

-
高铁酸盐(Fe(Ⅵ))具有较高的氧化电位[1],对含酚类和胺类等官能团的微污染物(micropollutants, MPs)具有较好的去除效果。Fe(Ⅵ)反应后的副产物Fe3+可水解成Fe(OH)3 (pKa1=2.2)[2-3],具有一定的絮凝吸附能力[4-5]。虽然Fe(Ⅵ)在水和废水处理的预氧化工艺中具有较大的应用潜力[6-7],但其作为一种选择性的氧化剂,对苯扎贝特、布洛芬等物质降解效果较差[8-10]。
碳纳米管(carbon nanotubes, CNT)作为一种典型一维纳米材料,具有结构稳定[11]、比表面积大等优点[12-13]。相较于活性炭等无序结构碳材料,CNTs具有较大的催化活化潜力和效率,可通过吸附协同氧化过程提高Fe(Ⅵ)对溴酚等的去除效能[14]。苯扎贝特(bezafibrate, BZF)作为广泛使用的降血脂药物,在水环境中的质量浓度可达57.2~4 600 ng·L−1[15-16],饮用水中的质量浓度可达0.8~270 ng·L−1[17],经由氧化途径降解BZF可降低其毒性效应并有效保障水质安全[18]。虽然BZF具有酰胺基团,但Fe(Ⅵ)难以有效降解BZF,其二级反应速率常数小于1 L·(mol·s)−1 [8,19]。
由此,本研究选取环境中广泛存在的、难以被Fe(Ⅵ)降解的BZF作为模型化合物,研究在CNT存在下,Fe(Ⅵ)对BZF的降解效果及反应动力学,鉴定了CNT对Fe(Ⅵ)的强化氧化过程中的活性物种。
-
本研究使用的碳材料有活性炭粉末(activated carbon, AC)、碳纳米管(CNTs)和氧化石墨烯(graphene oxide, GO)。其中,4种不同类型CNT的相关指标如表1所示。GO的单层率为99%,片径为0.5~5 μm,厚度为0.8~1.2 nm。所有溶液均使用来自Duna纯化系统的超纯水(18.2 MΩ·cm−1)配制。液相色谱使用的甲醇、甲酸和乙腈均为色谱纯。
AC(200目)、高铁酸钾(K2FeO4,Fe(Ⅵ),95%)、苯扎贝特(BZF,99%)、甲基苯基亚砜(PMSO,98%)、甲基苯基砜(PMSO2,98%)、2,2'-联氨-双(3-乙基苯并噻唑啉-6-磺酸)二胺盐(ABTS)、硼酸、硫代硫酸钠、十水合四硼酸钠、硫代硫酸钠、磷酸二氢钠、氯化钠、硫酸钠、硝酸钠、硅酸钠和碳酸氢钠均为分析纯。
-
本研究使用硼酸盐缓冲液(pH=7.6,5 mmol·L−1)测定Fe(Ⅵ)降解BZF的反应速率常数。反应条件为BZF(1 µmol)、CNT(4 mg·L−1)和Fe(Ⅵ)(20 μmol·L−1),在(21±1) °C下反应15min并使用Na2S2O3(60 mmol·L−1, 50 μL)和NaH2PO3(50 mmol·L−1, 50 μL)淬灭,通过0.22 μm 聚醚砜滤膜过滤后使用液相色谱进行分析(HPLC,Vanquish,赛默飞)和Supersil ODS2 色谱柱(100 mm×2.1 mm,3 μm,依利特)测定MPs的浓度。Fe(Ⅵ)储备液溶液使用硼酸盐溶液(5 mmol·L−1,pH=9.2)配制,在510 nm波长下使用紫外可见分光光度计(Cary 60,安捷伦)标定Fe(Ⅵ)浓度,并在5 min内使用。反应后剩余Fe(Ⅵ)使用ABTS法在415 nm下测定[20]。所有实验重复2次。
-
有机微污染物采用液相色谱进行分析(HPLC,Vanquish,赛默飞)配有紫外检测器和Supersil ODS2 色谱柱(100 mm×2.1 mm,3 μm,依利特)。其中BZF浓度在甲醇∶水=60∶40,流速0.4 mL·min−1,230 nm波长下测定。PMSO和PMSO2浓度在乙腈∶水=10∶90,流速0.2 mL·min−1,220 nm波长下测定。
-
降解产物采用飞行时间质谱LC-QToF-MS/MS(X500R,AB SCIEX)和C18色谱柱(2.1 mm×100 mm,3 μm,艾杰尔)进行鉴定。飞行时间质谱采用信息依赖采集(IDA)和ESI负离子模式。实验条件为BZF(10 μmol·L−1)、CNT(0或4 mg·L−1)、Fe(Ⅵ)(20 μmol·L−1)、Na2S2O3(600 mmol·L−1,500 μL)、NaH2PO3(5 mmol·L−1,500 μL)和喷雾电压为4500 V,m/z为50~400。
