



</td><td class="table_top_border table_bottom_border2" rowspan="2" align="center" valign="middle">吸收负荷/(mol∙mol<sup>−1</sup>)</td><td class="table_top_border table_bottom_border2" rowspan="2" align="center" valign="middle">再生效率</td></tr><tr><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">上层</td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">下层</td><td class="table_bottom_border2" style="width:0.5%"></td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">上层</td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">下层</td><td class="table_bottom_border2" style="width:0.5%"></td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">上层</td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">下层</td></tr></thead>
<tbody><tr><td align="center" valign="middle">2.5 mol·L<sup>−1</sup> AMP</td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">6.23</td><td align="center" valign="middle">0.83</td><td align="center" valign="middle">91.7%</td></tr><tr><td align="center" valign="middle">2.4 mol·L<sup>−1</sup> AMP+0.1 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">9.57</td><td align="center" valign="middle">0.85</td><td align="center" valign="middle">86.5%</td></tr><tr><td align="center" valign="middle">2.3 mol·L<sup>−1</sup> AMP+0.2 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">12.11</td><td align="center" valign="middle">0.88</td><td align="center" valign="middle">79.2%</td></tr><tr><td align="center" valign="middle">2.2 mol·L<sup>−1</sup> AMP+0.3 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">21%</td><td align="center" valign="middle">79%</td><td style="width:0.5%"></td><td align="center" valign="middle">1.6%</td><td align="center" valign="middle">98.4%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">15.43</td><td align="center" valign="middle">0.96</td><td align="center" valign="middle">75.4%</td></tr><tr><td align="center" valign="middle">2.1 mol·L<sup>−1</sup> AMP+0.4 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">40%</td><td align="center" valign="middle">60%</td><td style="width:0.5%"></td><td align="center" valign="middle">2.1%</td><td align="center" valign="middle">97.9%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">18.12</td><td align="center" valign="middle">1.01</td><td align="center" valign="middle">66.2%</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">2.0 mol·L<sup>−1</sup> AMP+0.5 mol·L<sup>−1</sup> TEPA</td><td class="table_bottom_border" align="center" valign="middle">48%</td><td class="table_bottom_border" align="center" valign="middle">52%</td><td class="table_bottom_border" style="width:0.5%"></td><td class="table_bottom_border" align="center" valign="middle">2.4%</td><td class="table_bottom_border" align="center" valign="middle">97.6%</td><td class="table_bottom_border" style="width:0.5%"></td><td class="table_bottom_border" align="center" valign="middle">—</td><td class="table_bottom_border" align="center" valign="middle">20.29</td><td class="table_bottom_border" align="center" valign="middle">1.04</td><td class="table_bottom_border" align="center" valign="middle">57.1%</td></tr></tbody>
<tfoot><tr><td colspan="11" align="left" valign="middle"> 注:DGDE/ H<sub>2</sub>O: 1/1; <i>V</i><sub>solution</sub>: 25 mL; <i>T</i><sub>absorption</sub>: 40 ℃; <i>T</i><sub>desorption</sub>: 120 ℃; <inline-formula><tex-math id="Z-20230913142141">$Q_{{\rm{CO}}_2} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border table_bottom_border2" rowspan="2" align="center" valign="middle">AMP/TEPA</td><td class="table_top_border" colspan="2" align="center" valign="middle">体积比</td><td class="table_top_border" style="width:0.5%"></td><td class="table_top_border" colspan="2" align="center" valign="middle">CO<sub>2</sub>分布</td><td class="table_top_border" style="width:0.5%"></td><td