


-
汞具有长距离迁移性和生物富集性,可以在大气中停留数月至1 a,随气流长距离传输,造成远离污染源地区环境汞污染。我国是全球大气汞排放量最大的国家,大约占全球大气汞排放量的30%,所以我国汞控制排放对全球汞减排具有重要意义[1-2]。因此,我国作为全球大气汞排放量最大的国家,汞排放控制也得到全球的广泛关注,面临巨大的履约压力。2021年,WU等[2-3]探究了2021年我国重点行业大气汞排放量为314 t,其中燃煤电厂和工业锅炉排放量分别为36 t和24 t,占总排放量的19.1%。因此,煤炭燃烧是我国汞排放控制的重点行业。
相较于国外燃煤行业专门汞控制技术(活性炭喷射技术),我国燃煤行业普遍采用污控措施协同控制措施降低烟气汞排放浓度。以燃煤电厂为例,目前燃煤电厂的污控措施基本都具有选择性催化还原(selective catalytic reduction,SCR)催化剂、除尘设备、湿法脱硫设备(wet fuel gas desulfurization device,WFGD)。汞在烟气中主要存在3种形态:气态元素汞(Hg0)、气态氧化汞(Hg2+)和颗粒态汞(Hgp)[4-6]。Hg2+和Hgp分别被WFGD和除尘设备控制捕集下来,捕集效率高达80%和99%以上。Hg0不易溶于水和被除尘捕集,所以燃煤行业烟气汞主要以Hg0形式存在。因此,燃煤行业汞减排关键在于通过催化剂将Hg0转化为Hg2+,进而被脱硫设备捕集。
目前,燃煤电厂普遍采用钒钨钛催化剂作为商业脱硝催化剂,但V2O5的高毒性、致癌性等特点限制了钒钨钛催化剂的使用。氧化铈(CeO2)具有良好储氧能力和氧化还原特性,可以作为催化剂V2O5的替代活性组分[7-9]。以往的研究表明,Ce基催化剂表现出良好的NOx还原能力或Hg0氧化能力,但单一的CeO2难以满足NOx还原和Hg0氧化双重催化需求,构建多活性位点多反应区域是满足NOx和Hg0协同控制的重要条件。CeMnTi催化剂具备良好的Hg0氧化性能和NOx低温还原性能,但CeO2高氧化性使SO2转化为Ce(SO4)2或Ce2(SO4)3,造成催化剂失活[10-12]。基于以上研究,开发SCR烟气条件下具备Hg0高效氧化和良好抗硫性能的催化剂尤为必要。
基于以上研究结果与不足,考虑到NH3、SO2和Hg0酸碱性的差异,本研究以CeO2为活性位点、TiO2为载体、WO3和CuO为改性剂,制备多种Ce基催化剂,构建酸碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力,探究了催化剂协同催化脱除和抗硫反应机制,为燃煤行业多污染协同控制和多设备协同控汞提供理论依据。
-
本研究采用德固赛公司生产的商业TiO2 (P25) 作为催化剂的载体。基于已有研究[13]结果,将一定量的Ce(NO3)3·6H2O和草酸溶解于去离子水,按照Ce:W:Cu:Ti比例添加一定量的(NH4)10H2(W2O7)6和Cu(NO3)2,再添加一定量的P25形成混合溶液,微波处理15 min。将混合溶液搅拌24h,然后再放入110 ℃烘箱中干燥12 h,最后在马弗炉中500 ℃焙烧4h后,自然降温到25 ℃,制备出不同Ce基催化剂。将催化剂研磨压片过筛得到40~60目催化剂样品用于汞氧化实验。所得催化剂包括Ce5Ti(Ce∶Ti=5∶95)、Ce5W9Ti(Ce∶W∶Ti=5∶9∶86)以及Cu5Ce5W9Ti(Cu∶Ce∶W∶Ti=5∶5∶9∶81)。
-
本研究采用X射线衍射(XRD)、X射线光电子能谱(XPS)、H2程序升温还原测试(H2-TPR)、CO2程序升温脱附分析(CO2-TPD)和X射线透射电镜(TEM)等表征手段对反应前后的催化剂进行表征,分析催化剂表面理化性质,探究汞氧化反应机制。XRD 物相分析采用Bruker D8 (德国布鲁克公司),管电流30 mA,管电压40 kV,扫描角度2θ为10°~90°,扫描速度为2 °·min−1。XPS分析采用X射线光电子能谱仪(Thermo ESCALAB 250XI)分析催化剂表面Cu、Ce、W、Ti、O、S、N、Cl和Hg元素的价态。H2-TPR实验采用AutoChem II
2920 型化学吸附仪 (Micrometritics Co.),实验步骤如下:取 50 mg 样品(40~60 目),在纯氧气氛下 400 ℃ 预处理 30 min;降温到50 ℃后用He吹扫15 min;然后通入体积分数为10% H2+90%Ar混合气,待仪器基线平稳后,以10 ℃·min−1速率升温至600 ℃,采用TCD 检测耗氢量。CO2-TPD实验步骤如下:取100 mg 样品 (40~60 目),在氦气氛下经500 ℃预处理30 min;冷却至50 ℃,再通入30 mL·min−1的CO2吸附至饱和,氦气吹扫1h 后开始TPD 实验,以10 ℃·min−1 速率升温至800 ℃,采用TCD 检测。 -
本研究采用固定床实验装置探究催化剂汞氧化性能。该固定床实验装置包括四部分:模拟烟气配气系统、汞源发生系统、催化反应系统和汞浓度与形态测试系统。催化反应系统通过加热系统精准调控反应温度,温度误差≤2 ℃。模拟烟气配气系统包括高纯氮气(N2)、氧气(O2)、标准混合气(HCl、SO2、NO和NH3等)和80 μg·m−3 Hg0,标准混合气均采用N2作为平衡气。以上标准气体均采用质量流量控制器控制气体添加,混合气体总流量为1L·min−1。空速是催化剂催化氧化性能的重要实验参数,本实验催化剂空速约为100 000 h−1。研究中各种模拟烟气组分见表1。反应前后烟气中Hg0和Hg2+的浓度通过烟气分析仪分析(Thermo Fisher 80i)。汞氧化率通过式(1)计算。
其中:η为汞氧化率,%;CHg2+和CHgt 分别代表反应管出口烟气Hg2+浓度和入口烟气总汞浓度,μg·m−3。所有数据为3组平行实验的平均值,实验数据相对误差不超过10%。
-
在基本烟气组分条件下,本研究对比了Ce5Ti、Ce5W9Ti和Cu5Ce5W9Ti催化剂的汞氧化性能。如图1所示,Ce作为一种中温活性组分,Ce5Ti和Ce5W9Ti在中温区间(250~400 ℃)表现出最佳的汞氧化性能,Ce5Ti和Ce5W9Ti催化剂汞氧化率分别达到58.4%~69.1%和68.8%~81.3%。Ce5Ti和Ce5W9Ti在低温区间(100~200 ℃)的汞氧化率普遍较低,分别低于53.1%和62.5%,表现出较差的低温活性。CuO掺杂的Cu5Ce5W9Ti表现出最佳的汞氧化性能,在300 ℃下汞氧化率达到最高,为88.8%。在中温区间,Cu5Ce5W9Ti的汞氧化率稳定在86.3%~88.8%。反应温度进一步上升到450 ℃,3种催化剂的汞氧化率均降低,说明温度过高使CeO2的催化氧化活性降低。与Ce5Ti和Ce5W9Ti相比,Cu5Ce5W9Ti在低温区间(100~200 ℃)表现出较高的汞氧化率,为76.3%~85.5%,表明CuO的掺杂可显著提升Ce基催化剂低温氧化活性。
