





-
氨基三亚甲基膦酸(amino trimethylene phosphonic acid,ATMP)是工业生产中常用的缓蚀阻垢剂[1],含有三个膦酸基团侧链,具有良好的螯合、低限抑制等作用,可有效阻止水中成垢盐形成水垢[2]。在工业冷却水中投加ATMP能够提高冷却水的浓缩倍数,提高循环水利用率的同时以减少结垢风险。然而,这些经过多次循环的冷却水中含磷污染物会逐渐累积,成为处理难题。ATMP是一种溶解性有机磷(dissolved organic phosphorus,DOP),在无机磷匮乏的水体环境下,是生物可利用性磷的潜在来源,易造成水体富营养化[3-4]。此外,ATMP还具有强螯合能力,能够再活化重金属,提高重金属在水环境中的浓度与生物可利用性,从而使重金属达到毒害水生生物的浓度,或是通过河岸过滤进入到饮用水中产生风险[5-7]。
ATMP化学结构中含有稳定的C-P键且具备很强的抗生物降解性和抗吸附性,现有化学沉淀法、生化法等去除ATMP普遍存在药剂利用率低、难以去除等问题[7]。目前,含ATMP废水的处理方法主要包括吸附法和高级氧化法。在传统污水处理设施中,活性污泥可以吸附去除80%~90%的ATMP[8]。铁盐、铝盐混凝在特定的工艺条件下可以去除80%以上的ATMP[9]。LIU等[10]制备了La/FeOOH@PAC吸附材料, 对膦酸盐羟基亚乙基二膦酸(1-Hydroxyethylidene-1,1-diphosphonic acid,HEDP)吸附率达到85%以上。FAN等[11]使用高铁酸钾作为氧化剂处理膦酸盐,总磷(total phosphorus,TP)的去除率可达90%。LEI等[8]使用电化学的方法诱导ATMP氧化,以钛钌铱阳极和未涂覆钛阴极作为电极材料,48 h内TP去除率达到85%,其中羟基自由基(·OH)是氧化ATMP的主要活性物种。
过氧化氢(H2O2)常作为产生·OH的前驱体被广泛应用于AOPs中[12]。电化学、紫外光(ultraviolet,UV)、超声波、金属离子等都能活化H2O2[8, 13-14],从而产生·OH等能够促使ATMP中有机磷氧化降解为无机磷的活性物种。在紫外光活化过氧化氢(UV/H2O2)体系中,H2O2吸收UV辐射能量,破坏O—O键并产生·OH,该体系具有以无污染的紫外光为能源,·OH的产量高且无其它副产物等优点,得到了广泛应用[15-16]。因此,使用UV/H2O2产生·OH活性物种对ATMP进行氧化降解具有可行性,是较为绿色环保的方法。
本研究以工业冷却废水中典型含磷阻垢剂ATMP为目标污染物,采用UV/H2O2体系将有机磷氧化降解。在UV照射下,向溶液中投加过氧化氢(H2O2),通过UV催化活化H2O2产生活性物种,将有机磷氧化降解为正磷酸盐(orthophosphate,PO43−)和小分子有机物。本研究比较了单独投加H2O2、单独UV和UV/H2O2体系对ATMP氧化降解为PO43−的效果。考察了不同UV/H2O2操作参数(UV波长、初始pH、H2O2浓度、反应温度、共存底物)对UV/H2O2氧化降解有机磷的影响,并研究了体系中ATMP氧化的反应机制,以期为含ATMP废水的处理提供参考。
-
主要试剂:氨基三亚甲基膦酸(ATMP,50%);过氧化氢(H2O2,30%);叔丁醇(TBA,C4H10O);无水硫酸钠;超纯水(18.2 MΩ·cm);浓硫酸(98%,H2SO4);氢氧化钠(NaOH)。实验中所有使用药品均为分析纯;其中过氧化氢和浓硫酸购自北京市通广精细化工公司;其余试剂购自上海麦克林生化科技有限公司。
-
本实验以工业冷却废水中的ATMP为目标污染物,采用UV/H2O2法降解ATMP。ATMP用作工业循环冷却水的缓蚀剂使用时,投加量一般为20~60 mg·L−1[12]。因此选择配置0.1 mmol·L−1 ATMP(其中P浓度为0.3 mmol·L−1)溶液,取500 mL上述溶液至于烧杯中。反应开始前使用0.1 mmol·L−1 H2SO4或NaOH调节溶液初始pH,然后投加设定浓度的H2O2,随后开启紫外灯启动反应。反应期间使用恒温磁力搅拌器将控制温度至设定值并保持恒定,磁力搅拌转速为400 r·min−1。总反应时间为90 min,分别在0、10、20、30、40、50、60、70、80和90 min时从反应器中抽取1 mL样品待测,取出样品后立即加入100 μL硫代硫酸钠(50 mmol·L−1)终止反应。PO43−浓度的测定方法为国标钼酸铵分光光度法。总磷(TP)浓度的测定方法为国标过硫酸钾氧化−钼酸铵分光光度法,分光光度计的工作波长为700 nm。所有实验均重复3次取平均值。
-
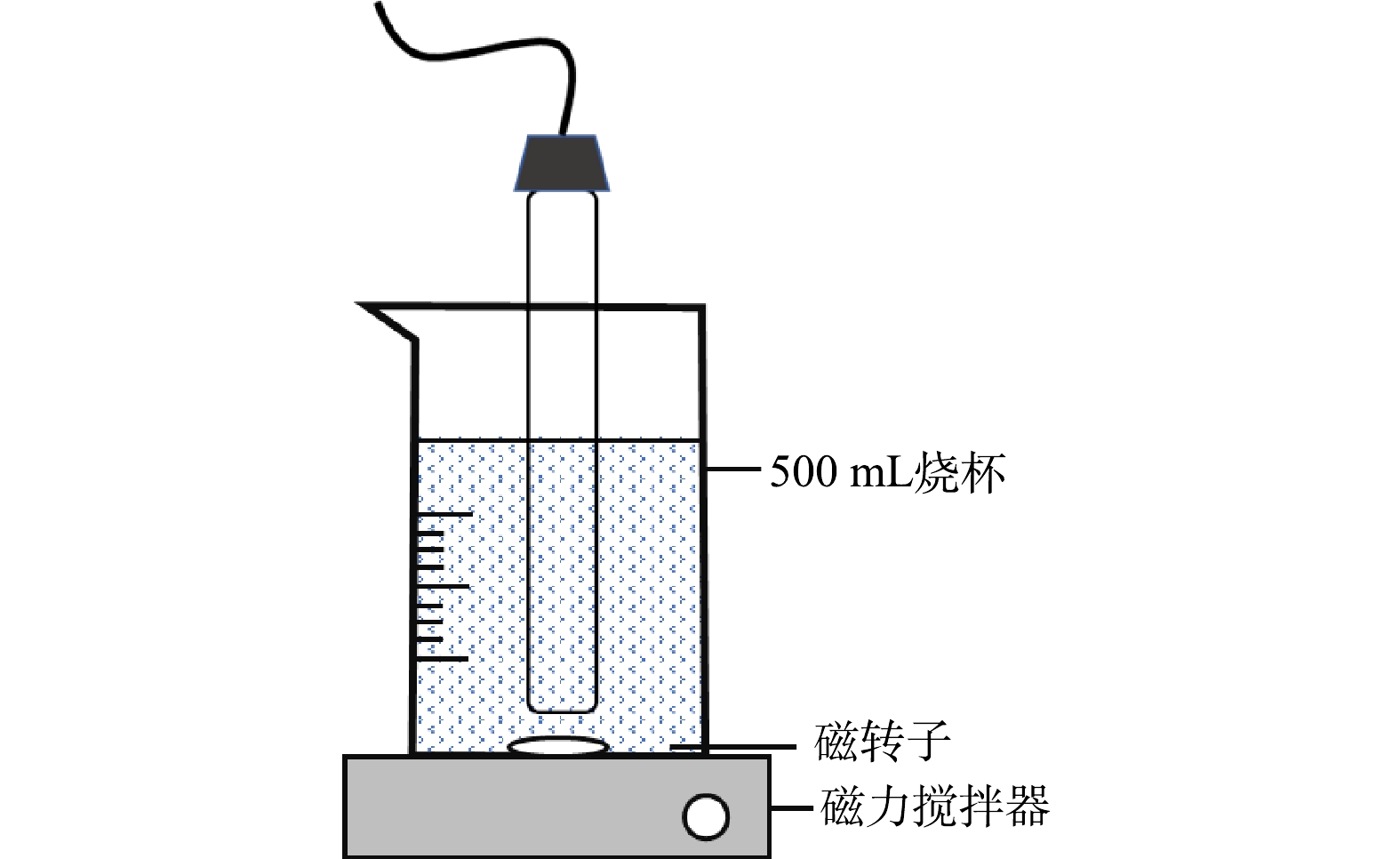
实验装置如图1所示,主要包括恒温磁力搅拌器(T09−1S,上海司乐仪器有限公司,中国)及不同波长紫外灯(λ=185、254、365 nm,10 W,海洛斯特殊光源,中国)。
其他主要仪器:紫外−可见分光光度计(T−N5000 Plus,上海佑科仪器仪表有限公司,中国)和pH计(PHS−3E,上海仪电科学仪器股份有限公司,中国)。
-
首先对比了单独施加UV(λ=185 nm)、单独投加H2O2(5.0 mmol·L−1)和UV/H2O2(λ=185 nm,H2O2投加量为5.0 mmol·L−1)体系对ATMP氧化降解为PO43−的效果。结果如图2所示,当溶液初始pH为7.0,反应90 min时,单独H2O2和单独UV体系中基本没有PO43−生成,其伪一级反应动力学常数均为0.001 min−1,不能实现高效率的ATMP氧化降解。然而,当UV和H2O2联合使用,反应90 min时,体系中PO43−生成量为0.24 mmol·L−1,约80%的ATMP被氧化降解为PO43−,一级反应动力学常数为0.015 min−1,远大于单独UV或单独H2O2。进一步将反应时间延长至190 min,此时PO43−生成量达到0.3 mmol·L−1,表明ATMP中的有机磷被完全矿化。UV和H2O2联合使用可以实现ATMP的高效氧化降解,这是因为UV可以活化H2O2,从而反应产生·OH等活性物种,大大提高反应氧化降解污染物的效率[17]。
-
1)紫外光波长。考察了典型紫外光波长(λ=185、254、365 nm)对ATMP氧化降解为PO43−的影响,结果如图3所示。当溶液中ATMP初始浓度为0.1 mmol·L−1,初始pH为7.0,H2O2投加量为5.0 mmol·L−1,紫外灯功率均为10 W时,不同紫外波长光源对于PO43−生成量大小比较结果为185 nm >254 nm >365 nm, 185 nm和254 nm能实现ATMP的高效氧化降解,365 nm则几乎不能氧化降解ATMP。这是因为短波UV相较于长波UV具有较高的能量, 185 nm和254 nm的光子能量分别为6.70 eV和4.88 eV[18],能活化H2O2产生·OH等活性物种,从而实现ATMP的氧化降解。185 nm相较于 254 nm具有更强的PO43−生成效果,可能归因于O2被185 nm的UV照射氧化为O3,O3本身就是常见氧化剂,能够促进反应体系的氧化效率[19]。因此后续实验使用UV 185 nm作为紫外光源。