class="table_top_border" colspan="2" align="center" valign="middle">粘度/(mPa·s)</td><td class="table_top_border table_bottom_border2" rowspan="2" align="center" valign="middle">吸收负荷/(mol∙mol<sup>−1</sup>)</td><td class="table_top_border table_bottom_border2" rowspan="2" align="center" valign="middle">再生效率</td></tr><tr><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">上层</td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">下层</td><td class="table_bottom_border2" style="width:0.5%"></td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">上层</td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">下层</td><td class="table_bottom_border2" style="width:0.5%"></td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">上层</td><td class="table_top_border2 table_bottom_border2" align="center" valign="middle">下层</td></tr></thead>
<tbody><tr><td align="center" valign="middle">2.5 mol·L<sup>−1</sup> AMP</td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">6.23</td><td align="center" valign="middle">0.83</td><td align="center" valign="middle">91.7%</td></tr><tr><td align="center" valign="middle">2.4 mol·L<sup>−1</sup> AMP+0.1 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">9.57</td><td align="center" valign="middle">0.85</td><td align="center" valign="middle">86.5%</td></tr><tr><td align="center" valign="middle">2.3 mol·L<sup>−1</sup> AMP+0.2 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">100%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">12.11</td><td align="center" valign="middle">0.88</td><td align="center" valign="middle">79.2%</td></tr><tr><td align="center" valign="middle">2.2 mol·L<sup>−1</sup> AMP+0.3 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">21%</td><td align="center" valign="middle">79%</td><td style="width:0.5%"></td><td align="center" valign="middle">1.6%</td><td align="center" valign="middle">98.4%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">15.43</td><td align="center" valign="middle">0.96</td><td align="center" valign="middle">75.4%</td></tr><tr><td align="center" valign="middle">2.1 mol·L<sup>−1</sup> AMP+0.4 mol·L<sup>−1</sup> TEPA</td><td align="center" valign="middle">40%</td><td align="center" valign="middle">60%</td><td style="width:0.5%"></td><td align="center" valign="middle">2.1%</td><td align="center" valign="middle">97.9%</td><td style="width:0.5%"></td><td align="center" valign="middle">—</td><td align="center" valign="middle">18.12</td><td align="center" valign="middle">1.01</td><td align="center" valign="middle">66.2%</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">2.0 mol·L<sup>−1</sup> AMP+0.5 mol·L<sup>−1</sup> TEPA</td><td class="table_bottom_border" align="center" valign="middle">48%</td><td class="table_bottom_border" align="center" valign="middle">52%</td><td class="table_bottom_border" style="width:0.5%"></td><td class="table_bottom_border" align="center" valign="middle">2.4%</td><td class="table_bottom_border" align="center" valign="middle">97.6%</td><td class="table_bottom_border" style="width:0.5%"></td><td class="table_bottom_border" align="center" valign="middle">—</td><td class="table_bottom_border" align="center" valign="middle">20.29</td><td class="table_bottom_border" align="center" valign="middle">1.04</td><td class="table_bottom_border" align="center" valign="middle">57.1%</td></tr></tbody>
<tfoot><tr><td colspan="11" align="left" valign="middle"> 注:DGDE/ H<sub>2</sub>O: 1/1; <i>V</i><sub>solution</sub>: 25 mL; <i>T</i><sub>absorption</sub>: 40 ℃; <i>T</i><sub>desorption</sub>: 120 ℃; <inline-formula><tex-math id="Z-20230913142141">$Q_{{\rm{CO}}_2} $</tex-math><alternatives><img class="graphic" src="202305103-liuchao-zhouxiaobin_Z-20230913142141.jpg"><img class="graphic" src="202305103-liuchao-zhouxiaobin_Z-20230913142141.png"></alternatives></inline-formula>: 80 mL·min<sup>−1</sup>。</td></tr></tfoot>