-
本研究对比了3种催化剂在基本烟气组分和SCR烟气组分条件下的汞氧化活性的差异性。如图2所示,在300 ℃下,相较于基本烟气组分,Ce5Ti催化剂在SCR烟气组分下汞氧化率明显降低,由62.7%下降到37.2%。在SCR烟气组分条件下,Ce5W9Ti催化剂汞并没有表现出明显的降低,汞氧化率由73.8%下降到62.4%,表明WO3掺杂明显提高了Ce5W9Ti催化剂对SCR反应条件的适应性。有研究[14-15]表明,SCR烟气组分可降低催化剂的汞氧化活性,这主要归咎于NH3与Hg0对活性位点的竞争吸附作用,高浓度的NH3分子明显降低了Hg0与活性位点接触的可能性。Cu5Ce5W9Ti催化剂SCR烟气组分条件下汞氧化表现与Ce5W9Ti相似,汞氧化率略微降低,由88.8%下降到81.3%,依然表现出较好的SCR烟气组分的适应性。
在200 ℃和400 ℃的条件下,SCR烟气组分对催化剂具有相似的的影响。在200 ℃下,SCR烟气组分对Ce5Ti和Ce5W9Ti催化剂汞氧化抑制作用较为明显,而在400 ℃下,SCR烟气组分的抑制作用相对减少。这可能是因为Ce5Ti和Ce5W9Ti催化剂缺失低温活性组分,低温条件下NH3吸附量增加,汞氧化性能较低;而高温条件下NH3吸附量降低,减少了对Hg0抑制吸附作用,提高催化剂汞氧化性能。
-
SO2和HCl是燃煤烟气普遍存在的酸性气体,对催化剂汞氧化性能具有重要影响。通过以上实验筛选,本研究对比了Ce5W9Ti和 Cu5Ce5W9Ti催化剂在SCR烟气组分、SCR+SO2烟气组分、SCR+HCl烟气组分以及SCR+SO2+HCl烟气组分中的汞氧化性能变化,分析SO2和HCl对Ce5W9Ti和Cu5Ce5W9Ti催化剂影响。
如图3所示,在SCR烟气组分(300 ℃)下,1 429 mg·m−3 SO2 的添加明显降低了Ce5W9Ti催化剂的汞氧化性能,汞氧化率从62.4%降低到41.4%,表明SO2严重抑制了Ce5W9Ti催化剂汞氧化活性。有研究表明:高浓度SO2极容易吸附在CeO2活性位点,形成稳定的硫酸铈,使Ce基活性位丧失储氧能力[14]。SO2同样降低了Cu5Ce5W9Ti催化剂的汞氧化性能,但汞氧化率下降幅度较小,由81.3%下降到78.8%。该结果表明CuO的掺杂明显提高了催化剂的抗硫性能,可能是CuO的掺杂增加了反应活性位点或是保护了Ce基活性位点,使得Cu5Ce5W9Ti在SO2存在条件下依然具有较高的汞氧化性能。在200 ℃下,SO2对Ce5W9Ti和Cu5Ce5W9Ti的汞氧化性能抑制作用进一步提高。常化振等[16]研究表明,在低温条件下SO2与NH3形成大量的亚硫酸氢铵,覆盖了催化剂表面的活性位点,导致催化剂活性较低。在400 ℃条件下,SO2的抑制作用减弱,可能因为高温抑制了SO2吸附和亚硫酸氢铵的形成[16]。
HCl的添加明显提高了Ce5W9Ti和 Cu5Ce5W9Ti催化剂汞氧化性能,汞氧化率分别提高到96.2%和98.4%。该结果表明HCl的添加可能增加了新的活性位点,促进了汞氧化率。进一步添加1 429 mg·m−3 SO2到模拟烟气,Ce5W9Ti和 Cu5Ce5W9Ti催化剂出现下降,但Ce5W9Ti和 Cu5Ce5W9Ti催化剂在SCR+HCl+SO2烟气组分下汞氧化率(76.8%和89.7%)依然明显高于SCR组分下汞氧化率(62.4%和81.3%)。该结果表明相较于SO2,HCl更具有竞争力,在催化剂表面依然形成新反应活性位,促进了Hg0的吸附和氧化。在200 ℃和400 ℃下,HCl显著提高了Ce5W9Ti和 Cu5Ce5W9Ti的汞氧化率和抗硫性能。
-
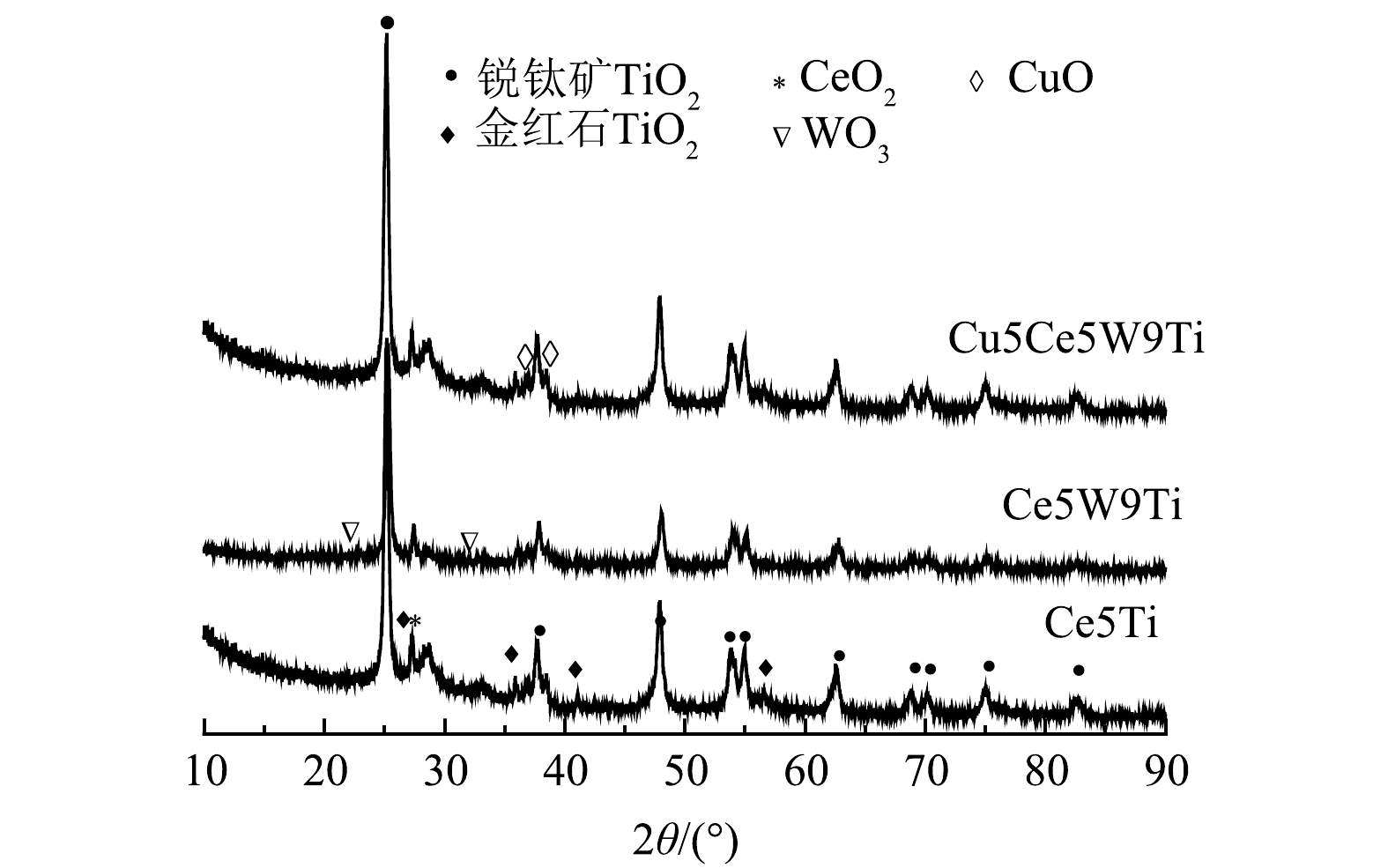
1) XRD分析。采用XRD技术分析了不同Ce基催化剂的晶相(图4)。3种催化剂XRD谱图主要以锐钛矿和红金石2种形态TiO2特征峰为主(JCPDS 21-
1272 ),符合载体P25组分80.0%锐钛矿和20.0%红金石组成特征。3种催化剂在2θ=28.35°位置出现一个微弱的CeO2晶峰,表明Ce在载体表面高度分散,具备较高的催化氧化活性。Ce5W9Ti和 Cu5Ce5W9Ti催化剂都没有出现明显的WO3晶峰,说明元素W在催化剂表面同样高度分散。Cu5Ce5W9Ti催化剂在35.5o和38.6o位置出现了微弱的CuO(111)特征峰(JCPDS 48-1548 ),证明Cu活性组分绝大部分均匀分布在催化剂表面。基于以上结果,Ce、W和Cu元素在载体表面高度均匀分散[13]。2) TEM分析。采用TEM分析了不同Ce基催化剂表面的元素晶体形态,结果见图5。如图5(a)所示,催化剂载体P25主要以立方体形态存在。如图5(b)和图5(c)所示,Ce5Ti和Ce5W9Ti催化剂表面CeO2以(111)晶型存在,晶格间距为0.31 nm,与XRD的分析结果一致。Ce5W9Ti催化剂表面没有发现WO3晶体,但Ce与Ti掺杂形成钛铀矿CeTi2O6(110)[17],晶体间距为0.34 nm,说明W元素的高度分散有利于Ce与Ti原子的螯合掺杂,可形成稳定的氧空位。如图5(d)所示,Cu5Ce5W9Ti催化剂表面出现间距为0.28 nm的CuO(110)晶格条纹和间距为0.31 nm的CeO2(111)晶格条纹,CuO与CeO2界面形成掺杂晶体界面。在界面位置,CuO、CeO晶体中的原子被相互替代,形成晶体错位,晶体条纹边界发生扭曲转移,促进大量Cu+和Ce3+形成,使催化剂表面形成氧空位,从而提高催化剂的储氧和氧化还原能力[18]。
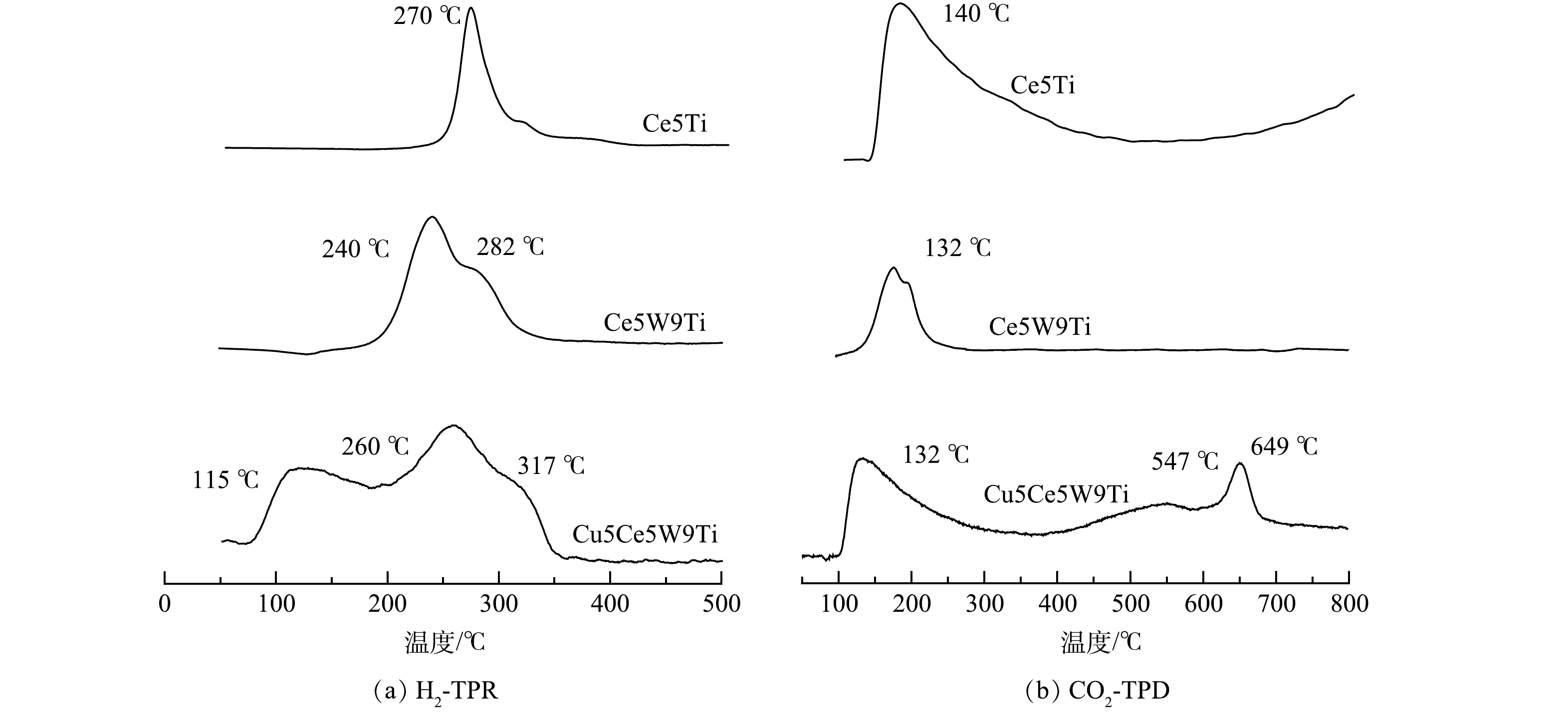
3) TPR和TPD分析。本研究采用H2-TPR和CO2-TPD技术分析了Ce5Ti、Ce5W9Ti和 Cu5Ce5W9Ti催化剂氧化位点和活性位点酸碱性。如图6(a)所示,Ce5Ti催化剂H2-TPR图谱出现一个270 ℃的主峰,代表CeO2形成的氧空位或活性位点。Ce5W9Ti催化剂的H2-TPR图谱中出现240 ℃和282 ℃ 2个主峰,结合TEM分析结果,除了CeO2活性位点,Ce与Ti掺杂形成的钛铀矿CeTi2O6(110)建 立新的活性位点[17]。因此,Ce5Ti、Ce5W9Ti催化剂在中温区间(250~400 ℃)表现出良好的汞氧化性能。如图6(a)所示,Cu5Ce5W9Ti催化剂的H2-TPR图谱中出现115、260和317 ℃ 3个主峰。260 ℃和317 ℃代表中温CeO2活性位点和Ce-Ti活性位点,115 ℃代表低温CuO活性位点,表明CuO的掺杂可提高催化剂在低温区域活性位点的形成。因此,Cu5Ce5W9Ti催化剂在图1表现出较高的低温(100~200 ℃)汞氧化性能。