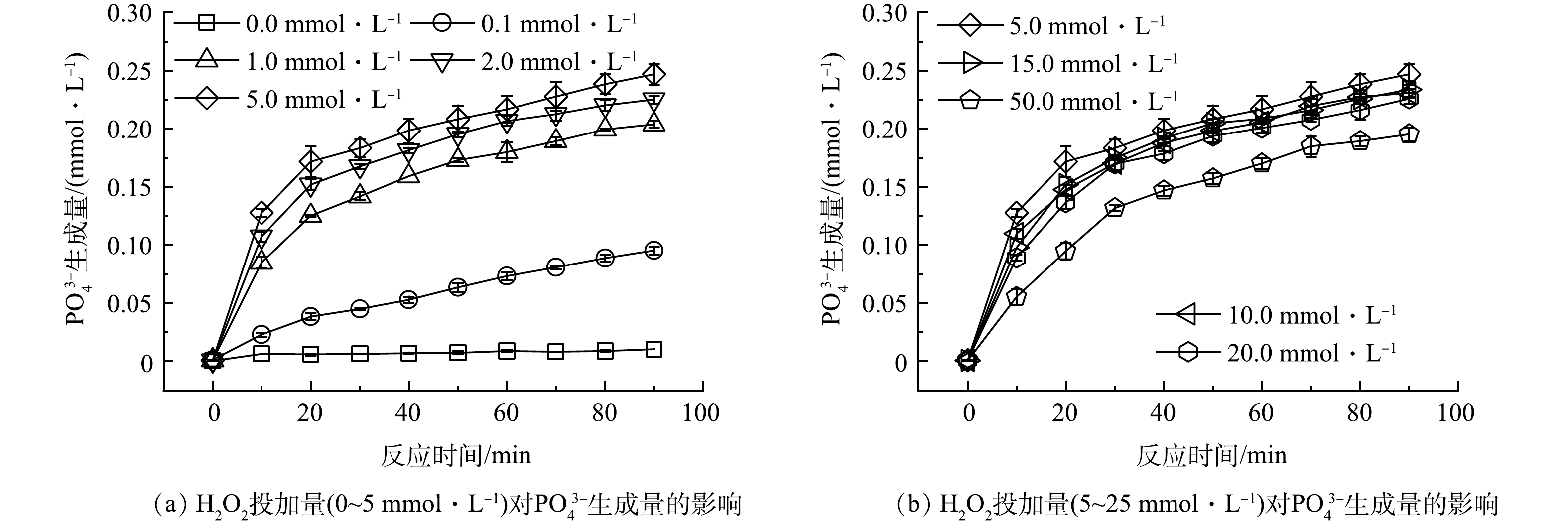
2) H2O2投加量。考察了不同H2O2投加量对ATMP氧化降解为PO43−的影响,结果如图4所示。当溶液中ATMP初始浓度为0.1 mmol·L−1,初始pH为7.0,紫外光源为UV 185 nm不同H2O2投加量(0.0、0.1、1.0、2.0、5.0、10.0、15.0、20.0和50.0 mmol·L−1)在反应90 min后生成的PO43−最高为0.24 mmol·L−1,此时对应的H2O2投加量为5.0 mmol·L−1。实验结果表明,当H2O2投加量小于5.0 mmol·L−1时,PO43−的生成量随着H2O2投加量的增加而逐渐升高;而当H2O2投加量超过5.0 mmol·L−1时,H2O2投加量继续增加反而使PO43−的生成量下降。此前研究表明H2O2本身对ATMP不存在氧化效果,因此,H2O2投加量增加导致的PO43−的生成量增加是因为H2O2浓度增加强化了体系内·OH等活性物种的生成,而当H2O2过量时,又可以成为·OH的清除剂,过量H2O2与生成的·OH进一步反应生成HO2−,从而降低了PO43−的生成量[15]。因此后续实验选择投加H2O2投加量为5.0 mmol·L−1。
3)溶液初始pH。溶液初始pH对ATMP氧化降解起到重要的影响作用。在紫外灯照射下,H2O2投加量为5.0 mmol·L−1,使用H2SO4和NaOH调节溶液pH(3.0、5.0、7.0、9.0、11.0),对UV/H2O2体系氧化降解ATMP的影响如图5(a)所示。初始pH由3.0升高至7.0时,溶液中ATMP的氧化降解效果逐渐上升。这是因为ATMP作为六元弱酸,溶液pH逐步升高的同时其去质子化程度也进一步加深,从而对·OH表现出更高的反应性[1,20]。当初始pH为7.0时,90 min的PO43−生成量最高,对应的ATMP去除率约为80%;此后再增加pH则会抑制PO43−生成,pH为11.0时90 min的PO43−生成量仅为0.10 mmol·L−1,这是因为H2O2在碱性条件下是极易分解成为氧化能力较弱的HO2·[20-21],进而影响ATMP的氧化降解。反应过程中的pH变化如图5(b)所示。pH在反应的前30 min内明显下降,而在在随后的60 min保持稳定。这是因为在ATMP氧化降解为PO43−的过程中会释放H+[20]。当初始溶液pH为3.0、5.0、7.0、9.0和11.0,反应30 min时,PO43−的生成量分别达到0.09、0.12、0.21、0.15和0.05 mmol·L−1,故此期间溶液pH明显下降;而在反应90 min时,不同初始溶液pH条件下对应的PO43−生成量分别为0.16、0.19、0.24、0.21和0.10 mmol·L−1,较反应前30 min相比PO43−生成量增加不显著,故此期间溶液pH趋于平缓。这与HUO等[22]的研究结果一致,溶液pH中性有利于UV/H2O2体系反应过程中污染物的降解。因此后续实验选择溶液初始pH为7.0。
4)反应温度。考察了在不同反应温度(25~65 ℃)对ATMP氧化降解为PO43−的效果,结果如图6所示。当溶液中ATMP初始浓度为0.1 mmol·L−1,初始pH为7.0,紫外光源为UV 185 nm,H2O2投加量为5.0 mmol·L−1时,随着反应温度从25 ℃上升到65 ℃,ATMP的氧化降解效率逐步降低。这可能是因为,H2O2容易受热分解,产生H2O和O2,从而降低了其利用率,减少体系中活性物种的生成,最终导致ATMP的氧化降解效率随温度的升高而降低[23]。鉴于提高的反应温度会导致反应效率的降低,因此确定实验均在室温(25±3) ℃下进行。
-
在工业循环冷却水中存在着各种与ATMP共存的环境底物,在UV/H2O2过程中,这些底物可能会影响ATMP的氧化降解。因此考察了工业循环冷却水中常见的无机阴离子(SO42−、Cl−、NO3−、CH3COO−)浓度以及腐殖酸(HA)对ATMP氧化降解为PO43−效果的影响。在UV 185 nm照射下,ATMP初始浓度为0.1 mmol·L−1,溶液初始pH为7.0,H2O2投加量为5.0 mmol·L−1条件下,分别考察了无机阴离子SO42−、Cl−、NO3−、CH3COO−(0.0、0.1、10.0、100.0 mmol·L−1)以及HA(0.00、0.05、0.10、0.50、1.00 mmol·L−1)对反应体系的影响,结果如图7、图8所示。
SO42−和Cl−对PO43−生成量的影响如图7(a)和图7(b)所示。SO42−和Cl−投加量增多时,PO43−生成量也随之提高,PO43−生成量由未投加时的0.24 mmol·L−1分别提升至0.27和0.26 mmol·L−1。原因可能是在UV/H2O2体系中加入SO42−和Cl−,体系中可能会产生·SO4−或是活性氯等多种活性物种,从而对ATMP的氧化降解产生强化效果[24-26]。工业生产中常使用地下水作为冷却水源,其中固有的SO42−和Cl−可以起到强化UV/H2O2氧化降解ATMP的作用。
CH3COO−和NO3−对PO43−生成的影响结果如图7(c)和图7(d)所示。CH3COO−和NO3−投加量增多时,PO43−生成量随之下降,90 min的PO43−生成量由未投加时的0.24 mmol·L−1分别降低至0.05 mmol·L−1和0.16 mmol·L−1。分析原因为,CH3COO−能与H2O2反应,生成过氧乙酸和H2O,降低了UV/H2O2体系中H2O2的浓度,从而抑制了ATMP氧化降解为PO43−的过程。NO3−具有较强的光化学活性,在紫外线范围内具有很强的吸收能力,从而阻止了紫外光的透射[27]。此外,NO3−在紫外光解体系能够产生活泼的NO2·,这些NO2·能与有机分子结合产生硝基副产物,从而可抑制ATMP氧化降解为PO43−的过程[28]。
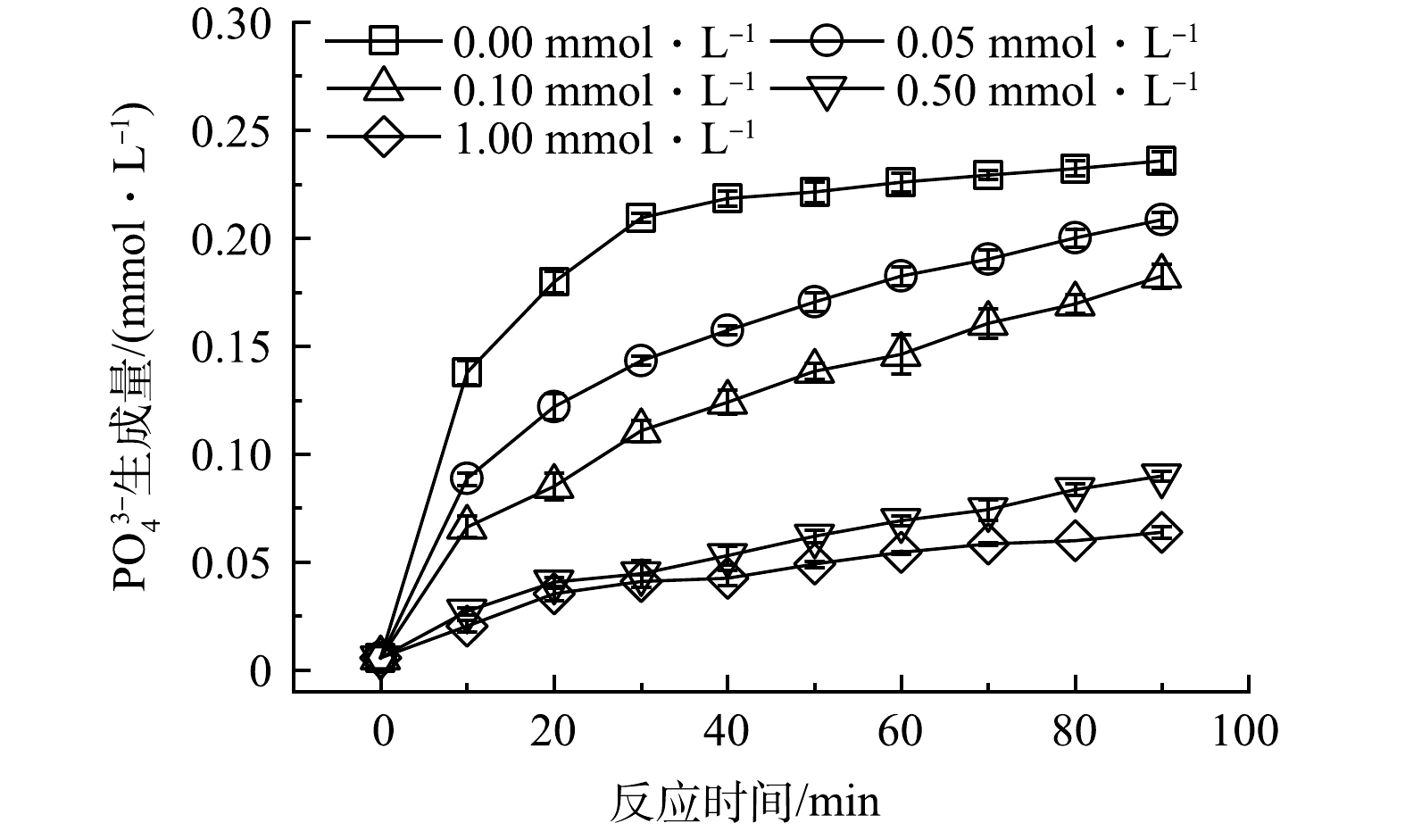
HA能显著抑制ATMP的氧化降解(图8),HA投加量为1.0 mmol·L−1时,PO43−生成量由0.24 mmol·L−1降低至了0.06 mmol·L−1。其它浓度的HA也都表现出抑制PO43−生成的效果。这是因为HA能够与ATMP竞争消耗反应体系产生的·OH等活性物种,抑制了PO43−的生成[29]。
-
为了探究UV/H2O2体系氧化降解ATMP的氧化机制,采用自由基淬灭实验定性分析体系中主要存在的活性物种,并进一步通过ESR测试进行验证。
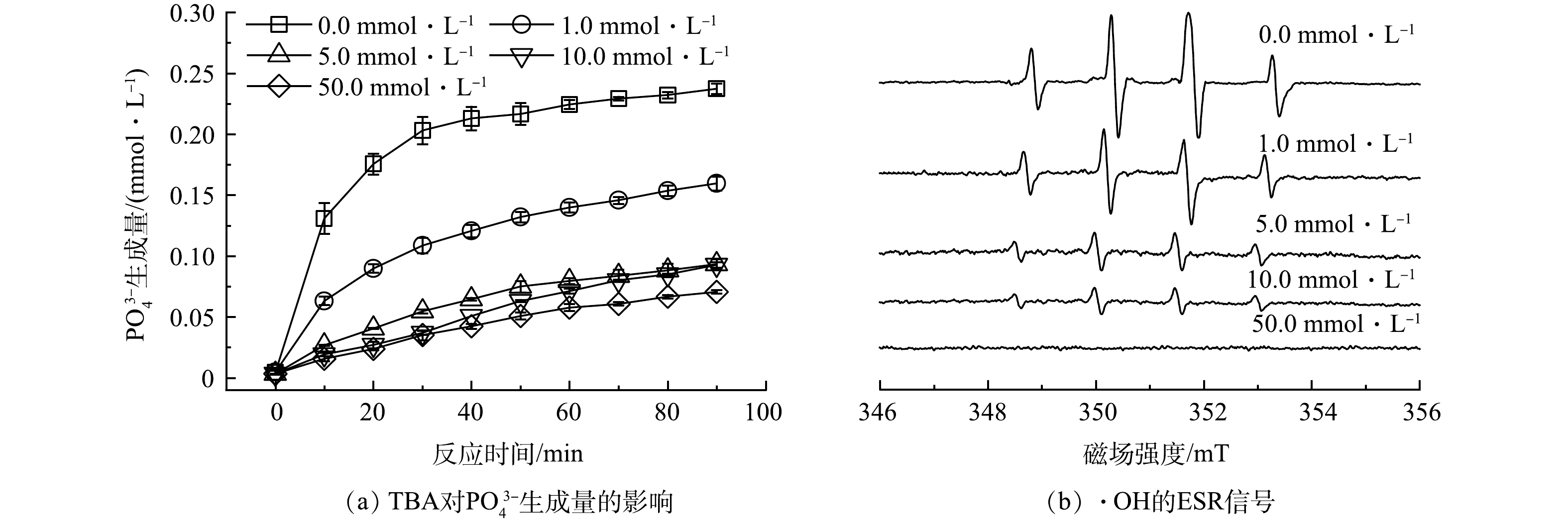
叔丁醇(TBA)是常用的·OH淬灭剂[30]。向反应溶液中加入不同浓度的TBA(0.0、1.0、5.0、10.0、50.0 mmol·L−1),观察其对ATMP氧化降解为PO43−的影响,结果如图9(a)所示。可见,不同浓度的TBA均对ATMP的氧化降解产生了抑制作用,在加入50.0 mmol·L−1的TBA淬灭剂时,90 min对应的PO43−生成量由未加入时的0.24 mmol·L−1降低至0.06 mmol·L−1。这表明UV/H2O2体系中的·OH是促使ATMP氧化降解的主要活性物种。