-
碳材料质量浓度为200 mg·L−1、BZF浓度为50 μmol·L−1、温度为(21±1) °C时,考察了碳纳米管(CNTs)、氧化石墨烯(GO)和粉末活性炭(AC)对Fe(Ⅵ)降解效能的影响。从图1(a)可以看出,CNT、AC、GO对BZF的吸附分别在5、15、10 min时达到平衡,此时CNT、AC、GO对BZF的去除率分别为7%、25%、94%。在CNT、AC、GO存在条件下,Fe(Ⅵ)对BZF的去除率分别为69%、35%、12%(图1(b)),表明CNT可强化Fe(Ⅵ)对BZF的去除。尽管GO对BZF的吸附去除率较高,但是在Fe(Ⅵ)氧化条件下GO对BZF的去除效果较差。这是由于GO 对BZF的吸附取决于GO表面官能团(如:环氧、羟基、酮类、酯类、有机硫酸盐和乳酸结构)与BZF之间的相互作用,Fe(Ⅵ)的强氧化性改变了GO的表面形貌与官能团[21],进而降低了GO对BZF的吸附效能。已有研究表明,碳材料(如AC、CNT)具有还原性,可还原金属离子(Fe(Ⅲ)、Cu(Ⅱ))[22-24]。鉴于CNT的还原特性,CNT会与Fe(Ⅵ)快速反应生成Fe(Ⅴ)[25],从而导致MPs的降解增强。所以CNT对Fe(Ⅵ)降解BZF的促进效果,可能是由于Fe(Ⅵ)在CNT表面生成了氧化性更强的Fe(Ⅴ)。
-
如图2(a)所示,在1 μmol·L−1 BZF、4 mg·L−1 CNTs、20 μmol·L−1 Fe(Ⅵ)以及pH=7.6、温度为(21±1) ℃的条件下,相似孔径的多壁碳纳米管(CNT)、羧基多壁碳纳米管(CNT-COOH)、羟基多壁碳纳米管(CNT-OH)和石墨化多壁碳纳米管(GCNT)对Fe(Ⅵ)降解BZF的去除率分别为69%、17%、47%、28%。在未加入CNT时,Fe(Ⅵ)降解BZF的表观反应速率常数小于1 L·(mol·s)−1,与文献报道的结果[8,19]一致。CNT-OH对Fe(Ⅵ)强化降解BZF的反应速率(115 L·(mol·s)−1)与CNT(147 L·L·(mol·s)−1)类似,说明强化降解过程与CNT表面的羟基官能团有关。此外,CNTs表面的官能团可以促进Fe(Ⅵ)的消耗(图2(b)),未加入CNTs时,测得反应终点的剩余Fe(Ⅵ)浓度为10.9 μmol·L−1。加入CNTs后,测得剩余Fe(Ⅵ)浓度均有不同程度的下降。说明 CNTs均可以促进Fe(Ⅵ)的消耗,并可能生成具有更强氧化能力的物种(如Fe(Ⅴ)),从而提高了BZF的降解速率。GCNT经过高温处理后其表面的活性官能较少,CNT-COOH表面主要为羧基官能团,CNT-COOH和GCNT较差的强化效果说明降解效果与CNTs表面官能团类型相关。未经过特定官能团修饰的和经过羟基修饰的CNTs对Fe(Ⅵ)的强化效果最好,说明强化氧化作用与羟基官能团有关,并且与其他官能团存在耦合协同作用。
-
1) pH的影响。在实际水处理条件下(pH=6~8),多壁碳纳米管(CNT)协同Fe(Ⅵ)对BZF的去除率随pH的降低而升高(图3(a))。在pH=8.0和pH=6.0下,CNT协同Fe(Ⅵ)对BZF的去除率从46%提高到88%,增长了1.91倍。根据图3(b)可知,pH从8.0下降到6.0,反应速率常数从89 L·(mol·s)−1提升到415 L·(mol·s)−1,增长了4.7倍。随着pH的降低,CNT协同Fe(Ⅵ)降解BZF的反应速率常数逐渐增大,酸性条件下HFeO4−比FeO42−具有更强的氧化能力,有利于Fe(Ⅵ)在CNT的协同下对BZF快速氧化。
2)磷酸盐的影响。由图3(c)可知,在5 mmol·L−1的磷酸盐和硼酸盐缓冲溶液中,Fe(Ⅵ)对BZF的去除率分别为0.9%和1.4%,反应速率常数均小于1 L·(mol·s)−1,这与其他研究结果基本一致[26]。相比于使用磷酸盐缓冲溶液,硼酸盐中Fe(Ⅵ)对BZF的去除率更高。CNT存在条件下,在硼酸盐和磷酸盐缓冲溶液中CNT协同Fe(Ⅵ)对BZF的去除率分别为69%和38%,此时二者的反应速率常数分别为147 L·(mol·s)−1和76 L·(mol·s)−1,使用硼酸盐缓冲溶液的反应速率常数是磷酸盐缓冲溶液的1.9倍。由于磷酸盐缓冲液中PO43−与Fe(Ⅲ)的络合作用,导致Fe(Ⅵ)氧化性的降低[27-28],所以PO43−对CNT协同Fe(Ⅵ)降解BZF存在明显的抑制作用,PO43−和Fe3+(Ksp= 9.91×10−16)络合导致Fe3+诱导生成的活性物种Fe(Ⅴ)减少,抑制Fe(Ⅵ)的氧化性,不利于Fe(Ⅵ)氧化去除有机污染物[29]。
