

 下载:
下载:
-
空气污染物中的挥发性有机化合物 (volatile organic chemicals,VOCs) 是形成对流层臭氧、PM2.5和光化学烟雾等二次污染物的主要前驱物。针对室内净化及工业生产中面临的诸多问题,建立并发展了“存储-氧化”循环净化VOCs新方法[1]。“存储-氧化”循环净化法为VOCs的脱除提供了一种新颖、有效的途径,即在室温下将VOCs存储,然后进行原位催化氧化再生[1-3]。适用于“存储-原位氧化”循环再生净化的催化剂应具有双功能性,一是“存储”VOCs分子,二是将富集的VOCs完全氧化为CO2和H2O。即在再生过程中无VOCs脱附逃逸,保证催化氧化速率要快于解吸速率[1, 4];同时催化剂需要具有良好的稳定性,实现重复循环利用[3, 5-7]。因此,增强目标VOCs分子与催化剂之间的键合,同时加快氧化再生速率成为该领域研究的关键。
甲苯 (C7H8) 是一种有机化合物,由一个苯环和一个甲基构成,动力学直径约为0.59 nm。β分子筛的孔径大小为0.75 nm×0.57 nm和0.65 nm×0.56 nm,与甲苯的分子动力学直径 (0.59 nm) 相近,有利于对甲苯的择形吸附[8-10]。基于此,本研究以β-25分子筛为载体,制备一系列不同Ag含量的Agx/β-25分子筛催化剂,用于“存储-原位氧化”循环净化甲苯,并采用UV-vis、H2-TPR和TEM表征Ag在β-25分子筛中的落位和存在状态,结合C7H8-TPD研究甲苯在不同Ag物种上的吸附强度,以探索其催化反应的控制工艺条件,为实现“存储-原位氧化”循环再生提供参考。
-
硝酸银 (AgNO3,分析纯) 采购于国药集团化学试剂有限公司。β分子筛采购于山东和发环保科技有限公司。实验中所用气体N2 (99.999%) 和O2 (99.999%) 均购于大连科纳科学技术开发有限责任公司,C7H8标准气(0.044 6 mmol·L−1 C7H8/N2)购于大连光明特种气体有限公司。
-
使用等体积浸渍的方法负载银,将质量 (a) 为15.7 g、78.5 g、157 g的硝酸银分别溶解于0.78 mL水中。称取1 g的β-25(m (SiO2) /m (Al2O3) =25)分子筛,用滴管将配置好的硝酸银溶液逐滴加入分子筛中,边滴加边搅拌,使之呈牙膏状;静置12 h后在真空干燥箱中以65 ℃干燥2 h;最后于马弗炉中以5 ℃·min−1的速率升温至450 ℃,并保持4 h,以期制备Agx/β-25(x=1/5/10)催化剂。
-
催化氧化脱除甲苯或“存储-原位氧化”循环脱除甲苯均在常压微型固定床石英微反应器中进行。将65 mg催化剂 (20~40目) 置于直径6 mm的固定床石英微反应器中。在活性测试前,催化剂在400 ℃的N2中预处理1 h。反应气组成:浓度为8.92×10−4 mmol·L−1的C7H8;体积分数21%的O2;相对湿度(relative humidity,RH) 为50% (25 ℃下)的N2,总流量为100 mL·min−1,空速(space velocity,SV)为36 000 h−1。在室温下,将气态H2O通过N2鼓泡法携带入反应气中,以RH表示其体积分数。甲苯通过入口处后经VOCs催化燃烧炉 (CuMn/γ-Al2O3,450 ℃) 时被完全氧化为CO2和H2O,并由气体分析仪 (S710) 监测CO2浓度而计算得出。甲苯存储容量(NC7H8,mmol·gcat−1)、C7H8氧化为CO2的转化率(X C7H8→CO2)和碳平衡(Bc)分别定义如式 (1) 和 (2) 所示。
式中:CC7H8in为原料气中C7H8的浓度,mmol·L−1;F1为总流量,mL·min−1;t1为穿透时间,min;Wcat是催化剂的质量,g。穿透时间定义为出口C7H8浓度达到入口甲苯浓度10%时的时间。
-
X射线衍射:采用日本Rigaku,型号为D/Max 2400的X射线衍射仪对催化剂晶型结构进行表征,扫描范围 (2 theta) 10 o~50 o,扫描速度10 o·min−1。紫外-可见光谱:使用UV-550光谱仪检测在190~800 nm的紫外-可见光谱。程序升温脱附:程序升温脱附实验在内径为4 mm的石英管反应器中装入65 mg催化剂,经氮气在400 ℃下预处理1 h,待冷却到室温后,于特定气氛中进行定量吸附。在吸附完成后切换回N2气氛,经短时间吹扫后,从室温以10 ℃·min−1的升温速率至500 ℃,使用质谱分析仪对脱附物种进行在线监测。
-
对负载了不同含量Ag的Agx/β-25催化剂的晶相结构进行XRD表征,结果如图1所示。当Ag的负载量 (质量分数) 由1%增至5%,仅观察到归属于β分子筛自身的衍射峰,未观察到归属于银物种的特征衍射峰。这可能是由于Ag在催化剂表面高度分散,或者Ag的负载量较低未被检测到所致[11]。当Ag负载量增至10%时,出现了属于金属Ag的特征衍射峰 (2θ=38.115 °) ,但峰强很弱。该结果说明催化剂表面的Ag发生聚集而出现金属银。