以DMPO为捕获剂,对体系中产生的含氧自由基进行捕获并进行ESR测试,结果如图9(b)所示。不同浓度的TBA投加量条件下,均仅出现了DMPO−·OH的特征峰信号,这表明体系中主要的活性物种为·OH。因此,在构建的UV/H2O2体系氧化降解ATMP的过程中,·OH是氧化ATMP的主要活性物种。
-
1)对比UV、H2O2和UV/H2O2体系对ATMP氧化降解为PO43−的效果,构建的UV/H2O2体系对ATMP具有较好氧化降解效果。
2)室温条件下,在UV 185 nm照射下,H2O2投加量为5.0 mmol·L−1,初始溶液pH为7.0时,反应90 min后,初始浓度为0.1 mmol·L−1的ATMP被氧化为PO43−的生成量可达0.24 mmol·L−1,对应的ATMP去除率为80%。
3)共存底物SO42−和Cl−对ATMP氧化降解存在促进作用;共存底物CH3COO−、NO3−和HA对ATMP氧化降解有显著抑制作用。
4)自由基淬灭和ESR分析证明,构建的UV/H2O2体系中·OH是实现ATMP氧化降解的主要活性物种。
5) ATMP氧化后形成正磷,可将该正磷废水进行pH调节,然后加入钙盐或铁盐等沉淀剂与正磷反应生成沉淀,将磷在废水中去除同时回收磷。以上研究结果可为含ATMP废水的处理提供参考。
UV/H2O2氧化降解废水中氨基三亚甲基膦酸性能及机理
Performance and mechanism of UV/H2O2 synergistic system on oxidation and degradation of amino trimethylene phosphonic acid
-
摘要: 本研究构建了以185 nm紫外光(ultraviolet,UV)为光源的UV活化过氧化氢(H2O2)体系(UV/H2O2),对工业废水中阻垢剂氨基三亚甲基膦酸(amino trimethylene phosphonic acid,ATMP)进行氧化降解。比较了UV、H2O2及UV/H2O2体系对ATMP的氧化降解效果。结果表明,UV/H2O2体系表现出较优的处理效果,ATMP被有效氧化降解为正磷酸盐(orthophosphate,PO43−)。考察了UV波长、溶液初始pH、H2O2浓度、反应温度、共存底物(SO42−、Cl−、NO3−、CH3COO−和腐殖酸(HA))对反应体系氧化ATMP为PO43−效率的影响。结果表明,室温条件下,初始浓度为0.1 mmol·L−1的ATMP(其中P浓度为0.3 mmol·L−1)在UV 185 nm照射下,H2O2 投加量为5.0 mmol·L−1,初始溶液pH为7.0时,反应90 min后,体系PO43−生成量可达0.24 mmol·L−1 ,对应的ATMP去除率为80%。共存底物SO42−、Cl−在较高浓度(>10.0 mmol·L−1)时对AMTP的氧化降解存在促进作用。共存底物NO3−、CH3COO−和HA对ATMP的氧化降解存在抑制作用,且这种抑制作用随其浓度增加而逐步增强。进一步研究了UV/H2O2体系氧化降解ATMP的反应机制。自由基淬灭实验证明,羟基自由基(·OH)是体系中氧化ATMP的主要活性物种,电子自旋共振波谱测试(ESR)分析结果进一步证明了这一点。以上研究结果可为含ATMP废水的处理提供参考。Abstract: In this study, an ultraviolet (UV) activated hydrogen peroxide (H2O2) system (UV/H2O2) with UV 185 nm as the light source was constructed to oxidize and degrade the scale inhibitor of amino trimethyl phosphonic acid (ATMP) in industrial wastewater. The oxidation and degradation of ATMP by UV, H2O2, and UV/H2O2 synergistic system were compared. The results showed that the UV/H2O2 system had a good performance on ATMP treatment and could effectively oxidize it to PO43−. The effects of UV wavelength, initial solution pH value, H2O2 concentration, reaction temperature, and coexisting substrates (SO42−, Cl−, NO3−, CH3COO−, and humic acid (HA)) on the oxidation efficiency of ATMP to PO43− were investigated. The results indicated that when the initial concentration of ATMP was 0.1 mmol·L−1, the dosage of H2O2 was 5.0 mmol·L−1, and the initial solution pH was 7.0, UV/H2O2 synergistic system could lead to the production of 0.24 mmol·L−1 PO43−, as well as 80% ATMP removal at ambient temperature and 90 min reaction. Coexisting substrates of SO42− and Cl− had a promoting effect on the oxidation of AMTP at high concentrations (>10.0 mmol·L−1). Coexisting substrates of NO3−, CH3COO−, and HA had inhibitory effects on the oxidation of ATMP, and this inhibitory effect gradually increased with increasing concentration. Furthermore, the reaction mechanism of UV/H2O2 oxidation of ATMP was studied. The radical quenching experiments proved that hydroxyl radicals (·OH) were the main active species for the oxidation of ATMP in the UV/H2O2 system, which was further confirmed by the analysis results of electron spin resonance (ESR) spectroscopy. The above research results can provide a reference for the treatment of wastewater containing ATMP.
