

-
受汽车行业发展的影响,每年报废汽车数量逐年增加,汽车破碎残余物 (automobile shredder residue,ASR) 作为汽车报废行业产生的工业固体废弃物,据估计,到2025年国内ASR产生量可达每年7×106 t[1],且呈现逐年增长趋势,若未能得到有效处置反而加重环境污染。为深入贯彻落实“十四五”规划,加强资源集约和再利用,需大力推进固体废弃物“资源化”、“减量化”和“无害化”处理,强化循环经济体系闭环,寻求ASR妥善的处置方法成为当前研究的热点之一。
研究表明采用传统填埋方式处理ASR,在填埋场浸出物中含有大量有毒化合物和重金属[2-3],ASR热值与褐煤热值相当[4],可作为垃圾焚烧发电厂原料,但焚烧产生大量飞灰以及二噁英等污染性物质,限制了焚烧处理ASR的进一步发展[5]。此外,ASR中Zn (2.10%) 、Cu (1.85%) 、Pb (0.26%) 、Cr (0.16%) 和Ni (0.12%) 等重金属含量高于一般工业固废[6],这使焚烧处置ASR时重金属污染控制问题比垃圾焚烧过程更加严重。相比之下,热解在缺氧条件下进行,产生的NOx、SOx及二噁英等二次污染物大幅减少。ASR主要包括塑料、橡胶、纤维、木材、纸张和泡绵等含碳物质[7],以及少量玻璃、污垢、岩石、沙石和残余金属碎屑等低价值组分[8],是一种典型的含碳基质固体废弃物。ANZANO等[9]通过基础理化性质分析,结果显示ASR热值为10~27 MJ·kg−1。ASR在500 ℃时完成主体分解[10],每千克ASR产生的热解气热值高达11 MJ,焦油产物中较高的碳含量使其能量回收价值颇高[11]。
目前,有关ASR热解的研究多集中在ASR不同物料组分对热解的影响上。HAYDARY等[12]发现ASR中橡胶含量的增加可提高焦炭产率,塑料含量的增加会导致气体产率的增加。YANG等[13]研究不同聚合物组分共热解过程时发现,塑料改变了纺织品和泡沫的分解机理,但对橡胶和皮革影响不大。为更深入诠释不同组分间的热解特性,HAN等[14]从ASR中分离出塑料、纤维、海绵和橡胶4种组分,并按恒定比例得到混合样品。结果显示,海绵和塑料在400 ℃发生较大程度反应,橡胶和纤维在该温度下反应程度较小。且热重分析结果表显示,橡胶和纤维有3个失重峰,海绵和塑料呈单一失重峰,说明后者化合物更加简单。SANTINI等[15]采用密度浮选的方式获得10种轻、重质ASR混合样品,热解实验结果显示浮选方式有效聚集了ASR中的聚合物,轻质有机组分更适合热解回收,且金属/重金属在热解过程中被释放出来,更易从热解焦中分离。但组分构成不同的ASR经热解后对热解气、焦油和热解焦的成分影响以及成分分析的研究尚未见报道,其热解特性和热解动力学分析需要进行深入研究。
因此,本研究从某报废汽车流水线以机械破碎结合比重筛选产生的5种典型预分选ASR物料为研究对象,在实验室固定床反应器中展开热解实验,通过气相色谱 仪(GC) 、气质联用仪 (GC-MS) 、拉曼光谱仪 (Raman) 、傅里叶红外光谱仪 (FTIR) 以及等离子体发射光谱仪 (ICP-OES) 分别对热解气组成、焦油组成、热解焦微观结构及其重金属分布等热解产物特性进行系统深入研究,揭示预分选后不同物料组成和温度对热解的影响规律,同时采用热重分析仪 (TGA) 对ASR热解过程动力学进行分析,为ASR热解技术开发提供重要理论支撑。
-
实验样品来源于某工厂报废汽车整车破碎拆解流水生产线上不同出料口产出的物料,根据物料比重和粒径进行筛选。本研究选择5个典型样品,依次编号为ASR1、ASR2、ASR3、ASR4和ASR5。ASR1与ASR2分别以纤维和橡胶为主;ASR3主要为含尘土的块状泡棉、皮革、纺织物和丝织品等,是块状的垃圾混合物;ASR4主要含小颗粒木屑、塑料片及纤维绒毛及沙土等;ASR5主要是金属碎屑、碎砂石、飞灰与细碎泡沫、木屑等小粒径物质混合物。样品工业分析和元素分析如表1所示。总体来看,ASR1和ASR2灰分低、有机物含量高、热值高,ASR3、4和5灰分含量逐渐增加、热值逐渐降低。对5种ASR中Cd、Cr、Pb、Ni四种重金属含量进行分析,重金属含量如表2所示。因Cd元素含量较低,在后续重金属分布中不再深入分析。
-
ASR热重实验采用同步热分析仪法 (STA449F3 TG-DSC,德国NETASCH公司) 进行。在实验过程中,选择升温速率为5、15和25 K·min−1,从室温升至900 ℃。使用直径约为8 mm、高度约为3 mm的氧化铝坩埚进行实验,为了减小样品内部传热和扩散影响并保证样品的均质性,实验前将样品烘干粉碎以充分混合,并筛分至120目 (0.125 mm) ,控制氮气流量在100 mL·min−1内,并控制样品用量在 (10±0.5) mg。实验时,将ASR原料放置于坩埚中,在设定的升温速率下将样品升温至900 ℃,持续观察失重情况,直至无明显失重。
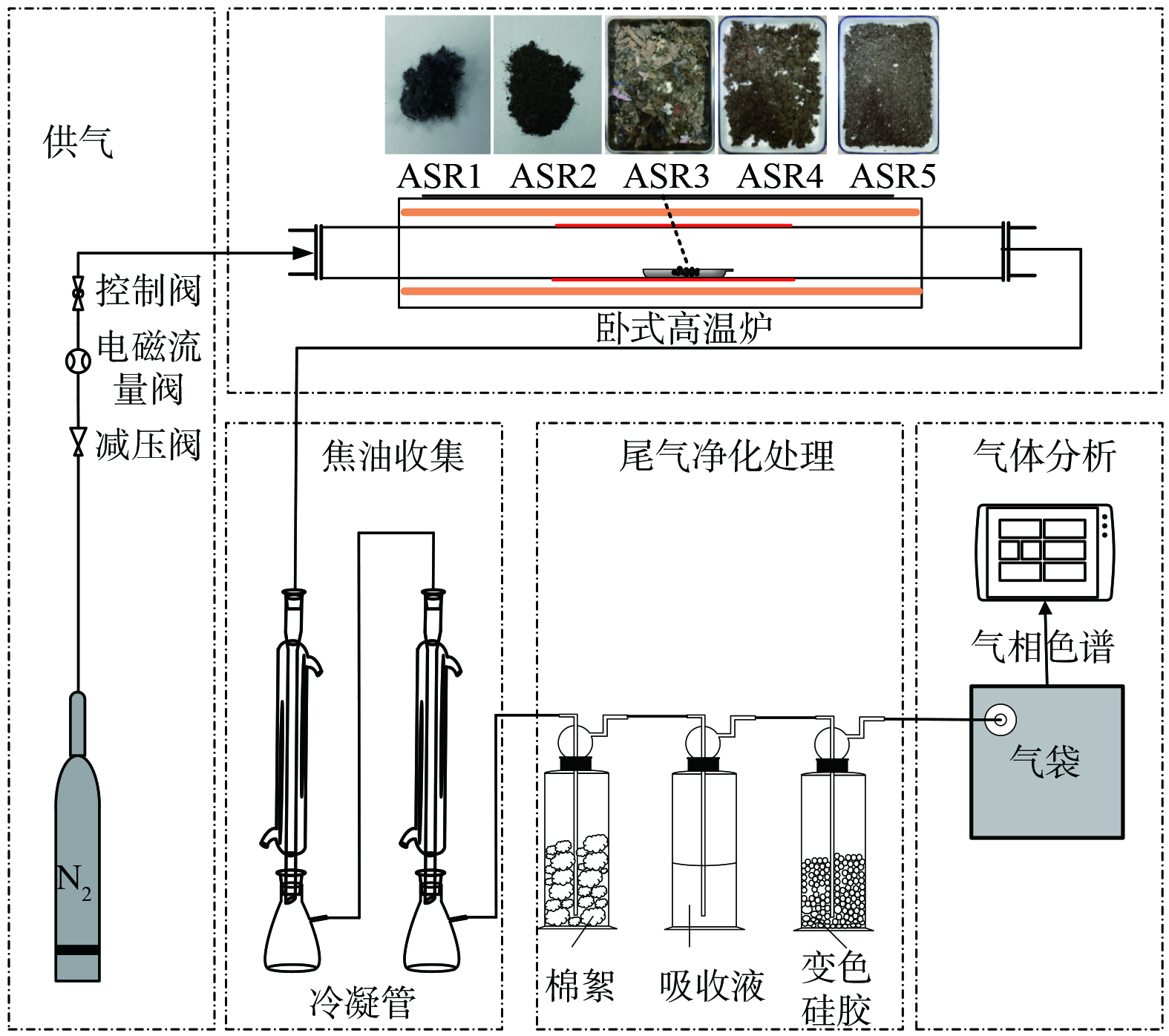
如图1所示,为ASR固定床热解实验装置示意图。实验时炉体以5 K·min−1的速率升温至设定温度 (500~900 ℃) 后稳定15 min,选用N2作为载气,以450 mL·min−1的速度向炉内通入载气,维持石英管内惰性气氛。每次实验称取 (5±0.5) g样品,反应持续45 min。通过铝箔气袋收集冷却和过滤干燥后的热解气。反应炉冷却至室温后,为保证实验数据准确性,每个实验条件下至少进行3次平行实验,计算平均值。取出焦样称重,热解焦油通过冷凝管进行冷凝收集,使用二氯甲烷溶剂 (分析纯,99.99%) 溶解蛇形冷凝管管壁粘附的热解焦油,获取的热解焦油溶液存储在−10 ℃的冷冻柜中。
-
1) 动力学分析方法。分布式活化能法 (DAEM) 是一个多反应模型,其模型假设为:热解过程由许多相互独立的一级不可逆反应组成;每个反应有确定的活化能,所有反应的活化能值呈某种连续分布。该模型对固相反应具有良好的适应性[16],不仅能较好地描述热解失重的真实过程,而且能够在较宽温度及升温速率范围内对反应过程进行准确描述,并在煤、生物质和固体废弃物等热解研究中得到广泛运用[17-19],DAEM法在热解动力学研究中的适用性得到较好的验证。ASR作为一种典型的工业固体废弃物,在本研究中,采用DAEM进行动力学分析,ASR的非等温热解过程中转化率x与时间t的关系可用DAEM模型表示[20],如式(1)所示。
式中:T是绝对温度,K;R是通用气体常数,J·mol−1·K−1;k0是频率因子,s−1;E是活化能,kJ·mol−1;f (E) 是活化能的分布函数。
x是样品转化率,其定义如式(2)所示。
式中:w0、wt、wf分别指煤样初始重量、t时刻样品重量和反应结束时刻样品重量,mg。
为估算动力学参数,MIURA等[21]基于DAEM方程提出了一种简易积分法,如式(3)所示。
式中:β是升温速率,K·min−1;R是通用气体常数,8.314 J·mol−1·K−1。