为探讨银在分子筛上的存在状态,对不同质量分数Ag的Agx/β-25催化剂进行了UV-vis表征。图2表明,对于Ag1.0/β-25催化剂,其谱图主要出现2个吸收峰,分别位于220 nm、310 nm处。220 nm处的吸收峰可归属为分子筛离子交换位上的Ag+,310 nm处的吸收峰可归属为带有一定电荷的Agnδ+团簇[12-13]。该结果表明在Ag1.0/β-25催化剂中,Ag主要以Ag+状态存在,同时含有少量Agnδ+团簇。随着银负载量由1%增至5%,归属于Ag+的吸收峰强度显著增强,这说明有更多的银被交换到分子筛的离子交换位上。继续增加Ag负载量至10%,310 nm归属于Agnδ+团簇的吸附峰峰强减弱[12-13],并且在400~500 nm处出现了归属为金属银颗粒的吸收峰[14]。该结果说明Ag开始聚集长大,这与XRD结果相一致。
XRD和UV-vis表征分析结果表明,Agx/β-25催化剂表面Ag物种的主要存在状态为Ag+和Agnδ+团簇。当Ag的负载量低于5%时,催化剂表面的银物种分散较好,均以Ag+和Agnδ+的形式存在;当Ag的负载量为10%时,催化剂表面的Agnδ+减少,最后伴随银纳米粒子的出现。
-
图3表现了不同质量分数Ag的Agx/β-25分子筛及β-25分子筛本身对甲苯的存储能力。β-25分子筛本身对甲苯的存储量为0.19 mmol·g−1。当负载1%的Ag后,催化剂对甲苯的存储提升较小;当负载量增至5%时,甲苯的存储量是Ag1.0/β-25的2倍,继续增加银负载量至10%,甲苯的存储量可达0.61 mmol·g−1。因此,当催化剂中Ag质量分数增大,甲苯存储量有显著提升,推测Ag引入后会与甲苯产生键合作用,从而提供新的甲苯存储位点,使存储量较分子筛本身有所增加。
-
催化剂的氧化能力是“存储-原位氧化”循环过程中的另一重要因素,因此,对Ag改性的β-25催化剂的甲苯氧化能力进行考察,结果如图4所示。Ag1.0/β-25催化剂的甲苯氧化能力最差,甲苯完全氧化为CO2的温度是420 ℃,其次是Ag5.0/β-25催化剂 (320 ℃) ,而Ag10.0/β-25催化剂的甲苯氧化能力最优。此时,甲苯完全氧化为CO2的温度低至260 ℃,这说明银的引入使催化剂的氧化能力显著提升。
-
实现“存储-原位氧化”循环脱除甲苯的关键在于保证再生过程中VOCs不会发生脱附逃逸而产生二次污染物。这就要求VOCs的催化氧化速率要大于脱附速率,基于此研究了程序升温过程中存储于催化剂的甲苯的脱附规律 (图5) 。甲苯的脱附曲线分为3个温区:低温区 (< 250 ℃) ,中温区 (250~500 ℃) 和高温区 (> 500 ℃) 。β-25催化剂上存储的甲苯脱附峰温为200 ℃,这属于吸附在分子筛Bronsted位点上的脱附[15]。在负载Ag后,不仅在低温区 (211 ℃) 存在脱附峰,在250~500 ℃之间还存在一个宽泛的脱附峰。随着Ag负载量的增加,210 ℃的甲苯脱附峰逐渐升高,这属于Ag纳米粒子上吸附甲苯的脱附。Ag纳米粒子与甲苯间依靠范德华力形成物理键合,故在低温下会发生脱附[16-17]。当Ag负载量 (质量分数) 由1%提升至5%,250~500 ℃的宽泛脱附峰峰强逐渐增强。根据UV-vis结果分析发现,当温度为250~500 ℃时,重叠解吸主要为吸附在Agnδ+团簇上而产生的甲苯脱附,因为大多数Agnδ+团簇的成核数是不固定的 (一般为2~8组成),这与温度为250~500 ℃时的宽泛脱附峰相一致。由于Ag独特的 (n-1) d10ns0电子构型,Agnδ+容易接受电子,从而与甲苯间形成反馈π键这种较强的键合,导致了甲苯的脱附温度升高(250~500 ℃)[16, 18-20]。当Ag负载量 (质量分数) 达到10%时,这个宽泛的脱附峰开始变得尖锐并向低温区移动,这说明过量引入的Ag使小粒径Agnδ+团簇团聚长大,故吸附强度逐渐减弱。在脱附过程中,还观察到一个有意义的现象,即当温度升至500 ℃,在Ag改性的β-25分子筛催化剂上,仍存在一定量甲苯未脱附。将未脱附的甲苯浓度与Ag负载量进行关联分析 (图6) 发现,在500 ℃时仍未脱附的甲苯浓度随Ag负载量增加而逐渐增大,与图2 中属于Ag+吸收峰峰强逐渐增大的规律相吻合。由此推断,当温度大于500 ℃时,仍未脱附的甲苯是与离子交换位上的Ag+强键合那部分甲苯。Ag+能吸引甲基电子发生部分氧化反应,从而形成强化学键合,在500 ℃时仍未脱附[11, 21-24]。通过以上甲苯升温脱附研究,得出Ag改性β分子筛催化剂表面存在3种不同强度甲苯吸附位点。根据甲苯的脱附温度高低,可将其分为强吸附位点、中强吸附位点和弱吸附位点。强吸附位点上的甲苯在500 ℃时仍未脱附;中强吸附位点上的甲苯在250~500 ℃发生脱附;弱吸附位点上的甲苯在低于250 ℃时即发生脱附。
将甲苯吸附位点与Ag存在状态进行关联分析后发现,强吸附位点对应于离子交换位上的Ag+,中强吸附位点对应于Agnδ+团簇,弱吸附位点对应于金属银颗粒和分子筛的B酸位。上述结果表明,引入Ag可有效增强甲苯与催化剂之间的键合,最优Ag负载量为5%。此时,Ag在催化剂表面高度分散,并主要以键合甲苯能力强的Ag+和小尺寸Agnδ+的形式存在。
-
对比各催化剂上的甲苯完全氧化温度与脱附温度后发现,即使在Ag10.0/β-25催化剂上,甲苯完全氧化的温度 (260 ℃) 仍高于甲苯的脱附温度 (吸附在Ag纳米粒子上的甲苯脱附峰温为211 ℃)。该结果表明,存储的甲苯在被完全氧化前即发生了脱附逃逸。因此,应通过控制甲苯的存储位点,使其有选择性地优先存储在强存储位上,从而实现甲苯脱附前的完全氧化再生。通过控制存储时间来调控甲苯在催化剂上的吸附位点的实验结果如图7所示。对于Ag5.0/β-25来说,当存储时间为60 min时,仅出现强吸附在Agnδ+处的甲苯脱附峰 (475 ℃) ,这说明甲苯是优先选择吸附在Ag+位点和Agnδ+上。随着存储时间从60 min延长至120 min、180 min,强吸附在Agnδ+的脱附峰峰强增大,且未观察到甲苯的低温脱附峰,这说明控制存储时间在180 min内时,甲苯仅选择性地吸附在Ag+和Agnδ+上,且键合能力较强。继续延长存储时间至240 min,才会逐渐出现吸附在分子筛B酸位和Ag纳米粒子上的低温脱附峰。这说明甲苯在Agx/β-25催化剂上的存储吸附顺序为Ag+→Agnδ+→Ag纳米粒子+B酸位。