如图6(b)所示,通过CO2-TPD分析了3种催化剂表面活性位点的酸碱性。有研究[19-20]表明,根据CO2脱附温度可将活性位点酸碱性分为弱碱(100~250 ℃)、中强碱(250~600 ℃)和强碱(600~800 ℃)。在Ce5Ti的CO2-TPD图谱中出现一个脱附主峰(140 ℃),表明CO2主要为弱碱活性位点;Ce5W9Ti的脱附主峰温度进一步降低到132 ℃,表明酸性氧化物WO3在催化剂表面的高度分散可进一步降低催化剂碱性强度。Cu5Ce5W9Ti的CO2-TPD图谱包含3个特征峰,即132、547和649 ℃ [21-22]。在SCR+SO2烟气条件下,强酸性气体SO2优先与碱性活性位(Cu—O—Ce和CuO)反应,从而保护了CeO2活性位依然保持高活性和氧化性,因此,Cu5Ce5W9Ti在SCR+SO2烟气条件下依然表现出良好的汞氧化性能(图3)。
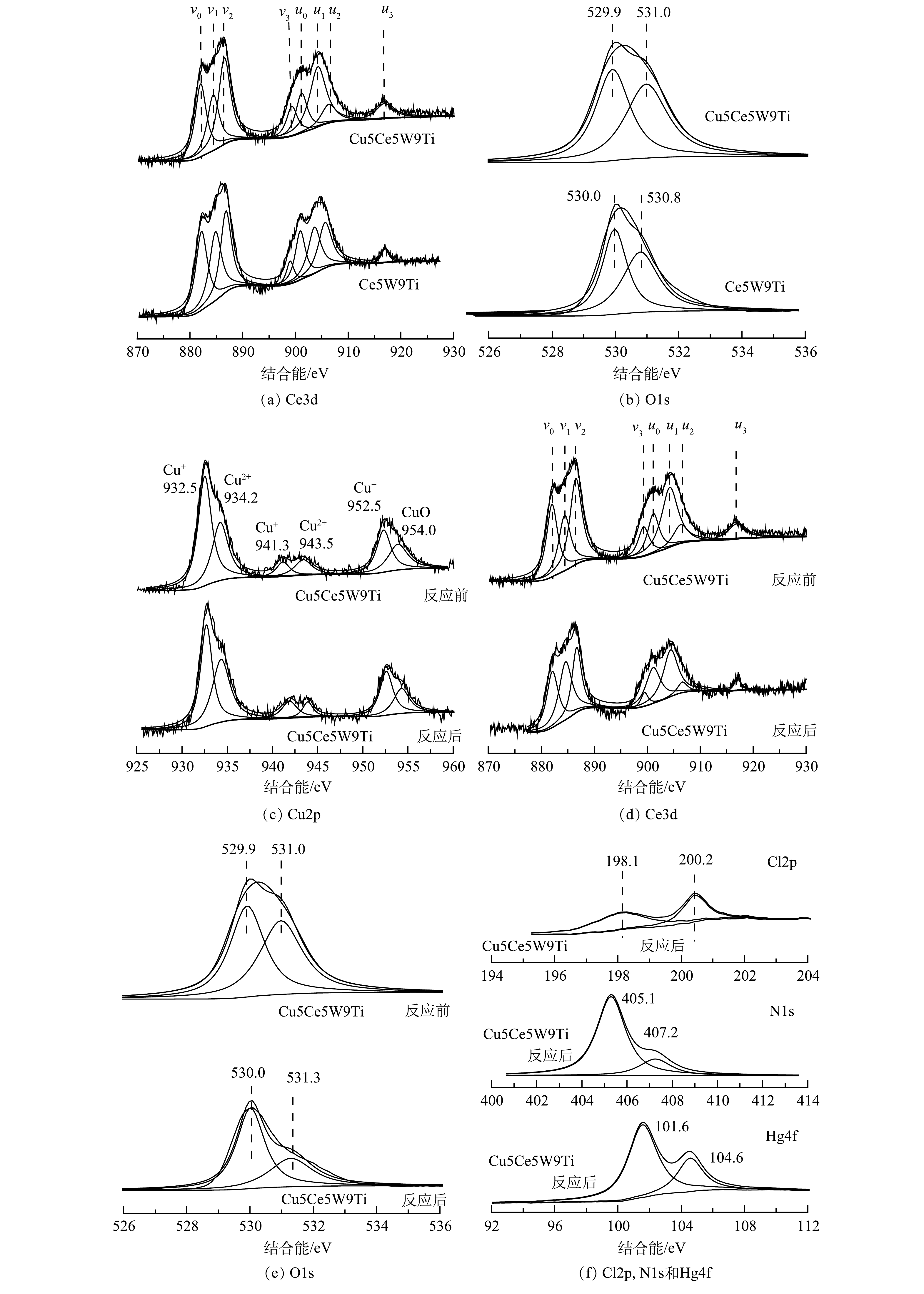
4) XPS分析。本研究采用XPS技术分析催化剂表面各种元素价态,探究催化剂汞氧化机制。如图7(a)所示,Ce元素能谱图包含8个分峰:u0、u1、u2、u3、v0、v1、v2和v3。其中u1和v1代表Ce3+;u0、u2、u3、v0、v2和v3代表Ce4+,Ce3+一般作为氧空位或是吸附氧位点[23]。Ce5W9Ti催化剂Ce3+峰面积和Ce4+峰面积比例(Ce3+/Ce4+)为31.2%,Cu5Ce5W9Ti 的Ce3+/Ce4+面积比例上升到39.2%。该结果与TEM分析结果一致,说明CuO与CeO2掺杂形成晶体错位,形成更多Ce3+和氧空位。
如图7(b)所示,Ce5W9Ti和Cu5Ce5W9Ti催化剂表面O元素2个特征峰:529.9~530.0 eV特征峰代表晶格氧,530.8~531.0 eV特征峰代表活性氧[24-25]。活性氧一般是氧空位吸附的高活性吸附氧,为Hg0氧化的关键活性位点。Ce5W9Ti催化剂活性氧特征峰面积占比为38.7%,Cu5Ce5W9Ti的活性氧特征峰面积占比为45.8%,该结果与Ce3+/Ce4+面积比例上升一致。如图7(c)所示,Cu5Ce5W9Ti催化剂表面在932.5 eV和934.2 eV的2个主要特征峰分别代表Cu+和CuO[26]。Cu5Ce5W9Ti催化剂表面Cu主要以Cu+形式存在,这与Cu、Ce原子掺杂形成氧空位界面保持一致。Cu5Ce5W9Ti催化剂反应前后Cu+/CuO比例由69.4%下降到57.2%,这可能是部分Cu+形成的活性氧与SO2反应形成大量的CuSO4,因此,Cu2+比例明显上升。如图7(d)所示,Cu5Ce5W9Ti 的Ce3+/Ce4+面积比例由反应前的39.2%下降到反应后的35.4%,说明部分Ce3+活性位在反应过程中转化为稳定的Ce4+,降低了催化剂的吸氧释氧能力。如图7(e)所示,Cu5Ce5W9Ti催化剂反应前后表面O元素2个特征峰发生明显变化。Cu5Ce5W9Ti催化剂活性氧特征峰面积占比有反应前的45.8%下降到反应后的35.3%,说明大量活性氧参与了Hg0氧化和NOx还原反应。
XPS表征没有发现反应前的Cu5Ce5W9Ti催化剂表面存在Cl、N和Hg原子。如图7(f)所示,SCR+SO2+HCl条件反应后的Cu5Ce5W9Ti催化剂表面Cl、N和Hg元素的价态。Cu5Ce5W9Ti催化剂表面Cl存在Cl−离子(198.1 eV)和活性Cl*(200.2 eV)2个特征峰[17, 23]。大面积活性Cl*特征峰的出现说明HCl在Cu5Ce5W9Ti催化剂表面吸附并转化为活性Cl*,促进Hg0转化为HgCl2。NO在催化剂表面XPS能谱图出现硝酸根(407.2 eV)和NO2(405.1 eV)2个特征峰。NO2特征峰面积要远大于硝酸根特征峰面积,说明NO在催化剂上主要以更高活性的NO2或是亚硝酸盐官能团形式存在。反应后的Cu5Ce5W9Ti催化剂表面存在Hg元素101.6 eV和104.6 eV两个特征峰,表明吸附在催化剂表明的汞主要以 Hg2+形式存在。
-
基于3种Ce基催化剂汞氧化实验和理化性质表征结果,本研究发现单一活性位点的Ce5Ti和Ce5W9Ti催化剂中温区间(250~400 ℃)表现出较高汞催化氧化活性(图1),但在SCR烟气组分下Ce5Ti的汞氧化率明显下降(图2),这归咎于NH3与Hg0竞争Ce基活性位点。WO3的掺杂提高了Ce5W9Ti在SCR烟气组分下的汞氧化率,为62.4%。CO2-TPD分析结果说明WO3的掺杂增加了Ce5W9Ti催化剂酸性(图6),增加了强碱性气体NH3的吸附位点,降低了NH3对Hg0吸附氧化竞争。同时XRD和TEM分析结果表明高度分散的WO3可促进Ce与Ti的耦合掺杂形成钛铀矿CeTi2O6(110),提高氧空位的比例。CuO的掺杂提高了Cu5Ce5W9Ti催化剂在低温区间(100~200 ℃)的汞氧化率(76.3%~85.5%),并且在SCR和SCR+SO2烟气条件下表现出较高的汞氧化率(81.3%和78.8%)。H2-TPR分析结果说明Cu掺杂增加Cu5Ce5W9Ti催化剂低温(115 ℃)活性位点,因此,提高催化剂的低温氧化活性;CO2-TPD分析结果表明W、Cu掺杂使Cu5Ce5W9Ti催化剂具备弱碱、中强碱、强碱3种活性位,可有效地分离酸性气体SO2、碱性气体NH3和Hg0吸附氧化反应区域,从而提高催化剂的抗硫性(图6)。TEM分析说明CuO和CeO2晶体掺杂的界面,发生原子替代和晶体错位,形成更多的Ce3+和Cu+作为氧空位,吸附更多的活性氧作为氧化位点,这一结论也在H2-TPR和XPS分析中得到证实(图6(a)和图7)。HCl和NO可与催化剂表面的活性氧反应,形成新的活性位点Cl*和NO2,促进Hg0氧化。综上所述,单一的活性位点难以满足NOx还原和Hg0协同控制;基于NH3、SO2和Hg0酸碱性的差异,构建强弱碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力,是提高催化剂协同污控性能和抗硫性重要途径。
-
1)单一活性位点的Ce5Ti和Ce5W9Ti催化剂在中温区间(250~400 ℃)表现出较高的汞氧化率,分别为58.4%~69.1%和68.8%~81.3%,但在低温区间(100~200 ℃)下的汞氧化率较低(低于53.1%和62.5%)。CuO掺杂提高了Cu5Ce5W9Ti催化剂低温活性,提高低温区间(100~200 ℃)汞氧化率至76.3%~85.5%。