</table></div></foreignObject></svg>"></inline-formula>: 80 mL·min<sup>−1</sup>。</td></tr></tfoot>
</table></div></foreignObject></svg>)
-
在众多的碳减排技术中,基于有机胺的化学吸收法具有吸收迅速、选择性好等特点,在CO2捕集应用领域深受青睐[1-2]。然而,传统有机胺吸收剂普遍存在再生能耗高的问题。如质量分数为30%乙醇胺 (MEA) 的再生能耗高达3.7 GJ∙t−1 CO2,其能耗约占碳捕获总能耗的50%~80%[3-5]。高再生能耗限制了有机胺化学吸收法的推广和应用。
近年来,相变吸收剂因其巨大的节能潜力而受到广泛关注。该吸收剂在吸收CO2之前是为均相溶液,吸收CO2后会分离成不相溶的两相,CO2主要富集在其中一相[6-8]。因此,只需将CO2富相用于再生,从而可大幅减少再生溶液的体积,有效降低再生能耗[9-10]。目前,多元伯胺/仲胺和叔胺的混合水溶液是相变吸收剂的主要类型,如二乙烯三胺 (DETA) /五甲基二乙烯三胺 (PMDETA) /H2O、三乙烯四胺 (TETA) /二乙醇胺 (DEA) /H2O、TETA/二甲基乙酰胺 (DMCA) /H2O等[11-14]。由于仅需将富相再生,这些吸收剂的再生能耗可降低至2.5 GJ∙t−1 CO2以下。然而,高活性的多元伯胺或仲胺吸收CO2后形成稳定的氨基甲酸盐,通过常规热再生难以分解,从而导致相变吸收剂再生效率低、再生性能差的问题。
2-氨基-2-甲基-1-丙醇 (AMP) 是一种具有独特空间位阻结构的伯胺,因其CO2吸收容量大、可再生性好,得到了广泛研究[15-17]。KHAN等[18]发现在95 ℃下AMP水溶液的再生效率可达90%,当再生温度提高到109 ℃,再生效率可达97.85%,优于相同条件下MEA的再生效果 (79.9%~85.4%) 。李清方等[19]在MEA水溶液中加入AMP,制备了MEA/AMP复合溶液。研究结果显示,加入AMP后,复合物溶液的再生率高达97.33%,远远优于相同浓度下MEA水溶液的再生效率 (89%) 。鉴于此,以AMP作为主吸收剂构建液-液相变吸收剂,有望大幅提高相变吸收剂的再生性能。然而,AMP特殊的空间位阻结构能提高溶液各组分的互溶度,使吸收剂难以发生相变[20-21]。因此,需添加相变调控剂促使AMP基吸收剂吸收CO2后发生液-液相变。鉴于多元伯胺吸收CO2后能使吸收剂易于分相,可引入少量多元胺作为分相调控剂,促使AMP基吸收剂能相变吸收CO2。
本研究以高活性多元胺四乙烯五胺 (TEPA) 作为相变调控剂,再生性能好的AMP作为主吸收剂,有机溶剂二乙二醇二甲醚 (DGDE) 作为相分离剂,水作为助溶剂,构建新型液-液相变吸收剂用于捕集。通过CO2鼓泡吸收反应器全面考察吸收剂的相变行为及CO2吸收-解吸特性,采用核磁共振碳谱 (13C NMR) 分析吸收剂捕集CO2反应产物种类,利用量子化学计算研究不同物质的极性和相互作用,阐释新型相变吸收剂捕集CO2的反应机理和TEPA调控相变机制,最后通过计算再生过程中的潜热和显热,评估该体系的节能潜力。
-
四乙烯五胺 (TEPA,纯度99%) 、羟乙基乙二胺 (AEEA,纯度99%) 、3-甲氨基丙胺 (MAPA,纯度98%) 、二乙烯三胺 (DETA,纯度99%) 、哌啶 (PZ,纯度99%) 、三乙烯四胺 (TETA,纯度70%) 、2-氨基-2-甲基-1-丙醇 (AMP,纯度99%) 、二乙二醇二甲醚 (DGDE,纯度99%) 、重水 (D2O) 、高纯二氧化碳 (CO2,纯度99.999%) ,实验用水为超纯水。
-
1) CO2吸收。将AMP (2.5~2.0 mol·L−1) 和多元胺/二元胺 (MAPA、AEEA、PZ、DETA、TETA、TEPA) (0.0~0.5 mol·L−1) 溶解在DGDE和水的混合液中 (体积比,4/6、5/5、6/4) 制备不同的吸收剂。随后将25 mL吸收剂置于鼓泡吸收管中,水浴加热至40 ℃。向吸收管通入CO2 (80 mL·min−1) 。通过皂膜流量计记录不同时刻吸收管出口流速。待吸收剂吸收饱和并液-液分相后,依次分析两相的体积百分比、CO2含量和粘度。基于吸收管进、出口流速,计算吸收速率,对吸收速率与吸收时间进行积分得到溶液的吸收负荷。
2) CO2解吸。仅将CO2富相放入油浴锅中进行再生,温度控制为120 ℃。通过皂膜流量计记录不同时刻下溶液的CO2释放速率。在解吸后,将贫相与再生溶液混合,用于下一个吸收循环。再生效率通过再生后溶液的吸收负荷与新鲜溶液的吸收负荷之比计算得到。
3) 13C NMR表征。13C NMR谱图通过Bruker AVANCE 500 MHz核磁共振波谱仪获得。为了锁定信号,将氧化氘D2O溶解在测试溶液的混合物中制备测试样品,即将0.2 mL D2O和0.4 mL溶液充分混合后引入直径为5 mm的核磁管。通过MestReNova软件分析13C NMR谱图中碳原子的化学位移。
4) 量子化学计算。基于密度泛函理论 (DFT) ,利用Gaussian 16软件包中的B3LYP/ 6-311++G (d, p) 基组对不同反应产物的几何构型进行优化计算。研究了溶剂和产物的最佳氢键构型及偶极矩,并使用GaussView 6.0进行可视化分析。
5) 再生能耗估算。再生能耗主要包括三部分:解吸反应热 (Qdes) 、显热 (Qsen) 和潜热 (Qlatent) 。胺类吸收剂间的再生能耗差异主要体现在溶剂的显热和潜热。显热可根据溶液的比热容、再生溶液质量等参数求得,潜热可基于溶剂的蒸发量和蒸发焓计算。
-
吸收过程中CO2吸收率和负荷可通过式 (1) 计算得到。
式中:rab表示CO2的吸收速率,mol·mol−1·min−1;Qin和Qout分别为吸收器入口和出口流速下的CO2流速,m3·min−1;Pact和P0分别表示实际状态和标准状态下的大气压力,kPa;Tact和T0分别是实际状态和标准状态下的温度,K;namine是吸收剂中胺的摩尔量,mol;L0是CO2吸收负载量,mol·mol−1。
再生效率可通过式 (2) 获得。
式中:η表示溶液的再生效率;Ln和L0分别表示第n次再生后的溶液和新鲜溶液的吸收负荷,mol·mol−1。
显热可通过式 (3) 求得。
式中:∆T为吸收塔底部与解吸塔底部解吸溶液的温差 (10 K) ;Cp指溶液的比热容,kJ·K−1·kg−1;
Δmco2 和msolution分别表示再生过程中CO2解吸的质量和吸收剂的质量,kg。潜热可以通过式 (4) 计算。
式中:Δmi为挥发性成分的质量,g;Hivap表示挥发性组分的蒸发焓,kJ·kg−1;TR、TB和TC分别指挥发性组分的解吸温度、正常沸点和临界沸点,K。
-
1) 相变调控剂的筛选。使用6种二元胺/多元胺 (MAPA、AEEA、PZ、DETA、TETA、TEPA) 作为相变调控剂,研究吸收剂在不同调控剂调控下的相变情况。其中,吸收剂AMP和相变调控剂的总浓度为2.5 mol·L−1,H2O与DGDE的体积比为1/1。吸收剂吸收饱和后的相变情况如图1所示。当吸收饱和后,所有吸收剂均能发生液-液相分离,但不同调控剂对相变的调控性能有所差异。当调控剂为PZ时,吸收剂的下层体积分数高达94%,而使用TEPA作为调控剂,下层体积仅占总体积52%。这可能与调控剂的氨基数量有关。氨基数量越多 (如TEPA为5个氨基) ,反应产物间的氢键数量越多,相互作用越强,产物越容易浓缩聚集,而氨基数量越少 (如PZ为2个氨基) ,产物不易浓缩聚集,则下层体积越大。此外,对6种吸收剂上下层的CO2分布进行测定,发现96%以上的CO2都富集于下层,因此,下层为CO2富相。下层体积越小,则吸收剂所需再生的体积越小,越有利于降低再生能耗。
此外,考察了几种相变吸收剂的CO2吸收性能 (吸收速率、吸收负荷) 和CO2解吸性能 (循环负荷) ,并以质量分数30% 的MEA溶液作为对比。如图2所示,在初始吸收阶段 (前20 min) ,质量分数30% 的MEA的CO2吸收速率较高,但几种相变吸收剂的最终吸收负荷均高于质量分数30% 的MEA,且其循环负荷也显著高于质量分数30% 的MEA,这表明几种相变吸收剂均具有良好的吸收和再生性能。不同相变调控剂的引入对吸收剂的CO2吸收和解吸性能影响不同。其中,TEPA调控的吸收剂的CO2吸收速率、吸收负荷和循环负荷均高于其他吸收剂。这主要是因为TEPA具有多个活性氨基基团 (2个伯氨基和3个仲氨基) ;氨基数量越多,越有利于CO2的吸收;吸收的CO2量越多,解吸时能释放的CO2量也越多。因此,基于相变吸收剂的CO2吸收-解吸性能,选择TEPA作为最佳的相变调控剂。
