

 下载:
下载:
-
四环素(tetracycline,TC)作为常用的广谱抗生素之一,被大量广泛应用于医药、水产养殖以及畜牧业等行业中。而以生化技术为主的传统污水处理工艺无法将其有效去除,致使其大量进入天然水体。目前,在地表水、地下水甚至饮用水等水体中均可检测到四环素,即使在微量水平上,四环素类抗生素也对生态系统和人类健康造成极大的威胁[1]。近来,基于过硫酸盐的高级氧化技术在去除水中抗生素方面表现出色。通过对过一硫酸盐(peroxymonosulfate,PMS)或过二硫酸盐(peroxydisulfate,PDS)进行活化,可原位产生强氧化性的硫酸根自由基等活性物质对污染物进行氧化分解甚至矿化[2]。其中,碳基催化剂在活化PMS去除废水中抗生素方面引起广泛关注[3]。特别是,氮改性碳材料在活化PMS方面展现出优异的性能,通过在碳材料结构中引入氮原子,可改变碳材料局部的电子浓度分布及配位结构,有利其对PMS的吸附与活化,从而表现出较高的催化活性[4-6]。如WANG等[4]利用壳聚糖作为氮源和碳源,通过先水热碳化再热解的方法制备了氮掺杂碳球,发现该碳材料具备较好的活化PMS产SO4·−能力,可高效去除水中多种污染物。另外,QU等[6]以三聚氰胺为氮源,以L-谷氨酸为碳源,通过高温热解制备了氮掺杂碳纳米片,该催化剂可活化PMS产生1O2和·O2−进而实现对BPA的去除。因此,开发具有活化PMS性能的新型氮掺杂碳材料至关重要。
塑料污染是世界范围内的一个重大环境问题[7]。其中,聚氯乙烯(polyvinyl chloride,PVC)作为世界第三大通用塑料,每年产量约4 000×104 t,仅次于聚乙烯和聚丙烯,且50%以上产自中国[8]。得益于塑料分子内高的碳含量,研究人员以废弃塑料作为碳源,制备了多种高附加值的碳材料[9],如石墨烯、碳纳米管和碳纳米片等,并将其应用于吸附[10-11]、催化和传感等领域[12]。MA等[8]利用低温好氧处理将PVC脱氯后,进一步裂解成碳纳米管,为废塑料再利用开发了新思路。YANG等[13]利用水热法对PVC进行脱氯回收,并利用KOH活化的方法制备了高效捕集CO2的微孔碳材料。近来,将废弃塑料转变为具有活化PMS能力的碳纳米材料,并将其应用于去除废水中难降解污染物的研究备受关注。如MIAO等[14]利用废弃旧丙烯粉末通过一锅热解法合成了氮掺杂碳纳米管,通过活化PMS的方式在高盐的环境中实现了对磺胺甲恶唑的有效降解。REN等[15]开发了一种基于盐模板的碳化方法,将高密度聚乙烯催化转化成碳材料活化PMS实现了苯酚的高效降解。最近WEI等[16]发现将三聚氰胺与PVC在室温下研磨,再经热处理后可制备出具有较好催化活性的氮改性碳催化剂。与PVC直接进行热处理过程中常伴有含氯的有机有毒污染物不同,该方法可在常温条件下先进行脱氯反应,从而避免后续热处理过程中有毒物质的产生。该方法是一种温和环保的将废弃PVC转变为高值碳纳米材料的方法,而所制备的氮掺杂碳材料对PMS的活化及对污染物的去除性能值得期待。
鉴于此,本研究以废弃聚氯乙烯塑料为碳源,三聚氰胺为氮源,采用隔氧高温热处理方法制备了氮掺杂的碳纳米片,并将其应用于活化PMS以去除水中四环素。系统地考察了催化剂的制备、催化反应条件及影响因素。此外,推测了该材料催化活化PMS去除四环素的反应机理。
-
过一硫酸盐(2KHSO5·KHSO4·K2SO4,PMS)、三聚氰胺(C3H6N6)、N,N-二甲基甲酰胺(DMF,C3H7NO)、无水硫酸钠(Na2SO4)、碳酸氢钠(NaHCO3)、磷酸二氢钠(NaH2PO4)、无水乙醇(CH3CH2OH)、无水甲醇(CH3OH,MeOH)、L-组氨酸(C6H9N3O2,L-his)、叔丁醇(C4H10O,TBA)、对苯醌(C6H4O2,p-BQ)、碘化钾(KI)均为分析纯,PVC塑料袋产自于苍南县龙港峰尚制袋厂。
-
采用在惰性气氛下高温热处理的方法制备氮掺杂碳纳米片[16]。首先,称取一定量废弃PVC塑料袋溶于10 mL DMF中,搅拌1 h使其充分溶解。将2 g三聚氰胺粉末加入上述溶液,并继续搅拌2 h反应得到白色胶状物。所得胶状物经离心水洗3次后,置于70 ℃鼓风干燥箱中干燥12 h,研磨得到固体粉末。随后,将所制备的固体粉末置于管式炉中,在氮气氛围下700 ℃下煅烧2 h,冷却后收集所得粉末即为制备的氮掺杂碳纳米片,标记为PVCN。另外,通过改变PVC与三聚氰胺的质量比(30%、60%和100%),分别制备不同质量比的PVCN(分别标记为30 PVCN、60 PVCN和100 PVCN)。
-
采用X射线衍射(XRD,日本smart Lab)在10°~90°内以6°·min−1的速率扫描图像,对催化剂的晶体结构进行表征。采用扫描电子显微镜(SEM,德国ZEISS Gemini SEM 300)和透射电子显微镜(TEM,美国FEI Talos F200S)对催化剂的微观形貌进行分析。采用X射线衍射仪(XPS,美国Thermo Scientific K-Alpha)分析样品中元素的组成形态。通过拉曼光谱(Raman,日本Horiba LabRAM HR Evolution)分析催化剂的结构缺陷与石墨化程度。采用电子顺磁共振谱仪(EPR,英国Edinburgh FLS1000)检测了反应体系中产生的活性物种种类,其中以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)和2,2,6,6-四甲基哌啶(TEMP)作为自旋捕获剂。使用电化学工作站(CHI 660 B,中国上海辰华),在传统的三电极体系中考察催化剂的电化学性质。其中,铂丝和饱和甘汞电极(SCE)分别作为对电极和参比电极,反应液电解质为0.05 mol·L−1的Na2SO4水溶液。
-
选择四环素作为模拟目标污染物对催化剂活性进行考察。首先,称取一定量的催化剂分散于100 mL 10 mg·L−1四环素溶液中,并保持持续搅拌。待催化剂与四环素达到吸附脱附平衡后,加入一定浓度PMS开始降解反应,其间间隔一定时间取样3 mL并立即与甲醇混合停止反应。悬浮液经0.22 μm尼龙膜过滤后,采用紫外-可见分光光度计分析所得溶液中TC浓度变化,TC的吸收波长为358 nm。为了探究材料的重复利用效果,将反应后的催化剂经0.22 μm滤膜过滤和水洗后并置于70 ℃烘箱烘干,收集所得固体粉末进行重复降解实验。使用总有机碳分析仪(TOC,德国Jena Multi N/C 3100)测定湖水中TOC。使用离子色谱仪(Thermo Scientific ICS-600)检测湖水中常见阴离子的浓度。反应过程中PMS的测定方法为[17]:将0.05 g的碳酸氢钠与1 g的碘化钾溶于5 mL去离子水,加水至9 mL,按照降解实验取样时间采集1 mL四环素溶液过0.22 μm滤膜,与上述9 mL溶液混合,静置15 min后用紫外分光光度计检测395 nm的吸光度。
-
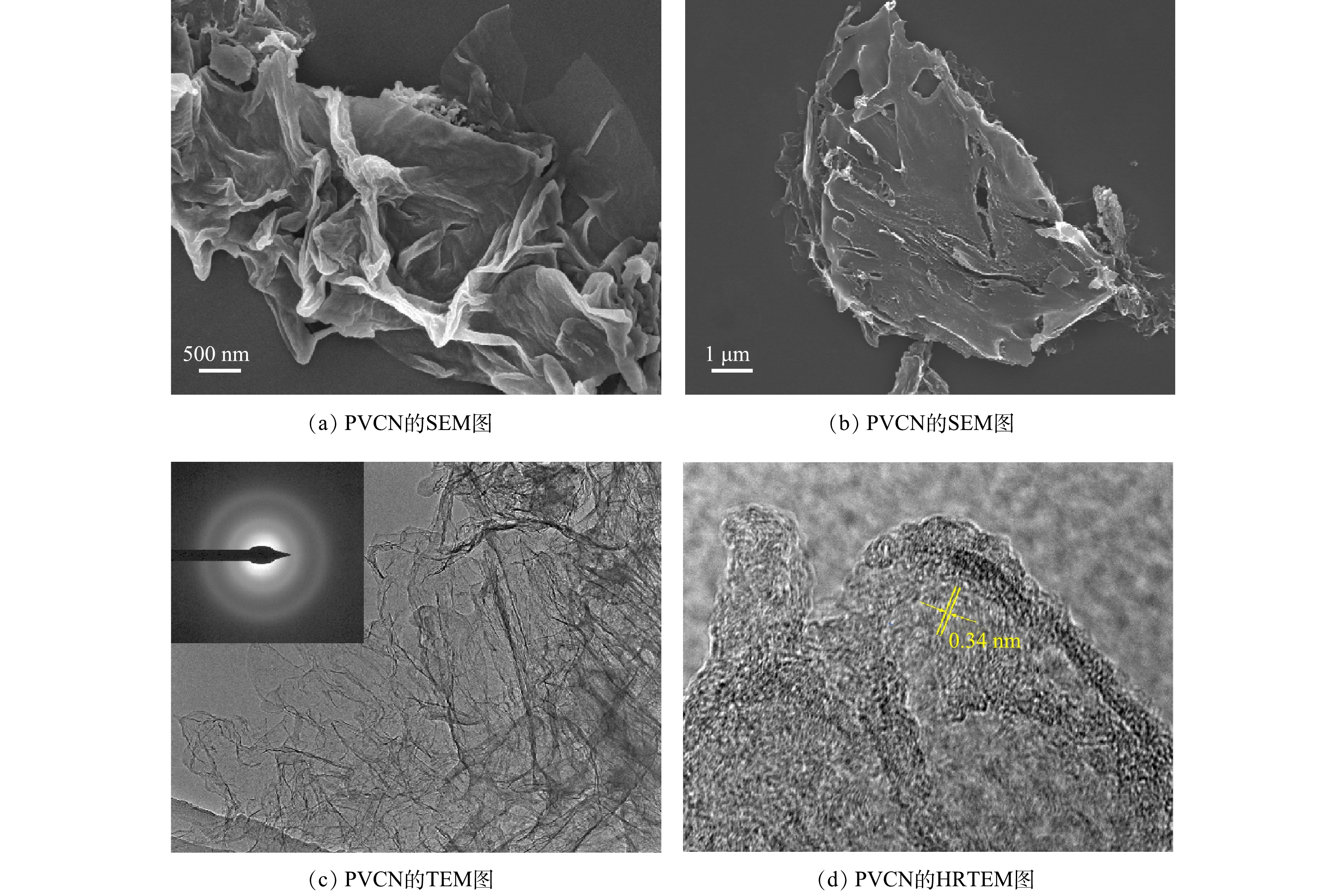
图1为经高温热解法制备的PVCN的SEM和TEM图。从图1(a)和图1(b)可以发现,所制备的PVCN为二维层状纳米片形貌且片层表面光滑。与石墨烯等二维碳材料类似,该纳米片堆叠在一起,并且可明显发现其表面存在为减少表面能而形成的褶皱。透射电镜图(图1(c))证实了该碳材料由较薄的纳米片层组成,且选区电子衍射图显示该碳材料主要为无定型结构。但从PVCN材料的高分辨透射图(图1(d))可清晰的发现短程有序的晶格条纹,表明其具有一定的结晶性。其中,0.34 nm的晶格间距对应于石墨碳的(002)晶面[18],进一步表明所制备的PVCN中含有石墨碳结构。另外,PVCN的石墨化区域表现出大的石墨化晶格条纹间距和高曲率,表明其可能存在丰富缺陷和活性位点[19]。
采用XRD对所制备的PVCN的晶体结构进行分析。如图2(a)所示,在PVCN的XRD谱图中,均可发现位于26.3°和42.5° 2个较宽衍射峰,分别对应石墨碳的(002)晶面和(101)晶面[20],较宽的(002)表明其具有石墨碳结构[14],但结晶度不高,这与TEM的表征结果相对应。图2(b)为材料的拉曼光谱。可见,在1 354 cm−1和1 585 cm−1处均出现2个特征峰,分别为D峰和G峰。其中,D峰与碳缺陷或石墨相碳中异质原子掺杂导致的无序排列有关,而G峰则对应于sp2杂化的结晶性石墨化碳原子,通过两者的比值(ID/IG)可评价碳材料缺陷度或石墨化度。由图2(b)可知,3种掺杂比的ID/IG值从小到大的排列顺序为:100 PVCN<60 PVCN<30 PVCN,100 PVCN的缺陷最少,30 PVCN的缺陷最多。ID/IG的增加是由于N掺杂引起的碳材料中sp2杂化向sp3杂化的转变。随着N掺杂量的增大,材料的缺陷程度增加,说明在高温热解过程中,形成了石墨化缺陷[16]。之前研究表明,在碳材料活化PMS过程中,碳材料结构中石墨化缺陷可为催化剂和PMS之间提供更多的电子转移路径[21]。利用XPS对材料的元素组成及价态进行分析。如图2(c)所示,不同比例的PVCN材料均由碳、氮和氧组成。同时,随着PVC含量的增加,样品中的碳和氮元素的相对比例也逐渐升高,该结果证实了氮原子已成功掺杂碳材料中。另外,由图2(d)可见,在g=2.003处可观察到明显的特征峰信号,该信号与碳原子外围未成对电子含量有关[6],表明PVCN样品中含有氮空位。催化剂中的N来源于三聚氰胺,这种情况可能是由于样品在高温煅烧过程中,部分N转化成氨气后逃逸从而形成了氮空位[5],有研究[4]表明,氮空位的引入可促进碳材料对PMS的吸附,从而提升其活化PMS性能。
