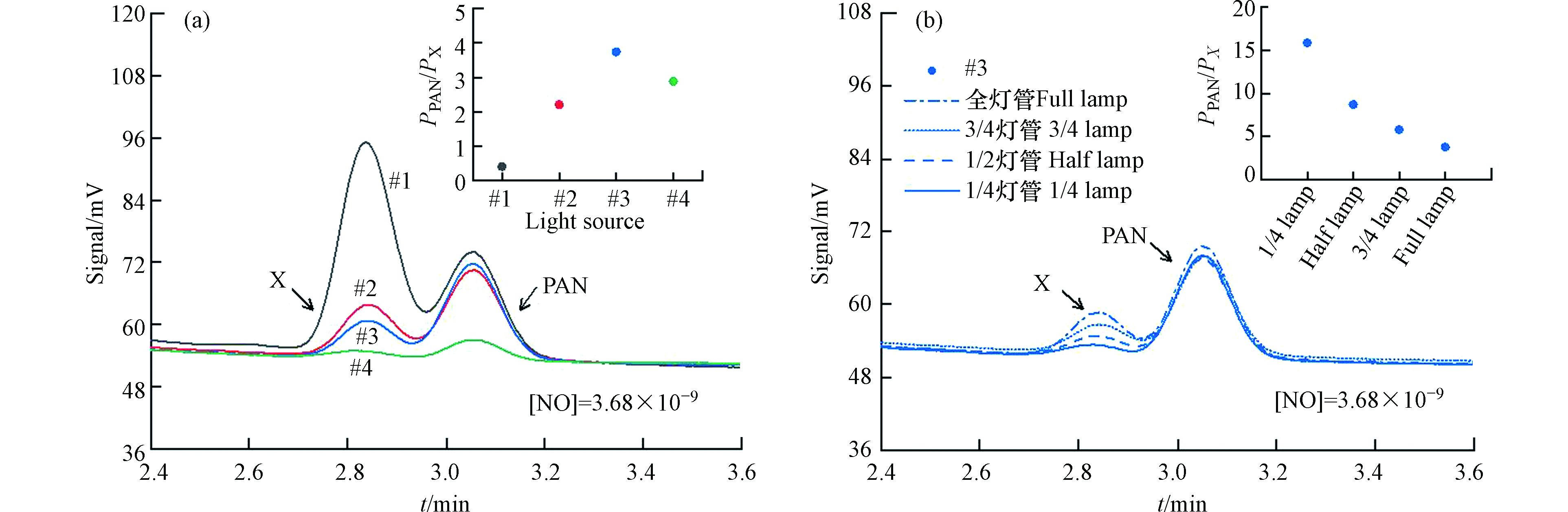
不同紫外光源和光照强度条件下PAN合成情况色谱结果如图3所示. 4种紫外灯照射条件下均在(2.83 ± 0.01) min和(3.05 ± 0.01) min产生两个峰(如图3(a)所示),其中后峰为PAN,前峰为丙酮光化学反应产生的未知副产物X,表明4种光源照射条件下均有PAN和X生成,且副产物在ECD上存在明显的响应. 254 nm(#1和#2)和312 nm(#3)下合成PAN的色谱峰面积相当(101151 μV·s、108767 μV·s及124185 μV·s),但365 nm(#4)光源条件下合成PAN色谱峰面积明显较小,仅为30578 μV·s. 表明254 nm和312 nm紫外光源条件下均可产生充足的过氧乙酰自由基并实现NOx向PAN的高效转化,而365 nm光源相对较弱,丙酮光解产生的过氧自由基明显不足,NO向PAN的转换效率远低于254 nm和312 nm光源. 对于未知副产物X而言,X产量在254 nm(#1)光源下最多,在254 nm(#2)和312 nm(#3)光源下相对较少,在365 nm(#4)光源下最少,其中254 nm(#1)光源下X与PAN的色谱峰产生较为严重的峰叠加问题,直接影响到PAN色谱峰的准确积分. 本研究以PAN与X的峰面积比值(PPAN/PX)作为衡量光化学合成的信噪比参数,其比值越高,光化学合成过程中未知副产物X对PAN的标定影响越小. 从图3(a)可见,312 nm(#3)光源条件下的PPAN/PX最大(3.7),254 nm(#1)光源条件下的PPAN/PX最小(0.4),表明312 nm(#3)光源条件下的未知副产物X对PAN的影响最小. 综上可知,312 nm(#3)光源下丙酮光解可产生充足的过氧自由基保障NOx向PAN的高效率转化,且未知副产物提供的干扰最小.