2) 吸收剂组分配比优化。根据筛选结果,选择TEPA为最佳相变调控剂。不同相变调控剂浓度对吸收剂的分相特性、吸收性能和再生性能也会产生影响[22]。因此,考察了不同TEPA浓度下吸收剂的分相特性、吸收负荷及下层 (CO2富相) 的再生性能。控制H2O与DGDE的体积比为1∶1,AMP和TEPA的总浓度为2.5 mol·L−1,制备了6种吸收剂:2.5 mol·L−1 AMP、2.4 mol·L−1 AMP+0.1 mol·L−1 TEPA、2.3 mol·L−1 AMP+0.2 mol·L−1 TEPA,2.2 mol·L−1 AMP+0.3 mol·L−1 TEPA、2.1 mol·L−1 AMP+0.4 mol·L−1 TEPA和2.0 mol·L−1 AMP+0.5 mol·L−1 TEPA。表1列出了6种吸收剂吸收CO2后上下层体积比、CO2分布、粘度、吸收负荷和再生效率等数据。结果表明,当加入的TEPA浓度低于0.2 mol·L−1时,吸收剂吸收CO2后仍为均相溶液。当TEPA浓度为0.3 mol·L−1时,吸收剂开始分相,且随着TEPA浓度的增加,吸收负荷逐渐增大,同时下层体积逐渐减小。下层体积越小,需再生的溶液量越少,越有利于节能。然而,随着下层体积越小,粘度则越大。此外,吸收剂的再生效率随TEPA浓度的增加而降低,这可能是因为TEPA能和CO2反应生成稳定的氨基甲酸盐产物,该产物较难分解。为确保吸收剂发生相变时下层 (CO2富相) 有更低的粘度和更好的吸收、再生性能,TEPA的浓度选择为0.3 mol·L−1。
当TEPA浓度为0.3 mol·L−1,吸收剂可发生相变,但此时富相比例高达79%,不利于减少再生体积。通过改变DGDE与H2O的体积比可调整上下层体积比例。如图3(a)所示,当DEDG/H2O的比例从4∶6变为7∶3时,下层的体积百分比从100%下降到30.0%。图3(b)给出了不同DGDE/H2O比值下CO2吸收速率和CO2负荷随时间变化的曲线。增加DEDG的比例有利于提高初始吸收率,但也会导致最终的CO2负荷轻微下降。前者是因为DGDE浓度增加促进了CO2在吸收剂中的物理溶解和扩散,因而提高了CO2吸收速率[23]。后者是由于水的减少可能限制CO2的水合反应,使CO2吸收量降低[24]。理论上,下层体积越小,送入再生塔的再生溶剂就越少,对降低再生能耗越有利[25]。但下层过小的体积会导致其粘度大增。综合考虑上下层比例、粘度、吸收量和再生效率,最终确定AMP和TEPA的浓度分别为2.2 mol·L−1和0.3 mol·L−1,DEDG与H2O的最佳比例为6∶4。在该配比下,吸收剂的吸收负荷高达0.88 mol∙mol−1,下层体积仅占总体积51%。
3) CO2吸收-解吸循环。CO2循环吸收-解吸性能是评估吸收剂重复使用稳定性的重要指标。由于AMP-DGDE-TEPA吸收CO2后发生分相,CO2富集在其中一相,故仅将CO2富相用于解吸,解吸后再与先前的贫相混合进行CO2循环吸收。如图4所示,AMP-DGDE-TEPA的吸收负荷在第一次循环吸收后由0.88降至0.65 mol∙mol−1。这可能是由于TEPA与CO2反应生成了较难分解的氨基甲酸盐,使TEPA难以再生,从而降低了吸收剂后续的CO2吸收能力。随着循环次数的增加,CO2吸收负荷仅略微下降。在第7次循环吸收,AMP-DGDE-TEPA的CO2吸收负荷仍能保持0.63 mol∙mol−1,约为质量分数30%MEA水溶液的3倍。这表明该相变吸收剂具有良好的CO2循环吸收稳定性。
-
1) 13C NMR分析。为明确AMP-DGDE-TEPA相变吸收剂的CO2吸收机理和TEPA调控相变机制,采用13C NMR分析有、无TEPA条件下吸收剂吸收CO2过程产物的变化情况,结果如图5(a)、(b)所示。图5(c)给出了吸收剂中主要物质的分子结构。
由图5(a)可见,AMP-DGDE吸收剂 (无TEPA) 在吸收CO2前,其谱图中检测到AMP和DGDE的碳信号。AMP的碳信号会发生化学位移,ppm为核磁共振中物质吸收峰的化学相对位移量。在吸收CO2后,其谱图中的160.4 ppm处出现一个新的峰信号,该信号为碳酸氢盐/碳酸盐 (HCO3−/CO32−) 物质的碳信号[26]。HCO3−/CO32−物质的生成是由于AMP与CO2反应形成的不稳定产物水解所致。同时,AMP的碳信号发生了轻微的偏移,这可能是质子化AMP产物形成的结果[27]。AMP-DGDE解吸CO2后,160.4 ppm处碳信号消失,且AMP的碳信号又回到CO2吸收前时的位置。这说反应产物很容易分解、AMP易于再生。对于A-D-T吸收剂,在CO2吸收前,除了AMP和DGDE的碳信号,还检测到微弱的TEPA碳信号,这可能是TEPA浓度较低的缘故。在吸收CO2后,AMP-DGDE-TEPA发生分相。吸收剂上层谱图中只检测到强烈的DGDE碳信号峰,而在下层谱图中除了存在AMP碳信号和较弱的DGDE碳信号外,在163.9和160.2 ppm处出现2个新的峰信号,分别对应氨基甲酸盐和HCO3−/CO32−物质的羰基碳信号[26, 28]。此结果表明,AMP-DGDE-TEPA分相后,上层为CO2贫相,主要包含DGDE,而吸收的CO2主要以氨基甲酸盐和HCO3−/CO32−物质的形式富集在下层中。由图5(a)可知,AMP吸收CO2的反应产物以HCO3−/CO32−形式存在,则氨基甲酸盐为TEPA与CO2的反应产物。当CO2解吸后,AMP-DGDE-TEPA下层谱图中的HCO3−/CO32−信号几乎完全消失,而氨基甲酸盐的碳信号仍然清晰可见。这表明HCO3−/CO32−比氨基甲酸盐更容易解吸,最后仍有部分CO2以氨基甲酸盐的形式存在未被解吸。
2) 反应机理。根据13C NMR结果,可推测AMP-DGDE-TEPA相变吸收剂吸收CO2的反应机理。溶液中存在两种胺,即AMP和TEPA。AMP和TEPA均能与CO2反应,首先形成两性离子,随后两性离子脱质子反应转化为基甲酸盐[29-30]。
式中B为溶剂中的碱 (如H2O、AMP或TEPA) ,能使不稳定的两性离子脱质子生成氨基甲酸盐。但由于AMP分子具有空间位阻结构,AMP-氨基甲酸盐在水溶液中很不稳定,容易水解生成HCO3−[31-33] (式 (12)),TEPA吸收CO2后可形成稳定的TEPA-氨基甲酸盐。
AMP-DGDE-TEPA相变吸收剂解吸CO2时,HCO3−类物质最先开始分解。此时,HCO3−作为质子受体,夺取质子化胺 (AmineH+,即AMPH+、TEPAH+) 中的质子后分解产生CO2。解吸反应如式 (13) 所示。
AMP作为主吸收剂,与CO2反应生成大量HCO3−。HCO3−浓度越高,吸收剂的CO2解吸速率越快。此外,本研究组之前的工作表明,相比于氨基甲酸盐,HCO3−分子彼此之间不易形成氢键,因而吸收剂中CO2产物以HCO3−离子的形式存在也有利于降低体系的粘度[34]。
在解吸后期,CO2的释放主要来自于TEPA-氨基甲酸盐的分解。氨基甲酸盐在解离过程中,首先接受来自质子化胺的自由质子形成两性离子,随后分解为胺和CO2[35-36]。其反应式如式 (14) 所示。
3) 相变机理。基于13C NMR分析结果,相比于A-D体系,A-D-T体系吸收CO2后新增TEPA的衍生产物。因而,可推测TEPA的衍生产物可能是引起相变的主要因素。根据已有研究,吸收剂相变的发生与组分极性的转变有密切关系,而物质的极性与其偶极矩呈正相关[37-38]。鉴于此,本研究通过量子化学计算研究新鲜溶液和CO2吸收饱和时吸收体系中各组分的偶极矩。由于TEPA具有多个氨基位点,吸收CO2后可形成多种不同结构的产物,本文仅讨论伯氨基位点反应后的情况 (即伯氨基位点形成的质子化胺和氨基甲酸盐)。由图6可知,在新鲜溶液中,AMP、TEPA、DGDE和H2O的偶极矩较为接近。CO2吸收后,AMP和TEPA转化为质子化胺和氨基甲酸盐产物 (AMPH+、TEPAH+和
TEPACO−2 ) ,同时生成HCO3−/CO32−。除CO32−外,其他反应产物与新鲜溶液各组分相比都表现出较高的偶极矩,尤其是TEPAH+和TEPACO−2 。物质的偶极矩越大,极性越高。高极性物质的形成可打破吸收剂原有的均相状态,从而引起分相。此外,通过量子化学计算研究了溶剂 (DGDE和H2O) 与产物之间的氢键作用 (图7) 。除了TEPACOO−与DGDE无法形成氢键,各反应产物均可与H2O和DGDE形成氢键。总体而言,反应产物与H2O间的氢键长度均比与DGDE之间的氢键短。氢键长度越短,分子间的相互作用越强[39]。
由此总结AMP-DGDE-TEPA吸收剂的相变机制。在反应前,各组分的极性相似,溶液处于均化状态。吸收CO2后,各产物的极性与它们的前体物质相比明显提高。其中,TEPA与CO2反应生成了高极性的TEPAH+和
TEPACO−2 ,打破了溶液原有的均相状态,促使溶液发生相变。相比于DGDE,H2O与各反应产物具有更强的相互作用力,其更倾向于聚集形成CO2富相,而DGDE则单独形成一相 (CO2贫相) 。由于反应产物和H2O的混合物密度比DGDE大,因此,DGDE迁移至溶液上层,而反应产物和H2O停留在溶液下层。A-D-T吸收剂的相变过程如图8所示。 -