-
图3为不同反应体系对TC去除率的影响。由图3(a)可知,单独PVCN对TC具有一定的吸附性能,经30 min吸附反应后基本达到吸附平衡。在反应阶段,当只有PMS存在时,TC在30 min内的去除率仅为25.8%,表明PMS具有一定的氧化分解TC能力,这可归因于PMS自身较弱的氧化能力。当只有PVCN存在时,经30 min反应后,溶液中TC的浓度基本保持不变。与之对比,当同时加入PVCN和PMS时,可明显发现TC的浓度急剧下降,PVCN对TC的去除率高达98%,其表观反应动力学常数k(0.106 min−1)为单独PMS(0.01 min−1)的10倍(图3(b))。同时,考察了不同PVC/三聚氰胺质量比的PVCN对TC去除效率的影响。随着PVC含量的增加,虽然所制备PVCN对TC的去除率呈现一定下降趋势(反应动力学常数由0.106 min−1衰减至0.66 min−1),但经30 min反应后,所有催化剂对TC去除效率仍然可维持在80%以上。该结果一方面表明催化剂中氮的相对含量对其活性有影响。另一方面也说明所制备的PVCN材料具有优异的活化PMS去除污染物TC的能力。
-
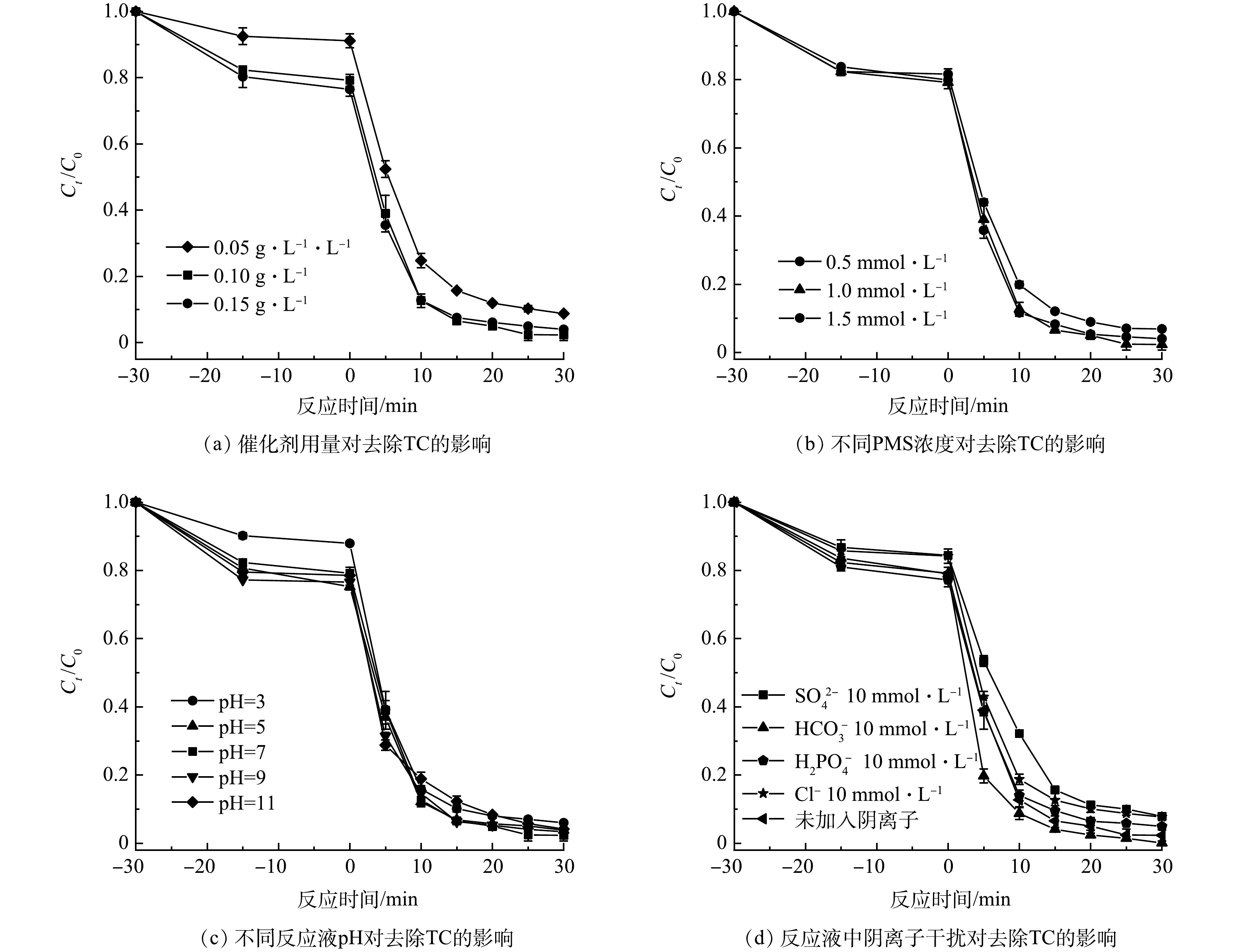
研究了不同催化剂投加量、pH、PMS浓度、阴离子干扰对四环素降解的影响。图4(a)为不同催化剂投加量对TC降解的影响。催化剂浓度一方面直接影响吸附性能,另一方面也会影响催化PMS活性位点的数量。随着催化剂投加量的增大,TC去除率呈现先增加后趋于稳定的趋势。当催化剂投加量为0.05 g·L−1时,反应30 min后,TC去除率为90%。而当催化剂投加量增至0.1 g·L−1时,TC去除率可达到98%。进一步增加催化剂投加量至1.5 g·L−1时,TC去除率稍微降至96%。这主要归因于随着催化剂投加量的增大,可提供更多的PMS活性点位,促使体系中产生更多的活性自由基[22],从而加速TC的降解。
由图3的对比实验结果可知,引入PMS后,PVCN对TC的降解效果得到了明显提升。因此,考察了不同PMS浓度对TC降解活性的影响。如图4(b)所示,随着PMS浓度的增加,TC的去除率呈现先增大后减小的趋势。当PMS浓度由0 mmol·L−1增至1 mmol·L−1时,TC的降解效果逐渐提高,去除率从92%增加至98%。而当PMS浓度由1 mmol·L−1进一步增至1.5 mmol·L−1时,去除率提高有限,这主要是由于PMS浓度由1 mmol·L−1增至1.5 mmol·L−1时,反应体系中产生了过多SO4·−,过量的SO4·−自由基之间会发生自淬反应或与HSO5−发生反应,生成氧化能力较弱的SO5·−自由基,从而导致参与反应的SO4·−减少,并使反应活性减弱[23]。
进一步考察了不同pH对TC降解的影响。如图4(c)所示,其中,当反应液初始pH为7时,TC去除率最高可达98%;当pH=3时,TC去除率相对有所减弱,为94%。这主要归因于在酸性条件下,HSO5−与过量的H+可形成更稳定的H2SO5,不利于催化剂与其反应生成SO4·−[24];当反应液初始pH为碱性时(pH=11),催化剂对TC去除效率为95.9%,这可能是由于在碱性条件下,一些SO4·−与OH−反应生成寿命较短、氧化还原电位较低、选择性较低的·OH。因此,高浓度的OH−可能会淬灭SO4·−并降低TC的去除效率[25]。总之,由图4(c)中可发现反应液pH对TC降解效果无明显影响,经过30 min反应后,对TC的去除率均达到90%以上。该结果表明所制备的PVCN碳材料具有在较宽的pH范围内活化PMS的能力,有利于其在实际工况中的应用。
实际水体环境中,常存在多种的无机阴离子,他们的存在会影响催化剂对PMS的活化以及自由基的生成,从而抑制或促进污染物的降解,因此,考察了不同阴离子对反应体系的影响。由图4(d)可知,当向反应液中分别投加10 mmol·L−1阴离子时,可以观察到除HCO3−和H2PO4−外,其他阴离子均对催化剂的活性表现出一定的抑制作用。其中,Cl−可能与SO4·−反应生成Cl·、Cl2·−和HClO,导致SO4·−的消耗,从而抑制TC的降解[26-27]。HCO3−是过硫酸盐氧化过程中极好的自由基清除剂,可同时消除反应体系中的SO4·−和·OH,然而,本实验中HCO3−的存在促进了TC的降解,这可能是因为HCO3−具有缓冲作用,可使反应pH保持在7左右,并且HCO3−与SO4·−能反应生成稳定性更高的·CO3−,从而提高了PVCN对TC的氧化还原能力[25-26]。SO42−对TC的降解产生了轻微的抑制作用,表明PVCN/PMS对SO42−有较好的耐受性。
-
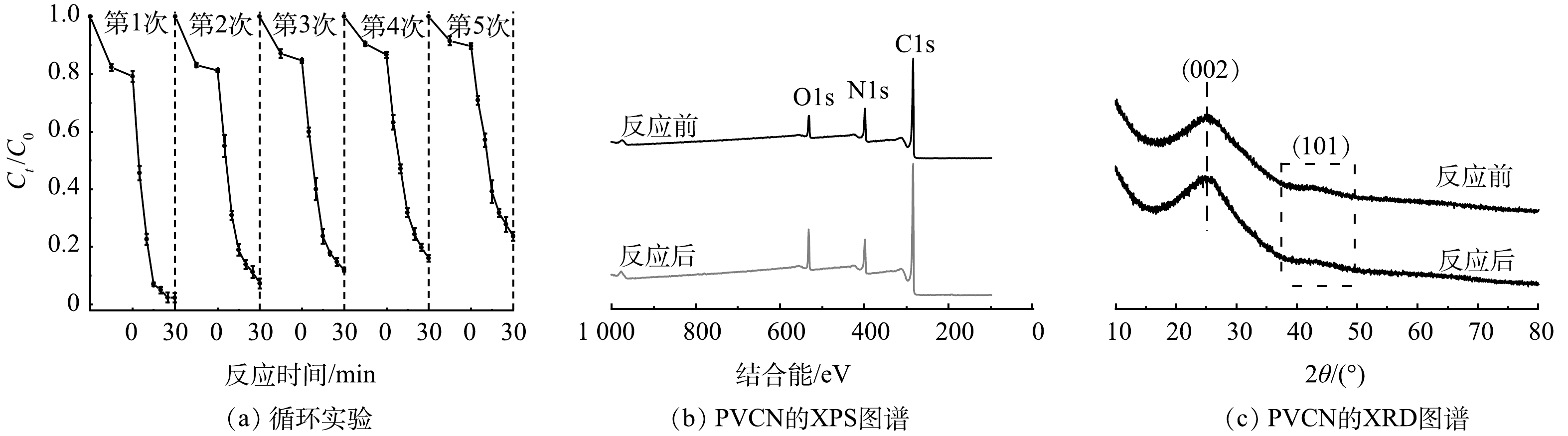
催化剂的稳定性是催化剂性能的一个重要指标。本研究中,采用重复性实验对PVCN的稳定性进行考察。如图5(a)所示,经5次重复实验后,PVCN催化剂对TC的降解性仍可维持在80%以上。此外,从图5(b)和图5(c)的催化剂反应前后XPS和XRD图谱可知,PVCN在反应后元素组成和晶体结构均未改变,表明该复合催化剂具备较高稳定性,具备一定的实际应用前景。
-
考察了在不同水质的实际水体中催化剂活化PMS去除TC性能的情况。如图6所示,在湖水和自来水中,所制备的氮掺杂碳纳米片催化剂均表现出较好的活化PMS去除TC的性能,对TC的去除率分别为94.7%和94.1%。由于湖水中的共存物质可能会对影响降解效果,因此,对湖水水质进行分析,湖水中TOC为15.48 mg·L−1,SO4·−、Cl− 、NO3−、PO43−和HCO3−的质量浓度分别为76.91、65.66、1.03、0.87和4.03 mg·L−1。上述实验结果证明SO4·−和Cl−均能对TC的降解产生干扰,导致催化剂对TC的降解性能有所减弱。尽管如此,该催化剂在湖水和自来水中仍具有优异的活化PMS去除TC的性能,表明其具有一定的实际应用前景。
-
为了确定体系中所产生自由基的种类,首先采用自由基淬灭实验,考察不同自由基对降解反应的贡献。有研究[28]表明,在羟基自由基(·OH)与硫酸根自由基(SO4·−)同时存在的条件下,由于叔丁醇(TBA)与羟基自由基(·OH)反应比硫酸根自由基(SO4·−)快,可选择性地淬灭体系中的·OH。而甲醇(MeOH)则可与·OH与SO4·−快速反应。因此,采用MeOH与TBA作为自由基淬灭剂来考察反应体系中·OH与SO4·−对TC去除的贡献情况。对苯醌(p-BQ)和L-组氨酸(L-his)作为超氧自由基(·O2−)和单线态氧(1O2)的淬灭剂[29]。由图7(a)可知,当向体系中加入5 mmol·L−1TBA后,TC的去除率由98%减至89%,表明在该反应体系中·OH的贡献较小。同时,5 mmol·L−1 MeOH的加入对催化活性无明显抑制作用,图7(c)显示了反应体系中PMS的消耗量,反应30 min后,PMS浓度仅利用了42.7%,说明体系内生成了较少的SO4·−[30],由此可推断在该反应体系中,·OH和SO4·−不是降解TC的主要活性物种。另一方面,对比分别加入5 mmol·L−1 p-BQ与L-his的结果可知,p-BQ与L-his对TC的降解均起到了明显的抑制作用,表明体系中发挥主要作用的活性物种为1O2及·O2−。以上结果表明,所制备氮掺杂碳材料活化PMS起主导作用的活性物种为1O2和·O2−,这与前人报道的氮掺杂石墨化碳活化PMS降解污染物过程类似[31]。
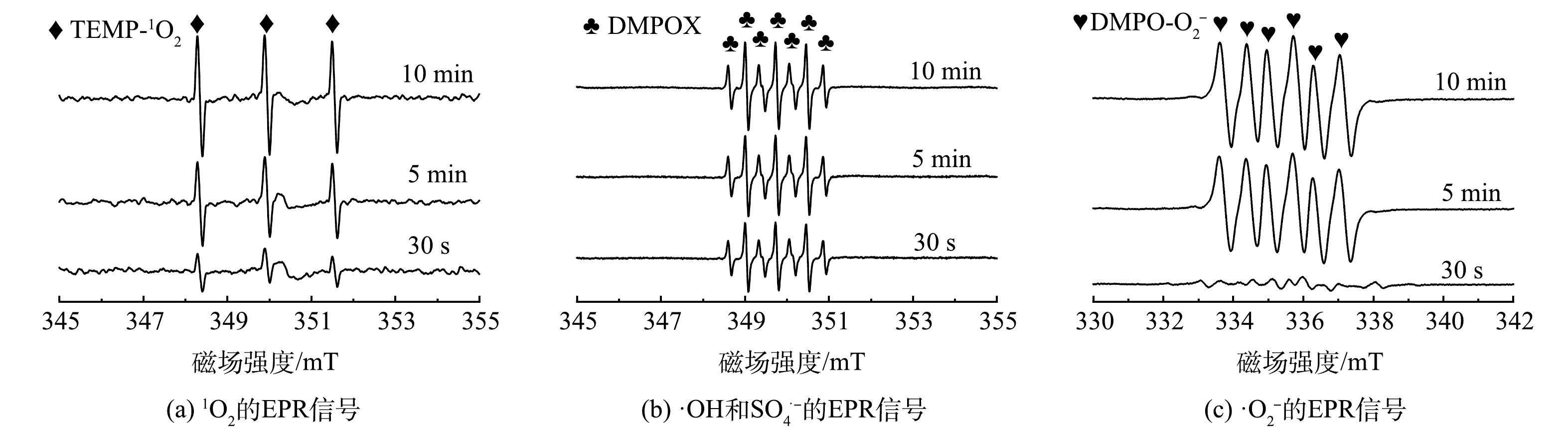
另外,采用EPR对反应过程中产生的活性物种进行分析。由图8(a)可知,当催化剂和PMS共同存在时,在反应体系中能明显检测到归属于TEMP-1O2加合物的三重特征峰信号(1:1:1),进一步证明了反应过程中有1O2的产生。同时,随着反应进行,该特征峰信号逐渐加强,间接表明所产生1O2的量不断增多。另外,由图8(b)中可明显观察到属于DMPOX的特征峰(1:2:1:2:1:2:1),而未捕获到归属于DMPO-·OH和DMPO-SO4·−加合物的特征峰。这可归因于催化剂与PMS反应可直接将DMPO氧化成DMPOX,这与之前报告的氮掺杂碳材料活化PMS的结果一致,说明在该过程中存在着电子转移方式的非自由基活化PMS过程[22]。图8(c)反映了超氧自由基的EPR信号。反应5 min后,可以观察到明显的DMPO-O2·−加合物的六重特征峰,且随着反应的进行(10 min后),该峰强度进一步提高,表明体系中不断有超氧自由基的生成[32]。以上测试进一步验证了自由基捕获实验结果,在反应过程中产生了单线态氧和超氧自由基。