2) 产物分析方法。利用气相色谱仪 (GC7890A,Agilent) 定量测定不同温度下热解前后释放的气体组分,分析其中的N2、H2、CO、CO2及CH4等组分含量,利用N2示踪法来进行定量计算[22]。热解焦油样品采用气相色谱质谱联用仪 (GC-MS,7890A/5975C,Agilent) 进行检测,色谱柱为Agilent HP-5MS毛细管柱,进样口温度为280 ℃,初始温度40 ℃保持2 min,以10 K·min−1升至280 ℃。气质联用仪器可对测试所得时域图进行图谱比对分析,得到焦油中各成分物质化学式以及相对含量大小。采用压片法借助红外光谱仪 (FTIR,Nicolet iS50,Thermo Fisher) 分析热解焦的官能团,采用拉曼光谱仪 (Raman,DRX,Thermo Fisher) 分析碳结构特性,实验每次随机选取若干热解焦颗粒,使用455 nm波长激光,采集800~2 000 cm−1范围内的光谱,分辨率为0.5 cm−1。采用微波消解仪和等离子体发射光谱仪 (ICP-OES,Agilent 5110) 测定物料原样及热解焦中重金属含量,微波消解过程中,在强酸环境下对样品进行加热,消解液中硝酸 (HNO3) 与氢氟酸 (HF) 按3∶1体积比混合,将重金属转化为离子形态的可溶性盐,取所得消解液检测重金属元素含量。
-
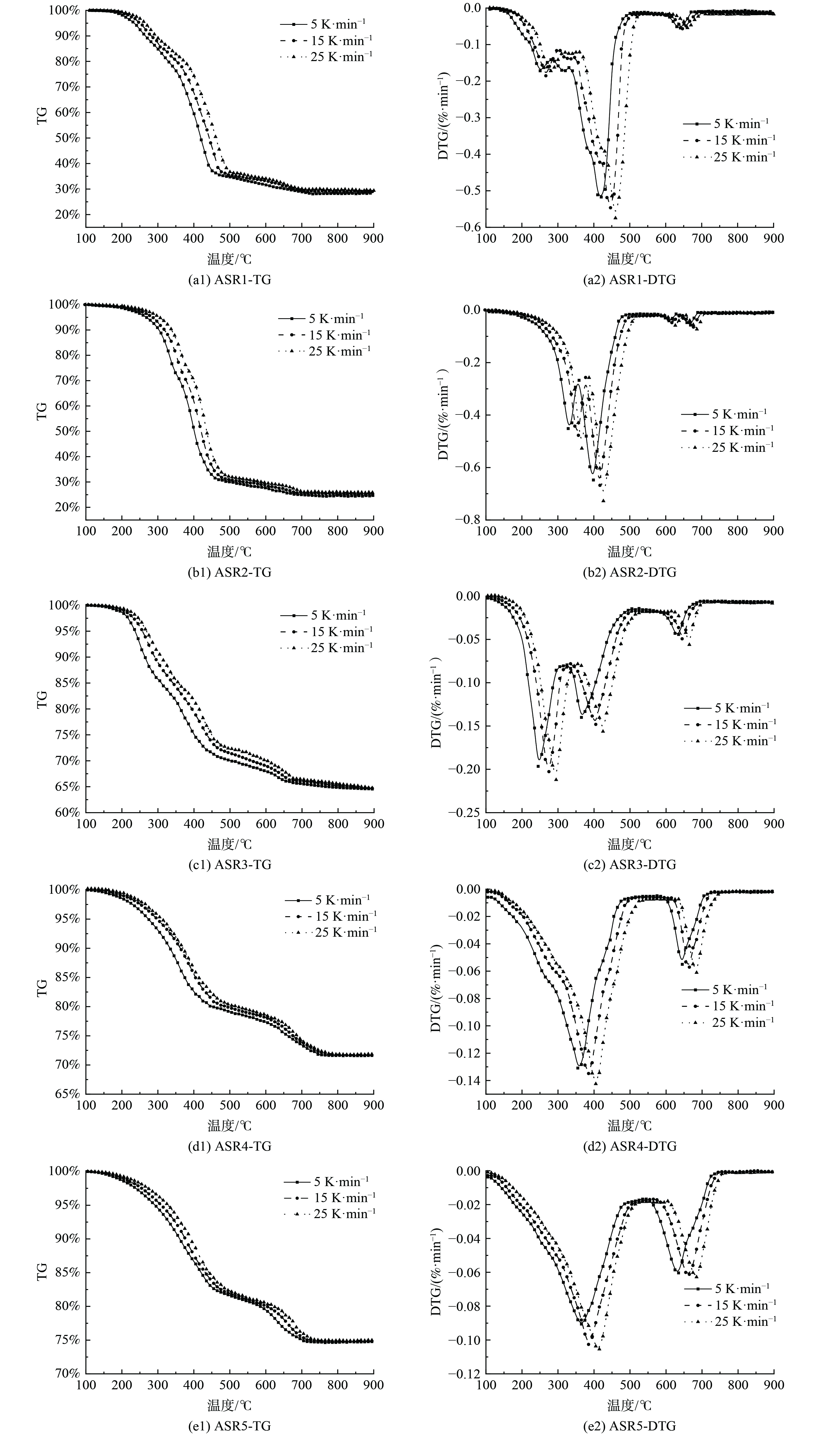
1) ASR热失重过程分析。图2为不同ASR物料在不同升温速率下的热解TG和DTG曲线。实验结果表明,5种ASR样品均经历了3个阶段的失重过程:200~500、500~600和600~800 ℃,其中主要的热解反应温度区间为200~500 ℃。在主反应温度区,ASR中部分含碳有机物分子因其键能较低开始发生裂解,同时部分含氧官能团支链也会发生热裂解分解成小分子化合物,以挥发分形式脱离物料,从而引起物料失重。500~600 ℃为衡定速率失重区间,在此温度区间内5种样品均未出现失重峰,表明在该温度范围内并未发生明显反应。600~700 ℃为高温下的失重温度区间,其中ASR5样品持续至750 ℃。在此温度范围内,剩余有机物与半挥发性物质基本反应完全,同时推测存在部分无机物受热分解,导致仍有小程度失重。此外,高温下热解气中的CO2与少量固定碳发生气化反应,进一步增加样品失重。
图2显示,经预分选处理后5种ASR物料仍在500 ℃完成主要热解过程,与DE MARCO等[10]研究结论相符,说明预分选并不影响ASR热解主反应温度区间。但区别在于,ASR1、2和3在500 ℃以下均有2个失重峰,表明有2个主反应,超过500 ℃后有小程度失重;ASR4与ASR5在500 ℃以下主失重峰相似,均为单峰,在600~750 ℃内两种物料均出现第二阶段反应,对应失重率达5%。分析认为,预分选改变了样品中主要成分占比,导致失重峰发生较大变化。根据Ni的研究[23],ASR样品成分复杂,包括海绵和泡沫在内的纺织品热解反应速率峰值在330 ℃左右,塑料和橡胶有2个热解反应速率峰,第一个在410 ℃左右,第二个在660 ℃左右。因此,纺织品、塑料和橡胶在330~500 ℃主要占失重的第一个峰。此外,ASR1主要成分为纤维,在240 ℃左右出现热解反应速率峰,低于纺织品、塑料和橡胶等组分热解反应速率峰对应温度,说明在低温下纤维类组分更易发生热解。
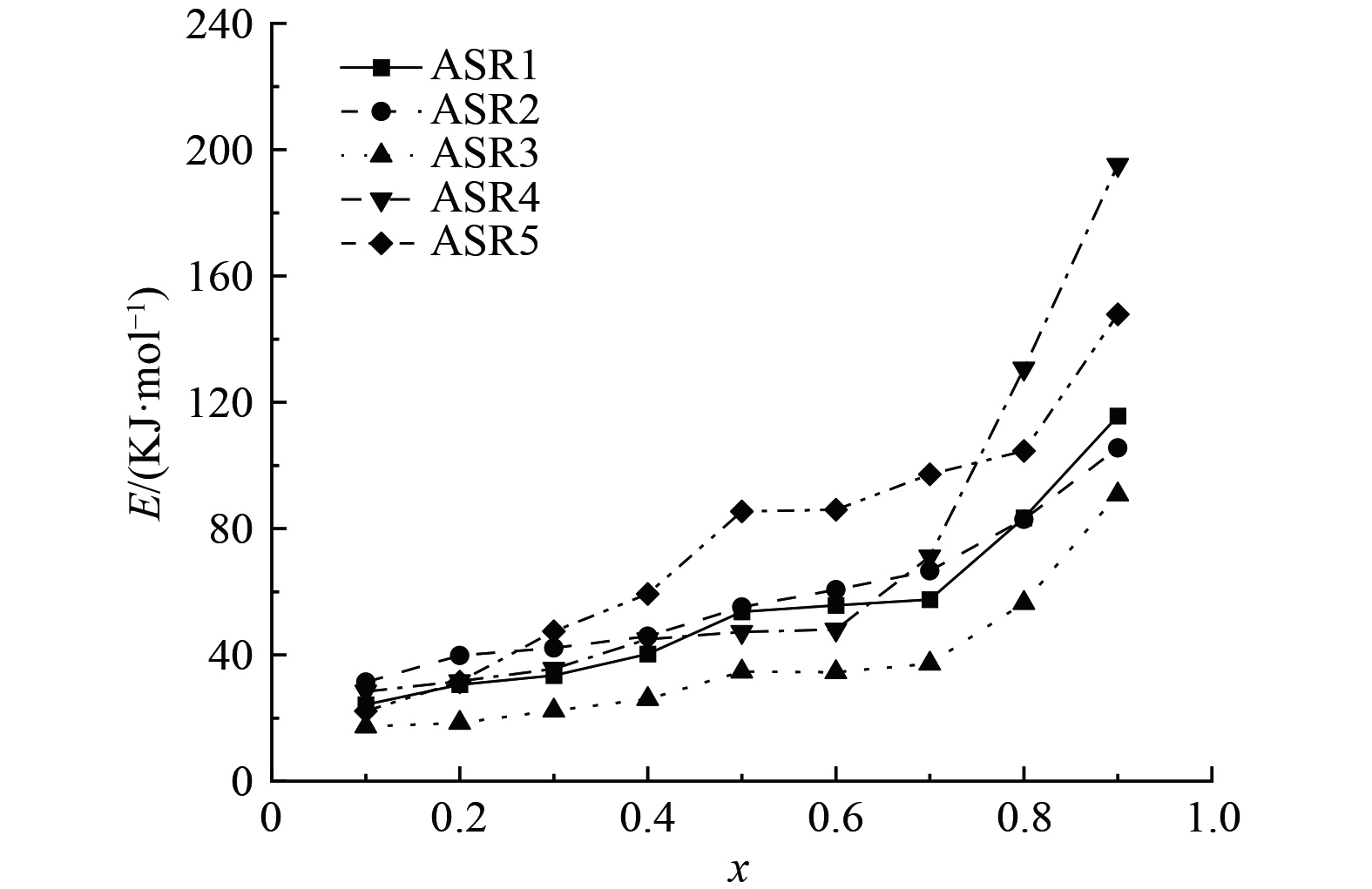
2) ASR热解动力学分析。根据热失重曲线和式(2)与式(3),在3种不同升温速率下选定同一转化率x,选择ln (β/T2) 对1/T作图,相同转化率x条件下ln (β/T2) 与1/T应为线性关系,其斜率即为活化能E。图3为拟合得到的ln (β/T2) 对1/T的关系曲线。根据ln (β/T2) 对1/T的线性回归拟合数据,图4为不同ASR样品计算活化能E随转化率x的关系曲线。如图所示,由于不同的热解温度范围内不同官能团分解所需的活化能存在差异,以ASR1为例,转化率从10%增至90%,热解活化能由31.39 kJ·mol−1升至105.56 kJ·mol−1。随着转化率增大,活化能逐渐增大,分析认为反应前期主要是大分子结构中成键强度弱的官能团先分解,不稳定的官能团断裂所需能量更少,随着反应的进行,不稳定的官能团已分解,剩余官能团更加稳定,此时化学键断裂需要更多能量[21]。
对比5种典型ASR物料,在相同转化率下,ASR3在各转化率下均有最低活化能,表明其在5种样品中反应难易程度最低。ASR4在转化率小于60%时,活化能增长趋势同余下4种样品;当转化率大于60%时,活化能大幅度增加,并在转化率到达90%时增至195.21 kJ·mol−1。分析认为ASR4在650 ℃以上的仍有较大质量损失,且TG曲线显示失重持续到750 ℃,其结果导致在更高的温度下进行反应需要更大的活化能,在活化能上体现为E值陡增。根据热失重过程特性可以得知,预分选不仅对5个ASR样品的组分和理化性质影响显著,而且改变了不同温度下的本征反应过程[12-13]。
-
图5(a)、(b)、(c)分别为5个样品在不同温度下热解焦产率、热解焦油产率和热解气产率分布规律。在500~900 ℃范围内,随温度升高,产气量呈现增长趋势。在热解过程中,弱化学键率先发生断裂形成气体,然后大分子物质裂解形成焦油,官能团也随之破裂生成气体[24]。因此,在整个热解进程中气体产率逐渐增加。当热解温度高于500 ℃,随反应器温度逐步升高至900 ℃,热解焦产率持续减小。在相同温度下,ASR1和ASR2的焦产率比ASR3、4和5小30%,这主要受预分选对ASR主要成分的影响,结合表1数据可知,ASR3、4和5含较多灰分,因其不参与热解反应,导致热解焦残留占比较大。此外,ASR1和ASR2样品含碳有机物比值更高,在整个热解过程中热解生成挥发分占比更大,导致热解焦产率小于ASR3、4和5。热解温度在500~900 ℃时,ASR1和ASR2在900 ℃气相产率相比于500 ℃提升约5倍,ASR3、4和5热解气产率提高约4倍,ASR1在900 ℃下热解气产率高达76.52%,ASR4仅占33.52%。可以看到,预分选可有效提升ASR原料能源转化效率。同时,高温还会引发焦油二次裂解,焦油量进一步减少,其中ASR1和ASR2焦油产率高于其他3种物料,且受温度影响较大,随热解温度提升焦油产率减少约45%,但不同的是,ASR2在900 ℃焦油产率维持在11.5%的较高水平。以上结果表明,提高温度有利于物料热解,产生更多的热解气体,且促使热解进行更加彻底,同时抑制焦油产生。预分选后不同ASR样品热解产物分布差异明显,ASR1和2产焦少、热解气和焦油生成量高。
-
图6是不同ASR样品热解气成分随温度变化曲线。在较低热解温度下,按照组成的体积占比,相对含量较高的为CO2、H2、CO以及CH4。这是由于ASR中聚合物热解产生较小的分子,分子进行环化反应或分解成轻质烃,形成各种不同长度的分子链。随着温度升高,反应产生的环状化合物和烃类化合物进一步裂解生成CH4、CO2和CO等分子[25]。而C2H2因成键困难,形成条件严苛,故含量最低,且基本不受温度的影响。从变化趋势中可以看到,除CO2和C2H2外,其它气体随温度的上升含量均有所提升,其中ASR3、4的CH4含量随温度的提高出现明显的先升后降趋势,这是由于高温下热解气中发生水煤气变换反应 (CO+H2O=CO2+H2、CH4+H2=CO+3H2) [26],以致CH4含量出现波动。而在5种ASR样品热解气中CO2含量随温度的升高反有明显地下降,这是因为热解阶段析出的CO2主要来自于低温下参与芳化缩聚的C=O和C-O自由基团发生重整等反应[27],当温度上升受自由基数量的限制,CO2生成量降低。而焦油中的组分在更高温度下发生二次裂解,生成小分子气态物质,再次降低了CO2在热解气中的占比[28]。此外,高温下开始发生反应 (C+CO2→2CO) ,即CO2与热解焦中的固定碳发生反应,在宏观上表现为CO2向CO转变,也会降低CO2的生成量,但该反应有利于热解气有效气成分的增加。在不考虑CO2占比的前提下,在ASR2中500、600和700 ℃下主要为CO2和H2,800和900 ℃下以CH4和CO为主,在ASR1中主要为CO;ASR3、ASR4和ASR5中800 ℃以下H2占比最高,900 ℃时CO有显著提升。可以看到,预分选后不同物料对热解气组成有较大影响,ASR3、ASR4与ASR5气体组成更加相近,ASR1和ASR2热解气在最主要成分上有所不同。