-
随着市政污水处理量逐年递增,导致污泥产量及污泥处理压力也迅速增加。目前污泥的主要处置方式有卫生填埋、焚烧、建材利用和土地利用等。然而,污泥中含有大量有机质、氮磷钾等营养物质,有利于其资源化、能源化处理。利用城市污水厂污泥制备污泥活性炭是上世纪80年代出现的一种新型污泥资源化利用途径[1]。相比于传统的污泥处理处置方法,市政污泥经高温碳化和活化制备污泥炭,在热解过程中能够杀死污泥中的病原体、固定污泥中的重金属和碳元素,具有良好的环境效益和经济效益[2-3]。在过去十几年里,污泥碳化制备生物炭应用于有机污染物的吸附与降解已取得一些研究进展[4-6]。
厌氧颗粒污泥法能有效处理高浓度有机废水,例如啤酒废水、制药废水和煤化工废水等,其工艺具有效率高、成本低、操作方便等优势[7-8]。厌氧颗粒污泥是微生物自絮凝的结果,为疏松结构且含大量的营养物质。由于厌氧颗粒污泥中含有大量的微生物,且种类丰富,故通过微生物新陈代谢作用可以将金属很好的分散在颗粒污泥中。以无定型污泥(活性污泥、脱水污泥等)作为底物制备污泥炭,其成型过程费用较高[9]。而颗粒污泥在厌氧反应过程中自然成型,并在热解后保持颗粒状态。因此,厌氧颗粒污泥是制备污泥炭的一种潜在原料[10],但还缺乏相关研究报道。
抗生素广泛应用于人类和动物的疾病预防与治疗,抗生素废水含有多种难降解且具有生物毒性的物质,污水处理厂对抗生素的最高转化率仅为81%,低浓度的抗生素也可能对环境造成潜在的影响。而催化湿式过氧化氢氧化技术(catalytic wet peroxide oxidation,CWPO)是一种处理难降解有机物废水的有效方法,其具有反应条件温和、试剂无毒[5]的特点。
本研究以厌氧颗粒污泥为原料制备污泥炭催化剂,以第1类头孢类抗生素——头孢氨苄为模型污染物,在CWPO体系中对其进行了降解实验,在此过程中考察了颗粒污泥炭的催化性能和稳定性,同时分析了颗粒污泥炭的理化性质,检测了中间产物并提出了可能的降解途径。本研究可为污泥的资源化、能源化利用和抗生素废水的高效治理提供参考。
1. 材料与方法
1.1 颗粒污泥炭的制备
厌氧颗粒污泥取自山西省某淀粉废水厂的废水处理厌氧反应器,粒径2~3 mm。依次用超纯水和乙醇冲洗颗粒污泥,再自然晾干,之后置于烘箱中105 ℃处理3 h,待冷却后,用管式炉进行炭化处理,实验装置示意图参考文献中的方法[11]建立。制备条件为:在80 mL·min−1的N2气氛下,以3 ℃·min−1速率升温至800 ℃,焙烧3 h,待冷却后,记为未改性颗粒污泥炭GSC-O。用53.4% (质量分数)的H3PO4在25 ℃对GSC-O进行改性24 h,之后用超纯水冲洗至中性,记为改性颗粒污泥炭GSC-P。
1.2 实验方法
配置头孢氨苄废水模拟溶液100 mL,加入250 mL锥形瓶中,再投加颗粒污泥炭,置于设置好温度的水浴振荡器上,以120 r·min−1的速度振荡混合进行吸附实验。待吸附平衡后,加入一定的过氧化氢,进行CWPO催化降解反应,每隔一段时间进行取样并立即加入Na2SO3抑制反应进行,用0.45 μm滤膜过滤后分析头孢氨苄的转化率。分别考察温度、pH、过氧化氢投加量、催化剂投加量、反应物初始浓度和反应时间等因素对头孢氨苄降解过程的影响。反应条件为:温度60 ℃、pH为3、过氧化氢投加量为1.0 g·L−1、催化剂投加量为1.0 g·L−1、反应物初始浓度为100 mg·L−1和反应时间300 min。实验过程中,改变其中一种条件,研究其对头孢氨苄转化率和TOC去除率的影响。
1.3 检测方法
使用元素分析仪(EURO EA3000)和电感耦合等离子体发射光谱法(ICP-OES,Horiba Jobin-Y von)测定颗粒污泥炭的元素组成;使用美国麦克ASAP 2460型物理吸附仪测定污泥炭的比表面积和孔容积;使用Rigaku Utima IV型X射线衍射仪(XRD)分析晶型结构;使用ZEISS MERLIN型扫描电子显微镜(SEM)结合X射线能谱(EDS)和元素面分布技术(EDS-mapping)观察颗粒污泥及颗粒污泥炭的表观形貌特征和元素组成;使用电子顺磁共振技术(EPR)技术并以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO,C6H11NO)为自旋捕捉剂进行自由基检测;头孢氨苄采用液相色谱法(伍丰LC100高效液相色谱仪)进行测定,色谱柱:Bioband GP120-C18(250 mm×4.6 mm,5 μm),流动相A为甲醇,流动相B为超纯水,A∶B=40∶60 (v∶v),检测波长λ为254 nm,流速为1.0 mL·min−1,柱温为35 ℃,进样量20 μL;用Bruker Solarix 15T 傅立叶变换离子回旋共振质谱(FT-ICR MS)探测鉴定头孢氨苄降解中间产物,数据处理由软件DataAnalysis 4.2(Bruker, Daltonics GmbH, Bremen, Germany)完成。
2. 结果与讨论
2.1 颗粒污泥性质
1)颗粒污泥炭组成。由表1可知,颗粒污泥炭的产率较低,经过磷酸改性后,部分灰分被去除,产率进一步降低。颗粒污泥在灰分去除的同时形成多孔结构,颗粒污泥炭的比表面积由10.03 m2·g−1增加到96.21 m2·g−1。由此可见,经过改性后,颗粒污泥炭的比表面积和孔容积均得到改善。
表 1 颗粒污泥炭的元素组成、比表面积和孔隙特征Table 1. Element composition, specific surface area and porous structure of granular sludge based biochar样品 产率/% 元素含量/% 比表面积/(m2·g−1) 孔容积/(cm3·g−1) C H O N S Si Al Fe Ca GSC-O 36.95 35.01 1.88 15.17 1.75 0.86 12.28 2.35 6.06 9.87 10.03 0.035 GSC-P 17.32 29.68 2.33 16.34 2.03 1.32 15.19 1.74 5.44 1.36 96.21 0.116 | Show Table DownLoad:
CSV
DownLoad:
CSV
颗粒污泥中金属类物质较丰富,因此,其热解产物污泥炭中金属种类较多。由表1可知,GSC-O和GSC-P的含铁量分别为6.06%和5.44%。与脱水污泥制备的污泥炭含铁量(0.978%)相比[12-13],颗粒污泥炭的含铁量很高,而Fe又是催化剂中重要的活性组分,这有利于后续进行的头孢氨苄降解实验。
2)颗粒污泥炭表征。颗粒污泥炭的XRD分析结果如图1(a)所示。GSC-O具有一些明显的特征峰,位于28.73°、34.17°、47.25°和50.90°的峰属于Ca(OH)2(JCPDS 72-0156)的(100)、(011)、(012)和(110)晶面,位于32.24°、37.40°和53.93°的衍射峰是CaO(JCPDS 82-1690),位于18.29°、21.15°、37.07°和47.16°衍射峰是Fe3O4(磁性,JCPDS 79-0416)。TU等[14]的研究也表明污泥炭中存在Fe3O4,这是其具有磁性的重要原因。另外,位于31.13°和33.82°的峰是Fe2O3(JCPDS 40-1139)的晶面(113)和(116)。然而,GSC-P污泥炭的XRD谱图几乎没有明显特征峰,在13°~35°处的宽峰是一种典型的无定型结构[15]。这说明改性后污泥炭的晶体结构被破坏[14]。由图1(b)可知,虽然改性后磁性稍有降低,但GSC-P仍具有磁性,可被磁铁吸引,故GSC-P易实现回收和利用。