3)共存阴离子的影响。除磷酸盐以外,其他共存阴离子同样对Fe(Ⅵ)的氧化效能会产生影响[30]。Cl−、SO42−、NO3−、SiO32−、HCO3−等是水处理过程中常见的阴离子。由图3(d)可知,Cl−、SO42−、NO3−、SiO32−、HCO3−均会不同程度地抑制CNT协同Fe(Ⅵ)对BZF的降解,在这些离子存在时,BZF的去除率分别降低至59%、67%、63%、48%、55%。共存阴离子对CNT协同Fe(Ⅵ)体系的影响由大到小为PO43−>SiO32−>HCO3−>Cl−>NO3−>SO42−。类似于PO43−对Fe3+的络合作用,SiO32−也可络合Fe3+,HCO3−可促进Fe3+水解,这些阴离子可促进溶液中的Fe3+向含铁沉淀转化,抑制Fe(Ⅴ)的生成并导致氧化能力降低[31-32]。
-
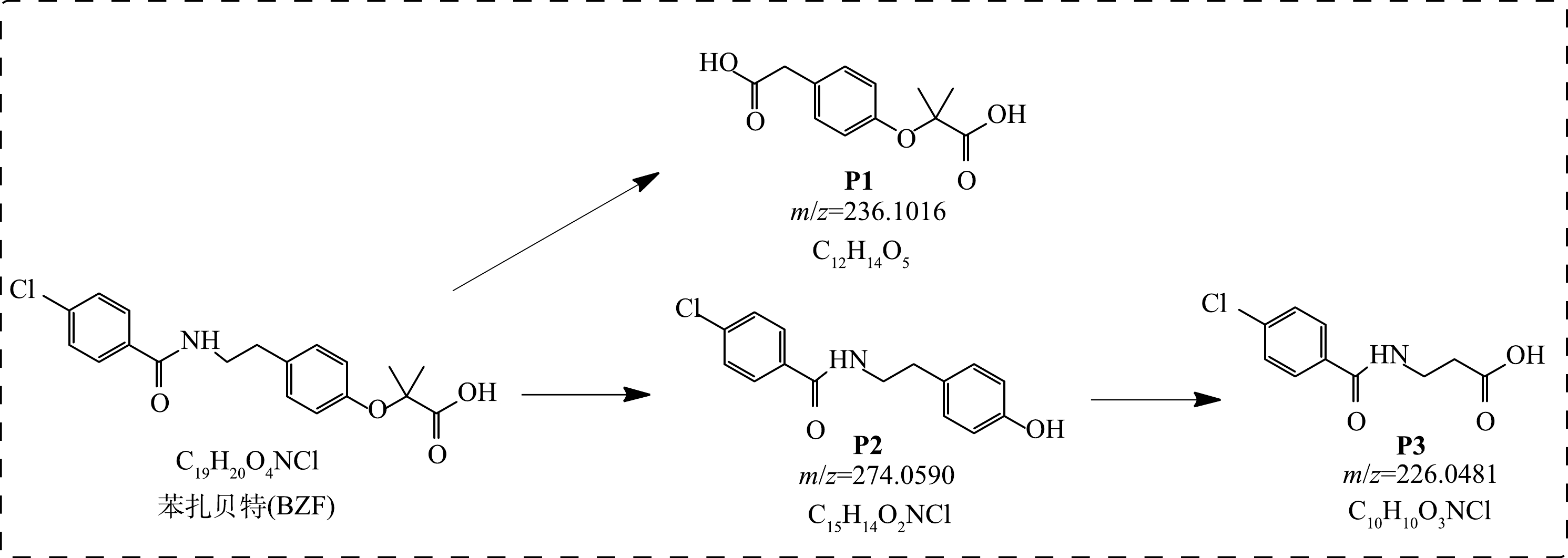
本节对CNT协同Fe(Ⅵ)去除BZF的机理进行了研究。使用LC-QToF-MS基于IDA模式鉴别出7种BZF的转化产物,其中P1、P2、P3是Fe(Ⅵ)降解BZF的产物,如图4所示,Fe(Ⅵ)降解BZF主要有以下2种途径:一是BZF酰胺水解的胺基产物进一步氧化生成产物P1,二是BZF通过羟基化形成酚类产物P2,并进一步经过苯环氧化成羧基产物P3。如图5所示,不同于单独使用Fe(Ⅵ)氧化降解BZF,CNT协同Fe(Ⅵ)降解BZF会生成4种产物P4、P5、P6、P7:BZF经过羟基化和甲基丙酸基团断裂产生3种同分异构体,即产物P4。产物P4经过羟基化、苯环氧化开环和羟基消去反应形成氧化产物P5。同时,P4也会通过羰基化、脱羰形成产物P6,再经过碳氮键裂解生成产物P7。在O3氧化降解BZF的过程中也发现了类似的氧化降解产物P4、P5、P6、P7[29],说明CNT强化了Fe(Ⅵ)的氧化性,将先水解后氧化的反应途径改变为氧化开环的反应途径,进而加快了其反应速率。
-
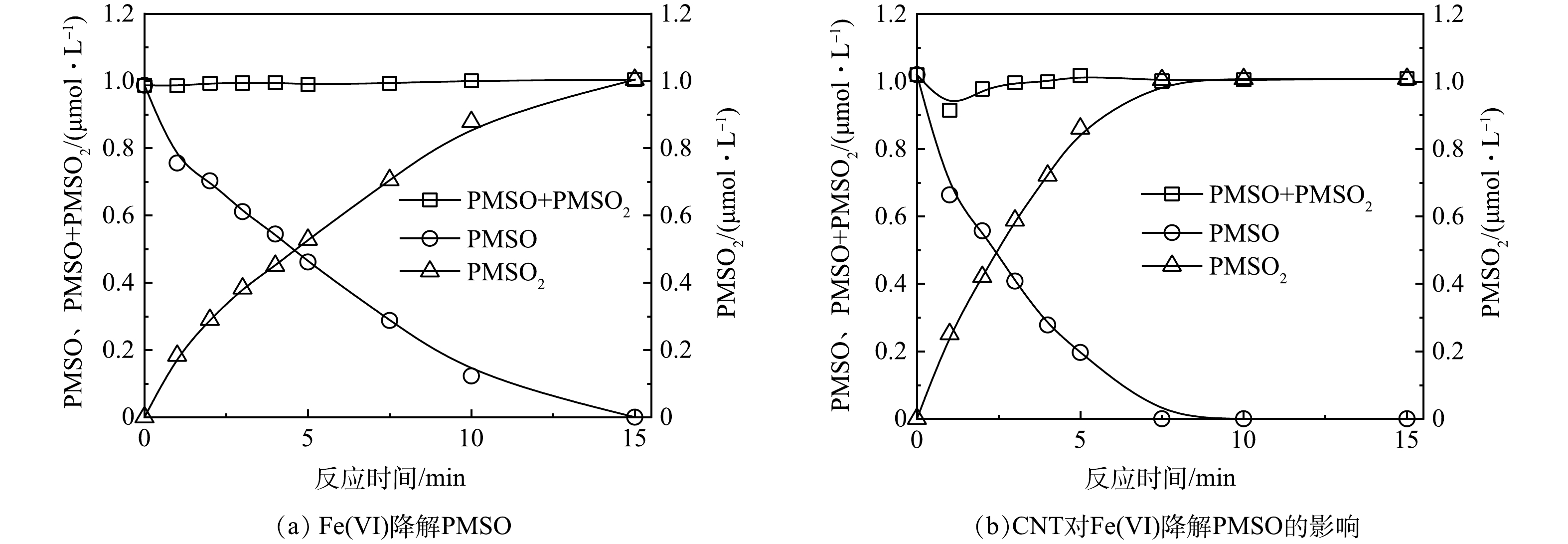
甲基苯基亚砜(PMSO)不易被Fe(Ⅵ)氧化,但易被Fe(Ⅴ)氧化生成甲基苯基砜(PMSO2)。在硼酸盐缓冲液中,Fe(Ⅴ)对PMSO氧化生成PMSO2的贡献率高达99%[29]。在pH为8.0的硼酸盐缓冲溶液中,Fe(Ⅵ)氧化PMSO的反应速率常数约为30 L·(mol·s)−1,远低于Fe(Ⅴ) (kapp = 1.52×107 L·(mol·s)−1)[33]。因此,探针化合物甲基苯基亚砜(PMSO)可用于识别Fe(Ⅵ)在氧化过程中的铁物种贡献。由图6(a)可知,在硼酸盐缓冲溶液中使用Fe(Ⅵ)氧化PMSO,5 min时ΔPMSO与ΔPMSO2的比值为1,即PMSO的半衰期约5 min。在CNT协同下(图6(b)),2.5 min时ΔPMSO与ΔPMSO2的比值为1,即PMSO的半衰期缩短至约2.5 min,说明CNT促进了Fe(Ⅴ)主导的氧化过程。且PMSO与PMSO2的总浓度均为1 μmol·L−1,说明PMSO仅被氧化为PMSO2,没有其他降解过程存在。由于Fe(Ⅴ)是PMSO氧化为PMSO2的关键活性物质[14, 34-35],所以CNT促进了Fe(Ⅵ)向Fe(Ⅴ)的转化,可有效提高Fe(Ⅵ)与BZF的反应速率。
-
1) CNT可协同Fe(Ⅵ)强化降解BZF,将表观二级反应速率由低于1 L·(mol·s)−1提升至147 L·(mol·s)−1,强化降解效应受CNT表面羟基官能团诱导。
2)酸碱度、磷酸盐以及阴离子均会影响CNT对Fe(Ⅵ)氧化能力的强化效应。pH从8.0下降到6.0,反应速率常数从89 L·(mol·s)−1提升到415 L·(mol·s)−1;水中阴离子络合或水解Fe3+则会抑制CNT的强化效果,当PO43−、Cl−、SO42−、NO3−、SiO32−、HCO3−的浓度为1 mmol·L−1时,对BZF去除率分别降低至38%、59%、67%、63%、48%、55%。
