










-
电镀工业是我国经济发达地区的重要加工行业,由于其在工业中适用性高,广泛分布于各个工业部门,电镀生产在耗费大量工艺用水的同时,也产生大量的电镀废水[1]。电镀废水具有重金属含量高、毒性大、污染物杂、环境危害严重等特点[2-4],属于难处理的工业废水。目前电镀废水的处理工艺主要采用物化预处理+生化处理+深度处理的组合工艺,涉及中水回用的主要采用膜分离技术[5],淡水回用于生产漂洗工段.常规的废水深度处理工艺不能彻底将其从水中去除,出水无法稳定达到电镀污染物排放标准(GB 21900-2008)的排放标准,开发高效的深度处理工艺已成为水处理领域的关注点。
臭氧氧化作为一种绿色处理工艺具有易操作、污染物去除效率高、无二次污染等优点已被广泛运用于饮用水和废水深度处理领域[6-9] 。由于臭氧氧化具有一定的选择性[10-11],研究人员开发了多相催化臭氧化技术克服了上述缺点,通过在臭氧氧化过程中加入非均相催化剂,使水中溶解性臭氧在催化剂表面发生链式反应产生羟基自由基(·OH)[12-13],从而提高水中有机物的去除率。
常见的非均相臭氧催化剂有金属氧化物(MnO2、FeOOH、TiO2等)[14-16]、多金属负载催化剂(RuO2/Al2O3、MnO2/Al2O3、TiO2/Al2O3等)[17-19]、矿物(Cu/堇青石、Mn/蜂窝陶瓷[20]等)和活性炭(MnOx/GAC[21]、多壁碳纳米管等)。目前工程项目中应用较广的催化剂多以球形陶瓷颗粒为载体负载多金属氧化物,常装填于固定床形式的反应器[22]内进行进行臭氧催化反应,但其传质效率低,水流易产生局部短流,影响臭氧催化氧化对水中有机物的去除效率。为了开发更为高效、稳定和经济的臭氧催化剂,研究人员除了在催化剂表面负载的活性组进行改进外,对催化剂的结构也进行了研究及优化。
基于陶瓷膜具有优异的化学性能,研究人员就臭氧预氧化+陶瓷膜工艺去除有机物和陶瓷膜改性催化臭氧氧化等开展了相关研究。2003年SCHLICHTER等[23]首次将臭氧氧化和陶瓷膜过滤相结合处理地表水和微污染原水,之后臭氧与陶瓷膜结合的相关研究开始逐渐增多。BYUN等[24]对陶瓷膜进行了改性,将氧化锰或氧化铁负载于陶瓷膜制备成催化膜,发现有机物的去除取决于陶瓷膜被金属氧化物纳米粒子包覆的类型,而且氧化锰膜的性能优于其他测试膜。我国对臭氧/陶瓷膜工艺的研究起步较晚,2011年清华大学的张锡辉课题组首先在国内使用臭氧预氧化/陶瓷膜组合工艺处理水中甲硫醚[25],此后该课题组使用该技术在饮用水处理、微污染水净化等领域的研究。
本研究以陶瓷膜为载体,采用浸渍-焙烧工艺制备了多组分臭氧催化过滤膜,实现了膜分离技术与催化臭氧氧化技术的同步耦合,利用陶瓷过滤膜的微米级孔道过滤废水,可实现污染物的定向移动,有效地促进了扩散传质,同时微米级孔道内部负载的催化剂,增大了催化模块的有效催化比表面积。利用XRD、SEM、DEX等技术对催化过滤膜进行了表征,并以电镀园区物化预处理后的混合废水为研究对象,考察催化过滤膜在常温下臭氧氧化过程中的催化活性。
-
陶瓷膜,购自江苏久吾高科技股份有限公司;La(NO3)3、Ce(NO3)3,均为分析纯,购自济宁天亿新材料有限公司;Mn(CH3COO)2、氢氧化钠、硝酸、浓硫酸、重铬酸钾,均为分析纯,购自国药集团化学试剂有限公司。
-
采用浸渍-焙烧法制备催化过滤膜:选用无机陶瓷膜作为催化过滤膜基体,用8%的氢氧化钠溶液浸泡,之后用质量百分比为13%的稀硝酸溶液浸渍1~2 h,再用去离子水洗净至出水中性后烘干备用;选用La(NO3)3、Ce(NO3)3和Mn(CH3COO)2作为催化剂活性组分的前驱物,配置不同质量比(La3+、Ce3+和Mn2+)的浸渍液,将烘干后的陶瓷膜在浸渍液中浸泡24 h后,在105 ℃真空烘4 h, 反复浸渍-烘干步骤5次;置于马弗炉中,在400~950 ℃条件下焙烧2~6 h即得陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜。
通过JEOL JSM-6480LV型扫描电镜(SEM)观察催化过滤膜的表面和断面形貌;催化过滤膜活性层的组成和晶体结构由电子能量色散X射线光谱(EDS)和X射线衍射(XRD, Bruker D8)进行表征,采用Cu Ka(λ=0.154 nm)辐射,扫描范围为10°~80°;利用干湿重量法测定催化过滤膜的孔隙率[26];测定陶瓷膜对不同尺寸聚苯乙烯微球的截留率,确定陶瓷膜的孔径大小[27]。
-
1)目标废水水质。实验所用废水为常州某电镀园区物化预处理后的混合废水,用去离子水稀释4倍后用作实验废水。目前该园区共有28个电镀车间,32条生产线,镀种涉及镀镍、镀铜、镀铬、镀锌、镀银和镀金。稀释后的废水COD为135 mg·L−1,总镍为0.04 mg·L−1,总铬为0.09 mg·L−1,浊度为6 NTU,pH为7.8,废水中有机物主要来源于镀液中添加的稳定剂、络合剂和光亮剂,包含聚乙二醇、柠檬酸、十二烷基磺酸钠和未知名称含氮杂环类物质等。
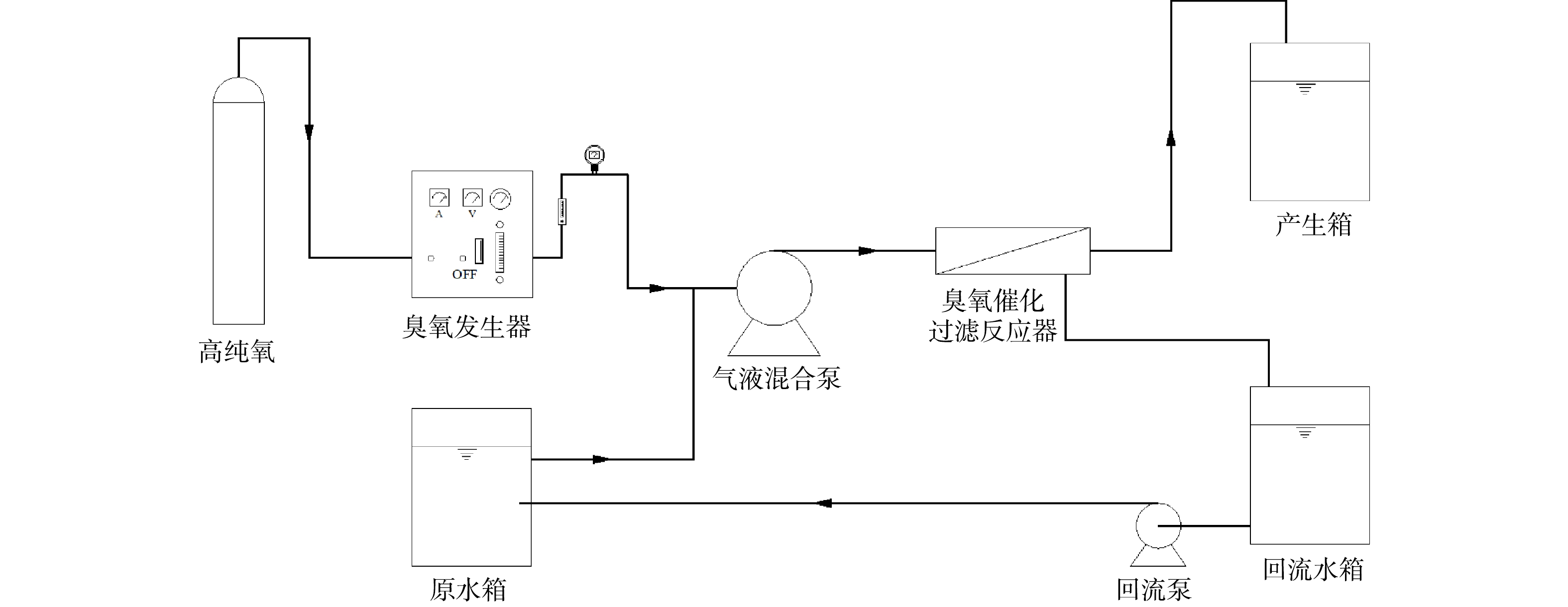
2)实验装置及运行参数。本实验所用装置如图1所示,实验在室温(25±2) °C下进行,实验装置处理规模为200 L·h−1,不锈钢材质。选用最优条件下制备得到的臭氧催化过滤膜处理废水,采用两支臭氧催化过滤膜并联的形式。通过控制臭氧投加量、回流比、跨膜压差、错流速率等反应条件,具体运行参数为:臭氧投加量1~5 mg·L−1;回流比90%;跨膜压差0.15 Mpa;错流速率1.0 m·s−1。观察废水COD降解情况考察催化剂的活性。
-
COD采用重铬酸钾标准方法[28]测定;pH采用酸度计(pHB-2,上海雷磁仪器厂)测定;镍和铬浓度采用焰原子吸收分光光度法测定(GB/T5750.6-2006),所用仪器型号为(TAS-990MFG,北京普析通用仪器厂);浊度采用便携式浊度仪(2100P,HACH)测定。
-
臭氧催化过滤膜的制备主要分为浸渍和焙烧2个主要步骤。笔者在前面的研究中发现催化剂的催化活性与浸渍液活性组分的配比、焙烧温度及焙烧时间有关[29]。本实验以电镀园区物化预处理出水进行一定比例稀释后的废水的COD去除率和臭氧催化过滤膜的膜通量为指标,对陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的制备工艺进行了优化。
1)活性组分比例的影响。配制以活性组分La3+、Ce3+、Mn2+不同质量比的浸渍液,保持其他条件相同,煅烧温度为700 ℃,煅烧时间 3 h,制备陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜,以一定比例稀释后的电镀园区物化预处理出水为污染物,考察浸渍液中金属离子的质量比与COD去除率的关系,结果如图2所示。可见,随着La3+和Ce3+含量的增加,催化剂的催化活性随之增加,在Ce3+、La3+、Mn2+的质量比到2:2:1,COD的去除率最高。一方面由于随着浸渍液中硝酸铈浓度的增加,煅烧后CeO2的结构更加有规律,晶核更成熟,镧作为同系元素性状与铈类似。另一方面,有研究[30-31]表明,金属氧化物的催化活性由高到低依次为La2O3>CeO2>MnO2。
但当浸渍液中Ce3+和La3+的比例进一步增加时,催化效率反而有所下降,一方面随着浓度的增加,催化活性物质的晶核增大,比表面积减小,不利于增加催化反应活性;另一方面由于尾水中特征有机污染物种类较多,MnO2对部分有机污染物的催化效果明显。因此,将浸渍液中金属离子Ce3+、La3+、Mn2+的质量比确定为2:2:1。
2)焙烧温度的影响。选择适宜的焙烧温度是臭氧催化过滤膜制备中的关键步骤。在浸渍液活性组分Ce3+、La3+、Mn2+的质量比为2:2:1,煅烧时间3 h的条件下,不同焙烧温度下制得的臭氧催化过滤膜在臭氧催化氧化下对废水COD的降解影响如图3所示。由图3可知,随着温度的逐渐升高,催化剂的催化活性先升高后呈现下降趋势。当焙烧温度为800 ℃时,陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的催化活性最高。这是由于硝酸镧在500 ℃下焙烧得到的产物主要成分为La5O7NO3和La2CO5,在650 ℃下焙烧得到的产物主要是La2O3,还含有少量杂质,当温度达到780 ℃时焙烧产物均是纯度较高的La2O3。当焙烧温度低于500 ℃时,La2O3和CeO2催化剂尚未完全形成,且催化剂的结晶差,颗粒粒径小,催化剂活性低。随着焙烧温度升高,催化剂结晶逐渐变好,颗粒粒径变大,催化效果增强。900 ℃以上高温焙烧时,CeO2催化剂内部结构坍塌,活性位减少,催化剂活性降低。另一方面温度过高也容易导致催化剂烧结,使催化活性组分在载体表面团聚,降低催化剂的比表面和导致活性位点缺失,催化效率降低。因此,本实验中将催化剂的焙烧温度确定为800 ℃。