-
基于图6数据,在“存储-原位氧化”循环净化甲苯实验中,将存储时间控制为180 min,此时甲苯全部存储在Ag+和Agnδ+位点上,其存储量可达0.32 mmol·gcat−1;在原位氧化再生阶段,存储的甲苯全部被氧化成为COx。图7表明,在3次循环测试中,碳平衡均大于98%,没有甲苯的提前脱附,这说明Ag5.0/β-25催化剂在“存储-原位氧化”循环再生净化甲苯过程中稳定性很好,也充分说明了通过调控存储位点实现“存储-原位氧化”循环再生净化甲苯这一思路的可行性。
-
1) 通过制备不同Ag负载量的β分子筛催化剂,研究了催化剂中Ag的状态。通过将甲苯的脱附温度与甲苯在不同存储位点上的键合强度相关联,将催化剂表面存储位点分为3个级别。强存储位点即离子交换位上为Ag+,对于甲苯有最强的键合能力,程序升温至500 ℃时仍未脱附。中强存储位点即离子交换位为Agnδ+团簇,其与甲苯之间可形成较稳定的π键合,在温度为250~500 ℃时出现脱附。弱存储位点即金属Ag纳米粒子和分子筛的酸性位,与甲苯间键合最弱 (范德华力) ,在210 ℃左右出现脱附。2) 依据甲苯在Agx/β-25分子筛上的存储强度,通过控制存储时间来调控甲苯在催化剂上的优先吸附顺序,使甲苯优先吸附在Ag+和Agnδ+强存储位点上,使脱附温度高于氧化温度,即可实现原位氧化再生。3) 通过优化Ag的最佳负载量为5%,Ag在催化剂表面高度分散,并主要以Ag+和小尺寸的Agnδ+的形式存在。此时吸附在Ag+和Agnδ+上的甲苯可达0.32 mmol·gcat−1,且存储的甲苯可全部被原位热氧化为COx。4) 在3次循环测试中,碳平衡均大于98%,再生过程中未发生甲苯的提前脱附,这说明Ag5.0/β-25催化剂在“存储-原位氧化”循环再生净化甲苯过程中有很好的稳定性及重现性,证实了这种再生思路的可行性。
基于调控存储位点实现甲苯的“存储-原位氧化”循环净化
Toluene removal through “storage - in situ oxidation” cycling via modulating the storage sites
-
摘要: “存储-原位氧化”循环净化气态污染物 (VOCs) 的方法是利用存储材料对VOCs的选择吸附,在室温先将VOCs富集并存储在催化剂上。当存储饱和后,通过升温使催化剂催化氧化活性提高,短时间内将存储的VOCs完全氧化为CO2和H2O,使得催化剂得以原位氧化再生。选择分子筛材料用于“存储-原位氧化”循环净化甲苯的关键问题是:大部分的甲苯以物理吸附的形式存储于分子筛催化剂的孔道中,脱附温度低,导致原位催化氧化过程中甲苯未被氧化前就脱附逃逸,造成二次污染。以不同银负载量的Agx/β-25催化剂作为研究对象,利用XRD和UV-vis对分子筛催化剂表面Ag物种的存在状态进行识别。将Ag物种状态与甲苯程序升温脱附结果定性关联,明确了甲苯在不同状态的Ag物种上的存储强弱。位于离子交换位上的Ag+与甲苯键合最强,其次为Agnδ+团簇,键合最弱的为金属银粒子。通过控制存储时间,可调控甲苯在Agx/β-25催化剂上的存储位点,使甲苯优先选择吸附在位于离子交换位上的Ag+或Agnδ+团簇上,当脱附温度高于氧化温度,即可实现低浓度甲苯的“存储-原位氧化”循环再生净化。通过该研究,可初步得出调控分子筛表面存储位点强弱的方法,并构建适宜的催化剂,为指导设计低浓度甲苯脱除的双功能催化材料提供参考。
-
关键词:
- 甲苯 /
- 银物种存在状态 /
- 存储位点调控 /
- “存储-原位氧化”循环
Abstract: “Storage-in situ oxidation” cycling process includes storing low concentration of volatile organic chemicals (VOCs) on the catalyst at room temperature and complete oxidation the stored species into CO2 and H2O at elevated temperature in a short time, and the catalyst can be oxidized and regenerated in situ. The key issue for application of this approach over zeolite materials is in situ regeneration of the zeolite materials without any release of the toluene or generation of secondary pollutants. Therefore, in this study, a series of Agx/β-25 catalysts were