


</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M15">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{3}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M15.jpg"><img class="graphic" src="202307113-wanghengxin_M15.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M16">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{2}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M16.jpg"><img class="graphic" src="202307113-wanghengxin_M16.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M17">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{1}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M17.jpg"><img class="graphic" src="202307113-wanghengxin_M17.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M18">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{2}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M18.jpg"><img class="graphic" src="202307113-wanghengxin_M18.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">5</td>
<td align="center" valign="middle">335.16</td>
<td align="center" valign="middle">79.35</td>
<td align="center" valign="middle">74.87</td>
<td align="center" valign="middle">91.31</td>
<td align="center" valign="middle">243.84</td>
</tr>
<tr>
<td align="center" valign="middle">24</td>
<td align="center" valign="middle">394.68</td>
<td align="center" valign="middle">101.39</td>
<td align="center" valign="middle">88.34</td>
<td align="center" valign="middle">128.47</td>
<td align="center" valign="middle">266.2</td>
</tr>
<tr>
<td align="center" valign="middle">36</td>
<td align="center" valign="middle">468.91</td>
<td align="center" valign="middle">103.59</td>
<td align="center" valign="middle">94.3</td>
<td align="center" valign="middle">124.77</td>
<td align="center" valign="middle">344.14</td>
</tr>
<tr>
<td align="center" valign="middle">48</td>
<td align="center" valign="middle">407.25</td>
<td align="center" valign="middle">102.71</td>
<td align="center" valign="middle">93.03</td>
<td align="center" valign="middle">124.46</td>
<td align="center" valign="middle">282,79</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">72</td>
<td align="center" valign="middle" class="table_bottom_border">376.14</td>
<td align="center" valign="middle" class="table_bottom_border">96.54</td>
<td align="center" valign="middle" class="table_bottom_border">84.47</td>
<td align="center" valign="middle" class="table_bottom_border">121.76</td>
<td align="center" valign="middle" class="table_bottom_border">254.38</td>
</tr>
</tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M15">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{3}^{-}} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">饥饿时长/h</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M14">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{0}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M14.jpg"><img class="graphic" src="202307113-wanghengxin_M14.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M15">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{3}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M15.jpg"><img class="graphic" src="202307113-wanghengxin_M15.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M16">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{2}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M16.jpg"><img class="graphic" src="202307113-wanghengxin_M16.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M17">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{1}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M17.jpg"><img class="graphic" src="202307113-wanghengxin_M17.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M18">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{2}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M18.jpg"><img class="graphic" src="202307113-wanghengxin_M18.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">5</td>
<td align="center" valign="middle">335.16</td>
<td align="center" valign="middle">79.35</td>
<td align="center" valign="middle">74.87</td>
<td align="center" valign="middle">91.31</td>
<td align="center" valign="middle">243.84</td>
</tr>
<tr>
<td align="center" valign="middle">24</td>
<td align="center" valign="middle">394.68</td>
<td align="center" valign="middle">101.39</td>
<td align="center" valign="middle">88.34</td>
<td align="center" valign="middle">128.47</td>
<td align="center" valign="middle">266.2</td>
</tr>
<tr>
<td align="center" valign="middle">36</td>
<td align="center" valign="middle">468.91</td>
<td align="center" valign="middle">103.59</td>
<td align="center" valign="middle">94.3</td>
<td align="center" valign="middle">124.77</td>
<td align="center" valign="middle">344.14</td>
</tr>
<tr>
<td align="center" valign="middle">48</td>
<td align="center" valign="middle">407.25</td>
<td align="center" valign="middle">102.71</td>
<td align="center" valign="middle">93.03</td>
<td align="center" valign="middle">124.46</td>
<td align="center" valign="middle">282,79</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">72</td>
<td align="center" valign="middle" class="table_bottom_border">376.14</td>
<td align="center" valign="middle" class="table_bottom_border">96.54</td>
<td align="center" valign="middle" class="table_bottom_border">84.47</td>
<td align="center" valign="middle" class="table_bottom_border">121.76</td>
<td align="center" valign="middle" class="table_bottom_border">254.38</td>
</tr>
</tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M16">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{2}^{-}} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">饥饿时长/h</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M14">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{0}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M14.jpg"><img class="graphic" src="202307113-wanghengxin_M14.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M15">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{3}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M15.jpg"><img class="graphic" src="202307113-wanghengxin_M15.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M16">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{2}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M16.jpg"><img class="graphic" src="202307113-wanghengxin_M16.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M17">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{1}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M17.jpg"><img class="graphic" src="202307113-wanghengxin_M17.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M18">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{2}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M18.jpg"><img class="graphic" src="202307113-wanghengxin_M18.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">5</td>
<td align="center" valign="middle">335.16</td>
<td align="center" valign="middle">79.35</td>
<td align="center" valign="middle">74.87</td>
<td align="center" valign="middle">91.31</td>
<td align="center" valign="middle">243.84</td>
</tr>
<tr>
<td align="center" valign="middle">24</td>
<td align="center" valign="middle">394.68</td>
<td align="center" valign="middle">101.39</td>
<td align="center" valign="middle">88.34</td>
<td align="center" valign="middle">128.47</td>
<td align="center" valign="middle">266.2</td>
</tr>
<tr>
<td align="center" valign="middle">36</td>
<td align="center" valign="middle">468.91</td>
<td align="center" valign="middle">103.59</td>
<td align="center" valign="middle">94.3</td>
<td align="center" valign="middle">124.77</td>
<td align="center" valign="middle">344.14</td>
</tr>
<tr>
<td align="center" valign="middle">48</td>
<td align="center" valign="middle">407.25</td>
<td align="center" valign="middle">102.71</td>
<td align="center" valign="middle">93.03</td>
<td align="center" valign="middle">124.46</td>
<td align="center" valign="middle">282,79</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">72</td>
<td align="center" valign="middle" class="table_bottom_border">376.14</td>
<td align="center" valign="middle" class="table_bottom_border">96.54</td>
<td align="center" valign="middle" class="table_bottom_border">84.47</td>
<td align="center" valign="middle" class="table_bottom_border">121.76</td>
<td align="center" valign="middle" class="table_bottom_border">254.38</td>
</tr>
</tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M17">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{1}} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">饥饿时长/h</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M14">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{0}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M14.jpg"><img class="graphic" src="202307113-wanghengxin_M14.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M15">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{3}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M15.jpg"><img class="graphic" src="202307113-wanghengxin_M15.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M16">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{2}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M16.jpg"><img class="graphic" src="202307113-wanghengxin_M16.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M17">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{1}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M17.jpg"><img class="graphic" src="202307113-wanghengxin_M17.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M18">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{2}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M18.jpg"><img class="graphic" src="202307113-wanghengxin_M18.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">5</td>
<td align="center" valign="middle">335.16</td>
<td align="center" valign="middle">79.35</td>
<td align="center" valign="middle">74.87</td>
<td align="center" valign="middle">91.31</td>
<td align="center" valign="middle">243.84</td>
</tr>
<tr>
<td align="center" valign="middle">24</td>
<td align="center" valign="middle">394.68</td>
<td align="center" valign="middle">101.39</td>
<td align="center" valign="middle">88.34</td>
<td align="center" valign="middle">128.47</td>
<td align="center" valign="middle">266.2</td>
</tr>
<tr>
<td align="center" valign="middle">36</td>
<td align="center" valign="middle">468.91</td>
<td align="center" valign="middle">103.59</td>
<td align="center" valign="middle">94.3</td>
<td align="center" valign="middle">124.77</td>
<td align="center" valign="middle">344.14</td>
</tr>
<tr>
<td align="center" valign="middle">48</td>
<td align="center" valign="middle">407.25</td>
<td align="center" valign="middle">102.71</td>
<td align="center" valign="middle">93.03</td>
<td align="center" valign="middle">124.46</td>
<td align="center" valign="middle">282,79</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">72</td>
<td align="center" valign="middle" class="table_bottom_border">376.14</td>
<td align="center" valign="middle" class="table_bottom_border">96.54</td>
<td align="center" valign="middle" class="table_bottom_border">84.47</td>
<td align="center" valign="middle" class="table_bottom_border">121.76</td>
<td align="center" valign="middle" class="table_bottom_border">254.38</td>
</tr>
</tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M18">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{2}} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">饥饿时长/h</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M14">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{0}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M14.jpg"><img class="graphic" src="202307113-wanghengxin_M14.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M15">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{3}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M15.jpg"><img class="graphic" src="202307113-wanghengxin_M15.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M16">$ \Delta{M}_{{\mathrm{N}\mathrm{O}}_{2}^{-}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M16.jpg"><img class="graphic" src="202307113-wanghengxin_M16.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M17">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{1}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M17.jpg"><img class="graphic" src="202307113-wanghengxin_M17.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2"><inline-formula><tex-math id="M18">$ \Delta{M}_{\mathrm{H}\mathrm{A}{\mathrm{c}}_{2}} $</tex-math><alternatives><img class="graphic" src="202307113-wanghengxin_M18.jpg"><img class="graphic" src="202307113-wanghengxin_M18.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">5</td>
<td align="center" valign="middle">335.16</td>
<td align="center" valign="middle">79.35</td>
<td align="center" valign="middle">74.87</td>
<td align="center" valign="middle">91.31</td>
<td align="center" valign="middle">243.84</td>
</tr>
<tr>
<td align="center" valign="middle">24</td>
<td align="center" valign="middle">394.68</td>
<td align="center" valign="middle">101.39</td>
<td align="center" valign="middle">88.34</td>
<td align="center" valign="middle">128.47</td>
<td align="center" valign="middle">266.2</td>
</tr>
<tr>
<td align="center" valign="middle">36</td>
<td align="center" valign="middle">468.91</td>
<td align="center" valign="middle">103.59</td>
<td align="center" valign="middle">94.3</td>
<td align="center" valign="middle">124.77</td>
<td align="center" valign="middle">344.14</td>
</tr>
<tr>
<td align="center" valign="middle">48</td>
<td align="center" valign="middle">407.25</td>
<td align="center" valign="middle">102.71</td>
<td align="center" valign="middle">93.03</td>
<td align="center" valign="middle">124.46</td>
<td align="center" valign="middle">282,79</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">72</td>
<td align="center" valign="middle" class="table_bottom_border">376.14</td>
<td align="center" valign="middle" class="table_bottom_border">96.54</td>
<td align="center" valign="middle" class="table_bottom_border">84.47</td>
<td align="center" valign="middle" class="table_bottom_border">121.76</td>
<td align="center" valign="middle" class="table_bottom_border">254.38</td>
</tr>
</tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">5</td>
<td align="center" valign="middle">335.16</td>
<td align="center" valign="middle">79.35</td>
<td align="center" valign="middle">74.87</td>
<td align="center" valign="middle">91.31</td>
<td align="center" valign="middle">243.84</td>
</tr>
<tr>
<td align="center" valign="middle">24</td>
<td align="center" valign="middle">394.68</td>
<td align="center" valign="middle">101.39</td>
<td align="center" valign="middle">88.34</td>
<td align="center" valign="middle">128.47</td>
<td align="center" valign="middle">266.2</td>
</tr>
<tr>
<td align="center" valign="middle">36</td>
<td align="center" valign="middle">468.91</td>
<td align="center" valign="middle">103.59</td>
<td align="center" valign="middle">94.3</td>
<td align="center" valign="middle">124.77</td>
<td align="center" valign="middle">344.14</td>
</tr>
<tr>
<td align="center" valign="middle">48</td>
<td align="center" valign="middle">407.25</td>
<td align="center" valign="middle">102.71</td>
<td align="center" valign="middle">93.03</td>
<td align="center" valign="middle">124.46</td>
<td align="center" valign="middle">282,79</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">72</td>
<td align="center" valign="middle" class="table_bottom_border">376.14</td>
<td align="center" valign="middle" class="table_bottom_border">96.54</td>
<td align="center" valign="middle" class="table_bottom_border">84.47</td>
<td align="center" valign="middle" class="table_bottom_border">121.76</td>
<td align="center" valign="middle" class="table_bottom_border">254.38</td>
</tr>
</tbody>
</table></div></foreignObject></svg>)
-
循环活性污泥法(cyclic activated sludge technology, CAST)作为一种基于序批式反应器(sequencing batch reactor, SBR)工艺改良的新技术,因其运行简便、结构紧凑及运行费用低等优点而被广泛应用于城镇污水处理厂中[1-2]。CAST反应池通常由选择池、缺氧池和好氧池3部分构成。在生物选择池内,将原水和混合液按体积比为5∶1的比例混合,使回流污泥处于高负荷条件下,这能有效抑制丝状菌的增殖,避免污泥膨胀[3-4]。由于污泥回流过程中会带有一定量的硝态氮(NO3−-N),反硝化菌在选择池和缺氧池中利用有机物将NO3−-N还原为氮气(N2)的同时,也将原水中的有机物储存为细胞内碳源[5],这为好氧池内的同步硝化-反硝化脱氮(simultaneous nitrification and denitrification, SND)创造了条件。BIAN等[6]同时研究CAST工艺的脱碳与脱氮性能,结果表明,TN主要通过以亚硝酸盐为间歇产物的同步硝化-反硝化机制去除,曝气期间同步硝化-反硝化的发生是由于反应器悬浮载体中存在微缺氧环境所致。WANG等[7]指出通过改变关键操作条件(温度,DO等)可以控制脱氮路径,控制同步硝化-反硝化成为主要脱氮路径,并在不同温度下通过调整有机负荷改善脱氮效率。WANG等[8]在对CAST系统进行脱氮性能的评估中,同样证实同步硝化-反硝化脱氮贡献与操作条件有关,通过优化操作条件可提高同步硝化-反硝化过程的脱氮性能,推测DO在1 mg·L−1时亚硝酸盐氧化菌的抑制作用会影响亚硝酸盐的积累。然而,目前关于CAST工艺的脱氮路径及脱氮特性的报道不多。基于此,对CAST工艺的脱氮性能进行评价十分必要。
为此,本研究以某污水处理厂CAST工艺为研究对象,通过对典型周期内氮组分、污泥浓度等进行测定,结合污泥活性、同步硝化-反硝化速率和饱食-饥饿(feast-famine)等批次实验,评价了该工艺的脱氮性能,以期为我国城市污水处理厂CAST工艺的设计、运行及水厂提质增效提供参考。
-
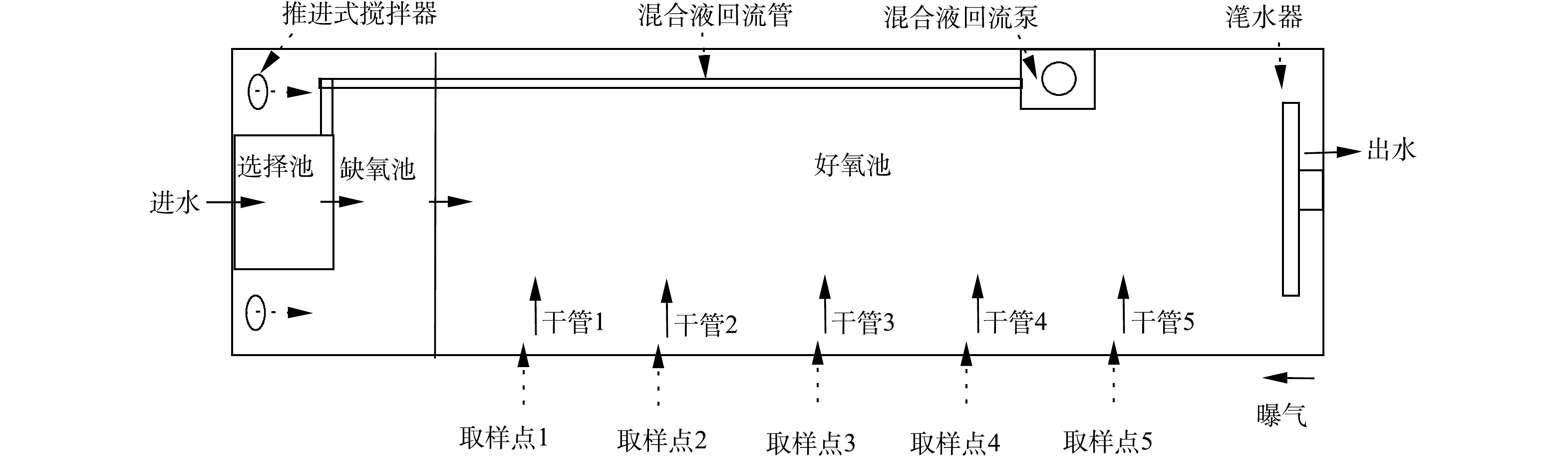
该污水处理厂处理规模为3.5×104 m3·d−1,其中一期工程2.0×104 m3·d−1,采用CAST工艺;二期工程1.5×104 m3·d−1,采用A2/O工艺。CAST工艺由4组反应池构成,每一组反应池的尺寸为76 m×11.6 m×6 m,选择池、缺氧池和好氧池体积比为1∶4∶25。该工艺水力停留时间(hydraulic retention time, HRT)为18.8 h,污泥龄(sludge retention time, SRT)为14.7 d,污泥回流比为20%。CAST工艺包括4个运行步骤:进水曝气、曝气、沉淀和出水,每步骤时长为1 h。缺氧池中安装潜水混合器,保证缺氧池内活性污泥处于悬浮状态;好氧池内安装微孔曝气器,以维持所需溶解氧(dissolved oxygen, DO)浓度及搅拌。在好氧池末端设有混合液回流泵,每运行周期前2 h进行回流,流量为80 m3·d−1。厌氧池和缺氧池设有氧化还原电位(oxidation-reduction potential, ORP)测定仪,在线显示氧化还原电位。好氧池中设有溶解氧仪和污泥浓度仪,在线检测DO浓度和污泥浓度,并反馈至中控室,随时调节鼓风机送风量和排泥量。
-
氨氮(NH4+-N)、亚硝氮(NO2−-N)和硝氮(NO3−-N)等的测定参照标准分析方法[9]。NH4+-N的测定采用纳氏试剂分光光度法,NO2−-N的测定采用N-(1-萘基)-乙二胺分光光度法,NO3−-N的测定采用紫外分光光度法,MLSS和MLVSS的测定采用重量法。DO由便携式溶氧仪(SevenGo pro SG6)测定。本研究中以乙酸钠作为批次实验的碳源,其测定方法为气象色谱法,测定仪器为SP-3420A型气象色谱仪。
-
为研究CAST工艺好氧池同步硝化-反硝化脱氮特性,沿池长设定5个取样点(图1),在进水曝气和曝气段每间隔15 min取样且测定氮组分浓度和记录DO浓度。