3)焙烧时间的影响。催化剂的焙烧时间对活性组分的前驱体能否转变成活性组分及最终催化剂活性有较大影响. 在浸渍液活性组分Ce3+、La3+、Mn2+的质量比为2:2:1条件下,分别在700 ℃下煅烧0.5、1、2、3、4、6 h制得的臭氧催化过滤膜在臭氧催化氧化下对废水COD的降解影响如图4所示。初始时催化剂的活性随着焙烧时间的延长而增加,当焙烧时间达到3 h时,催化剂的活性趋于稳定,当焙烧时间进一步增加至6 h后,催化效果反而出现小幅下降。因为焙烧时间较短时反应尚未全部完成,催化剂强度达不到要求,无法满足催化剂高温定型的要求,活性组分易出现粉末化;若焙烧时间过长,容易造成已形成的孔道结构塌陷,表面形貌改变,活性组分出现烧结或被掩蔽。因此,本实验中将陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的最佳焙烧时间定为3 h。
根据以上的研究结果,将陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的最佳制备条件确定为:将预处理后的无机陶瓷膜浸渍于含有La(NO3)3、Ce(NO3)3和Mn(CH3COO)2的溶液中,其中Ce3+、La3+、Mn2+的质量比为2:2:1。室温下浸渍24 h,然后在105 ℃真空烘4 h,反复浸渍-烘干步骤5次,在马弗炉中800 ℃条件下焙烧3 h,最后得到负载型金属氧化物臭氧催化剂。
-
1) SEM和EDS测试结果。图5无机陶瓷膜载体及陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的SEM图像。由图5(a)可以看出陶瓷膜表面呈疏松多孔结构,孔道彼此交错贯通,具有较大的比表面积孔体积,能提供较多的负载点位。图5(b)呈现了负载后催化过滤膜的形貌,可以明显看出在陶瓷膜表面和孔道内存在大量负载物,活性组分的晶粒大小匀称且分布均匀,未在表面发生团簇现象。
EDS通常被运用于分析物体表面涂层的元素组成。选取浸渍液的金属离子Ce3+、La3+、Mn2+的质量比为2:2:1,焙烧时间3 h,焙烧温度800 ℃的催化剂样品,进行EDS表征,结果如图6所示。由图6可知,陶瓷膜载体主要成分为Al2O3,载体经浸渍、高温煅烧后,催化剂表层的EDS谱图中增加了La、Ce和Mn的特征峰。结合表1可知,表层活性组分金属元素占总质量的11.85%,从而推测在催化剂的表层形成了Ti、Mn和Fe的金属氧化物,且活性组分负载率较高。
为进一步探究催化活性组分在陶瓷膜载体上孔道内的负载情况,分析了负载后的陶瓷膜截面元素分布情况。对陶瓷膜外部膜层的截面做了元素面分布图(EDS mapping)分析。由图7中可以清晰发现除了陶瓷膜基材元素Al和O外,Mn、La、Ce 3种元素在陶瓷膜孔道内呈现均匀分布。
2)催化过滤膜性能参数与分析。选用孔径为50 nm的陶瓷膜为载体,选取浸渍液的金属离子Ce3+、La3+、Mn2+的质量比为2:2:1,焙烧时间3 h,焙烧温度800 ℃的催化剂样品进行测试。负载前后膜孔径由原来的50 nm变为30 nm(表2),膜通量下降33%。这是因为经负载后,膜孔道被氧化生成的MnO2、CeO2和La2O3活性组分所占居,导致膜孔隙率和膜孔径的减少,从催化剂的SEM表征结果也可以证实这一点,膜通量的下降与孔径的下降有直接关系。负载后陶瓷膜的盐酸可溶率值为0.6%,说明了活性组分与陶瓷膜结合紧密,催化过滤膜稳定性强。
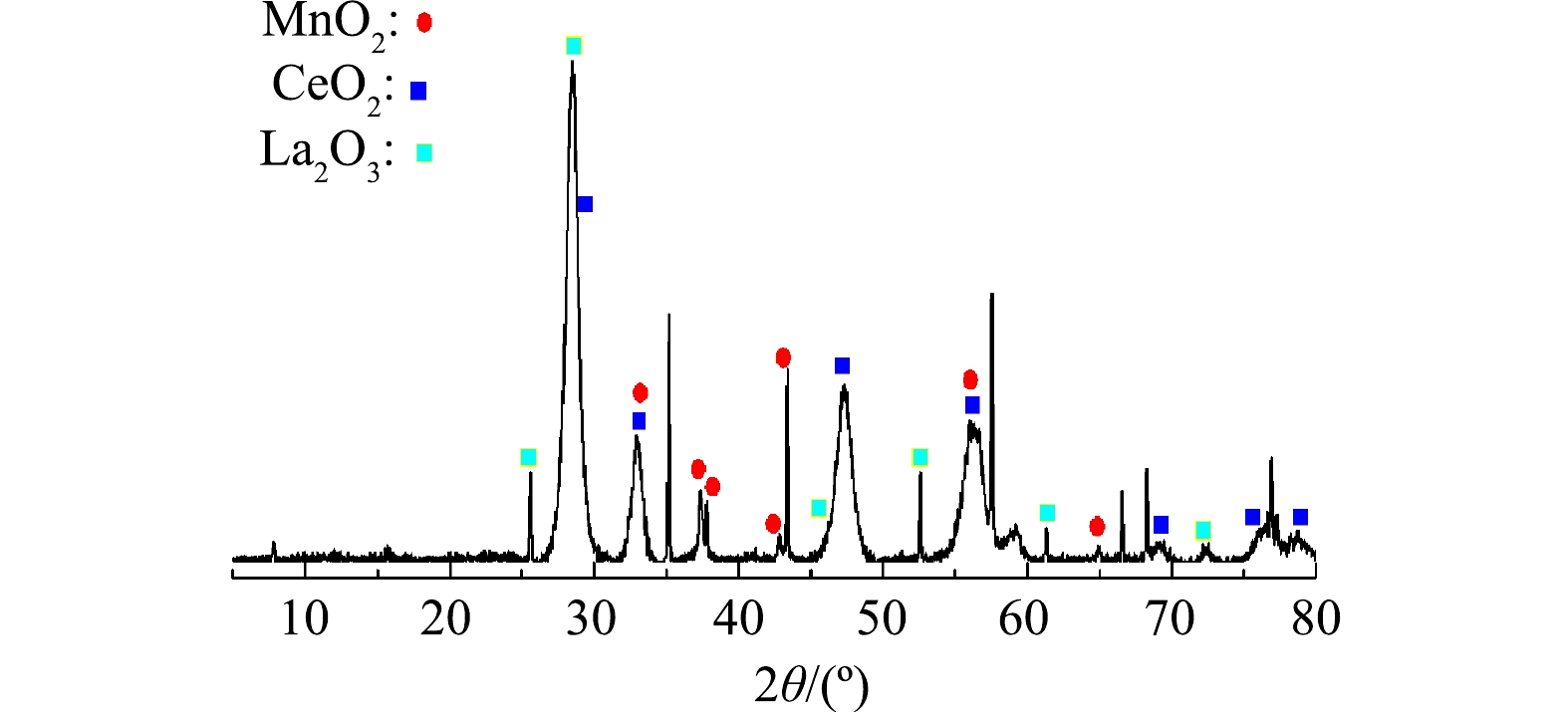
3) XRD表征结果。将负载后的催化过滤膜表层和孔道内的活性组分刮下进行XRD分析,结果如图8所示。可见,对照JCPDS标准卡,所得物质主相为Al2O3、CeO2、La2O3、MnO2,所得产物粉体为灰色,在2θ=28.8°、33.1°、47.5°、56.8°、69.4°、76.7°、79.1°附近出现的吸收峰属于立方萤石晶相结构的CeO2。在2θ=26.1°、27.9°、46.1°、53.7°、62.2°、73.4°出现的次强峰属于六方晶型La2O3,在2θ=33.3°、36.9°、37.9°、42.8°、44.1°、56.4°、64.8°出现的次强峰属于四方晶系的α-MnO2,未发现其他晶型是由于反应时间延长可使δ-MnO2 向α-MnO2转化。在2θ=35.1°、57.9°、59.1°、66.7°等处出现的一些强峰主要为Al2O3。这是由于Al2O3基体陶瓷膜的主要成分。以上表征结果表明,通过浸渍、焙烧可有效地将CeO2、La2O3、MnO2活性组分负载于陶瓷膜表面及孔道内。
-
1)催化过滤膜的催化活性。为了评估臭氧催化过滤膜的催化活性,将其与未负载的普通陶瓷膜进行对比实验,在臭氧投加量为5 mg·L−1,跨膜压差为0.15 MPa,回流比为90%的条件下进行实验,观察其膜通量变化和对水中COD的去除情况。
图9为膜通量的变化趋势。由图9可知,随着反应时间的增加,普通陶瓷膜和臭氧催化过滤膜的膜通量呈下降趋势,在臭氧条件下臭氧催化过滤膜的膜通量下降较为缓慢。经过4 h的分离后,在不投加臭氧的情况下,陶瓷膜的膜通量降低了63.8%;在臭氧条件下,普通陶瓷膜和臭氧催化过滤膜的膜通量分别降低了57.2%和46.2%,说明臭氧氧化可以缓解膜污染,同时在催化活性物质的作用下,膜污染的缓解作用更为明显。究其原因是上述反应为过滤和臭氧氧化反应的同步过程,在膜分离的过程中大部分有机物被截止在膜表面和孔道内,臭氧氧化可以降解膜表面的有机物,从而在一定程度上减缓膜污染。臭氧氧化具有一定的选择性[10],因而对膜污染的缓解效果不明显,而催化臭氧氧化产生的活性氧物种[32-33]能够无选择性的氧化有机物,从而更好地缓解膜污染。
图10反映了当臭氧投加量由0 mg·L−1逐步增加至5 mg·L−1,其余实验参数不变的条件下,水中COD的变化趋势。由图10可知,随着臭氧投加量的增加,催化过滤膜对水中COD的去除率不断升高,当臭氧投加量增加到5 mg·L−1时,污水COD的去除率达到51.2%。采用普通陶瓷过滤膜和催化过滤膜进行对比实验,在不投加臭氧的情况下,2种过滤膜对污水COD的去除率分别为5.6%和8.4%。这是由于在膜表面发生了过滤的物理反应,水中部分大分子有机污染物被截留,催化过滤膜由于负载了活性氧化物导致膜孔径更小,对有机污染物截留效率更高。当臭氧投加量逐步增高,催化过滤膜对水中COD的去除率明显高于普通陶瓷过滤膜;在臭氧投加量为5 mg·L−1时,2种过滤膜对污水COD的去除率分别为27.4%和51.2%,由于催化过滤膜上负载的CeO2、La2O3和MnO2活性组分能够有效地催化臭氧反应生产羟基自由基等活性基团,进而无选择性地氧化降解污水中的有机物污染物。
2)催化过滤膜稳定性测试。催化过滤膜的稳定性是评价其性能的一个重要指标,是决定催化剂使用寿命和产业化应用的关键因素。在臭氧投加量为5 mg·L−1,跨膜压差为0.15 MPa,回流比为90%的条件下进行实验,每次反应结束后的催化过滤膜不变,将实验废水更换为初始质量浓度的新鲜实验废水,连续使用5次为1个循环,每次使用1 h。经过6个循环后(每个循环催化剂应用5次),催化过滤膜多次使用对实验废水中COD去除率的影响如图11所示。由图11可知,随着使用次数的增加,催化过滤膜的活性并未有明显减弱,经多次循环后对水中COD的去除率稳定在50%以上,表现出较好的稳定性。对经臭氧催化过滤膜处理后的水样进行多次检测,均未检测出上述负载的金属离子,表明陶瓷膜载体上负载的金属氧化物在此实验条件下不易溶出。
-
1)以陶瓷膜为载体,La(NO3)3、Ce(NO3)3和Mn(CH3COO)2为催化活性成分的前躯体,采用浸渍-焙烧法制备了MnO2-CeO2-La2O3臭氧催化过滤膜,实现膜分离技术与催化臭氧氧化技术的耦合。浸渍液中Ce3+、La3+、Mn2+的质量比为2:2:1,焙烧时间为3 h,焙烧温度为800 ℃条件下制备出的催化过滤膜在臭氧催化氧化工艺中的催化活性最佳。