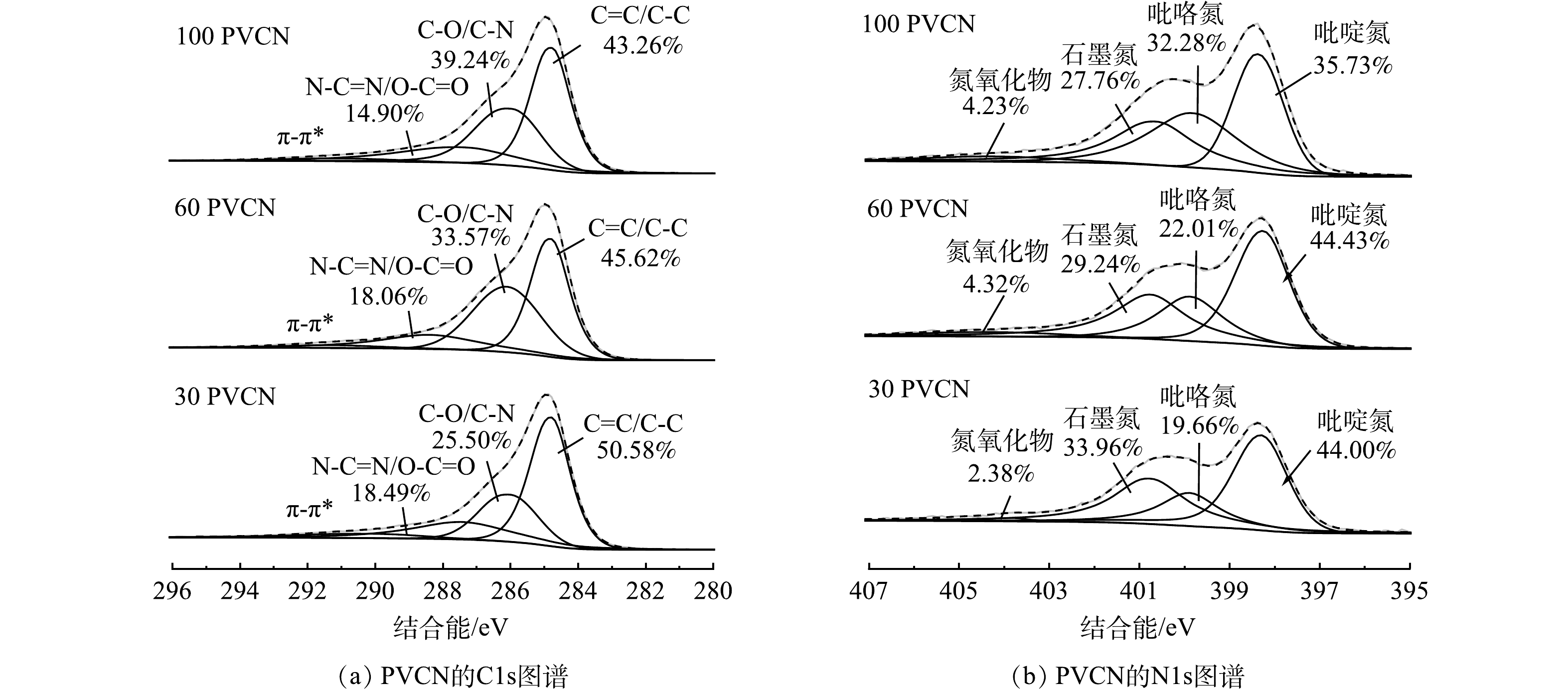
基于实验结果可知,催化剂中碳氮相对含量对其活化PMS去除TC效果有明显影响。因此,利用XPS对比分析了不同比例的PVCN中C和N成键情况。图9(a)为C1s的高分辨图,从中可以看出,30 PVCN中C=O或C=N键的相对占比要高于60 PVCN样品和100 PVCN,说明其含有更多C=O或C=N。有研究表明,碳材料中的C=O和C=N键可以作为PMS的活性位点[7],将PMS活化成1O2从而达到对污染物的降解[10,33]。因此,30 PVCN可提供更多活化PMS的位点。另外,由N1s的高分辨图谱(图9(b))可发现位于398.7、399.7、400.9和404.2 eV的4个拟合峰,分别对应于吡啶氮、吡咯氮、石墨氮和氮氧化物[34]。之前报道证实在氮掺杂碳材料中,吡啶氮和石墨氮可作为PMS的活性位点[35]。其中,石墨氮通过削弱碳电子云密度,改善四环素分子与π电子之间的相互作用,促进对四环素的吸附[17],而吡啶氮可作为PMS的吸附位点[4,36]。相比于60 PVCN和100 PVCN,30 PVCN中吡啶氮和石墨氮的占比最高,说明其可提供更多的活性位点,从而表现出更高的催化活性。另外,采用电化学计时电流方法,对所制备的PVCN材料与PMS和TC之间的电子转移行为进行研究。由图10可知,当加入PMS时会立即引起电流的增加,而加入TC后也会导致电流增加。这说明TC可通过PVCN将电子转移到PMS[37],该结果证明在PVCN/PMS体系中,电子转移作用也参与到TC的降解过程中。综上所述,30 PVCN活化PMS的途径主要通过以1O2和电子转移的非自由基以及·O2−的自由基方式进行,最终实现高效去除TC。
-
1)采用高温热处理法,以废弃PVC和三聚氰胺制备了氮掺杂碳纳米片催化剂,该催化剂具备较优的活化PMS降解TC的能力。在PMS浓度为1 mmol·L−1,30 min反应后,该PVCN/PMS体系对TC的去除率达到98%。
2)考察了PVC相对含量、PMS浓度、催化剂投加量和反应液pH等实验条件对催化活性的影响。当PVC/三聚氰胺为30%、PMS浓度为1 mmol·L−1及催化剂投加量为0.1 g·L−1时,该复合催化剂催化活性达到最优。同时,催化剂在宽pH范围内均具有较好的活化PMS去除TC的活性。
3)反应机理研究结果表明,该催化剂活化PMS去除TC反应过程中,其活化PMS的方式主要通过以1O2和电子转移为主的非自由基过程以及·O2−的自由基过程实现对TC的高效去除。同时,该复合催化剂具备较好的稳定性,且能适应不同水体环境,具备较好的应用前景。
废弃聚氯乙烯塑料衍生的氮掺杂碳纳米片活化过一硫酸盐去除水中四环素
Nitrogen doped carbon nanosheet derived from waste PVC plastic activated peroxymonosulfate for tetracycline removal
-
摘要: 本研究分别以废弃聚氯乙烯塑料和三聚氰胺作为碳源和氮源,采用高温热处理方法制备了氮掺杂薄层纳米碳片材料,并对其活化过一硫酸盐(PMS)去除水中抗生素四环素(TC)的性能进行研究。考察了PMS浓度、催化剂投加量、pH及不同离子对TC去除的影响。多种物化表征结果表明,所制备材料为薄层纳米片结构且主要由碳、氮和氧元素组成。降解实验结果表明,该催化剂具有较优的活化过一硫酸盐(PMS)去除TC的性能。在投加PMS浓度为1 mmol·L−1、催化剂投加量为0.1 g·L−1条件下,催化剂去除TC活性最佳,反应30 min后,TC的去除率达到98%。自由基捕获实验和电子自旋共振测试结果表明,该催化剂活化PMS降解TC过程中,起主导作用的为单线态氧、超氧自由基。重复性实验结果表明该催化剂具备较好的稳定性。本研究可为新型高效活化PMS碳基催化体系的开发及其在环境污染控制领域的应用提供参考。Abstract: In this work, nitrogen doped carbon nanosheet catalyst was synthesized via a thermal treatment method by employing waste polyvinyl chloride (PVC) plastic as carbon source and melamine as nitrogen source. The performance of tetracycline (TC) removal from water by the catalyst activated peroxymonosulfate (PMS) was evaluated. The effects of the operational parameters including PMS concentration, catalyst dosage, pH and different inorganic ions on TC degradation were studied. The results of various characterizations revealed that the prepared catalyst had a thin nanosheet structure and consisted of carbon, nitrogen and oxygen elements. Moreover, the catalyst displayed an excellent PMS activation ability for TC degradation. The optimum TC removal happened at the catalyst dosage of 0.1 g·L−1 and PMS content of 1 mmol·L−1, 98% of TC could be removed after 30 min reaction. As confirmed by results of the radical trapping experiments and EPR tests, the formed singlet oxygen and superoxide radicals played a vital role during TC degradation by catalyst/PMS. The result of cycling tests revealed that the catalyst had a relatively robust stability. This work paves the way for the rational design of a novel carbon-based catalytic system with high-efficiency PMS activation and application in the field of environmental pollution control.
-
Key words:
- peroxymonosulfate /
- nitrogen doped carbon /
- PVC /
- tetracycline
-
四环素(tetracycline,TC)作为常用的广谱抗生素之一,被大量广泛应用于医药、水产养殖以及畜牧业等行业中。而以生化技术为主的传统污水处理工艺无法将其有效去除,致使其大量进入天然水体。目前,在地表水、地下水甚至饮用水等水体中均可检测到四环素,即使在微量水平上,四环素类抗生素也对生态系统和人类健康造成极大的威胁[1]。近来,基于过硫酸盐的高级氧化技术在去除水中抗生素方面表现出色。通过对过一硫酸盐(peroxymonosulfate,PMS)或过二硫酸盐(peroxydisulfate,PDS)进行活化,可原位产生强氧化性的硫酸根自由基等活性物质对污染物进行氧化分解甚至矿化[2]。其中,碳基催化剂在活化PMS去除废水中抗生素方面引起广泛关注[3]。特别是,氮改性碳材料在活化PMS方面展现出优异的性能,通过在碳材料结构中引入氮原子,可改变碳材料局部的电子浓度分布及配位结构,有利其对PMS的吸附与活化,从而表现出较高的催化活性[4-6]。如WANG等[4]利用壳聚糖作为氮源和碳源,通过先水热碳化再热解的方法制备了氮掺杂碳球,发现该碳材料具备较好的活化PMS产SO4·−能力,可高效去除水中多种污染物。另外,QU等[6]以三聚氰胺为氮源,以L-谷氨酸为碳源,通过高温热解制备了氮掺杂碳纳米片,该催化剂可活化PMS产生1O2和·O2−进而实现对BPA的去除。因此,开发具有活化PMS性能的新型氮掺杂碳材料至关重要。