CAST反应池的脱氮主要由3部分贡献:缺氧池反硝化脱氮(DEN)、同步硝化-反硝化脱氮(SND)、沉淀阶段的内源反硝化脱氮(ED)。总脱氮量根据式(1)计算,同步硝化-反硝化脱氮量根据式(2)计算,内源反硝化脱氮量根据式(3)计算,选择池及缺氧池脱氮量根据式(4)计算,每周期开始时池内总氮量根据式(5)计算,每周期进水总氮量根据式(6)计算,每周期出水总氮量根据式(7)计算,曝气结束时池内总氮量根据式(8)计算。
式中:M 为每周期总脱氮量,mg(以N计);Minf为每周期进水总氮量,mg;Meff为每周期出水总氮量,mg;Msnd为每周期同步硝化-反硝化脱氮量,mg;Mst为每周期起始时CAST池内总氮量,mg;Mhd为每周期缺氧池脱氮量,mg;Med为每周期内源反硝化脱氮量,mg;Mend为每周期曝气结束时CAST池总氮量,mg; V1为混合液回流量,80 m3·h−1;T为回流时间,2 h;C1为回流混合液平均NO3−-N浓度,mg·L−1;V2为每周期开始时池内混合液体积,m3;C2为每周期开始时池内混合液总氮浓度,mg·L−1;V3为进水体积,m3;C3为进水总氮浓度,mg·L−1;C4为出水总氮浓度,mg·L−1;C5为曝气结束时池内混合液总氮浓度,mg·L−1。
-
为研究CAST反应池内不同DO浓度下的同步硝化-反硝化脱氮特性,本研究在CAST工艺进水结束时取1号采样点的泥水混合物于5 L试验装置内,在DO浓度分别为1.0、1.5、2.0和2.5 mg·L−1下进行同步硝化-反硝化率测定。试验装置内设有搅拌器,在反应器顶端设有取样口。测定过程中,每隔15 min取样并测定NH4+-N和NO3−-N浓度,随后根据浓度变化拟合得出NH4+-N消耗速率和NO3−-N生成速率,最终根据两速率计算得出同步硝化-反硝化率(式(9))。
式中:
R 为同步硝化反硝化率,%;RNH+4-N 为NH4+-N消耗速率,mg·(g·h)−1(mg以氮计、g以VSS计);RNH+4-N 为NO3−-N生成速率,mg·(g·h)−1。 -
为确定CAST工艺中饥饿时长对微生物吸收乙酸的影响,本研究将活性污泥等分为5份,置于1 L血清瓶中,活性污泥(SS)浓度为3 608 mg·L−1,VSS·SS−1为0.55。向每一份污泥加入足量的NO3−-N使其处于饥饿状态,饥饿时长依次为5、24、36、48和72 h。饥饿结束后加入足量的乙酸、NO3−-N和NH4+-N,间隔15 min进行取样,测定基质浓度变化,根据式(10)~式(12)计算最大乙酸吸收量。
式中:
ΔMHAc1 为将NO3−-N还原为NO2−-N及N2所消耗的乙酸量,mg·L−1;ΔMNO−3 为反应过程中浓度减少量,mg·L−1;ΔMNO−2 为反应过程中浓度增加量,mg·L−1;ΔMHAc0 为反应过程中消耗的乙酸量,mg·L−1;ΔMHAc2 为反应过程中吸收的乙酸量,mg·L−1;CVSS 为污泥浓度,g·L−1;ΔMHAc 为单位质量微生物吸收的乙酸量,g·g−1(以VSS计)。 -
活性污泥的内源反硝化活性采用基质消耗速率法测定,具体操作如下:CAST工艺每周期曝气结束时,在好氧池末端取一定量的活性污泥,用水淘洗去除残余基质,将淘洗过的活性污泥加入密闭的反应器内,向其中加入硝酸钠,控制NO3−-N为20 mg·L−1,每隔30 min取泥水混合物进行过滤,之后测定滤液中的NO3−-N、NO2−-N和NH4+-N的浓度,活性通过基质浓度变化线性拟合得出。
-
CAST工艺自2013年投入运行以来,各项水质指标均满足《城镇污水处理厂污染物排放标准》(GB 18918-2002)一级B标准。2021年10月—2022年5月,CAST工艺的主要进出水水质见表1。可以看出,CAST工艺出水COD、TN和PO43--P浓度分别小于30、15和0.3 mg·L−1,均达到A标准的水质要求。而对于出水NH4+-N,尽管略高于A标准所规定的1.5 mg·L−1,但在二期A2/O工艺出水稀释下,污水厂总出水可达标排放。
-
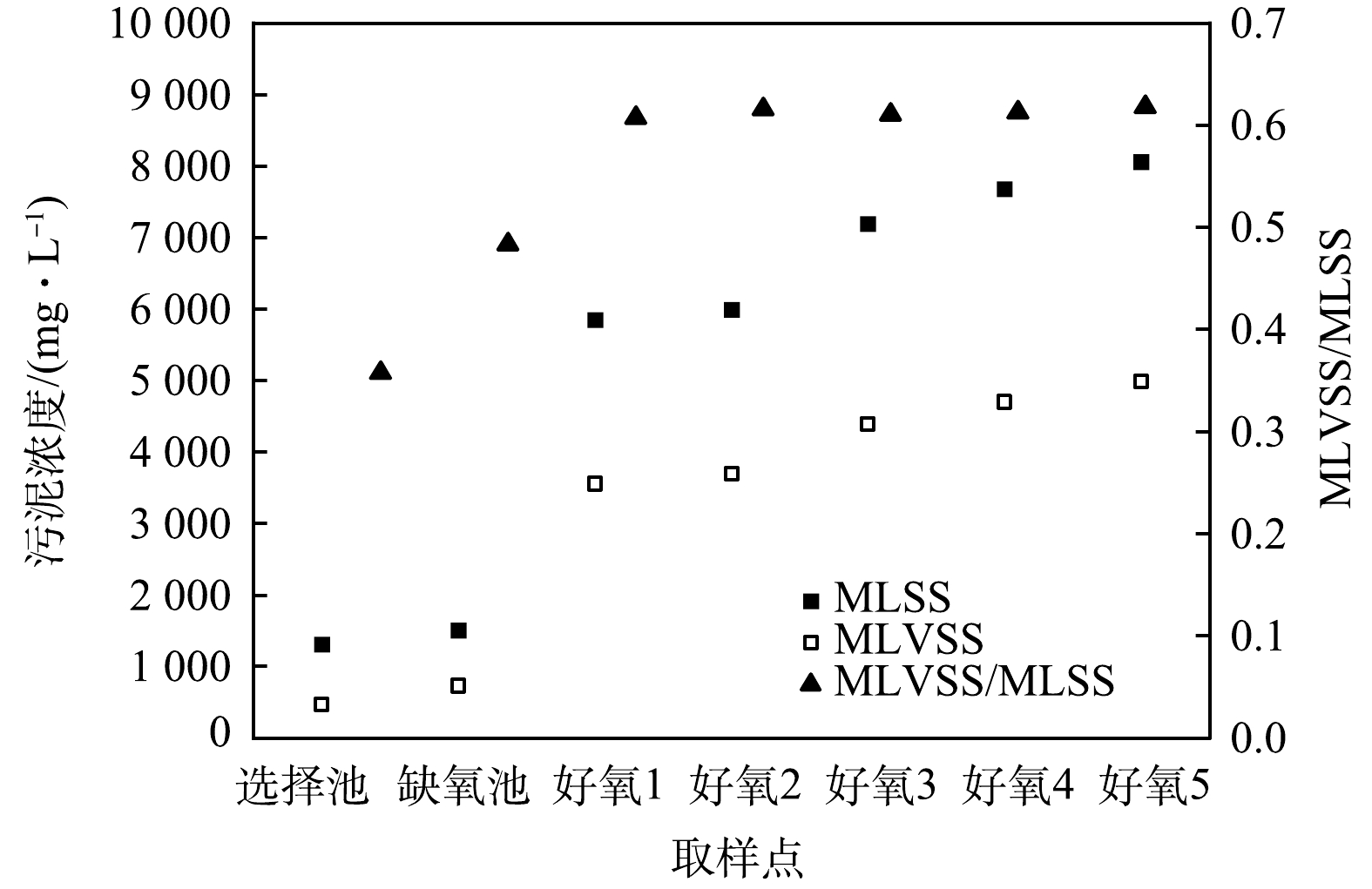
CAST工艺属于SBR工艺的一种演变,其运行方式也与SBR工艺相同,但CAST的推流式进水方式使得好氧池的反应并非完全混合式。考虑到CAST工艺的脱氮主要在好氧池。因此,首先对好氧池流态进行分析,结合选择池、缺氧池和好氧池的污泥浓度测定,判断各反应区的流体力学特性。由测定结果可知(图2),各反应区的污泥浓度分布并不均匀,在选择池和缺氧池内污泥浓度基本不变,说明这2个反应区的流态呈完全混合式;而好氧池进水端污泥浓度最低,约6 000 mg·L−1;出水端最高,约8 000 mg·L−1,说明回流混合液和原水混合进入好氧池后,以推流式方式前进,由于混合液回流比较低,使得污泥浓度沿程增加。
CAST工艺序批式的运行方式及推流式进水方式使其具有独特的脱氮特性。在周期开始时,回流混合液和原水在选择池内混合,此时混合液中的NO3−-N被反硝化菌利用原水中的有机物还原成N2(反硝化脱氮)。值得注意的是,CAST工艺的回流比仅为20%,使得回流至缺氧池的NO3−-N含量有限,继而选择池和缺氧池对TN脱除的贡献率较低;其次,回流混合液中的活性污泥在选择池和缺氧池中将原水中的有机物以胞内聚合物的形式储存,之后进入好氧池内,进而作为同步硝化-反硝化脱氮的碳源;最后,当曝气结束后,好氧池内活性污泥开始沉淀,池中剩余DO首先被消耗殆尽,此时好氧池部分区域进入缺氧状态,反硝化菌开始利用胞内聚合物及内碳源进行内源反硝化脱氮。综上所述,CAST工艺脱氮途径包括选择池及缺氧池反硝化脱氮、好氧池同步硝化-反硝化脱氮及内源反硝化脱氮。
为明确3种脱氮途径对TN脱除的贡献率,结合典型周期水质指标,利用式(1)~(8)对其进行计算,结果见表2。可以看出,选择池和缺氧池中反硝化脱氮贡献率最低,仅为(1.64±0.05)%,这一结果主要和混合液回流比较低(20%)有关,其可通过增大回流比提升,但需对回流泵进行更换;沉淀出水阶段的内源反硝化脱氮贡献最大,可达(62.86±4.13)%,类似结果在二沉池中也有观察到[10-11],但受限于沉淀时间短等因素,其脱氮贡献率难以进一步提升;除去反硝化和内源反硝化脱氮,TN的脱除全由好氧池同步硝化-反硝化途径完成,贡献率可达(35.50±4.15)%,对其进行优化,或成为进一步提升CAST工艺脱氮性能的突破口。基于此,本研究从如何优化同步硝化-反硝化过程入手,通过探究DO浓度和饥饿时长对同步硝化-反硝化脱氮性能的影响机制,从而确定适宜CAST工艺的运行策略。
-
为分析DO浓度对同步硝化-反硝化过程的影响特性,测定了不同DO浓度下好氧池曝气阶段DO、NH4+-N及NO3−-N浓度随时间的变化情况,结果如图3所示。图3(a)~(b)为低DO浓度(0.5~1 mg·L−1)下同步硝化-反硝化的脱氮特性。由图3(a)可知,反应过程中NH4+-N仅很少一部分被硝化,曝气结束时仍有约12 mg·L−1NH4+-N,而NO3−-N浓度几乎为零,不能为同步硝化-反硝化过程提供足够的电子受体,进而导致脱氮效率极低。
图3(c)~(d)为中等DO浓度(1~4 mg·L−1)下同步硝化-反硝化脱氮特性。由图3(c)可知,取样点1、3和5的NH4+-N和NO3−-N浓度随反应的进行呈现先升后降的趋势。在进水曝气阶段,取样点1 NH4+-N浓度由3.80 mg·L−1升至10.72 mg·L−1,而NO3−-N浓度仅由1.85 mg·L−1升至2.94 mg·L−1,这一结果由原水和硝化反应联合贡献。原水会带入一定量的NH4+-N,但此时硝化能力有限,无法及时将NH4+-N氧化为NO3−-N,于是NH4+-N浓度不断升高。对NO3−-N而言,原水会对其进行稀释,但伴随着硝化反应的进行,NO3−-N会不断积累,因而NO3−-N浓度略有增加,但会明显低于NH4+-N浓度的增加;在曝气阶段,若仅存在硝化反应,NH4+-N的下降将同NO3−-N的升高相匹配。但实际情况是,NH4+-N和 NO3−-N浓度同时下降,在曝气结束时分别降至4.09 mg·L−1和5.29 mg·L−1,此结果证实了同步硝化-反硝化脱氮的存在。由于取样点3和5距进水口有一定距离,推流式进水方式使得其起始NH4+-N和COD较低,进而DO浓度快速升高。而当进水逐渐推移到这两点时(取样点3为20~40 min;取样点5为40~60 min),由于COD降低和硝化反应的进行,DO浓度开始下降,之后随着NH4+-N和COD的降低,DO浓度开始缓慢回升。依据计算可以得出,同步硝化-反硝化、内源反硝化和反硝化途径对TN的脱除贡献率分别为65.7%、32.5%和1.80%。依据计算可得,同步硝化-反硝化、内源反硝化和反硝化途径对TN的脱除贡献分别为65.7%、32.5%和1.80%(进水和出水TN分别为38.80 mg·L−1和6.01 mg·L−1, TN去除率为84.51%)。
图3(e)~(f)为高DO浓度(1~6 mg·L−1)下同步硝化-反硝化脱氮特性。由图3(e)和图3(f)可知,3个取样点的 DO 浓度随时间变化规律与中 DO 水平下的变化规律基本一致,但其浓度均高于后者,取样点1、3、5的DO平均浓度分别为3.41、4.48和6.07 mg·L−1。在高DO浓度条件下,氧扩散能力增强,活性污泥絮体内部难以维持缺氧环境,同步硝化-反硝化脱氮过程所需环境遭到破坏,致使NO3−-N不断积累,曝气结束时3个取样点的NH4+-N和NO3−-N浓度依次达到1.41、1.27、0.58 mg·L−1和13.92、12.48、9.88 mg·L−1。依据计算可得,同步硝化-反硝化、内源反硝化和反硝化途径对TN的脱除贡献分别为39.2%、58.6%和2.20%(进水和出水TN分别为40.01和10.20 mg·L−1,TN去除率为74.51%)。
综上所述,中DO浓度下可观察到明显的同步硝化-反硝化过程,同步硝化-反硝化脱氮效果最理想,而高DO和低DO浓度下无法观察到明显的同步硝化-反硝化过程,脱氮效果较差。因此,CAST工艺好氧池DO应控制为中等浓度,这对同步硝化-反硝化过程进行更有利,从而提高TN去除率。
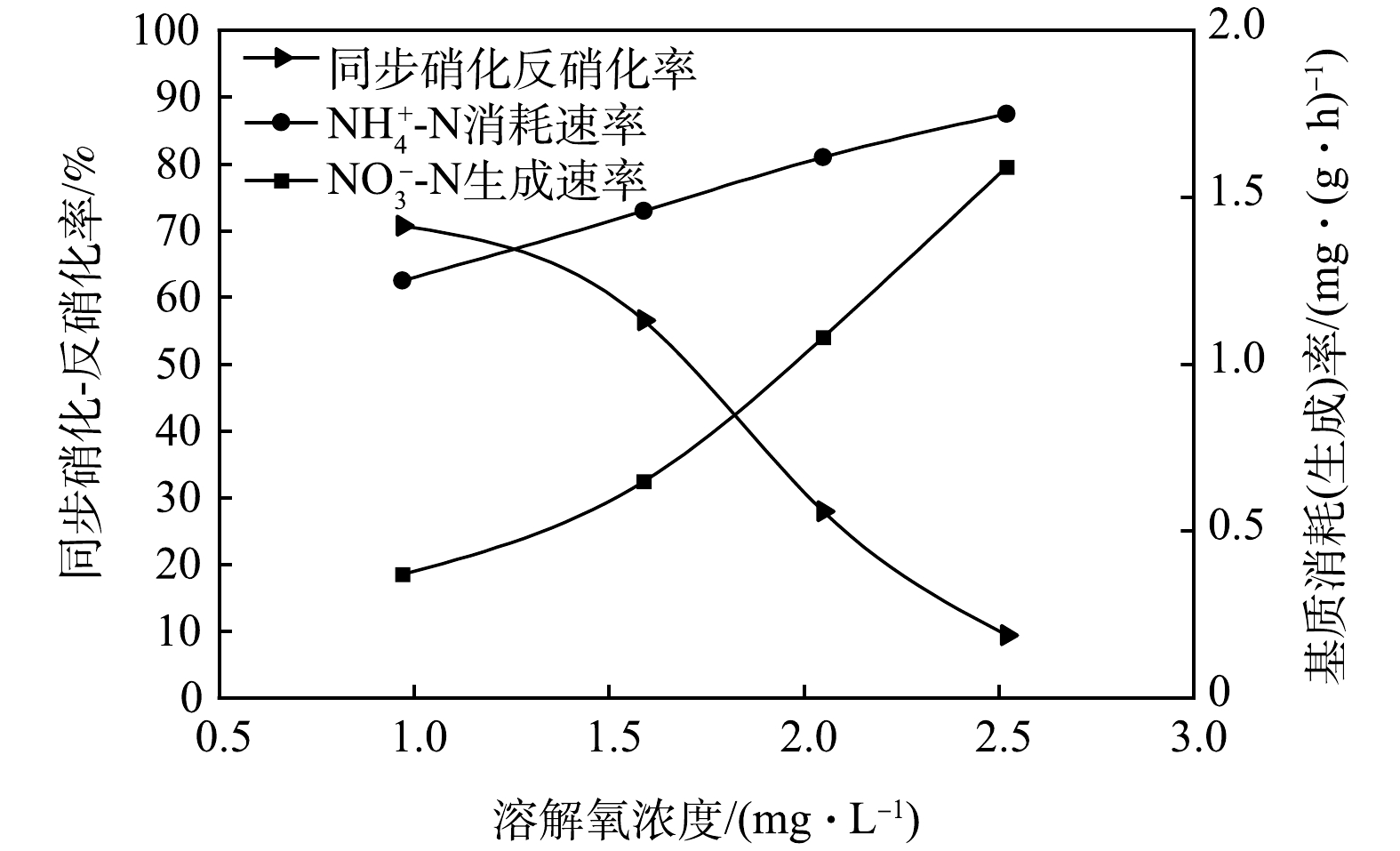
基于不同DO浓度下典型周期内同步硝化-反硝化对TN脱除的贡献变化,特进行同步硝化-反硝化率测定,以确定同步硝化-反硝化脱氮的最适宜DO浓度。在CAST反应池进水结束后取取样点1的混合液,利用序批式反应器对不同DO浓度(1.0~2.5 mg·L−1)下的同步硝化-反硝化率进行测定,结果如图4所示。由图4可知,随着DO浓度升高,同步硝化-反硝化率逐渐下降。当DO浓度控制在 1 mg·L−1 时,同步硝化-反硝化率达到最高值(70.66%),NH4+-N消耗速率和NO3−-N生成速率分别为1.25 mg·(g·h)−1和0.37 mg·(g·h)−1。而当DO浓度控制在2.52 mg·L−1时,同步硝化-反硝化率降为9.34%,NH4+-N消耗速率和NO3−-N生成速率分别为1.75 mg·(g·h)−1和1.59 mg·(g·h)−1。相比较而言,随着DO浓度的升高,NH4+-N消耗速率增幅并不明显,仅为40%,但NO3−-N生成速率增幅明显,达到329.73%。这表明在DO浓度为2.52 mg·L−1时同步硝化-反硝化过程已严重受限。因此,CAST工艺DO浓度应控制在1.0~1.5 mg·L−1,此时同步硝化-反硝化性能最强,这一结果与实际运行过程内中等DO浓度下所得结果一致,同样符合徐凤等[12]认为的最佳溶解氧浓度(0~2mg·L−1)的观点。
-
CAST工艺中20%的混合液回流比使得回流至CAST反应池前端的污泥有限,且污泥在好氧池内以推流式方式前进,污泥从好氧池末端回流至前端理论需要3~5 d,于是微生物将长期处于饥饿状态,即CAST工艺运行具有饱食-饥饿的特性,即非平衡增长[13-14]。饱食-饥饿理论[15]认为污泥所处饥饿时长会影响胞内聚合物的储存量,而同步硝化-反硝化脱氮能力又取决于胞内聚合物的量。因此,确定合适的饥饿时长,进而明确合适的污泥回流比对提高CAST工艺脱氮效率尤为重要。
为探究饥饿时长对微生物吸收有机物的影响,进行批次实验,结果见表3。由表3可知,随着饥饿时长增加,活性污泥对HAc的吸收量呈现出先上升后下降的趋势。在饥饿时长为5 h时
ΔMHAc 最小,仅为0.123 g·g−1;随着饥饿时长的增加,活性污泥内原有细胞聚合物被逐渐消耗,使得活性污泥对HAc的吸收能力和储存胞内聚合物的空间得以提升,当饥饿时长为36 h时ΔMHAc 达到最大,为0.173 g·g−1;随着饥饿时长的进一步增加,微生物开始利用细胞体进行反硝化,导致参与吸收的微生物数量有所减少(可检测到NH4+-N浓度升高),进而吸收量减小,当饥饿时长为72 h时ΔMHAc 仅有0.173 g·g−1。综上所述,最佳饥饿时长应控制为36 h,此时的HAc吸收量最大,更有利于同步硝化-反硝化脱氮。本研究中CAST工艺好氧池有效容积为4 254 m3,若活性污泥停留时间为36 h,则每天由好氧池末端回流至选择池的污泥量为3 016 m3,CAST工艺每天运行6个周期,每周期回流量为502 m3,基于每周期进水量为1 125 m3,CAST工艺的最佳回流比应控制为45%,这对好氧池的同步硝化-反硝化和内源反硝化脱氮过程更有利。 -
为了解CAST工艺好氧池内源反硝化脱氮特性,在曝气结束时取1、3和5取样点的泥水混合物,对其内源反硝化活性进行了测定,结果见图5。由图5可知,取样点1泥水混合物内源反硝化活性最大,达到1.48 mg·(g·h)−1,明显高于取样点3(1.08 mg·(g·h)−1)和取样点5 (1.05 mg·(g·h)−1)。造成这一结果的原因是取样点1靠近进水端,活性污泥所吸附的有机物及微生物储存的胞内聚合物较多,所以可用于内源反硝化反应的电子供体较多。而对于取样点3和取样点5的活性污泥,较长的污泥龄导致外源有机物和胞内聚合物已被大量消耗,内源反硝化反应只能以细胞作为碳源进行,致使内源反硝化速率较低。内源反硝化主要是利用内碳源(细胞体)进行反硝化,细胞体的化学结构通式为C5H7NO2,化学计量反应如式(13)所示。
由式(13)可知,每去除1 g的NO3−-N理论上会产生0.25 g的NH4+-N。而在CAST工艺中,取样点1、3和5每去除1 g的NO3−-N依次产生0.09、0.13和0.20 g NH4+-N,均低于利用细胞体进行反硝化的理论值,但沿着推流方向逐渐接近理论值。此结果证实内源反硝化过程不单以细胞体作为碳源,还包括污泥所吸附的有机物及微生物储存的胞内聚合为碳源。
-
表4列举了CAST、A2/O和氧化沟工艺的脱氮指标。由表4可知,3种工艺出水COD、NH4+-N和TN浓度相当,均满足相关排放要求。而对于出水TN,CAST、A2/O和氧化沟工艺的去除量分别为29.1、36.06和39.03 mg,每去除1 mg TN所需COD依次为5.77、6.41和15.94 mg,此结果说明CAST工艺更具脱氮潜力,但考虑到CAST工艺出水TN明显高于A2/O和氧化沟工艺,因而对其运行提出以下调控建议:1)DO浓度调控:依据现场测定和同步硝化-反硝化率测定实验,为获得良好的同步硝化-反硝化脱氮性能(较高同步硝化-反硝化率)及降低运行费用(减少供氧量),在CAST工艺好氧池内实行阶梯曝气,沿泥水混合物流动方向不断升增加曝气量,控制好氧池前段、中段和后段的DO质量浓度依次处于0.6~1.0、1.0~2.0和2~3 mg·L−1。2)污泥回流比调控:依据饱食-饥饿实验,为保证微生物将进水有机物尽可能多的以内碳源的形式储存,从而增强同步硝化-反硝化脱氮性能,将CAST工艺的回流比由当下的20%(饥饿时长为80 h)提升至45%(饥饿时长为36 h)。
-
1) CAST工艺好氧池内同步硝化-反硝化和沉淀过程中内源反硝化对总氮去除的贡献占据主导作用,贡献率分别为 (35.50±4.15)%和(62.86±4.13)%,而缺氧池反硝化脱氮贡献仅为(1. 64±0.05)%。
2) DO浓度对同步硝化-反硝化脱氮性能有重要影响,控制DO浓度在1.0~1.5 mg·L−1时,可获得最大同步硝化-反硝化脱氮效果。
3)饥饿时长对微生物吸收HAc有重要影响,当饥饿时长为36 h时,可获得最大HAc吸收量,达到每1 g VSS消耗0.173 g HAc,可为同步硝化-反硝化脱氮提供更多碳源。
某污水处理厂CAST工艺的脱氮性能评价
Evaluation of nitrogen removal performance of the CAST process in a wastewater treatment plant
-
摘要: 通过对某污水处理厂循环活性污泥法工艺(cyclic activated sludge technology, CAST)中选择池、缺氧池和好氧池中氮组分和污泥浓度进行测定,结合污泥活性、同步硝化-反硝化(simultaneous nitrification and denitrification, SND)速率及饱食-饥饿(feast-famine)批次实验,评价该处理工艺的脱氮性能。结果表明,好氧池内同步硝化-反硝化和沉淀过程中的内源反硝化(endogenous denitrification, ED)脱氮对总氮去除的贡献占据主导,分别为(35.50±4.15)%和(62.86±4.13)%,而缺氧池反硝化(DEN)脱氮贡献仅为(1.64±0.05)%;溶解氧(dissolved oxygen, DO)浓度对CAST工艺脱氮性能有极大影响,控制好氧池中DO浓度为1~1.5 mg·L−1时可获得最佳脱氮效果,CAST工艺的TN去除率可达84.51%;饱食-饥饿批次实验证明,饥饿时长为36 h时对乙酸(HAc)的吸收能力最强,可达每1 g VSS消耗0.173 g HAc,依此可推算出CAST工艺的最佳回流比为45%。Abstract: The nitrogen removal performance of CAST process in a wastewater treatment was evaluated by measuring the nitrogen components and suspended solids in the selective, anoxic and aerobic tanks, combined with the simultaneous nitrification and denitrification (SND) rate and feast-famine batch tests. The results showed that the contribution to total nitrogen(TN) removal through SND in aerobic tank and endogenous denitrification at sedimentation stage were dominated, which were (35.50±4.15)% and (62.86±4.13)%, respectively, while the denitrification in anoxic tank only accounted for (1.64±0.05)%. The Dissolved oxygen (DO) concentration had a great influence on the nitrogen removal performance of CAST process. The best nitrogen removal effect happened when the DO concentration in aerobic pool was controlled at 1~1.5 mg·L−1, and the TN removal rate of CAST process could reach 84.51%. The Feast-Famine batch tests proved that the maximum absorption of HAc occurred at the famine duration of 36 h, being up to 0.173 g HAc per 1 g VSS consumption. Based on this result, the optimal reflux ratio could be estimated to be 45%.