从图3(a)中可以看出,尽管312 nm(#3)光源条件未知副产物的干扰较小,但副产物的色谱峰仍会与PAN峰出现一定程度的叠加而影响PAN的准确积分. 由于副产物的量会随着紫外辐射减弱而降低,为此本研究拟通过遮挡发光灯管长度改变到达反应池的总紫外辐射量,从而寻找保证PAN高转化率并且降低副产物影响的辐射条件. 图3(b)给出了在312 nm(#3)光源不同光照条件下PAN和X的色谱分离检测情况. 由图可知,PAN峰面积基本不随光照强度变化而变化(全灯管至1/4灯管分别为124185 μV·s、112799 μV·s、117954 μV·s及127857 μV·s),而X的色谱峰随灯管发光长度减小而减小. 从PPAN/PX可以更加清楚看出,1/4灯管照射条件下PPAN/PX最大(15.9),副产物对PAN影响最小. 此外,在1/4灯管照射条件下X峰基本与基线齐平,对PAN的积分基本不会产生影响,已满足实验需求,无需开展更短灯管实验. 因此,选取312 nm(#3)光源1/4灯管作为光化学合成系统的光源条件,可以保障NOx向PAN的高效率转化,且未知副产物基本不会干扰PAN的积分.
-
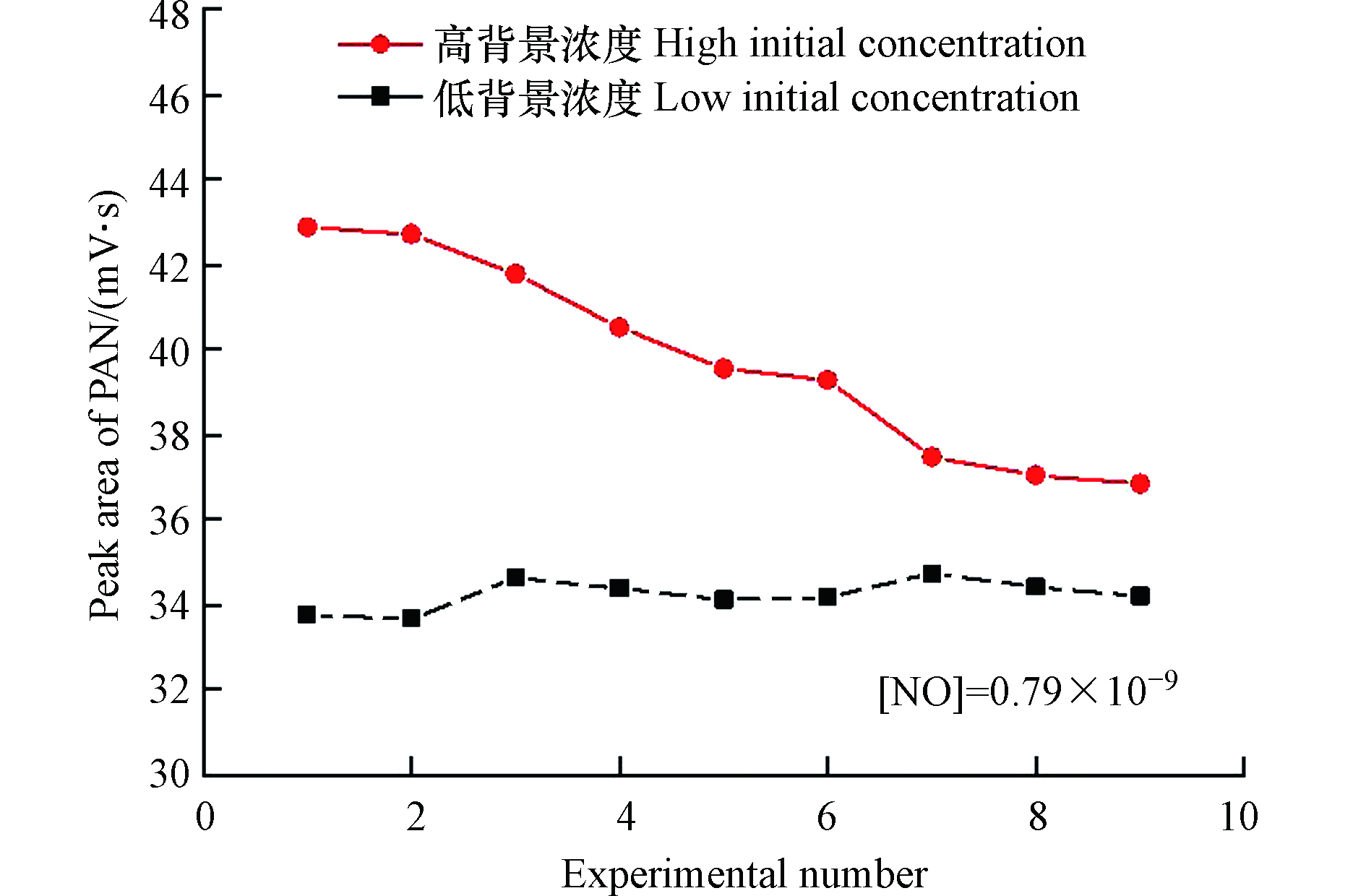