塑料污染是世界范围内的一个重大环境问题[7]。其中,聚氯乙烯(polyvinyl chloride,PVC)作为世界第三大通用塑料,每年产量约4 000×104 t,仅次于聚乙烯和聚丙烯,且50%以上产自中国[8]。得益于塑料分子内高的碳含量,研究人员以废弃塑料作为碳源,制备了多种高附加值的碳材料[9],如石墨烯、碳纳米管和碳纳米片等,并将其应用于吸附[10-11]、催化和传感等领域[12]。MA等[8]利用低温好氧处理将PVC脱氯后,进一步裂解成碳纳米管,为废塑料再利用开发了新思路。YANG等[13]利用水热法对PVC进行脱氯回收,并利用KOH活化的方法制备了高效捕集CO2的微孔碳材料。近来,将废弃塑料转变为具有活化PMS能力的碳纳米材料,并将其应用于去除废水中难降解污染物的研究备受关注。如MIAO等[14]利用废弃旧丙烯粉末通过一锅热解法合成了氮掺杂碳纳米管,通过活化PMS的方式在高盐的环境中实现了对磺胺甲恶唑的有效降解。REN等[15]开发了一种基于盐模板的碳化方法,将高密度聚乙烯催化转化成碳材料活化PMS实现了苯酚的高效降解。最近WEI等[16]发现将三聚氰胺与PVC在室温下研磨,再经热处理后可制备出具有较好催化活性的氮改性碳催化剂。与PVC直接进行热处理过程中常伴有含氯的有机有毒污染物不同,该方法可在常温条件下先进行脱氯反应,从而避免后续热处理过程中有毒物质的产生。该方法是一种温和环保的将废弃PVC转变为高值碳纳米材料的方法,而所制备的氮掺杂碳材料对PMS的活化及对污染物的去除性能值得期待。
鉴于此,本研究以废弃聚氯乙烯塑料为碳源,三聚氰胺为氮源,采用隔氧高温热处理方法制备了氮掺杂的碳纳米片,并将其应用于活化PMS以去除水中四环素。系统地考察了催化剂的制备、催化反应条件及影响因素。此外,推测了该材料催化活化PMS去除四环素的反应机理。
1. 材料与方法
1.1 实验原料
过一硫酸盐(2KHSO5·KHSO4·K2SO4,PMS)、三聚氰胺(C3H6N6)、N,N-二甲基甲酰胺(DMF,C3H7NO)、无水硫酸钠(Na2SO4)、碳酸氢钠(NaHCO3)、磷酸二氢钠(NaH2PO4)、无水乙醇(CH3CH2OH)、无水甲醇(CH3OH,MeOH)、L-组氨酸(C6H9N3O2,L-his)、叔丁醇(C4H10O,TBA)、对苯醌(C6H4O2,p-BQ)、碘化钾(KI)均为分析纯,PVC塑料袋产自于苍南县龙港峰尚制袋厂。
1.2 催化剂制备方法
采用在惰性气氛下高温热处理的方法制备氮掺杂碳纳米片[16]。首先,称取一定量废弃PVC塑料袋溶于10 mL DMF中,搅拌1 h使其充分溶解。将2 g三聚氰胺粉末加入上述溶液,并继续搅拌2 h反应得到白色胶状物。所得胶状物经离心水洗3次后,置于70 ℃鼓风干燥箱中干燥12 h,研磨得到固体粉末。随后,将所制备的固体粉末置于管式炉中,在氮气氛围下700 ℃下煅烧2 h,冷却后收集所得粉末即为制备的氮掺杂碳纳米片,标记为PVCN。另外,通过改变PVC与三聚氰胺的质量比(30%、60%和100%),分别制备不同质量比的PVCN(分别标记为30 PVCN、60 PVCN和100 PVCN)。
1.3 催化剂表征
采用X射线衍射(XRD,日本smart Lab)在10°~90°内以6°·min−1的速率扫描图像,对催化剂的晶体结构进行表征。采用扫描电子显微镜(SEM,德国ZEISS Gemini SEM 300)和透射电子显微镜(TEM,美国FEI Talos F200S)对催化剂的微观形貌进行分析。采用X射线衍射仪(XPS,美国Thermo Scientific K-Alpha)分析样品中元素的组成形态。通过拉曼光谱(Raman,日本Horiba LabRAM HR Evolution)分析催化剂的结构缺陷与石墨化程度。采用电子顺磁共振谱仪(EPR,英国Edinburgh FLS1000)检测了反应体系中产生的活性物种种类,其中以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)和2,2,6,6-四甲基哌啶(TEMP)作为自旋捕获剂。使用电化学工作站(CHI 660 B,中国上海辰华),在传统的三电极体系中考察催化剂的电化学性质。其中,铂丝和饱和甘汞电极(SCE)分别作为对电极和参比电极,反应液电解质为0.05 mol·L−1的Na2SO4水溶液。
1.4 降解实验与分析方法
选择四环素作为模拟目标污染物对催化剂活性进行考察。首先,称取一定量的催化剂分散于100 mL 10 mg·L−1四环素溶液中,并保持持续搅拌。待催化剂与四环素达到吸附脱附平衡后,加入一定浓度PMS开始降解反应,其间间隔一定时间取样3 mL并立即与甲醇混合停止反应。悬浮液经0.22 μm尼龙膜过滤后,采用紫外-可见分光光度计分析所得溶液中TC浓度变化,TC的吸收波长为358 nm。为了探究材料的重复利用效果,将反应后的催化剂经0.22 μm滤膜过滤和水洗后并置于70 ℃烘箱烘干,收集所得固体粉末进行重复降解实验。使用总有机碳分析仪(TOC,德国Jena Multi N/C 3100)测定湖水中TOC。使用离子色谱仪(Thermo Scientific ICS-600)检测湖水中常见阴离子的浓度。反应过程中PMS的测定方法为[17]:将0.05 g的碳酸氢钠与1 g的碘化钾溶于5 mL去离子水,加水至9 mL,按照降解实验取样时间采集1 mL四环素溶液过0.22 μm滤膜,与上述9 mL溶液混合,静置15 min后用紫外分光光度计检测395 nm的吸光度。
2. 结果与讨论
2.1 催化剂表征
图1为经高温热解法制备的PVCN的SEM和TEM图。从图1(a)和图1(b)可以发现,所制备的PVCN为二维层状纳米片形貌且片层表面光滑。与石墨烯等二维碳材料类似,该纳米片堆叠在一起,并且可明显发现其表面存在为减少表面能而形成的褶皱。透射电镜图(图1(c))证实了该碳材料由较薄的纳米片层组成,且选区电子衍射图显示该碳材料主要为无定型结构。但从PVCN材料的高分辨透射图(图1(d))可清晰的发现短程有序的晶格条纹,表明其具有一定的结晶性。其中,0.34 nm的晶格间距对应于石墨碳的(002)晶面[18],进一步表明所制备的PVCN中含有石墨碳结构。另外,PVCN的石墨化区域表现出大的石墨化晶格条纹间距和高曲率,表明其可能存在丰富缺陷和活性位点[19]。
采用XRD对所制备的PVCN的晶体结构进行分析。如图2(a)所示,在PVCN的XRD谱图中,均可发现位于26.3°和42.5° 2个较宽衍射峰,分别对应石墨碳的(002)晶面和(101)晶面[20],较宽的(002)表明其具有石墨碳结构[14],但结晶度不高,这与TEM的表征结果相对应。图2(b)为材料的拉曼光谱。可见,在1 354 cm−1和1 585 cm−1处均出现2个特征峰,分别为D峰和G峰。其中,D峰与碳缺陷或石墨相碳中异质原子掺杂导致的无序排列有关,而G峰则对应于sp2杂化的结晶性石墨化碳原子,通过两者的比值(ID/IG)可评价碳材料缺陷度或石墨化度。由图2(b)可知,3种掺杂比的ID/IG值从小到大的排列顺序为:100 PVCN<60 PVCN<30 PVCN,100 PVCN的缺陷最少,30 PVCN的缺陷最多。ID/IG的增加是由于N掺杂引起的碳材料中sp2杂化向sp3杂化的转变。随着N掺杂量的增大,材料的缺陷程度增加,说明在高温热解过程中,形成了石墨化缺陷[16]。之前研究表明,在碳材料活化PMS过程中,碳材料结构中石墨化缺陷可为催化剂和PMS之间提供更多的电子转移路径[21]。利用XPS对材料的元素组成及价态进行分析。如图2(c)所示,不同比例的PVCN材料均由碳、氮和氧组成。同时,随着PVC含量的增加,样品中的碳和氮元素的相对比例也逐渐升高,该结果证实了氮原子已成功掺杂碳材料中。另外,由图2(d)可见,在g=2.003处可观察到明显的特征峰信号,该信号与碳原子外围未成对电子含量有关[6],表明PVCN样品中含有氮空位。催化剂中的N来源于三聚氰胺,这种情况可能是由于样品在高温煅烧过程中,部分N转化成氨气后逃逸从而形成了氮空位[5],有研究[4]表明,氮空位的引入可促进碳材料对PMS的吸附,从而提升其活化PMS性能。
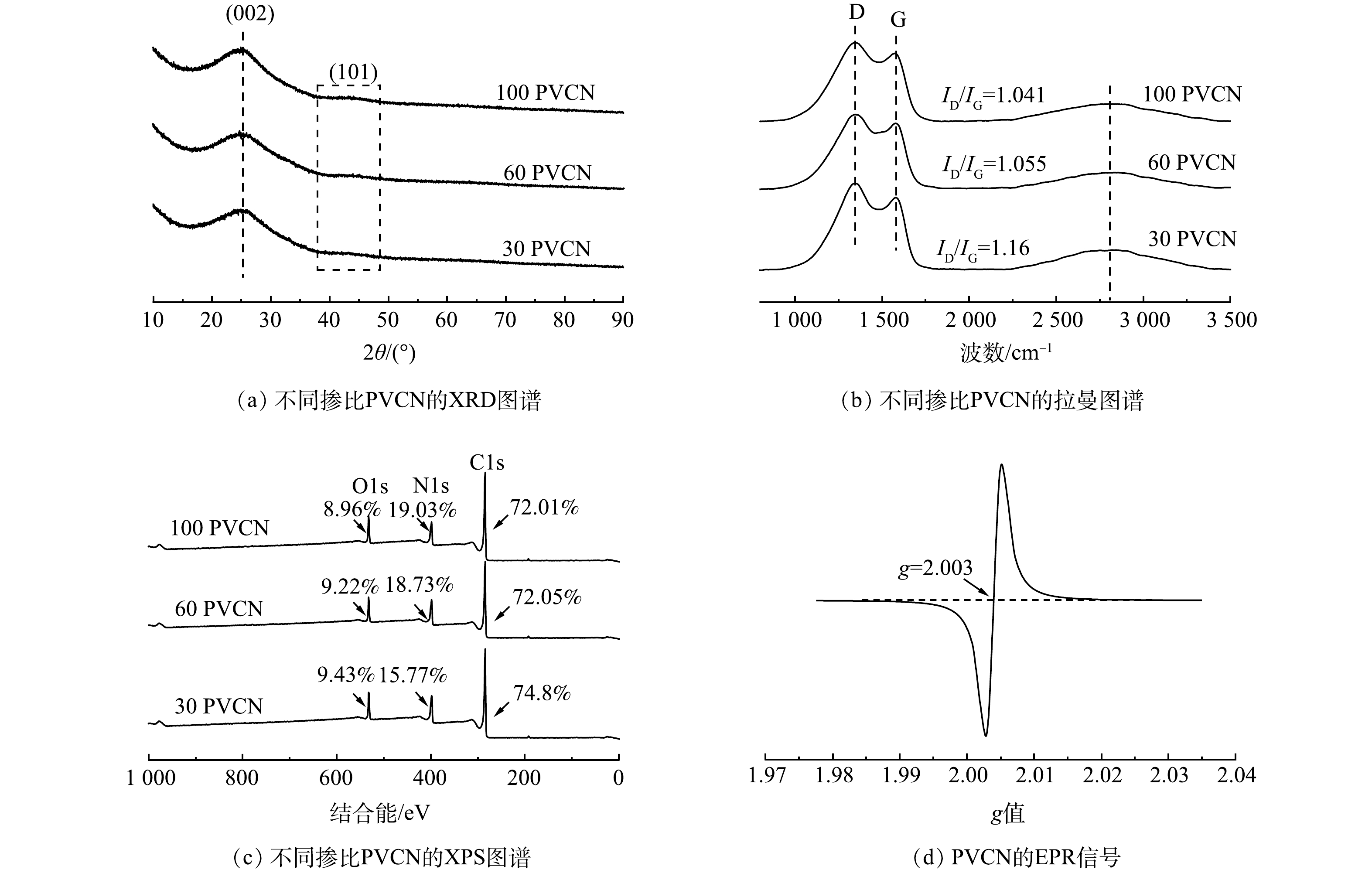 图 2 PVCN的XRD图谱、拉曼图谱、XPS图谱和EPR信号Figure 2. XRD patterns, Raman shift, XPS spectra and EPR signal of PVCN
图 2 PVCN的XRD图谱、拉曼图谱、XPS图谱和EPR信号Figure 2. XRD patterns, Raman shift, XPS spectra and EPR signal of PVCN2.2 PVCN活化PMS降解TC性能评价