2)浸渍后的陶瓷膜经高温煅烧后生成的活性组分均匀地附着在表面及孔道内,陶瓷膜的孔隙率、孔径和通量均有所减小,XRD分析结果表明,催化剂表面形成了活性组分La2O3、CeO2和MnO2,表层活性组分金属元素占总质量的比例达到11.85%。
3)以MnO2-CeO2-La2O3臭氧催化过滤膜为研究对象,在常温和常压下,采用同步臭氧催化和过滤工艺处理电镀园区物化预处理后的混合废水取得了良好的处理效果。在臭氧投加量为5 mg·L−1、跨膜压差为0.15 MPa、回流比为90%的条件下,水中COD的去除率达到51.2%。催化过滤膜经过6次循环后对水中COD的去除率稳定在50%以上,表现出较好的稳定性。
陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的制备及表征
Preparation and characterization of MnO2-CeO2-La2O3 ozone catalytic ceramic filtration membranes
-
摘要: 以陶瓷膜为载体,采用浸渍-焙烧法制备了MnO2-CeO2-La2O3臭氧催化过滤膜,使用SEM、EDS和XRD等分析方法对其形貌和结构进行了表征分析。以电镀园区预处理混合废水为处理对象,考察了所制备的催化过滤膜在同步臭氧催化氧化和膜过滤作用下的催化活性。结果表明,在浸渍液中Ce3+、La3+、Mn2+的质量比为2:2:1、焙烧时间为3 h和焙烧温度为800 ℃下制备出的催化过滤膜对电镀园区废水有较好的催化性能。在臭氧投加量为5 mg·L−1,跨膜压差为0.15 MPa,回流比为90%的条件下,废水COD的去除率可达到51.2%,与未负载催化剂的陶瓷膜相比,COD去除率提高了23.8%,稳定性实验测试结果表明,以上制备的催化过滤膜具有良好的稳定性能。以上研究结果可实现膜分离技术与催化臭氧氧化技术的耦合,提高出水水质,增强催化过滤膜的抗污染能力。Abstract: MnO2-CeO2-La2O3 ozone catalytic ceramic filtration membranes were prepared by impregnation-calcination method using ceramic membranes as carriers, and the morphology and structure were characterized by SEM, EDS and XRD. The catalytic activity of the membranes prepared in this work was explored under the synchronous processes of catalytic ozonation and membrane filtration when the pretreatment mixed wastewater from an electroplating park was taken as the treating object. The results indicated that the catalytic filter membranes prepared at the mass ratio of 2:2:1 for Ce3+, La3+ and Mn2+ in the impregnation solution, calcination time of 3 h and 800 °C had a good catalytic performance. At the ozone dosage of 5 mg·L−1, transmembrane pressure of 0.15 MPa, and reflux ratio of 90%, the COD removal rate of wastewater could reach 51.2%, it increased by 23.8% compared with the ceramic membranes without loading catalysts. In addition, the stability tests demonstrated that the ozone catalytic ceramic filtration membranes had a good stability. Above all, the coupling of the membrane separation technology and catalytic ozonation was realized, and the effluent water quality was improved, and the anti-pollution ability of the catalytic filtration membranes were enhanced.
-
煤炭燃烧会产生氮氧化物、二氧化硫和烟尘等污染物,对大气环境和人群健康造成危害[1]。2014年,国家发展改革委、环境保护部、国家能源局三部委印发《煤电节能减排升级与改造行动计划(2014—2020年)》。该计划旨在推进现役燃煤发电机组大气污染物达标排放环保改造,确保重点地区燃煤发电机组污染物排放达到燃气轮机机组排放限值[2],即排放的NOx质量浓度低于50 mg·m−3、SO2质量浓度低于35 mg·m−3、PM质量浓度低于5 mg·m−3。
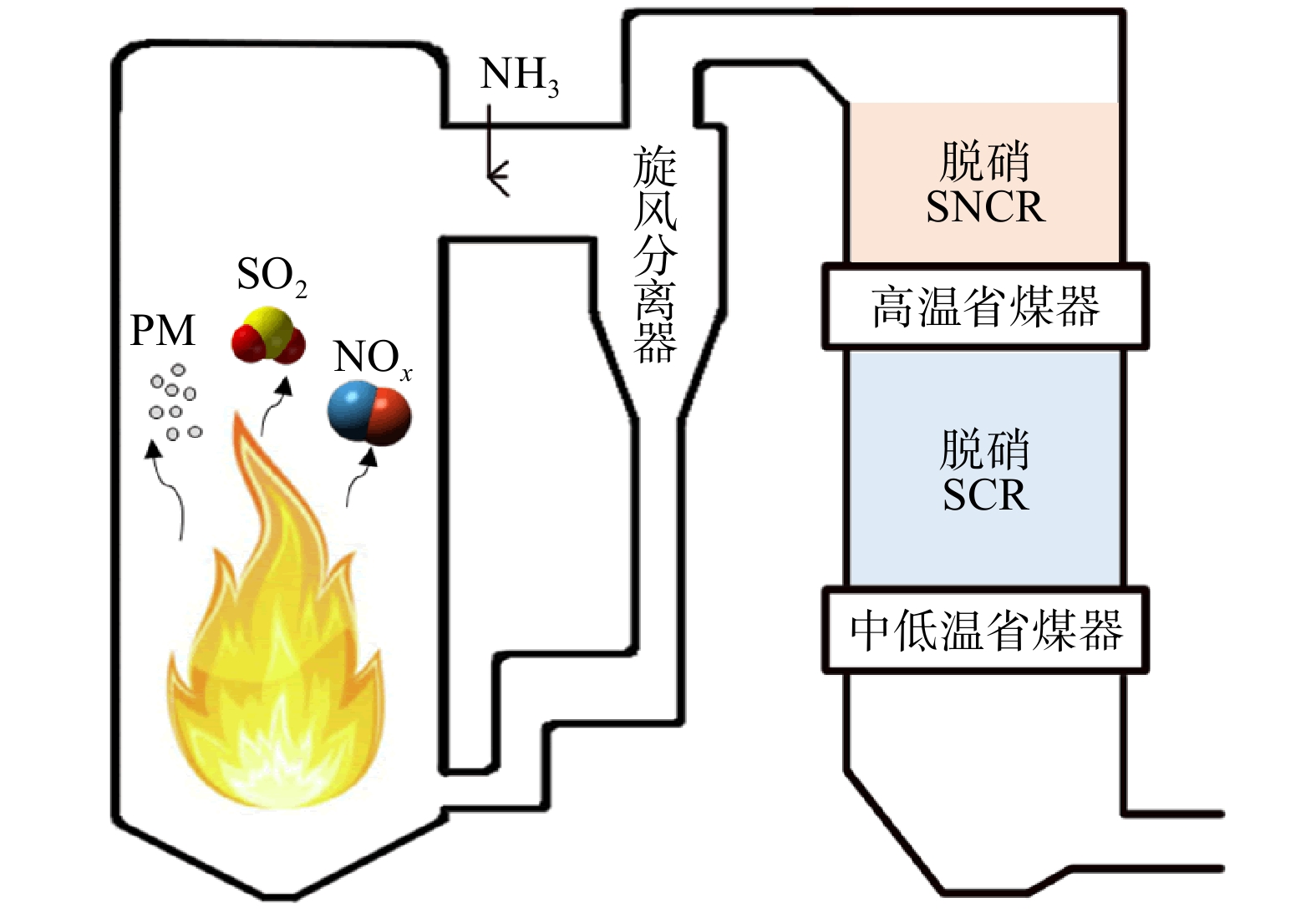
近年来,循环流化床(circulating fluidized bed, CFB)技术得到发展并广泛应用。该技术具有燃料适应性强、燃料循环利用率较高、NOx排放量低的优点[3]。 为满足燃煤机组NOx排放限值,国内多家电厂对CFB机组开展了选择性非催化还原(selective non-catalytic reduction, SNCR)和选择性催化还原(selective non-catalytic reduction, SCR)联合脱硝技术改造。为满足NOx排放要求,河北省某330 MW循环流化床机组在原有低氮燃烧器+SNCR脱硝基础上,增设单层SCR催化剂,达到NOx排放低于50 mg·m−3的目标,且改造后还原剂使用量低于改造前,同时锅炉效率基本不受影响,实现了SNCR与SCR脱硝技术良好耦合[4]。邓志鹏等[5]采用SNCR/SCR联合脱硝技术对某220 t·h−1循环流化床锅炉进行改造,改造后机组可达到50 mg·m−3的排放标准,氨逃逸也控制在3×10−6以内。然而,SNCR/SCR联合脱硝系统存在入口NOx测量滞后性大、准确性差、系统非线性强[6],以及出口浓度测量值对于入口喷氨量反应迟等问题,导致现有控制系统难以精准控制喷氨量[7]。喷氨量过少会导致出口NOx质量浓度超出排放限制;喷氨量过多会出现氨逃逸并生成硫酸盐导致空预器、催化剂严重堵塞,对设备安全造成影响[8]。为保证出口烟气中NOx质量浓度的达标、降低氨逃逸量、提高脱硝系统运行的安全性和经济性,亟需研究入口NOx质量浓度滞后修正与喷氨量精准控制的方法。
在入口NOx质量浓度滞后修正方面,软测量作为一种间接测量技术发展迅速。孙璐培[9]利用最小二乘支持向量机(least square support vector machine, LSSVM)建立了多尺度分析的NOx排放软测量模型;ZHAI等[10]在考虑系统内各参数的延迟关系的基础上,采用自适应LSSVM模型实现入口NOx质量浓度的预测;TAN等[11]采用长短期记忆神经网络,搭建了电厂NOx质量浓度预测模型,通过敏感性方法分析影响NOx质量浓度变化的因素,但尚未摸清变工况下入口处NOx的快速变化情况。在喷氨量控制方面,石铙桥[12]研究了通过电厂历史运行数据辨识状态空间矩阵,实现SNCR脱硝系统喷氨量控制,但并未考虑入口NOx变化对系统的影响。刘博文[13]建立了带入口软测量的多模型预测控制器,但入口软测量模型并未与控制系统很好地结合起来。
本研究针对某CFB型机组的SNCR/SCR联合脱硝系统入口NOx质量浓度表计测量滞后,对锅炉总给煤量的典型历史运行工况进行数据聚类,建立了适应锅炉变工况下的全局LSTM神经网络预测模型,对入口NOx质量浓度进行滞后修正。在此基础上,提出基于入口NOx质量浓度修正的多模型预测控制策略,并进行现场工程应用验证,以期平稳控制脱硝系统出口的NOx质量浓度,减少氨逃逸,使脱硝系统运行更加经济、安全。