2) WO3以不定晶型的形式高度分散于催化剂的表面,可促进Ce与Ti原子的替代和掺杂,形成钛铀矿CeTi2O6(110),提高氧空位的暴露;同时酸性氧化物WO3的掺杂可增加催化剂表面酸性活性位点,促进强碱性气体NH3的吸附,进而降低NH3和Hg0对Ce基氧化位点的竞争吸附,因此,Ce5W9Ti催化剂在SCR烟气组分下依然保持较高的汞氧化性能。
3) CuO、WO3引入在Cu5Ce5W9Ti催化剂表面形成强弱碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力和抗硫性能,为多污控措施协同控制汞排放提供关键技术理论。
SCR气氛条件下Ce基催化剂汞氧化性能与机制
Mercury oxidation performance and mechanism of Ce-based catalyst under SCR flue gas
-
摘要: 燃煤电厂和工业锅炉为我国最大的大气汞固定排放源,我国采用多种污控措施协同控制技术降低燃煤行业烟气汞排放。该控制技术的关键是通过协同脱硝脱汞催化剂将Hg0氧化为Hg2+,继而被湿法脱硫技术吸收捕集。针对Ce基催化剂在SCR组分下汞氧化率低和抗硫性差的问题,本研究采用浸渍方法以CeO2为活性位点、TiO2为载体、WO3和CuO为改性剂,制备了Ce5Ti、Ce5W9Ti、Cu5Ce5W9Ti 3种Ce基催化剂,并探究了各种催化剂在不同烟气条件下汞氧化性能。Ce5Ti和Ce5W9Ti催化剂在中温(250~400 ℃)条件下表现出较高的汞催化氧化效率,分别达到58.4%~69.1%和68.8%~81.3%,但在低温(100~200 ℃)条件下的汞氧化率较低(低于53.1%和62.5%)。CuO的掺杂提高了Cu5Ce5W9Ti催化剂在低温区间(100~200 ℃)的汞氧化率,可达到76.3%~85.5%。WO3的掺杂提高了Ce5W9Ti催化剂在SCR烟气组分下的汞氧化率,依然可达到62.4%以上;CuO的掺杂提高了Cu5Ce5W9Ti催化剂在SCR和SCR+SO2烟气组分下汞氧化率,分别达到81.3%和78.8%。CuO、WO3引入使Cu5Ce5W9Ti表面形成强弱碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力和抗硫性能,为多污控措施协同控制汞排放提供关键催化理论。Abstract: Coal-fired power plants and industrial boilers are the largest sources of atmospheric mercury emission in China. Multi pollution synergistic control measures are adopted to reduce flue gas mercury emission in coal-fired industries. The key of synergistic control technology is to oxidizing Hg0 to Hg2+ through synergistical denitrification and mercury removal catalyst(SCR catalyst), and be captured by the wet desulfurization technology. In view of the low mercury oxidation efficiency and poor sulfur resistance of Ce-based SCR catalysts, three Ce-based catalysts of Ce5Ti, Ce5W9Ti and Cu5Ce5W9Ti were prepared by the impregnation method with CeO2 as the active site, TiO2 as the carrier, and WO3 and CuO as the modifying agent. The mercury oxidation efficiency of various catalysts were investigated at different flue gas components. Ce5Ti and Ce5W9Ti catalysts showed higher mercury oxidation efficiency (58.4%~69.1% and 68.8%~81.3%, respectively) in the middle temperature range (250~400°C), but lower mercury oxidation efficiency (lower than 53.1% and 62.5%) in the low temperature range (100~200 ℃). CuO doping improved the mercury oxidation efficiency (76.3%~85.5%) of Cu5Ce5W9Ti catalyst in the low temperature range (100~200°C); WO3 doping increased the mercury efficiency of Ce5W9Ti catalyst to 62.4% under SCR flue gas component. The mercury oxidation efficiencies of Cu5Ce5W9Ti in SCR and SCR+SO2 flue gases increased to 81.3% and 78.8%, respectively. The introduction of CuO and WO3 to Cu5Ce5W9Ti catalyst led to form strong and weak alkaline multi-reactive zones on its surface, separate NH3, SO2 and Hg0 adsorption zones, reduce their competitive adsorption and improve the Hg0 oxidation capacity and sulfur resistance of catalysts. This study provides key technical theories for the mercury emission reduction of multi-pollution synergistic control measures.