-
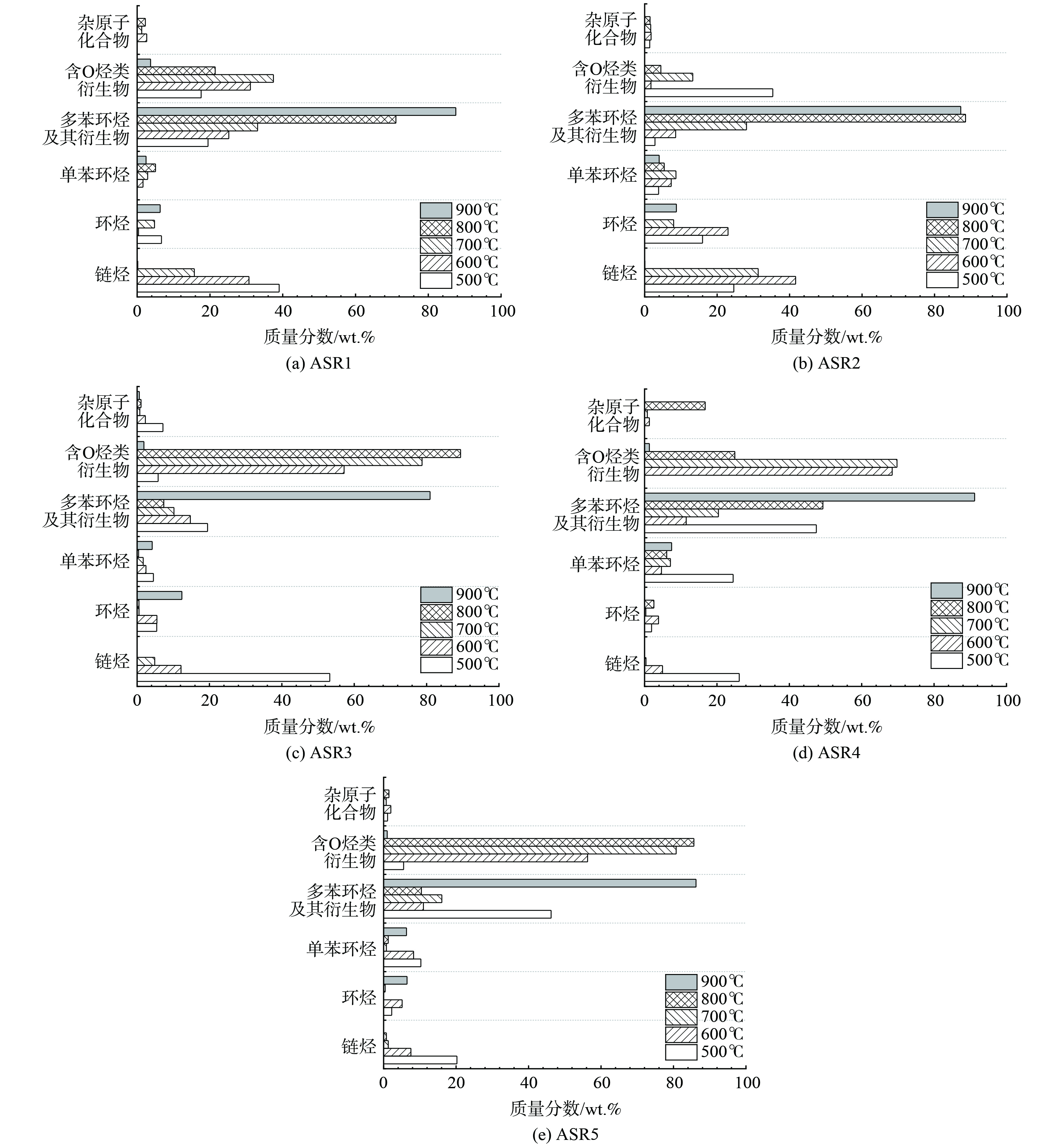
以ASR1样品为例,图7为不同热解温度下热解焦油GC-MS时域图,分析采用了面积归一法进行积分定量,并结合NIST标准质谱图库进行组分定性。为了保证实验数据的可信度,选择与焦油中已知组分分子式相似度达到90%以上的物质作为焦油组分。由于焦油中包含许多种不同的组分,为简化研究,仅统计含量超过2%的组分。对ASR在不同热解温度下收集到的焦油成分分类统计,结果如图8所示。统计结果表明,焦油随温度升高,成分从低温的链烃、环烃转向生成多苯环类及其衍生物。将焦油成分进行分类,主要分为链烃、环烃、单苯环烃、多苯环烃及其衍生物、含O烃类衍生物以及杂原子化合物 (如N、Si、S和Cl等) 共6大类物质。在600 ℃以下ASR1、2和3焦油中物质更多以链烃和含氧烃类衍生物为主,ASR4和ASR5以链烃和多苯环烃及其衍生物为主。当温度上升至900 ℃,5种样品中烃类和含氧烃类衍生物等含量急剧下降,以多苯环烃及其衍生物为主要成分。在YANG等[29]采用了TG-FTIR-GC/MS分析技术对ASR热解焦油分析后发现,焦油中许多物质具有不稳定或较弱的化学键,如=CH-,=CH2,-C=C-和-C=CH2等,这些化学键在高温下更易发生转化,结合焦油中物质种类的转变趋势,表明焦油成分在温度提升后发生了弱键断裂和苯环化变化[24,30]。
-
1) 官能团表征分析。傅里叶红外光谱 (FTIR) 可反映出物质表面所含官能团信息,对同种ASR物料不同温度下所得谱图进行对比,可从官能团角度分析热解焦结构变化,结果如图9所示。特征峰变化主要为:醇、酚类中的-OH伸缩振动 (3 650-3 600 cm−1) ,为尖锐吸收峰,分子间-OH伸缩振动 (3 500-3 200 cm−1) ,为宽吸收峰;胺类中-NH的伸缩振动 (3 500-3 300 cm−1) ;羧酸中-COOH伸缩振动 (3 400-2 500 cm−1) ;-C=C-烯烃类伸缩振动 (1 610-1 590 cm−1) [31-32];脂肪链末端-CH3对称弯曲振动 (1 460-1 380 cm−1) ;3H取代的伸缩振动 (850-720 cm−1) [33-34]。在3 450 cm−1波段出现了1个宽且强的吸收峰,这表明ASR中存在醇、酚、羧酸等游离态羟基基团伸缩振动以及可能的胺类中-NH的伸缩振动,从而使得伸缩振动范围较宽。在500~900 ℃内,随温度的升高峰强均有所减弱,说明在热解过程中,热解焦中有醇、酚分子间羟基或者羧基发生了脱羟基反应或胺类物质中-NH脱除。值得注意的是,ASR1热解焦随温度的上升出现了3 650 cm−1对应的醇类、酚类中的-OH伸缩振动。ASR1、2、3和4热解焦中1 600 cm−1吸收振动峰随温度上升而消失,说明对应该范围的-C=C-官能团在反应中发生转变,分析认为是-C=C-向苯环化转变导致。ASR1、2、4和5热解焦中1 430 cm−1吸收振动峰随温度的升高而消失,表明在此范围内对应烷烃上-CH3脱除,脱离的-CH3自由基或生成气相中的小分子物质。780 cm−1吸收振动峰对应的H取代,表明热解焦中H自由基的脱除,有利于热解气中小分子物质的产生,例如CH4、H2和碳氢化合物的生成。可以看到,随热解温度的升高,热解焦表面部分分子基团脱除,尤其以官能团为主的支链发生断裂,例如-OH、-CH3、-C=C-以及-COOH等。小分子官能团的消失是热解气中轻质物质和一碳化合物等产生的原因,升高温度破坏小分子官能团加剧热解焦进一步碳化,同时促使热解焦碳晶结构向更有序化方向转变[28]。
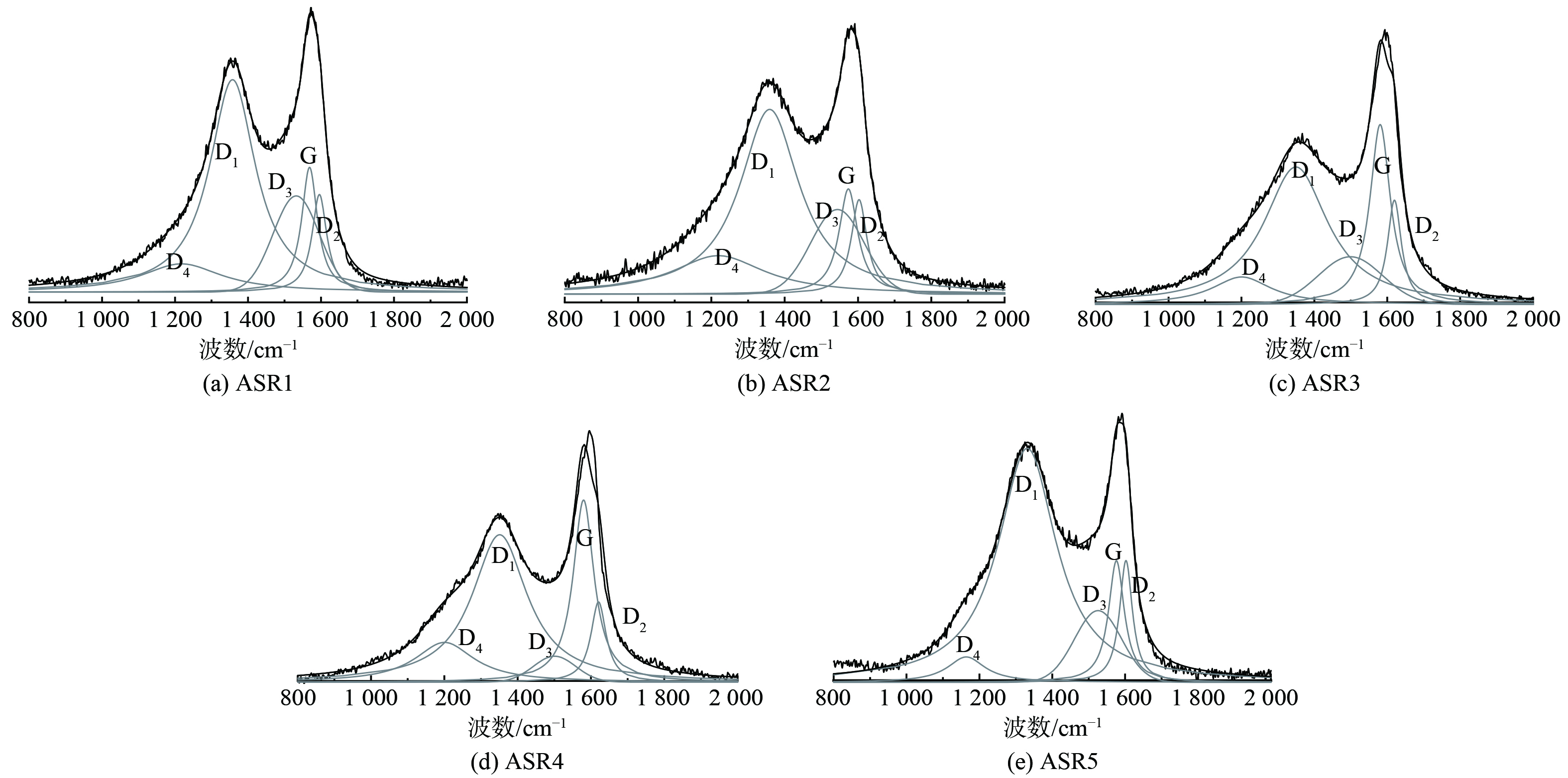
2) 碳结构分析。原始拉曼谱图通常是由多个特征峰叠加而成,通过分峰拟合可分解成4个洛伦兹子峰 (D4、D1、G、D2) 和1个高斯子峰[35]。以500 ℃热解焦为例,拉曼图谱分峰拟合结果如图10所示。D1峰与G峰的积分面积之比 (ID1/IG) 通常被用于定量表征热解焦结构在气化过程中的变化,表示碳结构的晶格度或石墨化程度,研究发现,ID1/IG与晶体的平面尺寸成反比,即ID1/IG比值的降低表明煤焦有序化程度升高[36]。表3为热解温度为500~900 ℃下热解焦拉曼峰面积比ID1/IG。如表所示,5种ASR样品的热解焦ID1/IG值均随热解温度的升高而降低,ID1/IG值越小表明半焦的有序化程度越高,也即热解焦石墨化程度越高,结合热解焦FTIR表征结果,官能团的消失使自由基脱除,从而使碳晶结构向更有序化发展。研究还发现,在500~900 ℃内,ASR3和ASR5热解焦ID1/IG值始终低于ASR1、2和4,说明除温度对ID1/IG值有影响外,物料组成不同也是影响热解焦结构的重要原因素之一。
3) 重金属分布。已有研究表明,ASR含较高含量重金属[11],在热解过程中重金属会进一步向热解焦中富集。对热解半焦中Cr、Pb和Ni含量进行分析,参考LI等[37]对热解半焦中重金属含量的分析方法,以热解半焦中重金属的富集率 (ER) 和残留率 (RR) 作为评价热解后重金属富集和固定程度的参数,计算公式如(4)~(5)所示。
式中:Cx为生物炭中重金属的质量分数,mg·kg−1;C为原料中重金属的质量分数,mg·kg−1。
式中:y为热解半焦收率,由热解半焦质量与热解原料质量之比计算得出。
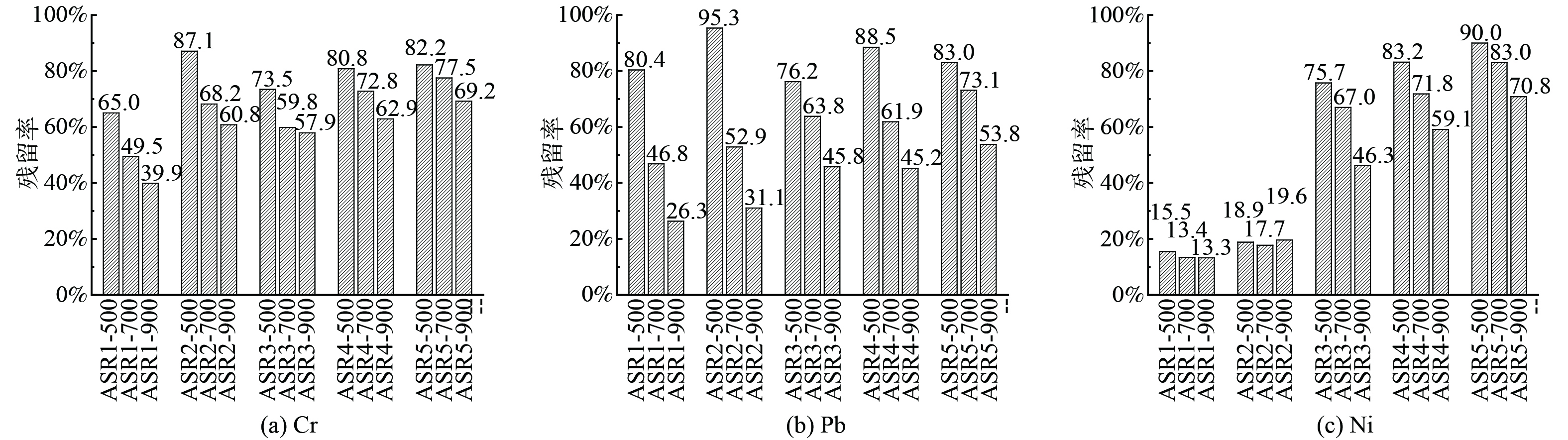
含量测定选取500、700、900 ℃下所得焦样,热解焦重金属富集率结果如图11所示。随温度的提高,5个样品热解焦中重金属的残留率均有不同程度的下降,表明温度的提高,重金属向热解气和焦油中转移,但固存在焦中的含量在800 ℃以下仍能保持50%以上,多数重金属保留在热解焦中。Ni在物料ASR1和ASR2中残留率表现不同于ASR3、4和5,残留率基本不受温度的影响。这种差异性可能的解释是由ASR物料组成的差异造成的。但在ASR3、4和5中Ni元素残留率随温度的升高而降低,这与Cr、Pb没有差异。
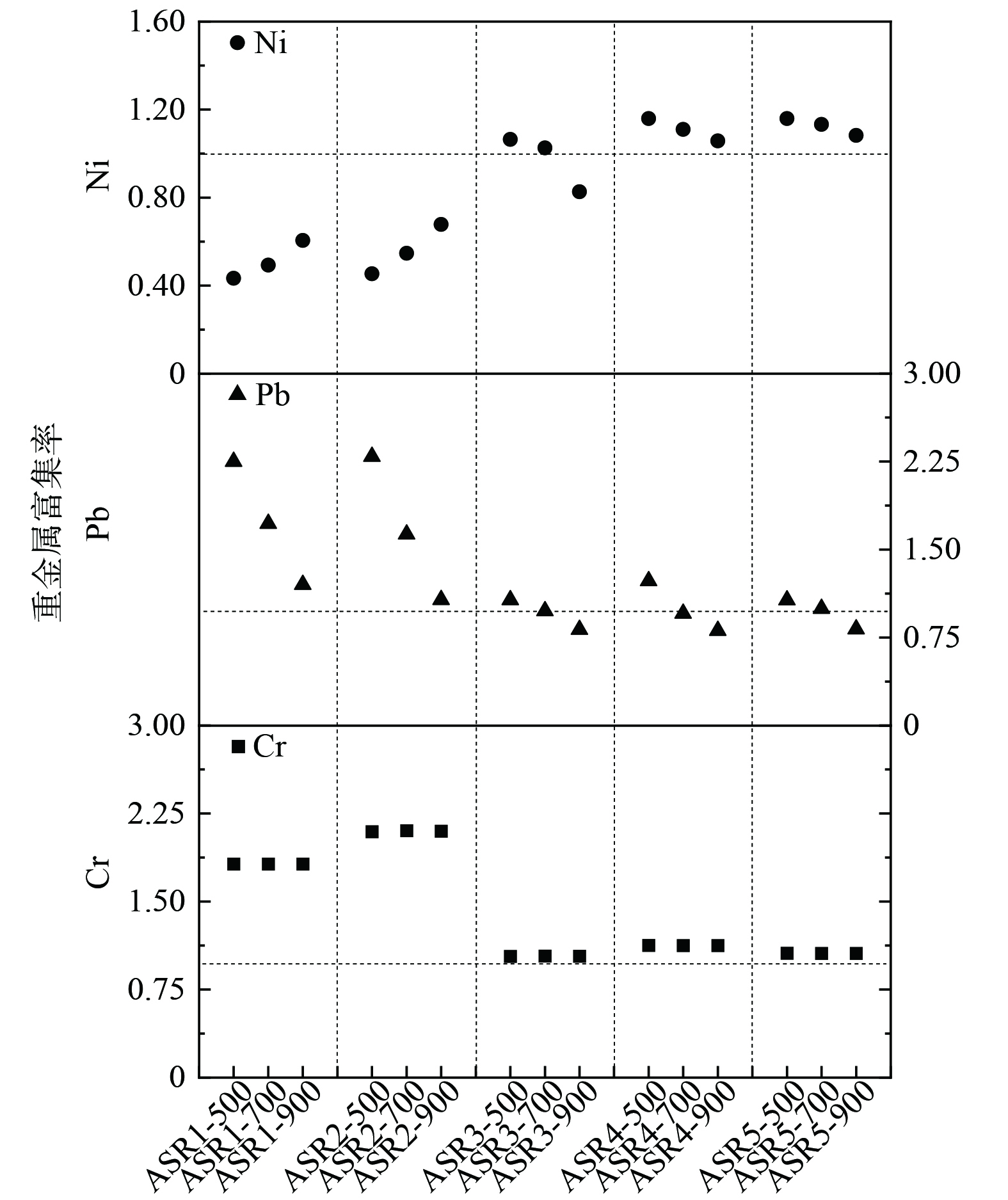