1. 建模数据分析
1.1 CFB锅炉测点及数据采集
本文研究对象为一台燃煤循环流化床锅炉,供应的煤种煤质较为稳定,满负荷下蒸发量为220 t·h−1,采用SNCR与SCR耦合的脱硝系统。还原剂采用液氨,经过液氨蒸发装置蒸发后喷入炉内,与烟气中NOx进行还原反应,从而达到脱硝的目的。循环流化床锅炉结构如图1所示,建模所采用的主要参数如表1所示。
表 1 入口NOx质量浓度测量修正模型输入特征Table 1. Parameters of NOx mass concentration参数名称 单位 测点个数 磨煤机瞬时给煤量 t·h−1 4 一次风体积流量 m3·h−1 2 二次风体积流量 m3·h−1 2 沸下温度 ℃ 4 炉膛出口烟气温度 ℃ 2 喷氨量体积流量 L·h−1 1 脱硝入口NOx质量浓度 mg·m−3 1 烟气含氧量 % 1 催化剂入口烟气温度 ℃ 2 脱硝出口NOx质量浓度 mg·m−3 1 脱硝出口氨逃逸质量浓度 mg·m−3 1 | Show Table DownLoad:
CSV
DownLoad:
CSV
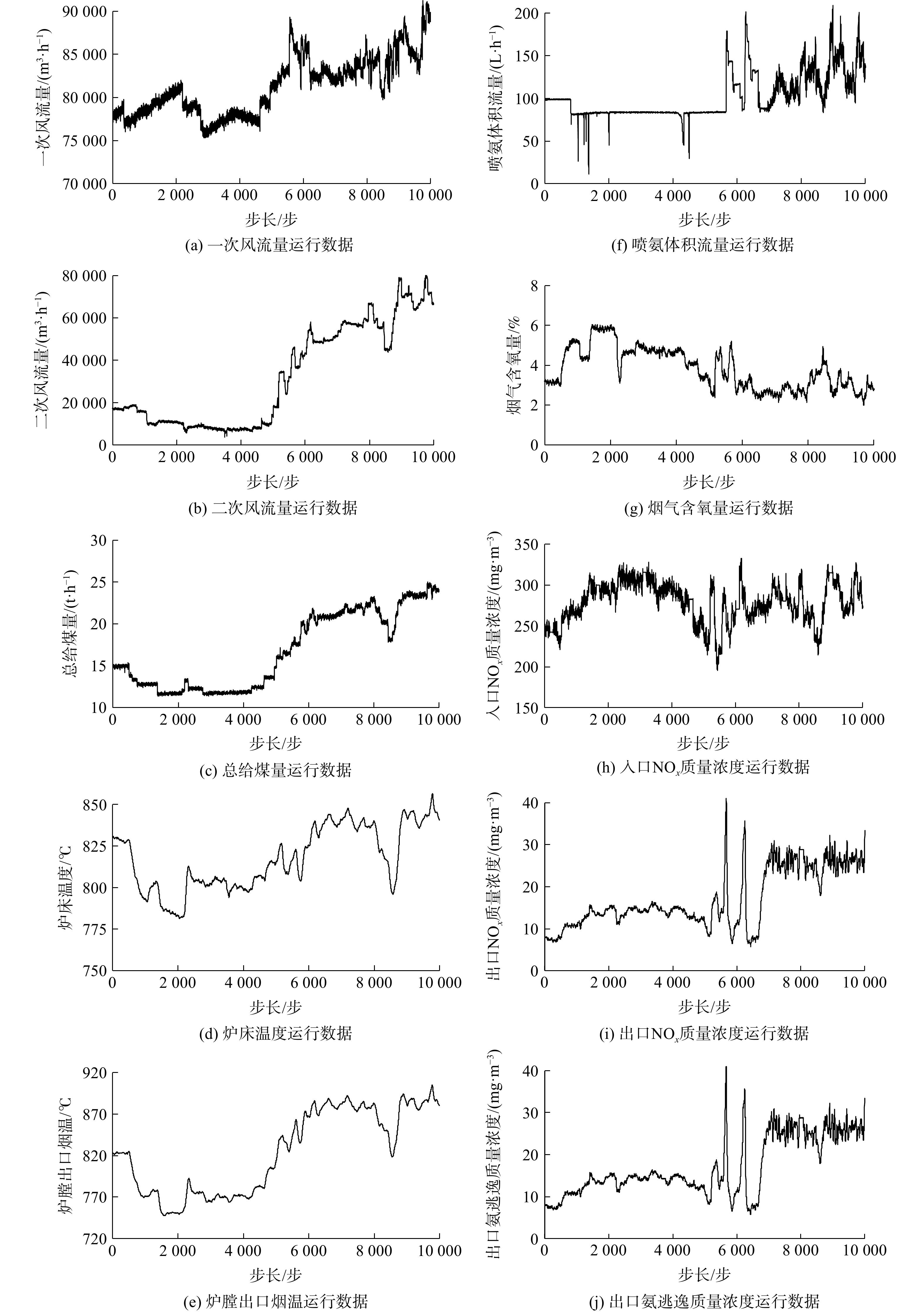
建立模型所需的数据由电厂信息(Plant Information, PI)系统采集。采样间隔5 s,采样周期20天,包含低、中、高多个负荷段,升、降、平稳负荷多种运行工况,数据一定程度上可覆盖电厂运行典型工况特征。部分现场数据如图2所示。
1.2 NOx质量浓度测量滞后分析
CFB锅炉脱硝系统入口NOx质量浓度通常由污染物排放连续监测系统(continuous emission monitoring system, CEMS)检测。从炉中抽气至就地分析腔中,通过CEMS分析烟气污染物成分信息。由于在抽气过程中,存在较长的抽气管路,再加上入口仪表分析反应时长,因此该系统滞后性较强,需要进行入口NOx质量浓度的滞后修正,再作为喷氨量控制的前馈信号,以提高系统在变工况下的响应速度。分析滞后时间的方法通常有计算法和信号分析法[14]。本研究采用信号分析方法,通过分析吹扫信号结束信号与NOx质量浓度测量值回升的时间差,得到CEMS测量滞后时间约为72 s(图3)。
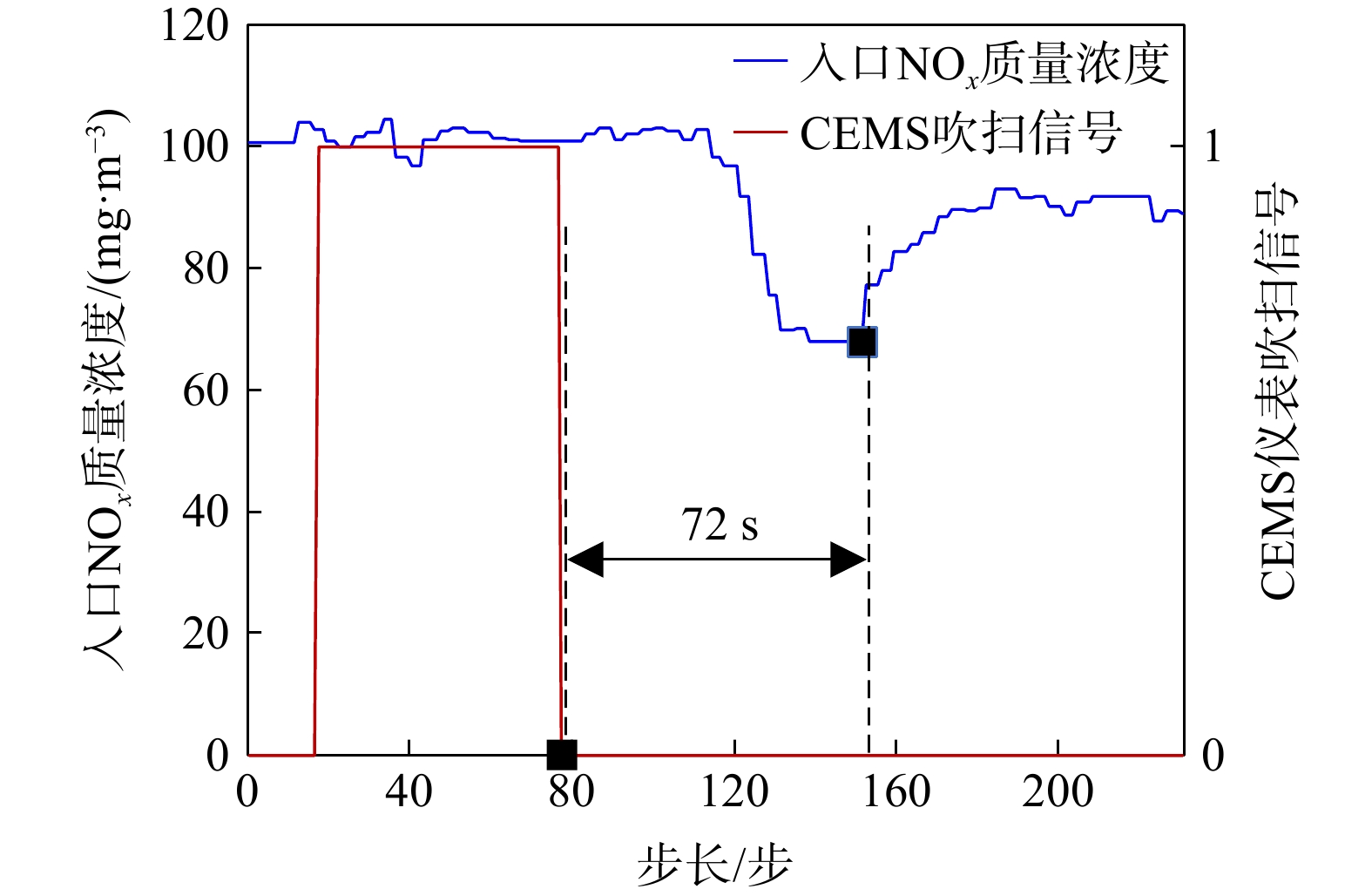 图 3 入口NOx测量CEMS滞后时间分析Figure 3. CEMS lag time analysis for inlet NOx mass concentration注:图中横坐标每步的采样周期为1 s。
图 3 入口NOx测量CEMS滞后时间分析Figure 3. CEMS lag time analysis for inlet NOx mass concentration注:图中横坐标每步的采样周期为1 s。1.3 运行典型工况划分
由于脱硝系统具有很强的非线性特征[15],而动态矩阵的预测模型采用阶跃响应线性叠加的方式,仅通过一个动态矩阵模型去预测系统在整个工况区间上的响应较为困难。因此,本研究通过核密度估计方法确定系统运行的若干典型工况,并针对多个典型工况进行建模,以提高控制精度。核密度估计方法如文献[16]所述,分析对象为可代表CFB锅炉运行特征的瞬时总给煤量。对20 d总给煤量的采样数据进行核密度估计,结果如图4所示。11.8、17.7和25.3 t·h−1为历史运行工况中瞬时总给煤量核密度统计的3个概率密度极大值点,可代表此台锅炉运行的3个典型工况。后续入口NOx质量浓度预测模型与多模型控制器模型均建立在这3个典型工况点上。
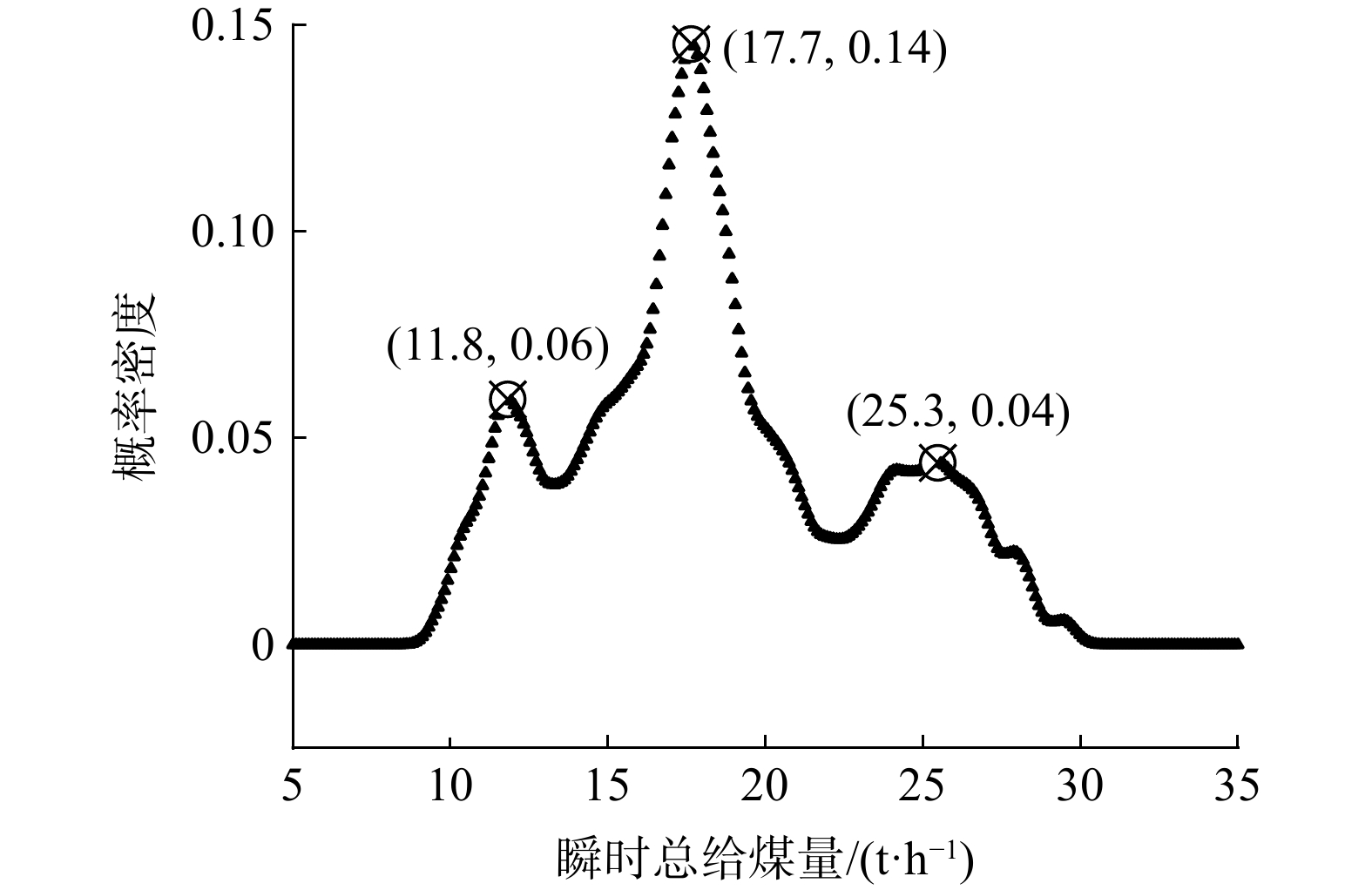 图 4 基于核密度分析的给煤量典型运行工况Figure 4. Analysis of typical operation conditions of coal feed based on kernel density estimation注:图中纵坐标数值代表相应总给煤量工况在总样本数据中出现的频率,3个标注点为概率密度曲线的极大值点,作为3个典型工况段划分的参考中心点。
图 4 基于核密度分析的给煤量典型运行工况Figure 4. Analysis of typical operation conditions of coal feed based on kernel density estimation注:图中纵坐标数值代表相应总给煤量工况在总样本数据中出现的频率,3个标注点为概率密度曲线的极大值点,作为3个典型工况段划分的参考中心点。2. 脱硝系统入口NOx质量浓度修正
2.1 修正模型建模方法
有学者研究了脱硝系统入口NOx质量浓度建模的方法,包括机理建模[17-18]、CFD建模[19-20]和机器学习[11, 21-22]等。本研究采用长短期记忆(long short-term memory, LSTM)神经网络,预测未来70 s的脱硝入口NOx质量浓度与CEMS当前测量浓度值的差值。该方法优势有2点:一是可在神经网路训练过程中避免当前测量值作为输入值时权重占比过大的影响;二是可避免因CEMS仪表吹扫时仪表测量值缺失带来的影响。
以总给煤量11.8、17.7和25.3 t·h−1为聚类中心,对历史数据进行欧式距离聚类,得到3个数据集,并分别采用LSTM进行训练,欧氏距离公式见式(1)。
d(x,y)=√n∑i=1(xi−yi)2 (1) 式中:x、y分别表示2个样本点数据;d表示欧氏距离;n表示样本数量。
2.2 修正结果与测量值的对比
LSTM网络训练数据采样间隔为20 s。采用2个隐藏层结构,回看时间步长(look-back timesteps)为24步,预测时间步长(prediction timesteps)为4步,训练数据与测试数据比例为0.8:0.2。为验证LSTM神经网络预测效果,与几种主流的机器学习预测方法的结果进行了对比,(表2)。LSTM在RMSE指标及相关性指标(R squared, R2)上结果均优于其他模型,且满足作为控制前馈的要求。
表 2 不同模型在测试集上预测效果对比Table 2. Comparison of prediction effects of different models on test datasets模型名称 RMSE/(mg·m−3) R2/% LSTM 3.53 89 ANN 17.11 40 RFR 17.06 40 | Show TableDownLoad:
CSV
3. 喷氨量的控制及仿真
由于传统控制手段控制的脱硝系统在工况波动较大情况下时,会出现喷氨响应滞后、出口NOx质量浓度波动较大的问题,控制品质难以得到保证,故本研究基于上述入口NOx质量浓度测量修正为前馈的多模型预测控制(multi-model predict control, MMPC)策略,结果如图5所示。基于Simulink开发了脱硝系统仿真,并与无入口浓度修正的多模型预测控制及原有控制策略效果进行对比。
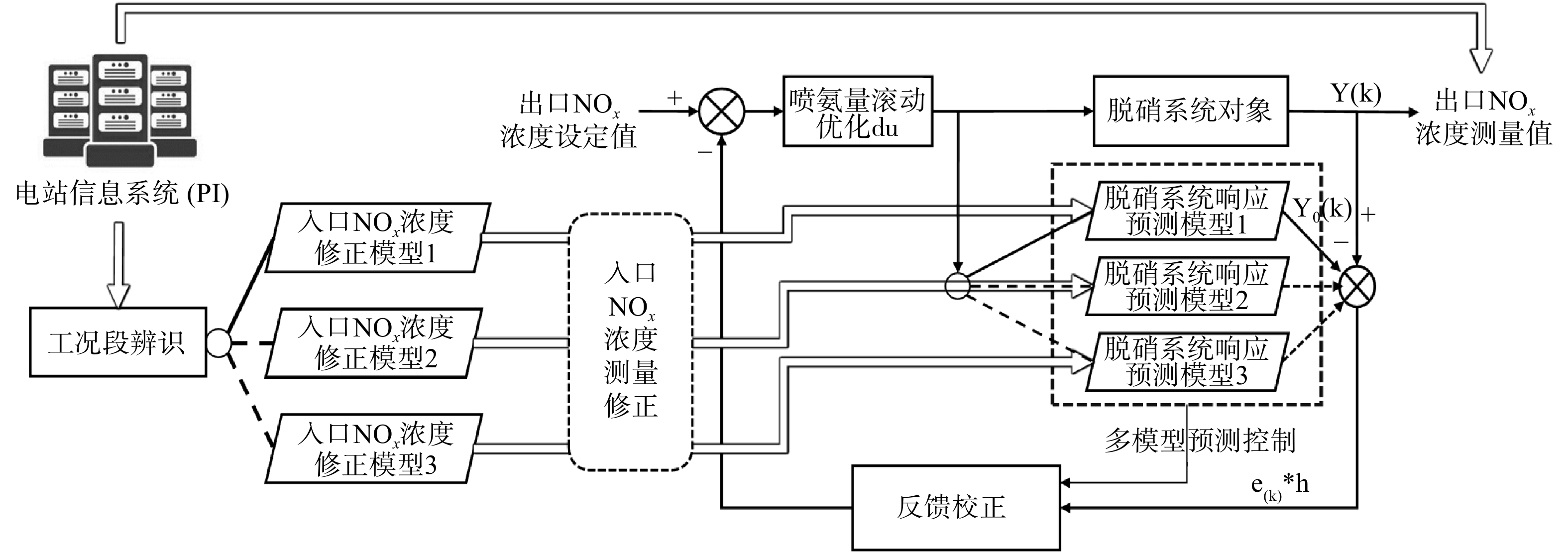 图 5 基于入口NOx质量浓度修正的多模型预测控制策略示意图Figure 5. Schematic diagram of multi-model predictive control strategy based on modification of inlet NO x massconcentration
图 5 基于入口NOx质量浓度修正的多模型预测控制策略示意图Figure 5. Schematic diagram of multi-model predictive control strategy based on modification of inlet NO x massconcentration3.1 控制预测模型辨识及拟合结果
动态矩阵模型预测控制(dynamic matrix control, DMC)为模型预测控制的一种,主要分为模型预测、滚动优化和反馈校正3个部分[23]。其中,模型预测采用阶跃响应模型,可用一阶惯性系统模型(式(2))。
Y(s)=∑mi=1Gi(s)·Xi(s)=∑mi=1(KpiTpis+1eTdis)·Xi(s) (2) 式中:Kp代表比例环节,Tp代表响应环节,Td代表纯滞后环节。采用动态惯性权重[24]的改进型粒子群寻优算法(improved particle swarm optimization, IPSO),寻找在11.8、17.7和25.3 t·h−1这3种典型工况下的脱硝系统预测模型。动态惯性权重调整后的公式为式(3)。