3) CNT强化Fe(Ⅵ)降解去除BZF主要通过羰基化、羟基化、丙酸甲酯的脱失以及脱羧等途径实现。
4) CNT强化Fe(Ⅵ)转化为Fe(Ⅴ),并促进PMSO氧化为PMSO2,反应半衰期由5 min缩短为2.5 min。
碳纳米管强化高铁酸盐降解苯扎贝特的效能与机理
The efficiency and mechanism of carbon nanotube-enhanced bezafibrate degradation by ferrate
-
摘要: 高铁酸盐具有较强的氧化性,是一种在水和废水处理中有较大应用潜力的绿色氧化剂,但对水中部分微污染物降解效果较差。本文研究了碳纳米管对高铁酸盐降解苯扎贝特的强化效果,探究了强化降解反应的途径,考察了水质因素对降解效果的影响,鉴定了强化降解的关键活性物种。结果表明,碳纳米管的表面羟基对强化高铁酸盐降解有促进作用,在碳纳米管存在且质量浓度为4 mg·L−1时,苯扎贝特的去除率由2%升至69%。酸性条件有利于碳纳米管强化高铁酸盐降解苯扎贝特;当水pH由8.0降至6.0时,表观反应速率常数由89 L·(mol·s)−1提升至415 L·(mol·s)−1。碳纳米管可强化Fe(Ⅵ)向Fe(Ⅴ)的转化,并使Fe(Ⅴ)成为提高苯扎贝特强化降解效果的关键活性物种;常见阴离子对强化氧化有抑制作用,影响程度为PO43−>SiO32−>HCO3−>Cl−>NO3−>SO42−。Abstract: Ferrate (Fe(Ⅵ)) has a great application potential as a green oxidant in water and wastewater treatment due to its strong oxidation capacity, but it is less effective on degrade some recalcitrant organic micropollutants in water. In this study, the enhancement effect of carbon nanotubes (CNTs) on the degradation of bezafibrate (BZF) by Fe(Ⅵ) was investigated, the enhanced degradation reaction pathway was explored, the influence of water quality factors on the degradation effect was evaluated, and the key active species for enhanced degradation were identified. The results show that the surface hydroxyl groups on CNTs could promote the BZF degradation by Fe(Ⅵ). At CNTs concentration of 4 mg·L−1, the removal rate of BZF increased from 2% to 69%. Acidic conditions were conducive to CNTs-enhanced BZF degradation by Fe(Ⅵ); The apparent reaction rate constant increased from 89 L·(mol·s)−1 to 415 L·(mol·s)−1 when pH decreased from 8.0 to 6.0. CNTs could enhance the conversion of Fe(Ⅵ) to Fe(Ⅴ), Fe(Ⅴ) became a crucial substance to increase the degradation rate of BZF; Common anions had inhibitory effects on enhanced oxidation, the corresponding impact level was PO43−>SiO32−>HCO3−>Cl−>NO3−>SO42−.