光化学合成系统中的石英反应池及气路内壁均可能对PAN以及副产物X存在一定的吸附,导致该系统存在产物残留现象(图4),并可能对PAN的标定造成影响. 本研究开展了不同初始背景浓度下PAN合成的对比实验,结果如图4所示. 实验发现,在高初始背景浓度(> 0.11 × 10−9)情况下,合成PAN的色谱响应随着连续合成的进行会出现持续下降现象,即在90 min内从43 mV·s下降至38 mV·s,下降约12%;在低初始背景浓度(0.03 × 10−9)情况下,合成PAN的色谱响应能够快速稳定到34 mV·s并在随后的依次合成中基本保持不变(RSD = 1.06%),这有可能归因于PAN与X在反应池及气路内壁表面与系统内合成气体之间存在吸附和脱附的动态平衡. 当系统残留浓度较高时,系统内发生PAN与X由反应池及气路内壁向合成气体的脱附释放过程并最终达到动态平衡,导致合成气体中PAN的浓度逐渐降低并最后达到稳定;反之,系统内发生PAN与X由合成气体向反应池及气路内壁吸附过程并快速达到动态平衡,引起合成气体中PAN浓度开始快速抬升并达到稳定. 由此可见,光化学产物的残留使得合成系统达到稳定所需时间变长,而低残留条件下合成系统能够快速达到稳定. 因此,在开始PAN的合成实验时,应首先采用大流量零级空气对合成系统进行清洗以降低产物残留的影响,并检测确定背景浓度,以保证标定数据的准确性.