w=wmax−R(wmax−wmin)Rmax (3) 式中:w为惯性权重;R为当前迭代次数;Rmax为最大迭代次数。
各参数辨识结果及模型预测效果分别如表3和图6所示。这表明在各典型工况段上,预测模型均有较好的拟合效果,可满足系统仿真要求。
表 3 各典型工况段模型参数IPSO算法辨识结果Table 3. Identification results by IPSO algorithm for model parameters under typical operation conditionsons给煤量/(t·h−1) 辨识参数 喷氨量模型参数 烟气流量模型参数 入口NOx模型参数 给煤量模型参数 均方误差/(mg m−3) 11.8 kp -3.66e-01 4.04e-05 1.48e-01 5.17e-01 2.28 Tp 2.99e+00 3.97e+00 2.68e+00 2.23e+00 Td 2.02e+00 2.00e+00 3.09e+00 1.00e+00 17.7 kp -2.40e-01 6.66e-05 1.21e-01 5.22e-01 1.89 Tp 1.33e+00 3.13e+00 3.67e+00 2.33e+00 Td 1.91e+00 4.10e+00 3.43e+00 1.13e+00 25.3 kp -2.13e-01 3.96e-04 1.24e-01 9.04e-01 2.08 Tp 1.33e+00 4.79e-01 3.72e+00 2.73e+00 Td 2.02e+00 4.10e+00 3.10e+00 1.00e+00 | Show TableDownLoad:
CSV
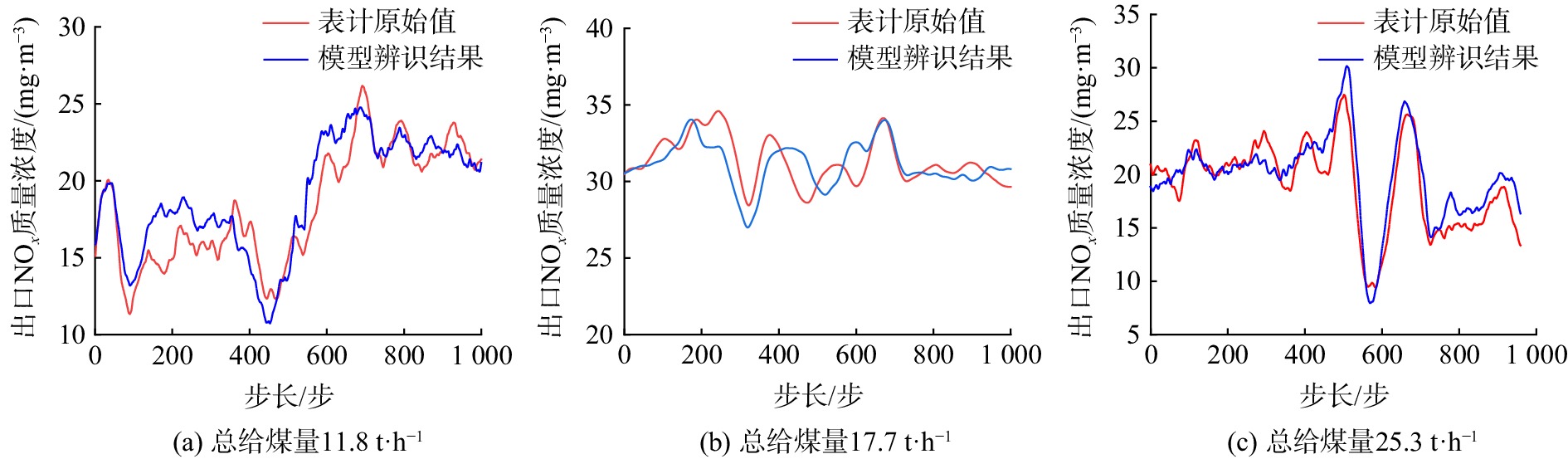 图 6 各典型工况段上模型辨识效果Figure 6. Model identification effects on typical operation conditions注:图中横坐标每步的采样周期为5 s。
图 6 各典型工况段上模型辨识效果Figure 6. Model identification effects on typical operation conditions注:图中横坐标每步的采样周期为5 s。3.2 脱硝系统仿真及控制效果对比
基于Simulink仿真软件,采用上述辨识模型的参数结果,搭建了脱硝系统仿真平台,仿真系统如图7所示。该系统分为4个部分,电厂运行数据部分为从现场采集的总风量、入口NOx质量浓度、总给煤量数据按采样周期5 s输入仿真系统;脱硝系统仿真部分为基于脉冲响应模型的脱硝系统仿真模块,可计算出实际出口NOx质量浓度;DMC控制器部分为经典的DMC控制器,红色部分则是入口NOx质量浓度测量修正模块,给入上述LSTM神经网络所预测出的入口NOx质量浓度差值。
 图 7 基于Simulink软件的脱硝系统仿真框图Figure 7. Diagram of denitration system based on Simulink software
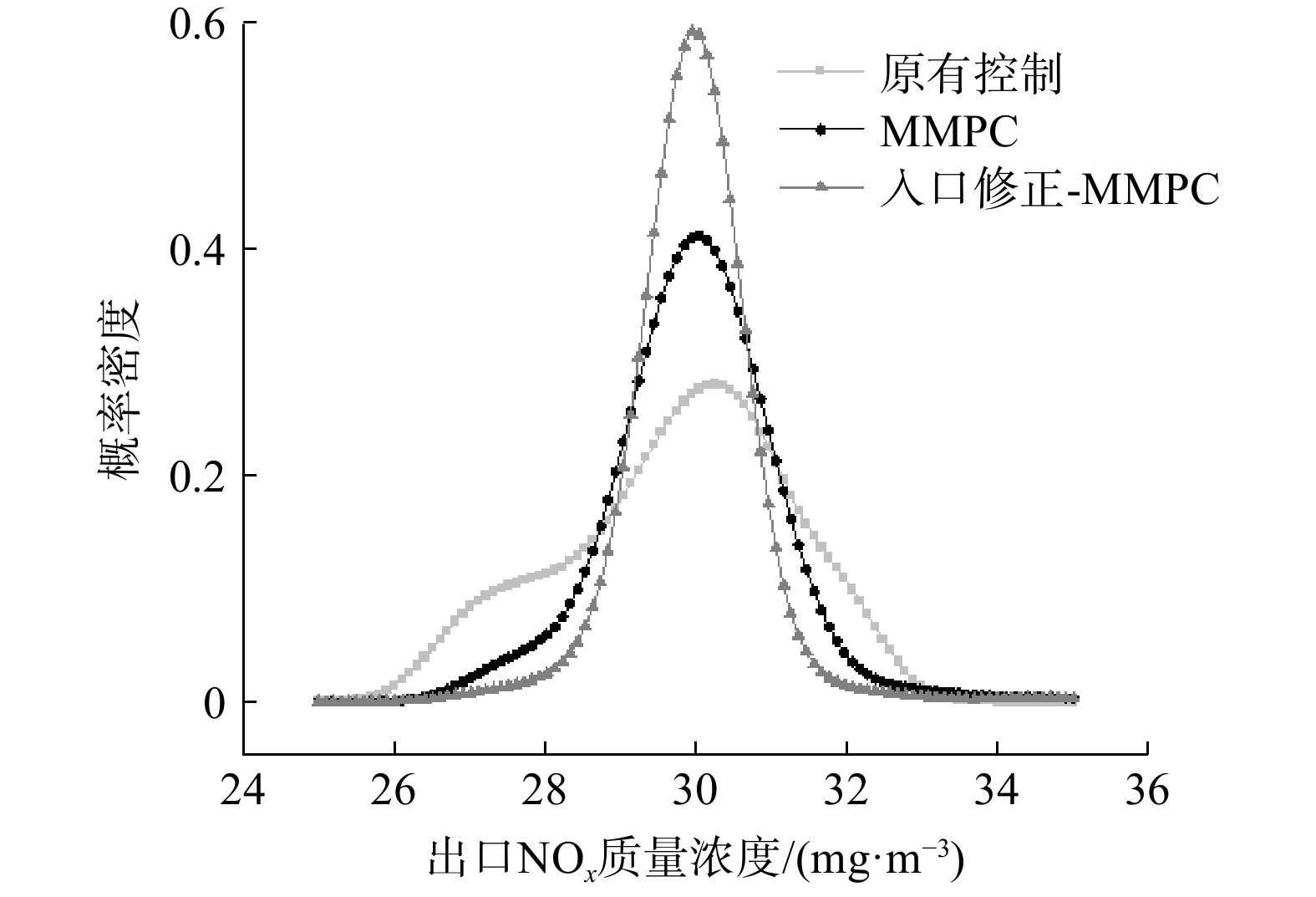
图 7 基于Simulink软件的脱硝系统仿真框图Figure 7. Diagram of denitration system based on Simulink software在控制出口NOx质量浓度为30 mg·m−3的目标下,采用实际现场运行数据进行脱硝系统喷氨量控制仿真,仿真时间步长为800步。控制效果对比如图8所示。当仿真时间步长为100~ 200步时,原有控制在入口NOx质量浓度下降时喷氨量响应速率较慢,导致出口NOx质量浓度远离目标值,而MMPC控制器反应迅速。在仿真时间为400步和570步时,入口NOx质量浓度变化幅度较大,MMPC控制平稳性优于原有控制,而带入口NOx质量浓度修正的MMPC控制器波动幅度更小。将上述控制结果绘成频率折线图(图9)。结果表明,有入口浓度修正的MMPC控制平稳度优于普通MMPC控制,更优于原有控制。原有控制、MMPC与入口修正MMPC出口NOx质量浓度标准差分别为1.617、0.955、与0.584 mg·m−3。较原有控制,MMPC控制波动减少40.6%,入口修正MMPC控制波动减少63.7%。
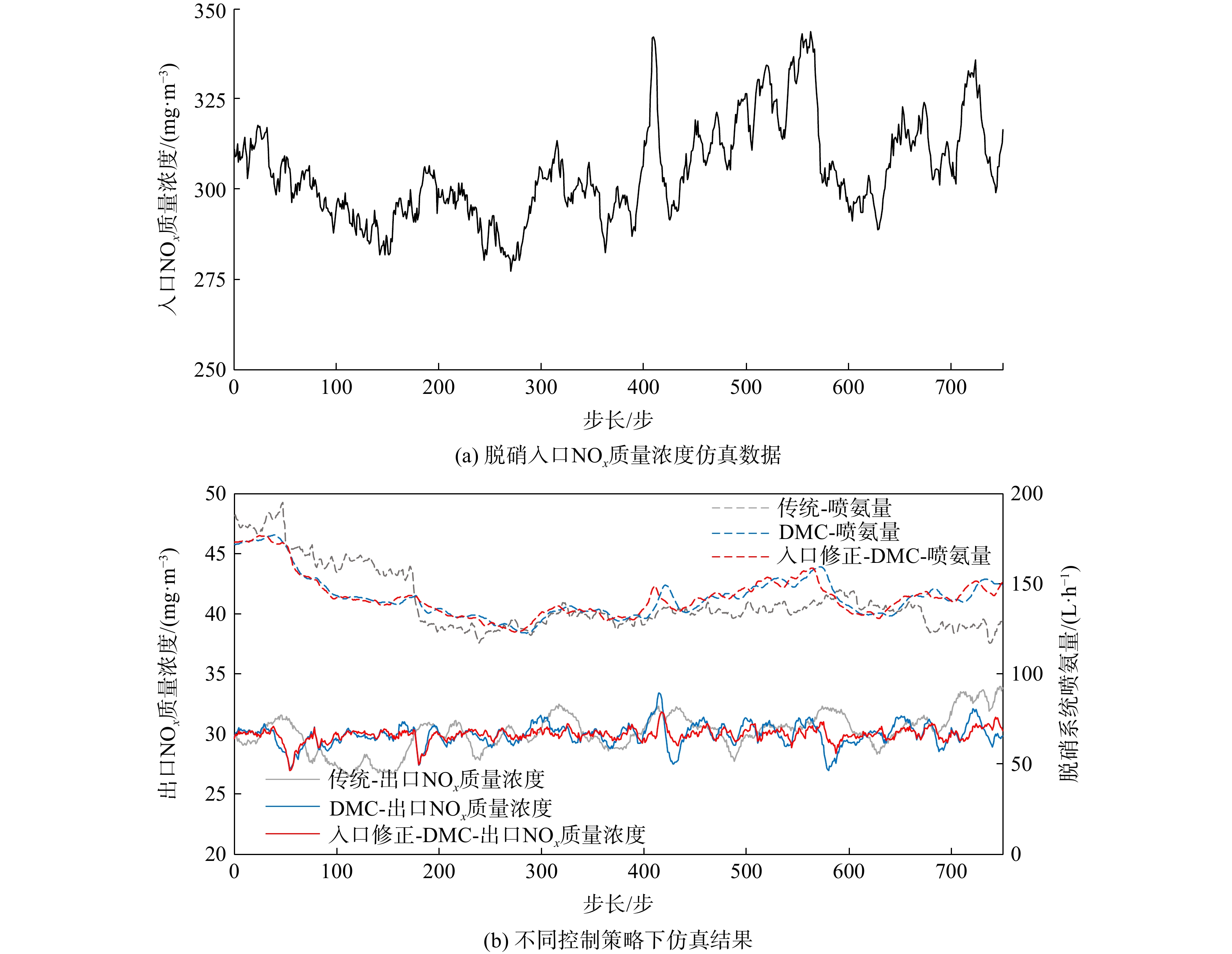 图 8 不同控制策略喷氨量控制效果仿真对比Figure 8. Simulation comparion of ammonia injection control effect with different control strategies注:图中横坐标每步的采样周期为5 s。
图 8 不同控制策略喷氨量控制效果仿真对比Figure 8. Simulation comparion of ammonia injection control effect with different control strategies注:图中横坐标每步的采样周期为5 s。3.3 入口NOx测量修正对控制效果的影响
控制出口NOx质量浓度平稳的目的有2点:一是尽量减少脱硝系统出口氨逃逸,降低出口氨腐蚀对设备造成破坏的风险;二是减少喷氨量,提高运行经济性。在该前提下,将出口NOx质量浓度设定值提高至40 mg·m−3,以观察系统响应。
图10表明,前80步为目标值30~40 mg·m−3的过渡过程,在出口NOx质量浓度抬高至40 mg·m−3后便不存在太多超调过程即往回回落,并稳定在40 mg·m−3附近。在480步入口NOx质量浓度有较大变化时,不带入口NOx质量浓度修正的MMPC控制波动范围为37.5 ~ 43 mg·m−3,而带入口NOx质量浓度修正的MMPC控制波动范围为39 ~ 42 mg·m−3,范围更窄。在入口NOx质量浓度波动较大的情况下,带入口NOx质量浓度修正的MMPC控制出口NOx质量浓度稳定在40 mg·m−3左右,表现出良好的控制品质。
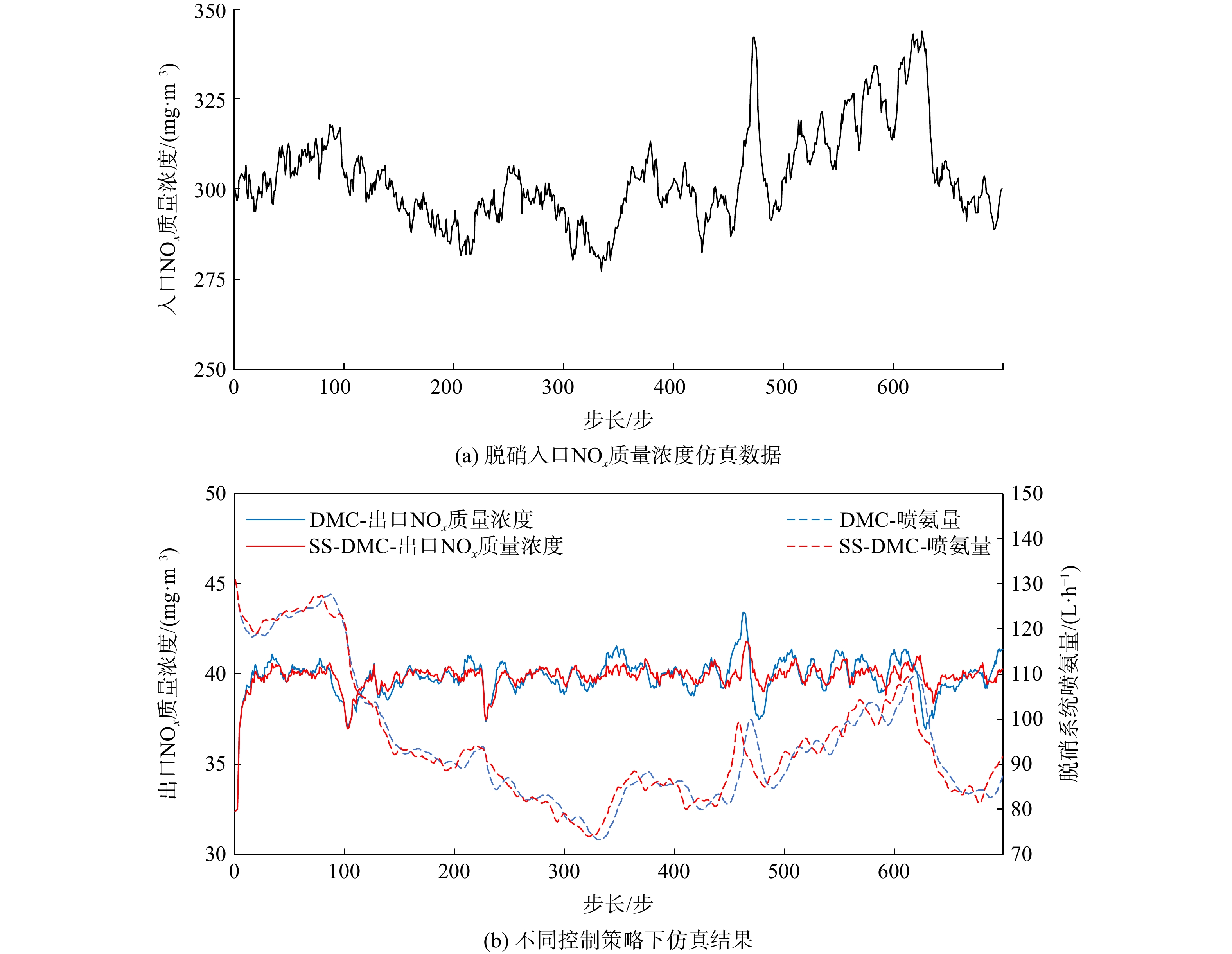 图 10 出口NOx质量浓度40 mg·m-3目标值时控制效果对比Figure 10. Control quality comparison on 40 mg·m-3 target of outlet NOx concentration注:图中横坐标每步的采样周期为5 s。
图 10 出口NOx质量浓度40 mg·m-3目标值时控制效果对比Figure 10. Control quality comparison on 40 mg·m-3 target of outlet NOx concentration注:图中横坐标每步的采样周期为5 s。4. 工程应用案例
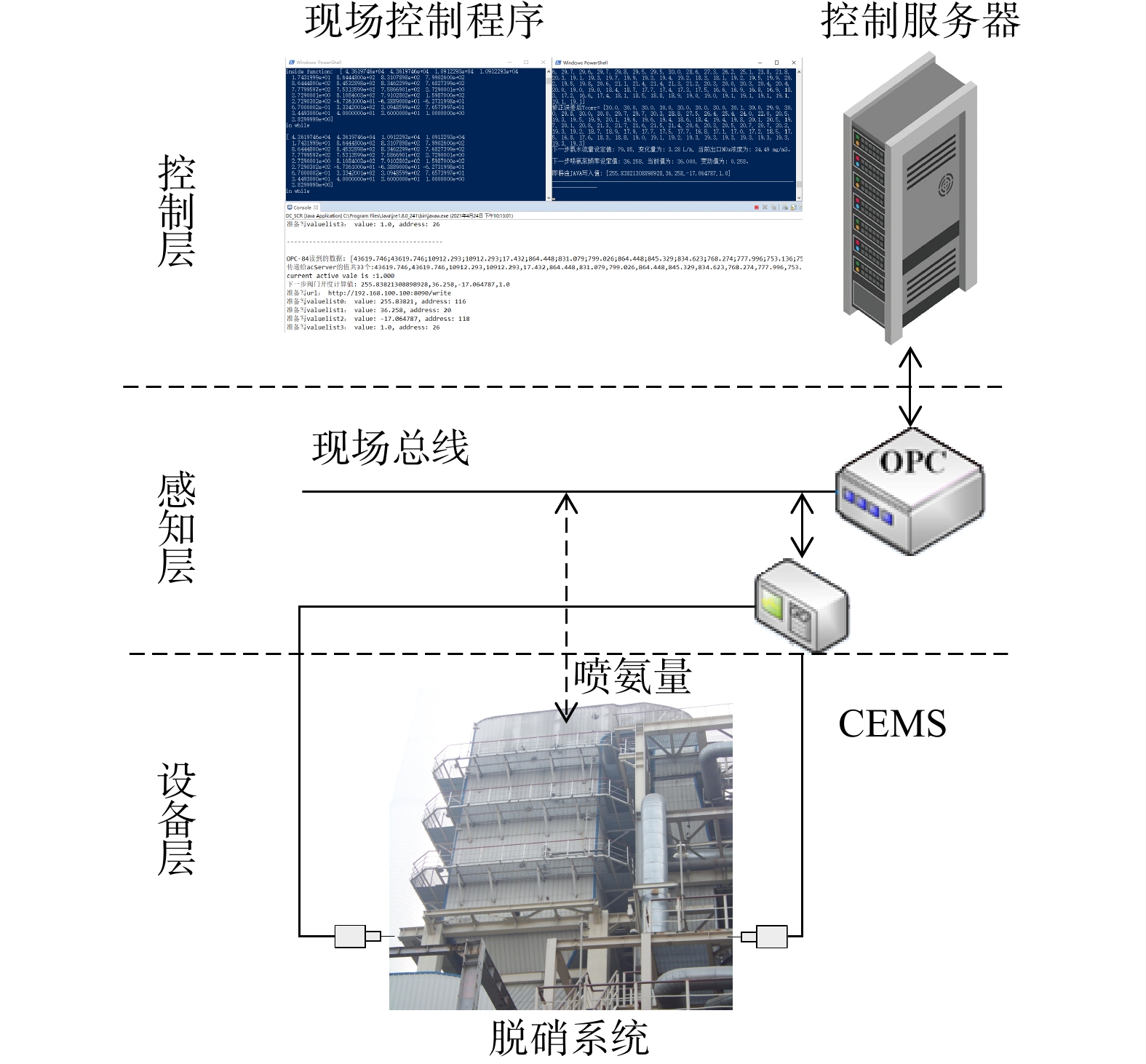
为检验控制策略在实际应用中的效果,开发了与燃煤CFB机组现场生产环境兼容的脱硝控制系统,并在浙江嘉兴某热电厂开展了48 h的现场测试,期间经历了快速升、降负荷及高、中、低不同负荷的各种典型工况。现有控制系统部署在独立的网络系统中,电厂原有控制系统通过OPC交换机向现有控制系统传输数据,优化计算后的控制指令和相关数据再通过Modbus协议传输给DCS系统,实现智能控制。图11为现场部署的脱硝控制系统硬链接示意图。
在脱硝入口NOx质量浓度修正方面,当风、煤等参数发生较大波动时,入口NOx质量浓度预测修正可提示预判变化趋势,作为前馈使喷氨量提前动作。图12表明,在第1 000 步和第2 500 步左右时,模型分别判断出较大的上升趋势和下降趋势。但在较为平稳工况下的修正效果仍有待改进,这是由于输入模型的关键参数煤质信息有缺失,且修正模型无法完全反应生成的细节变化、未考虑流场中NOx分布不匀等问题。
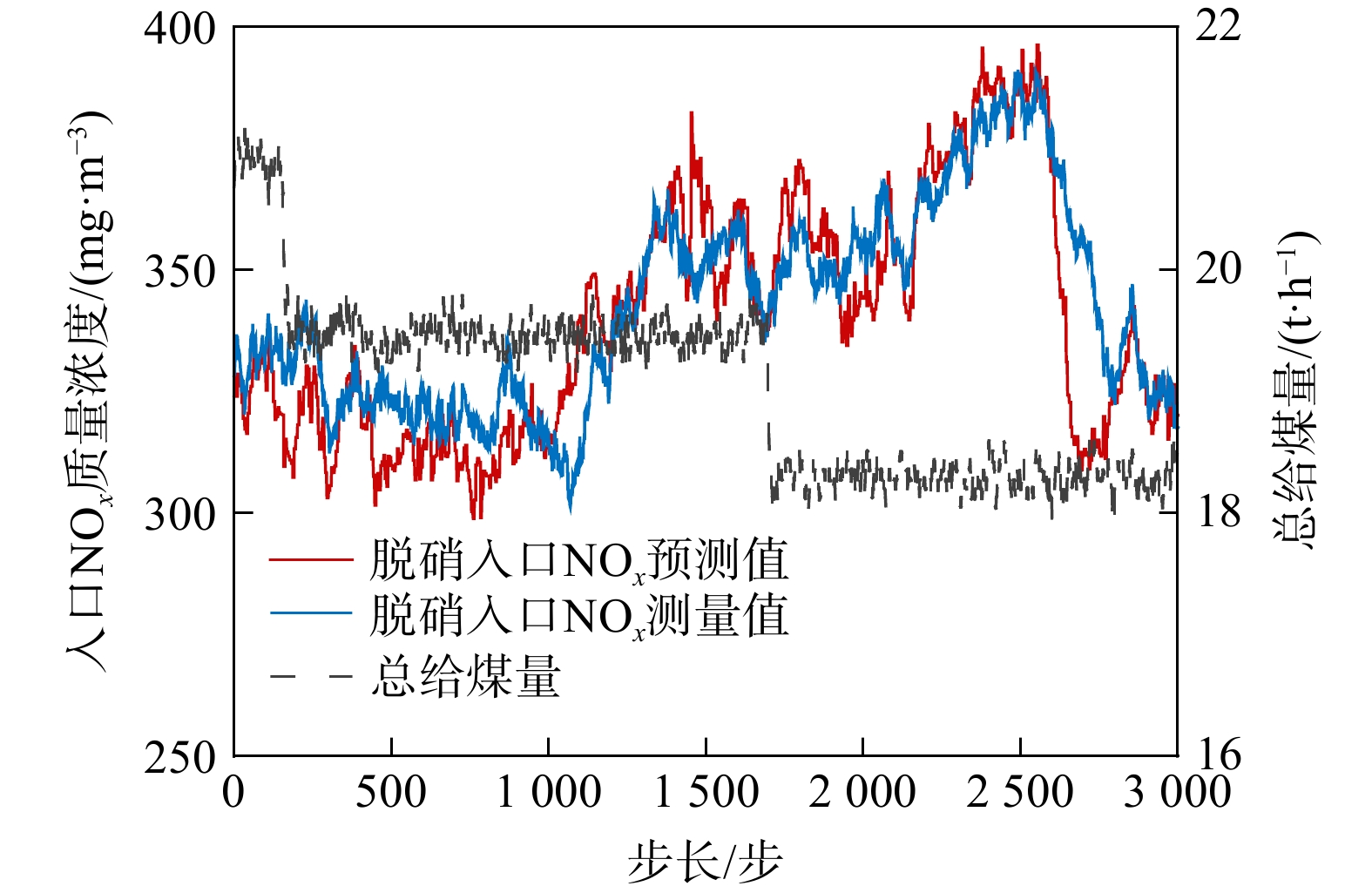 图 12 现场脱硝入口NOx质量浓度修正效果Figure 12. Correction of inlet NOx concentration on scene注:图中横坐标每步的采样周期为5 s。