prepared and the status of surface Ag species were characterized through XRD and UV-vis. By qualitative analysis, the C7H8-temperature-programmed desorption results with the status of Ag species, the different storage sites and bond strength for C7H8 storage were clarified. Ag+ at the ion exchange site had the strongest bond to toluene, followed by Agnδ+ clusters, and the weakest bond was silver. The storage site could be modulated by controlling the storage time, so that toluene could be preferentially adsorbed on strong adsorption sites (Ag+ and Agnδ+), which could enable the in situ thermal oxidation regeneration of toluene when the desorption temperature was higher than oxidation temperature. The method of regulating the strength of storage sites on the surface of molecular sieve was preliminarily mastered, and a suitable catalyst was constructed in this study, which can provide a promising strategy to synthesis and design bifunctional catalyst for toluene removal through “storage - in situ oxidation” cycling. -
空气污染物中的挥发性有机化合物 (volatile organic chemicals,VOCs) 是形成对流层臭氧、PM2.5和光化学烟雾等二次污染物的主要前驱物。针对室内净化及工业生产中面临的诸多问题,建立并发展了“存储-氧化”循环净化VOCs新方法[1]。“存储-氧化”循环净化法为VOCs的脱除提供了一种新颖、有效的途径,即在室温下将VOCs存储,然后进行原位催化氧化再生[1-3]。适用于“存储-原位氧化”循环再生净化的催化剂应具有双功能性,一是“存储”VOCs分子,二是将富集的VOCs完全氧化为CO2和H2O。即在再生过程中无VOCs脱附逃逸,保证催化氧化速率要快于解吸速率[1, 4];同时催化剂需要具有良好的稳定性,实现重复循环利用[3, 5-7]。因此,增强目标VOCs分子与催化剂之间的键合,同时加快氧化再生速率成为该领域研究的关键。
甲苯 (C7H8) 是一种有机化合物,由一个苯环和一个甲基构成,动力学直径约为0.59 nm。β分子筛的孔径大小为0.75 nm×0.57 nm和0.65 nm×0.56 nm,与甲苯的分子动力学直径 (0.59 nm) 相近,有利于对甲苯的择形吸附[8-10]。基于此,本研究以β-25分子筛为载体,制备一系列不同Ag含量的Agx/β-25分子筛催化剂,用于“存储-原位氧化”循环净化甲苯,并采用UV-vis、H2-TPR和TEM表征Ag在β-25分子筛中的落位和存在状态,结合C7H8-TPD研究甲苯在不同Ag物种上的吸附强度,以探索其催化反应的控制工艺条件,为实现“存储-原位氧化”循环再生提供参考。
1. 材料与试剂
1.1 实验材料
硝酸银 (AgNO3,分析纯) 采购于国药集团化学试剂有限公司。β分子筛采购于山东和发环保科技有限公司。实验中所用气体N2 (99.999%) 和O2 (99.999%) 均购于大连科纳科学技术开发有限责任公司,C7H8标准气(0.044 6 mmol·L−1 C7H8/N2)购于大连光明特种气体有限公司。
1.2 催化剂制备
使用等体积浸渍的方法负载银,将质量 (a) 为15.7 g、78.5 g、157 g的硝酸银分别溶解于0.78 mL水中。称取1 g的β-25(m (SiO2) /m (Al2O3) =25)分子筛,用滴管将配置好的硝酸银溶液逐滴加入分子筛中,边滴加边搅拌,使之呈牙膏状;静置12 h后在真空干燥箱中以65 ℃干燥2 h;最后于马弗炉中以5 ℃·min−1的速率升温至450 ℃,并保持4 h,以期制备Agx/β-25(x=1/5/10)催化剂。
1.3 催化活性评价