-
Key words:
- ferrate /
- carbon nanotubes /
- bezafibrate /
- enhanced oxidation /
- kinetics
-
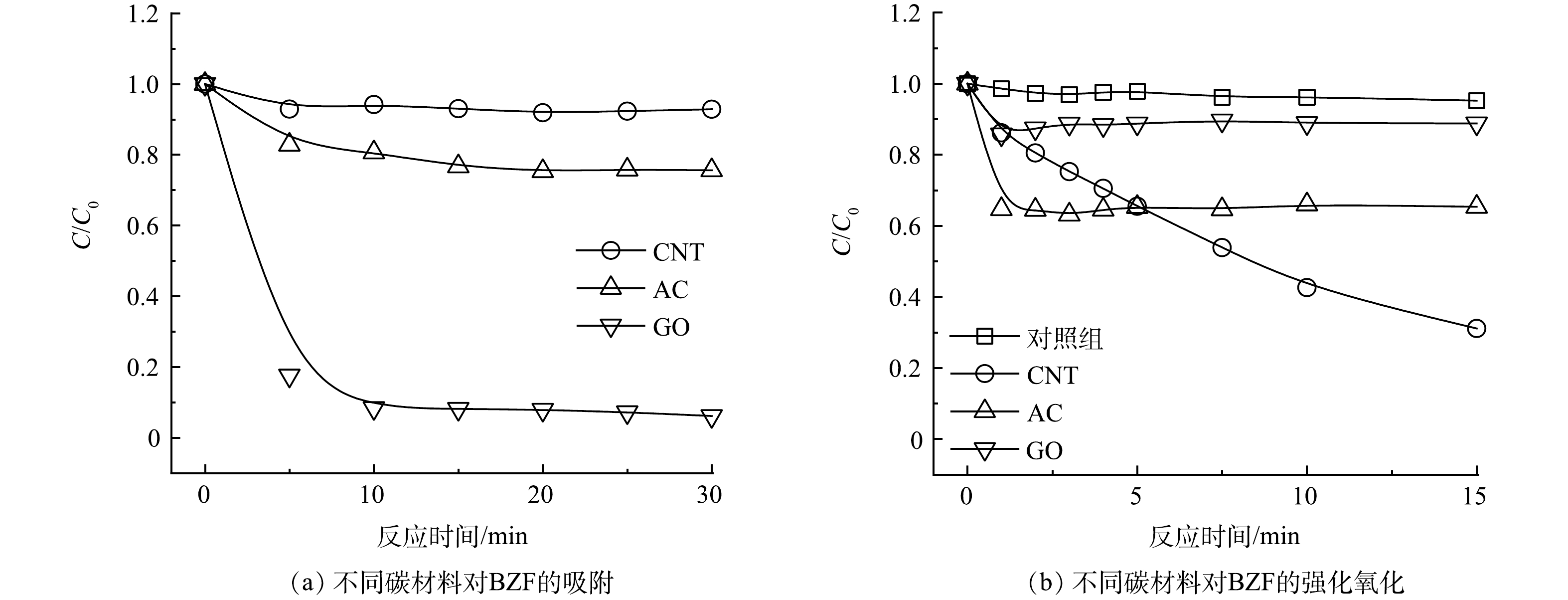
图 1 不同碳材料对BZF的吸附和强化氧化
Figure 1. Adsorption of BZF on different carbon materials and the effect on the degradation of BZF by Fe(Ⅵ).
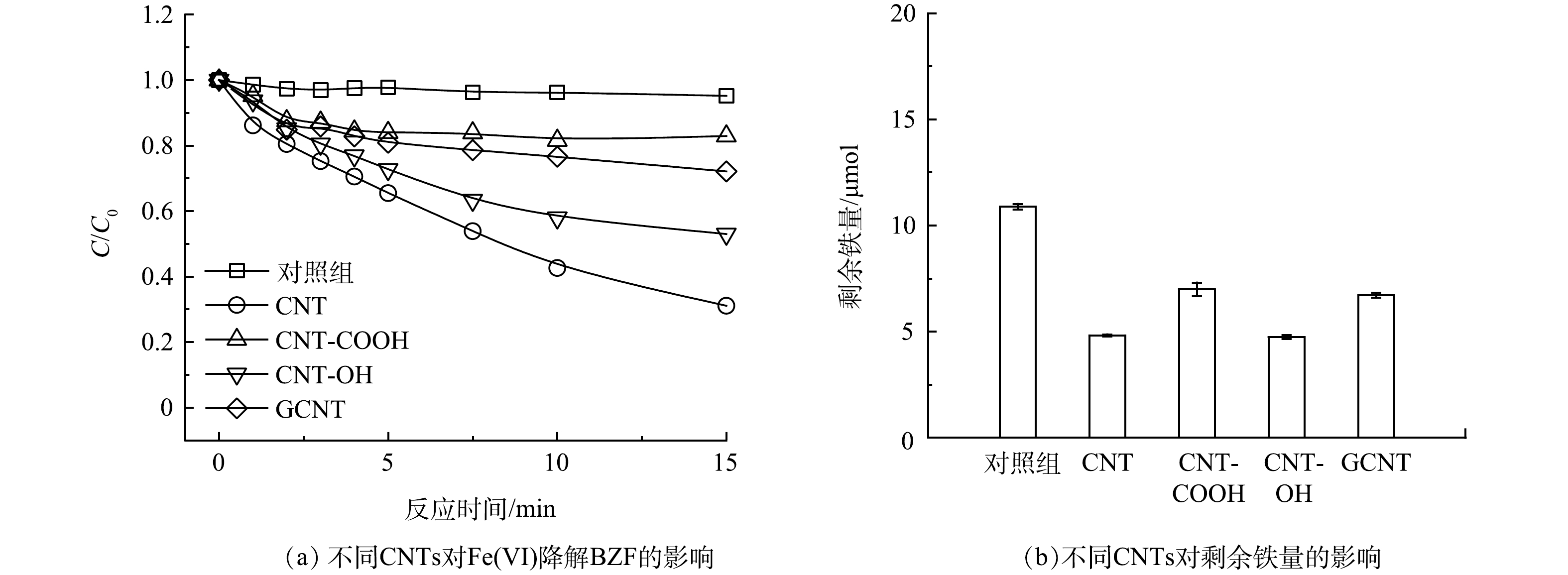
图 2 不同CNTs对Fe(Ⅵ)降解BZF和剩余铁量的影响
Figure 2. Effects of different types of CNTs on BZF degradation by Fe(Ⅵ) and residual Fe(Ⅵ).