-
Key words:
- catalysts /
- CeO2 /
- mercury oxidation /
- acid-base property /
- synergistic control
-
汞具有长距离迁移性和生物富集性,可以在大气中停留数月至1 a,随气流长距离传输,造成远离污染源地区环境汞污染。我国是全球大气汞排放量最大的国家,大约占全球大气汞排放量的30%,所以我国汞控制排放对全球汞减排具有重要意义[1-2]。因此,我国作为全球大气汞排放量最大的国家,汞排放控制也得到全球的广泛关注,面临巨大的履约压力。2021年,WU等[2-3]探究了2021年我国重点行业大气汞排放量为314 t,其中燃煤电厂和工业锅炉排放量分别为36 t和24 t,占总排放量的19.1%。因此,煤炭燃烧是我国汞排放控制的重点行业。
相较于国外燃煤行业专门汞控制技术(活性炭喷射技术),我国燃煤行业普遍采用污控措施协同控制措施降低烟气汞排放浓度。以燃煤电厂为例,目前燃煤电厂的污控措施基本都具有选择性催化还原(selective catalytic reduction,SCR)催化剂、除尘设备、湿法脱硫设备(wet fuel gas desulfurization device,WFGD)。汞在烟气中主要存在3种形态:气态元素汞(Hg0)、气态氧化汞(Hg2+)和颗粒态汞(Hgp)[4-6]。Hg2+和Hgp分别被WFGD和除尘设备控制捕集下来,捕集效率高达80%和99%以上。Hg0不易溶于水和被除尘捕集,所以燃煤行业烟气汞主要以Hg0形式存在。因此,燃煤行业汞减排关键在于通过催化剂将Hg0转化为Hg2+,进而被脱硫设备捕集。
目前,燃煤电厂普遍采用钒钨钛催化剂作为商业脱硝催化剂,但V2O5的高毒性、致癌性等特点限制了钒钨钛催化剂的使用。氧化铈(CeO2)具有良好储氧能力和氧化还原特性,可以作为催化剂V2O5的替代活性组分[7-9]。以往的研究表明,Ce基催化剂表现出良好的NOx还原能力或Hg0氧化能力,但单一的CeO2难以满足NOx还原和Hg0氧化双重催化需求,构建多活性位点多反应区域是满足NOx和Hg0协同控制的重要条件。CeMnTi催化剂具备良好的Hg0氧化性能和NOx低温还原性能,但CeO2高氧化性使SO2转化为Ce(SO4)2或Ce2(SO4)3,造成催化剂失活[10-12]。基于以上研究,开发SCR烟气条件下具备Hg0高效氧化和良好抗硫性能的催化剂尤为必要。
基于以上研究结果与不足,考虑到NH3、SO2和Hg0酸碱性的差异,本研究以CeO2为活性位点、TiO2为载体、WO3和CuO为改性剂,制备多种Ce基催化剂,构建酸碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力,探究了催化剂协同催化脱除和抗硫反应机制,为燃煤行业多污染协同控制和多设备协同控汞提供理论依据。
1. 材料及方法
1.1 催化剂的制备
本研究采用德固赛公司生产的商业TiO2 (P25) 作为催化剂的载体。基于已有研究[13]结果,将一定量的Ce(NO3)3·6H2O和草酸溶解于去离子水,按照Ce:W:Cu:Ti比例添加一定量的(NH4)10H2(W2O7)6和Cu(NO3)2,再添加一定量的P25形成混合溶液,微波处理15 min。将混合溶液搅拌24h,然后再放入110 ℃烘箱中干燥12 h,最后在马弗炉中500 ℃焙烧4h后,自然降温到25 ℃,制备出不同Ce基催化剂。将催化剂研磨压片过筛得到40~60目催化剂样品用于汞氧化实验。所得催化剂包括Ce5Ti(Ce∶Ti=5∶95)、Ce5W9Ti(Ce∶W∶Ti=5∶9∶86)以及Cu5Ce5W9Ti(Cu∶Ce∶W∶Ti=5∶5∶9∶81)。
1.2 催化剂的表征分析
本研究采用X射线衍射(XRD)、X射线光电子能谱(XPS)、H2程序升温还原测试(H2-TPR)、CO2程序升温脱附分析(CO2-TPD)和X射线透射电镜(TEM)等表征手段对反应前后的催化剂进行表征,分析催化剂表面理化性质,探究汞氧化反应机制。XRD 物相分析采用Bruker D8 (德国布鲁克公司),管电流30 mA,管电压40 kV,扫描角度2θ为10°~90°,扫描速度为2 °·min−1。XPS分析采用X射线光电子能谱仪(Thermo ESCALAB 250XI)分析催化剂表面Cu、Ce、W、Ti、O、S、N、Cl和Hg元素的价态。H2-TPR实验采用AutoChem II
2920 型化学吸附仪 (Micrometritics Co.),实验步骤如下:取 50 mg 样品(40~60 目),在纯氧气氛下 400 ℃ 预处理 30 min;降温到50 ℃后用He吹扫15 min;然后通入体积分数为10% H2+90%Ar混合气,待仪器基线平稳后,以10 ℃·min−1速率升温至600 ℃,采用TCD 检测耗氢量。CO2-TPD实验步骤如下:取100 mg 样品 (40~60 目),在氦气氛下经500 ℃预处理30 min;冷却至50 ℃,再通入30 mL·min−1的CO2吸附至饱和,氦气吹扫1h 后开始TPD 实验,以10 ℃·min−1 速率升温至800 ℃,采用TCD 检测。1.3 烟气模拟实验
本研究采用固定床实验装置探究催化剂汞氧化性能。该固定床实验装置包括四部分:模拟烟气配气系统、汞源发生系统、催化反应系统和汞浓度与形态测试系统。催化反应系统通过加热系统精准调控反应温度,温度误差≤2 ℃。模拟烟气配气系统包括高纯氮气(N2)、氧气(O2)、标准混合气(HCl、SO2、NO和NH3等)和80 μg·m−3 Hg0,标准混合气均采用N2作为平衡气。以上标准气体均采用质量流量控制器控制气体添加,混合气体总流量为1L·min−1。空速是催化剂催化氧化性能的重要实验参数,本实验催化剂空速约为100 000 h−1。研究中各种模拟烟气组分见表1。反应前后烟气中Hg0和Hg2+的浓度通过烟气分析仪分析(Thermo Fisher 80i)。汞氧化率通过式(1)计算。
表 1 汞催化氧化实验模拟烟气组分Table 1. The components of simulated flue gas in mercury oxidation experiments序号 组分 温度 元素比例 1 基本烟气组分 100~450 ℃ 6%O2+N2 2 SCR烟气组分 200、300和400 ℃ 6%O2+134 mg·m−3 NO+76 mg·m−3 NH3+N2 3 SCR+HCl烟气组分 200、300和400 ℃ 6%O2+134 mg·m−3 NO+76 mg·m−3 NH3+16 mg·m−3 HCl+N2 4 SCR+SO2烟气组分 200、300和400 ℃ SCR组分+1 429 mg·m−3 SO2 5 SCR+HCl+SO2烟气组分 200、300和400 ℃ SCR组分+16 mg·m−3 HCl+1 429 mg·m−3 SO2 | Show Table DownLoad:
CSV
DownLoad:
CSV
η=CHg2+CHgt×100% (1) 其中:η为汞氧化率,%;CHg2+和CHgt 分别代表反应管出口烟气Hg2+浓度和入口烟气总汞浓度,μg·m−3。所有数据为3组平行实验的平均值,实验数据相对误差不超过10%。
2. 结果与讨论
2.1 不同Ce基催化剂的汞氧化性能
在基本烟气组分条件下,本研究对比了Ce5Ti、Ce5W9Ti和Cu5Ce5W9Ti催化剂的汞氧化性能。如图1所示,Ce作为一种中温活性组分,Ce5Ti和Ce5W9Ti在中温区间(250~400 ℃)表现出最佳的汞氧化性能,Ce5Ti和Ce5W9Ti催化剂汞氧化率分别达到58.4%~69.1%和68.8%~81.3%。Ce5Ti和Ce5W9Ti在低温区间(100~200 ℃)的汞氧化率普遍较低,分别低于53.1%和62.5%,表现出较差的低温活性。CuO掺杂的Cu5Ce5W9Ti表现出最佳的汞氧化性能,在300 ℃下汞氧化率达到最高,为88.8%。在中温区间,Cu5Ce5W9Ti的汞氧化率稳定在86.3%~88.8%。反应温度进一步上升到450 ℃,3种催化剂的汞氧化率均降低,说明温度过高使CeO2的催化氧化活性降低。与Ce5Ti和Ce5W9Ti相比,Cu5Ce5W9Ti在低温区间(100~200 ℃)表现出较高的汞氧化率,为76.3%~85.5%,表明CuO的掺杂可显著提升Ce基催化剂低温氧化活性。
 图 1 Ce基催化剂对汞的氧化性能(基本烟气组分)Figure 1. Mercury oxidation efficiency of various Ce-based catalysts (basic flue gas)
图 1 Ce基催化剂对汞的氧化性能(基本烟气组分)Figure 1. Mercury oxidation efficiency of various Ce-based catalysts (basic flue gas)2.2 SCR烟气组分对Ce基催化剂汞氧化性能影响