图3为不同反应体系对TC去除率的影响。由图3(a)可知,单独PVCN对TC具有一定的吸附性能,经30 min吸附反应后基本达到吸附平衡。在反应阶段,当只有PMS存在时,TC在30 min内的去除率仅为25.8%,表明PMS具有一定的氧化分解TC能力,这可归因于PMS自身较弱的氧化能力。当只有PVCN存在时,经30 min反应后,溶液中TC的浓度基本保持不变。与之对比,当同时加入PVCN和PMS时,可明显发现TC的浓度急剧下降,PVCN对TC的去除率高达98%,其表观反应动力学常数k(0.106 min−1)为单独PMS(0.01 min−1)的10倍(图3(b))。同时,考察了不同PVC/三聚氰胺质量比的PVCN对TC去除效率的影响。随着PVC含量的增加,虽然所制备PVCN对TC的去除率呈现一定下降趋势(反应动力学常数由0.106 min−1衰减至0.66 min−1),但经30 min反应后,所有催化剂对TC去除效率仍然可维持在80%以上。该结果一方面表明催化剂中氮的相对含量对其活性有影响。另一方面也说明所制备的PVCN材料具有优异的活化PMS去除污染物TC的能力。
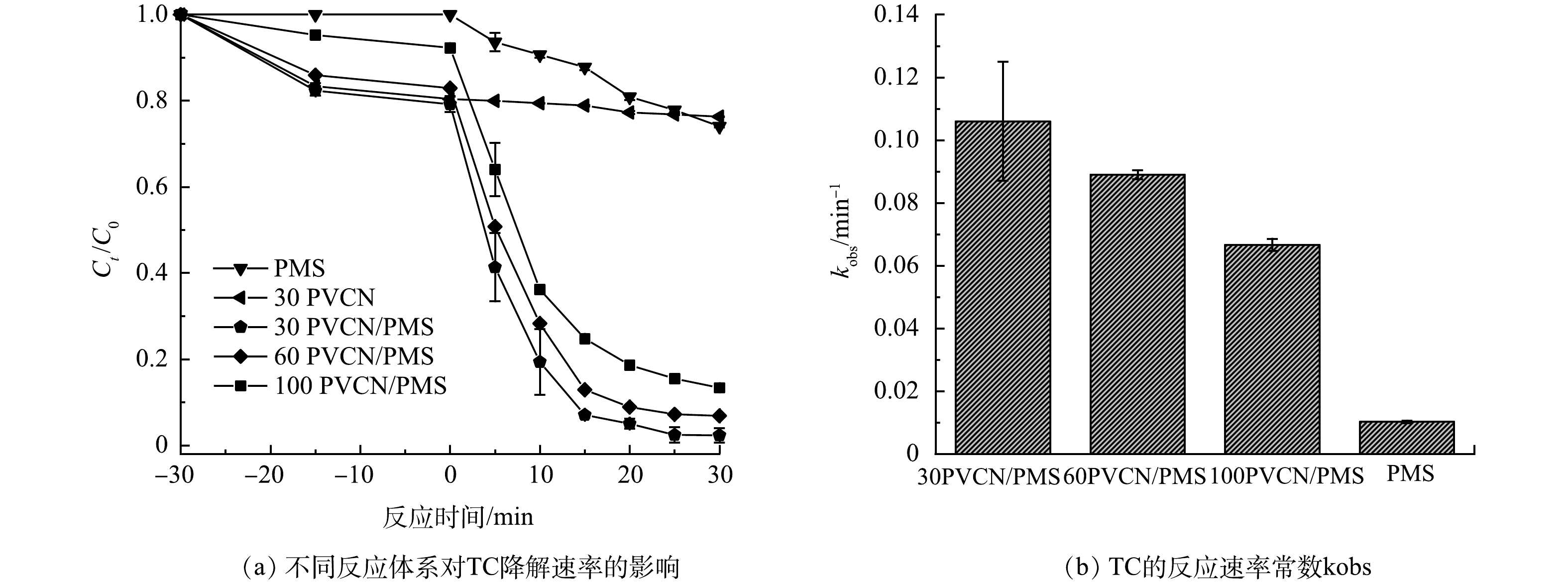 图 3 不同质量比PVCN和不同反应体系对TC降解效果的影响及一级动力学常数Figure 3. Effects of different catalytic reaction systems on TC degradation and first-order kinetic constants
图 3 不同质量比PVCN和不同反应体系对TC降解效果的影响及一级动力学常数Figure 3. Effects of different catalytic reaction systems on TC degradation and first-order kinetic constants2.3 不同条件对去除TC的影响
研究了不同催化剂投加量、pH、PMS浓度、阴离子干扰对四环素降解的影响。图4(a)为不同催化剂投加量对TC降解的影响。催化剂浓度一方面直接影响吸附性能,另一方面也会影响催化PMS活性位点的数量。随着催化剂投加量的增大,TC去除率呈现先增加后趋于稳定的趋势。当催化剂投加量为0.05 g·L−1时,反应30 min后,TC去除率为90%。而当催化剂投加量增至0.1 g·L−1时,TC去除率可达到98%。进一步增加催化剂投加量至1.5 g·L−1时,TC去除率稍微降至96%。这主要归因于随着催化剂投加量的增大,可提供更多的PMS活性点位,促使体系中产生更多的活性自由基[22],从而加速TC的降解。
由图3的对比实验结果可知,引入PMS后,PVCN对TC的降解效果得到了明显提升。因此,考察了不同PMS浓度对TC降解活性的影响。如图4(b)所示,随着PMS浓度的增加,TC的去除率呈现先增大后减小的趋势。当PMS浓度由0 mmol·L−1增至1 mmol·L−1时,TC的降解效果逐渐提高,去除率从92%增加至98%。而当PMS浓度由1 mmol·L−1进一步增至1.5 mmol·L−1时,去除率提高有限,这主要是由于PMS浓度由1 mmol·L−1增至1.5 mmol·L−1时,反应体系中产生了过多SO4·−,过量的SO4·−自由基之间会发生自淬反应或与HSO5−发生反应,生成氧化能力较弱的SO5·−自由基,从而导致参与反应的SO4·−减少,并使反应活性减弱[23]。
进一步考察了不同pH对TC降解的影响。如图4(c)所示,其中,当反应液初始pH为7时,TC去除率最高可达98%;当pH=3时,TC去除率相对有所减弱,为94%。这主要归因于在酸性条件下,HSO5−与过量的H+可形成更稳定的H2SO5,不利于催化剂与其反应生成SO4·−[24];当反应液初始pH为碱性时(pH=11),催化剂对TC去除效率为95.9%,这可能是由于在碱性条件下,一些SO4·−与OH−反应生成寿命较短、氧化还原电位较低、选择性较低的·OH。因此,高浓度的OH−可能会淬灭SO4·−并降低TC的去除效率[25]。总之,由图4(c)中可发现反应液pH对TC降解效果无明显影响,经过30 min反应后,对TC的去除率均达到90%以上。该结果表明所制备的PVCN碳材料具有在较宽的pH范围内活化PMS的能力,有利于其在实际工况中的应用。
实际水体环境中,常存在多种的无机阴离子,他们的存在会影响催化剂对PMS的活化以及自由基的生成,从而抑制或促进污染物的降解,因此,考察了不同阴离子对反应体系的影响。由图4(d)可知,当向反应液中分别投加10 mmol·L−1阴离子时,可以观察到除HCO3−和H2PO4−外,其他阴离子均对催化剂的活性表现出一定的抑制作用。其中,Cl−可能与SO4·−反应生成Cl·、Cl2·−和HClO,导致SO4·−的消耗,从而抑制TC的降解[26-27]。HCO3−是过硫酸盐氧化过程中极好的自由基清除剂,可同时消除反应体系中的SO4·−和·OH,然而,本实验中HCO3−的存在促进了TC的降解,这可能是因为HCO3−具有缓冲作用,可使反应pH保持在7左右,并且HCO3−与SO4·−能反应生成稳定性更高的·CO3−,从而提高了PVCN对TC的氧化还原能力[25-26]。SO42−对TC的降解产生了轻微的抑制作用,表明PVCN/PMS对SO42−有较好的耐受性。
2.4 催化剂稳定性
催化剂的稳定性是催化剂性能的一个重要指标。本研究中,采用重复性实验对PVCN的稳定性进行考察。如图5(a)所示,经5次重复实验后,PVCN催化剂对TC的降解性仍可维持在80%以上。此外,从图5(b)和图5(c)的催化剂反应前后XPS和XRD图谱可知,PVCN在反应后元素组成和晶体结构均未改变,表明该复合催化剂具备较高稳定性,具备一定的实际应用前景。
2.5 在实际水体中的应用
考察了在不同水质的实际水体中催化剂活化PMS去除TC性能的情况。如图6所示,在湖水和自来水中,所制备的氮掺杂碳纳米片催化剂均表现出较好的活化PMS去除TC的性能,对TC的去除率分别为94.7%和94.1%。由于湖水中的共存物质可能会对影响降解效果,因此,对湖水水质进行分析,湖水中TOC为15.48 mg·L−1,SO4·−、Cl− 、NO3−、PO43−和HCO3−的质量浓度分别为76.91、65.66、1.03、0.87和4.03 mg·L−1。上述实验结果证明SO4·−和Cl−均能对TC的降解产生干扰,导致催化剂对TC的降解性能有所减弱。尽管如此,该催化剂在湖水和自来水中仍具有优异的活化PMS去除TC的性能,表明其具有一定的实际应用前景。
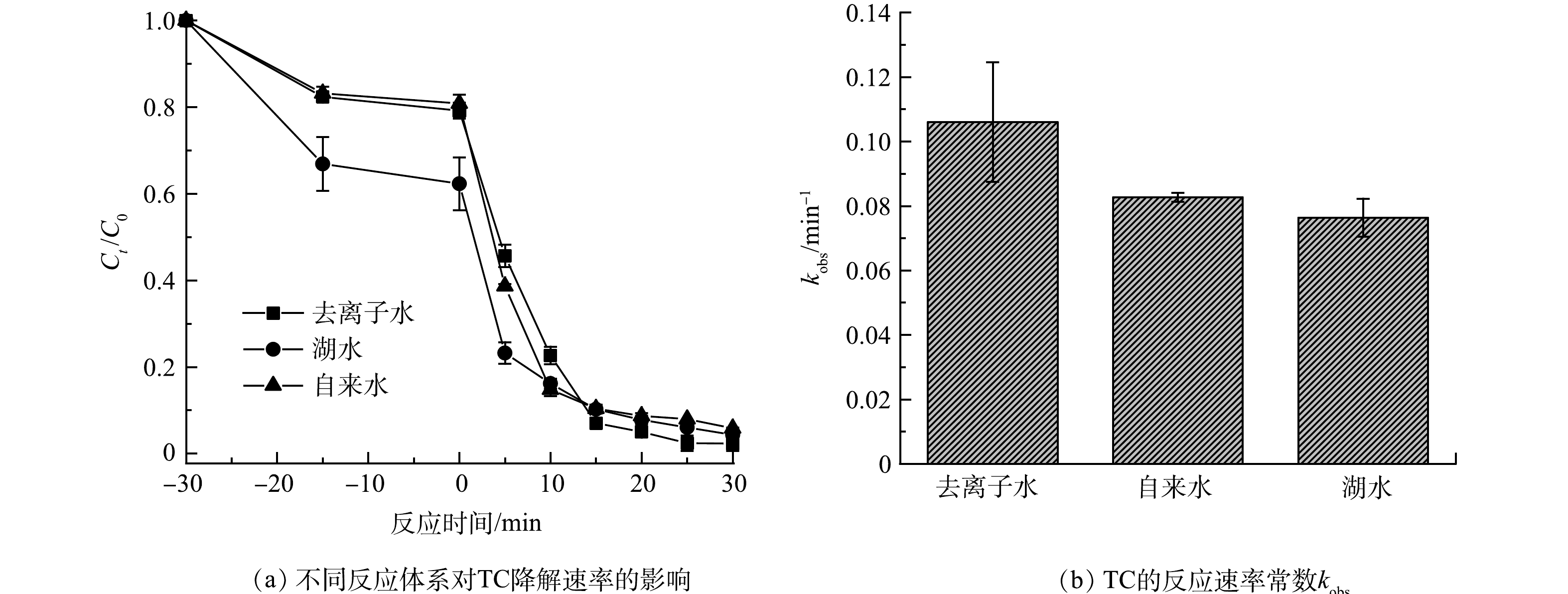 图 6 不同水质对降解TC效能的影响Figure 6. Effect of different water quality on the efficacy of TC degradation
图 6 不同水质对降解TC效能的影响Figure 6. Effect of different water quality on the efficacy of TC degradation2.6 催化机理分析
为了确定体系中所产生自由基的种类,首先采用自由基淬灭实验,考察不同自由基对降解反应的贡献。有研究[28]表明,在羟基自由基(·OH)与硫酸根自由基(SO4·−)同时存在的条件下,由于叔丁醇(TBA)与羟基自由基(·OH)反应比硫酸根自由基(SO4·−)快,可选择性地淬灭体系中的·OH。而甲醇(MeOH)则可与·OH与SO4·−快速反应。因此,采用MeOH与TBA作为自由基淬灭剂来考察反应体系中·OH与SO4·−对TC去除的贡献情况。对苯醌(p-BQ)和L-组氨酸(L-his)作为超氧自由基(·O2−)和单线态氧(1O2)的淬灭剂[29]。由图7(a)可知,当向体系中加入5 mmol·L−1TBA后,TC的去除率由98%减至89%,表明在该反应体系中·OH的贡献较小。同时,5 mmol·L−1 MeOH的加入对催化活性无明显抑制作用,图7(c)显示了反应体系中PMS的消耗量,反应30 min后,PMS浓度仅利用了42.7%,说明体系内生成了较少的SO4·−[30],由此可推断在该反应体系中,·OH和SO4·−不是降解TC的主要活性物种。另一方面,对比分别加入5 mmol·L−1 p-BQ与L-his的结果可知,p-BQ与L-his对TC的降解均起到了明显的抑制作用,表明体系中发挥主要作用的活性物种为1O2及·O2−。以上结果表明,所制备氮掺杂碳材料活化PMS起主导作用的活性物种为1O2和·O2−,这与前人报道的氮掺杂石墨化碳活化PMS降解污染物过程类似[31]。
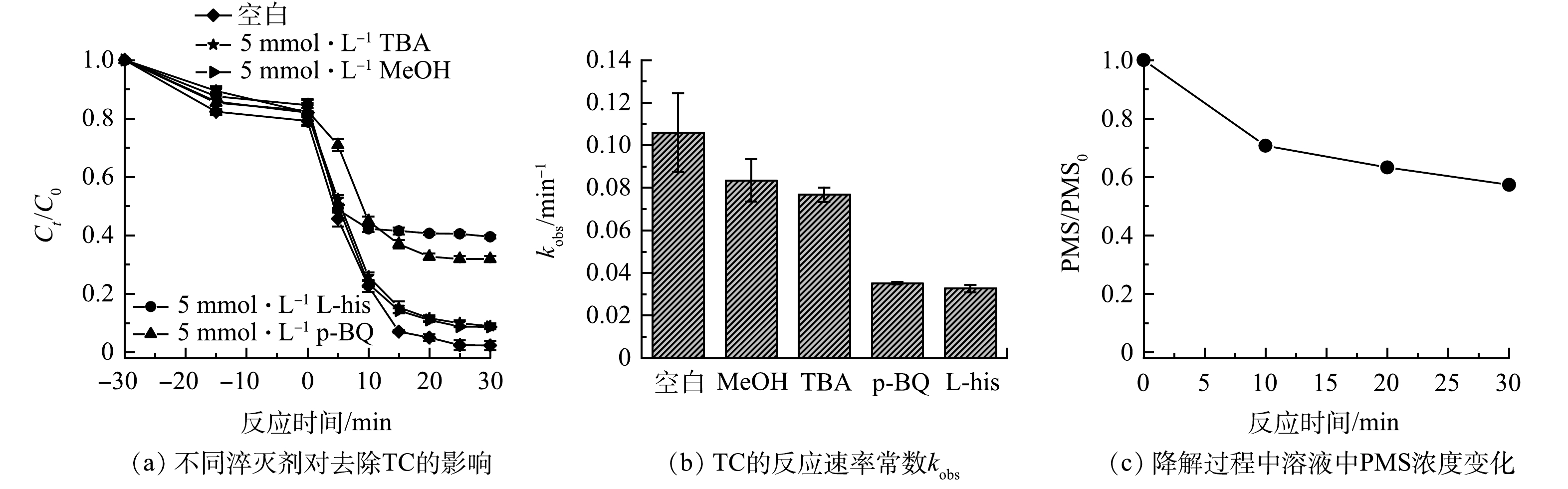 图 7 不同淬灭剂对降解TC效能的影响Figure 7. Influence of the degradation of TC in different radical scavengers