图 12 现场脱硝入口NOx质量浓度修正效果Figure 12. Correction of inlet NOx concentration on scene注:图中横坐标每步的采样周期为5 s。在脱硝系统出口NOx质量浓度控制方面,主要从2个方面进行验证:一是控制系统在机组快速升降负荷情况下的控制效果,检验控制的快速性、平稳性;二是检验在不同负荷段的控制效果,检验多模型策略的可行性和系统的鲁棒性。图13为快速升负荷下的控制效果对比,原有控制出口NOx波动范围为±21 mg·m−3,而入口修正-MMPC出口NOx波动范围为±6 mg·m−3。而且原有控制滞后,在出口NOx超过50 mg·m−3后才调大喷氨量,导致了喷氨过量,氨逃逸达到8 mg·m−3,而入口修正-MMPC喷氨量提前动作,将氨逃逸维持在了3 mg·m−3以内。图14为快速降负荷时控制效果的对比,原有控制的出口NOx波动达到了±12 mg·m−3,而入口修正-MMPC出口NOx波动控制在±5 mg·m−3内,故氨逃逸也较低。
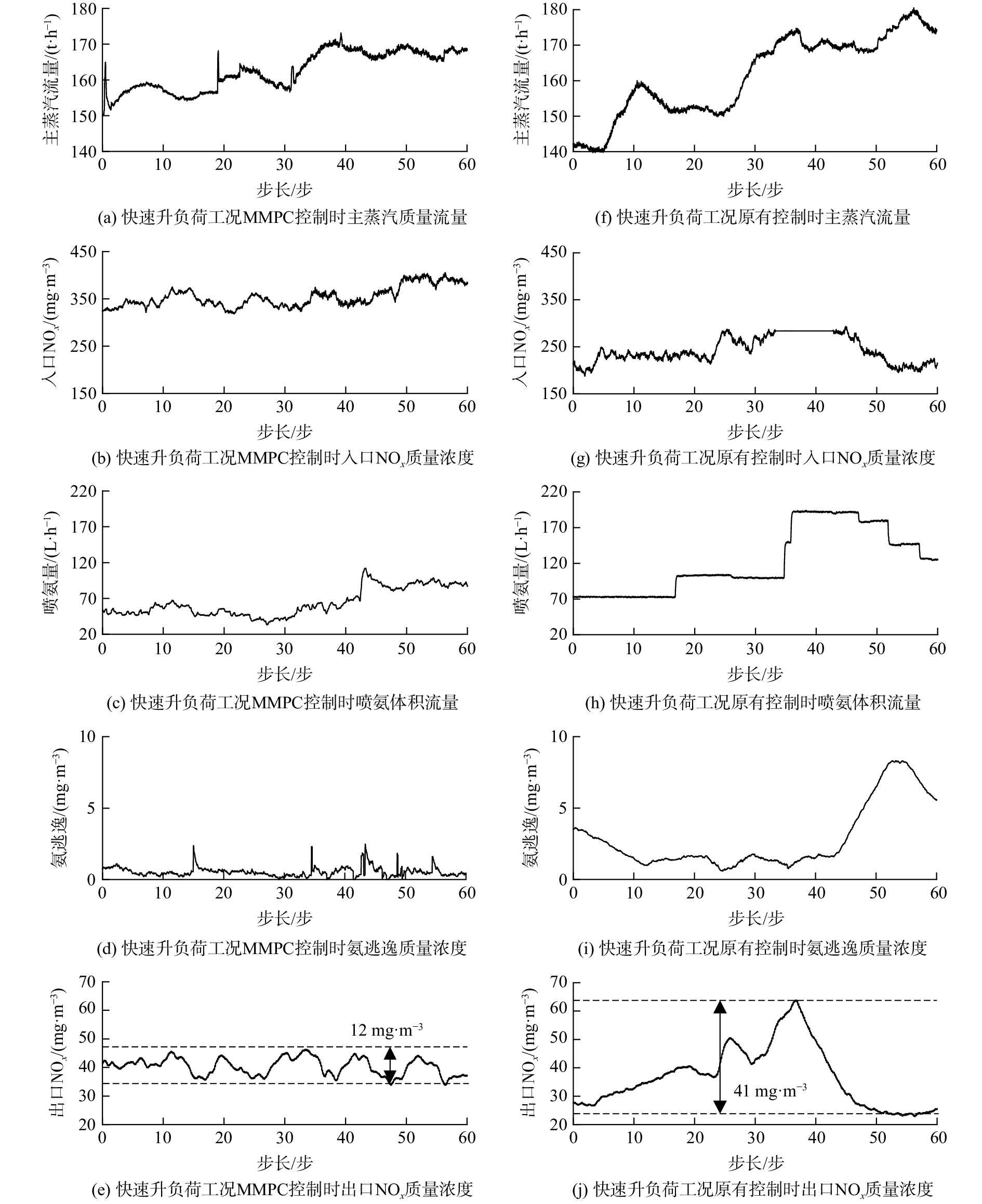 图 13 现场快速升负荷工况下控制效果对比Figure 13. Comparison of control effects under the condition of rapid load increase on scene注:虚线表示现场实验过程中出口NOx质量浓度波动的上下限范围,波动范围越小证明控制品质越好。图中横坐标每步的采样周期为1 min。
图 13 现场快速升负荷工况下控制效果对比Figure 13. Comparison of control effects under the condition of rapid load increase on scene注:虚线表示现场实验过程中出口NOx质量浓度波动的上下限范围,波动范围越小证明控制品质越好。图中横坐标每步的采样周期为1 min。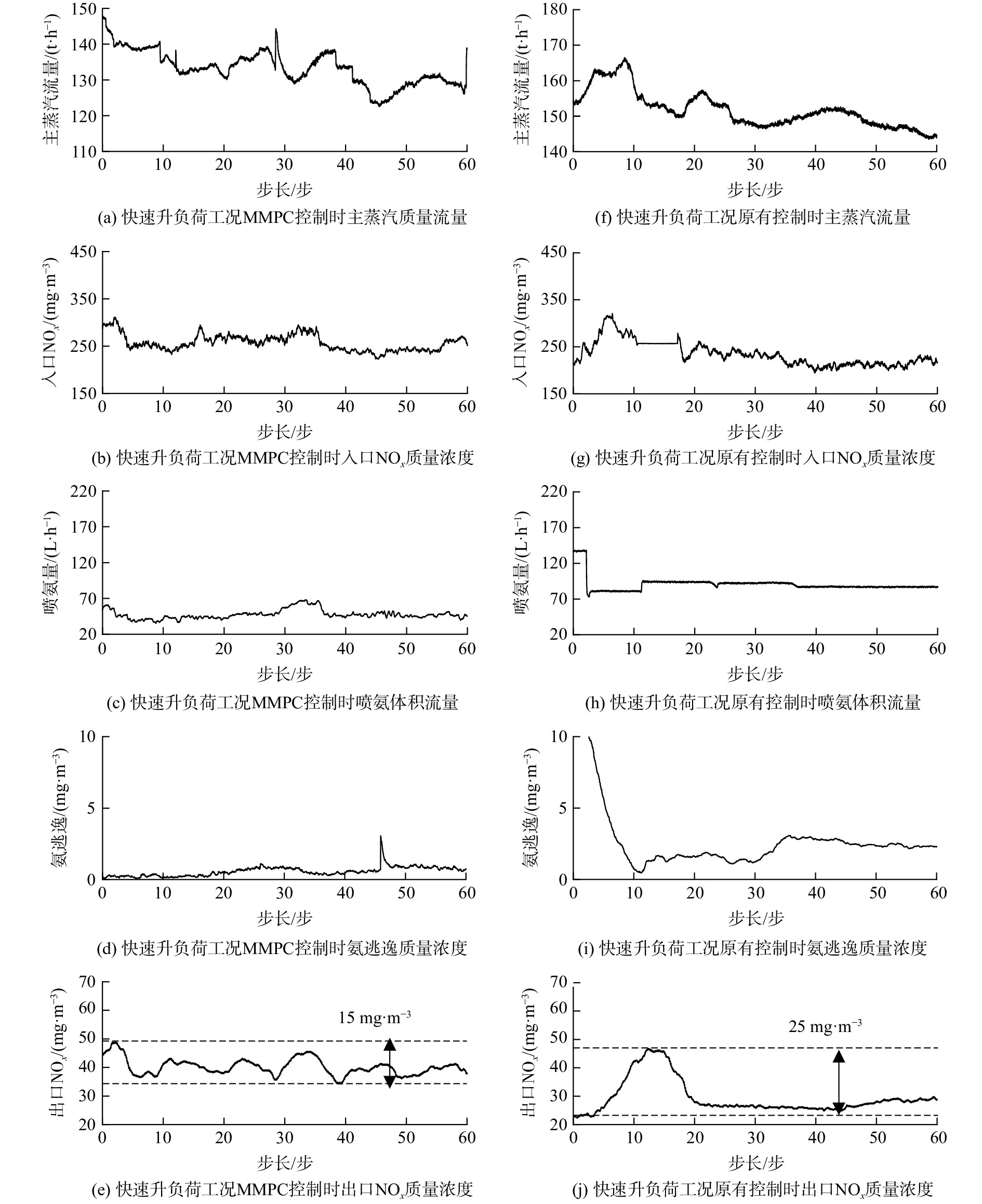 图 14 现场快速降负荷工况下控制效果对比Figure 14. Comparison of control effects under the condition of rapid load decrease on scene注:虚线表示现场实验过程中出口NOx质量浓度波动的上下限范围,波动范围越小证明控制品质越好。图中横坐标每步的采样周期为1 min。
图 14 现场快速降负荷工况下控制效果对比Figure 14. Comparison of control effects under the condition of rapid load decrease on scene注:虚线表示现场实验过程中出口NOx质量浓度波动的上下限范围,波动范围越小证明控制品质越好。图中横坐标每步的采样周期为1 min。为验证控制模型在全局工况下的鲁棒性,对高、中、低负荷工况下的控制效果进行对比(表4)。统计现场实验数据和历史数据在相应负荷段的1 h内,出口NOx质量浓度及氨逃逸的最大值、最小值和标准差统计量,用以表征控制效果好坏。在出口NOx质量浓度控制方面,在同样设定值35 mg·m−3的情况下,入口修正-MMPC控制在各工况段始终控制出口浓度低于40 mg·m−3,实现了较好地卡边控制。而原有控制波动较大,甚至有超标的风险。在氨逃逸控制方面,入口修正-MMPC控制氨逃逸最大值也小于原有控制水平,标准差上更是有巨大差距。这也说明入口修正-MMPC控制更为平稳,氨逃逸值波动较小,使脱硝系统的平稳性和经济性均有明显改善。
表 4 不同负荷段工况下控制效果对比Table 4. Comparison of control effects under different load segments锅炉蒸发量 控制策略 出口NOx质量浓度/(mg·m−3) 氨逃逸的质量浓度/(mg·m−3) 最大值 最小值 标准差 最大值 最小值 标准差 120 t·h−1 入口修正-MMPC 39.9 32.1 1.7 1.08 0.73 0.06 120 t·h−1 原有控制 48.8 30.6 4.8 2.16 0.87 0.24 150 t·h−1 入口修正-MMPC 38.0 29.5 2.2 0.95 0.59 0.07 150 t·h−1 原有控制 54.9 21.0 8.7 15.38 0.77 5.10 200 t·h−1 入口修正-MMPC 39.8 24.4 3.4 4.69 0.18 0.65 200 t·h−1 原有控制 47.2 22.1 7.3 5.94 0.40 1.48 注:最大值代表在相应控制策略下出现的波动最大值,最小值代表在相应控制策略下出现的波动最小值。 | Show TableDownLoad:
CSV
综上所述,入口修正-MMPC策略在升降负荷工况下响应较快,出口NOx质量浓度控制平稳,氨逃逸较低。由于多模型的应用建立了准确的全局预测模型,在高中低典型工况上预测模型均有较高的准确性、鲁棒性好,因此可满足长期运行要求。相较于原有控制,入口修正-MMPC可有效降低出口NOx质量浓度的波动,减少过量喷氨次数,降低氨逃逸,节约氨水使用量,从而提升脱硝系统运行经济性、安全性。
5. 结论
1)由于不同工况聚类的长短期记忆神经网络模型的准确度要高于常见的回归模型人工神经网络和随机森林回归,故在脱硝入口NOx质量浓度修正方面,可证明长短期记忆神经网络在处理时间序列数据上的优势。模型在测试集上的均方根误差为3.53 mg·m−3,相关性系数为89%。
2)在喷氨量控制方面,采用多模型预测控制策略,可克服脱硝系统非线性特性强的问题,增强控制系统在不同工况下的鲁棒性。采用入口浓度修正的多模型预测控制策略,可使控制系统在入口NOx质量浓度发生突变时,喷氨量提前动作响应,相比没有入口修正的多模型预测控制策略,出口NOx质量浓度控制更平稳。
3)现场应用表明,脱硝系统采用入口浓度修正的多模型预测控制策略后,在变工况和高、中、低多负荷区间条件下,出口NOx质量浓度波动明显降低。这表明多模型预测控制策略下系统的运行优于原有控制策略,该策略亦可提高脱硝系统的经济性。
-
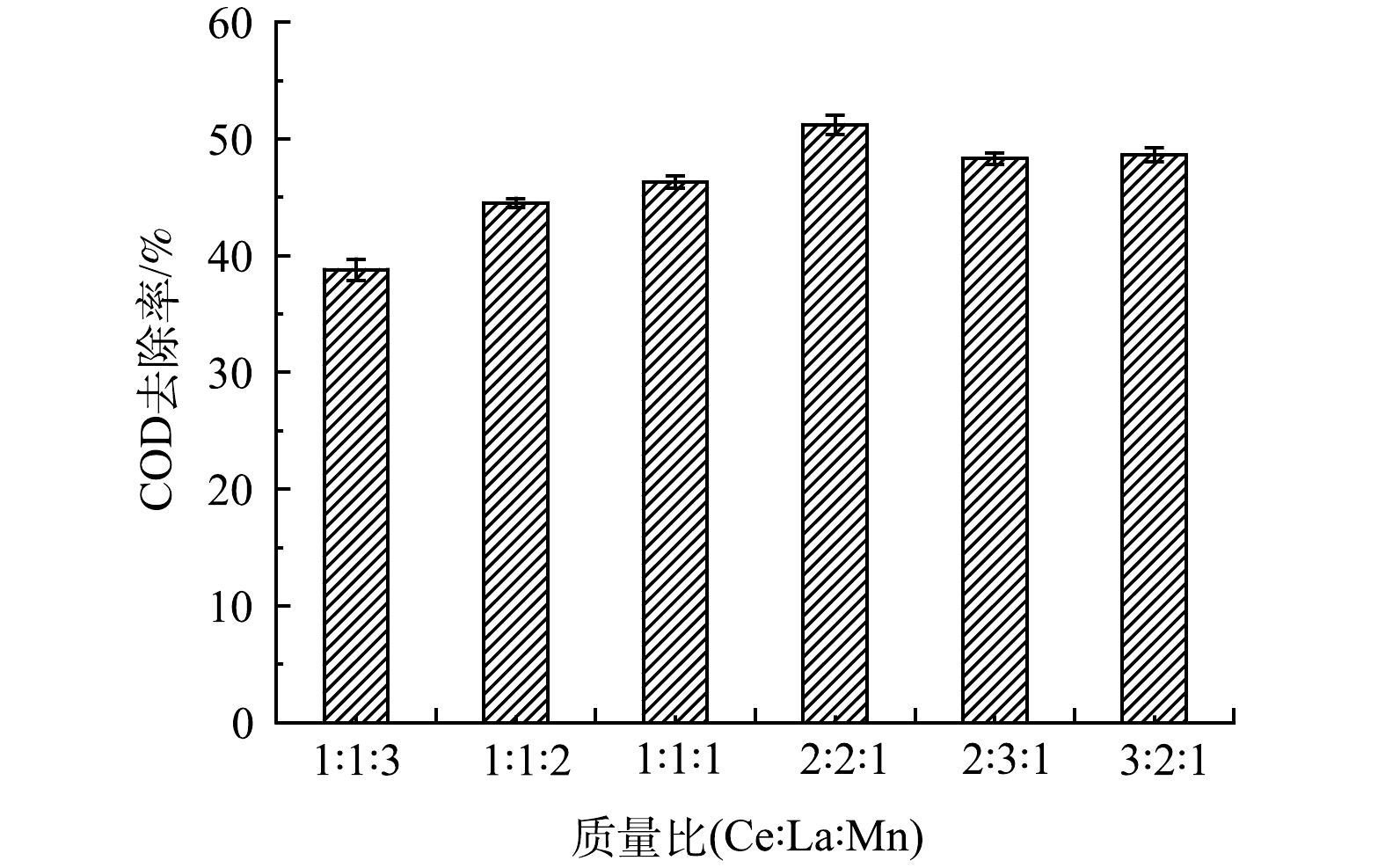
图 2 活性组分比例对催化过滤膜催化活性的影响
Figure 2. Effect of the ratio of active ingredient on the catalytic activity of the catalytic filtration membranes
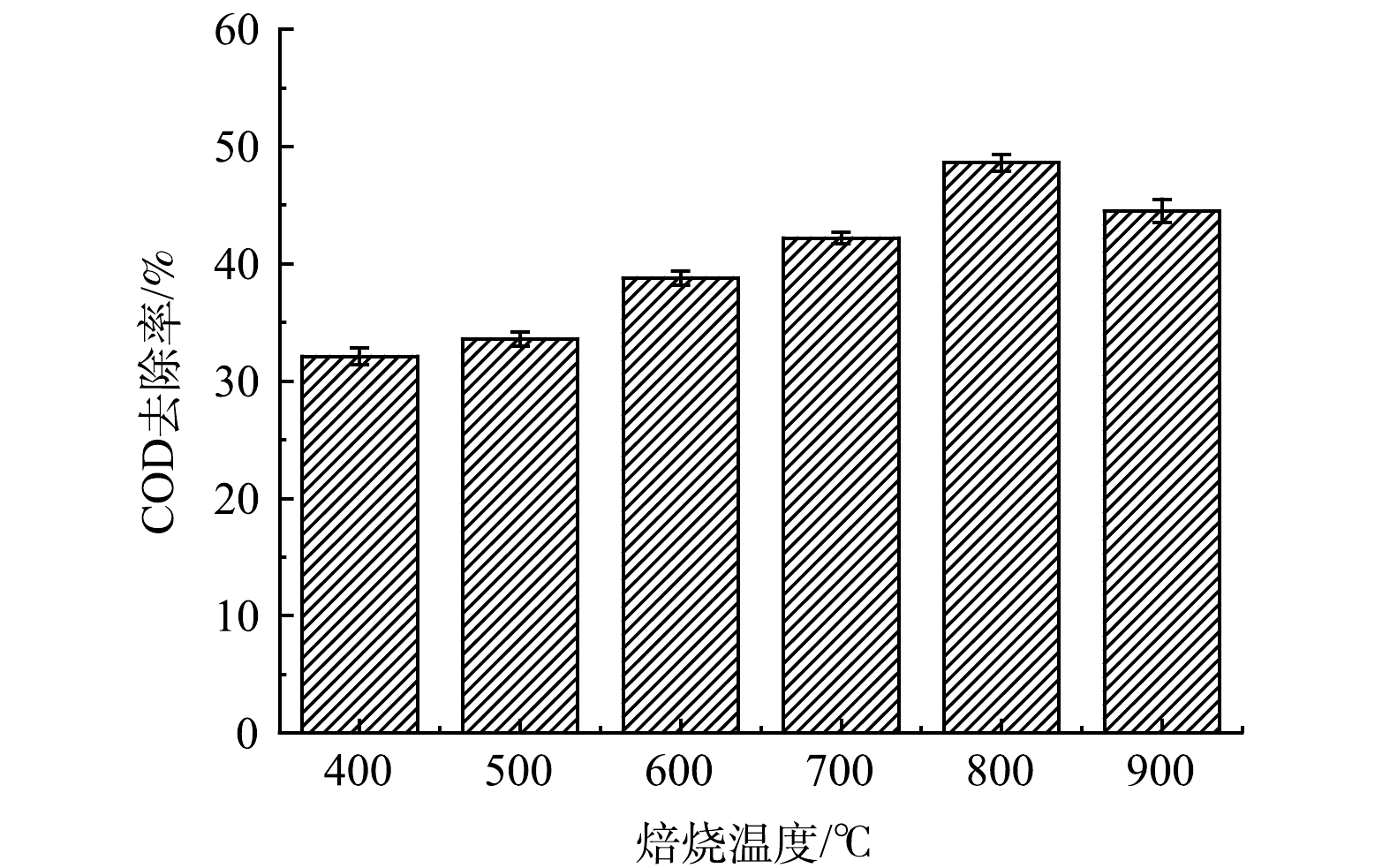
图 3 焙烧温度对催化过滤膜催化活性的影响
Figure 3. Effect of calcination temperature on the catalytic activity of the catalytic filtration membranes
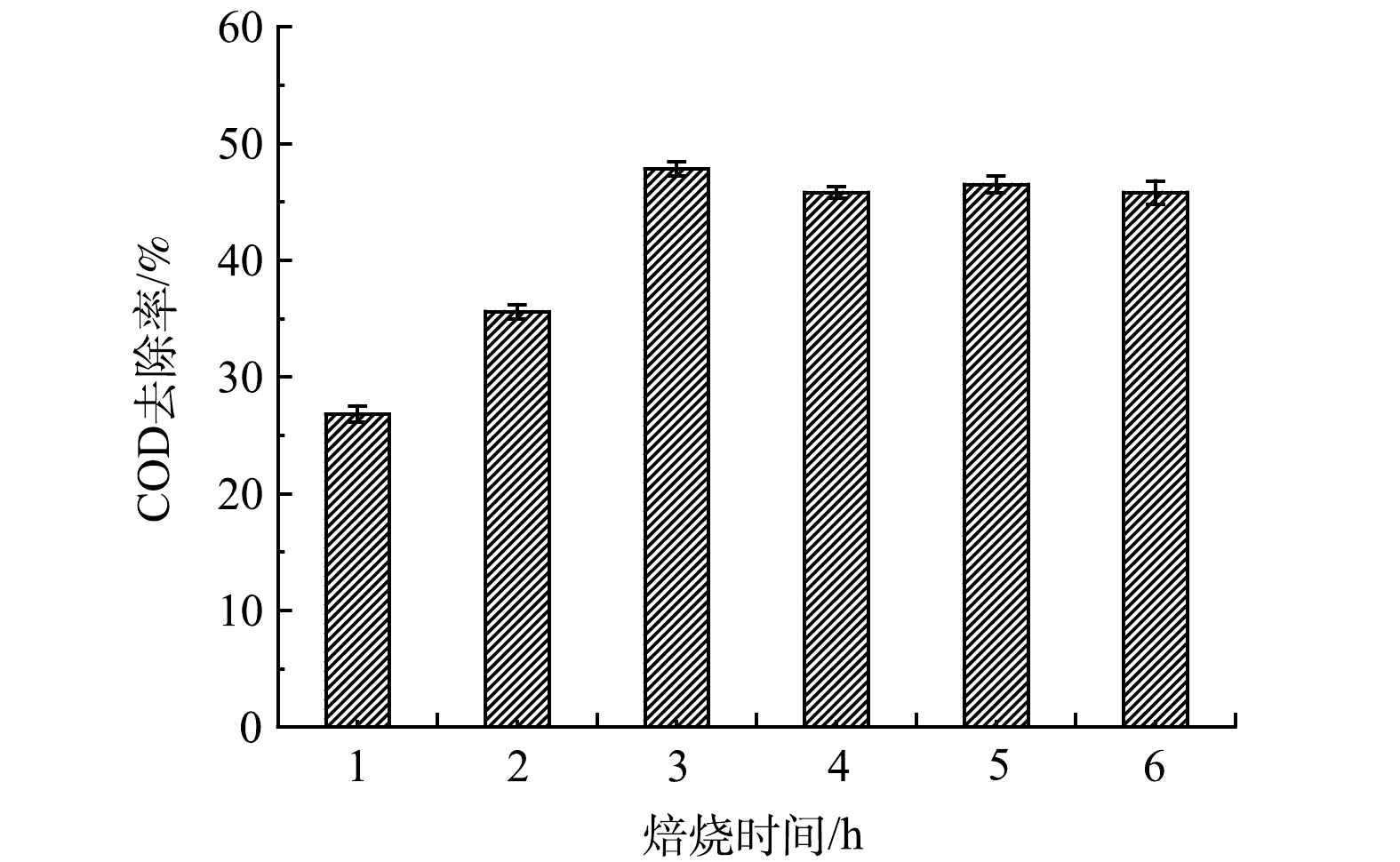
图 4 焙烧时间对催化过滤膜催化活性的影响
Figure 4. Effect of calcination time on the catalytic activity of the catalytic filtration membranes

图 5 陶瓷膜载体及催化过滤膜的SEM分析
Figure 5. SEM images of ceramic membrane carrier and catalytic ceramic filtration membranes
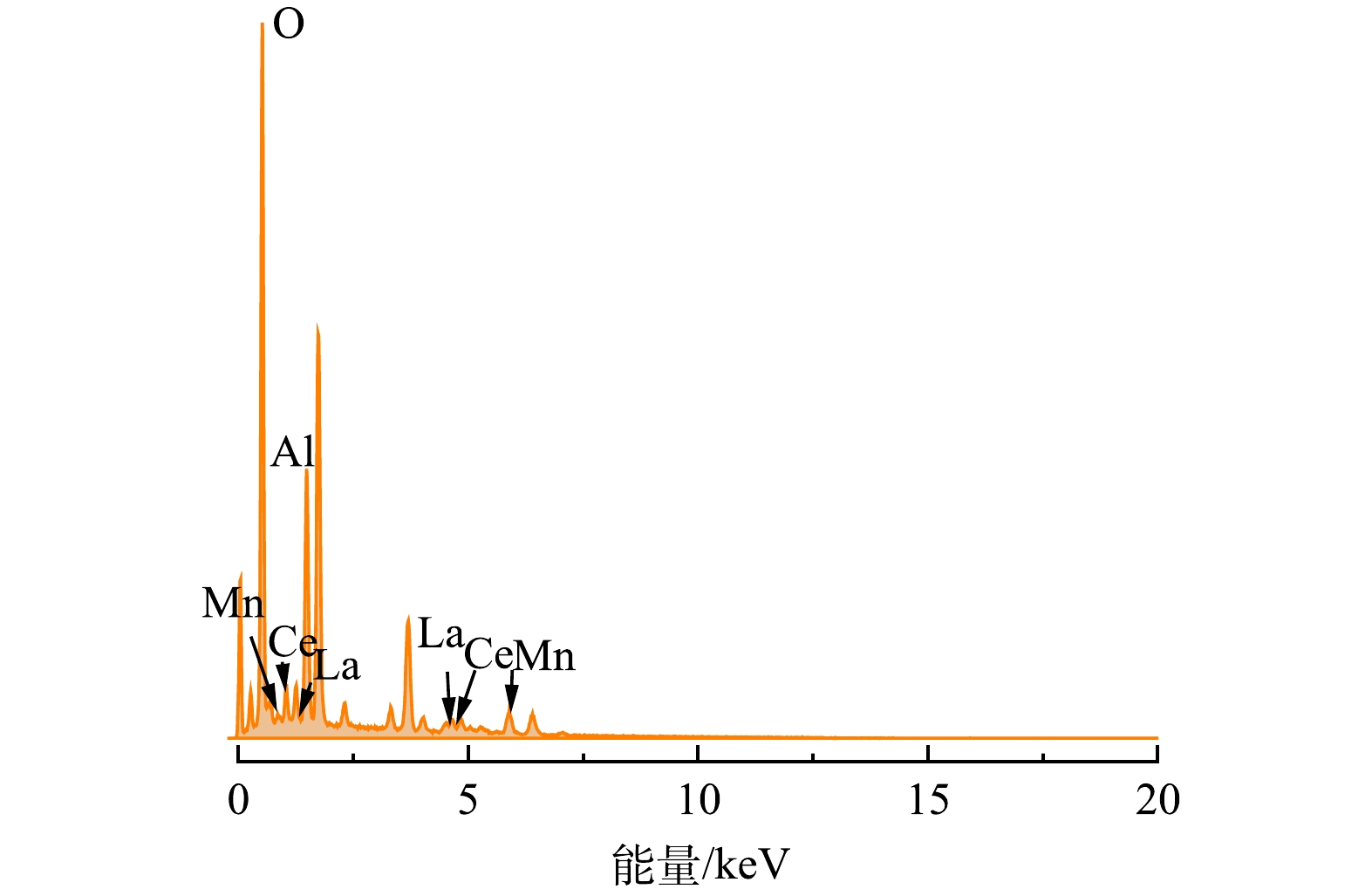
图 6 陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的EDS谱图
Figure 6. EDS spectrums of MnO2-CeO2-La2O3 catalytic ceramic filtration membranes

图 7 陶瓷负载型MnO2-CeO2-La2O3臭氧催化过滤膜的元素分布图
Figure 7. The element distribution of MnO2-CeO2-La2O3 catalytic ceramic filtration membranes
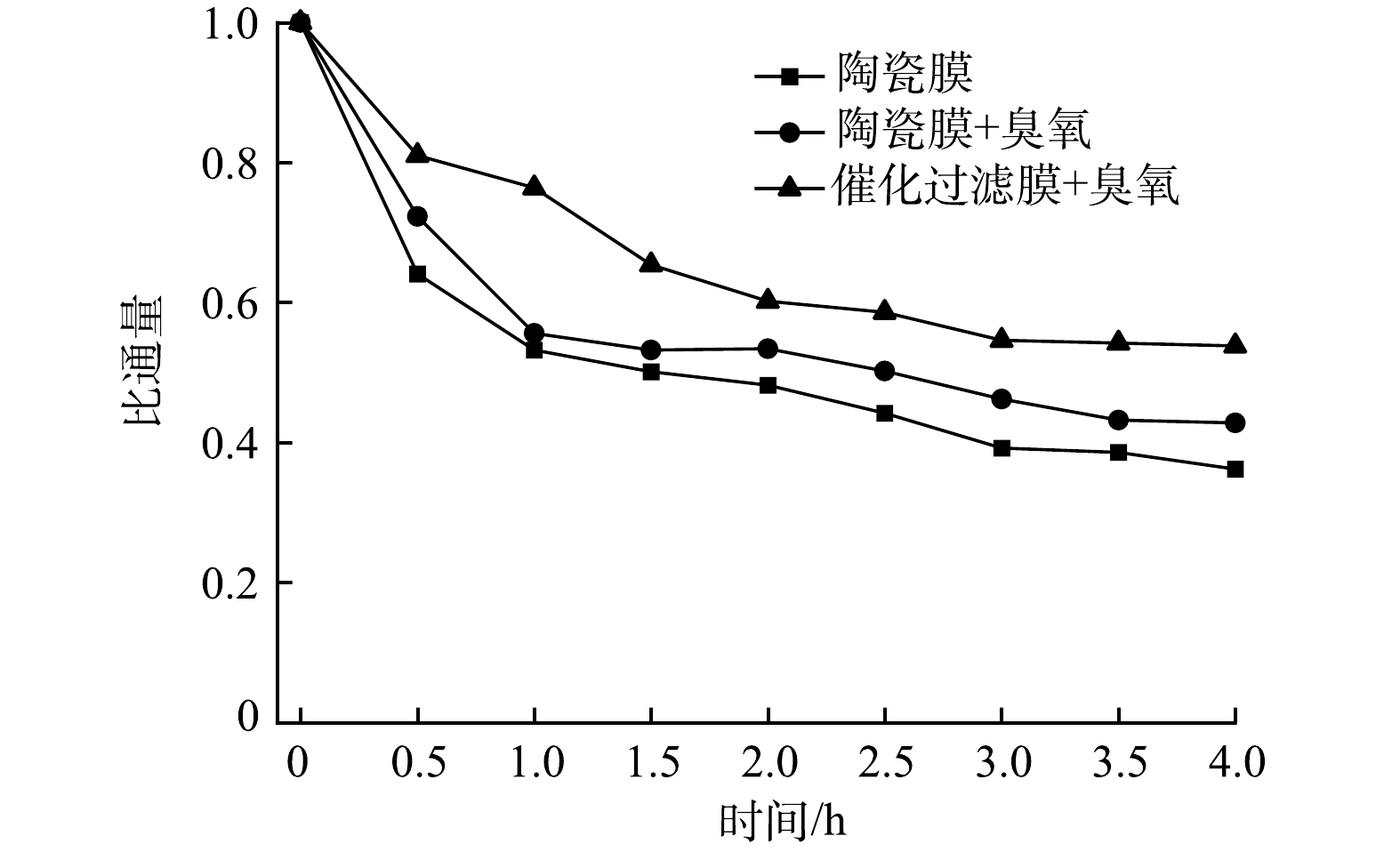
图 9 陶瓷膜催化过滤膜在臭氧催化氧化条件下膜通量变化趋势