本研究对比了3种催化剂在基本烟气组分和SCR烟气组分条件下的汞氧化活性的差异性。如图2所示,在300 ℃下,相较于基本烟气组分,Ce5Ti催化剂在SCR烟气组分下汞氧化率明显降低,由62.7%下降到37.2%。在SCR烟气组分条件下,Ce5W9Ti催化剂汞并没有表现出明显的降低,汞氧化率由73.8%下降到62.4%,表明WO3掺杂明显提高了Ce5W9Ti催化剂对SCR反应条件的适应性。有研究[14-15]表明,SCR烟气组分可降低催化剂的汞氧化活性,这主要归咎于NH3与Hg0对活性位点的竞争吸附作用,高浓度的NH3分子明显降低了Hg0与活性位点接触的可能性。Cu5Ce5W9Ti催化剂SCR烟气组分条件下汞氧化表现与Ce5W9Ti相似,汞氧化率略微降低,由88.8%下降到81.3%,依然表现出较好的SCR烟气组分的适应性。
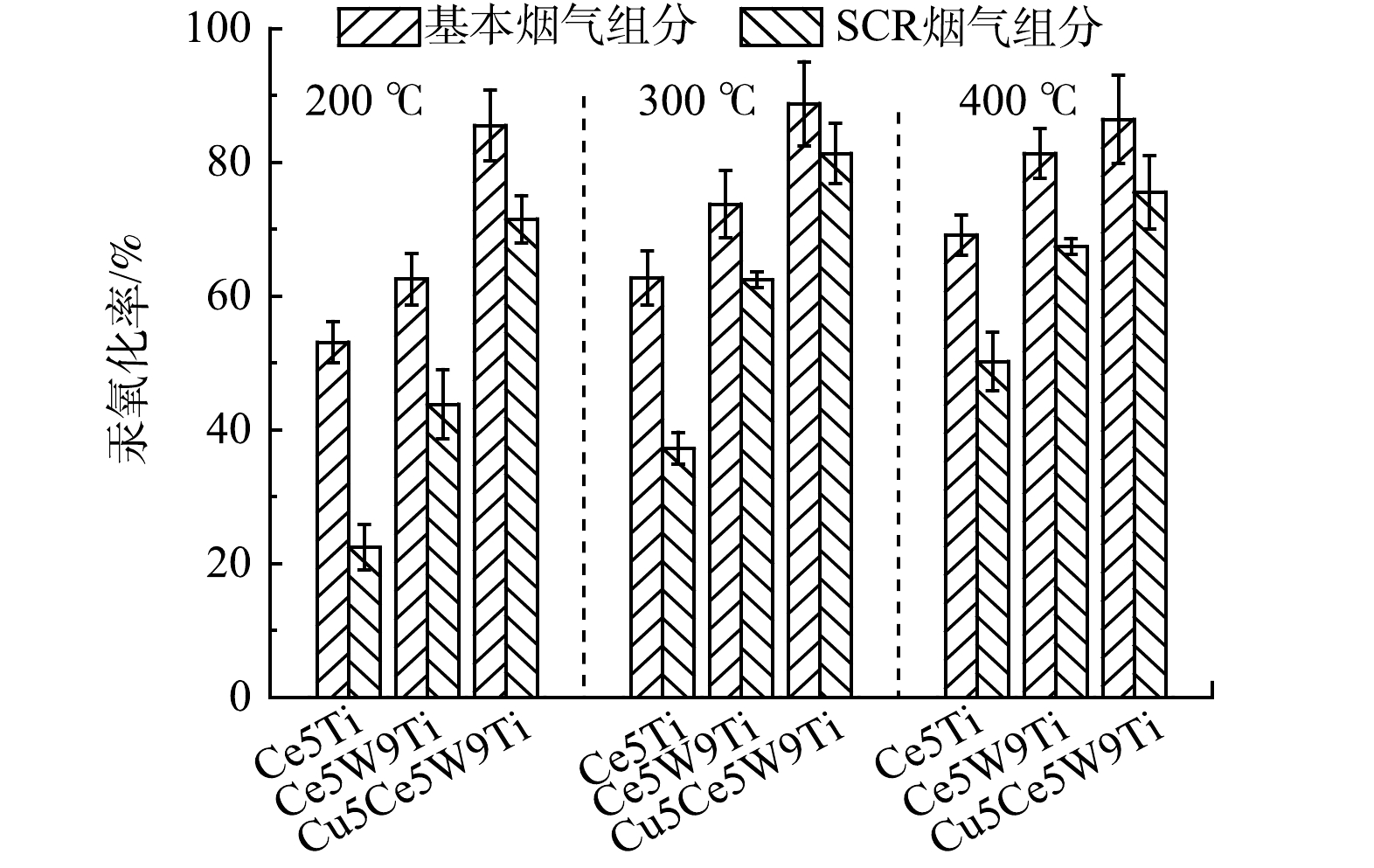 图 2 SCR烟气组分对Ce基催化剂汞氧化性能影响Figure 2. Mercury oxidation efficiency of various Ce-based catalysts at SCR flue gases
图 2 SCR烟气组分对Ce基催化剂汞氧化性能影响Figure 2. Mercury oxidation efficiency of various Ce-based catalysts at SCR flue gases在200 ℃和400 ℃的条件下,SCR烟气组分对催化剂具有相似的的影响。在200 ℃下,SCR烟气组分对Ce5Ti和Ce5W9Ti催化剂汞氧化抑制作用较为明显,而在400 ℃下,SCR烟气组分的抑制作用相对减少。这可能是因为Ce5Ti和Ce5W9Ti催化剂缺失低温活性组分,低温条件下NH3吸附量增加,汞氧化性能较低;而高温条件下NH3吸附量降低,减少了对Hg0抑制吸附作用,提高催化剂汞氧化性能。
2.3 SO2和HCl组分对Ce5W9Ti和 Cu5Ce5W9Ti催化剂汞氧化性能影响
SO2和HCl是燃煤烟气普遍存在的酸性气体,对催化剂汞氧化性能具有重要影响。通过以上实验筛选,本研究对比了Ce5W9Ti和 Cu5Ce5W9Ti催化剂在SCR烟气组分、SCR+SO2烟气组分、SCR+HCl烟气组分以及SCR+SO2+HCl烟气组分中的汞氧化性能变化,分析SO2和HCl对Ce5W9Ti和Cu5Ce5W9Ti催化剂影响。
如图3所示,在SCR烟气组分(300 ℃)下,1 429 mg·m−3 SO2 的添加明显降低了Ce5W9Ti催化剂的汞氧化性能,汞氧化率从62.4%降低到41.4%,表明SO2严重抑制了Ce5W9Ti催化剂汞氧化活性。有研究表明:高浓度SO2极容易吸附在CeO2活性位点,形成稳定的硫酸铈,使Ce基活性位丧失储氧能力[14]。SO2同样降低了Cu5Ce5W9Ti催化剂的汞氧化性能,但汞氧化率下降幅度较小,由81.3%下降到78.8%。该结果表明CuO的掺杂明显提高了催化剂的抗硫性能,可能是CuO的掺杂增加了反应活性位点或是保护了Ce基活性位点,使得Cu5Ce5W9Ti在SO2存在条件下依然具有较高的汞氧化性能。在200 ℃下,SO2对Ce5W9Ti和Cu5Ce5W9Ti的汞氧化性能抑制作用进一步提高。常化振等[16]研究表明,在低温条件下SO2与NH3形成大量的亚硫酸氢铵,覆盖了催化剂表面的活性位点,导致催化剂活性较低。在400 ℃条件下,SO2的抑制作用减弱,可能因为高温抑制了SO2吸附和亚硫酸氢铵的形成[16]。
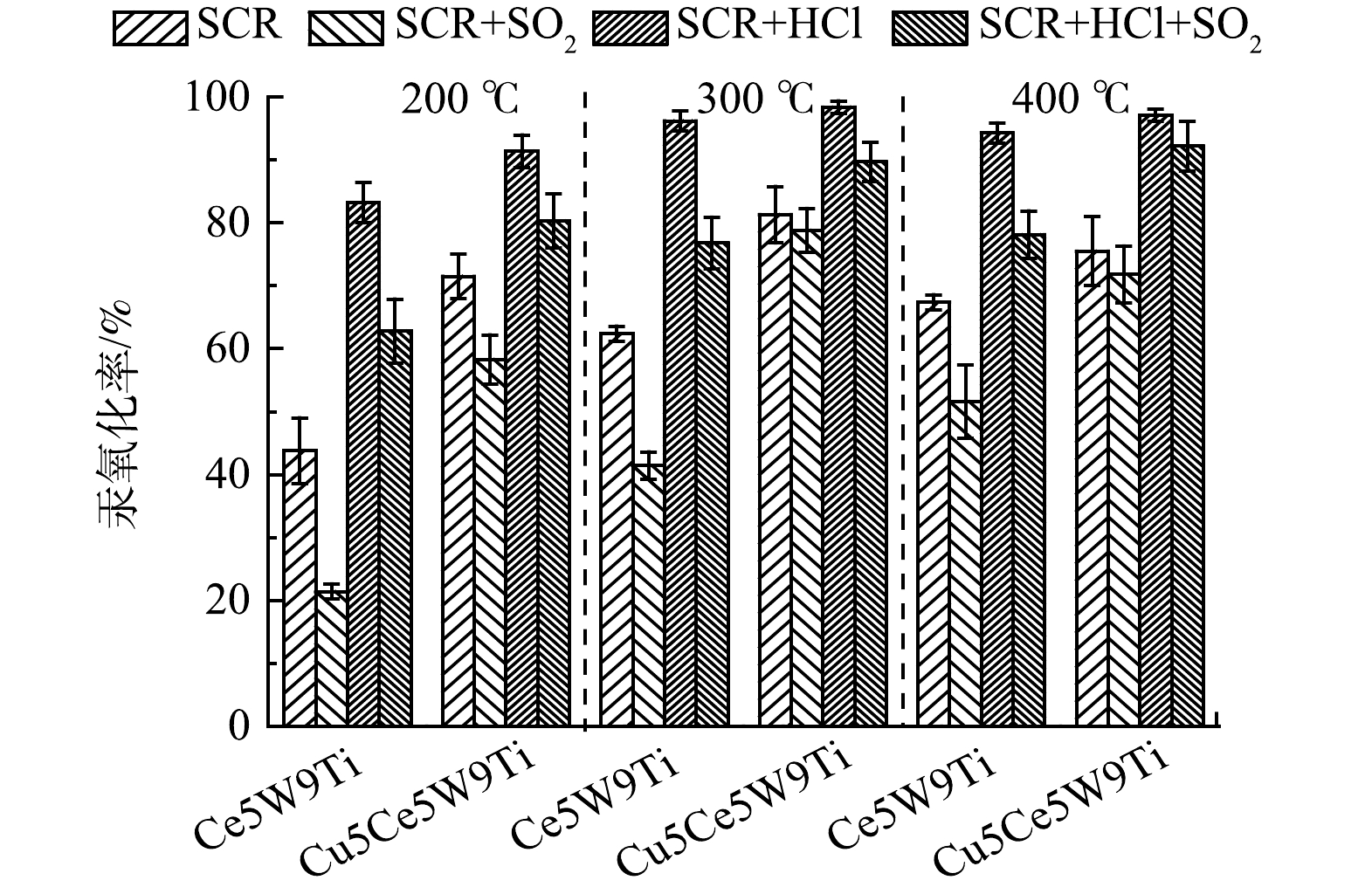 图 3 SO2和HCl对Ce基催化剂汞氧化性能影响Figure 3. Mercury oxidation efficiency of various Ce-based catalysts at the presence of SO2 and HCl
图 3 SO2和HCl对Ce基催化剂汞氧化性能影响Figure 3. Mercury oxidation efficiency of various Ce-based catalysts at the presence of SO2 and HClHCl的添加明显提高了Ce5W9Ti和 Cu5Ce5W9Ti催化剂汞氧化性能,汞氧化率分别提高到96.2%和98.4%。该结果表明HCl的添加可能增加了新的活性位点,促进了汞氧化率。进一步添加1 429 mg·m−3 SO2到模拟烟气,Ce5W9Ti和 Cu5Ce5W9Ti催化剂出现下降,但Ce5W9Ti和 Cu5Ce5W9Ti催化剂在SCR+HCl+SO2烟气组分下汞氧化率(76.8%和89.7%)依然明显高于SCR组分下汞氧化率(62.4%和81.3%)。该结果表明相较于SO2,HCl更具有竞争力,在催化剂表面依然形成新反应活性位,促进了Hg0的吸附和氧化。在200 ℃和400 ℃下,HCl显著提高了Ce5W9Ti和 Cu5Ce5W9Ti的汞氧化率和抗硫性能。
2.4 理化表征
1) XRD分析。采用XRD技术分析了不同Ce基催化剂的晶相(图4)。3种催化剂XRD谱图主要以锐钛矿和红金石2种形态TiO2特征峰为主(JCPDS 21-
1272 ),符合载体P25组分80.0%锐钛矿和20.0%红金石组成特征。3种催化剂在2θ=28.35°位置出现一个微弱的CeO2晶峰,表明Ce在载体表面高度分散,具备较高的催化氧化活性。Ce5W9Ti和 Cu5Ce5W9Ti催化剂都没有出现明显的WO3晶峰,说明元素W在催化剂表面同样高度分散。Cu5Ce5W9Ti催化剂在35.5o和38.6o位置出现了微弱的CuO(111)特征峰(JCPDS 48-1548 ),证明Cu活性组分绝大部分均匀分布在催化剂表面。基于以上结果,Ce、W和Cu元素在载体表面高度均匀分散[13]。2) TEM分析。采用TEM分析了不同Ce基催化剂表面的元素晶体形态,结果见图5。如图5(a)所示,催化剂载体P25主要以立方体形态存在。如图5(b)和图5(c)所示,Ce5Ti和Ce5W9Ti催化剂表面CeO2以(111)晶型存在,晶格间距为0.31 nm,与XRD的分析结果一致。Ce5W9Ti催化剂表面没有发现WO3晶体,但Ce与Ti掺杂形成钛铀矿CeTi2O6(110)[17],晶体间距为0.34 nm,说明W元素的高度分散有利于Ce与Ti原子的螯合掺杂,可形成稳定的氧空位。如图5(d)所示,Cu5Ce5W9Ti催化剂表面出现间距为0.28 nm的CuO(110)晶格条纹和间距为0.31 nm的CeO2(111)晶格条纹,CuO与CeO2界面形成掺杂晶体界面。在界面位置,CuO、CeO晶体中的原子被相互替代,形成晶体错位,晶体条纹边界发生扭曲转移,促进大量Cu+和Ce3+形成,使催化剂表面形成氧空位,从而提高催化剂的储氧和氧化还原能力[18]。
3) TPR和TPD分析。本研究采用H2-TPR和CO2-TPD技术分析了Ce5Ti、Ce5W9Ti和 Cu5Ce5W9Ti催化剂氧化位点和活性位点酸碱性。如图6(a)所示,Ce5Ti催化剂H2-TPR图谱出现一个270 ℃的主峰,代表CeO2形成的氧空位或活性位点。Ce5W9Ti催化剂的H2-TPR图谱中出现240 ℃和282 ℃ 2个主峰,结合TEM分析结果,除了CeO2活性位点,Ce与Ti掺杂形成的钛铀矿CeTi2O6(110)建 立新的活性位点[17]。因此,Ce5Ti、Ce5W9Ti催化剂在中温区间(250~400 ℃)表现出良好的汞氧化性能。如图6(a)所示,Cu5Ce5W9Ti催化剂的H2-TPR图谱中出现115、260和317 ℃ 3个主峰。260 ℃和317 ℃代表中温CeO2活性位点和Ce-Ti活性位点,115 ℃代表低温CuO活性位点,表明CuO的掺杂可提高催化剂在低温区域活性位点的形成。因此,Cu5Ce5W9Ti催化剂在图1表现出较高的低温(100~200 ℃)汞氧化性能。
如图6(b)所示,通过CO2-TPD分析了3种催化剂表面活性位点的酸碱性。有研究[19-20]表明,根据CO2脱附温度可将活性位点酸碱性分为弱碱(100~250 ℃)、中强碱(250~600 ℃)和强碱(600~800 ℃)。在Ce5Ti的CO2-TPD图谱中出现一个脱附主峰(140 ℃),表明CO2主要为弱碱活性位点;Ce5W9Ti的脱附主峰温度进一步降低到132 ℃,表明酸性氧化物WO3在催化剂表面的高度分散可进一步降低催化剂碱性强度。Cu5Ce5W9Ti的CO2-TPD图谱包含3个特征峰,即132、547和649 ℃ [21-22]。在SCR+SO2烟气条件下,强酸性气体SO2优先与碱性活性位(Cu—O—Ce和CuO)反应,从而保护了CeO2活性位依然保持高活性和氧化性,因此,Cu5Ce5W9Ti在SCR+SO2烟气条件下依然表现出良好的汞氧化性能(图3)。
4) XPS分析。本研究采用XPS技术分析催化剂表面各种元素价态,探究催化剂汞氧化机制。如图7(a)所示,Ce元素能谱图包含8个分峰:u0、u1、u2、u3、v0、v1、v2和v3。其中u1和v1代表Ce3+;u0、u2、u3、v0、v2和v3代表Ce4+,Ce3+一般作为氧空位或是吸附氧位点[23]。Ce5W9Ti催化剂Ce3+峰面积和Ce4+峰面积比例(Ce3+/Ce4+)为31.2%,Cu5Ce5W9Ti 的Ce3+/Ce4+面积比例上升到39.2%。该结果与TEM分析结果一致,说明CuO与CeO2掺杂形成晶体错位,形成更多Ce3+和氧空位。