图 7 不同淬灭剂对降解TC效能的影响Figure 7. Influence of the degradation of TC in different radical scavengers另外,采用EPR对反应过程中产生的活性物种进行分析。由图8(a)可知,当催化剂和PMS共同存在时,在反应体系中能明显检测到归属于TEMP-1O2加合物的三重特征峰信号(1:1:1),进一步证明了反应过程中有1O2的产生。同时,随着反应进行,该特征峰信号逐渐加强,间接表明所产生1O2的量不断增多。另外,由图8(b)中可明显观察到属于DMPOX的特征峰(1:2:1:2:1:2:1),而未捕获到归属于DMPO-·OH和DMPO-SO4·−加合物的特征峰。这可归因于催化剂与PMS反应可直接将DMPO氧化成DMPOX,这与之前报告的氮掺杂碳材料活化PMS的结果一致,说明在该过程中存在着电子转移方式的非自由基活化PMS过程[22]。图8(c)反映了超氧自由基的EPR信号。反应5 min后,可以观察到明显的DMPO-O2·−加合物的六重特征峰,且随着反应的进行(10 min后),该峰强度进一步提高,表明体系中不断有超氧自由基的生成[32]。以上测试进一步验证了自由基捕获实验结果,在反应过程中产生了单线态氧和超氧自由基。
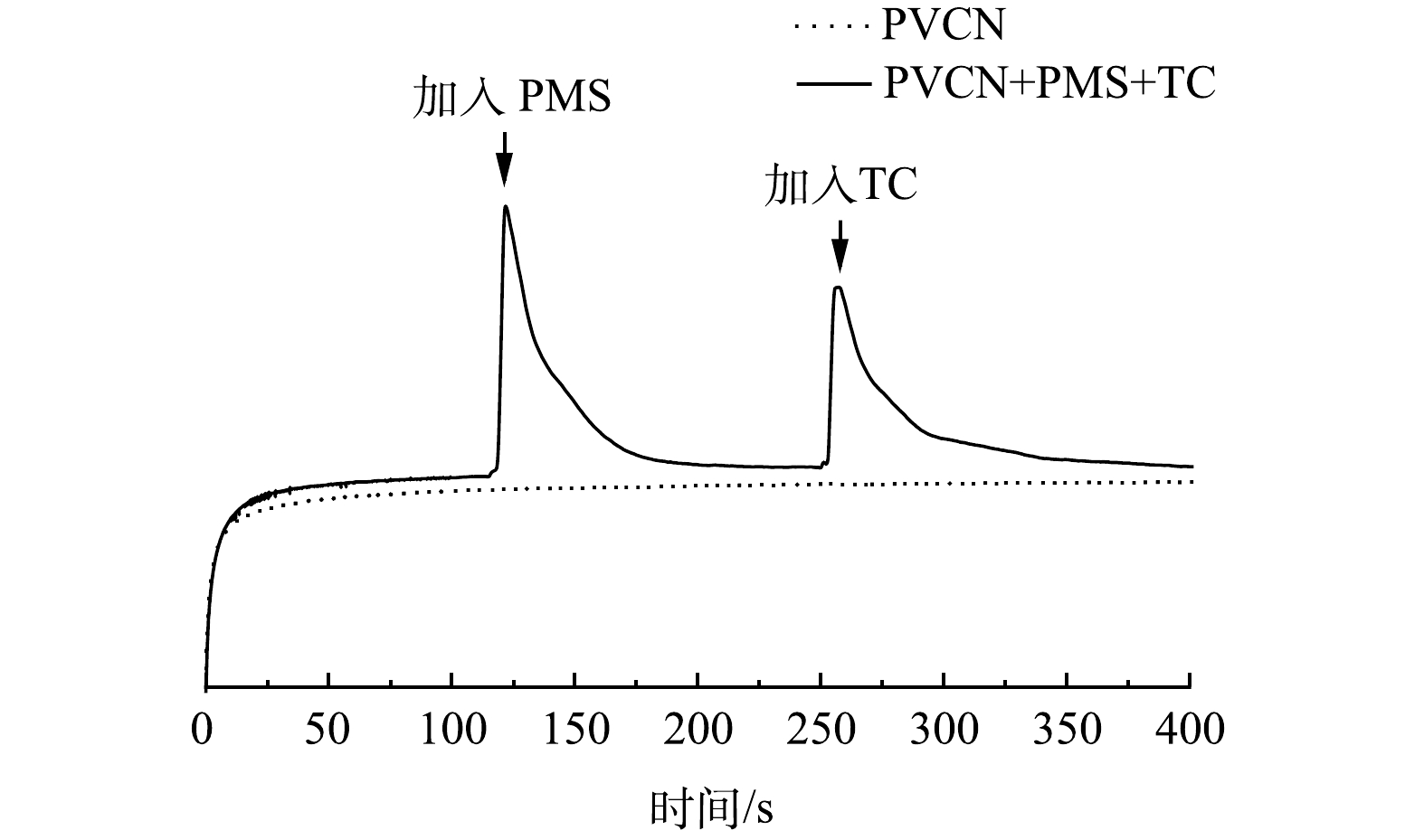
基于实验结果可知,催化剂中碳氮相对含量对其活化PMS去除TC效果有明显影响。因此,利用XPS对比分析了不同比例的PVCN中C和N成键情况。图9(a)为C1s的高分辨图,从中可以看出,30 PVCN中C=O或C=N键的相对占比要高于60 PVCN样品和100 PVCN,说明其含有更多C=O或C=N。有研究表明,碳材料中的C=O和C=N键可以作为PMS的活性位点[7],将PMS活化成1O2从而达到对污染物的降解[10,33]。因此,30 PVCN可提供更多活化PMS的位点。另外,由N1s的高分辨图谱(图9(b))可发现位于398.7、399.7、400.9和404.2 eV的4个拟合峰,分别对应于吡啶氮、吡咯氮、石墨氮和氮氧化物[34]。之前报道证实在氮掺杂碳材料中,吡啶氮和石墨氮可作为PMS的活性位点[35]。其中,石墨氮通过削弱碳电子云密度,改善四环素分子与π电子之间的相互作用,促进对四环素的吸附[17],而吡啶氮可作为PMS的吸附位点[4,36]。相比于60 PVCN和100 PVCN,30 PVCN中吡啶氮和石墨氮的占比最高,说明其可提供更多的活性位点,从而表现出更高的催化活性。另外,采用电化学计时电流方法,对所制备的PVCN材料与PMS和TC之间的电子转移行为进行研究。由图10可知,当加入PMS时会立即引起电流的增加,而加入TC后也会导致电流增加。这说明TC可通过PVCN将电子转移到PMS[37],该结果证明在PVCN/PMS体系中,电子转移作用也参与到TC的降解过程中。综上所述,30 PVCN活化PMS的途径主要通过以1O2和电子转移的非自由基以及·O2−的自由基方式进行,最终实现高效去除TC。
3. 结论
1)采用高温热处理法,以废弃PVC和三聚氰胺制备了氮掺杂碳纳米片催化剂,该催化剂具备较优的活化PMS降解TC的能力。在PMS浓度为1 mmol·L−1,30 min反应后,该PVCN/PMS体系对TC的去除率达到98%。
2)考察了PVC相对含量、PMS浓度、催化剂投加量和反应液pH等实验条件对催化活性的影响。当PVC/三聚氰胺为30%、PMS浓度为1 mmol·L−1及催化剂投加量为0.1 g·L−1时,该复合催化剂催化活性达到最优。同时,催化剂在宽pH范围内均具有较好的活化PMS去除TC的活性。
3)反应机理研究结果表明,该催化剂活化PMS去除TC反应过程中,其活化PMS的方式主要通过以1O2和电子转移为主的非自由基过程以及·O2−的自由基过程实现对TC的高效去除。同时,该复合催化剂具备较好的稳定性,且能适应不同水体环境,具备较好的应用前景。
-
图 2 PVCN的XRD图谱、拉曼图谱、XPS图谱和EPR信号
Figure 2. XRD patterns, Raman shift, XPS spectra and EPR signal of PVCN
图 3 不同质量比PVCN和不同反应体系对TC降解效果的影响及一级动力学常数
Figure 3. Effects of different catalytic reaction systems on TC degradation and first-order kinetic constants
图 6 不同水质对降解TC效能的影响
Figure 6. Effect of different water quality on the efficacy of TC degradation
图 7 不同淬灭剂对降解TC效能的影响
Figure 7. Influence of the degradation of TC in different radical scavengers
-
[1] LEICHTWEIS J, VIEIRA Y, WELTER N, et al. A review of the occurrence, disposal, determination, toxicity and remediation technologies of the tetracycline antibiotic[J]. Process Safety and Environmental Protection, 2022, 160: 25-40. doi: 10.1016/j.psep.2022.01.085 [2] LIN D, FU Y, LI X, et al. Application of persulfate-based oxidation processes to address diverse sustainability challenges: A critical review[J]. Journal of Hazardous Materials, 2022, 440: 129722. doi: 10.1016/j.jhazmat.2022.129722 [3] GAO Y, LI T, ZHU Y, et al. Highly nitrogen-doped porous carbon transformed from graphitic carbon nitride for efficient metal-free catalysis[J]. Journal of Hazardous Materials. 2020, 393: 121280. [4] WANG G, LIU Y, DONG X, et al. Transforming radical to non-radical pathway in peroxymonosulfate activation on nitrogen doped carbon sphere for enhanced removal of organic pollutants: Combined effect of nitrogen species and carbon structure[J]. Journal of Hazardous Materials, 2022, 437: 129357. doi: 10.1016/j.jhazmat.2022.129357 [5] KHAN A, GOEPEL M, COLMENARES J C, et al. Chitosan-based N-doped carbon materials for electrocatalytic and photocatalytic applications[J]. ACS Sustainable Chemistry Engineering, 2020, 8: 4708-4727. doi: 10.1021/acssuschemeng.9b07522 [6] QU G, JIA P, TANG S, et al. Enhanced peroxymonosulfate activation via heteroatomic doping defects of pyridinic and pyrrolic N in 2D N-doped carbon nanosheets for BPA degradation[J]. Journal of Hazardous Materials, 2024, 461: 132626. doi: 10.1016/j.jhazmat.2023.132626 [7] HU S, JOHNSON D M, JIANG M, et al. The effect of polyvinyl chloride (PVC) color on biofilm development and biofilm-heavy metal chemodynamics in the aquatic environment[J]. Science of the Total Environment, 2023, 905: 166924. doi: 10.1016/j.scitotenv.2023.166924 [8] MA W, ZHU Y, CAI N, et al. Preparation of carbon nanotubes by catalytic pyrolysis of dechlorinated