吸收剂的再生能耗和溶剂成本均是重要的CO2捕集经济指标。吸收剂的再生能耗由解吸反应热、显热和潜热三部分组成[40]。其中,解吸反应热由胺与CO2的反应特性决定,但不同胺类吸收剂的反应热差异不大。吸收剂间再生能耗差异主要体现在溶剂的显热和潜热。表2比较了AMP-DGDE-TEPA与已报道的几种CO2吸收剂 (包括相变吸收剂[41-42]、单原子溶剂[43]和离子液体[44]) 的显热、潜热和试剂价格,并以质量分数为30%的MEA水溶液作为参考。AMP-DGDE-TEPA的显热和潜热分别为0.33和0.43 GJ∙t−1 CO2,较质量分数为30% 的MEA降低57%和65%,同时也略低于其他CO2吸收剂,这说明AMP-DGDE-TEPA具有较大的节能潜力。从溶剂成本来看,AMP-DGDE-TEPA的价格稍高于其他吸收剂。但AMP-DGDE-TEPA具有较低的再生能耗和良好的循环使用稳定性,在实际使用中有利于降低CO2捕集成本。
-
1) 以AMP为主吸收剂,DGDE和H2O为溶剂,TEPA为相变调控剂,设计了一种具有良好再生性能的新型相变CO2吸收剂 (AMP-DGDE-TEPA) 。经过体系优化,AMP-DGDE-TEPA的最佳配方为2.2 mol·L−1 AMP+0.3 mol·L−1 TEPA,DGDE∶H2O=6∶4。在该配方下,吸收剂的CO2吸收负荷高达0.88 mol∙mol−1,富相体积仅占吸收剂总体积51%。经7次循环吸收,AMP-DGDE-TEPA的吸收负荷仍能保持0.63 mol∙mol−1,再生效率为71.6%。2) 13C NMR结果表明,AMP与CO2反应产物主要为碳酸氢盐,易于分解,而TEPA转化为稳定的质子化胺和氨基甲酸盐。量子化学计算表明,引入TEPA后生成的质子化TEPA和氨基甲酸盐具有高极性,打破了溶液原有的均相状态,从而促使溶液发生相变。相比于DGDE,H2O和极性反应产物具有更强的相互作用力,这些物质聚集形成CO2富相,而DGDE则单独形成CO2贫相。3) 能耗计算结果表明,AMP-DGDE-TEPA的再生显热和潜热仅为0.33和0.43 GJ∙t−1 CO2,说明其具有良好的节能潜力。
TEPA调控AMP-DGDE水溶液相变吸收CO2性能及机理
Performance of phase change CO2 absorption in TEPA-regulated AMP-DGDE aqueous solution and its mechanism
-
摘要: 相变吸收剂在降低CO2捕集能耗方面具有较大优势,但现有吸收剂普遍存在再生性能差的问题。基于2-氨基-2-甲基-1-丙醇 (AMP) 的吸收剂再生性能优异,但通常难以发生相变。利用四乙烯五胺 (TEPA) 作为相变调控剂引入AMP-二乙二醇二甲醚 (DGDE) 水溶液,构建了具有良好再生性能的新型相变吸收剂AMP-DGDE-TEPA。在最佳配比下,AMP-DGDE-TEPA的吸收负荷可达0.88 mol∙mol−1,其中97.6%的CO2富集于溶液下层,下层体积仅占总体积51%。经7次吸收-解吸循环,吸收剂的吸收负荷仍能保持0.63 mol∙mol−1,再生效率为71.6%。13C核磁共振结果表明,AMP与CO2反应生成易于分解的碳酸氢盐,因而吸收剂具有良好的再生性能;而TEPA的引入可使系统中生成稳定的质子化TEPA和氨基甲酸盐。质子化TEPA和氨基甲酸盐具有高极性,可打破吸收剂原有的均相状态,促使吸收剂发生液-液相变。相比于DGDE,H2O和极性反应产物之间具有更强的相互作用力,这些物质聚集形成CO2富相,而DGDE则单独形成CO2贫相。此外,AMP-DGDE-TEPA的再生显热和潜热仅为0.33和0.43 GJ∙t−1 CO2,具有良好的节能潜力。该研究结果可为高效低能耗碳捕集材料的制备提供参考。
-
关键词:
- CO2捕集 /
- 相变吸收剂 /
- 2-氨基-2-甲基-1-丙醇 /
- 四乙烯五胺 /
- 相变机理
Abstract: Phase change absorbents have great advantages in reducing the energy consumption of CO2 capture, but the existing absorbers generally suffer disadvantages in regenerability. 2-Amino-2-methyl-1-propanol (AMP)-based absorbents have excellent regeneration performance but are generally difficult to undergo phase change. In this study, tetraethylenepentamine (TEPA) was introduced as a phase change regulator into AMP-diethylene glycol dimethyl ether (DGDE) aqueous solution, aiming to develop a new AMP-based phase change absorbent with good regeneration performance, namely, AMP-DGDE-TEPA. Under the optimal ratio, the absorption loading of AMP-DGDE-TEPA reached 0.88 mol∙mol-1, of which 97.6% of the absorbed CO2 was enriched in the lower phase with only 51% of the total volume. After 7 cycles of absorption-desorption, the absorption loading of AMP-DGDE-TEPA remained at 0.63 mol∙mol-1, and the corresponding regeneration efficiency was 71.6%. The species in the CO2-loaded A-D-T solution were identified by 13C nuclear magnetic resonance technique. AMP reacted with CO2 to produce bicarbonate species that was easy to decompose, ensuring the good regenerability of AMP-DGDE-TEPA. Meanwhile, with the introduction of TEPA, stable protonated TEPA and TEPA-carbamate formed in the AMP-DGDE-TEPA system. The results of quantum chemical calculations indicated that protonated TEPA and TEPA-carbamate were highly polar, which broke the original assimilation state of AMP-DGDE-TEPA and drove it to undergo phase change. Since H2O had strong affinity to polar reaction products, they gathered together to form the CO2-rich phase, while DGDE showed a relatively weak affinity to polar reaction products and was solely separated from the solution to form the CO2-lean phase. During desorption, the sensible and latent heat of AMP-DGDE-TEPA was calculated to be 0.33 and 0.43 GJ∙t-1 CO2, which indicated the good energy-saving potential of AMP-DGDE-TEPA. -

图 1 不同相变调控剂对AMP-DGDE水溶液的相变调控
Figure 1. Phase-change behavior of AMP-DGDE aqueous solution with different regulators