如图7(b)所示,Ce5W9Ti和Cu5Ce5W9Ti催化剂表面O元素2个特征峰:529.9~530.0 eV特征峰代表晶格氧,530.8~531.0 eV特征峰代表活性氧[24-25]。活性氧一般是氧空位吸附的高活性吸附氧,为Hg0氧化的关键活性位点。Ce5W9Ti催化剂活性氧特征峰面积占比为38.7%,Cu5Ce5W9Ti的活性氧特征峰面积占比为45.8%,该结果与Ce3+/Ce4+面积比例上升一致。如图7(c)所示,Cu5Ce5W9Ti催化剂表面在932.5 eV和934.2 eV的2个主要特征峰分别代表Cu+和CuO[26]。Cu5Ce5W9Ti催化剂表面Cu主要以Cu+形式存在,这与Cu、Ce原子掺杂形成氧空位界面保持一致。Cu5Ce5W9Ti催化剂反应前后Cu+/CuO比例由69.4%下降到57.2%,这可能是部分Cu+形成的活性氧与SO2反应形成大量的CuSO4,因此,Cu2+比例明显上升。如图7(d)所示,Cu5Ce5W9Ti 的Ce3+/Ce4+面积比例由反应前的39.2%下降到反应后的35.4%,说明部分Ce3+活性位在反应过程中转化为稳定的Ce4+,降低了催化剂的吸氧释氧能力。如图7(e)所示,Cu5Ce5W9Ti催化剂反应前后表面O元素2个特征峰发生明显变化。Cu5Ce5W9Ti催化剂活性氧特征峰面积占比有反应前的45.8%下降到反应后的35.3%,说明大量活性氧参与了Hg0氧化和NOx还原反应。
XPS表征没有发现反应前的Cu5Ce5W9Ti催化剂表面存在Cl、N和Hg原子。如图7(f)所示,SCR+SO2+HCl条件反应后的Cu5Ce5W9Ti催化剂表面Cl、N和Hg元素的价态。Cu5Ce5W9Ti催化剂表面Cl存在Cl−离子(198.1 eV)和活性Cl*(200.2 eV)2个特征峰[17, 23]。大面积活性Cl*特征峰的出现说明HCl在Cu5Ce5W9Ti催化剂表面吸附并转化为活性Cl*,促进Hg0转化为HgCl2。NO在催化剂表面XPS能谱图出现硝酸根(407.2 eV)和NO2(405.1 eV)2个特征峰。NO2特征峰面积要远大于硝酸根特征峰面积,说明NO在催化剂上主要以更高活性的NO2或是亚硝酸盐官能团形式存在。反应后的Cu5Ce5W9Ti催化剂表面存在Hg元素101.6 eV和104.6 eV两个特征峰,表明吸附在催化剂表明的汞主要以 Hg2+形式存在。
2.5 Ce基催化剂汞氧化机制分析
基于3种Ce基催化剂汞氧化实验和理化性质表征结果,本研究发现单一活性位点的Ce5Ti和Ce5W9Ti催化剂中温区间(250~400 ℃)表现出较高汞催化氧化活性(图1),但在SCR烟气组分下Ce5Ti的汞氧化率明显下降(图2),这归咎于NH3与Hg0竞争Ce基活性位点。WO3的掺杂提高了Ce5W9Ti在SCR烟气组分下的汞氧化率,为62.4%。CO2-TPD分析结果说明WO3的掺杂增加了Ce5W9Ti催化剂酸性(图6),增加了强碱性气体NH3的吸附位点,降低了NH3对Hg0吸附氧化竞争。同时XRD和TEM分析结果表明高度分散的WO3可促进Ce与Ti的耦合掺杂形成钛铀矿CeTi2O6(110),提高氧空位的比例。CuO的掺杂提高了Cu5Ce5W9Ti催化剂在低温区间(100~200 ℃)的汞氧化率(76.3%~85.5%),并且在SCR和SCR+SO2烟气条件下表现出较高的汞氧化率(81.3%和78.8%)。H2-TPR分析结果说明Cu掺杂增加Cu5Ce5W9Ti催化剂低温(115 ℃)活性位点,因此,提高催化剂的低温氧化活性;CO2-TPD分析结果表明W、Cu掺杂使Cu5Ce5W9Ti催化剂具备弱碱、中强碱、强碱3种活性位,可有效地分离酸性气体SO2、碱性气体NH3和Hg0吸附氧化反应区域,从而提高催化剂的抗硫性(图6)。TEM分析说明CuO和CeO2晶体掺杂的界面,发生原子替代和晶体错位,形成更多的Ce3+和Cu+作为氧空位,吸附更多的活性氧作为氧化位点,这一结论也在H2-TPR和XPS分析中得到证实(图6(a)和图7)。HCl和NO可与催化剂表面的活性氧反应,形成新的活性位点Cl*和NO2,促进Hg0氧化。综上所述,单一的活性位点难以满足NOx还原和Hg0协同控制;基于NH3、SO2和Hg0酸碱性的差异,构建强弱碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力,是提高催化剂协同污控性能和抗硫性重要途径。
3. 结论
1)单一活性位点的Ce5Ti和Ce5W9Ti催化剂在中温区间(250~400 ℃)表现出较高的汞氧化率,分别为58.4%~69.1%和68.8%~81.3%,但在低温区间(100~200 ℃)下的汞氧化率较低(低于53.1%和62.5%)。CuO掺杂提高了Cu5Ce5W9Ti催化剂低温活性,提高低温区间(100~200 ℃)汞氧化率至76.3%~85.5%。
2) WO3以不定晶型的形式高度分散于催化剂的表面,可促进Ce与Ti原子的替代和掺杂,形成钛铀矿CeTi2O6(110),提高氧空位的暴露;同时酸性氧化物WO3的掺杂可增加催化剂表面酸性活性位点,促进强碱性气体NH3的吸附,进而降低NH3和Hg0对Ce基氧化位点的竞争吸附,因此,Ce5W9Ti催化剂在SCR烟气组分下依然保持较高的汞氧化性能。
3) CuO、WO3引入在Cu5Ce5W9Ti催化剂表面形成强弱碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力和抗硫性能,为多污控措施协同控制汞排放提供关键技术理论。
-
图 1 Ce基催化剂对汞的氧化性能(基本烟气组分)
Figure 1. Mercury oxidation efficiency of various Ce-based catalysts (basic flue gas)
图 2 SCR烟气组分对Ce基催化剂汞氧化性能影响
Figure 2. Mercury oxidation efficiency of various Ce-based catalysts at SCR flue gases
图 3 SO2和HCl对Ce基催化剂汞氧化性能影响
Figure 3. Mercury oxidation efficiency of various Ce-based catalysts at the presence of SO2 and HCl
表 1 汞催化氧化实验模拟烟气组分
Table 1. The components of simulated flue gas in mercury oxidation experiments
序号 组分 温度 元素比例 1 基本烟气组分 100~450 ℃ 6%O2+N2 2 SCR烟气组分 200、300和400 ℃ 6%O2+134 mg·m−3 NO+76 mg·m−3 NH3+N2 3 SCR+HCl烟气组分 200、300和400 ℃ 6%O2+134 mg·m−3 NO+76 mg·m−3 NH3+16 mg·m−3 HCl+N2 4 SCR+SO2烟气组分 200、300和400 ℃ SCR组分+1 429 mg·m−3 SO2 5 SCR+HCl+SO2烟气组分 200、300和400 ℃ SCR组分+16 mg·m−3 HCl+1 429 mg·m−3 SO2
下载: 导出CSV
-
[1] United Nations Environment Programme. Global mercury assessment 2013: sources, emissions, releases and environmental transport[R]. Geneva: 2013. [2] WU Q, WANG S, LI G, et al. Temporal trend and spatial distribution of speciated atmospheric mercury emissions in China during 1978-2014[J], Environmental Science & Technology, 2016, 50(24): 13428-13435. [3] LIU K, WU Q, WANG S, et al. Highly resolved inventory of mercury release to water from anthropogenic sources in China[J], Environmental Science & Technology, 2021, 55(20): 13860-13868. [4] ZHAO S, QU Z, YAN N, et al. Ag-modified AgI–TiO2 as an excellent and durable catalyst for catalytic oxidation of elemental mercury[J]. RSC Advances, 2015, 5(39): 30841-30850. doi: 10.1039/C5RA00838G [5] 杨子文, 佟莉, 左朋莱, 等. 不同烟气组分对Cu2O改性V2O5-MoO3/TiO2脱硝催化剂汞氧化性能的影响[J]. 环境工程学报, 2022, 16(9): 2911-2920. doi: 10.12030/j.cjee.202205121 [6] 李述贤, 郑旭东, 龚建军, 等. 利用氯化锌和硫改性玉米秸秆生物炭稳定汞污染土壤[J]. 