-
磷(P)是现代农业和化学工业中广泛使用的重要资源,通常以磷酸盐的形式存在于水溶液中[1]. 然而,在磷化工的快速发展和过量施用磷肥下导致磷大量向淡水资源转移,水体磷浓度超标,藻类过度生长,引起水体富营养化,造成磷资源损失和环境污染[2 − 3]. 此外,磷是一种不可再生资源,由于每年对磷的需求不断增长[4],预计在未来100—400年内将完全消耗掉[5]. 因此,废水中磷的去除和回收对于缓解富营养化染和磷资源危机至关重要[6].
迄今为止,化学沉淀[7]、膜分离工艺[8]、生物处理[9]、阴离子交换[10]和吸附[11]等多种技术已被证明可以用于去除水体中磷酸盐. 而吸附法因其操作简单、效率高成本低、应用方便等优点,吸引了众多研究人员的关注. 已经发现多种吸附材料,如水和氧化铁[12]、多孔二氧化硅[13]和碳基材料[14]等,它们具有良好的吸附性,用于去除废水磷酸盐的吸附剂. 但相对而言,这些吸附剂多为粉末形式很难在水中回收,这可能会造成二次污染问题[15]. 并且一些材料存在苛刻的问题制备条件好,成本高,去除能力低,难以分离等缺点. 制备高效稳定、绿色环保可循环利用的新型磷酸盐吸附材料吸附和回收水体磷酸盐,这是近年来的研究热点之一[16].
近年来,合成纤维作为催化剂和吸附剂载体受到研究者的广泛关注. 其中,腈纶纤维(PANF)含有丰富的氰基和酯基等化学性质活泼的基团,在一定条件下可以进行特定的化学反应,并可以转化为具有多种官能团的新型功能化纤维,是一种优秀的载体材料. 腈纶纤维具有成本低、易获得、密度低、柔韧性好等特点,其在大规模机械化加工和环境保护方面也具有一定的优势[17]. 腈纶纤维可以在一定条件进行灵活改性,构建富含氨基、羟基、羧基、季铵盐等组分[18]. 例如,Xu等[19]通过简单的化学接枝反应合成了一种可回收的载铁胺化聚丙烯腈纤维(PANAF-Fe)去除废水中的磷酸盐;Zheng等[20]合成了富含氯离子的功能化聚丙烯腈纤维(PANAF-Cl),对废水中磷酸盐的去除率高达90%以上,最大吸附容量为15.49 mg·g−1. 丁天琦[21]以聚丙烯腈(PAN)作为基体,制备出多孔碳纳米纤维膜,对磷酸盐最大去除量可达131 mg·g−1.
铁、铜、镧等金属和磷酸盐具有较强的结合能力,其在水体磷酸盐的去除获得较多的应用. 然而,使用固载铜离子的胺化纤维对水中磷酸盐的吸附尚缺乏研究. 本研究以腈纶纤维为原料,将Cu2+固载在胺化改性腈纶纤维表面,制备了新型磷酸盐吸附剂(PANAF-Cu),探究了该吸附剂对水体中磷酸盐的吸附性能,为废水中磷的回收提供理论依据.
1. 实验部分(Experimental section)
1.1 试剂与仪器
腈纶纤维(抚顺石化公司,中国),使用前将其剪成长度约10 cm备用. 实验中所有化学试剂均为国内市售分析纯试剂,且未进一步纯化. 本实验中所用化学试剂:乙二胺、磷酸二氢钾(上海阿拉丁试剂有限公司)、三水合硝酸铜(上海麦克林生化科技股份有限公司);盐酸、硝酸、硫酸(西陇科技有限公司);碳酸钾、氢氧化钠、碳酸氢钠(上海素艺化学试剂有限公司);氯化钾、硫酸钾、硝酸钾、一水合柠檬酸(国药集团化学试剂有限公司)、乙二胺四乙酸二钠(EDTA,上海苏懿化学试剂有限公司)、氯化钠(上海中试化工有限公司). 本研究中的所有实验使用的皆为去离子水.
实验中主要用到的仪器为:集热式恒温加热磁力搅拌器(DF-101S),巩义市予华仪器有限责任公司;磁力搅拌器(84-1A),金坛区西城新瑞仪器厂;鼓风干燥箱(DHG-9070A型),常州海博仪器设备有限公司;pH计(FE20~standard),梅特勒-托利多仪器有限公司;循环水式多用真空泵(SHB-Ⅲ),郑州长城科工贸有限公司;分光光度计(722G),上海仪电分析仪器有限公司.
1.2 胺化腈纶纤维的合成
将600 mg干燥的腈纶纤维(PANF)、20 mL乙二胺和20 mL去离子水(DI)先在室温条件下混合搅拌,然后加入到高压反应釜中125°C反应100 min.反应结束后,将纤维从溶液中分离出来,后用70—80℃去离子水反复洗涤,再将纤维置于60℃烘箱中干燥过夜,得到淡黄色的胺化纤维(PANAF),即胺化腈纶纤维.
1.3 铜离子固载胺化腈纶纤维的设计合成
准备好50 mL 10 mmol·L−1的三水合硝酸铜(Cu(NO3)2·3H2O)配制的硝酸铜溶液,将50 mg干燥过夜后的胺化纤维加入到溶液中,室温下搅拌30 min,过滤后,进一步洗涤,反复冲洗,后将纤维放置在60 ℃的烘箱中干燥6 h,即可得到载铜的功能化纤维PANAF-Cu.
1.4 材料的表征
扫描电子显微镜(SEM,德国蔡司Sigma 300),傅里叶变换红外光谱仪(FTIR,美国PerkinElmer Spectrum One),全自动元素分析仪(EA,德国Vario EL Cube),X射线光电子能谱仪(XPS,美国Thermo Scientific K-Alpha),X射线衍射仪(XRD,德国Bruker D8 Advance),pH计(METTLER TOLEDO,FE20),可见分光光度计(722G,Shanghai).
1.5 铜离子固载腈纶纤维(PANAF-Cu)的表征
利用扫描电子显微镜(SEM)、元素分析(EA)、傅里叶红外光谱仪(FTIR)、X射线衍射光谱(XRD)和能量色散X射线谱仪(XPS)等表征功能化纤维的表面形貌、化学结构及内部晶体结构等,从而根据表征结果检验改性纤维是否初步制备成功.
1.6 吸附实验
分别探究不同吸附时间、不同pH值、不同温度以及不同初始磷酸盐浓度下PANAF-Cu对磷酸盐的吸附能力. 纤维吸附饱和后,用镊子将纤维吸附剂从溶液中分离出来,从剩余溶液中吸取1 mL溶液放入50 mL容量瓶中,后按钼酸铵分光光度法测定不同条件下待测液中磷酸盐的浓度,测定低浓度磷酸盐时,要先将剩余溶液用0.22 μm滤膜过滤,后使用ICP-OES测定. 所有吸附实验均进行3次重复. 计算吸附量和吸附率公式如下:
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) 式中,η为吸附率,%;qe为吸附P的量,mg·g−1;C0和Ce分别为无机磷初始质量浓度和平衡浓度,mg·L−1;V为溶液体积,L;W为吸附剂添加量,g.
1.7 吸附动力学
吸附动力学是用来描述吸附剂吸附溶质速率快慢的方式,采用拟一级动力学和拟二级动力学模型探究正在进行的吸附过程,以便讨论其合适的吸附机理. 不同动力学模型如下:
stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) 其中, qt和qe为t时刻和平衡时的吸附量(mg·g−1)K1和K2依次为拟一级动力学和拟二级动力学的吸附速率常数.
1.8 吸附等温学
吸附等温线是描述在一定温度下吸附剂达到吸附平衡状态时吸附量(qe)和吸附质浓度(Ce)之间关系的曲线,常见的模型有Freundlich模型、Langmuir模型、Temkin 模型和D-R方程,本文采用Langmuir和Freundlich模型探究PANAF-Cu对无机磷的吸附方式,拟合方程如下:
stringUtils.convertMath(!{formula.content}) (5) stringUtils.convertMath(!{formula.content}) (6) 其中,qm为Langmuir模型估算的理论最大吸附量,KL为Langmuir常数,KF和n分别是Freundlich常数和均匀性系数.
2. 结果与讨论(Results and discussion)
2.1 功能化纤维(PANAF-Cu)的制备
固载铜离子的功能化腈纶纤维(PANAF-Cu)的合成由两步法完成,首先腈纶纤维和乙二胺发生胺化反应,纤维表面富含伯氨功能基的胺化纤维(PANAF),进一步利用氮和铜的配位作用,成功构建铜离子螯合的胺化纤维(PANAF-Cu). 胺化纤维的改性程度可以通过增重法来计算,胺化后的纤维对比腈纶纤维质量增加地较为明显,增重为10%,且纤维颜色也发生了变化,说明乙二胺被成功接枝到纤维表面. 胺化的程度可以通过控制反应时间或者乙二胺的浓度,过低的胺化程度不利于铜离子的螯合,然而过高的胺化程度导致纤维机械强度的下降,因此本文选择增重为10%的PANAF作进一步研究.
此外,本文研究了胺化纤维与不同浓度硝酸铜溶液制备得到的PANAF-Cu对磷酸盐的吸附能力, PANAF-Cu功能化纤维在Cu(NO3)2浓度增加的同时,其对磷酸盐的去除能力也不断增加,并且在Cu(NO3)2浓度为10 mmol·L−1时达到最高值,最大吸附量为35.29 mg·g−1. 然而,随着硝酸铜浓度的继续增加,PANAF-Cu中铜的含量达到饱和,导致其对磷酸盐的吸附能力趋势放缓至平衡,这是因为PANAF对铜的固载量是有限的,表明当铜离子浓度大于10 mmol·L−1时,PANAF对铜离子的固载量达到饱和. 因此,选择浓度为10 mmol·L−1的硝酸铜溶液制备PANAF-Cu.
2.2 功能化纤维(PANAF-Cu)的表征
2.2.1 纤维微观形貌分析
如图1可知,样品都呈连续完整的纤维状,表明纤维在改性前后及使用过程中,其完整性得到了良好的保持,未遭受到破坏. 放大2000倍的电子显微镜图像看到,随着改性反应的进行,纤维表面出现了若干裂纹,可能是因为氨基的进入和增溶作用导致纤维直径增加,出现裂纹,在一定程度上增加了纤维的表面积. 放大20000倍后,在胺化反应和螯合Cu2+后,可以清楚地观察到纤维不同程度的变形和溶胀,纤维表面明显变得粗糙,同时还被一层颗粒状物所覆盖,这可能是因为纤维表面的薄膜形成了由若干Cu-N配位产生的片晶.
2.2.2 纤维表面组分分析
为进一步明确纤维结构,利用傅里叶红外光谱(FTIR)和能谱图[22]分析了纤维改性前后的纤维表面组分. 图2(a)显示了每个阶段纤维的EDS能谱图,发现与PANAF相比,PANAF-Cu出现了新的Cu元素的峰,这表明Cu2+成功的固载到了胺化纤维上. 图2(b)显示了每个阶段纤维的红外光谱,PANF在2242 cm−1处的吸收峰和在1732 cm−1处的吸收峰分别为腈纶纤维第一单体丙烯腈的C≡N的伸缩振动和第二单体丙烯酸甲酯的C=O伸缩振动[17]. 由于胺化反应消耗了PANF上的氰基,胺化反应后2242 cm−1处的吸收峰强度下降. 此外,由于PANF中的C≡N在强碱性EDA溶液中被水解,与EDA中的NH2基团在1640 cm−1处形成了酰胺C=O双键的伸缩震动峰,说明了胺基基团的成功接枝[23]. 在胺化改性纤维吸附Cu2+后,在538 cm−1处新峰的出现,可能归因为Cu-N的配位结构,表明Cu已被-NH所固化[24]. 上述结果表明了胺基的成功接枝,以及Cu2+在纤维上的固载.