催化氧化脱除甲苯或“存储-原位氧化”循环脱除甲苯均在常压微型固定床石英微反应器中进行。将65 mg催化剂 (20~40目) 置于直径6 mm的固定床石英微反应器中。在活性测试前,催化剂在400 ℃的N2中预处理1 h。反应气组成:浓度为8.92×10−4 mmol·L−1的C7H8;体积分数21%的O2;相对湿度(relative humidity,RH) 为50% (25 ℃下)的N2,总流量为100 mL·min−1,空速(space velocity,SV)为36 000 h−1。在室温下,将气态H2O通过N2鼓泡法携带入反应气中,以RH表示其体积分数。甲苯通过入口处后经VOCs催化燃烧炉 (CuMn/γ-Al2O3,450 ℃) 时被完全氧化为CO2和H2O,并由气体分析仪 (S710) 监测CO2浓度而计算得出。甲苯存储容量(NC7H8,mmol·gcat−1)、C7H8氧化为CO2的转化率(X C7H8→CO2)和碳平衡(Bc)分别定义如式 (1) 和 (2) 所示。
NC7H8 = CinC7H8F1t1Wcat. (1) XC7H8→CO2(%)=CoutCO27×CinC7H8×100% (2) Bc=nproducedCO+nproducedCO2+7×ndesorptionC7H87×nstoredC7H8×100% (3) 式中:CC7H8in为原料气中C7H8的浓度,mmol·L−1;F1为总流量,mL·min−1;t1为穿透时间,min;Wcat是催化剂的质量,g。穿透时间定义为出口C7H8浓度达到入口甲苯浓度10%时的时间。
1.4 催化剂表征方法
X射线衍射:采用日本Rigaku,型号为D/Max 2400的X射线衍射仪对催化剂晶型结构进行表征,扫描范围 (2 theta) 10 o~50 o,扫描速度10 o·min−1。紫外-可见光谱:使用UV-550光谱仪检测在190~800 nm的紫外-可见光谱。程序升温脱附:程序升温脱附实验在内径为4 mm的石英管反应器中装入65 mg催化剂,经氮气在400 ℃下预处理1 h,待冷却到室温后,于特定气氛中进行定量吸附。在吸附完成后切换回N2气氛,经短时间吹扫后,从室温以10 ℃·min−1的升温速率至500 ℃,使用质谱分析仪对脱附物种进行在线监测。
2. 结果与讨论
2.1 催化剂表征
对负载了不同含量Ag的Agx/β-25催化剂的晶相结构进行XRD表征,结果如图1所示。当Ag的负载量 (质量分数) 由1%增至5%,仅观察到归属于β分子筛自身的衍射峰,未观察到归属于银物种的特征衍射峰。这可能是由于Ag在催化剂表面高度分散,或者Ag的负载量较低未被检测到所致[11]。当Ag负载量增至10%时,出现了属于金属Ag的特征衍射峰 (2θ=38.115 °) ,但峰强很弱。该结果说明催化剂表面的Ag发生聚集而出现金属银。
 图 1 不同含量Ag改性的β-25催化剂XRD谱图Figure 1. XRD patterns of Ag/β-25 catalysts modified with different Ag contents
图 1 不同含量Ag改性的β-25催化剂XRD谱图Figure 1. XRD patterns of Ag/β-25 catalysts modified with different Ag contents为探讨银在分子筛上的存在状态,对不同质量分数Ag的Agx/β-25催化剂进行了UV-vis表征。图2表明,对于Ag1.0/β-25催化剂,其谱图主要出现2个吸收峰,分别位于220 nm、310 nm处。220 nm处的吸收峰可归属为分子筛离子交换位上的Ag+,310 nm处的吸收峰可归属为带有一定电荷的Agnδ+团簇[12-13]。该结果表明在Ag1.0/β-25催化剂中,Ag主要以Ag+状态存在,同时含有少量Agnδ+团簇。随着银负载量由1%增至5%,归属于Ag+的吸收峰强度显著增强,这说明有更多的银被交换到分子筛的离子交换位上。继续增加Ag负载量至10%,310 nm归属于Agnδ+团簇的吸附峰峰强减弱[12-13],并且在400~500 nm处出现了归属为金属银颗粒的吸收峰[14]。该结果说明Ag开始聚集长大,这与XRD结果相一致。
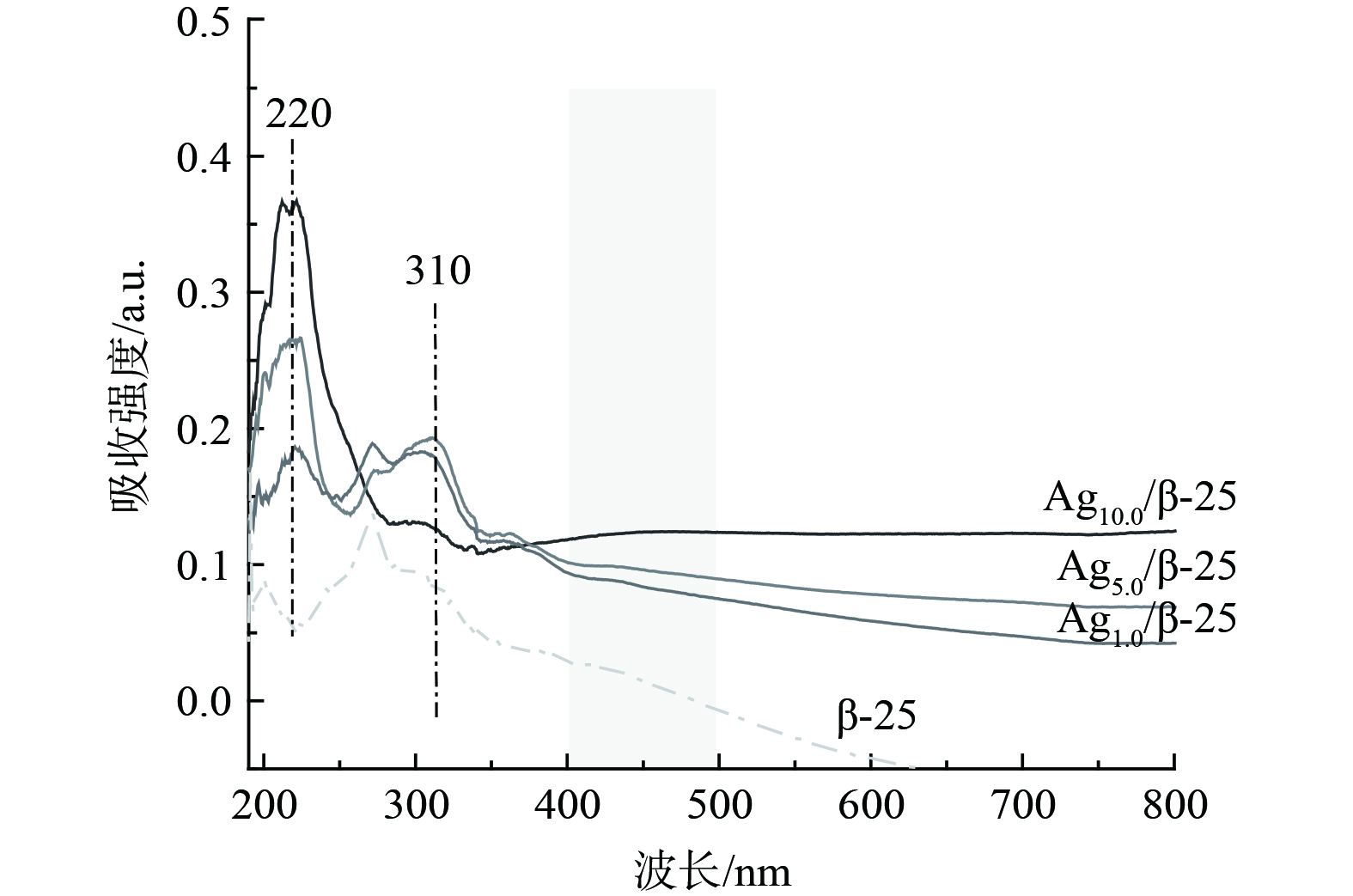 图 2 不同含量Ag改性的β-25催化剂UV-vis谱图Figure 2. UV-vis spectra of Ag/β-25 samples modified with different Ag contents
图 2 不同含量Ag改性的β-25催化剂UV-vis谱图Figure 2. UV-vis spectra of Ag/β-25 samples modified with different Ag contentsXRD和UV-vis表征分析结果表明,Agx/β-25催化剂表面Ag物种的主要存在状态为Ag+和Agnδ+团簇。当Ag的负载量低于5%时,催化剂表面的银物种分散较好,均以Ag+和Agnδ+的形式存在;当Ag的负载量为10%时,催化剂表面的Agnδ+减少,最后伴随银纳米粒子的出现。
2.2 银改性β-25分子筛上甲苯的存储和氧化
2.2.1 甲苯存储性能
图3表现了不同质量分数Ag的Agx/β-25分子筛及β-25分子筛本身对甲苯的存储能力。β-25分子筛本身对甲苯的存储量为0.19 mmol·g−1。当负载1%的Ag后,催化剂对甲苯的存储提升较小;当负载量增至5%时,甲苯的存储量是Ag1.0/β-25的2倍,继续增加银负载量至10%,甲苯的存储量可达0.61 mmol·g−1。因此,当催化剂中Ag质量分数增大,甲苯存储量有显著提升,推测Ag引入后会与甲苯产生键合作用,从而提供新的甲苯存储位点,使存储量较分子筛本身有所增加。