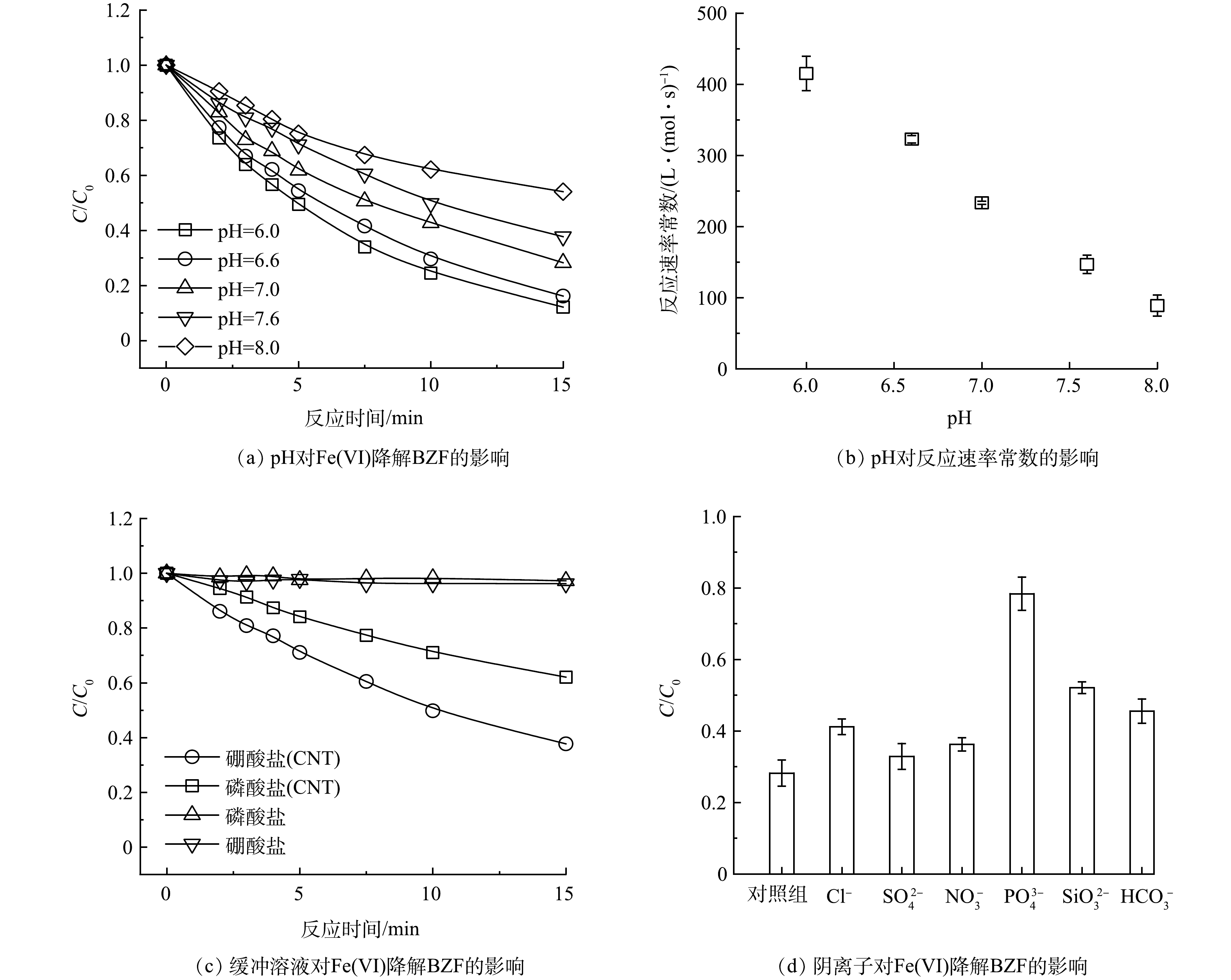
图 3 pH、缓冲液和阴离子对Fe(Ⅵ)降解BZF的影响
Figure 3. Effects of pH, buffer and anions Fe(Ⅵ) on BZF degradation
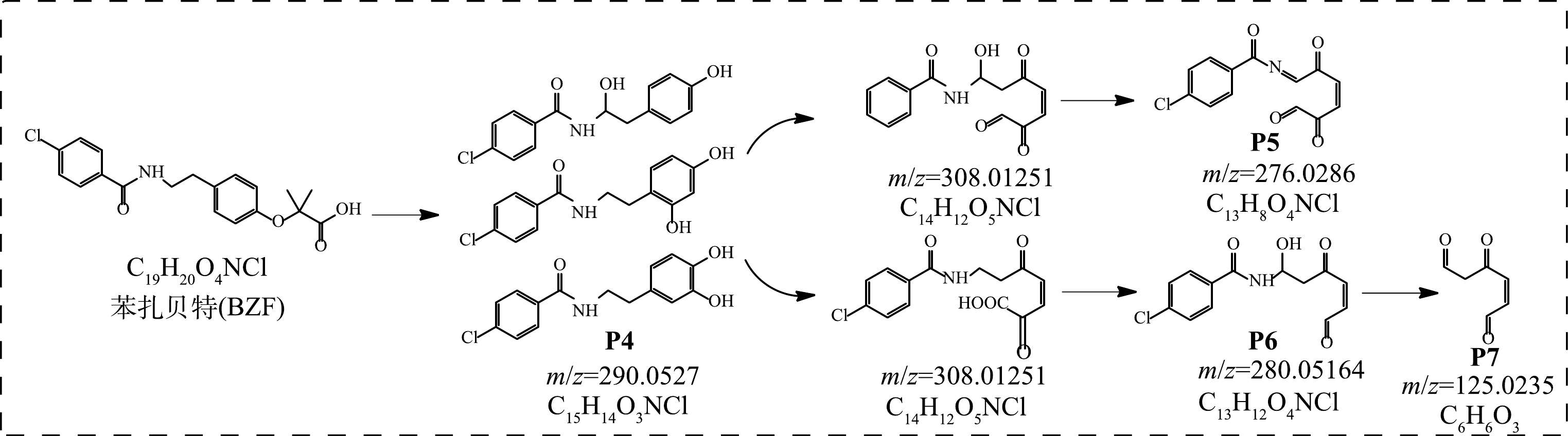
图 5 CNT对Fe(Ⅵ)降解BZF的降解产物分析与可能的反应途径
Figure 5. Proposed BZF degradation pathway by Fe(Ⅵ) in the presence of CNT
表 1 4种不同碳纳米管的相关指标
Table 1. Properties of four kinds of carbon nanotubes
材料类型 缩写 内径/nm 外径/nm 长度/μm 纯度/% 官能团含量/% 碳纳米管 CNT 5~10 10~20 10~30 98 / 石墨化碳纳米管 GCNT 5~10 10~20 5~30 99.9 / 羧基碳纳米管 CNT-COOH 5~10 10~20 10~30 98 2.0 羟基碳纳米管 CNT-OH 5~10 10~20 10~30 98 3.06  下载: 导出CSV
下载: 导出CSV
-
[1] JIANG J Q, STANFORD C, ALSHEYAB M. The online generation and application of ferrate for sewage treatment: A pilot scale trial[J]. Separation and Purification Technology, 2009, 68(2): 227-231. doi: 10.1016/j.seppur.2009.05.007 [2] RUSH J, BIELSKI B H. Kinetics of ferrate (Ⅴ) decay in aqueous solution: A pulse-radiolysis study[J]. Inorganic Chemistry, 1989, 28(21): 3947-3951. doi: 10.1021/ic00320a004 [3] DUAN J, GREGORY J. Coagulation by hydrolysing metal salts[J]. Advances in Colloid and Interface Science, 2003, 100-102: 475-502. doi: 10.1016/S0001-8686(02)00067-2 [4] 苑宝玲, 曲久辉. 高铁酸盐氧化絮凝去除藻类的机制[J]. 中国环境科学, 2002, 22(5): 397-399. doi: 10.3321/j.issn:1000-6923.2002.05.004 [5] 刘伟, 马军. 高铁酸盐预氧化对藻类细胞的破坏作用及其助凝机理[J]. 环境科学学报, 2002, 22(1): 24-28. doi: 10.3321/j.issn:0253-2468.2002.01.006 [6] LEE Y, CHO M, KIM J Y, et al. Chemistry of ferrate in aqueous solution and its applications as a green chemical[J]. Journal of Industrial and Engineering Chemistry, 2004, 10(1): 161-171. [7] SHARMA V K, ZBORIL R, VARMA R S. Ferrates: Greener oxidants with multimodal action in water treatment technologies[J]. Accounts of Chemical Research, 2015, 48(2): 182-191. doi: 10.1021/ar5004219 [8] DAR A A, PAN B, QIN J, et al. Sustainable ferrate oxidation: reaction chemistry, mechanisms and removal of pollutants in wastewater[J]. Environmental Pollution, 2021, 290: 117-127. [9] SHARMA V K, MISHRA S K. Ferrate(Ⅵ) oxidation of ibuprofen: A kinetic study[J]. Environmental Chemistry Letters, 2006, 3: 182-185. doi: 10.1007/s10311-005-0002-5 [10] WANG J, KIM J, ASHLEY D C, et al. Peracetic acid enhances micropollutant degradation by ferrate through promotion of electron transfer efficiency[J]. Environmental Science & Technology, 2022, 56(16): 11683-11693. [11] ZHOU O, SHIMODA H, GAO B, et al. Materials science of carbon nanotubes: Fabrication, integration, and properties of