-
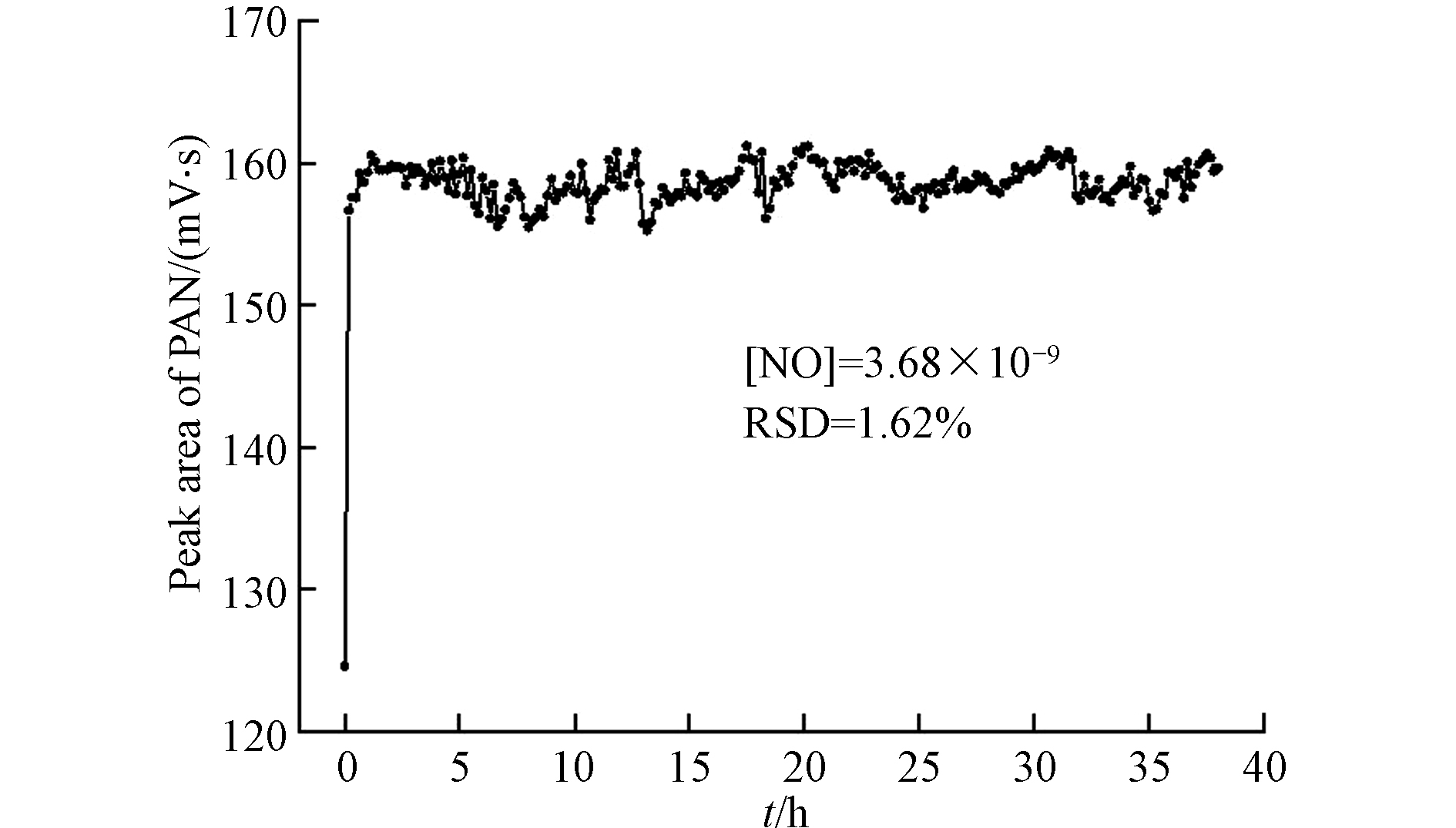
如前文所述,光化学合成系统存在产物残留现象,这些产物是否会在合成系统内随着合成时间的进行而累积并影响PAN标定的稳定性尚未清楚. 为此,本研究开展了连续长时间光化学合成实验,结果如图5所示. 实验发现,实验开始后半小时内PAN的合成基本趋于稳定,并在随后的38 h连续合成实验中基本保持稳定,其PAN峰面积的相对标准偏差RSD = 1.62%,表明本研究所搭建的同步稀释光化学合成系统基本不存在产物累积问题,并可在长时间连续稳定合成PAN.