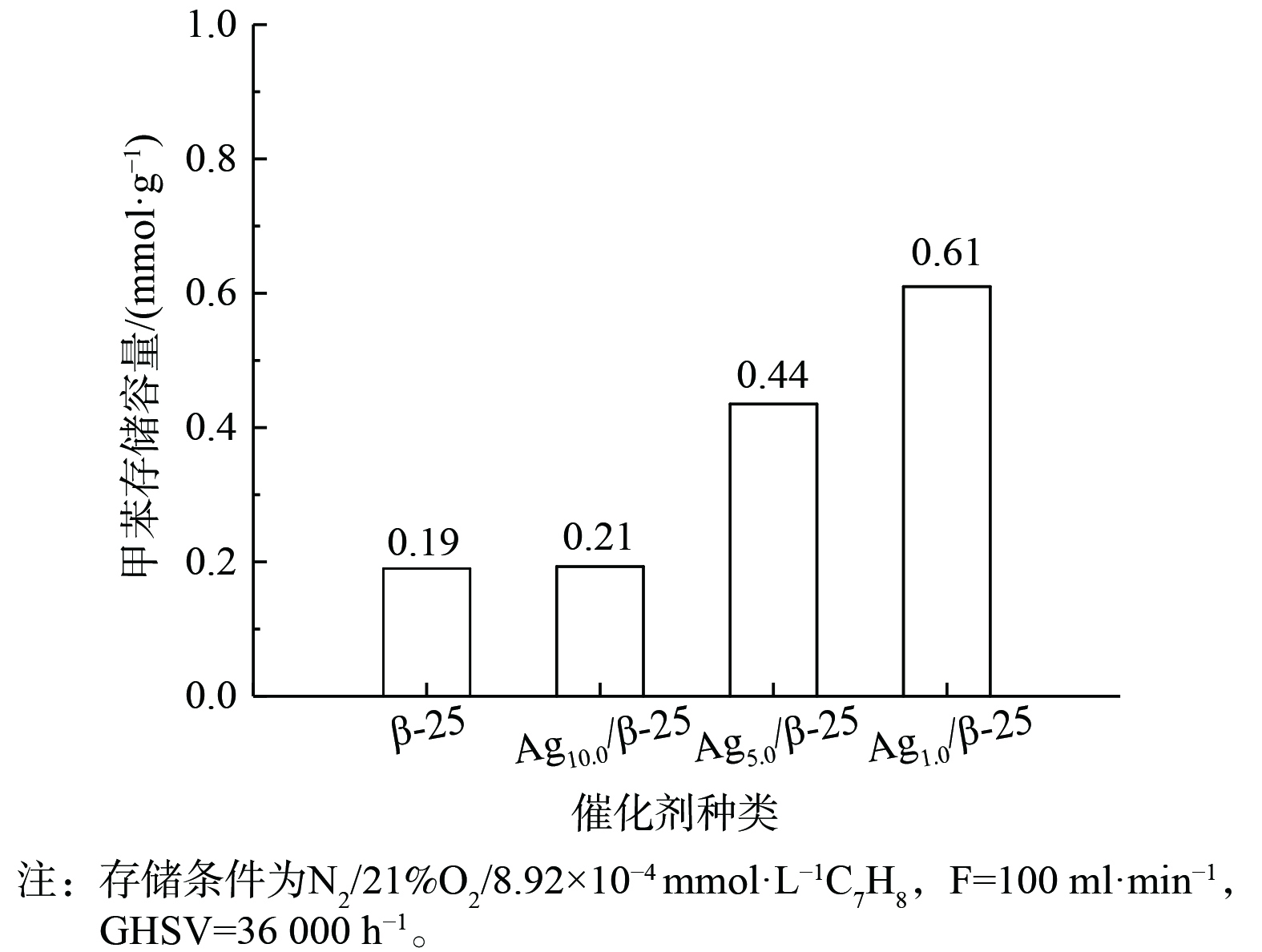 图 3 Agx/β-25系列催化剂上甲苯存储量对比Figure 3. Comparison of toluene storage on Agx/β-25 catalysts
图 3 Agx/β-25系列催化剂上甲苯存储量对比Figure 3. Comparison of toluene storage on Agx/β-25 catalysts2.2.2 甲苯氧化性能
催化剂的氧化能力是“存储-原位氧化”循环过程中的另一重要因素,因此,对Ag改性的β-25催化剂的甲苯氧化能力进行考察,结果如图4所示。Ag1.0/β-25催化剂的甲苯氧化能力最差,甲苯完全氧化为CO2的温度是420 ℃,其次是Ag5.0/β-25催化剂 (320 ℃) ,而Ag10.0/β-25催化剂的甲苯氧化能力最优。此时,甲苯完全氧化为CO2的温度低至260 ℃,这说明银的引入使催化剂的氧化能力显著提升。
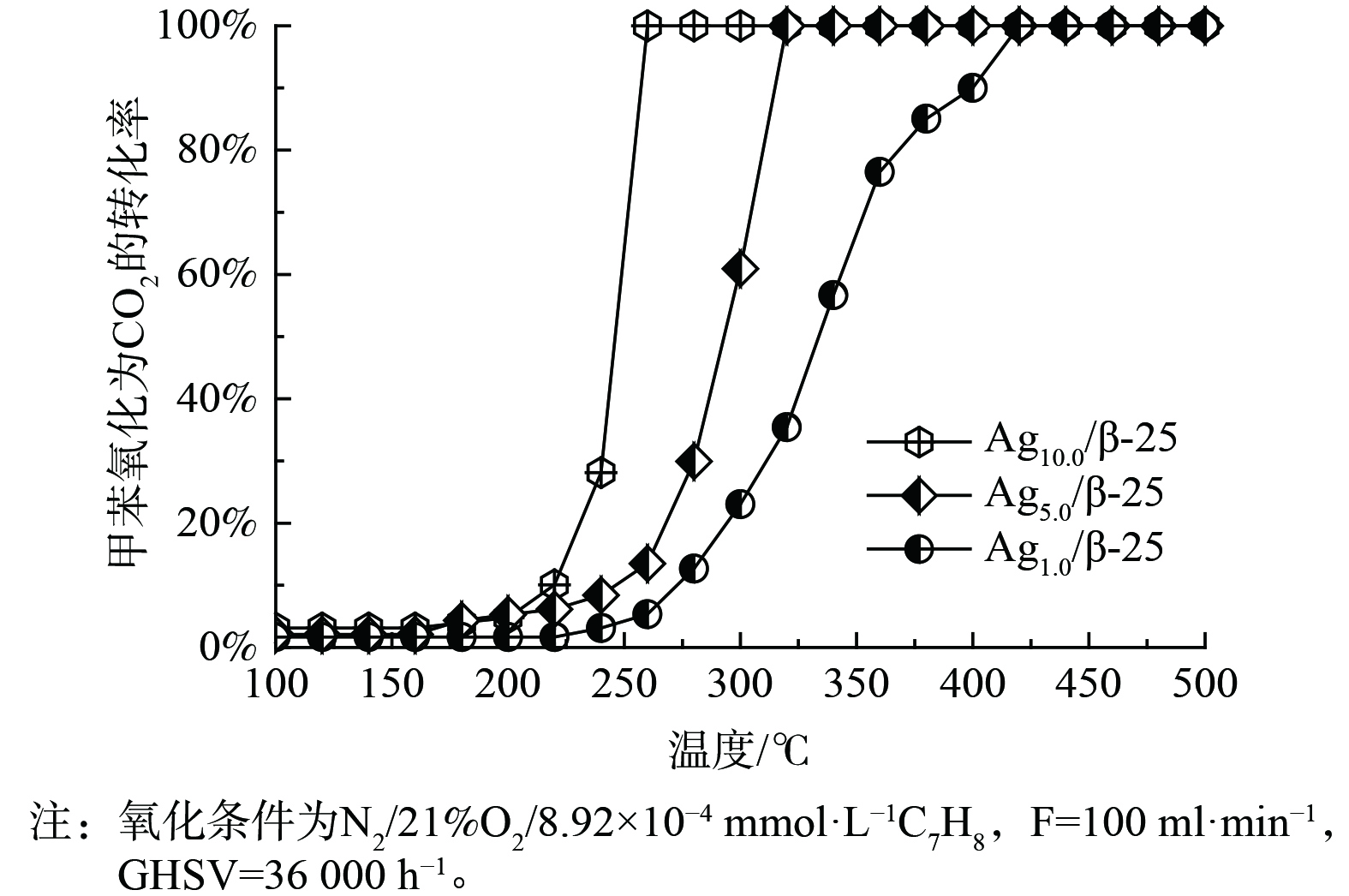 图 4 Agx/β-25系列催化剂上甲苯催化氧化活性Figure 4. Catalytic oxidation activity of toluene on Agx/β-25 catalysts
图 4 Agx/β-25系列催化剂上甲苯催化氧化活性Figure 4. Catalytic oxidation activity of toluene on Agx/β-25 catalysts2.2.3 存储的甲苯在程序升温过程中的脱附
实现“存储-原位氧化”循环脱除甲苯的关键在于保证再生过程中VOCs不会发生脱附逃逸而产生二次污染物。这就要求VOCs的催化氧化速率要大于脱附速率,基于此研究了程序升温过程中存储于催化剂的甲苯的脱附规律 (图5) 。甲苯的脱附曲线分为3个温区:低温区 (< 250 ℃) ,中温区 (250~500 ℃) 和高温区 (> 500 ℃) 。β-25催化剂上存储的甲苯脱附峰温为200 ℃,这属于吸附在分子筛Bronsted位点上的脱附[15]。在负载Ag后,不仅在低温区 (211 ℃) 存在脱附峰,在250~500 ℃之间还存在一个宽泛的脱附峰。随着Ag负载量的增加,210 ℃的甲苯脱附峰逐渐升高,这属于Ag纳米粒子上吸附甲苯的脱附。Ag纳米粒子与甲苯间依靠范德华力形成物理键合,故在低温下会发生脱附[16-17]。当Ag负载量 (质量分数) 由1%提升至5%,250~500 ℃的宽泛脱附峰峰强逐渐增强。根据UV-vis结果分析发现,当温度为250~500 ℃时,重叠解吸主要为吸附在Agnδ+团簇上而产生的甲苯脱附,因为大多数Agnδ+团簇的成核数是不固定的 (一般为2~8组成),这与温度为250~500 ℃时的宽泛脱附峰相一致。由于Ag独特的 (n-1) d10ns0电子构型,Agnδ+容易接受电子,从而与甲苯间形成反馈π键这种较强的键合,导致了甲苯的脱附温度升高(250~500 ℃)[16, 18-20]。当Ag负载量 (质量分数) 达到10%时,这个宽泛的脱附峰开始变得尖锐并向低温区移动,这说明过量引入的Ag使小粒径Agnδ+团簇团聚长大,故吸附强度逐渐减弱。在脱附过程中,还观察到一个有意义的现象,即当温度升至500 ℃,在Ag改性的β-25分子筛催化剂上,仍存在一定量甲苯未脱附。将未脱附的甲苯浓度与Ag负载量进行关联分析 (图6) 发现,在500 ℃时仍未脱附的甲苯浓度随Ag负载量增加而逐渐增大,与图2 中属于Ag+吸收峰峰强逐渐增大的规律相吻合。由此推断,当温度大于500 ℃时,仍未脱附的甲苯是与离子交换位上的Ag+强键合那部分甲苯。Ag+能吸引甲基电子发生部分氧化反应,从而形成强化学键合,在500 ℃时仍未脱附[11, 21-24]。通过以上甲苯升温脱附研究,得出Ag改性β分子筛催化剂表面存在3种不同强度甲苯吸附位点。根据甲苯的脱附温度高低,可将其分为强吸附位点、中强吸附位点和弱吸附位点。强吸附位点上的甲苯在500 ℃时仍未脱附;中强吸附位点上的甲苯在250~500 ℃发生脱附;弱吸附位点上的甲苯在低于250 ℃时即发生脱附。
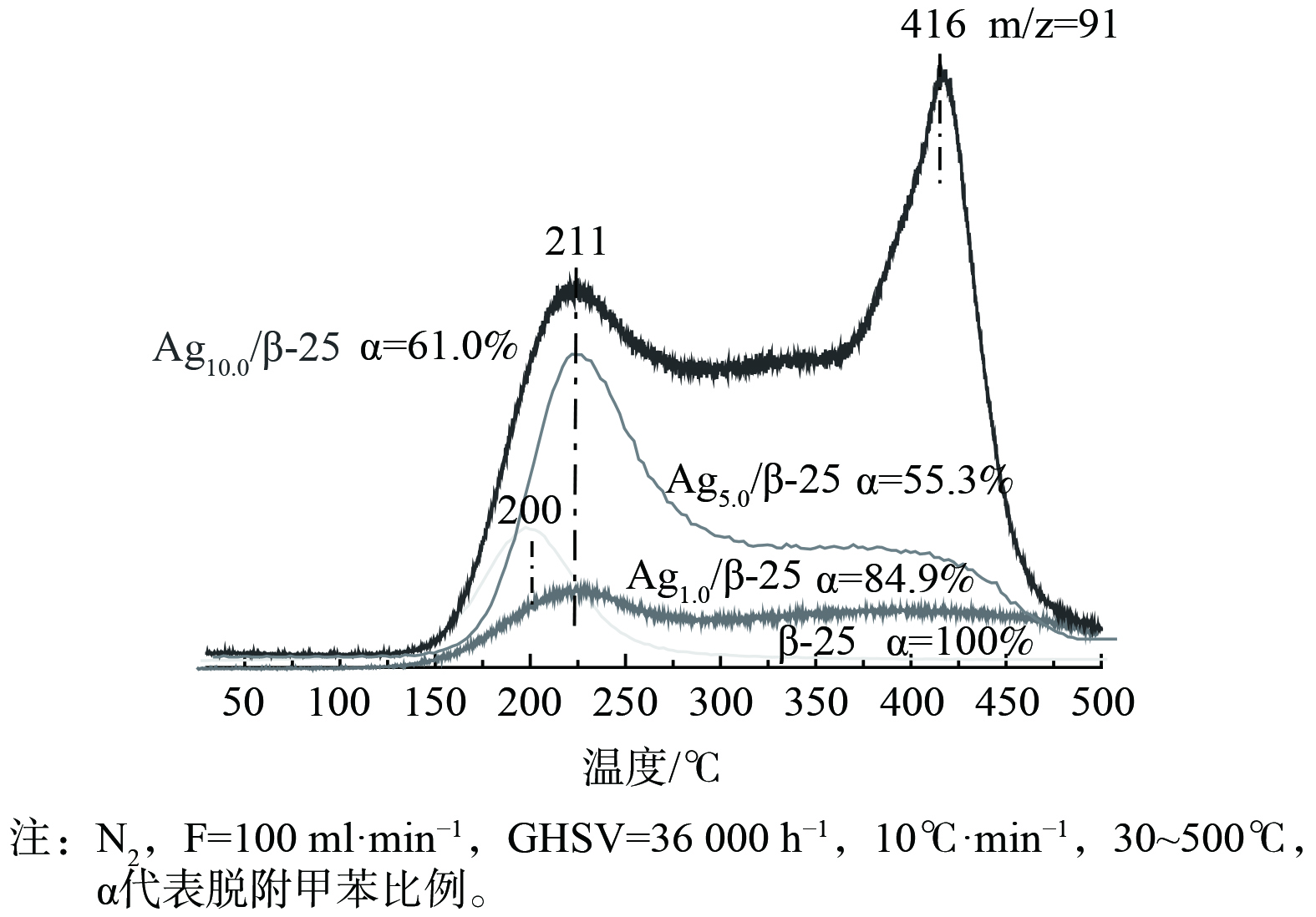 图 5 Agx/β-25系列催化剂上存储的甲苯在N2下程序升温脱附Figure 5. TPD experiments of adsorbed C7H8 under N2 atmosphere on Agx/β catalysts
图 5 Agx/β-25系列催化剂上存储的甲苯在N2下程序升温脱附Figure 5. TPD experiments of adsorbed C7H8 under N2 atmosphere on Agx/β catalysts将甲苯吸附位点与Ag存在状态进行关联分析后发现,强吸附位点对应于离子交换位上的Ag+,中强吸附位点对应于Agnδ+团簇,弱吸附位点对应于金属银颗粒和分子筛的B酸位。上述结果表明,引入Ag可有效增强甲苯与催化剂之间的键合,最优Ag负载量为5%。此时,Ag在催化剂表面高度分散,并主要以键合甲苯能力强的Ag+和小尺寸Agnδ+的形式存在。
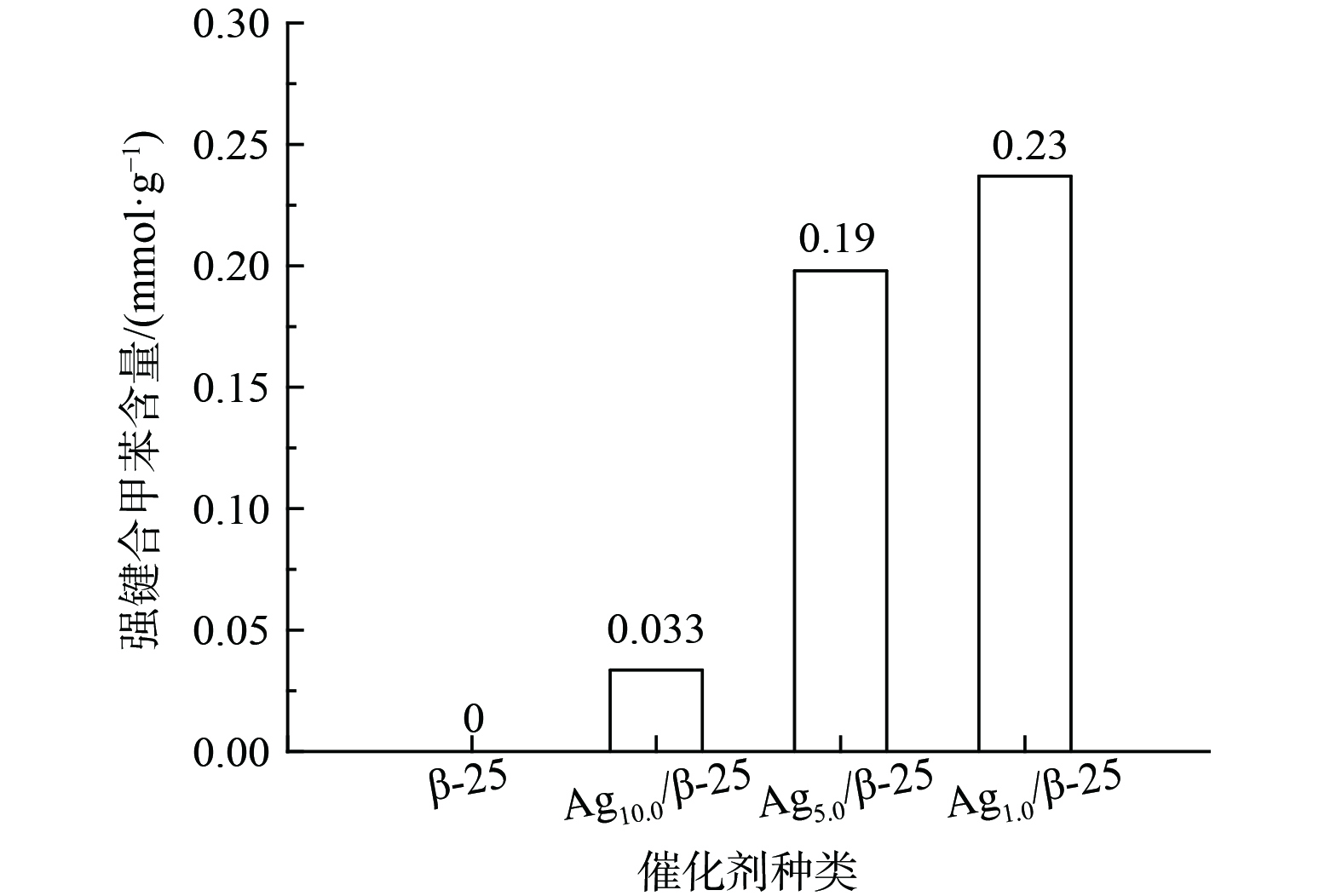 图 6 Agx/β-25系列催化剂上强键合甲苯含量Figure 6. Content of strongly bonded toluene on Agx/β -25 catalysts