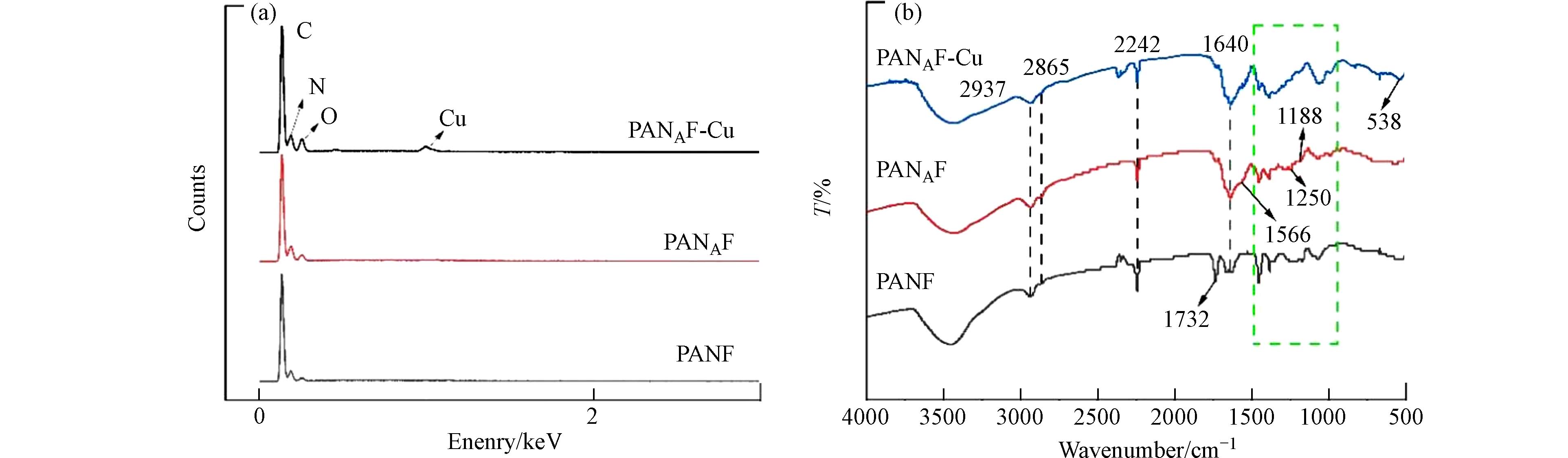 图 2 不同纤维的EDS能谱图(a) 和红外光谱图(b)Figure 2. EDS energy spectrum (a) and infrared spectra (b) of different fibers
图 2 不同纤维的EDS能谱图(a) 和红外光谱图(b)Figure 2. EDS energy spectrum (a) and infrared spectra (b) of different fibers2.2.3 纤维表面晶体分析
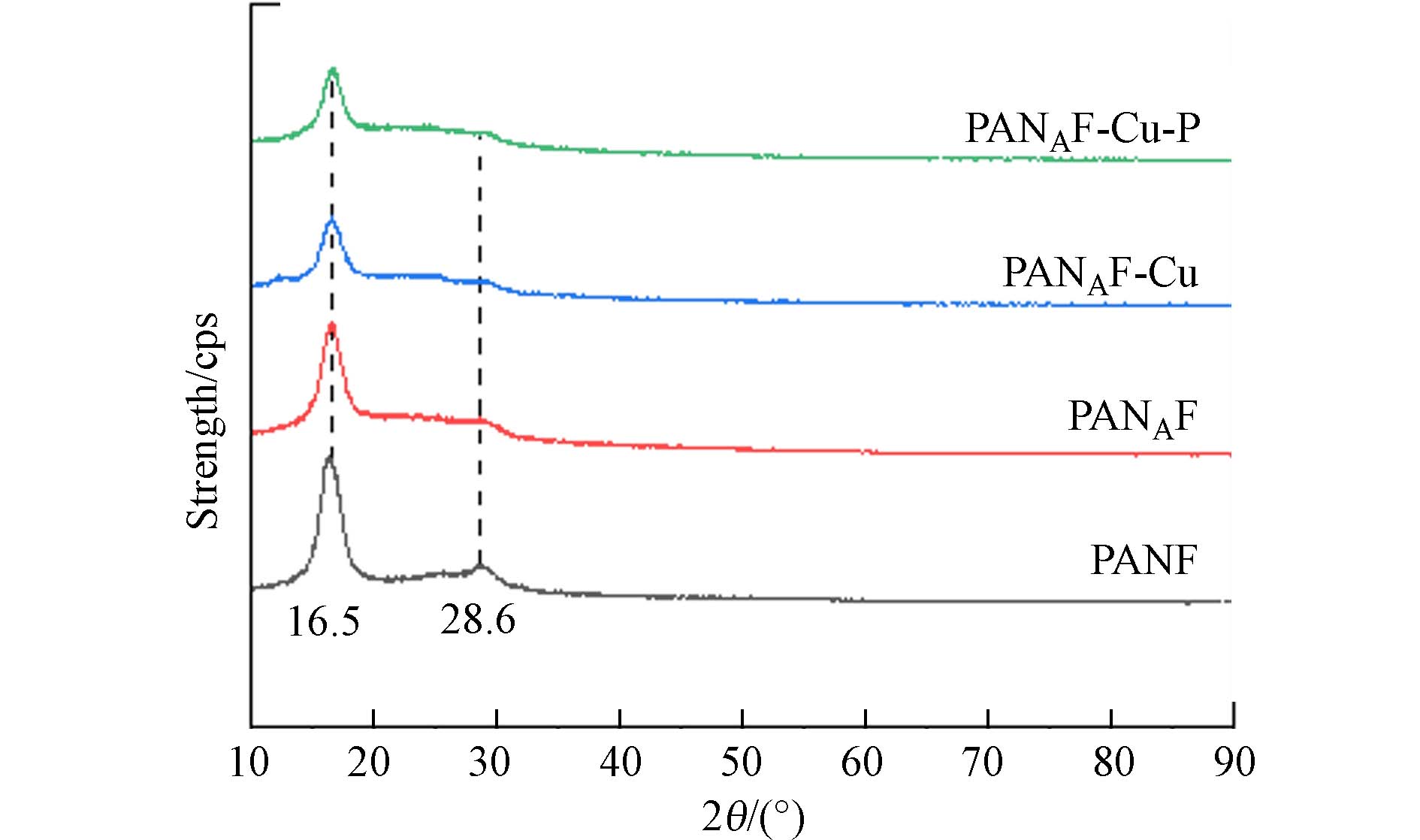
通过X射线衍射(XRD)表征了不同纤维的晶体结构. 图3显示了不同阶段的纤维的XRD图像,每个纤维都在2θ为16.5°处有一个共同的衍射峰,2θ = 28.6°处有一个不太强烈的衍射峰,表明PANAF、PANAF-Cu和PANAF-Cu-P与PANF具有相似的特征峰,表明纤维改性后仍保持了腈纶纤维原有的晶体结构,未被破坏,依然保持良好. 此外,PANAF、PANAF-Cu和PANAF-Cu-P较弱的衍射峰的强度明显降低,表明聚丙烯腈的聚合物链结构已经被胺分子部分溶解. 进一步固载Cu2+后,XRD光谱中并没有观察到结晶的Cu,这表明加载在纤维表面的固体Cu处于无定形状态,并可能通过Cu—N键与纤维骨架结合.
2.2.4 纤维元素含量分析
通过元素分析分析了不同阶段纤维的元素组成,相关数据见表1. 与PANF相比,PANAF中的C含量降低,H含量增加,因为乙二胺比原始腈纶纤维具有更少的C和更多的H,表明乙二胺被成功接枝到PANF上. 此外,N的存在也证实了不是所有的C≡N基团都被水解,参与EDA的官能化,所以改性过程可能仅发生在表面. 引入Cu元素后,PANAF-Cu中C、H含量的降低表明—NH2直接参与了Cu2+的吸附. 引入P元素后,相对于PANAF-Cu,PANAF-Cu-P中N含量的下降表明Cu与N形成配位键,被封存在纤维上,以上实验结果证明PANAF-Cu的成功改性.
表 1 不同纤维的元素分析数据Table 1. Elemental analysis data of different fibers样品Sample C/% H/% N/% 1 PANF 66.11 4.86 23.91 2 PANAF 60.30 5.89 22.77 3 PANAF-Cu 55.28 5.49 22.57 4 PANAF-Cu-P 55.76 5.65 21.91 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.3 PANAF-Cu对磷酸盐的吸附性能研究
2.3.1 pH对 PANAF-Cu吸附磷性能的影响
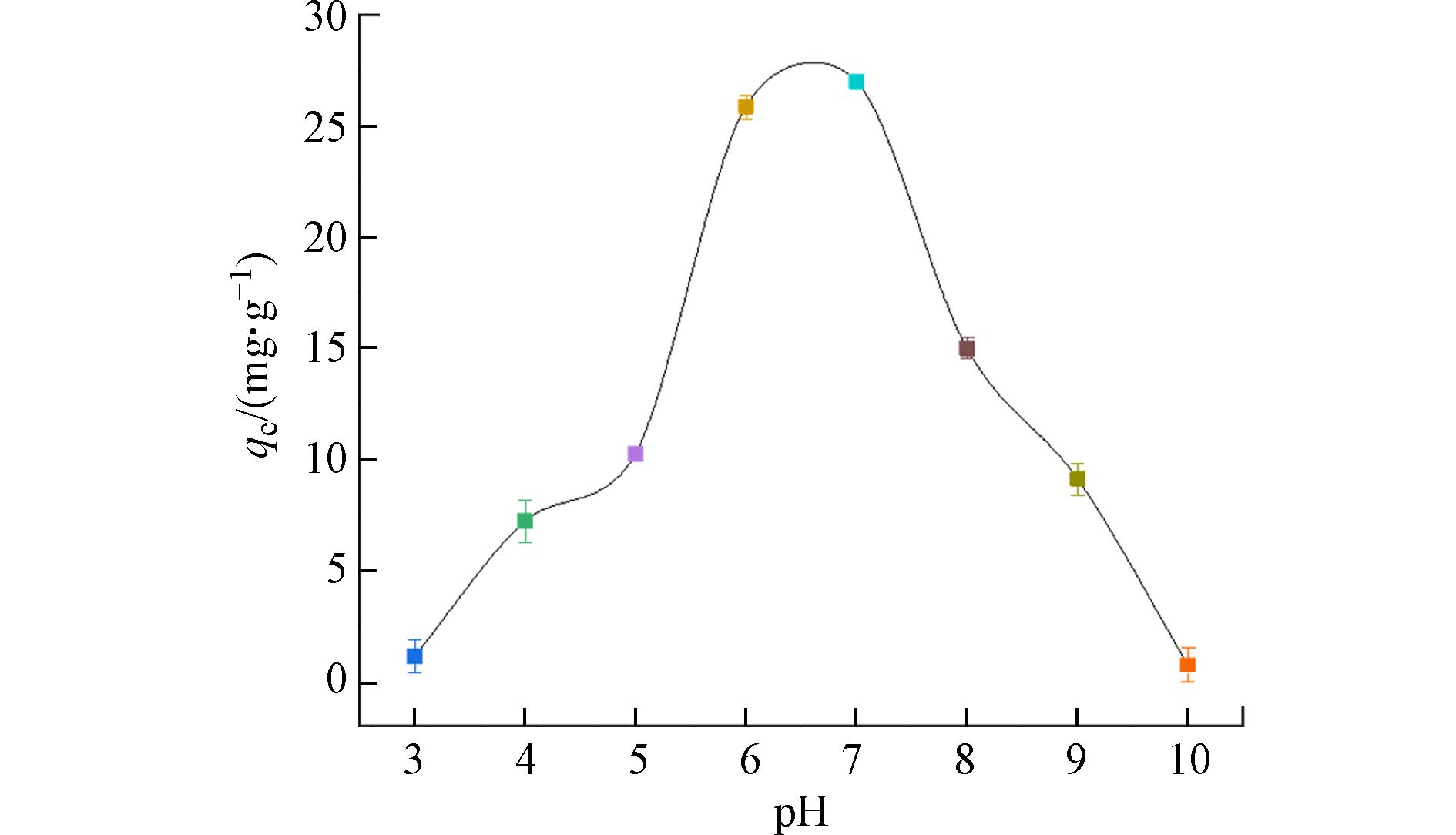
pH值是决定吸附效果的关键因素. 图4表明,pH值在3—7时,PANAF-Cu对磷酸盐的吸附量随着pH的升高而迅速增大,并在pH=7时达到最大值26.99 mg·g−1. 聚丙烯腈纤维的氨基的非选择性吸附是以静电作用形式产生的,因此,带正电的表面可以增强带负电的磷酸根阴离子的吸附. 其后通过配体的交换让表面负载的Cu2+对磷的进行选择性吸附. 在pH值在5—8的条件下,PANAF-Cu对磷酸盐的吸附量都能维持在较好的水平,吸附量仍能保持到10 mg·g−1以上. 表明固载铜离子的胺化改性纤维具有较高且稳定的磷酸盐去除效率.
2.3.2 吸附动力学分析
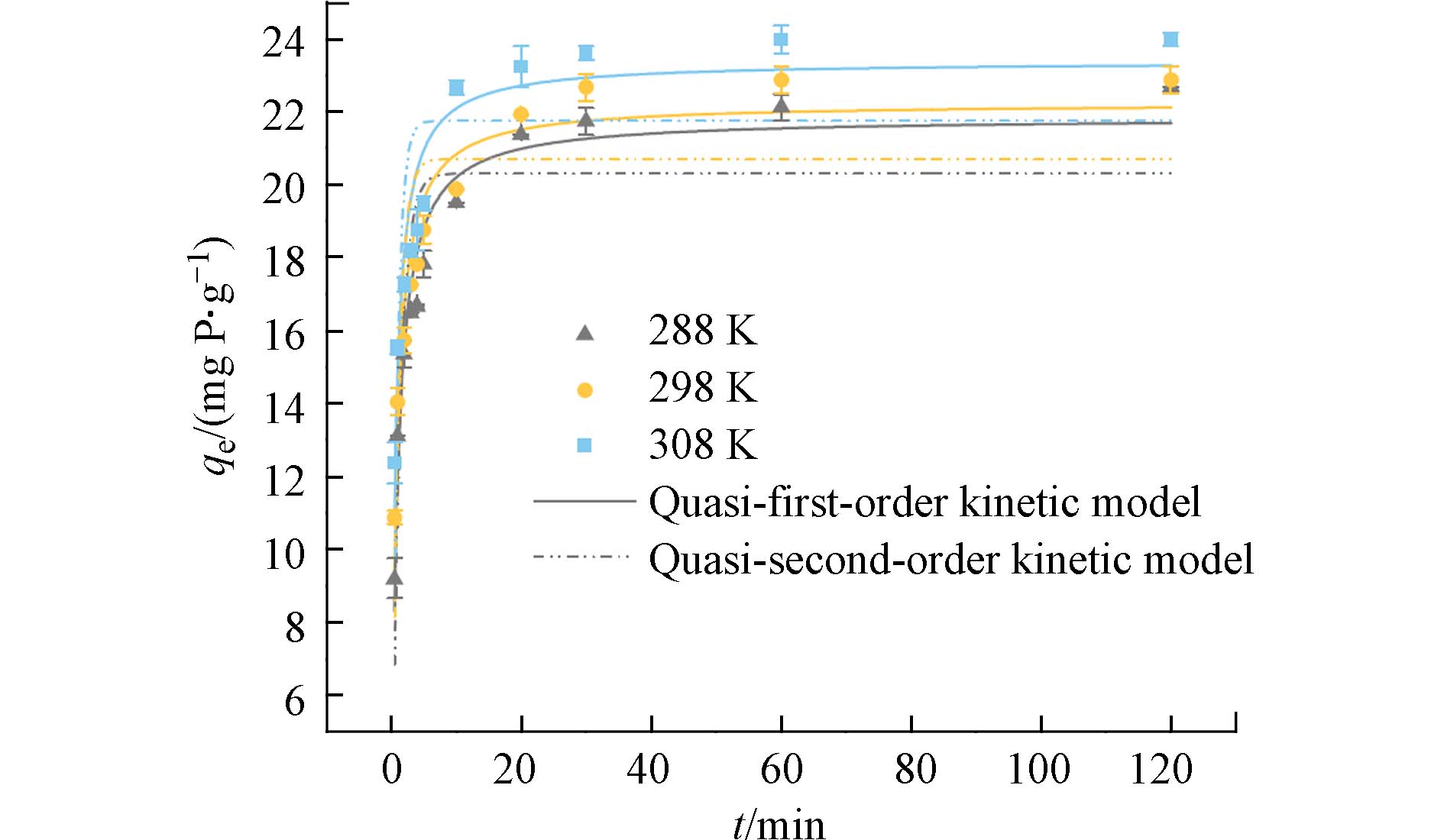
分别研究了在288、298 、308 K 的3种温度条件下,纤维吸附剂随时间改变对磷酸盐的吸附量的变化. 3条曲线的变化趋势大致相同,前10分钟内吸附速度较快,随后吸附速率减缓,在30 min左右基本达到吸附平衡. 这表明,在刚开始吸附阶段,吸附剂表面暴露的大量活性位点可以很迅速地捕获水中的磷酸盐. 随着时间的推移,吸附剂的表面吸附活性位点逐渐达到饱和,吸附过程随之减慢. 为深入了解PANAF-Cu对磷酸盐的吸附过程,分别通过拟一级动力学和拟二级动力学对吸附数据进行拟合. 拟合图和动力学参数分别如图5和表2所示,在288、298、308 K等3种温度下,拟二级动力学模型的R2均高于拟一级动力学模型;而且,用拟二级动力学模型拟合的qe值也更贴近于实际的吸附量. 因此,该吸附剂对磷酸盐的吸附过程更加倾向于化学吸附[25].