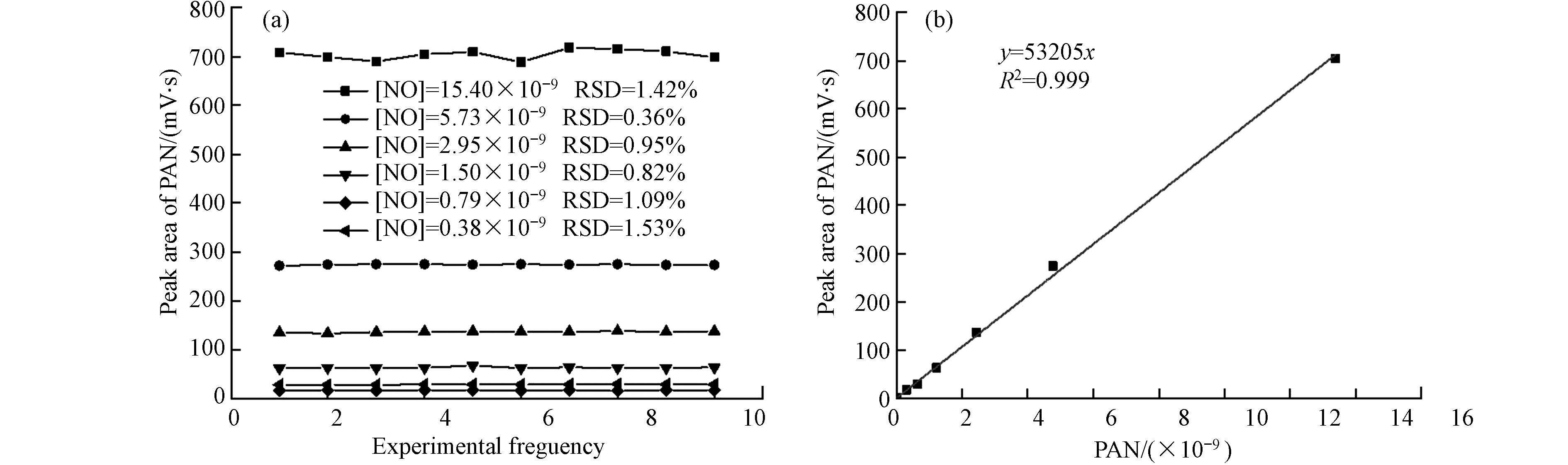
基于以上工作所确定的光化学合成条件,本研究在6个NO体积分数梯度(0.38 × 10−9、0.79 × 10−9、1.50 × 10−9、2.95 × 10−9、5.73 × 10−9、15.40 × 10−9)下进行了PAN的不同体积分数梯度的合成实验,每个梯度实验时间维持在2 h以上,且保证PAN的测试数据不少于12组. 实验结果发现,每个梯度的合成实验均在半小时以内达到稳定并随后保持稳定状态. 每个梯度合成稳定后的数据波动情况如图6(a)所示,对于NO体积分数0.38 × 10−9—15.4 × 10−9的范围内,每个梯度连续合成10组PAN的峰面积相对标准偏差值均小于2%,表明PAN的连续合成均非常稳定.
基于梯度合成实验数据,建立了PAN分析仪的标准工作曲线(图6(b)). 根据曲线,线性回归决定系数为R2 = 0.999,表明所搭建的同步稀释光化学合成系统性能稳定,并且本研究所使用的TGC-PAN在线分析仪对PAN具有非常好的线性响应关系.