热解过程中,重金属按照去向分为3个方向:热解气、焦油和热解焦。在一般情况下,期望在反应过程中的重金属应尽可能保留于固相中,以降低重金属伴随热解气和焦油发生逃逸,增加环境污染压力和后续工艺除杂的复杂程度。图12为5种ASR物料在不同温度下热解后重金属富集的结果。富集率以1作为标准:热解半焦中重金属含量与等量原料中的含量比值为1。从结果来看,按富集能力从大到小排列:Cr>Pb>Ni。Cr元素整体ER值均大于1,且基本不受温度变化的影响。从ER值的大小来看,ASR1和ASR2样品在Cr元素固定上更具优势,而ASR3、4和5也能保持Cr基本固定在半焦中。Pb元素的ER值在5种ASR物料中的表现类似,随温度的升高ER值减小,但在ASR1和ASR2中Pb元素的ER值变化幅度高于ASR3、4和5。Ni元素ER值在不同物料中的变化差异较大,在ASR1、ASR2中ER值随温度升高而增大,在ASR3、ASR4、ASR5 ER值随温度升高而略有减小。总体上,ASR中不同重金属的富集程度主要受物料种类和热解温度的影响。
-
1) ASR热解经历3个失重阶段,反应温度区间集中于200~500 ℃,不受预分选ASR样品组成影响。热解动力学分析显示不同预分选样品活化能差异显著,主要与物料组分热解难度相关。热解产物产率分布受ASR物料成分影响明显,但总体上随热解温度升高,热解气产率增大,焦油发生二次裂解,生成量减少,且随温度升高焦油向多苯环物质转化。
2) 热解焦主要发生-OH、-CH3、-C=C-以及-COOH等分子基团的脱除,温度升高热解焦石墨化程度增大,但不同预分选样品石墨化程度差异明显。热解半焦中重金属残留率与热解温度和样品组成密切相关。热解过程重金属固存在半焦的比例整体保持在40%以上,过高的热解温度不利于重金属的固定。
预分选汽车破碎残余物热解特性及其动力学分析
Pyrolysis characteristics and dynamic analysis of pre-separated automobile shredder residues
-
摘要: 我国每年产生的汽车破碎残余物 (automobile shredder residue,ASR) 数量巨大,亟需无害化处理和资源化利用。固废热解过程因产生的二次污染物少,且产品利用价值高,逐渐成为研究的热点。针对某报废汽车生产线产生的5种典型预分选ASR物料,在固定床反应器中考察了温度 (500~900 ℃) 和物料种类对ASR热解三相产物组成、热解焦微观结构及重金属分布规律的影响,通过热重 (TG) 分析ASR从常温到900 ℃热失重过程,并采用分布式活化能 (DAEM) 法计算其热解动力学。结果表明,ASR热解主要发生于200~500 ℃范围内,热解反应活化能为17.39~195.21 kJ·mol−1。提高热解温度,ASR样品可产生更多热解气,且H2、CO和CH4等成分占比增加,焦油产生量减少,同时促进焦油二次裂解以及组分向苯环化转化。红外和拉曼光谱分析表明,热解温度升高,热解焦中主要发生-OH、-CH3、-C=C-以及-COOH等分子基团的脱除,不同ASR热解焦ID1/IG值在3.77~6.57内,石墨化程度增大,热解焦中重金属分布受温度和物料种类影响显著。本研究结果可为开发ASR热解技术提供参考。Abstract: A huge amount of Automobile Shredder Residue (ASR) is produced in China every year, and a reasonable disposal method is urgently needed. Solid waste pyrolysis has gradually become a hot topic of research due to its ability to reduce and recycle ASR and its high fuel utilization value. In this study, the effects of temperature (500~900 °C) and material on the composition of ASR pyrolysis three-phase products, microstructure of pyrolytic char and distribution pattern of heavy metals were investigated in a fixed-bed reactor for five typical pre-sorted ASR materials generated from an end-of-life automobile production line, and the thermal weight loss of ASR from room temperature to 900 °C was analyzed by thermogravimetry (TG), and the distributed activation energy method (DAEM) to calculate its pyrolysis kinetics. The results showed that the ASR pyrolysis mainly occurred in the range of 200~500 °C, and the activation energy of pyrolysis reaction was 17.39~195.21 kJ·mol−1. Increasing the pyrolysis temperature, the ASR samples could produce more pyrolysis gas, and the percentage of components such as H2, CO and CH4 increased, and the amount of tar production decreased, while promoting the secondary cracking of tar and the conversion of components to benzene cyclization. FTIR and Raman spectroscopy analysis showed that the removal of molecular groups such as -OH, -CH3, -C=C- and -COOH mainly occurred in pyrolytic char with increasing pyrolysis temperature, and the ID1/IG values of different ASR pyrolytic char were within 3.77~6.57 with increasing graphitization, and the distribution of heavy metals in pyrolytic char was significantly affected by temperature and type of material. The results of this study can provide reference for the development of ASR pyrolysis technology.