PVC[J]. Waste Management, 2023, 169: 62-69. doi: 10.1016/j.wasman.2023.06.034 [9] SILVA D E P, FRAGAL V H, FRAGAL E H, et al. Sustainable energy and waste management: How to transform plastic waste into carbon nanostructures for electrochemical supercapacitors[J]. Waste Management, 2023, 171: 71-85. doi: 10.1016/j.wasman.2023.08.028 [10] LUO H, FU H, YIN H, et al. Carbon materials in persulfate-based advanced oxidation processes: The roles and construction of active sites[J]. Journal of Hazardous Materials, 2022, 426: 128044. doi: 10.1016/j.jhazmat.2021.128044 [11] YAO N, WANG X, YANG Z, et al. Characterization of solid and liquid carbonization products of polyvinyl chloride (PVC) and investigation of the PVC-derived adsorbent for the removal of organic compounds from water[J]. Journal of Hazardous Materials, 2023, 456: 131687. doi: 10.1016/j.jhazmat.2023.131687 [12] WANG W, CAO Y, HU X, et al. Granular reduced graphene oxide/Fe3O4 hydrogel for efficient adsorption and catalytic oxidation of p-perfluorous nonenoxybenzene sulfonate[J]. Journal of Hazardous Materials, 2020, 386: 121662. doi: 10.1016/j.jhazmat.2019.121662 [13] YANG F M, LIU X, LI M B, et al. Polyvinyl chloride (PVC) derived microporous carbons prepared via hydrothermal dechlorination and potassium hydroxide activation for efficient CO2 capture[J]. Renewable and Sustainable Energy Reviews, 2023, 180: 113279. doi: 10.1016/j.rser.2023.113279 [14] MIAO J, ZHU Y, WEI Y, et al. Plastic wastes-derived N-doped carbon nanotubes for efficient removal of sulfamethoxazole in high salinity wastewater via nonradical peroxymonosulfate activation[J]. Journal of Hazardous Materials, 2024, 465: 133344. doi: 10.1016/j.jhazmat.2023.133344 [15] REN S, XU X, ZHU Z S, et al. Catalytic transformation of microplastics to functional carbon for catalytic peroxymonosulfate activation: Conversion mechanism and defect of scavenging[J]. Applied Catalysis B:Environmental, 2024, 342: 123410. doi: 10.1016/j.apcatb.2023.123410 [16] WEI S, QIU Y, SUN X, et al. Sustainable nanoporous carbon catalysts derived from melamine assisted cross-linking of poly (vinyl chloride) waste for acetylene hydrochlorination[J]. ACS Sustainable Chemistry Engineering, 2022, 10: 10476-10485. doi: 10.1021/acssuschemeng.2c01183 [17] 罗宝坚. 氮掺杂及硼氮共掺杂碳材料活化过硫酸盐降解四环素研究[D]. 广州: 广州大学, 2023. [18] ZHAO C, MENG L, CHU H, et al. Ultrafast degradation of emerging organic pollutants via activation of peroxymonosulfate over Fe3C/Fe@N-C-x: Singlet oxygen evolution and electron-transfer mechanisms[J]. Applied Catalysis B:Environmental, 2023, 321: 122034. doi: 10.1016/j.apcatb.2022.122034 [19] CHENG D, WANG Z, CHEN C, et al. Defect-enriched, nitrogen-doped graphitic carbon microspheres within 3D interconnected super-macropores as efficient oxygen electrocatalysts for breathing Zn-Air battery[J]. Carbon, 2019, 145: 38-46. doi: 10.1016/j.carbon.2019.01.010 [20] QI M Y, ZHANG Y, LI Q, et al. Experimental and theoretical insights into metal-free catalytic reduction of nitrophenols over porous nitrogen-doped reduced graphene oxide[J]. Chemical Engineering Journal, 2023, 474: 145823. doi: 10.1016/j.cej.2023.145823 [21] TIAN H, CUI K, SUN S, et al. Sequential doping of exogenous iron and nitrogen to prepare Fe-N structured biochar to enhance the activity of OTC degradation[J]. Separation and Purification Technology, 2023, 322: 124249. doi: 10.1016/j.seppur.2023.124249 [22] GENG G, GAO Y, ZHANG Z, et al. Renewable and robust biomass waste-derived Co-doped carbon aerogels for PMS activation: Catalytic mechanisms and phytotoxicity assessment[J]. Ecotoxicology and Environmental Safety, 2021, 220: 112381. doi: 10.1016/j.ecoenv.2021.112381 [23] MA H, XU S, ZHANG X, et al. N-doped coal-based carbon membrane coupling peroxymonosulfate activation for bisphenol A degradation: The role of micro-carbon structure