环境工程学报, 2021, 15(4): 1403-1408. doi: 10.12030/j.cjee.202008083 [7] Chang H, Wu Q, Zhang T, et al. Design strategies for CeO2-MoO3 catalysts for deNOx and Hg0 oxidation in the presence of HCl: the significance of the Surface acid-base properties[J]. Environmental Science & Technology, 2015, 49(20): 12388-12394. [8] LI G, WANG S, WU Q, et al. Exploration of reaction mechanism between acid gases and elemental mercury on the CeO2WO3TiO2 catalyst via In situ DRIFTS[J]. Fuel, 2019, 239(1): 162-172. [9] LI J, WU Q, WANG Y, et al. Improvement of NH3 resistance over CuO/TiO2 catalysts for elemental mercury oxidation in a wide temperature range[J]. Catalysis Today, 2021, 376(1): 276-284. [10] HE C, SHEN B, CHI G, et al. Elemental mercury removal by CeO2/TiO2-PILCs under simulated coal-fired flue gas[J]. Chemical Engineering Journal, 2016, 300(1): 1-8. [11] 吴响, 段钰锋, 姚婷, 等. Ce-Mn/TiO2吸附剂的脱汞性能及SO2特性[J]. 中国环境科学, 2019, 39(6): 2336-2343. doi: 10.3969/j.issn.1000-6923.2019.06.013 [12] 谭增强, 牛国平, 陈晓文, 等. Mn-Ce/分子筛的脱汞特性研究[J]. 环境科学, 2015, 36(6): 1983-1988. [13] LI G, SHAO S, WANG S, et al. Flame synthesized nanoscale catalyst (CuCeWTi) with excellent Hg0 oxidation activity and hydrothermal resistance[J]. Journal of Hazardous Materials, 2021, 408(1): 124427-124435. [14] LI H, WU C, LI Y, et al. Impact of SO2 on elemental mercury oxidation over CeO2–TiO2 catalyst[J]. Chemical Engineering Journal, 2013, 219(1): 319-326. [15] LV Q, CAI M, WANG C, et al. Investigation on elemental mercury removal and antideactivation performance of modified SCR catalysts[J]. Asia-Pacific Journal of Chemical Engineering, 2018, 13(4): 1-14. [16] XU L, WANG C, CHANG H, et al. New insight into SO2 poisoning and regeneration of CeO2–WO3/TiO2 and V2O5–WO3/TiO2 catalysts for low-Temperature NH3–SCR[J]. Environmental Science & Technology, 2018, 52(12): 7064-7071. [17] MICHALOW K, LU Y, KOWALSKI K, et al. Flame-made WO3/CeOx-TiO2 catalysts for selective catalytic reduction of NOx by NH3[J]. ACS Catalysis, 2015, 5(10): 5657-5672. doi: 10.1021/acscatal.5b01580 [18] JOHNSTON A, SENANAYAKE S, PLATA J, et al. Nature of the Mixed-oxide interface in ceria–titania catalysts: Clusters, chains, and nanoparticles[J]. The Journal of Physical Chemistry C, 2013, 117(28): 14463-14471. doi: 10.1021/jp3125268 [19] WU J, SU T, JIANG Y, et al. In situ DRIFTS study of O3 adsorption on CaO, γ-Al2O3 , CuO, α-Fe2O3 and ZnO at room temperature for the catalytic ozonation of cinnamaldehyde[J]. Applied Surface Science, 2017, 412(7): 290-305. [20] TADA S, SHIMIZU T, KAMEYAMA H, et al. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures[J]. International Journal of Hydrogen Energy, 2012, 37(7): 5527-5531. doi: 10.1016/j.ijhydene.2011.12.122 [21] WITOON T, KACHABAN N, DONPHAI W, et al. Tuning of catalytic CO2 hydrogenation by changing composition of CuO–ZnO–ZrO2 catalysts[J]. Energy Conversion and Management, 2016, 118: 21-31. doi: 10.1016/j.enconman.2016.03.075 [22] JANGJOU Y, ALI M, CHANG Q, et al. Effect of SO2 on NH3 oxidation over a Cu-SAPO-34 SCR catalyst[J]. Catalysis Science & Technology, 2016, 6(8): 2679-2685. [23] WATANABE S, MA X and SONG C. Characterization of structural and surface properties of nanocrystalline TiO2−CeO2 mixed oxides by XRD, XPS, TPR and TPD[J]. The Journal of Physical Chemistry C, 2009, 113(32): 14249-14257. doi: 10.1021/jp8110309 [24] HE C, SHEN B and LI F. Effects of flue gas components on removal of elemental mercury over Ce-MnO x/Ti-PILCs[J]. Journal of Hazardous Materials, 2016, 304(1): 10-17. [25] CHEN W, PEI Y, HUANG W, et al. Novel effective catalyst for elemental mercury removal from coal-fired flue gas and the mechanism investigation[J]. Environmental Science & Technology, 2016, 50(5): 2564-2572. [26] CHEN C, JIA W, LIU S, et al. Simultaneous NO removal and Hg0 oxidation over CuO doped V2O5-WO3/TiO2 catalysts in simulated coal-fired flue gas[J]. Energy & Fuels, 2018, 32(6): 7025-7034. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 236
- HTML全文浏览数: 236
- PDF下载数: 10
- 施引文献: 0