-
受汽车行业发展的影响,每年报废汽车数量逐年增加,汽车破碎残余物 (automobile shredder residue,ASR) 作为汽车报废行业产生的工业固体废弃物,据估计,到2025年国内ASR产生量可达每年7×106 t[1],且呈现逐年增长趋势,若未能得到有效处置反而加重环境污染。为深入贯彻落实“十四五”规划,加强资源集约和再利用,需大力推进固体废弃物“资源化”、“减量化”和“无害化”处理,强化循环经济体系闭环,寻求ASR妥善的处置方法成为当前研究的热点之一。
研究表明采用传统填埋方式处理ASR,在填埋场浸出物中含有大量有毒化合物和重金属[2-3],ASR热值与褐煤热值相当[4],可作为垃圾焚烧发电厂原料,但焚烧产生大量飞灰以及二噁英等污染性物质,限制了焚烧处理ASR的进一步发展[5]。此外,ASR中Zn (2.10%) 、Cu (1.85%) 、Pb (0.26%) 、Cr (0.16%) 和Ni (0.12%) 等重金属含量高于一般工业固废[6],这使焚烧处置ASR时重金属污染控制问题比垃圾焚烧过程更加严重。相比之下,热解在缺氧条件下进行,产生的NOx、SOx及二噁英等二次污染物大幅减少。ASR主要包括塑料、橡胶、纤维、木材、纸张和泡绵等含碳物质[7],以及少量玻璃、污垢、岩石、沙石和残余金属碎屑等低价值组分[8],是一种典型的含碳基质固体废弃物。ANZANO等[9]通过基础理化性质分析,结果显示ASR热值为10~27 MJ·kg−1。ASR在500 ℃时完成主体分解[10],每千克ASR产生的热解气热值高达11 MJ,焦油产物中较高的碳含量使其能量回收价值颇高[11]。
目前,有关ASR热解的研究多集中在ASR不同物料组分对热解的影响上。HAYDARY等[12]发现ASR中橡胶含量的增加可提高焦炭产率,塑料含量的增加会导致气体产率的增加。YANG等[13]研究不同聚合物组分共热解过程时发现,塑料改变了纺织品和泡沫的分解机理,但对橡胶和皮革影响不大。为更深入诠释不同组分间的热解特性,HAN等[14]从ASR中分离出塑料、纤维、海绵和橡胶4种组分,并按恒定比例得到混合样品。结果显示,海绵和塑料在400 ℃发生较大程度反应,橡胶和纤维在该温度下反应程度较小。且热重分析结果表显示,橡胶和纤维有3个失重峰,海绵和塑料呈单一失重峰,说明后者化合物更加简单。SANTINI等[15]采用密度浮选的方式获得10种轻、重质ASR混合样品,热解实验结果显示浮选方式有效聚集了ASR中的聚合物,轻质有机组分更适合热解回收,且金属/重金属在热解过程中被释放出来,更易从热解焦中分离。但组分构成不同的ASR经热解后对热解气、焦油和热解焦的成分影响以及成分分析的研究尚未见报道,其热解特性和热解动力学分析需要进行深入研究。
因此,本研究从某报废汽车流水线以机械破碎结合比重筛选产生的5种典型预分选ASR物料为研究对象,在实验室固定床反应器中展开热解实验,通过气相色谱 仪(GC) 、气质联用仪 (GC-MS) 、拉曼光谱仪 (Raman) 、傅里叶红外光谱仪 (FTIR) 以及等离子体发射光谱仪 (ICP-OES) 分别对热解气组成、焦油组成、热解焦微观结构及其重金属分布等热解产物特性进行系统深入研究,揭示预分选后不同物料组成和温度对热解的影响规律,同时采用热重分析仪 (TGA) 对ASR热解过程动力学进行分析,为ASR热解技术开发提供重要理论支撑。
1. 实验与方法
1.1 实验原料
实验样品来源于某工厂报废汽车整车破碎拆解流水生产线上不同出料口产出的物料,根据物料比重和粒径进行筛选。本研究选择5个典型样品,依次编号为ASR1、ASR2、ASR3、ASR4和ASR5。ASR1与ASR2分别以纤维和橡胶为主;ASR3主要为含尘土的块状泡棉、皮革、纺织物和丝织品等,是块状的垃圾混合物;ASR4主要含小颗粒木屑、塑料片及纤维绒毛及沙土等;ASR5主要是金属碎屑、碎砂石、飞灰与细碎泡沫、木屑等小粒径物质混合物。样品工业分析和元素分析如表1所示。总体来看,ASR1和ASR2灰分低、有机物含量高、热值高,ASR3、4和5灰分含量逐渐增加、热值逐渐降低。对5种ASR中Cd、Cr、Pb、Ni四种重金属含量进行分析,重金属含量如表2所示。因Cd元素含量较低,在后续重金属分布中不再深入分析。
表 1 样品工业分析和元素分析Table 1. Proximate and ultimate analyses of samples样品 工业分析wad/% 元素分析wad/% 干基低位发热量/(MJ·kg−1) M Ash V FC C H N S O* ASR1 0.81±0.08 17.93±0.21 73.50±0.20 7.76±0.14 45.70±0.22 3.94±0.11 1.09±0.09 1.57±0.10 28.96±0.15 21.89±0.41 ASR2 0.99±0.07 24.51±0.25 61.53±0.21 12.97±0.18 48.60±0.21 8.03±0.15 1.54±0.11 1.28±0.11 15.05±0.18 20.46±0.23 ASR3 0.76±0.08 44.12±0.48 48.14±0.18 6.98±0.09 31.51±0.23 2.88±0.14 0.66±0.08 1.95±0.09 18.12±0.20 15.66±0.20 ASR4 1.35±0.10 49.91±0.45 43.21±0.18 5.53±0.11 31.50±0.18 3.25±0.17 0.70±0.09 1.94±0.07 11.35±0.21 12.69±0.18 ASR5 1.17±0.09 63.29±0.55 30.88±0.22 4.66±0.12 17.35±0.16 1.58±0.17 0.78±0.08 1.34±0.07 14.49±0.20 8.69±0.19 注:M代表水分,V代表挥发分,A代表灰分,FC代表固定碳,ad代表空气干燥基,*为通过差减法计算。 | Show Table DownLoad:
CSV
表 2 ASR样品重金属元素含量Table 2. Content of heavy metal elements in ASR
DownLoad:
CSV
表 2 ASR样品重金属元素含量Table 2. Content of heavy metal elements in ASRmg·kg−1 样品 元素种类 Cd Cr Pb Ni ASR1 1.21±0.05 509.74±0.11 101.10±0.07 187.47±0.08 ASR2 1.10±0.04 422.49±0.09 95.74±0.05 161.36±0.09 ASR3 20.11±0.10 884.11±0.15 612.49±0.14 514.51±0.15 ASR4 7.78±0.03 693.33±0.14 558.66±0.17 441.41±0.14 ASR5 15.22±0.06 841.40±0.18 562.43±0.15 461.10±0.13 | Show TableDownLoad:
CSV
1.2 实验装置
ASR热重实验采用同步热分析仪法 (STA449F3 TG-DSC,德国NETASCH公司) 进行。在实验过程中,选择升温速率为5、15和25 K·min−1,从室温升至900 ℃。使用直径约为8 mm、高度约为3 mm的氧化铝坩埚进行实验,为了减小样品内部传热和扩散影响并保证样品的均质性,实验前将样品烘干粉碎以充分混合,并筛分至120目 (0.125 mm) ,控制氮气流量在100 mL·min−1内,并控制样品用量在 (10±0.5) mg。实验时,将ASR原料放置于坩埚中,在设定的升温速率下将样品升温至900 ℃,持续观察失重情况,直至无明显失重。
如图1所示,为ASR固定床热解实验装置示意图。实验时炉体以5 K·min−1的速率升温至设定温度 (500~900 ℃) 后稳定15 min,选用N2作为载气,以450 mL·min−1的速度向炉内通入载气,维持石英管内惰性气氛。每次实验称取 (5±0.5) g样品,反应持续45 min。通过铝箔气袋收集冷却和过滤干燥后的热解气。反应炉冷却至室温后,为保证实验数据准确性,每个实验条件下至少进行3次平行实验,计算平均值。取出焦样称重,热解焦油通过冷凝管进行冷凝收集,使用二氯甲烷溶剂 (分析纯,99.99%) 溶解蛇形冷凝管管壁粘附的热解焦油,获取的热解焦油溶液存储在−10 ℃的冷冻柜中。
1.3 研究方法
1) 动力学分析方法。分布式活化能法 (DAEM) 是一个多反应模型,其模型假设为:热解过程由许多相互独立的一级不可逆反应组成;每个反应有确定的活化能,所有反应的活化能值呈某种连续分布。该模型对固相反应具有良好的适应性[16],不仅能较好地描述热解失重的真实过程,而且能够在较宽温度及升温速率范围内对反应过程进行准确描述,并在煤、生物质和固体废弃物等热解研究中得到广泛运用[17-19],DAEM法在热解动力学研究中的适用性得到较好的验证。ASR作为一种典型的工业固体废弃物,在本研究中,采用DAEM进行动力学分析,ASR的非等温热解过程中转化率x与时间t的关系可用DAEM模型表示[20],如式(1)所示。
x=1−∫∞0exp[−k0∫t0exp(−ERT)dt]f(E)dE (1) 式中:T是绝对温度,K;R是通用气体常数,J·mol−1·K−1;k0是频率因子,s−1;E是活化能,kJ·mol−1;f (E) 是活化能的分布函数。
x是样品转化率,其定义如式(2)所示。
x=w0−wtw0−wf (2) 式中:w0、wt、wf分别指煤样初始重量、t时刻样品重量和反应结束时刻样品重量,mg。
为估算动力学参数,MIURA等[21]基于DAEM方程提出了一种简易积分法,如式(3)所示。
ln(βT2)=ln(k0RE)+0.6075−ERT (3) 式中:β是升温速率,K·min−1;R是通用气体常数,8.314 J·mol−1·K−1。
2) 产物分析方法。利用气相色谱仪 (GC7890A,Agilent) 定量测定不同温度下热解前后释放的气体组分,分析其中的N2、H2、CO、CO2及CH4等组分含量,利用N2示踪法来进行定量计算[22]。热解焦油样品采用气相色谱质谱联用仪 (GC-MS,7890A/5975C,Agilent) 进行检测,色谱柱为Agilent HP-5MS毛细管柱,进样口温度为280 ℃,初始温度40 ℃保持2 min,以10 K·min−1升至280 ℃。气质联用仪器可对测试所得时域图进行图谱比对分析,得到焦油中各成分物质化学式以及相对含量大小。采用压片法借助红外光谱仪 (FTIR,Nicolet iS50,Thermo Fisher) 分析热解焦的官能团,采用拉曼光谱仪 (Raman,DRX,Thermo Fisher) 分析碳结构特性,实验每次随机选取若干热解焦颗粒,使用455 nm波长激光,采集800~2 000 cm−1范围内的光谱,分辨率为0.5 cm−1。采用微波消解仪和等离子体发射光谱仪 (ICP-OES,Agilent 5110) 测定物料原样及热解焦中重金属含量,微波消解过程中,在强酸环境下对样品进行加热,消解液中硝酸 (HNO3) 与氢氟酸 (HF) 按3∶1体积比混合,将重金属转化为离子形态的可溶性盐,取所得消解液检测重金属元素含量。
2. 结果与讨论
2.1 ASR热失重及动力学分析
1) ASR热失重过程分析。图2为不同ASR物料在不同升温速率下的热解TG和DTG曲线。实验结果表明,5种ASR样品均经历了3个阶段的失重过程:200~500、500~600和600~800 ℃,其中主要的热解反应温度区间为200~500 ℃。在主反应温度区,ASR中部分含碳有机物分子因其键能较低开始发生裂解,同时部分含氧官能团支链也会发生热裂解分解成小分子化合物,以挥发分形式脱离物料,从而引起物料失重。500~600 ℃为衡定速率失重区间,在此温度区间内5种样品均未出现失重峰,表明在该温度范围内并未发生明显反应。600~700 ℃为高温下的失重温度区间,其中ASR5样品持续至750 ℃。在此温度范围内,剩余有机物与半挥发性物质基本反应完全,同时推测存在部分无机物受热分解,导致仍有小程度失重。此外,高温下热解气中的CO2与少量固定碳发生气化反应,进一步增加样品失重。