macroscopic structures of carbon nanotubes[J]. Accounts of Chemical Research, 2002, 35(12): 1045-1053. doi: 10.1021/ar010162f [12] SONI S K, THOMAS B, KAR V R. A comprehensive review on carbon nanotubes and carbon nanotube-reinforced composites: Syntheses, characteristics and applications[J]. Materials Today Communications, 2020, 25: 1015-1046. [13] RAFIEE R, POURAZIZI R. Influence of carbon nanotube functionalization on the interphase region between carbon nanotube and polymer[J]. Computational Materials Science, 2015, 96: 573-578. doi: 10.1016/j.commatsci.2014.03.056 [14] SUN S, JIANG J, QIU L, et al. Activation of ferrate by carbon nanotube for enhanced degradation of bromophenols: kinetics, products, and involvement of ferrate (Ⅴ)/ferrate (Ⅳ)[J]. Water Research, 2019, 156: 1-8. doi: 10.1016/j.watres.2019.02.057 [15] TERNES T A. Occurrence of drugs in german sewage treatment plants and rivers[J]. Water Research, 1998, 32(11): 3245-3260. doi: 10.1016/S0043-1354(98)00099-2 [16] CALAMARI D, ZUCCATO E, CASTIGLIONI S, et al. Strategic survey of therapeutic drugs in the rivers and lambro in northern Italy[J]. Environmental Science & Technology, 2003, 37: 1241-1248. [17] IDO A, HIROMORI Y, MENG L, et al. Occurrence of fibrates and their metabolites in source and drinking water in shanghai and zhejiang, China[J]. Scientific Reports, 2017, 7(1): 459-471. doi: 10.1038/s41598-017-00540-x [18] DANTAS R F, CANTERINO M, MAROTTA R, et al. Bezafibrate removal by means of ozonation: primary intermediates, kinetics, and toxicity assessment[J]. Water Research, 2007, 41(12): 2525-2532. doi: 10.1016/j.watres.2007.03.011 [19] ZHOU Z, JIANG J Q. Treatment of selected pharmaceuticals by ferrate: Performance, kinetic studies and identification of oxidation products[J]. Journal of Pharmaceutical and Biomedical Analysis, 2015, 106: 37-45. doi: 10.1016/j.jpba.2014.06.032 [20] LEE Y, KISSNER R, VON G U. Reaction of ferrate (Ⅵ) with ABTS and self-decay of ferrate: Kinetics and mechanisms[J]. Environmental Science & Technology, 2014, 48(9): 5154-5162. [21] GAO W. Graphene oxide: reduction recipes, spectroscopy, and applications[J]. The Chemistry of Graphene Oxide, 2015: 61-95. [22] CHOI H C, SHIM M, SARUNYA B A, et al. Spontaneous reduction of metal ions on the sidewalls of carbon nanotubes[J]. Journal of the American Chemical Society, 2002, 124 31: 9058-9059. [23] SEO J, LEE H J, LEE H, et al. Enhanced production of reactive oxidants