图 6 Agx/β-25系列催化剂上强键合甲苯含量Figure 6. Content of strongly bonded toluene on Agx/β -25 catalysts2.2.4 甲苯存储位点的调控
对比各催化剂上的甲苯完全氧化温度与脱附温度后发现,即使在Ag10.0/β-25催化剂上,甲苯完全氧化的温度 (260 ℃) 仍高于甲苯的脱附温度 (吸附在Ag纳米粒子上的甲苯脱附峰温为211 ℃)。该结果表明,存储的甲苯在被完全氧化前即发生了脱附逃逸。因此,应通过控制甲苯的存储位点,使其有选择性地优先存储在强存储位上,从而实现甲苯脱附前的完全氧化再生。通过控制存储时间来调控甲苯在催化剂上的吸附位点的实验结果如图7所示。对于Ag5.0/β-25来说,当存储时间为60 min时,仅出现强吸附在Agnδ+处的甲苯脱附峰 (475 ℃) ,这说明甲苯是优先选择吸附在Ag+位点和Agnδ+上。随着存储时间从60 min延长至120 min、180 min,强吸附在Agnδ+的脱附峰峰强增大,且未观察到甲苯的低温脱附峰,这说明控制存储时间在180 min内时,甲苯仅选择性地吸附在Ag+和Agnδ+上,且键合能力较强。继续延长存储时间至240 min,才会逐渐出现吸附在分子筛B酸位和Ag纳米粒子上的低温脱附峰。这说明甲苯在Agx/β-25催化剂上的存储吸附顺序为Ag+→Agnδ+→Ag纳米粒子+B酸位。
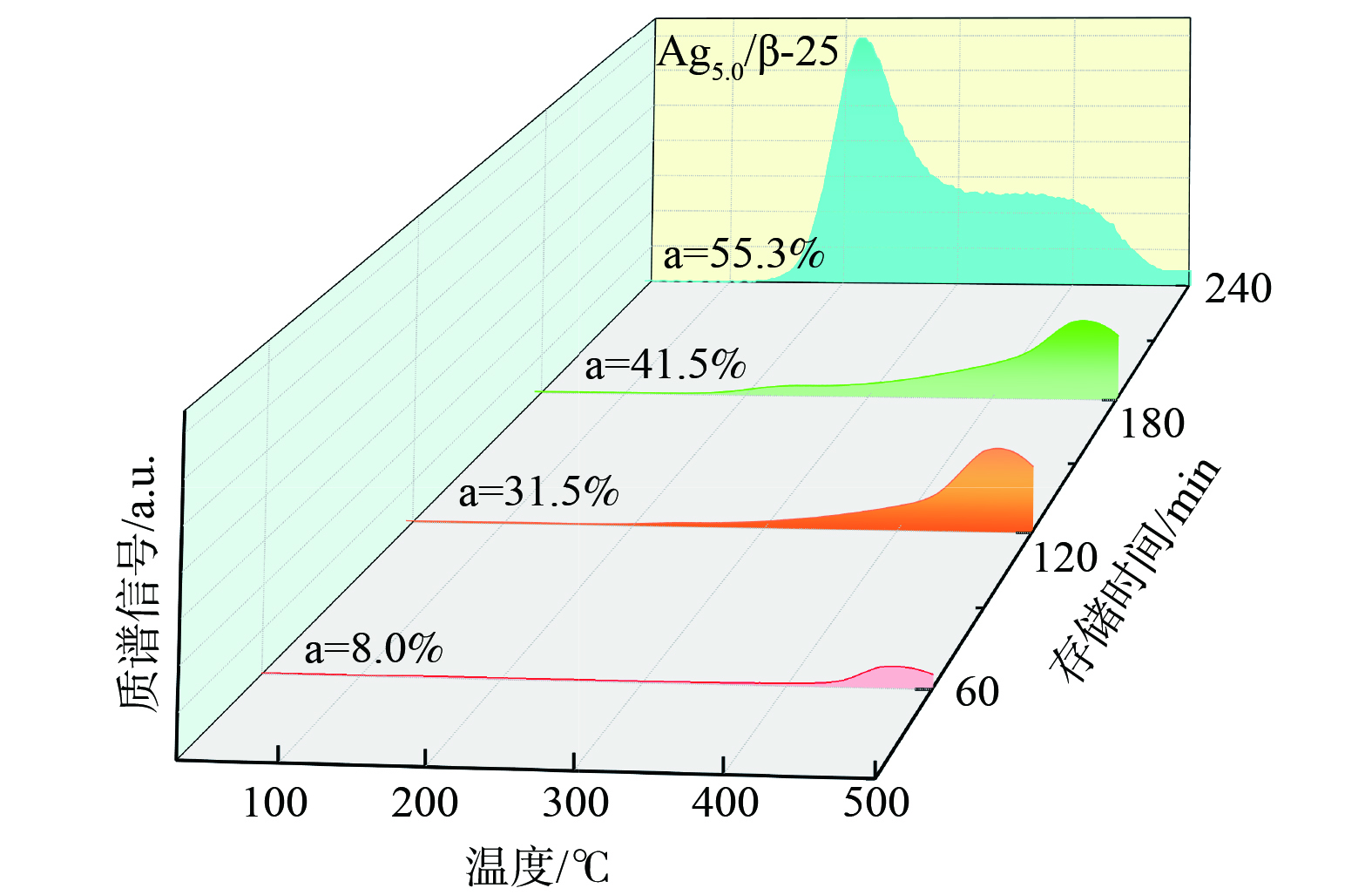 图 7 Ag5.0/β-25催化剂上连续3次“存储-原位再生”循环甲苯的性能Figure 7. Purification property of toluene during three consecutive “storage-in situ regeneration” cycles over Ag5.0/β-25 catalysts
图 7 Ag5.0/β-25催化剂上连续3次“存储-原位再生”循环甲苯的性能Figure 7. Purification property of toluene during three consecutive “storage-in situ regeneration” cycles over Ag5.0/β-25 catalysts2.3 “存储-原位氧化”循环净化甲苯
基于图6数据,在“存储-原位氧化”循环净化甲苯实验中,将存储时间控制为180 min,此时甲苯全部存储在Ag+和Agnδ+位点上,其存储量可达0.32 mmol·gcat−1;在原位氧化再生阶段,存储的甲苯全部被氧化成为COx。图7表明,在3次循环测试中,碳平衡均大于98%,没有甲苯的提前脱附,这说明Ag5.0/β-25催化剂在“存储-原位氧化”循环再生净化甲苯过程中稳定性很好,也充分说明了通过调控存储位点实现“存储-原位氧化”循环再生净化甲苯这一思路的可行性。
3. 结论
1) 通过制备不同Ag负载量的β分子筛催化剂,研究了催化剂中Ag的状态。通过将甲苯的脱附温度与甲苯在不同存储位点上的键合强度相关联,将催化剂表面存储位点分为3个级别。强存储位点即离子交换位上为Ag+,对于甲苯有最强的键合能力,程序升温至500 ℃时仍未脱附。中强存储位点即离子交换位为Agnδ+团簇,其与甲苯之间可形成较稳定的π键合,在温度为250~500 ℃时出现脱附。弱存储位点即金属Ag纳米粒子和分子筛的酸性位,与甲苯间键合最弱 (范德华力) ,在210 ℃左右出现脱附。2) 依据甲苯在Agx/β-25分子筛上的存储强度,通过控制存储时间来调控甲苯在催化剂上的优先吸附顺序,使甲苯优先吸附在Ag+和Agnδ+强存储位点上,使脱附温度高于氧化温度,即可实现原位氧化再生。3) 通过优化Ag的最佳负载量为5%,Ag在催化剂表面高度分散,并主要以Ag+和小尺寸的Agnδ+的形式存在。此时吸附在Ag+和Agnδ+上的甲苯可达0.32 mmol·gcat−1,且存储的甲苯可全部被原位热氧化为COx。4) 在3次循环测试中,碳平衡均大于98%,再生过程中未发生甲苯的提前脱附,这说明Ag5.0/β-25催化剂在“存储-原位氧化”循环再生净化甲苯过程中有很好的稳定性及重现性,证实了这种再生思路的可行性。
-
图 1 不同含量Ag改性的β-25催化剂XRD谱图
Figure 1. XRD patterns of Ag/β-25 catalysts modified with different Ag contents
图 2 不同含量Ag改性的β-25催化剂UV-vis谱图
Figure 2. UV-vis spectra of Ag/β-25 samples modified with different Ag contents
图 3 Agx/β-25系列催化剂上甲苯存储量对比
Figure 3. Comparison of toluene storage on Agx/β-25 catalysts
图 4 Agx/β-25系列催化剂上甲苯催化氧化活性
Figure 4. Catalytic oxidation activity of toluene on Agx/β-25 catalysts
图 5 Agx/β-25系列催化剂上存储的甲苯在N2下程序升温脱附
Figure 5. TPD experiments of adsorbed C7H8 under N2 atmosphere on Agx/β catalysts