图2显示,经预分选处理后5种ASR物料仍在500 ℃完成主要热解过程,与DE MARCO等[10]研究结论相符,说明预分选并不影响ASR热解主反应温度区间。但区别在于,ASR1、2和3在500 ℃以下均有2个失重峰,表明有2个主反应,超过500 ℃后有小程度失重;ASR4与ASR5在500 ℃以下主失重峰相似,均为单峰,在600~750 ℃内两种物料均出现第二阶段反应,对应失重率达5%。分析认为,预分选改变了样品中主要成分占比,导致失重峰发生较大变化。根据Ni的研究[23],ASR样品成分复杂,包括海绵和泡沫在内的纺织品热解反应速率峰值在330 ℃左右,塑料和橡胶有2个热解反应速率峰,第一个在410 ℃左右,第二个在660 ℃左右。因此,纺织品、塑料和橡胶在330~500 ℃主要占失重的第一个峰。此外,ASR1主要成分为纤维,在240 ℃左右出现热解反应速率峰,低于纺织品、塑料和橡胶等组分热解反应速率峰对应温度,说明在低温下纤维类组分更易发生热解。
2) ASR热解动力学分析。根据热失重曲线和式(2)与式(3),在3种不同升温速率下选定同一转化率x,选择ln (β/T2) 对1/T作图,相同转化率x条件下ln (β/T2) 与1/T应为线性关系,其斜率即为活化能E。图3为拟合得到的ln (β/T2) 对1/T的关系曲线。根据ln (β/T2) 对1/T的线性回归拟合数据,图4为不同ASR样品计算活化能E随转化率x的关系曲线。如图所示,由于不同的热解温度范围内不同官能团分解所需的活化能存在差异,以ASR1为例,转化率从10%增至90%,热解活化能由31.39 kJ·mol−1升至105.56 kJ·mol−1。随着转化率增大,活化能逐渐增大,分析认为反应前期主要是大分子结构中成键强度弱的官能团先分解,不稳定的官能团断裂所需能量更少,随着反应的进行,不稳定的官能团已分解,剩余官能团更加稳定,此时化学键断裂需要更多能量[21]。
 图 3 不同转化率下ASR热解动力学拟合曲线Figure 3. Fitting curve of ASR pyrolysis kinetics under different conversion rates
图 3 不同转化率下ASR热解动力学拟合曲线Figure 3. Fitting curve of ASR pyrolysis kinetics under different conversion rates对比5种典型ASR物料,在相同转化率下,ASR3在各转化率下均有最低活化能,表明其在5种样品中反应难易程度最低。ASR4在转化率小于60%时,活化能增长趋势同余下4种样品;当转化率大于60%时,活化能大幅度增加,并在转化率到达90%时增至195.21 kJ·mol−1。分析认为ASR4在650 ℃以上的仍有较大质量损失,且TG曲线显示失重持续到750 ℃,其结果导致在更高的温度下进行反应需要更大的活化能,在活化能上体现为E值陡增。根据热失重过程特性可以得知,预分选不仅对5个ASR样品的组分和理化性质影响显著,而且改变了不同温度下的本征反应过程[12-13]。
2.2 热解产物分布
图5(a)、(b)、(c)分别为5个样品在不同温度下热解焦产率、热解焦油产率和热解气产率分布规律。在500~900 ℃范围内,随温度升高,产气量呈现增长趋势。在热解过程中,弱化学键率先发生断裂形成气体,然后大分子物质裂解形成焦油,官能团也随之破裂生成气体[24]。因此,在整个热解进程中气体产率逐渐增加。当热解温度高于500 ℃,随反应器温度逐步升高至900 ℃,热解焦产率持续减小。在相同温度下,ASR1和ASR2的焦产率比ASR3、4和5小30%,这主要受预分选对ASR主要成分的影响,结合表1数据可知,ASR3、4和5含较多灰分,因其不参与热解反应,导致热解焦残留占比较大。此外,ASR1和ASR2样品含碳有机物比值更高,在整个热解过程中热解生成挥发分占比更大,导致热解焦产率小于ASR3、4和5。热解温度在500~900 ℃时,ASR1和ASR2在900 ℃气相产率相比于500 ℃提升约5倍,ASR3、4和5热解气产率提高约4倍,ASR1在900 ℃下热解气产率高达76.52%,ASR4仅占33.52%。可以看到,预分选可有效提升ASR原料能源转化效率。同时,高温还会引发焦油二次裂解,焦油量进一步减少,其中ASR1和ASR2焦油产率高于其他3种物料,且受温度影响较大,随热解温度提升焦油产率减少约45%,但不同的是,ASR2在900 ℃焦油产率维持在11.5%的较高水平。以上结果表明,提高温度有利于物料热解,产生更多的热解气体,且促使热解进行更加彻底,同时抑制焦油产生。预分选后不同ASR样品热解产物分布差异明显,ASR1和2产焦少、热解气和焦油生成量高。
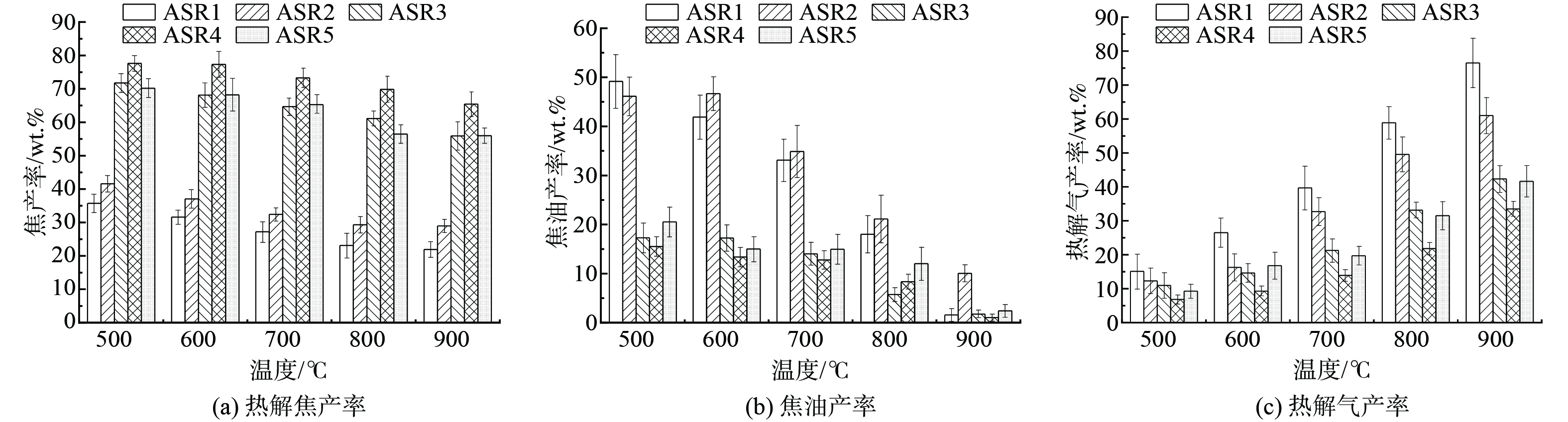 图 5 不同ASR样品在不同热解温度下三相产物分布规律Figure 5. Distribution of pyrolytic products in different ASR samples at different pyrolysis temperatures
图 5 不同ASR样品在不同热解温度下三相产物分布规律Figure 5. Distribution of pyrolytic products in different ASR samples at different pyrolysis temperatures2.3 热解气组成及分析
图6是不同ASR样品热解气成分随温度变化曲线。在较低热解温度下,按照组成的体积占比,相对含量较高的为CO2、H2、CO以及CH4。这是由于ASR中聚合物热解产生较小的分子,分子进行环化反应或分解成轻质烃,形成各种不同长度的分子链。随着温度升高,反应产生的环状化合物和烃类化合物进一步裂解生成CH4、CO2和CO等分子[25]。而C2H2因成键困难,形成条件严苛,故含量最低,且基本不受温度的影响。从变化趋势中可以看到,除CO2和C2H2外,其它气体随温度的上升含量均有所提升,其中ASR3、4的CH4含量随温度的提高出现明显的先升后降趋势,这是由于高温下热解气中发生水煤气变换反应 (CO+H2O=CO2+H2、CH4+H2=CO+3H2) [26],以致CH4含量出现波动。而在5种ASR样品热解气中CO2含量随温度的升高反有明显地下降,这是因为热解阶段析出的CO2主要来自于低温下参与芳化缩聚的C=O和C-O自由基团发生重整等反应[27],当温度上升受自由基数量的限制,CO2生成量降低。而焦油中的组分在更高温度下发生二次裂解,生成小分子气态物质,再次降低了CO2在热解气中的占比[28]。此外,高温下开始发生反应 (C+CO2→2CO) ,即CO2与热解焦中的固定碳发生反应,在宏观上表现为CO2向CO转变,也会降低CO2的生成量,但该反应有利于热解气有效气成分的增加。在不考虑CO2占比的前提下,在ASR2中500、600和700 ℃下主要为CO2和H2,800和900 ℃下以CH4和CO为主,在ASR1中主要为CO;ASR3、ASR4和ASR5中800 ℃以下H2占比最高,900 ℃时CO有显著提升。可以看到,预分选后不同物料对热解气组成有较大影响,ASR3、ASR4与ASR5气体组成更加相近,ASR1和ASR2热解气在最主要成分上有所不同。
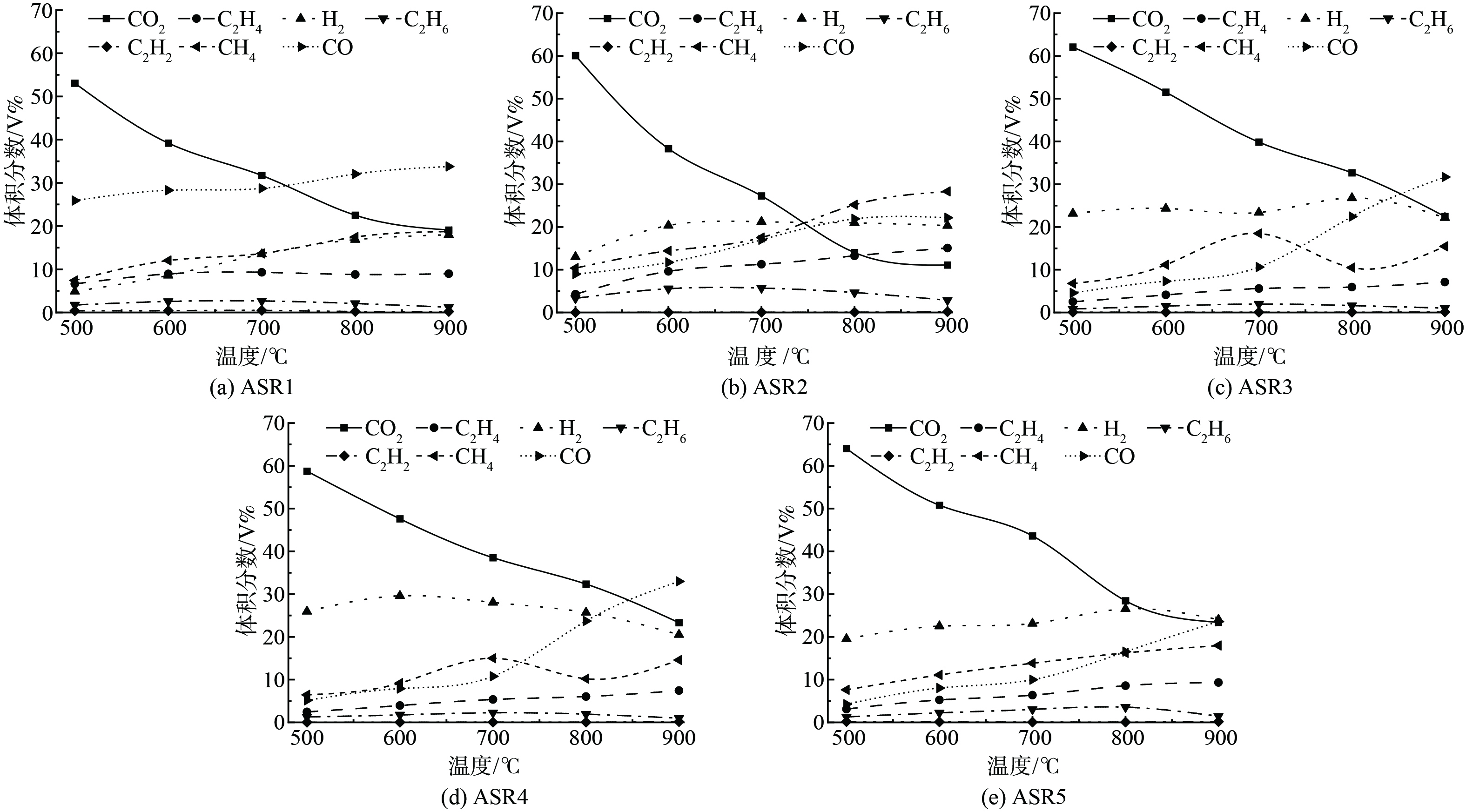 图 6 不同样品和温度对热解气组成的影响Figure 6. Effects of different samples and temperatures on gas composition
图 6 不同样品和温度对热解气组成的影响Figure 6. Effects of different samples and temperatures on gas composition2.4 焦油组成及分析
以ASR1样品为例,图7为不同热解温度下热解焦油GC-MS时域图,分析采用了面积归一法进行积分定量,并结合NIST标准质谱图库进行组分定性。为了保证实验数据的可信度,选择与焦油中已知组分分子式相似度达到90%以上的物质作为焦油组分。由于焦油中包含许多种不同的组分,为简化研究,仅统计含量超过2%的组分。对ASR在不同热解温度下收集到的焦油成分分类统计,结果如图8所示。统计结果表明,焦油随温度升高,成分从低温的链烃、环烃转向生成多苯环类及其衍生物。将焦油成分进行分类,主要分为链烃、环烃、单苯环烃、多苯环烃及其衍生物、含O烃类衍生物以及杂原子化合物 (如N、Si、S和Cl等) 共6大类物质。在600 ℃以下ASR1、2和3焦油中物质更多以链烃和含氧烃类衍生物为主,ASR4和ASR5以链烃和多苯环烃及其衍生物为主。当温度上升至900 ℃,5种样品中烃类和含氧烃类衍生物等含量急剧下降,以多苯环烃及其衍生物为主要成分。在YANG等[29]采用了TG-FTIR-GC/MS分析技术对ASR热解焦油分析后发现,焦油中许多物质具有不稳定或较弱的化学键,如=CH-,=CH2,-C=C-和-C=CH2等,这些化学键在高温下更易发生转化,结合焦油中物质种类的转变趋势,表明焦油成分在温度提升后发生了弱键断裂和苯环化变化[24,30]。
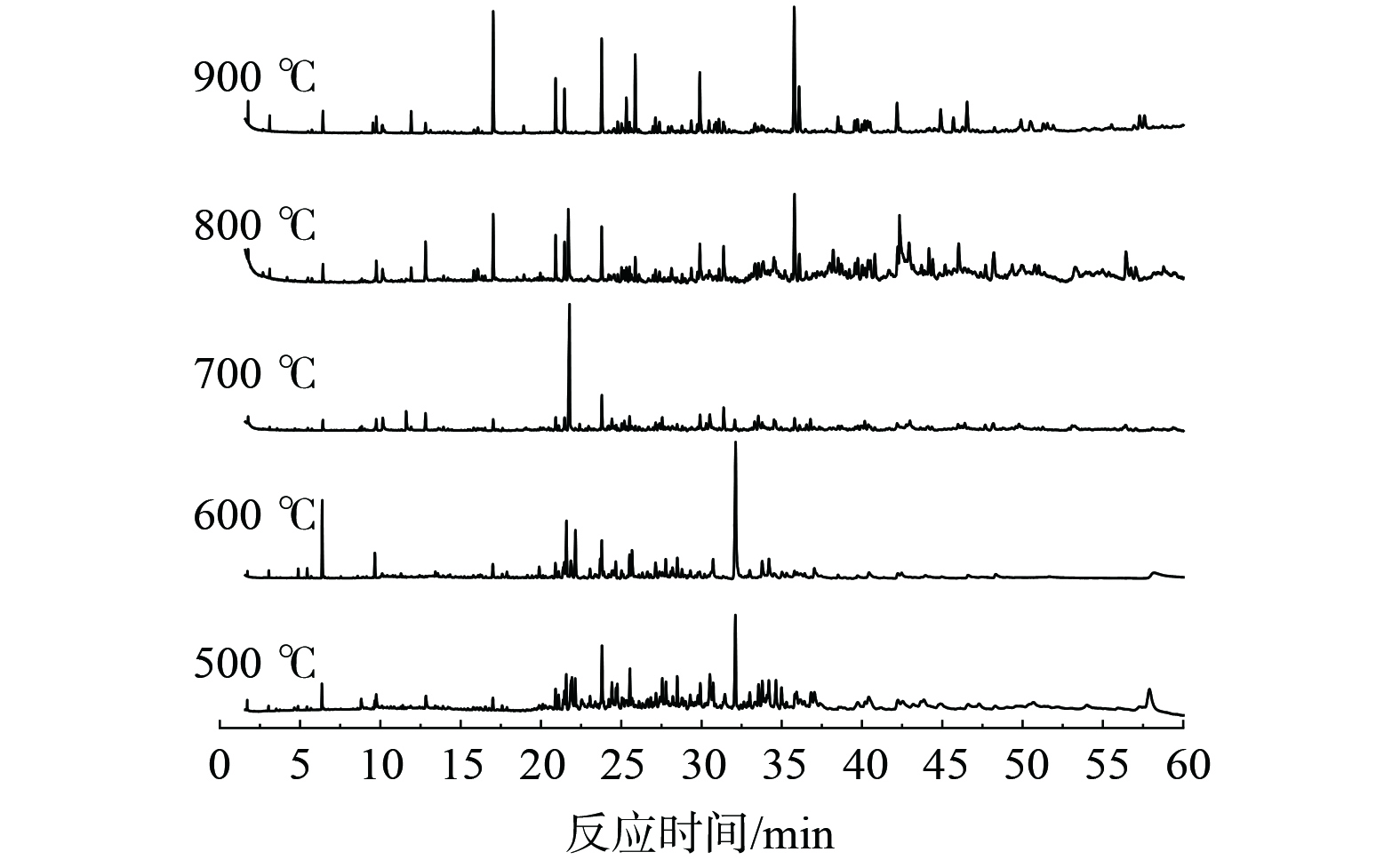 图 7 不同温度下热解焦油GC-MS时域图Figure 7. GC–MS spectra of pyrolytic tar with different temperatures
图 7 不同温度下热解焦油GC-MS时域图Figure 7. GC–MS spectra of pyrolytic tar with different temperatures2.5 ASR热解焦微观结构分析
1) 官能团表征分析。傅里叶红外光谱 (FTIR) 可反映出物质表面所含官能团信息,对同种ASR物料不同温度下所得谱图进行对比,可从官能团角度分析热解焦结构变化,结果如图9所示。特征峰变化主要为:醇、酚类中的-OH伸缩振动 (3 650-3 600 cm−1) ,为尖锐吸收峰,分子间-OH伸缩振动 (3 500-3 200 cm−1) ,为宽吸收峰;胺类中-NH的伸缩振动 (3 500-3 300 cm−1) ;羧酸中-COOH伸缩振动 (3 400-2 500 cm−1) ;-C=C-烯烃类伸缩振动 (1 610-1 590 cm−1) [31-32];脂肪链末端-CH3对称弯曲振动 (1 460-1 380 cm−1) ;3H取代的伸缩振动 (850-720 cm−1) [33-34]。在3 450 cm−1波段出现了1个宽且强的吸收峰,这表明ASR中存在醇、酚、羧酸等游离态羟基基团伸缩振动以及可能的胺类中-NH的伸缩振动,从而使得伸缩振动范围较宽。在500~900 ℃内,随温度的升高峰强均有所减弱,说明在热解过程中,热解焦中有醇、酚分子间羟基或者羧基发生了脱羟基反应或胺类物质中-NH脱除。值得注意的是,ASR1热解焦随温度的上升出现了3 650 cm−1对应的醇类、酚类中的-OH伸缩振动。ASR1、2、3和4热解焦中1 600 cm−1吸收振动峰随温度上升而消失,说明对应该范围的-C=C-官能团在反应中发生转变,分析认为是-C=C-向苯环化转变导致。ASR1、2、4和5热解焦中1 430 cm−1吸收振动峰随温度的升高而消失,表明在此范围内对应烷烃上-CH3脱除,脱离的-CH3自由基或生成气相中的小分子物质。780 cm−1吸收振动峰对应的H取代,表明热解焦中H自由基的脱除,有利于热解气中小分子物质的产生,例如CH4、H2和碳氢化合物的生成。可以看到,随热解温度的升高,热解焦表面部分分子基团脱除,尤其以官能团为主的支链发生断裂,例如-OH、-CH3、-C=C-以及-COOH等。小分子官能团的消失是热解气中轻质物质和一碳化合物等产生的原因,升高温度破坏小分子官能团加剧热解焦进一步碳化,同时促使热解焦碳晶结构向更有序化方向转变[28]。
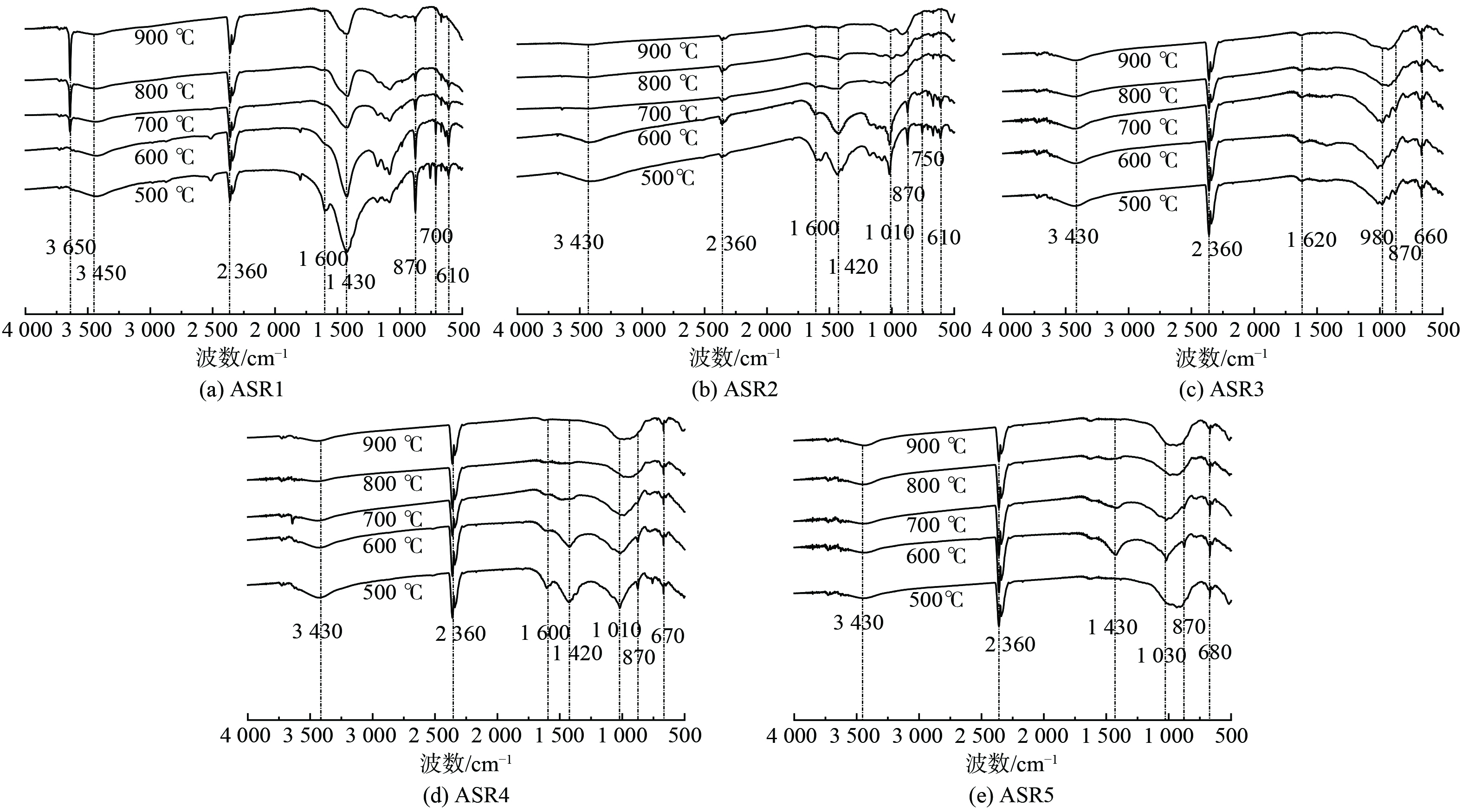 图 9 不同温度下热解焦FTIR光谱图Figure 9. FTIR spectra of pyrolytic char with different temperatures