by fenton-like reactions in the presence of carbon materials[J]. Journal of Chemical & Engineering Data, 2015, 273: 502-508. [24] PAN B, FENG M, MCDONALD T J, et al. Enhanced ferrate (Ⅵ) oxidation of micropollutants in water by carbonaceous materials: Elucidating surface functionality[J]. Chemical Engineering Journal, 2020, 398: 125-157. [25] WANG Z, ZHU J, ZHENG H. Improvement of duration-based water rights management with optimal water intake on/off events[J]. Water Resources Management, 2015, 29(8): 2927-2945. doi: 10.1007/s11269-015-0979-x [26] HUANG Z S, WANG L, LIU Y L, et al. Impact of phosphate on ferrate oxidation of organic compounds: An underestimated oxidant[J]. Environmental Science & Technology, 2018, 52(23): 13897-13907. [27] KOLAŘíK J, PRUCEK R, TUČEK J, et al. Impact of inorganic ions and natural organic matter on arsenates removal by ferrate (Ⅵ): Understanding a complex effect of phosphates ions[J]. Water Research, 2018, 141: 357-365. doi: 10.1016/j.watres.2018.05.024 [28] HUANG Z S, WANG L, LIU Y L, et al. Ferrate self-decomposition in water is also a self-activation process: Role of ferrate (Ⅴ) species and enhancement with ferrate (Ⅲ) in methyl phenyl sulfoxide oxidation by excess ferrate[J]. Water Research, 2021, 197: 117-134. [29] KHADHRAOUI M, TRABELSI H, KSIBI M, et al. Discoloration and detoxicification of a congo red dye solution by means of ozone treatment for a possible water reuse[J]. Journal of Hazardous Materials, 2009, 161(2/3): 974-981. [30] JIANG Y, GOODWILL J E, TOBIASON J E, et al. Effect of different solutes, natural organic matter, and particulate ferrate (Ⅲ) on ferrate decomposition in aqueous solutions[J]. Environmental Science & Technology, 2015, 49(5): 2841-2848. [31] LEUPIN O X, HUG S J. Oxidation and removal of arsenic from aerated groundwater by filtration through sand and zero-valent iron[J]. Water Research, 2005, 39(9): 1729-1740. doi: 10.1016/j.watres.2005.02.012 [32] ZHU J, YU F, MENG J, et al. Overlooked role of ferrate (Ⅳ) and ferrate (Ⅴ) in organic contaminant oxidation by Fe (Ⅵ)[J]. Environmental Science & Technology, 2020, 54(15): 9702-9710. [33] ZHANG H, LUO M, ZHOU P, et al. Enhanced ferrate oxidation of sulfamethoxazole in water by calcium peroxide: The role of ferrate (Ⅳ) and ferrate (Ⅴ)[J]. Journal of Hazardous Materials, 2022, 425: 128-145. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1523
- HTML全文浏览数: 1523
- PDF下载数: 70
- 施引文献: 0