表 2 PANAF-Cu对磷酸盐的动力学参数Table 2. Kinetic parameters of PANAF-Cu on phosphateT/K 拟一级动力学模型Quasi-first-order kinetic model 拟二级动力学模型Quasi-second-order kinetic model K1/ min qe /(mg·g−1) R2 K2 /min−1 qe/ (mg·g−1) R2 288 0.824 19.781 0.754 0.056 21.533 0.952 298 0.999 20.700 0.688 0.066 22.237 0.931 308 0.927 21.754 0.627 0.073 23.379 0.906 | Show TableDownLoad:
CSV
2.3.3 吸附等温线分析
为了阐明PANAF-Cu对磷酸盐的吸附模式和吸附性能,在室温下,对不同初始磷酸盐浓度下进行了吸附等温实验. 实验发现,吸附剂对磷酸盐的吸附能力伴随着磷酸盐初始浓度的升高而增强,直至吸附饱和. 这可能是因为在较低的磷酸盐浓度下,磷的配位位点更多,随着磷浓度的提高,活性吸附位点也逐渐达到饱和,吸附量不再升高. 利用Langmuir和Freundlich吸附等温线模型对数据进行拟合,通过拟合得到的图和相关数据分别位于图6和表3.
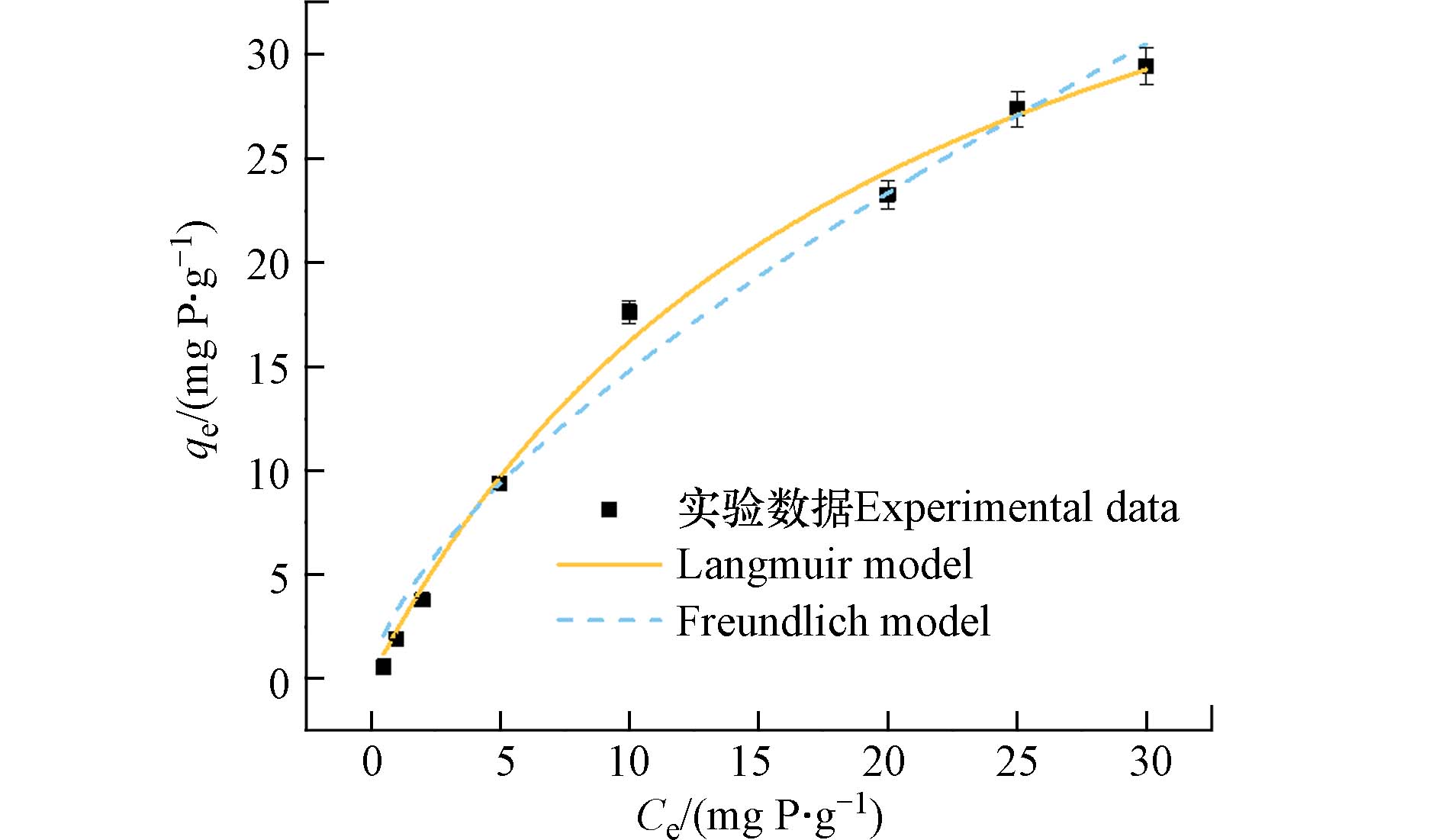 图 6 PANAF-Cu对磷酸盐的吸附等温线模型Figure 6. Isothermal model of phosphate adsorption by PANAF-Cu
图 6 PANAF-Cu对磷酸盐的吸附等温线模型Figure 6. Isothermal model of phosphate adsorption by PANAF-Cu从表3可以看出,Langmuir模型拟合曲线的回归参数大于Freundlich模型,说明PANAF-Cu对磷酸盐的吸附更适合于Langmuir模型. 因此,吸附剂对磷酸盐的吸附更趋向于均匀的单分子层化学吸附. 通过Langmuir模型估算PANAF-Cu对磷酸盐的最大吸附量为49.03 mg·g−1.
表 3 PANAF-Cu对磷酸盐的等温学参数Table 3. Isothermal parameters of PANAF-Cu on phosphateLangmuir Freundlich qe /(mg·g−1) KL/(L·mg−1) R2 KF /(mg·g−1·(L·mg−1)1/n) 1/n R2 49.026 20.258 0.995 3.242 0.659 0.982 | Show TableDownLoad:
CSV
2.3.4 共存阴离子影响
为测定PANAF-Cu对磷酸盐去除的选择性,选定硫酸盐、氯化物、硝酸盐和碳酸盐为代表性的共存阴离子. 在浓度为1×10−3 mol·L−1的磷酸盐中分别加入同等浓度的Cl−、NO3−、CO32-以及SO42-等共存离子溶液,探究PANAF-Cu对磷酸盐的吸附能力,结果见图7. 结果表明, 4种共存离子对PANAF-Cu吸附磷酸盐的影响如下:SO42-﹥CO32-﹥Cl−﹥NO3−. 其中NO3−对磷酸盐的吸附影响最小. SO42-和CO32-比Cl−和NO3−更有竞争力,可能是因为硫酸盐和碳酸盐作为二价离子具有更高的电荷密度,比单价阴离子能更迅速地被吸附. 以上实验说明PANAF-Cu在实际水体中也能在较多阴离子的影响下具有很好的除磷效果.
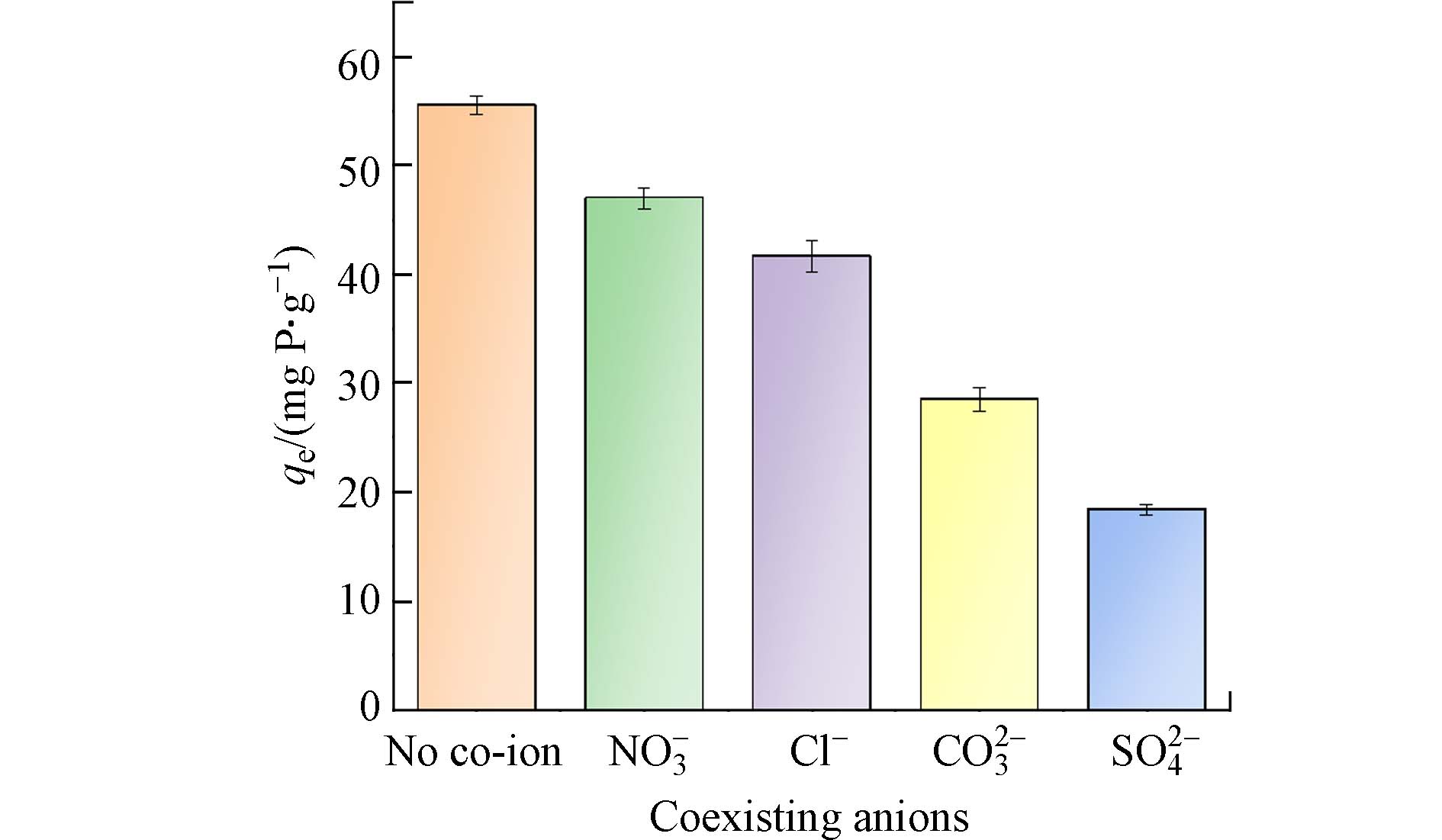 图 7 共存离子对PANAF-Cu吸附磷酸盐的影响Figure 7. Effect of coexisting ions on phosphate adsorption by PANAF-Cu
图 7 共存离子对PANAF-Cu吸附磷酸盐的影响Figure 7. Effect of coexisting ions on phosphate adsorption by PANAF-Cu2.3.5 吸附机理
XPS分析进一步揭示了吸附剂的化学状态和元素组成. 图8显示了吸附前后PANAF-Cu纤维的XPS光谱,与PANAF相比,PANAF-Cu出现了新的Cu元素的峰(图8a),这表明Cu2+成功的固载到了胺化纤维上,吸附后的纤维中出现了一个133.5 eV的信号峰,这是由P 2p轨道引起的,表明磷酸盐被纤维成功捕获(图8b). Cu2+对磷酸盐吸附的贡献由纤维吸附后Cu元素的高分辨率光谱证明(图8 c-d),对比吸附前的纤维,Cu的高分辨率光谱发现,PANAF-Cu-P的高分辨率Cu 2p光谱被分解成两个峰,最初位于934.73 eV的Cu—N键结合能下降到932.61 eV,这可以归因于来自磷酸盐的P原子为Cu提供了额外的电子,形成了新的N-Cu-P配位键,从而增加了Cu元素电子云密度,降低了结合能,这证实了改性纤维表面的Cu2+通过配位吸收起到了对P的固化作用. 即表明络合-配体相互作用是该吸附过程的主要机制[26]. 根据N的吸附前后的高分辨率光谱(图8 e-f)发现原本位于400.62 eV 的—NH2(—NH)键结合能下降到399.93 eV,氨基与Cu离子形成Cu-N的配位结构,纤维表面的Cu2+通过和磷酸盐中P—O键作用形成新的N-Cu-P配位去除磷酸盐,该基团的移动表明含氮官能团在PANAF-Cu对废水中的磷酸盐的吸附中起着重要作用. 因此根据上述结果,可以推断PANAF-Cu对磷酸盐的有效吸附主要依赖于纤维表面的Cu-N配位结构,从而形成新的N-Cu-P配位结构.