图 9 不同温度下热解焦FTIR光谱图Figure 9. FTIR spectra of pyrolytic char with different temperatures2) 碳结构分析。原始拉曼谱图通常是由多个特征峰叠加而成,通过分峰拟合可分解成4个洛伦兹子峰 (D4、D1、G、D2) 和1个高斯子峰[35]。以500 ℃热解焦为例,拉曼图谱分峰拟合结果如图10所示。D1峰与G峰的积分面积之比 (ID1/IG) 通常被用于定量表征热解焦结构在气化过程中的变化,表示碳结构的晶格度或石墨化程度,研究发现,ID1/IG与晶体的平面尺寸成反比,即ID1/IG比值的降低表明煤焦有序化程度升高[36]。表3为热解温度为500~900 ℃下热解焦拉曼峰面积比ID1/IG。如表所示,5种ASR样品的热解焦ID1/IG值均随热解温度的升高而降低,ID1/IG值越小表明半焦的有序化程度越高,也即热解焦石墨化程度越高,结合热解焦FTIR表征结果,官能团的消失使自由基脱除,从而使碳晶结构向更有序化发展。研究还发现,在500~900 ℃内,ASR3和ASR5热解焦ID1/IG值始终低于ASR1、2和4,说明除温度对ID1/IG值有影响外,物料组成不同也是影响热解焦结构的重要原因素之一。
表 3 热解焦拉曼光谱峰面积比Table 3. Raman band area ratio of pyrolytic char热解温度/ ℃ ID1/IG值 ASR1 ASR2 ASR3 ASR4 ASR5 500 5.17±0.09 3.71±0.15 5.02±0.13 6.57±0.12 4.24±0.11 600 5.05±0.11 3.61±0.17 4.94±0.16 6.33±0.17 4.13±0.16 700 4.92±0.10 3.52±0.12 4.88±0.12 6.10±0.09 4.03±0.14 900 4.77±0.11 3.33±0.16 4.82±0.15 5.89±0.09 3.92±0.16 800 4.38±0.15 3.12±0.12 4.78±0.11 5.74±0.08 3.77±0.12 | Show TableDownLoad:
CSV
3) 重金属分布。已有研究表明,ASR含较高含量重金属[11],在热解过程中重金属会进一步向热解焦中富集。对热解半焦中Cr、Pb和Ni含量进行分析,参考LI等[37]对热解半焦中重金属含量的分析方法,以热解半焦中重金属的富集率 (ER) 和残留率 (RR) 作为评价热解后重金属富集和固定程度的参数,计算公式如(4)~(5)所示。
ER=Cx/C×100\% (4) 式中:Cx为生物炭中重金属的质量分数,mg·kg−1;C为原料中重金属的质量分数,mg·kg−1。
RR=ER×y (5) 式中:y为热解半焦收率,由热解半焦质量与热解原料质量之比计算得出。
含量测定选取500、700、900 ℃下所得焦样,热解焦重金属富集率结果如图11所示。随温度的提高,5个样品热解焦中重金属的残留率均有不同程度的下降,表明温度的提高,重金属向热解气和焦油中转移,但固存在焦中的含量在800 ℃以下仍能保持50%以上,多数重金属保留在热解焦中。Ni在物料ASR1和ASR2中残留率表现不同于ASR3、4和5,残留率基本不受温度的影响。这种差异性可能的解释是由ASR物料组成的差异造成的。但在ASR3、4和5中Ni元素残留率随温度的升高而降低,这与Cr、Pb没有差异。
热解过程中,重金属按照去向分为3个方向:热解气、焦油和热解焦。在一般情况下,期望在反应过程中的重金属应尽可能保留于固相中,以降低重金属伴随热解气和焦油发生逃逸,增加环境污染压力和后续工艺除杂的复杂程度。图12为5种ASR物料在不同温度下热解后重金属富集的结果。富集率以1作为标准:热解半焦中重金属含量与等量原料中的含量比值为1。从结果来看,按富集能力从大到小排列:Cr>Pb>Ni。Cr元素整体ER值均大于1,且基本不受温度变化的影响。从ER值的大小来看,ASR1和ASR2样品在Cr元素固定上更具优势,而ASR3、4和5也能保持Cr基本固定在半焦中。Pb元素的ER值在5种ASR物料中的表现类似,随温度的升高ER值减小,但在ASR1和ASR2中Pb元素的ER值变化幅度高于ASR3、4和5。Ni元素ER值在不同物料中的变化差异较大,在ASR1、ASR2中ER值随温度升高而增大,在ASR3、ASR4、ASR5 ER值随温度升高而略有减小。总体上,ASR中不同重金属的富集程度主要受物料种类和热解温度的影响。
3. 结论
1) ASR热解经历3个失重阶段,反应温度区间集中于200~500 ℃,不受预分选ASR样品组成影响。热解动力学分析显示不同预分选样品活化能差异显著,主要与物料组分热解难度相关。热解产物产率分布受ASR物料成分影响明显,但总体上随热解温度升高,热解气产率增大,焦油发生二次裂解,生成量减少,且随温度升高焦油向多苯环物质转化。
2) 热解焦主要发生-OH、-CH3、-C=C-以及-COOH等分子基团的脱除,温度升高热解焦石墨化程度增大,但不同预分选样品石墨化程度差异明显。热解半焦中重金属残留率与热解温度和样品组成密切相关。热解过程重金属固存在半焦的比例整体保持在40%以上,过高的热解温度不利于重金属的固定。
-
图 3 不同转化率下ASR热解动力学拟合曲线
Figure 3. Fitting curve of ASR pyrolysis kinetics under different conversion rates
图 5 不同ASR样品在不同热解温度下三相产物分布规律
Figure 5. Distribution of pyrolytic products in different ASR samples at different pyrolysis temperatures
图 6 不同样品和温度对热解气组成的影响
Figure 6. Effects of different samples and temperatures on gas composition
图 7 不同温度下热解焦油GC-MS时域图
Figure 7. GC–MS spectra of pyrolytic tar with different temperatures
图 9 不同温度下热解焦FTIR光谱图
Figure 9. FTIR spectra of pyrolytic char with different temperatures
表 1 样品工业分析和元素分析
Table 1. Proximate and ultimate analyses of samples
样品 工业分析wad/% 元素分析wad/% 干基低位发热量/(MJ·kg−1) M Ash V FC C H N S O* ASR1 0.81±0.08 17.93±0.21 73.50±0.20 7.76±0.14 45.70±0.22 3.94±0.11 1.09±0.09 1.57±0.10 28.96±0.15 21.89±0.41 ASR2 0.99±0.07 24.51±0.25 61.53±0.21 12.97±0.18 48.60±0.21 8.03±0.15 1.54±0.11 1.28±0.11 15.05±0.18 20.46±0.23 ASR3 0.76±0.08 44.12±0.48 48.14±0.18 6.98±0.09 31.51±0.23 2.88±0.14 0.66±0.08 1.95±0.09 18.12±0.20 15.66±0.20 ASR4 1.35±0.10 49.91±0.45 43.21±0.18 5.53±0.11 31.50±0.18 3.25±0.17 0.70±0.09 1.94±0.07 11.35±0.21 12.69±0.18 ASR5 1.17±0.09 63.29±0.55 30.88±0.22 4.66±0.12 17.35±0.16 1.58±0.17 0.78±0.08 1.34±0.07 14.49±0.20 8.69±0.19 注:M代表水分,V代表挥发分,A代表灰分,FC代表固定碳,ad代表空气干燥基,*为通过差减法计算。
下载: 导出CSV
表 2 ASR样品重金属元素含量
Table 2. Content of heavy metal elements in ASR
mg·kg−1 样品 元素种类 Cd Cr Pb Ni ASR1 1.21±0.05 509.74±0.11 101.10±0.07 187.47±0.08 ASR2 1.10±0.04 422.49±0.09 95.74±0.05 161.36±0.09 ASR3 20.11±0.10 884.11±0.15 612.49±0.14 514.51±0.15 ASR4 7.78±0.03 693.33±0.14 558.66±0.17 441.41±0.14 ASR5 15.22±0.06 841.40±0.18 562.43±0.15 461.10±0.13
下载: 导出CSV
表 3 热解焦拉曼光谱峰面积比
Table 3. Raman band area ratio of pyrolytic char
热解温度/ ℃ ID1/IG值 ASR1 ASR2 ASR3 ASR4 ASR5 500 5.17±0.09 3.71±0.15 5.02±0.13 6.57±0.12 4.24±0.11 600 5.05±0.11 3.61±0.17 4.94±0.16 6.33±0.17 4.13±0.16 700 4.92±0.10 3.52±0.12 4.88±0.12 6.10±0.09 4.03±0.14 900 4.77±0.11 3.33±0.16 4.82±0.15 5.89±0.09 3.92±0.16 800 4.38±0.15 3.12±0.12 4.78±0.11 5.74±0.08 3.77±0.12
下载: 导出CSV
-
[1] JANG Y C, CHOI K, JEONG J, et al. Recycling and material-flow analysis of end-of-life vehicles towards resource circulation in South Korea[J]. Sustainability, 2022, 14(3): 1270. doi: 10.3390/su14031270 [2] TENG C Y, ZHOU K G, PENG C H, et al. Characterization and treatment of landfill leachate: A review[J]. Water Research, 2021, 203: 117525. doi: 10.1016/j.watres.2021.117525 [3] RABONI M, TORRETTA V, URBINI G, et al. Automotive shredder residue: A survey of the hazardous organic micro-pollutants spectrum in landfill biogas[J]. Waste Management & Research, 2015, 33(1): 48-54. [4] 宋斌. 中国报废汽车破碎残余物的理化特征与热重分析研究[D]. 上海: 上海交通大学, 2013. [5] MANCINI G, VIOTTI P, LUCIANO A, et al. On the ASR and ASR thermal residues characterization of full scale treatment plant[J]. Waste Management, 2014, 34(2): 448-457. doi: 10.1016/j.wasman.2013.11.002 [6] MALLAMPATI S R, LEE B H, MITOMA Y, et al. Sustainable recovery of precious metals from end-of-life vehicles shredder residue by a novel hybrid ball-milling and nanoparticles enabled froth flotation process[J]. Journal of Cleaner Production, 2018, 171: 66-75. doi: 10.1016/j.jclepro.2017.09.279 [7] 陈铭. 面向材料效率的汽车产品回收利用关键技术研究[J]. 