Figure 9. Flux variation trend of the ceramic membrane and the catalytic ceramic filtration membranes under the condition of ozone catalytic oxidation
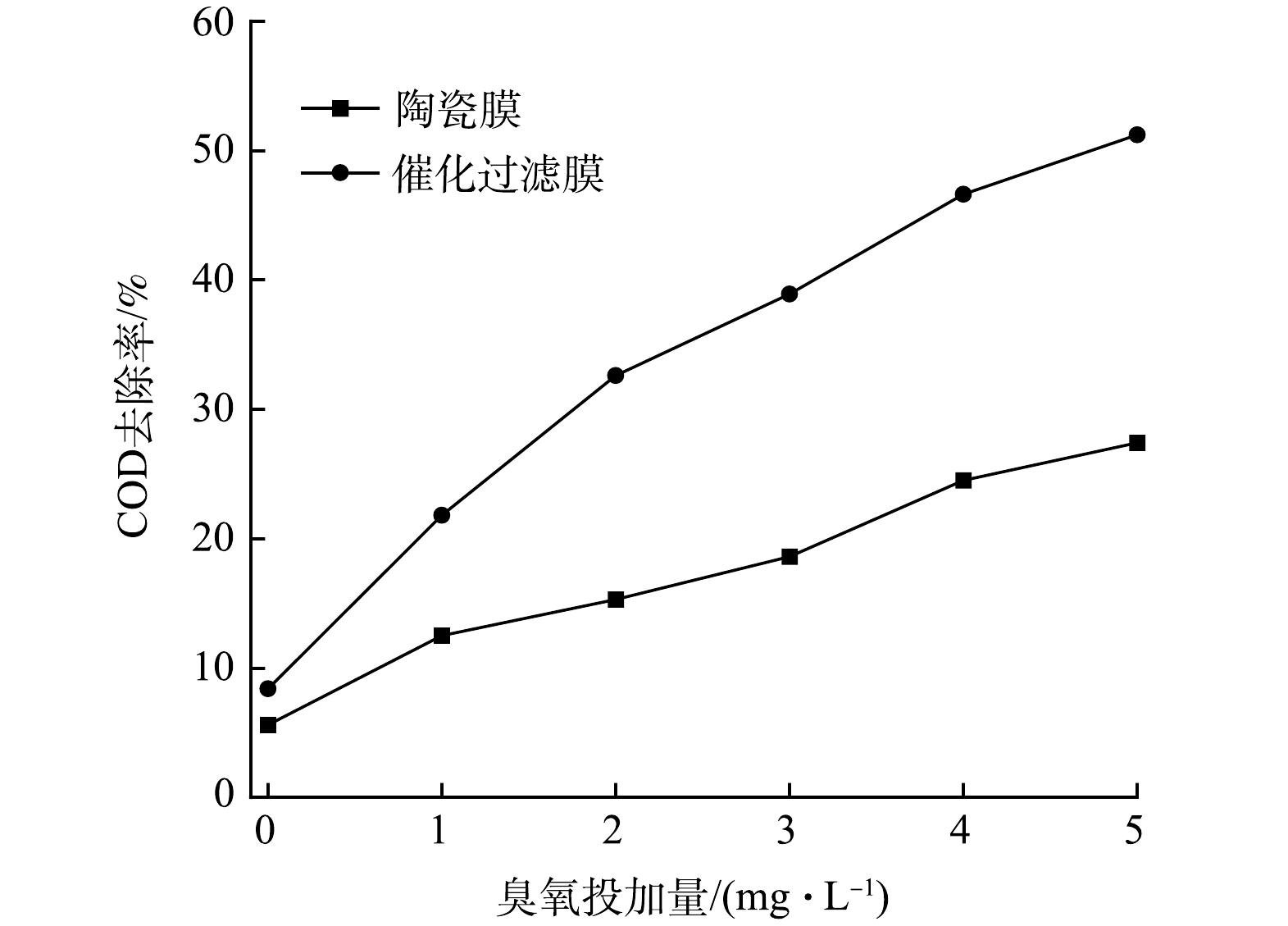
图 10 陶瓷膜和催化过滤膜臭氧催化下水中COD的去除率
Figure 10. COD removal efficiency from wastewater by the ceramic membrane and the catalytic filter membrane
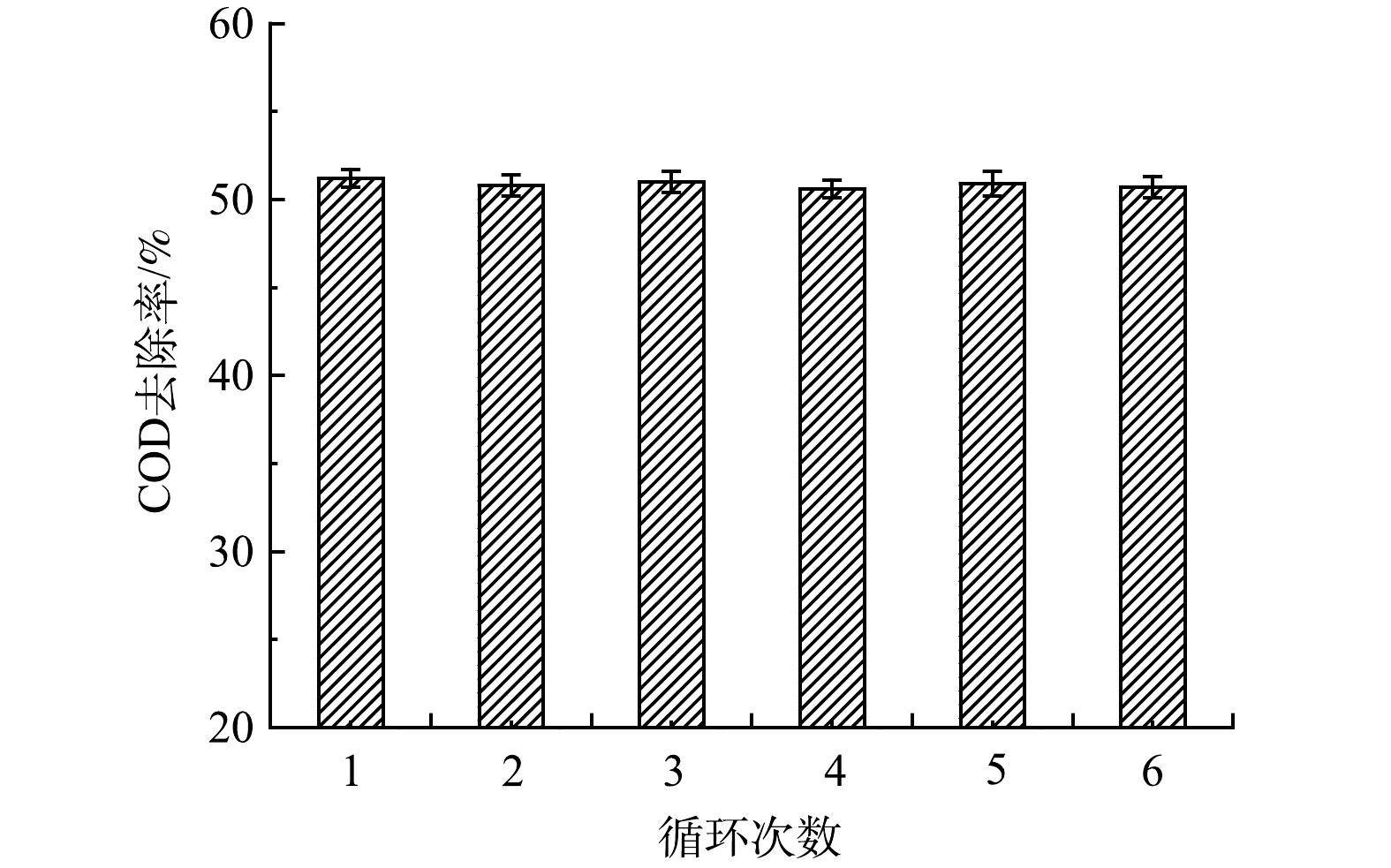
图 11 催化过滤膜的稳定性测试
Figure 11. Stability test of the catalytic ceramic filtration membranes
表 1 催化过滤膜各活性组分元素含量
Table 1. Element content of active components of the catalytic ceramic filtration membranes
元素 质量分数/% 原子分数/% O 48.07 65.11 Al 40.08 32.17 Mn 3.65 1.44 La 4.21 0.66 Ce 3.99 0.62
下载: 导出CSV
表 2 催化过滤膜的性能参数
Table 2. Parameters of the catalytic ceramic filtration membranes
陶瓷膜种类 孔隙率/% 孔径/nm 纯水通量/(L·(m2·h·Mpa)−1) 盐酸可溶率/% 陶瓷膜基体 33 50 2 000 — 负载后的陶瓷膜 26 30 1 260 0.6
下载: 导出CSV
-
[1] 聂颖. 电镀含镍废水的处理[D]. 大连: 大连理工大学, 2018. [2] VINAY K, DWIVEDI S K. Toxicity potential of electroplating wastewater and its bioremediation approaches: A review[J]. Environmental Technology Reviews, 2021, 10(1): 238-254. doi: 10.1080/21622515.2021.1983030 [3] WANG L, LUO Z, HONG Y, et al. The treatment of electroplating wastewater using an integrated approach of interior microelectrolysis and Fenton combined with recycle ferrite[J]. Chemosphere, 2022, 286: 131543. doi: 10.1016/j.chemosphere.2021.131543 [4] RAJORIA S, VASHISHTHA M, SANGAL V K. Treatment of electroplating industry wastewater: A review on the various techniques[J]. Environmental science and pollution research international, 2022, 48(29): 72196-72246. [5] ROXANNE E, JAN R, SOMAYEH K, et al. A reverse osmosis process to recover and recycle trivalent chromium from electroplating wastewater[J]. Membranes, 2022, 12(9): 853. doi: 10.3390/membranes12090853 [6] SUNGEUN L, JIAMING L S, URS V G, et al. Ozonation of organic compounds in water and wastewater: A critical review[J]. Water Research, 2022, 213: 118053. doi: 10.1016/j.watres.2022.118053 [7] BARBAR K H, MARIA Z, JACEK N. Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment[J]. Applied Catalysis B: Environmental, 2003, 46(4): 639-666. doi: 10.1016/S0926-3373(03)00326-6 [8] REBEKKA G, BAPTISTE C, MORENO R, et al. Oxidation of 51 micropollutants during drinking water ozonation: Formation of transformation products and their fate during biological post-filtration[J]. Water Research, 2021, 207: 117812. doi: 10.1016/j.watres.2021.117812 [9] WU Q Y, YANG Z W, DU Y, et al. The promotions on radical formation and micropollutant degradation by the synergies between ozone and chemical reagents (synergistic ozonation) : A review[J]. Journal of Hazardous Materials, 2021, 418: 126327. doi: 10.1016/j.jhazmat.2021.126327 [10] 涂 勇, 张耀辉, 徐 军, 等. 臭氧对化工园区废水厂二级出水的选择性氧化[J]. 环境工程学报, 2015, 9(11): 5295-5300. doi: 10.12030/j.cjee.20151126 [11] XIX Z M , SHENTU J L , LONG Y Y, et al. Effect of dissolved organic matter on selective oxidation of toluene by ozone micro-nano bubble water[J]. Chemosphere, 2023, 325: 138400. [12] WANG Y X, CHEN L L, CHEN C M, et al. Occurrence of both hydroxyl radical and surface oxidation pathways in N-doped layered nanocarbons for aqueous catalytic ozonation[J]. Applied Catalysis B: Environmental, 2019, 254: 283-291. doi: 10.1016/j.apcatb.2019.05.008 [13] 刘雨果, 金 鑫, 许建军, 等. 电凝聚臭氧化耦合工艺的二级出水处理特性与机理研究[J]. 