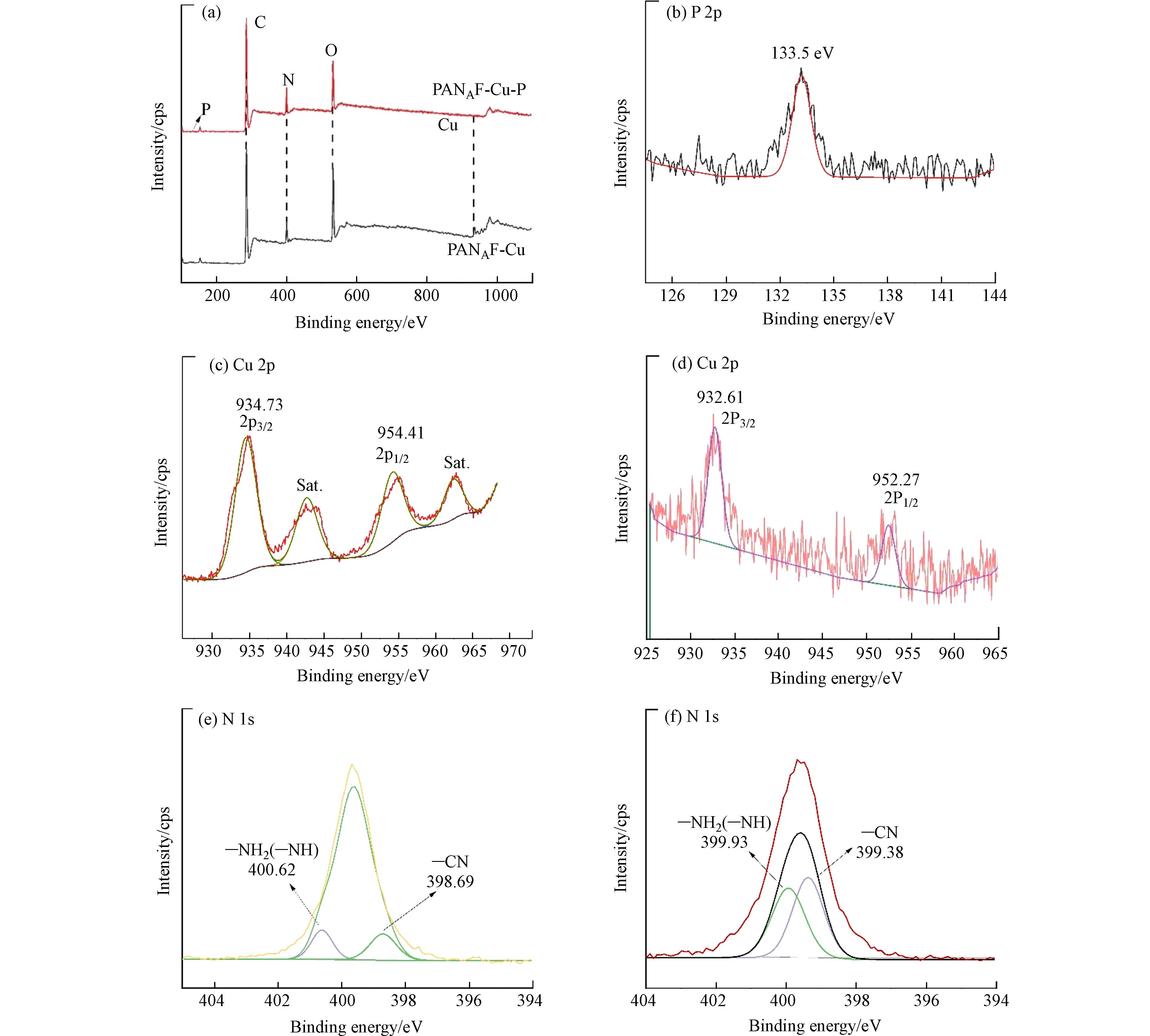 图 8 功能化纤维吸附磷酸盐前后的XPS谱图Figure 8. XPS spectra of functionalized fibers before and after phosphate adsorption(a)PANAF-Cu吸附磷酸盐前后的XPS总谱图,(b)P 2p的高分辨光谱图,(c-d)PANAF-Cu吸附磷酸盐前、后的Cu 2p的高分辨光谱图,(e-f)PANAF-Cu吸附磷酸盐前、后的N 1s的高分辨光谱图(a) XPS survey spectra of PANAF-Cu before and after phosphate adsorption, (b) High resolution XPS spectrum of P2p, (c-d) High resolution XPS spectrum of Cu2p before and after phosphate adsorption, (e-f) High resolution XPS spectrum of N1s before and after phosphate adsorption
图 8 功能化纤维吸附磷酸盐前后的XPS谱图Figure 8. XPS spectra of functionalized fibers before and after phosphate adsorption(a)PANAF-Cu吸附磷酸盐前后的XPS总谱图,(b)P 2p的高分辨光谱图,(c-d)PANAF-Cu吸附磷酸盐前、后的Cu 2p的高分辨光谱图,(e-f)PANAF-Cu吸附磷酸盐前、后的N 1s的高分辨光谱图(a) XPS survey spectra of PANAF-Cu before and after phosphate adsorption, (b) High resolution XPS spectrum of P2p, (c-d) High resolution XPS spectrum of Cu2p before and after phosphate adsorption, (e-f) High resolution XPS spectrum of N1s before and after phosphate adsorption2.3.6 循环性能实验
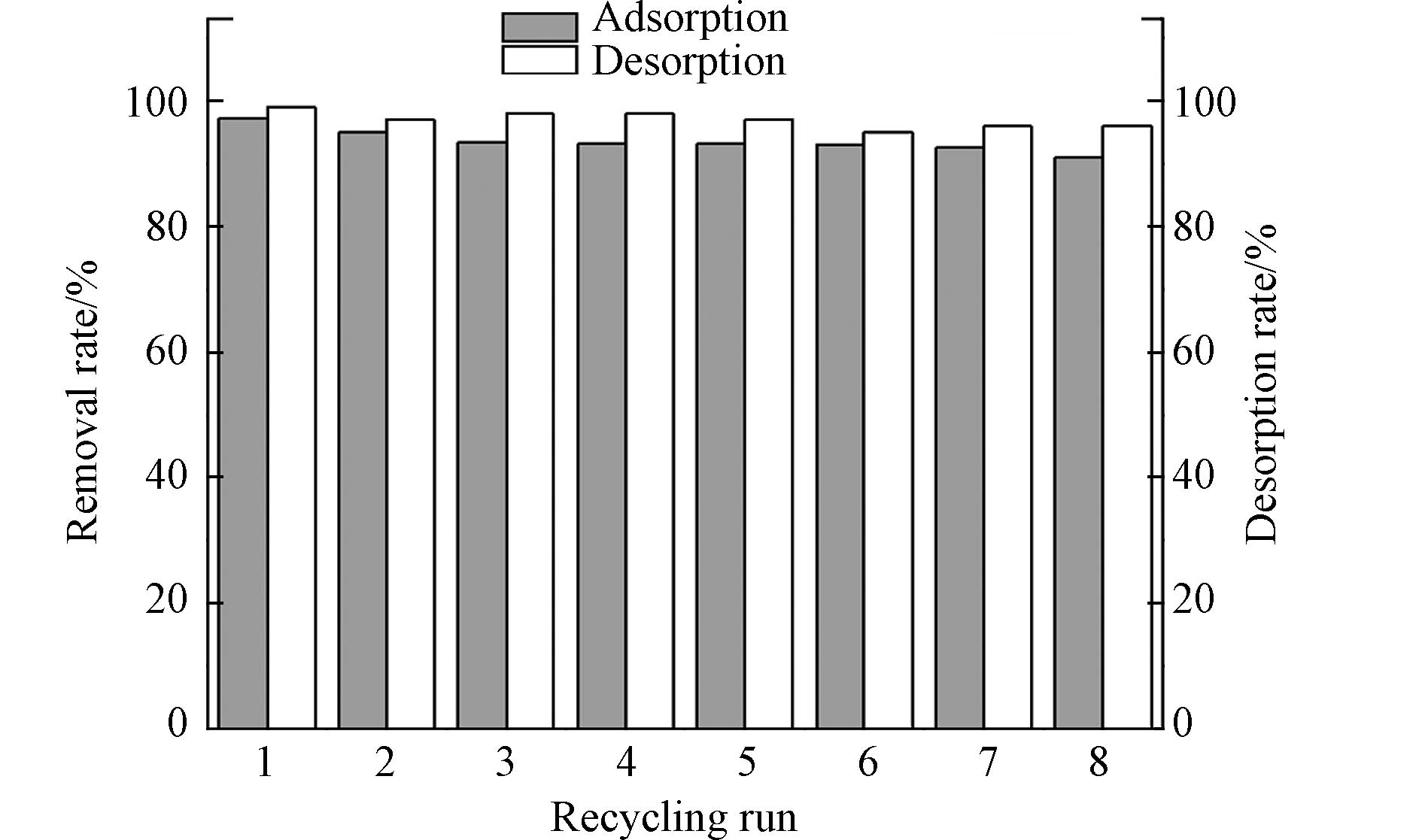
为探究不同解吸剂对磷酸盐的解吸率影响,进行对比实验(表4),发现HCl、NaCl、C6H8O7(柠檬酸)和EDTA(乙二胺四乙酸二钠)对磷酸盐的解吸率分别为52.50%、2.50%、87.50%和97.50%,EDTA的解吸率最高为97.5%. 从图9可以看出,经过8次循环后,PANAF-Cu对磷酸盐的解吸率基本稳定在90%以上,相应的解吸率也保持在95%以上,表明PANAF-Cu具有多重吸附和再生的优良特性.
表 4 不同洗脱液对PANAF-Cu吸附磷酸盐后的解吸率Table 4. Desorption rate of phosphate adsorbed by different eluents to PANAF-Cu洗脱剂Eluent 浓度/(mmol·L−1)Concentration V/mL T/ h 解吸率/%Desorption rate HCl 1 50 1 52.5 NaCl 1 50 1 2.5 C6H3O7 1 50 1 87.5 EDTA 1 50 1 97.5 | Show TableDownLoad:
CSV
2.3.7 改性纤维在连续流动实验中的应用
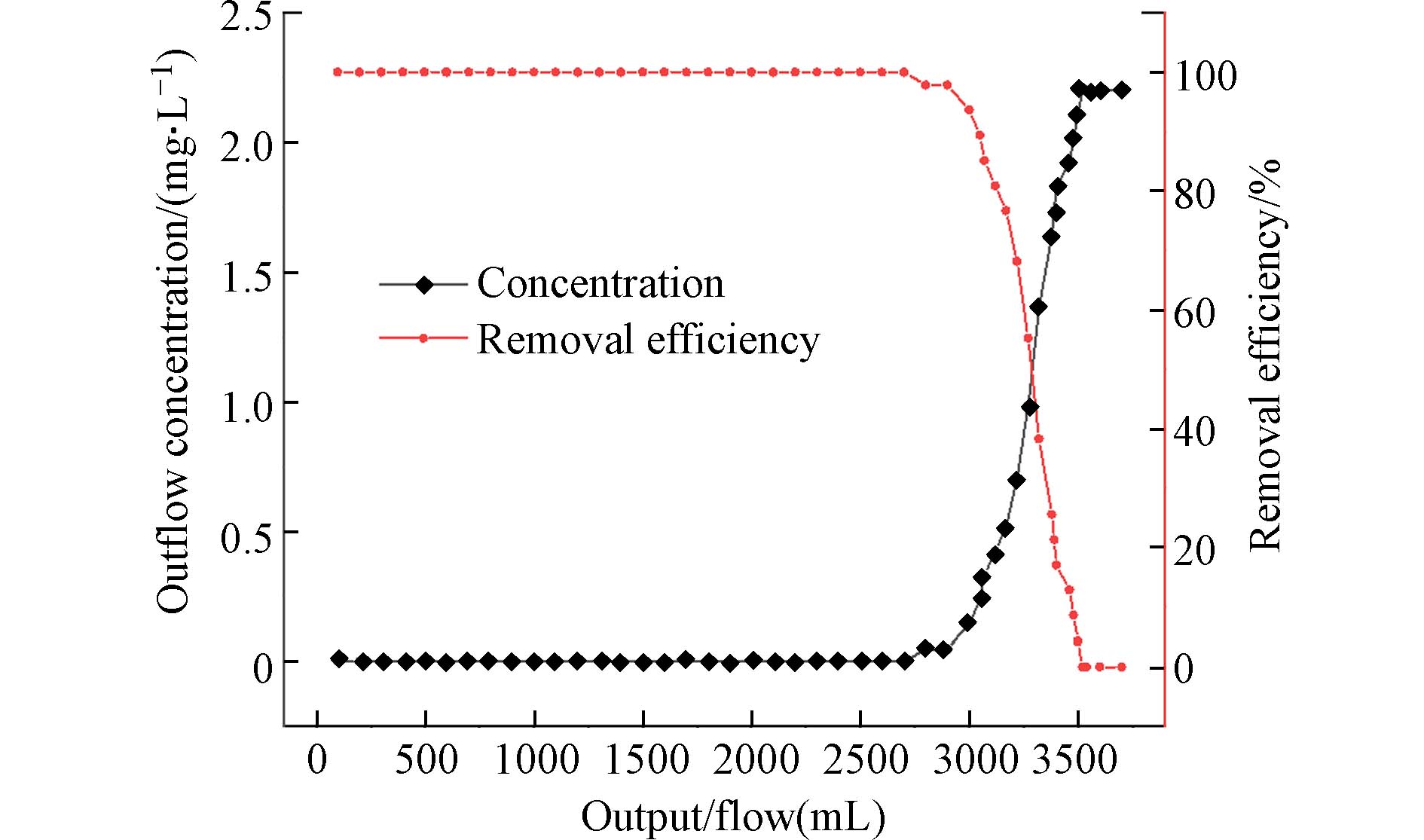
用初始浓度为2 mg·L−1的磷酸盐溶液利用恒流泵以0.5 mL·min−1的速率进行连续流动实验. 每隔50 mL测试出水中的磷酸盐浓度,就可以绘制出磷酸盐的穿透曲线,如图10所示. 实验发现,随着出水量的增加,出水中的磷酸盐浓度也随之增加. 当出水量小于2700 mL时,磷酸盐去除率维持在99%以上,大于2700 mL后,对硫酸盐的去除率大幅度下降. 这些结果皆说明PANAF-Cu在模拟自然水体和流动的水体条件下的具有优异除磷效果.
2.3.8 PANAF-Cu在实际废水中除磷性能研究
本研究进一步探究PANAF-Cu在实际水体的应用效果,测试了PANAF-Cu对巢湖(自然水体)磷污染水体中对磷的吸附能力. 为了更好地说明问题,将采集的水样的磷酸盐浓度调整为1000 µg· L−1 P. 分别将0、5、10、20、40、60 mg的PANAF-Cu置于10 mL上述水样中搅拌24h,利用ICP测定剩余磷酸盐浓度,测定结果见表5. 实验表明,当PANAF-Cu添加量超过20 mg时,磷酸盐浓度即可降至100 µg·L−1以下,已低于湖泊富营养化的最小磷浓度200 µg·L−1. 可见,本研究制备得到的少量PANAF-Cu在实际水体中也可以有效的降低水体的富营养化程度.
表 5 PANAF-Cu对磷酸盐的吸附极限测试Table 5. Adsorption limit test of phosphate by PANAF-CuPANAF-Cu的质量Quality of PANAF-Cu 体积/ mLVolume 时间/ hTime 磷酸盐浓度/(µg·L−1 P)Phosphate concentration 0 10 24 1113 5 10 24 759 10 10 24 393 20 10 24 98 40 10 24 92 60 10 24 90 | Show TableDownLoad:
CSV
2.3.9 与其他除磷吸附剂的比较
为了评估功能纤维吸附剂的吸附效果,表6列出了近年来报道的一些用于去除废水P的吸附剂材料. 值得注意的是,本工作中合成的铜胺负载纤维可以在较温和的反应条件下就可以获得较高的吸附能力. 此外,与表中列举的其他吸附剂有限的P吸附能力相比,接枝了Cu-N配体结构的功能化纤维表现出了更高的P容量和更快的吸附速率. 这可以归因于配位中心的铜离子和-NH2中的N原子之间的配位模式所引起的铜离子的高反应性以及形成的N-Cu-P之间的独特结合机制. 此外,PANAF-Cu在循环使用8次以后依然能够有效地去除磷酸盐. 总的来说,与目前报道的磷酸盐选择性吸附材料相比,本课题中报道的功能纤维吸附剂在常温下具有更高的反应活性和更好的选择性吸附性能.