中国机械工程, 2018, 29(21): 2615-2625. [8] KHODIER A, WILLIAMS K, DALLISON N. Challenges around automotive shredder residue production and disposal[J]. Waste Management, 2018, 73: 566-573. doi: 10.1016/j.wasman.2017.05.008 [9] ANZANO M, COLLINA E, PICCINELLI E, et al. Lab-scale pyrolysis of the automotive shredder residue light fraction and characterization of tar and solid products[J]. Waste Management, 2017, 64: 263-271. doi: 10.1016/j.wasman.2017.03.013 [10] DE MARCO I, CABALLERO B M, CABRERO M A, et al. Recycling of automobile shredder residues by means of pyrolysis[J]. Journal of Analytical and Applied Pyrolysis, 2007, 79(1): 403-408. [11] 倪飞箭. 报废汽车破碎残余物热裂解/气化回收机理与资源化初探[D]. 上海: 上海交通大学, 2015. [12] HAYDARY J, SUSA D, GELINGER V, et al. Pyrolysis of automobile shredder residue in a laboratory scale screw type reactor[J]. Journal of Environmental Chemical Engineering, 2016, 4(1): 965-972. doi: 10.1016/j.jece.2015.12.038 [13] YANG B, CHEN M. Influence of interactions among polymeric components of automobile shredder residue on the pyrolysis temperature and characterization of pyrolytic products[J]. Polymers, 2020, 12(8): 1682. doi: 10.3390/polym12081682 [14] HAN S, JANG Y C, CHOI Y S, et al. Thermogravimetric kinetic study of automobile shredder residue (ASR) pyrolysis[J]. Energies, 2020, 13(6): 1451. doi: 10.3390/en13061451 [15] SANTINI A, PASSARINI F, VASSURA I, et al. Auto shredder residue recycling: Mechanical separation and pyrolysis[J]. Waste Management, 2012, 32(5): 852-858. doi: 10.1016/j.wasman.2011.10.030 [16] 赵岩, 邱朋华, 谢兴, 等. 煤热解动力学分布活化能模型适用性分析[J]. 煤炭转化, 2017, 40(1): 13-18. doi: 10.19726/j.cnki.ebcc.2017.01.003 [17] DE CAPRARIIS B, SANTARELLI M L, SCARSELLA M, et al. Kinetic analysis of biomass pyrolysis using a double distributed activation energy model[J]. Journal of Thermal Analysis and Calorimetry, 2015, 121(3): 1403-1410. doi: 10.1007/s10973-015-4665-2 [18] XU D, CHAI M, DONG Z, et al. Kinetic compensation effect in logistic distributed activation energy model for lignocellulosic biomass pyrolysis[J]. Bioresource Technology, 2018, 265: 139-145. doi: 10.1016/j.biortech.2018.05.092 [19] BHAVANAM A, SASTRY R C. Kinetic study of solid waste pyrolysis using distributed activation energy model[J]. Bioresource Technology, 2015, 178: 126-131. doi: 10.1016/j.biortech.2014.10.028 [20] MIURA K. A new and simple method to estimate f(E) and k0(E) in the distributed activation energy model from three sets of experimental data[J]. Energy & Fuels, 1995, 9(2): 302-307. [21] MIURA K, MAKI T. A simple method for estimating f(E) and k0(E) in the distributed activation energy model[J]. Energy & Fuels, 1998, 12(5): 864-869. [22] 崔童敏. 快速热解过程中煤生物质初次破碎机理及其化学结构变化的研究[D]. 上海: 华东理工大学, 2017. [23] NI F J, CHEN M. Research on ASR in China and its energy recycling with pyrolysis method[J]. Journal of Material Cycles and Waste Management, 2015, 17(1): 107-117. doi: 10.1007/s10163-014-0232-3 [24] YUE C Y, GAO P P, TANG L F, et al. Effects of N2/CO2 atmosphere on the pyrolysis characteristics for municipal solid waste pellets[J]. Fuel, 2022, 315: 123233. doi: 10.1016/j.fuel.2022.123233 [25] GUO Q J, ZHANG X, LI C, et al. TG-MS study of the thermo-oxidative behavior of plastic automobile shredder residues[J]. Journal of Hazardous Materials, 2012, 209: 443-448. [26] 郭怡君, 李军, 黄宏宇, 等. 有机固体废弃物热解技术及热解气组成综述[J]. 新能源进展, 2023, 11(2): 106-122. [27] 李超, 刘新民, 耿启金, 等. 热重质谱联用研究废旧汽车高聚物热解特性[J]. 环境科学学报, 2011, 31(8): 1724-1729. doi: 10.13671/j.hjkxxb.2011.08.025 [28] ZHOU R S, CAO R, LIU Y Q, et al. Study on the characteristics and mechanism of fast co-pyrolysis of coal tar asphaltene and biomass[J]. Journal of Analytical and Applied Pyrolysis, 2022, 161: 105409. doi: 10.1016/j.jaap.2021.105409 [29] YANG B, CHEN M. Py-FTIR-GC/MS analysis of volatile products of automobile shredder residue pyrolysis[J]. Polymers, 2020, 12(11): 2734. doi: 10.3390/polym12112734 [30] CHEN B, LIU B, CHAO Y, et al. Formation mechanism of biomass aromatic hydrocarbon tar on quantum chemistry[J]. Journal of Renewable Materials, 2022, 10(12): 3491-3504. doi: 10.32604/jrm.2022.021302 [31] HE X Q, LIU X F, NIE B S, et al. FTIR and Raman spectroscopy characterization of functional groups in various rank coals[J]. Fuel, 2017, 206: 555-563. doi: 10.1016/j.fuel.2017.05.101 [32] XIE X, ZHAO Y, QIU P H, et al. Investigation of the relationship between infrared structure and pyrolysis reactivity of coals with different ranks[J]. Fuel, 2018, 216: 521-530. doi: 10.1016/j.fuel.2017.12.049 [33] XIE X, LIU L, LIN D, et al. Influence of different state alkali and alkaline earth metal on chemical structure of Zhundong coal char pyrolyzed at elevated pressures[J]. Fuel, 2019, 254: 115691. doi: 10.1016/j.fuel.2019.115691 [34] WANG R Z, YUE J F, JIANG J C, et al. Hydrothermal CO2-assisted pretreatment of wheat straw for hemicellulose degradation followed with enzymatic hydrolysis for glucose production[J]. Waste and Biomass Valorization, 2021, 12(3): 1483-1492. doi: 10.1007/s12649-020-01103-4 [35] 余俊钦, 卫俊涛, 丁路, 等. 生物质灰添加对无烟煤煤焦气化特性的影响[J]. 燃料化学学报, 2018, 46(10): 1161-1167. [36] SOLOMON P R, CARANGELO R M. FTIR analysis of coal. 1. techniques and determination of hydroxyl concentrations[J]. Fuel, 1982, 61(7): 663-669. doi: 10.1016/0016-2361(82)90014-X [37] LI Z Y, HUANG Y J, ZHU Z C, et al. Co-pyrolysis of sewage sludge with polyvinyl chloride (PVC)/CaO: Effects on heavy metals behavior and ecological risk[J]. Fuel, 2023, 333: 126281. doi: 10.1016/j.fuel.2022.126281 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2129
- HTML全文浏览数: 2129
- PDF下载数: 42
- 施引文献: 0