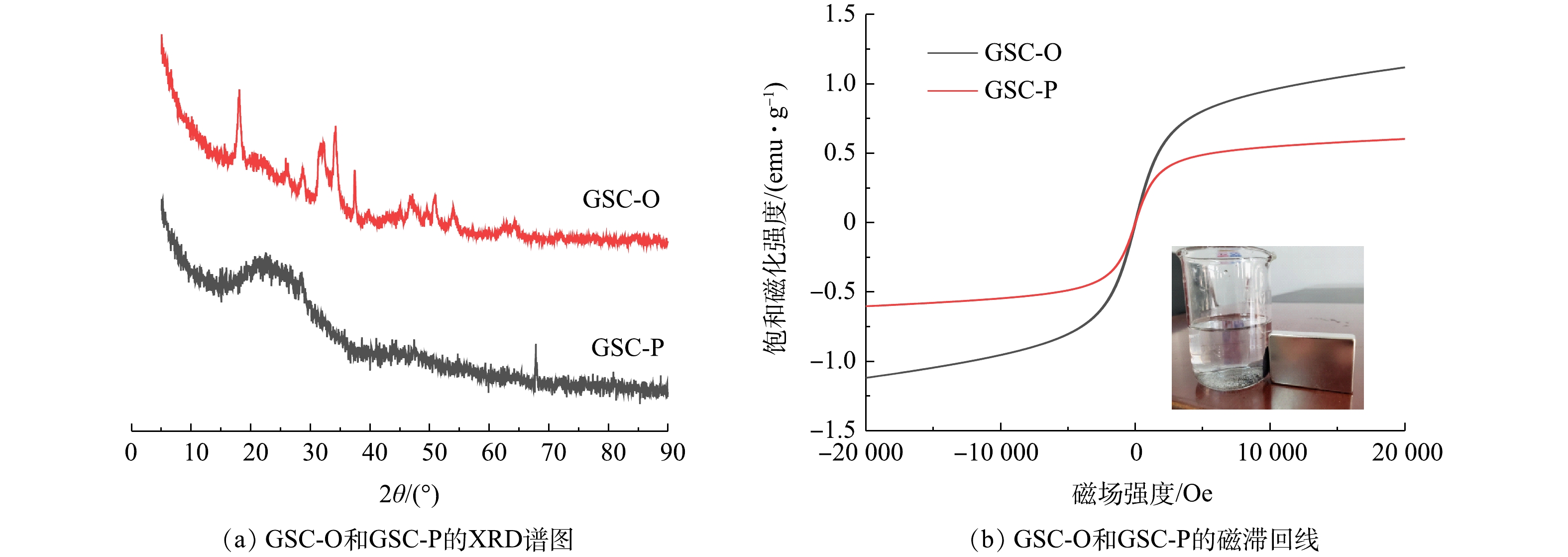 图 1 颗粒污泥炭GSC-O和GSC-P的XRD谱图和磁滞回线Figure 1. XRD patterns and Hysteresis loops of GSC-O and GSC-P
图 1 颗粒污泥炭GSC-O和GSC-P的XRD谱图和磁滞回线Figure 1. XRD patterns and Hysteresis loops of GSC-O and GSC-P颗粒污泥及污泥炭的表面特征如图2所示。与颗粒污泥外部(图2(a))相比,颗粒污泥内部(图2(b))更粗糙且微生物更丰富。颗粒污泥经过高温热解后,微生物破壁死亡,胞内有机物转化为生物炭基体。如图2(c)所示,未改性颗粒污泥炭GSC-O样品表面较光滑、孔结构不发达。有研究[16]采用物理改性和化学改性法对热解后的污泥炭进行改性处理,以提高污泥炭催化性能。由图2(d)可见,改性颗粒污泥炭GSC-P表面粗糙、疏松多孔,从而增大颗粒污泥炭的比表面积和孔隙率,这与表1中的结果一致。虽然GSC-P的含铁量略低于GSC-O,但对比表2可知,GSC-P污泥炭表面上的铁含量为GSC-O的2倍,有利于催化过氧化氢分解产生羟基自由基,从而提高头孢氨苄的降解效率。
 图 2 颗粒污泥和颗粒污泥炭的SEM和EDS元素面扫描图Figure 2. SEM and elemental surface scanning images of the granular sludge and granular sludge based biochar表 2 GSC-O和GSC-P的EDS元素面扫描结果Table 2. EDS elemental surface scanning results of GSC-O and GSC-P
图 2 颗粒污泥和颗粒污泥炭的SEM和EDS元素面扫描图Figure 2. SEM and elemental surface scanning images of the granular sludge and granular sludge based biochar表 2 GSC-O和GSC-P的EDS元素面扫描结果Table 2. EDS elemental surface scanning results of GSC-O and GSC-P样品 元素百分占比/% C N O Al Si P S Ca Fe GSC-O 55 1 13 2 2 8 3 13 3 GSC-P 47 2 14 2 2 8 6 13 6 | Show TableDownLoad:
CSV
图3是颗粒污泥炭的C1s和Fe2p的XPS谱图。为了分析污泥炭表面官能团的存在形态和含量,对XPS谱图进行了分峰处理。C1s可分为3种峰:石墨态碳(C—C,284.80 eV),酚羟基及醚类碳(C—O,286.00~286.20 eV),羧基及酯类碳(C=O,287.30~287.70 eV)[16]。由图3(a)可见,GSC-O中的C—C含量很高,占87.70%,而经过磷酸改性得到的GSC-P中,部分石墨态碳被氧化,C—C含量降低为55.29%,生成了C—O(36.47%)和C=O(8.25%)等含氧结构。污泥炭表面存在4种结合形态的Fe,即FeO(710.00~710.01 eV)、α-Fe2O3(711.50~712.04 eV)、γ-Fe2O3(722.95~723.47 eV)和Fe3O4(725.43~725.45 eV)[17]。虽然经磷酸改性得到的GSC-P中铁含量略低于GSC-O(表1),但其表面铁含量较多(图2(f)),在XPS谱图中峰强度较大(图3(b)),且Fe(II)所占比例较大(26.21%),有利于提高催化剂的催化活性。
2.2 催化降解过程中的影响因素
实验中探索了温度、pH、H2O2投加量、催化剂投加量和污染物浓度对头孢氨苄转化率的影响,结果如图4所示。前120 min进行吸附实验,不同条件下头孢氨苄的吸附去除率小于5%。颗粒污泥炭比表面积仅为10.03 m2·g−1和96.21 m2·g−1(表1),限制了其对污染物的吸附作用,因此,吸附作用对头孢氨苄的去除影响可忽略不计。
 图 4 温度、pH、H2O2、催化剂投加量和污染物浓度对头孢氨苄转化率和TOC去除率的影响Figure 4. Effects of temperature, pH, the dosage of H2O2 and catalyst and pollutant concentration on cephalexin conversion and TOC removal
图 4 温度、pH、H2O2、催化剂投加量和污染物浓度对头孢氨苄转化率和TOC去除率的影响Figure 4. Effects of temperature, pH, the dosage of H2O2 and catalyst and pollutant concentration on cephalexin conversion and TOC removal如图4(a)、图4(b)和图4(c)所示,随着温度的升高,2种催化剂对头孢氨苄的转化率逐渐升高。这是因为高温增加分子碰撞的概率,有利于·OH的产生[18]。当温度为60 ℃,GSC-P对头孢氨苄的转化率已高达90.2%,而GSC-O对头孢氨苄的转化率仅为23.9%。如图4(d)、图4(e)和图4(f)所示,随着pH由2升高至6,投加GSC-O和GSC-P的体系中头孢氨苄的转化率呈现下降趋势,说明酸性环境更有利于头孢氨苄的降解。当pH由4上升至6时,对于GSC-P催化剂,头孢氨苄的转化率由67.6%下降至43.7%。这是因为在酸性条件下,过氧化氢易分解产生羟基自由基,从而可促进头孢氨苄的降解[4, 19]。如图4(g)、图4(h)和图4(i)所示,当H2O2投加量为0.5 g·L−1时,GSC-O和GSC-P对头孢氨苄的转化率分别为19.5%和76.6%。当H2O2投加量增加为1.0 g·L−1时,GSC-P催化剂对头孢氨苄的转化率为90.2%,TOC去除率为53.9%,这是因为头孢氨苄降解生成了一些中间产物,并没有完全矿化。下文中的FT-ICR-MS结果也证实了这一结论。如图4(j)、图4(k)和图4(l)所示,随着GSC-P投加量增大,头孢氨苄的转化率增加。当GSC-P投加量达到1.0 g·L−1和1.5 g·L−1时,头孢氨苄转化率分别为90.2%和92.6%。这是因为催化剂投加量的提高,为反应提供更多的活性位点,促进·OH产生及头孢氨苄的降解[4]。如图4(m)、图4(n)和图4(o)所示,随着污染物浓度升高,头孢氨苄的转化率和TOC去除率降低。当污染物浓度为100 mg·L−1以下时,GSC-P对头孢氨苄的转化率高于89.6%,且TOC去除率大于53.5%。