and nitrogen species[J]. Journal of Cleaner Production, 2023, 423: 138713. doi: 10.1016/j.jclepro.2023.138713 [24] NI T, ZHANG H, YANG Z, et al. Enhanced adsorption and catalytic degradation of antibiotics by porous 0D/3D Co3O4/g-C3N4 activated peroxymonosulfate: An experimental and mechanistic study[J]. Journal of Colloid and Interface Science, 2022, 625: 466-478. doi: 10.1016/j.jcis.2022.06.057 [25] YIN C, LIU Y, KANG X, et al. Synergistic degradation of tetracycline by CDs decorated g-C3N4 under LED light irradiation combined with the persulfate-based advanced oxidation process[J]. Applied Catalysis A:General, 2022, 636: 118571. doi: 10.1016/j.apcata.2022.118571 [26] ZOU H, LIU Y, NI L, et al. Enhanced degradation of tetracycline via visible-light-assisted peroxymonosulfate activation over oxygen vacancy rich Fe2O3-CoFe2O4 heterostructures[J]. Separation and Purification Technology, 2023, 314: 123586. doi: 10.1016/j.seppur.2023.123586 [27] YU Y, CHEN D, XU W, et al. Synergistic adsorption-photocatalytic degradation of different antibiotics in seawater by a porous g-C3N4/calcined-LDH and its application in synthetic mariculture wastewater[J]. Journal of Hazardous Materials, 2021, 416: 126183. doi: 10.1016/j.jhazmat.2021.126183 [28] ZHANG C, QIN D, ZHOU Y, et al. Dual optimization approach to Mo single atom dispersed g-C3N4 photocatalyst: Morphology and defect evolution[J]. Applied Catalysis B:Environmental, 2022, 303: 120904. doi: 10.1016/j.apcatb.2021.120904 [29] ZHUO S N, SUN H, WANG Z Y, et al. A magnetic biochar catalyst with dual active sites of Fe3C and Fe4N derived from floc: The activation mechanism for persulfate on degrading organic pollutant[J]. Chemical Engineering Journal, 2023, 455: 140702. doi: 10.1016/j.cej.2022.140702 [30] LIU M, NING Y, REN M, et al. Internal electric field-modulated charge migration behavior in MoS2 /MIL‐53(Fe) S‐scheme heterojunction for boosting visible‐light‐driven photocatalytic chlorinated antibiotics degradation[J]. Small, 2023, 19: 2303876. doi: 10.1002/smll.202303876 [31] LI K, MA S L, XU S J, et al. The mechanism changes during bisphenol A degradation in three iron functionalized biochar/peroxymonosulfate systems: The crucial roles of iron contents and graphitized carbon layers[J]. Journal of Hazardous Materials, 2021, 404: 124145. doi: 10.1016/j.jhazmat.2020.124145 [32] LI J, NI Z B, GAO Q Z, et al. Core-shell structured cobalt–nickel bimetallic sulfide with dual redox cycles to activate peroxymonosulfate for glyphosate removal[J]. Chemical Engineering Journal, 2023, 453: 139972. doi: 10.1016/j.cej.2022.139972 [33] WANG L D, WANG Y N, WANG Z X, et al. Proton transfer triggered in-situ construction of C=N active site to activate PMS for efficient autocatalytic degradation of low-carbon fatty amine[J]. Water Research, 2023, 240: 120119. doi: 10.1016/j.watres.2023.120119 [34] WU D, SONG W Y, CHEN L L, et al. High-performance porous graphene from synergetic nitrogen doping and physical activation for advanced nonradical oxidation[J]. Journal of Hazardous Materials, 2020, 381: 121010. doi: 10.1016/j.jhazmat.2019.121010 [35] LAN J H, ZHANG Q, YANG G X, et al. Removal of tetracycline hydrochloride by N, P co-doped carbon encapsulated Fe2P activating PMS: A non-radical pathway dominated by singlet oxygen[J]. Journal of Environmental Chemical Engineering, 2023, 11: 110481. doi: 10.1016/j.jece.2023.110481 [36] 张朋朋, 氮掺杂碳材料活化过硫酸盐降解水中酚类有机污染物[D]. 长春: 吉林大学, 2019. [37] XIE J L, PAN X F, JIANG C M, et al. Enhanced conversion of superoxide radical to singlet oxygen in peroxymonosulfate activation by metal-organic frameworks derived heteroatoms dual-doped porous carbon catalyst[J]. Environmental Research, 2023, 236: 116745. doi: 10.1016/j.envres.2023.116745 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1889
- HTML全文浏览数: 1889
- PDF下载数: 67
- 施引文献: 0