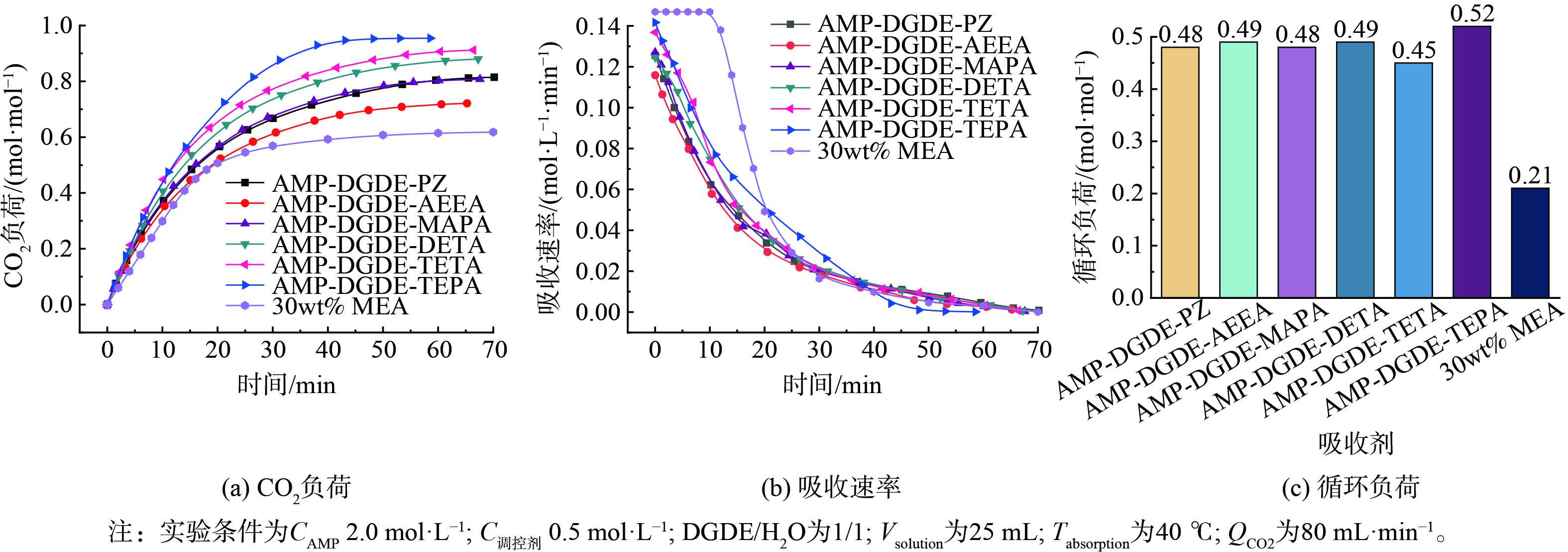
图 2 不同相变调控剂对AMP-DGDE水溶液CO2吸收和解吸性能的影响
Figure 2. The effect of different regulators on CO2 absorption and desorption performance of AMP-DGDE aqueous solution

图 3 不同DGDE/H2O体积比下吸收剂的CO2吸收性能
Figure 3. CO2 absorption performance of absorbents with different volume ratios of DGDE to H2O
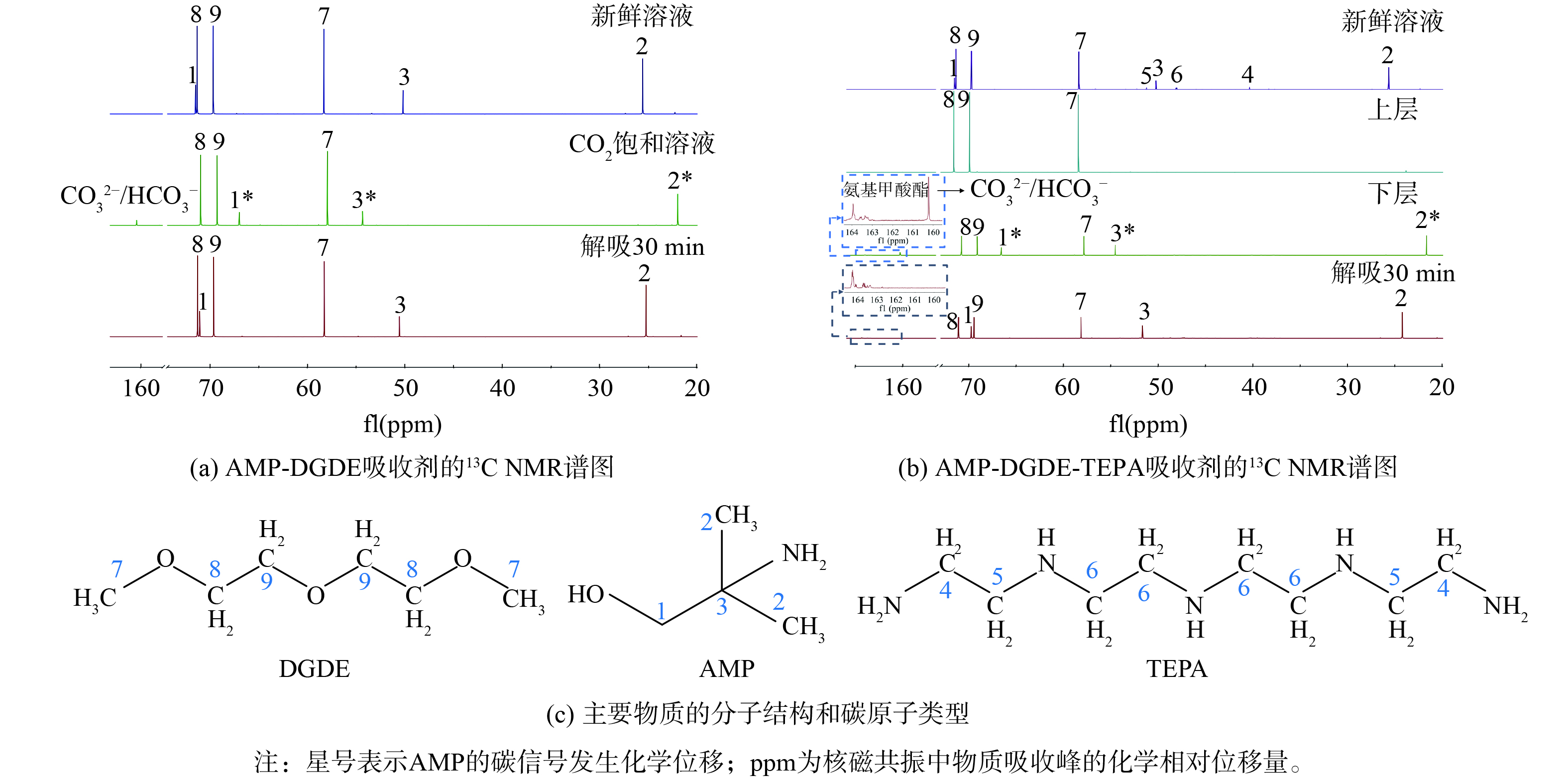
图 5 吸收剂的13C NMR谱图和主要物质的分子结构
Figure 5. 13C NMR spectra of the absorbent and molecular structures of main species
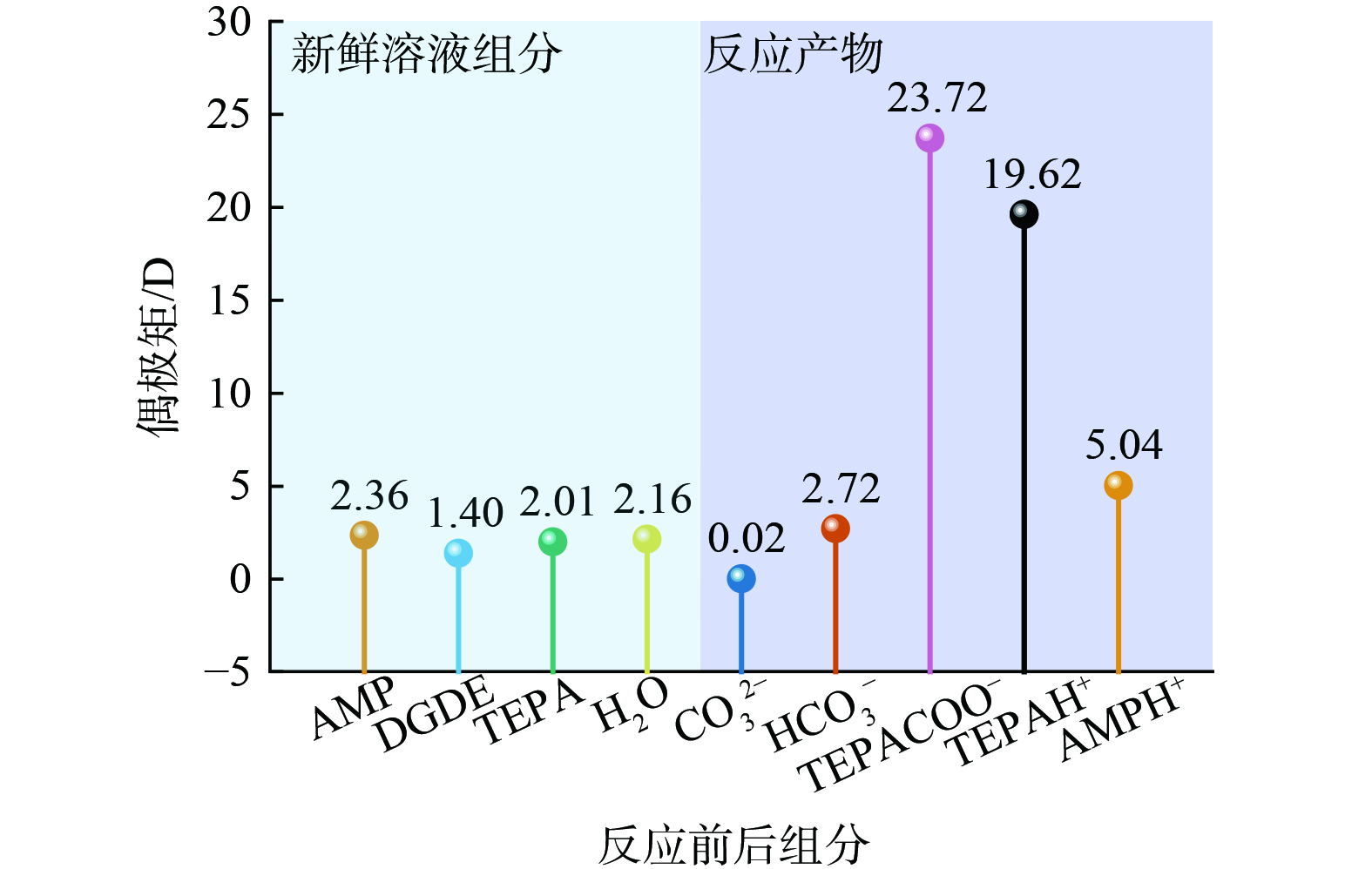
图 6 反应前后各组分的偶极矩
Figure 6. Dipole moments of different components before and after the reaction
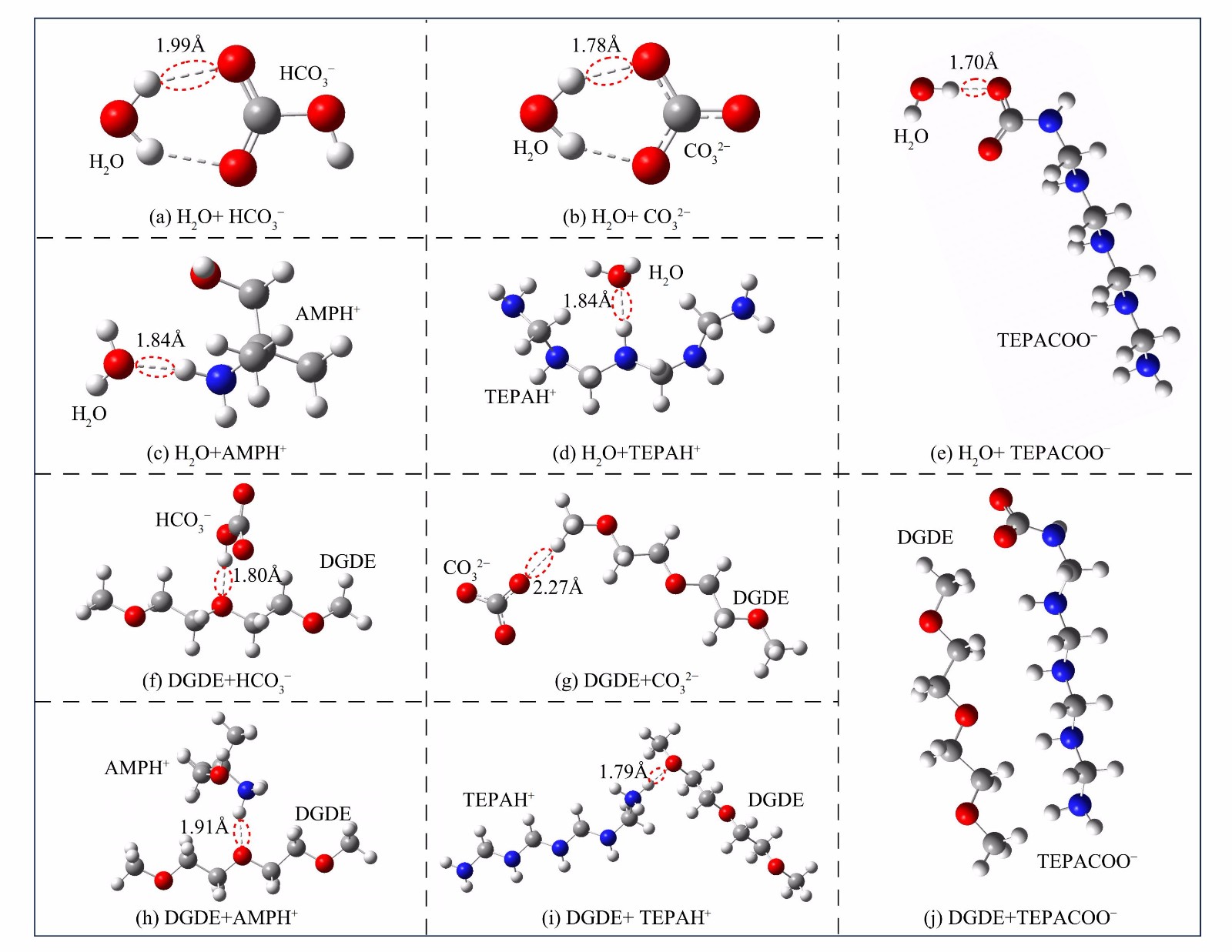
图 7 反应产物与H2O和DGDE的氢键作用
Figure 7. Hydrogen bonding of reaction products with H2O and DGDE

图 8 TEPA调控AMP-DGDE水溶液相变吸收CO2示意图
Figure 8. Schematic diagram of the phase separation for CO2 absorption in the TEPA-regulated AMP-DGDE solution
表 1 不同AMP与TEPA摩尔比例下吸收剂饱和时的分相特性、CO2负荷及再生效率
Table 1. Phase separation characteristics, CO2 loading and regeneration efficiency of the CO2-saturated solutions with different molar ratios of AMP to TEPA
AMP/TEPA 体积比 CO2分布 粘度/(mPa·s) 吸收负荷/(mol∙mol−1) 再生效率 上层 下层 上层 下层 上层 下层 2.5 mol·L−1 AMP — 100% — 100% — 6.23 0.83 91.7% 2.4 mol·L−1 AMP+0.1 mol·L−1 TEPA — 100% — 100% — 9.57 0.85 86.5% 2.3 mol·L−1 AMP+0.2 mol·L−1 TEPA — 100% — 100% — 12.11 0.88 79.2% 2.2 mol·L−1 AMP+0.3 mol·L−1 TEPA 21% 79% 1.6% 98.4% — 15.43 0.96 75.4% 2.1 mol·L−1 AMP+0.4 mol·L−1 TEPA 40% 60% 2.1% 97.9% — 18.12 1.01 66.2% 2.0 mol·L−1 AMP+0.5 mol·L−1 TEPA 48% 52% 2.4% 97.6% — 20.29 1.04 57.1% 注:DGDE/ H2O: 1/1; Vsolution: 25 mL; Tabsorption: 40 ℃; Tdesorption: 120 ℃; QCO2  下载: 导出CSV
下载: 导出CSV
表 2 不同类型吸收剂的再生显热、潜热及试剂价格的比较
Table 2. Comparison of the Qsen and Qlatent of different absorbents and the prices of chemicals used in absorbents
吸收剂 显热/ (GJ∙t−1 CO2) 潜热/ (GJ∙t−1 CO2) 化学试剂 价格/ (元∙t−1) 质量分数30%的 MEA (本研究) 0.77 1.24 MEA 8 500 AMP-DGDE-TEPA (本研究) 0.33 0.43 AMPDGDETEPA 65 00020 00063 000 MEA-环丁砜-H2O (相变吸收剂) [41] 0.32 0.63 MEA环丁砜 8 50020 000 TETA-四甲基丙二胺 (相变吸收剂) [42] 0.32 0.49 TETA四甲基丙二胺 56 00058 000 铜基单原子溶液 (单原子溶剂) [43] 0.25 - 三水合硝酸铜N,N-二甲基甲酰胺MEA 26 0004 5008 500 [DETAH][Tz] (离子液体) [44] 1.09 1.34 DETA三唑钠 (Tz) 29 00062 000
下载: 导出CSV
-
[1] BARZAGLI F, MANI F, PERUZZINI M. A comparative study of the CO2 absorption in some solvent-free alkanolamines and in aqueous monoethanolamine (MEA)[J]. Environmental Science & Technology, 2016, 50(13): 7239-7246. [2] 杨菲, 刘苗苗, 陆诗建, 等. 适用于烟气CO2捕集的相变吸收剂研究进展[J]. 低碳化学与化工, 2023, 48(02): 113-120. [3] LI J, YOU C, CHEN L, et al. Dynamics of CO2 absorption and desorption processes in alkanolamine with cosolvent polyethylene glycol[J]. Industrial & Engineering Chemistry Research, 2012, 51(37): 12081-12088. [4] 梁怀勇, 周小斌, 姚靖, 等. 羟乙基乙二胺/二甲基亚砜溶液高效捕集二氧化碳的性能及机理[J]. 环境化学, 2021, 40(06): 1895-1902. doi: 10.7524/j.issn.0254-6108.2020122102 [5] 周小斌. 新型两相胺吸收剂捕集二氧化碳研究[D]. 厦门: 华侨大学, 2019. [6] XU M, WANG S, XU L. Screening of physical-chemical biphasic solvents for CO2 absorption[J]. International Journal of Greenhouse Gas Control, 2019, 85: 199-205. doi: 10.1016/j.ijggc.2019.03.015 [7] ARONU U E, HOFF K A, SVENDSEN H F. CO2 capture solvent selection by combined absorption-desorption analysis[J]. Chemical Engineering Research & Design, 2011, 89(8A): 1197-1203. [8] RAYNAL L, BOUILLON P-A, GOMEZ A, et al. From MEA to demixing solvents and future steps, a roadmap for lowering the cost of post-combustion carbon capture[J]. Chemical Engineering Journal, 2011, 171(3): 742-752. doi: 10.1016/j.cej.2011.01.008 [9] LUO W, GUO D, ZHENG J, et al. CO2 absorption using biphasic solvent: Blends of diethylenetriamine, sulfolane, and water[J]. International Journal of Greenhouse Gas Control, 2016, 53: 141-148. doi: 10.1016/j.ijggc.2016.07.036 [10] 涂智芳, 魏建文, 周小斌. 固-液相变二氧化碳吸收剂的研究进展[J]. 