2.3 反应动力学分析
降解过程的动力学分析结果如图5所示。头孢氨苄的催化降解过程分多个阶段,每个阶段均符合一级反应动力学特征。在120~150 min内,降解速率较快,符合一种一级反应动力学,头孢氨苄转化率约为45%;在150~270 min内,降解速率降低,进入另一种一级反应动力学,头孢氨苄转化率为89.6%;在270~300 min内,降解速率趋于平缓,头孢氨苄的转化率仅增加10%左右。
因此,综合考虑降解效率和经济等因素,在CWPO体系中,用磷酸改性的颗粒污泥炭GSC-P催化降解头孢氨苄对最佳条件为:温度为60 ℃、pH为3、过氧化氢投加量为1.0 g·L−1、催化剂投加量为1.0 g·L−1、反应物初始浓度为100 mg·L−1和反应时间300 min,此时,头孢氨苄的转化率为89.6%。而未改性颗粒污泥炭GSC-O对头孢氨苄的转化率仅为21%。由表1可知,GSC-O和GSC-P中的含铁量均较高,但GSC-O的催化活性却远低于GSC-P。由颗粒污泥炭的表征结果(表2和图3(b))可知,与GSC-O相比,GSC-P表面上的铁含量较多,且Fe(II)所占比例较大,表面疏松多孔,官能团如羟基、羧基等较丰富,这有利于提供催化反应的活性组分和活性位点。
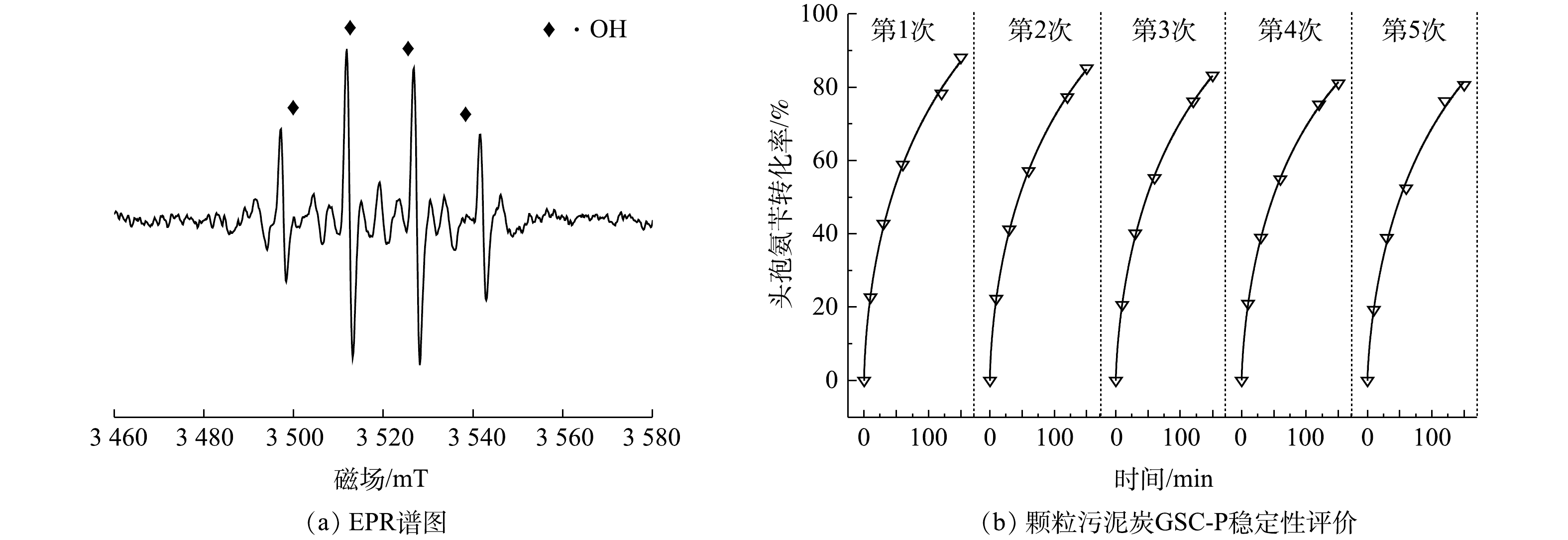
2.4 催化稳定性考察
以DMPO为自旋捕捉剂进行自由基检测,结果如图6(a)所示。可见,DMPO-OH特征峰比例为1:2:2:1,证明为·OH[20]。在最佳条件下,对颗粒污泥炭GSC-P进行了5次循环利用,基于催化作用得到的头孢氨苄转化率结果如图6(b)所示。头孢氨苄的转化率稳定维持在80%~88%。颗粒污泥炭的铁溶出率仅为0.83%,远低于一些文献中铁的溶出率(2.77%~11.5%)[12, 14, 17, 21]。上述结果表明,经磷酸常温改性的颗粒污泥炭具有较高的催化活性,可以重复使用,是一种催化性能和稳定性较高的非均相CWPO催化剂。与粉末状污泥炭相比,颗粒污泥炭有一定形状且呈现磁性更易回收,可以重复使用,从而避免了二次污染[5]。
2.5 头孢氨苄的降解途径
通过FT-ICR MS对头孢氨苄降解的中间产物进行了探测鉴定。根据检测的中间产物(表3),提出了降解途径。如图7所示,降解过程主要包括羟基化、去甲基化、脱羧和脱烷基等。头孢氨苄的甲基被·OH进攻氧化为羧基生成P1,再通过脱羧、羟基化使β-内酰胺环开环生成P2[22-23],P2上六元环连接的羧基碳失去1个氧原子得到P3,同时,P2通过脱羧和断键(C—N)反应生成P4和P5,之后生成苯甲酰甲酸(P6)、氨基乙酰胺(P7)、苯甲酸(P8)、丁烯(P9)、丁二酮(P10)等小分子有机物,由于TOC去除率为53.9%,所以部分中间产物进一步矿化生成无机小分子CO2、H2O等。HE等[24]的研究中也检测到P2、P3和P5。
表 3 FT-ICR MS分析头孢氨苄催化降解中间产物的结果Table 3. Intermediates identified by FT-ICR MS analysis during the cephalexin catalytic degradation产物 分子式 m/z检测值 m/z理论值 离子类型 — C16H17N3SO4 348.101 4 348.101 3 [M+H]+ P1 C16H15N3SO6 400.057 8 400.057 4 [M+Na]+ P2 C15H19N3SO7 386.101 9 386.101 6 [M+H]+ P3 C15H19N3SO6 370.107 3 370.106 7 [M+H]+ P4 C8H10N2O 151.086 7 151.086 6 [M+H]+ P5 C6H9NO3S 174.023 2 174.023 0 [M+H]+ P6 C8H6O3 173.020 3 173.020 9 [M+Na]+ P7 C2H6N2O 147.088 1 147.088 7 [2M-H]- P8 C7H6O2 123.044 4 123.044 1 [M+H]+ P9 C4H8 111.117 8 111.117 9 [2M-H]- P10 C4H6O2 171.066 3 171.066 3 [2M-H]- | Show TableDownLoad:
CSV
 图 7 颗粒污泥炭GSC-P催化头孢氨苄的降解途径Figure 7. Catalytic degradation pathway of cephalexin with GSC-P
图 7 颗粒污泥炭GSC-P催化头孢氨苄的降解途径Figure 7. Catalytic degradation pathway of cephalexin with GSC-P3. 结论
1)以厌氧颗粒污泥制备颗粒污泥炭后,通过磷酸改性可以有效提高其催化活性。改性后催化剂表面铁含量增大,催化剂比表面积和孔容积增大,表面官能团较为丰富,有利于催化湿式过氧化氢氧化反应。
2)综合考虑降解效率和经济等因素,在CWPO体系中其对头孢氨苄的反应条件宜为:温度60 ℃、pH为3、过氧化氢投加量为1.0 g·L−1、催化剂投加量为1.0 g·L−1、反应物初始浓度为100 mg·L−1和反应时间300 min,在此条件下头孢氨苄的转化率为89.6%。
3) GSC-P催化稳定性较高,在反复使用5次后,活性组分Fe的溶出率很低,仅为0.83%,头孢氨苄的转化率稳定在80%~88%。颗粒污泥炭具有一定形状且呈现磁性易回收,可以重复使用,可避免二次污染。
4)头孢氨苄的降解是通过羟基化、去甲基化、脱羧和脱烷基等过程生成小分子有机物,再进一步矿化生成CO2和H2O等无机物。
-
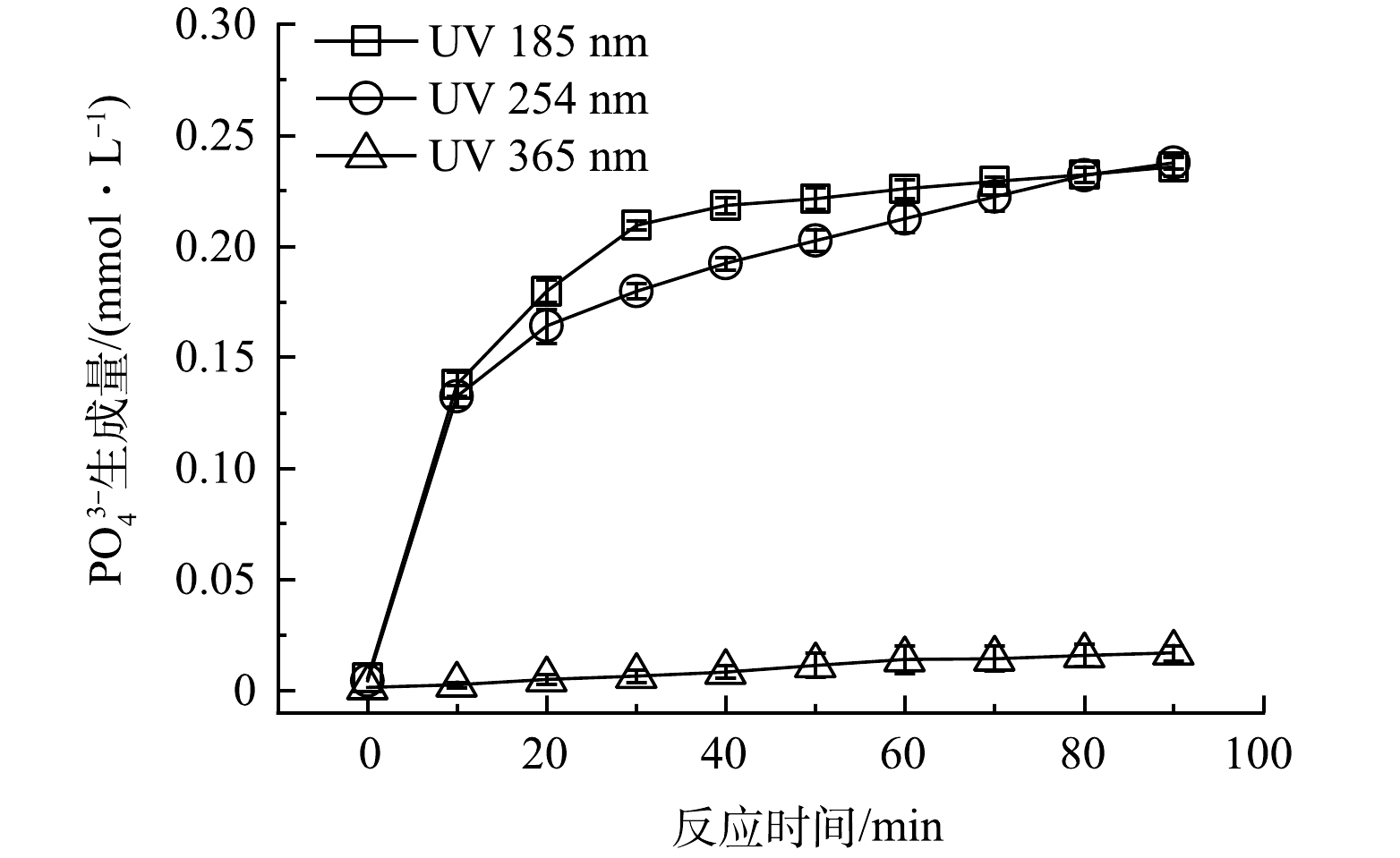
图 3 不同紫外光波长对PO43−生成量的影响
Figure 3. Effect of different ultraviolet wavelength on the production of PO43−
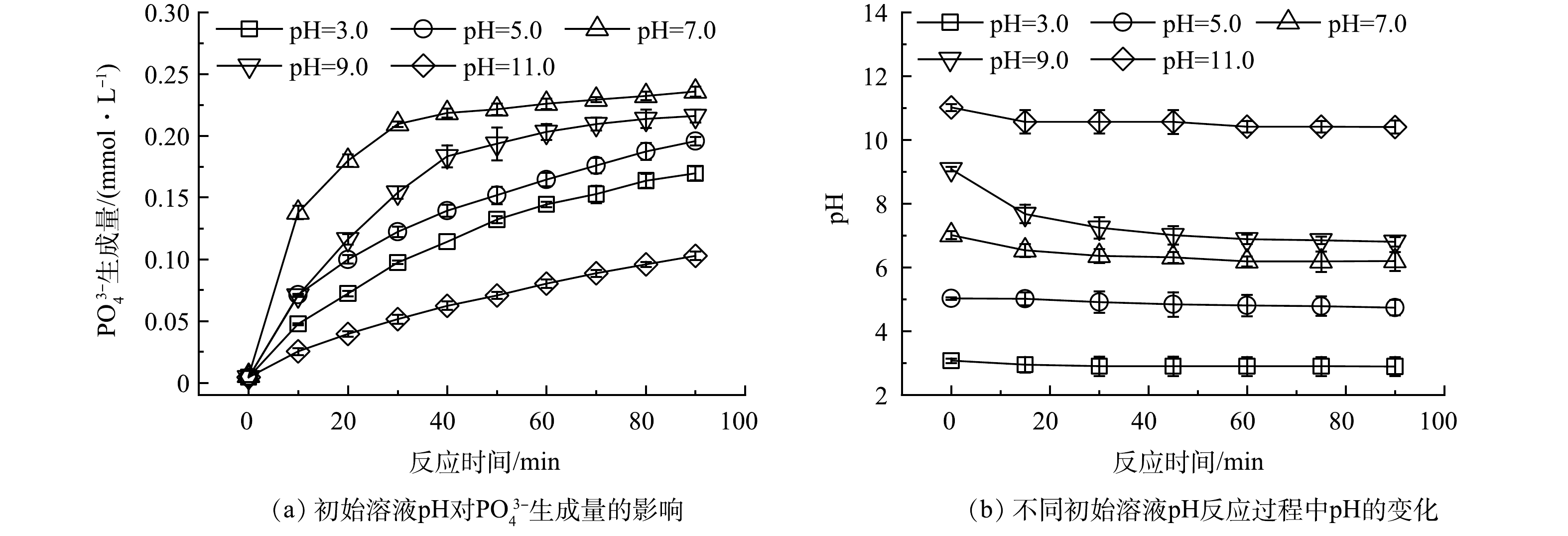
图 5 初始溶液pH对PO43−生成量的影响
Figure 5. Effect of initial solution pH on the production of PO43−
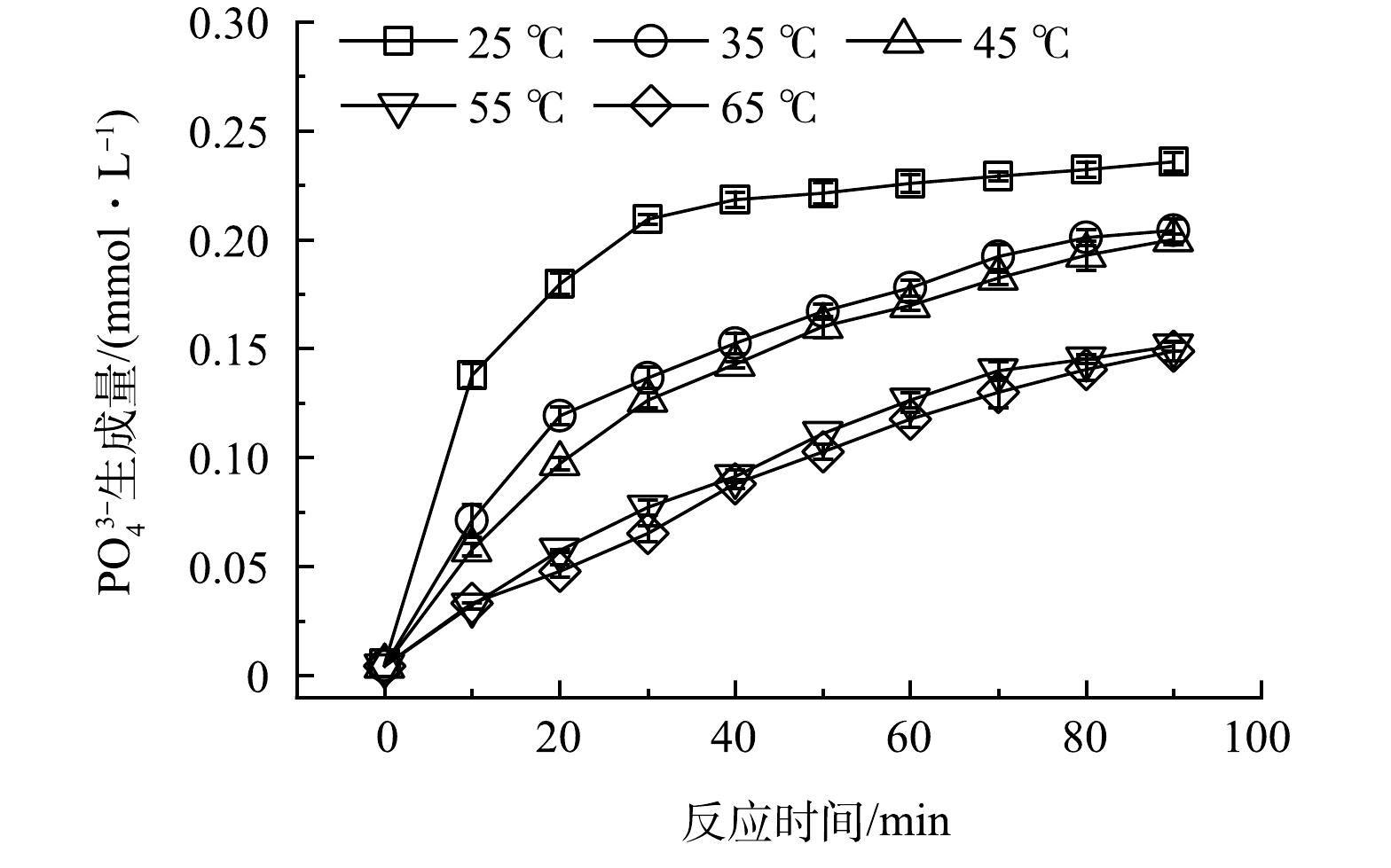
图 6 不同反应温度对PO43−生成量的影响
Figure 6. Effects of reaction temperature on the production of PO43−
-