-
本研究使用TGC-PAN在线分析仪于2019年3月在中国科学院生态环境研究中心开展了为期一个月的外场观测,并利用所搭建的同步稀释光化学合成系统每周对TGC-PAN在线分析仪进行校准. 2019年3月份北京市大气中PAN、PM2.5、O3、氮氧化物以及气象参数的时间序列如图7所示. 分析结果表明,2019年3月份北京市大气中PAN的体积分数范围为0.20 × 10−9—7.34 × 10−9,平均值为1.88 × 10−9 ± 1.63 × 10−9,明显高于2011年3月北京同期PAN体积分数平均值和最大值(0.69 × 10−9 ± 0.55 × 10−9,2.26 × 10−9)[26],而最小值与其基本一致(均为0.20 × 10−9),表明北京市2019年3月份大气氧化性平均水平出现了显著抬升,而大气氧化性的背景水平基本维持不变. 从图中可见,观测期间大气中PAN、PM2.5以及NOx和相对湿度的变化趋势相似,明显受到风速变化的影响. 风速相对较小时(如3月1—5日、3月7—11日、3月17—20日及3月26—28日),各类污染物以及相对湿度均出现了较大抬升;而当风速增大时,各类污染物以及相对湿度又快速降低至较低水平. 然而大气中O3的变化趋势却明显不同. 风速较低时,O3在白天的峰值出现了一定抬升,而在夜晚快速降至零点附近;风速较高时,O3浓度并没有出现显著降低,却呈现出昼夜浓度水平基本相当的现象,大约维持在40 × 10−9左右,这与已有北京春季大气臭氧报道一致[27-28]. 这可能由于较低风速有利于各类大气污染物积累,日间的光化学反应增加了臭氧生成,而夜间O3会被NO快速滴定消耗降至极低浓度水平. 当风速较大时,大气中PAN、PM2.5以及NOx等污染物被快速清除至较低水平,而O3则受到高空背景输入影响,因此臭氧处于相对较高浓度水平.
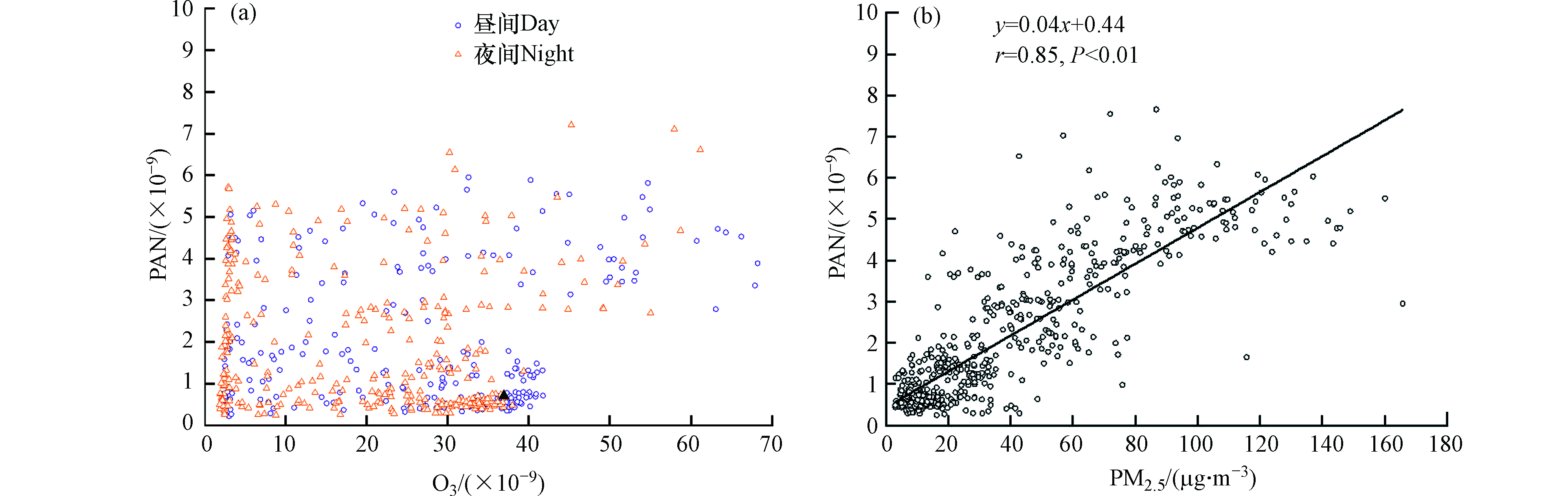
PAN与O3通常作为光化学污染的重要指示剂,一般具有较好的正相关性. 本研究发现,北京市3月大气中PAN与O3在观测期基本不存在相关性(昼间r = 0.06, P = 0.35;夜间r = −0.04, P = 0.50,见图8(a)),与以往的观测结果相一致[29]. 这可能主要由于大气中PAN和O3的源汇过程差异所致. PAN只能通过挥发性有机物(VOCs)和氮氧化物(NOx)光化学反应产生,主要通过热解去除;而O3的源汇更加复杂,其来源既可以由光化学生成,也存在高空背景O3的对流传输,其去除主要包括NO的滴定、光解以及还原物质消耗等多种途径. 北京市3月份太阳辐射相对较弱,气温较低,该时期大气光化学生成PAN和O3速率相对较低. 气温较低的相对静稳(通常低于15 ℃,风速通常小于3 m·s−1)气象条件有利于NO和PAN的积累,反而会加速O3与NO的滴定消耗;此外大风过境时,PAN会被快速清除,而此时高空O3成为北京市O3的主要来源,这些最终导致了北京市3月份大气中PAN和O3观测期间基本不存在相关性.