洁净煤技术, 2022, 28(09): 122-132. doi: 10.13226/j.issn.1006-6772.cru21111901 [11] ZHOU X B, LIU F, LV B H, et al. Evaluation of the novel biphasic solvents for CO2 capture: Performance and mechanism[J]. International Journal of Greenhouse Gas Control, 2017, 60: 120-128. doi: 10.1016/j.ijggc.2017.03.013 [12] ARSHAD M W, FOSBOL P L, VON SOLMS N, et al. Heat of absorption of CO2 in phase change solvents: 2-(diethylamino)ethanol and 3-(methylamino)propylamine[J]. Journal of Chemical and Engineering Data, 2013, 58(7): 1974-1988. doi: 10.1021/je400289v [13] PINTO D D D, KNUUTILA H, FYTIANOS G, et al. CO2 post combustion capture with a phase change solvent. Pilot plant campaign[J]. International Journal of Greenhouse Gas Control, 2014, 31: 153-164. doi: 10.1016/j.ijggc.2014.10.007 [14] ZHANG S, SHEN Y, SHAO P, et al. Kinetics, thermodynamics, and mechanism of a novel biphasic solvent for CO2 capture from flue gas[J]. Environmental Science & Technology, 2018, 52(6): 3660-3668. [15] MATIN N S, THOMPSON J, ONNEWEER F M, et al. Thermal degradation rate of 2-amino-2-methyl-1-propanol to cyclic 4, 4-dimethyl-1, 3-oxazolidin-2-one: Experiments and kinetics modeling[J]. Industrial & Engineering Chemistry Research, 2016, 55(36): 9586-9593. [16] 安山龙, 王以群, 吴子健. 空间位阻胺AMP捕集CO2技术研究进展[J]. 应用化工, 2020, 49(10): 2636-2640. doi: 10.3969/j.issn.1671-3206.2020.10.050 [17] BARBAROSSA V, BARZAGLI F, MANI F, et al. Efficient CO2 capture by non-aqueous 2-amino-2-methyl-1-propanol (AMP) and low temperature solvent regeneration[J]. RSC Advances, 2013, 3(30): 12349-12355. doi: 10.1039/c3ra40933c [18] KHAN A A, HALDER G N, SAHA A K. Carbon dioxide capture characteristics from flue gas using aqueous 2-amino-2-methyl-1-propanol (AMP) and monoethanolamine (MEA) solutions in packed bed absorption and regeneration columns[J]. International Journal of Greenhouse Gas Control, 2015, 32: 15-23. doi: 10.1016/j.ijggc.2014.10.009 [19] 李清方, 陆诗建, 张建, 等. MEA-AMP二元复配溶液吸收烟气中二氧化碳的实验研究[J]. 精细石油化工, 2010, 27(6): 19-23. doi: 10.3969/j.issn.1003-9384.2010.06.005 [20] PARK J Y, YOON S J, LEE H. Effect of steric hindrance on carbon dioxide absorption into new amine solutions: Thermodynamic and spectroscopic verification through solubility and NMR analysis[J]. Environmental Science & Technology, 2003, 37(8): 1670-1675. [21] ZHANG Z, YANG H, BI J, et al. The leading role of the steric hindrance effect and dipole-dipole interaction in superlattice nanostructures formed via the assembly of discotic liquid crystals[J]. New Journal of Chemistry, 2018, 42(24): 20087-20094. doi: 10.1039/C8NJ05405C [22] ZHOU X, LIU C, ZHANG J, et al. Novel 2-amino-2-methyl-1-propanol-based biphasic solvent for energy-efficient carbon dioxide capture using tetraethylenepentamine as a phase change regulator[J]. Energy, 2023, 270: 126930. doi: 10.1016/j.energy.2023.126930 [23] YUAN Y, ROCHELLE G T. CO2 absorption rate in semi-aqueous monoethanolamine[J]. Chemical Engineering Science, 2018, 182: 56-66. doi: 10.1016/j.ces.2018.02.026 [24] ZHANG W, JIN X, TU W, et al. Development of MEA-based CO2 phase change absorbent[J]. Applied Energy, 2017, 195: 316-323. doi: 10.1016/j.apenergy.2017.03.050 [25] JIANG C, CHEN H, WANG J, et al. Phase splitting agent regulated biphasic solvent for efficient CO2 capture with a low heat duty[J]. Environmental Science & Technology, 2020, 54(12): 7601-7610. [26] KIM Y E, MOON S J, YOON Y I, et al. Heat of absorption and absorption capacity of CO2 in aqueous solutions of amine containing multiple amino groups[J]. Separation and Purification Technology, 2014, 122: 112-118. doi: 10.1016/j.seppur.2013.10.030 [27] ZHANG S, SHEN Y, SHAO P, et al. Kinetics, thermodynamics, and mechanism of a novel biphasic solvent for CO2 capture from flue gas[J]. Environ Sci Technol, 2018, 52(6): 3660-3668. doi: 10.1021/acs.est.7b05936 [28] YANG