表 6 与其他除磷吸附剂的比较Table 6. Comparison with other phosphorus removal adsorbent吸附剂Adsorbent 吸附时间Adsorption time 最大吸附量(mg·g−1)Maximum adsorption capacity 循环次数Number of cycles 参考文献 黏土-牡蛎壳复合吸附材料 7 d 10 — [27] 鸟蛤壳粉 20 min 7.1 — [28] 掺杂淀粉的磷石膏和磷矿浮选尾矿 60 min 31.28 — [29] 淀粉包裹的Fe3O4纳米颗粒 90 min 7.73 3 [30] 磁性淀粉基Fe3O4黏土聚合物(CIONP) 2 h 3.12 3 [31] 炼钢渣 72 h 10.21 — [32] 掺杂SiO2的活性炭 1 h 0.65 — [33] 固载Fe2+的活性炭 4 h 14.12 — [34] 固载铜离子的功能化腈纶纤维(PANAF-Cu) 24 h 26.99 8 本研究 | Show TableDownLoad:
CSV
3. 结论(Conclusion)
以聚丙烯腈纤维为原料,通过胺化改性使得聚丙烯腈纤维表面固载胺基,在通过胺基与铜的配位作用,成功合成具有Cu-N配位结构的新型吸附剂(PANAF-Cu). 在pH=7时达到吸附量最大值26.99 mg·g−1,pH值在5—8的条件下,吸附量仍能保持到10 mg·g−1以上. 表明固载铜离子的胺化改性纤维具有较高且稳定的磷酸盐去除效率. PANAF-Cu的吸附过程更符合伪二级动力学吸附模型,吸附等温学拟合结果更适合Langmuir等温模型,表明P在PANAF-Cu属单分子层吸附,在一定温度内,对磷酸盐的去除效果随着温度升高而增加. 通过吸/脱附循环性能试验,经过8次循环后,PANAF-Cu对磷酸盐的吸附率基本稳定在90%以上,相应的解吸度效率也保持在95%以上,PANAF-Cu良好的除磷可循环性;吸附极限试验表明当出水量达到2700 mL时,对磷酸盐的去除率仍达到99%,具有较高的实际应用能力. XPS解析表明,PANAF-Cu对磷酸盐的吸附机理主要为纤维表面形成新的N-Cu-P配位结. PANAF-Cu对治理磷污染的废水具有很大的应用优势,是一种高效净化与回收水体磷酸盐的吸附材料.
-
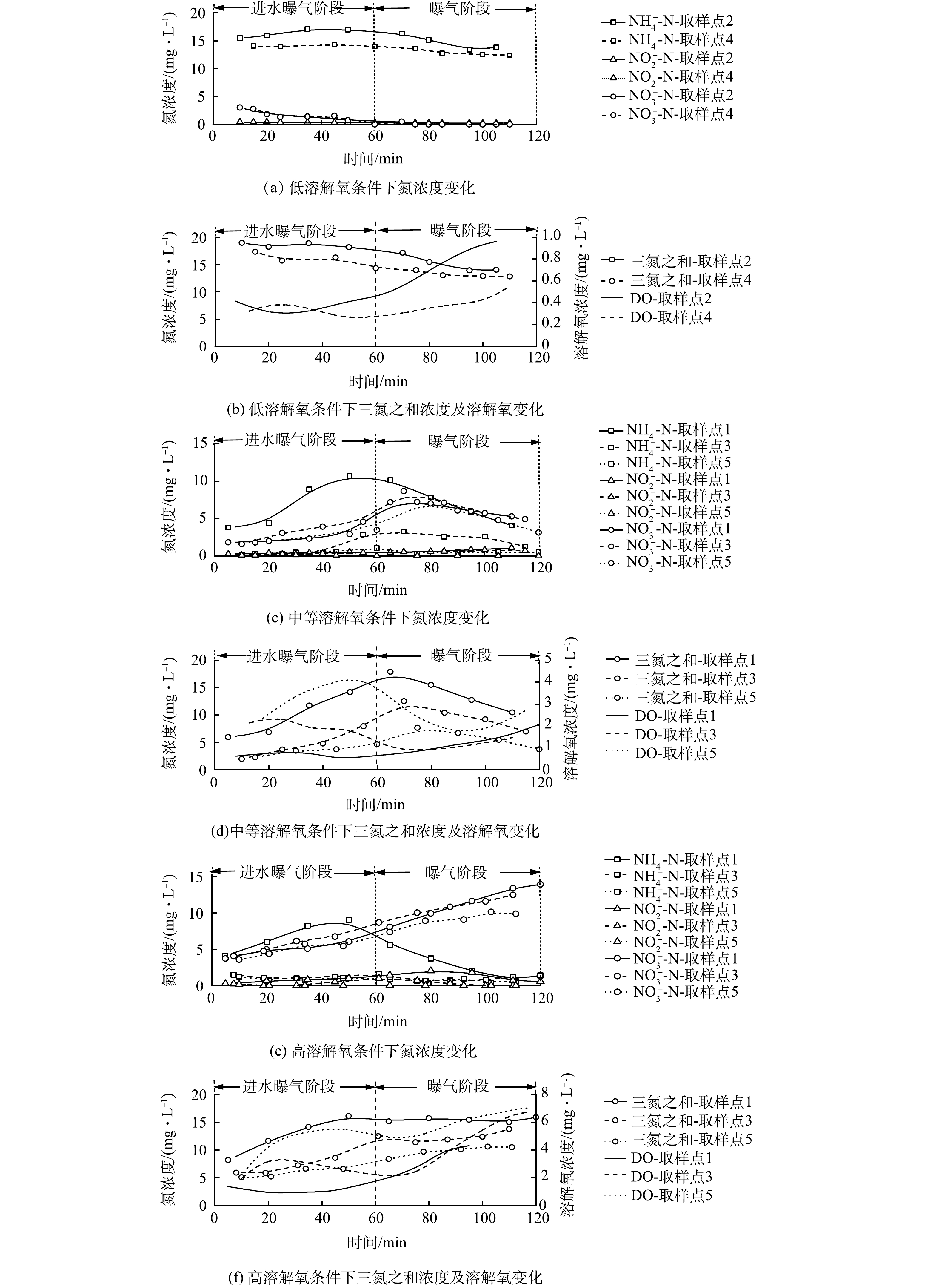
图 3 不同DO浓度下各取样点基质浓度变化
Figure 3. Variation of nitrogen concentration at each sampling point at different DO concentrations
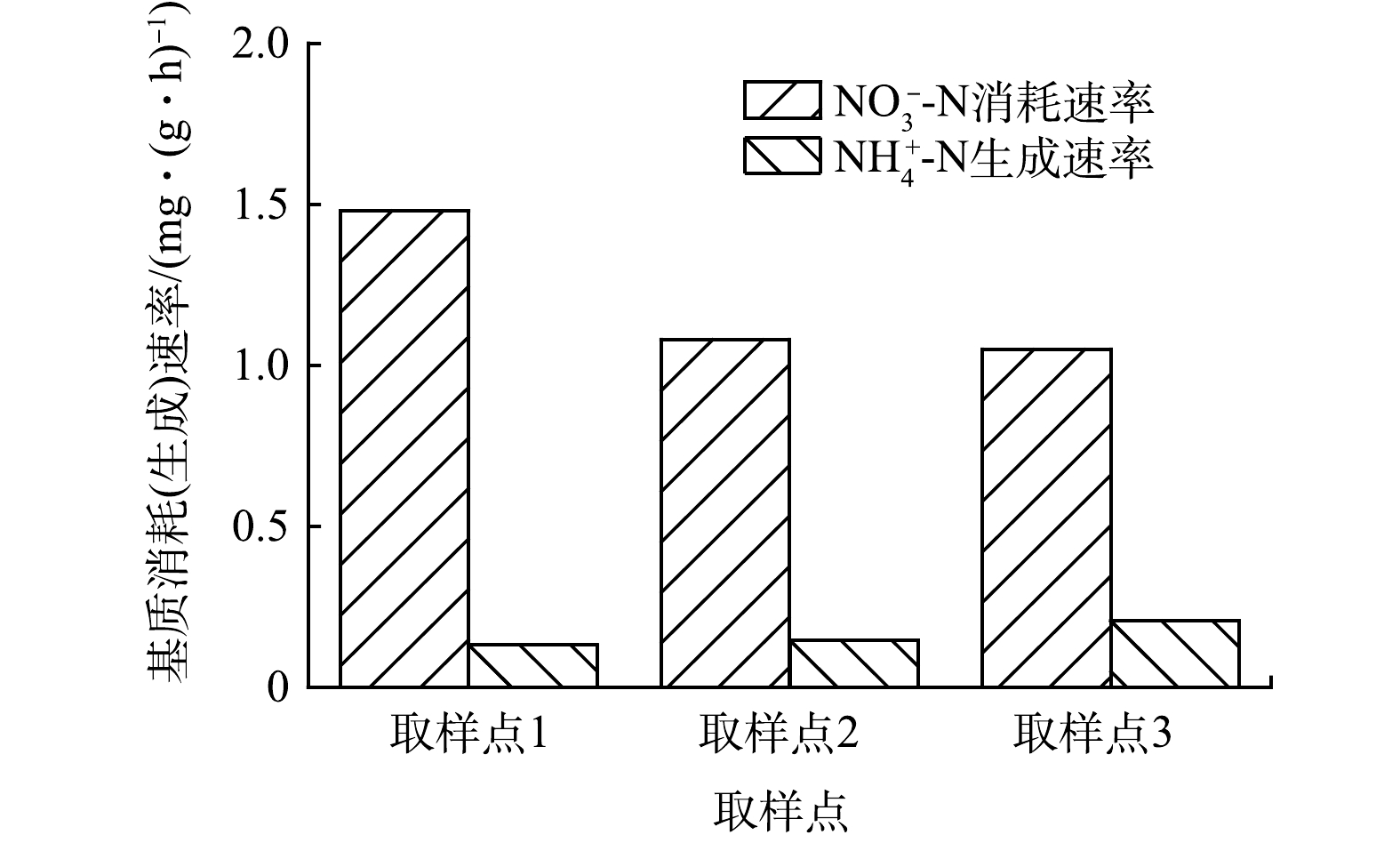
图 5 不同取样点内源反硝化过程中氮消耗(生成)速率
Figure 5. Rate of nitrogen consumption (production) during endogenous denitrification at different sampling points
表 1 进出水水质
Table 1. Quality of influent and effluent
mg·L−1 水样 COD NH4+-N NO3−- N TN PO43--P 进水 188±22 32.7±7.60 — 40.50±9.50 2.98±1.15 出水 20±3 1.60±1.30 6.74±3.59 11.30±2.20 0.07±0.06
下载: 导出CSV
表 2 CAST工艺典型周期各途径脱氮贡献占比
Table 2. Contribution of each pathway to nitrogen removal
脱氮方式 脱氮量/g 占比/% 反硝化脱氮 526.01±19.91 1.64±0.05 内源反硝化脱氮 20 093.13±1 291.55 62.86±4.13 同步硝化-反硝化脱氮 11 366.27±1 553.06 35.50±4.15 总脱氮 31 985.40±1 212.71 100
下载: 导出CSV
表 3 不同饥饿时长下活性污泥对HAc的吸收量
Table 3. Uptake of HAc by activated sludge at different famine durations
饥饿时长/h ΔMHAc0 ΔMNO−3 ΔMNO−2 ΔMHAc1 ΔMHAc2 5 335.16 79.35 74.87 91.31 243.84 24 394.68 101.39 88.34 128.47 266.2 36 468.91 103.59 94.3 124.77 344.14 48 407.25 102.71 93.03 124.46 282,79 72 376.14 96.54 84.47 121.76 254.38
下载: 导出CSV
表 4 CAST、A2/O和氧化沟工艺脱氮指标
Table 4. Denitrification indicators of CAST, A2/O and oxidation ditch processes
mg·L−1 工艺 进水COD 进水NH4+-N 进水TN 出水COD 出水NH4+-N 出水TN CAST 188±23 32.70±7.60 40.50±9.50 20±3 1.60±1.30 11.40±2.20 A2/O 249±38 36.12±5.38 44.36±4.87 18±3 0.32±0.26 8.30±1.26 氧化沟 643±136 35.12±4.89 47.50±5.33 21±3 1.07±0.57 8.47±1.01
下载: 导出CSV
-
[1] 邓伟斌. 南方某污水处理厂CAST工艺提标改造[J]. 中国给水排水, 2016, 32(16): 3-4. [2] 王俊, 王琴, 陈杰云, 等. CAST工艺处理小城镇污水的运行模式优化研究[J]. 中国给水排水, 2011, 27(19): 17-20. [3] MERVYN C, 朱明权. 循环式活性污泥法(CAST)的应用及其发展[J]. 中国给水排水, 1996, 12(6): 4-10. doi: 10.3321/j.issn:1000-4602.1996.06.001 [4] 孙剑辉, 闫怡新, 徐文刚. 循环式活性污泥法(CAST)的设计要点[J]. 中国给水排水, 2003, 19(1): 89-91. doi: 10.3321/j.issn:1000-4602.2003.01.028 [5] 孟栋. 利用剩余活性污泥合成聚羟基脂肪酸酯的研究进展[J]. 生物工程学报, 2019, 35(11): 2165-76. [6] BIAN D J, GUO H Y, ZHANG W H, et al. Simultaneous C and N removal in a hybrid CAST reactor[C]. 3rd International Conference on Bioinformatics and Biomedical Engineering, Beijing, 2009: 5145-5146. [7] WANG Y Y, ZHANG Z X, YAN M, et al. Impact of operating conditions on nitrogen removal using cyclic activated sludge technology (CAST)[J]. Journal of Science and Health Part A:Toxic/Hazardous Substances & Environmental Engineering, 2010, 45(3): 370-376. [8] WANG S P, YU J J, LIU Y H, et al, Achieving and maintaining of short-cut nitrification in a cyclic activated sludge system[J]. Water Science and Technology, 2011, 64(10): 2016-2022. [9] 国家环境保护局. 水和废水监测分析方法(第四版)[M]. 北京: 中国环境科学出版社, 2002. [10] 钱亮, 贺北平, 刘瑞东, 等. 西安市第四污水处理厂一期工程升级改造经验总结[J]. 中国给水排水, 2016, 32(2): 74-78. [11] 田敏, 崔涛, 吕恺, 等. 西安市第四污水处理厂A2/O工艺的脱氮性能评价[J]. 中国给水排水, 2020, 36(13): 1-6. [12] 徐凤, 李佩武, 刘艳辉. 曝气量对CAST工艺脱氮性能的影响[J]. 天津城市建设学院学报, 2010, 16(4): 281-284. [13] 彭党聪, 王志盈, 袁林江. 活性污泥系统非平衡增长理论及其应用[J]. 中国给水排水, 2001, 17(2): 19-21. doi: 10.3321/j.issn:1000-4602.2001.02.006 [14] MAJONE M, DIRCKS K, BEIM J. Aerobic storage under dynamic conditions in activated sludge processes. The state of the art[J]. Water Science & Technology, 1999, 39(1): 61-73. [15] 拉马尔奥, 王凤石. 废水处理概论[M]. 北京: 中国建筑工业出版社, 1982. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2406
- HTML全文浏览数: 2406
- PDF下载数: 151
- 施引文献: 0