如前文所述,观测期间大气中PAN和PM2.5浓度呈现非常相似的变化趋势,本研究利用线性回归分析对他们之间的相关性进行探究. 分析结果表明(图8(b)),两者呈显著的正相关(r = 0.85, P < 0.01),大气中PAN的浓度随PM2.5浓度的增加而增大. 这可能由于大气中PAN在气温相对较低的3月份具有较长的大气寿命,光化学产生的PAN可以在大气中相对稳定存在,其大气浓度会受到扩散条件改变而发生积累或清除,于是便表现出与颗粒物相似的变化趋势. 除了受气象条件这个外因影响以外,PAN和PM2.5某些相似性的形成机制也可能是该现象的内因. PAN来自于光化学二次生成,而PM2.5除了直接排放外,也存在复杂的二次生成,两者的二次生成均是大气氧化的产物. 观测期间大气中PAN的浓度较好地记录了气团所经受的氧化历程. 当风速较低时,大气中挥发性有机物和NOx等各类大气污染物浓度容易累积[27],并增强了大气氧化性[30],于是非常有利于大气中PAN的快速生成和积累,此时相对静稳的气象条件也加速了PM2.5前体物的累积,增强的大气氧化性进而促进了PM2.5的生成和积累,最终导致了大气中PAN和PM2.5出现同步抬升的现象. 而当大风过境时,大气中PAN、PM2.5及其前体物就会被快速清除至极低浓度. 综上所述,在北京市气温较低的3月份,相对静稳天气条件加速了大气污染物的积累,增强了大气的氧化性,促进了大气中PAN和PM2.5的生成与积累,使得两者浓度呈现显著正相关.
2.1. 不同光源及光照强度对PAN合成的影响
2.2. 产物残留对PAN标定的影响
2.3. 光化学合成标定方法评价
2.4. 实际外场大气观测
-
(1)同步稀释光化学合成系统中紫外光源是影响PAN合成与标定的重要影响因素之一,其中副产物对PAN的影响随着辐射强度减弱而降低,最终发现紫外光源为遮挡3/4的312 nm紫外灯管,可在保证PAN的高合成效率的情况下,最大程度上降低丙酮光解副产物对标定的干扰. 光化学合成系统的产物残留会直接影响PAN合成的稳定性,每次开始合成实验前应当采用零级空气清洗反应池,以降低残留的影响,以保证标定数据的准确性.
(2)优化后的同步稀释光化学合成条件可在半小时后实现PAN的连续稳定合成,相对标准偏差值均小于2%,且不同浓度合成梯度的PAN呈现非常显著的线性,完全满足PAN在线分析仪的标定需求.
(3)2019年3月北京市大气中PAN的体积分数范围为0.20 × 10−9—7.34 × 10−9,平均值为(1.88 × 10−9 )± (1.63 × 10−9),显著高于2011年同期平均浓度水平. 静稳天气下污染物的积累增强了大气氧化性,并促进了大气中PAN和PM2.5的生成与积累,使得北京春季PAN与PM2.5浓度呈现显著正相关.