D Z, LV M, CHEN J. Efficient non-aqueous solvent formed by 2-piperidineethanol and ethylene glycol for CO2 absorption[J]. Chemical Communications, 2019, 55(83): 12483-12486. doi: 10.1039/C9CC06320J [29] RINKER E B, ASHOUR S S, SANDALL O C. Absorption of carbon dioxide into aqueous blends of diethanolamine and methyldiethanolamine[J]. Industrial & Engineering Chemistry Research, 2000, 39(11): 4346-4356. [30] SUN W C, YONG C B, LI M H. Kinetics of the absorption of carbon dioxide into mixed aqueous solutions of 2-amino-2-methyl-l-propanol and piperazine[J]. Chemical Engineering Science, 2005, 60(2): 503-516. doi: 10.1016/j.ces.2004.08.012 [31] STOWE H M, VILCIAUSKAS L, PAEK E, et al. On the origin of preferred bicarbonate production from carbon dioxide (CO2) capture in aqueous 2-amino-2-methyl-1-propanol (AMP)[J]. Physical Chemistry Chemical Physics, 2015, 17(43): 29184-29192. doi: 10.1039/C5CP04876A [32] SHERMAN B J, ROCHELLE G T. Thermodynamic and mass-transfer modeling of carbon dioxide absorption into aqueous 2-amino-2-methyl-1-propanol[J]. Industrial & Engineering Chemistry Research, 2017, 56(1): 319-330. [33] 张婷婷. 空间位阻胺AMP(2-氨基-2-甲基-1-丙醇)吸收CO2的研究[D]. 上海: 上海师范大学, 2013. [34] ZHOU X, JING G, LV B, et al. Low-viscosity and efficient regeneration of carbon dioxide capture using a biphasic solvent regulated by 2-amino-2-methyl-1-propanol[J]. Applied Energy, 2019, 235: 379-390. doi: 10.1016/j.apenergy.2018.10.118 [35] ZHANG X W, ZHANG X, LIU H L, et al. Reduction of energy requirement of CO2 desorption from a rich CO2-loaded MEA solution by using solid acid catalysts[J]. Applied Energy, 2017, 202: 673-684. doi: 10.1016/j.apenergy.2017.05.135 [36] SHI H C, NAAMI A, IDEM R, et al. Catalytic and non catalytic solvent regeneration during absorption-based CO2 capture with single and blended reactive amine solvents[J]. International Journal of Greenhouse Gas Control, 2014, 26: 39-50. doi: 10.1016/j.ijggc.2014.04.007 [37] WANG L, ZHANG Y, WANG R, et al. Advanced monoethanolamine absorption using sulfolane as a phase splitter for CO2 capture[J]. Environ Sci Technol, 2018, 52(24): 14556-14563. doi: 10.1021/acs.est.8b05654 [38] LV J, LIU S, LING H, et al. Development of a promising biphasic absorbent for postcombustion CO2 capture: Sulfolane+2-(methylamino)ethanol+H2O[J]. Industrial & Engineering Chemistry Research, 2020, 59(32): 14496-14506. [39] ZHAN X, LV B, YANG K, et al. Dual-functionalized ionic liquid biphasic solvent for carbon dioxide capture: High-efficiency and energy saving[J]. Environmental Science & Technology, 2020, 54(10): 6281-6288. [40] BIHONG L, KEXUAN Y, XIAOBIN Z, et al. 2-Amino-2-methyl-1-propanol based non-aqueous absorbent for energy-efficient and non-corrosive carbon dioxide capture[J]. Applied Energy, 2020, 264: 114703. doi: 10.1016/j.apenergy.2020.114703 [41] WANG L, ZHANG Y, WANG R, et al. Advanced monoethanolamine absorption using sulfolane as a phase splitter for CO2 capture[J]. Environmental Science & Technology, 2018, 52(24): 14556-14563. [42] SHEN Y, JIANG C, ZHANG S, et al. Biphasic solvent for CO2 capture: Amine property-performance and heat duty relationship[J]. Applied Energy, 2018, 230: 726-733. doi: 10.1016/j.apenergy.2018.09.005 [43] ZHOU C, ZHANG C, ZHANG T, et al. Single-atom solutions promote carbon dioxide capture[J]. Applied Energy, 2023, 332: 120570. doi: 10.1016/j.apenergy.2022.120570 [44] LV B, WU J, LIN C, et al. Kinetic and heat duty study of aprotic heterocyclic anion-based dual functionalized ionic liquid solutions for carbon capture[J]. Fuel, 2020, 263: 116676. doi: 10.1016/j.fuel.2019.116676 期刊类型引用(1)
1. 白雨鑫,郑润芬,魏立新,赵福君,韩晓瑜. 用于捕集CO_2的相变吸收剂与乙醇胺吸收剂性能对比. 石油与天然气化工. 2024(04): 19-27+48 .  百度学术
百度学术
其他类型引用(0)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3315
- HTML全文浏览数: 3315
- PDF下载数: 74
- 施引文献: 1