[1] ADENUSI H, CHASS G, BODO E. Theoretical insights into the structure of the aminotris(methylenephosphonic acid) (ATMP) anion: A possible partner for conducting ionic media[J]. Symmoletry, 2020, 12(6): 920-935. [2] ROTT E, SCHöNBERGER H, MINKE R, et al. Batch studies of phosphonate adsorption on granular ferric hydroxides[J]. Water Science and Technology, 2020, 81(1): 10-20. doi: 10.2166/wst.2020.055 [3] 艾思洁. 典型有机磷阻燃剂对铜绿微囊藻生长的影响及机理研究[D]. 武汉: 中南民族大学, 2022. [4] ZHANG X, LI J, FAN W Y, SHENG G P. Photomineralization of effluent organic phosphorus to orthophosphate under simulated light illumination[J]. Environmental Science & Technology, 2019, 53(9): 4997-5004. [5] WANG S, QIAN J, ZHANG B, et al. Unveiling the occurrence and potential ecological risks of organophosphate esters in municipal wastewater treatment plants across china[J]. Environmental Science & Technology, 2023, 57(5): 1907-1918. [6] ROTT E, STEINMETZ H, METZGER J W. Organophosphonates: A review on environmental relevance, biodegradability and removal in wastewater treatment plants[J]. Science of the Total Environment, 2018, 615: 1176-1191. doi: 10.1016/j.scitotenv.2017.09.223 [7] WANG Z, CHEN G, PATTON S, et al. Degradation of nitrilotris-methylenephosphonic acid (NTMP) antiscalant via persulfate photolysis: Implications on desalination concentrate treatment[J]. Water Research, 2019, 159: 30-37. doi: 10.1016/j.watres.2019.04.051 [8] LEI Y, SAAKES M, VAN DER WEIJDEN R D, BUISMAN C J N. Electrochemically mediated calcium phosphate precipitation from phosphonates: Implications on phosphorus recovery from non-orthophosphate[J]. Water Research, 2020, 169: 115206. doi: 10.1016/j.watres.2019.115206 [9] ROTT E, MINKE R, STEINMETZ H. Removal of phosphorus from phosphonate-loaded industrial wastewaters via precipitation/flocculation[J]. Journal of Water Process Engineering, 2017, 17: 188-196. doi: 10.1016/j.jwpe.2017.04.008 [10] LIU Y, LU S, YANG Q, LI C. Adsorption of phosphonate antiscalant HEDP from reverse osmosis concentrates by La/FeOOH@PAC[J]. Journal of Inorganic Materials, 2021, 36(8): 841-846. doi: 10.15541/jim20200512 [11] FAN W Y, ZHANG X, GUO P C, SHENG G P. Highly efficient removal of phosphonates by ferrate-induced oxidation coupled with in situ coagulation[J]. Journal of Hazardous Materials, 2023, 451: 131104. doi: 10.1016/j.jhazmat.2023.131104 [12] MIKLOS D B, REMY C, JEKEL M, et al. Evaluation of advanced oxidation processes for water and wastewater treatment – A critical review[J]. Water Research, 2018, 139: 118-131. doi: 10.1016/j.watres.2018.03.042 [13] 沈文华. 基于电生过氧化氢的高级氧化技术降解典型新兴污染物[D]. 北京: 清华大学, 2017. [14] SUN S, SHAN C, YANG Z, et al. Self-enhanced selective oxidation of phosphonate into phosphate by Cu(II)/H2O2: Performance, mechanism, and validation[J]. Environmental Science & Technology, 2021, 56(1): 634-641. [15] XUE G, ZHENG M, QIAN Y, et al. Comparison of aniline removal by UV/CaO2 and UV/H2O2: Degradation kinetics and mechanism[J]. Chemosphere, 2020, 255: 126983. doi: 10.1016/j.chemosphere.2020.126983 [16] BOKHARI T H, AHMAD N, JILANI M I, et al. UV/H2O2, UV/H2O2/SnO2 and Fe/H2O2 based advanced oxidation processes for the degradation of disperse violet 63 in aqueous medium[J]. Materials Research Express, 2020, 7(1): 015531. doi: 10.1088/2053-1591/ab6c15 [17] ZHONG Q, CHEN F, LI X, et al. Optimal