环境科学学报, 2020, 40(11): 3877-3884. doi: 10.13671/j.hjkxxb.2020.0178 [14] SUI M H, SHENG L, LU K X, et al. FeOOH catalytic ozonation of oxalic acid and the effect of phosphate binding on its catalytic activity[J]. Applied Catalysis B: Environmental, 2010, 96: 94-110. doi: 10.1016/j.apcatb.2010.02.005 [15] GRACIA R, CORTES S, SARASA J, et al. TiO2-catalysed ozonation of raw Ebro river water[J]. Water Research, 2000, 34: 1525-1532. doi: 10.1016/S0043-1354(99)00297-3 [16] MOHAMMAD A A, AURELIO H M. Comparative study of ozone and MnO2/O3 effects on the elimination of TOC and COD of raw water at the Valmayor station[J]. Desalination, 2007, 207: 179-183. doi: 10.1016/j.desal.2006.07.010 [17] WANG J B, CHENG J Y, WANG C, et al. Catalytic ozonation of dimethyl phthalate with RuO2/Al2O3 catalysts prepared by microwave irradiation[J]. Catalysis Communications, 2013, 4: 1-5. [18] GAO E H, MENG R Y, JIN Q, et al. Highly effective mineralization of acetic acid wastewater via catalytic ozonation over the promising MnO2/γ-Al2O3 catalyst[J]. Chemical Physics Impact, 2023, 6: 100149. doi: 10.1016/j.chphi.2022.100149 [19] CHEN Y H, HSIEH D C, SHANG N C. Efficient mineralization of dimethyl phthalate by catalytic ozonation using TiO2/Al2O3 catalyst[J]. Journal of Hazardous Materials, 2011, 192: 1017-1025. doi: 10.1016/j.jhazmat.2011.06.005 [20] ZHAO L, MA J, SUN Z Z, et al. Catalytic ozonation for the degradation of nitrobenzene in aqueous solution by ceramic honeycomb supported manganese[J]. Applied Catalysis B: Environmental, 2008, 83: 256-264. doi: 10.1016/j.apcatb.2008.02.009 [21] JUN M J, SUN M H, ZHANG T, et al. Effect of pH on MnOx/GAC catalyzed ozonation for degradation of nitrobenzene[J]. Water Research, 2005, 39(5): 779-786. doi: 10.1016/j.watres.2004.11.020 [22] 邹思宇, 凌二锁, 乐淑荣, 等. 臭氧催化氧化反应器模拟与分析[J]. 化工进展, 2019, 38(9): 3969-3978. doi: 10.16085/j.issn.1000-6613.2018-2476 [23] SCHLICHTER B, MAVROV V, CHMIEL H. Study of a hybrid process combining ozonation and membrane filtration—filtration of model solutions[J]. Desalination, 2003, 156(1): 257-265. [24] BYUN S, DAVIES S H, ALPATOVA A L, et al. Mn oxide coated catalytic membranes for a hybrid ozonation-membrane filtration: Comparison of Ti, Fe and Mn oxide coated membranes for water quality[J]. Water Research, 2011, 45(1): 163-170. doi: 10.1016/j.watres.2010.08.031 [25] 谢宇铭, 张锡辉. 陶瓷膜组合工艺对水中甲硫醚去除效果研究[J]. 环境科学与技术, 2011, 34(8): 131-133. doi: 10.3969/j.issn.1003-6504.2011.08.029 [26] ZOU Y, MALZBENDER J. Development and optimization of porosity measurement techniques[J]. Ceramics International, 2016, 42: 2861-2870. doi: 10.1016/j.ceramint.2015.11.015 [27] 宋颖, 葛圆圆, 韩玉蓉, 等. GPs-PVA/MCE 多功能杂化膜的制备及性能[J]. 化工进展, 2021, 40(11): 6287-6294. [28] 国家环境保护总局. 水和废水监测分析方法. 第4版[M]. 北京: 中国环境科学出版社, 2002. [29] 张耀辉, 涂 勇, 唐 敏, 等. Fe2O3-TiO2-MnO2/Al2O3催化臭氧化催化剂的制备及表征[J]. 中国环境科学, 2016, 36(10): 3003-3009. doi: 10.3969/j.issn.1000-6923.2016.10.023 [30] 叶苗苗, 陈忠林, 沈吉敏, 等. La2O3和CeO2的制备及催化臭氧氧化对氯硝基苯[J]. 哈尔滨工业大学学报, 2009, 41(4): 77-80. doi: 10.3321/j.issn:0367-6234.2009.04.017 [31] FARIA P C, MONTEIRO D C, Pereira M F, et al. Cerium, manganese and cobalt oxides as catalysts for the ozonation of selected organic compounds[J]. Chemosphere, 2009, 74: 818-824. doi: 10.1016/j.chemosphere.2008.10.016 [32] CHENG Y Z, WANG B Y, YAN P W, et al. In-situ formation of surface reactive oxygen species on defective sites over N-doped biochar in catalytic ozonation[J]. Chemical Engineering Journal, 2023, 454: 140232. doi: 10.1016/j.cej.2022.140232 [33] OH S Y, NGUYEN T H. Ozonation of phenol in the presence of biochar and carbonaceous materials: The effect of surface functional groups and graphitic structure on the formation of reactive oxygen species[J]. Journal of Environmental Chemical Engineering, 2022, 10: 107386. doi: 10.1016/j.jece.2022.107386 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2103
- HTML全文浏览数: 2103
- PDF下载数: 64
- 施引文献: 0