degradation of typical phosphonate antiscalant in saline water in UV/electrochemical oxidation system: Kinetics and mechanism[J]. Journal of Water Process Engineering, 2023, 53: 103806. doi: 10.1016/j.jwpe.2023.103806 [18] ZHANG Y L, WANG W L, LEE M Y, et al. Promotive effects of vacuum-UV/UV (185/254 nm) light on elimination of recalcitrant trace organic contaminants by UV-AOPs during wastewater treatment and reclamation: A review[J]. Science of The Total Environment, 2022, 818: 151776. doi: 10.1016/j.scitotenv.2021.151776 [19] LIU Y, WU J, CHENG N, et al. The overlooked role of UV185 induced high-energy excited states in the dephosphorization of organophosphorus pesticide by VUV/persulfate[J]. Chemosphere, 2023, 334: 138993. doi: 10.1016/j.chemosphere.2023.138993 [20] Guo Z J, Wang K F, Liu M L, et al. Enhanced electrochemical harmless removal of ammolonia nitrogen and simultaneously recovery of phosphorus with peroxydisulfate activated by Fe inductive electrode[J]. Separation and Purification Technology, 2024, 343: 126918. doi: 10.1016/j.seppur.2024.126918 [21] 许盛彬. 活性炭负载纳米零价铁诱发芬顿反应降解甲基橙的研究[D]. 黑龙江: 哈尔滨工业大学, 2015. [22] HUO Y, LI M, AN Z, et al. Effect of pH on UV/H2O2-mediated removal of single, mixed and halogenated parabens from water[J]. Journal of Hazardous Materials, 2024, 462: 132818. doi: 10.1016/j.jhazmat.2023.132818 [23] SRITHEP S, PHATTARAPATTAMAWONG S. Kinetic removal of haloacetonitrile precursors by photo-based advanced oxidation processes (UV/H2O2, UV/O3, and UV/H2O2/O3)[J]. Chemosphere, 2017, 176: 25-31. doi: 10.1016/j.chemosphere.2017.02.107 [24] GUO K, WU Z, CHEN C, FANG J. UV/Chlorine process: An efficient advanced oxidation process with multiple radicals and functions in water treatment[J]. Accounts of Chemical Research, 2022, 55(3): 286-297. doi: 10.1021/acs.accounts.1c00269 [25] 刘永泽. 高级氧化过程中OH·和SO4·−定量分析及溴代副产物生成规律研究[D]. 黑龙江: 哈尔滨工业大学, 2015. [26] OH W D, DONG Z, LIM T T. Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: Current development, challenges and prospects[J]. Applied Catalysis B: Environmental, 2016, 194: 169-201. doi: 10.1016/j.apcatb.2016.04.003 [27] GU D M, GUO C S, FENG Q Y, et al. Degradation of ketamine and methamphetamine by the UV/H2O2 system: Kinetics, mechanisms and comparison[J]. Water, 2020, 12(11): 2999. doi: 10.3390/w12112999 [28] GAO X, ZHANG Q, YANG Z, et al. Formation of nitrophenolic byproducts during UV-Activated peroxydisulfate oxidation in the presence of nitrate[J]. ACS ES& T Engineering, 2022, 2(2): 222-231. [29] MUTKE X A M, DREES F, LUTZE H V, SCHMIDT T C. Oxidation of the N-containing phosphonate antiscalants NTMP and DTPMP in reverse osmosis concentrates: Reaction kinetics and degradation rate[J]. Chemosphere, 2023, 341: 139999. doi: 10.1016/j.chemosphere.2023.139999 [30] WANG L, LI B, DIONYSIOU D D, et al. Overlooked formation of H2O2 during the hydroxyl radical-scavenging process when using alcohols as scavengers[J]. Environmental Science & Technology, 2022, 56(6): 3386-3396. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1268
- HTML全文浏览数: 1268
- PDF下载数: 29
- 施引文献: 0