图 6 Agx/β-25系列催化剂上强键合甲苯含量
Figure 6. Content of strongly bonded toluene on Agx/β -25 catalysts
-
[1] CHEN B B, WU L, WU B, et al. Catalytic materials for low concentration VOCs removal through “storage-regeneration” cycling[J]. ChemCatChem, 2019, 11(16): 3646-3661. doi: 10.1002/cctc.201900581 [2] POSCHL U, SHIRAIWA M. Multiphase chemistry at the atmosphere-biosphere interface influencing climate and public health in the anthropocene[J]. Chemical Reviews, 2015, 115(10): 4440-4475. doi: 10.1021/cr500487s [3] HE C, CHENG J, ZHANG X, et al. Recent advances in the catalytic oxidation of volatile organic compounds: a review based on pollutant sorts and sources[J]. Chemical Reviews, 2019, 119(7): 4471-4568. doi: 10.1021/acs.chemrev.8b00408 [4] WANG Y D, CHEN B B, WU B, et al. “Storage-oxidation” cycling process for indoor benzene removal at room temperature[J]. Catalysis Today, 2017, 297: 193-200. doi: 10.1016/j.cattod.2017.04.054 [5] ZHANG J, XU X P, ZHAO S Z, et al. Recent advances of zeolites in catalytic oxidations of volatile organic compounds[J]. Catalysis Today, 2023, 410: 56-67. doi: 10.1016/j.cattod.2022.03.031 [6] MURINDABABISHA D, YUSUF A, SUN Y, et al. Current progress on catalytic oxidation of toluene: a review[J]. Environmental Science and Pollution Research International, 2021, 28(44): 62030-62060. doi: 10.1007/s11356-021-16492-9 [7] LIOTTA L F. Catalytic oxidation of volatile organic compounds on supported noble metals[J]. Applied Catalysis B: Environmental, 2010, 100(3/4): 403-412. [8] ROKICIŃSKA A, DROZDEK M, DUDEK B, et al. Cobalt-containing BEA zeolite for catalytic combustion of toluene[J]. Applied Catalysis B: Environmental, 2017, 212: 59-67. doi: 10.1016/j.apcatb.2017.04.067 [9] LI Y, LI L, YU J H. Applications of zeolites in sustainable chemistry[J]. Chem, 2017, 3(6): 928-949. doi: 10.1016/j.chempr.2017.10.009 [10] ILIĆ B, WETTSTEIN S G. A review of adsorbate and temperature-induced zeolite framework flexibility[J]. Microporous and Mesoporous Materials, 2017, 239: 221-234. doi: 10.1016/j.micromeso.2016.10.005 [11] MIKHEEVA N N, ZAIKOVSKII V I, LARICHEV Y V, et al. Toluene abatement on Ag-CeO2/SBA-15 catalysts: Synergistic effect of silver and ceria[J]. Materials Today Chemistry, 2021, 21: 100530. doi: 10.1016/j.mtchem.2021.100530 [12] DZWIGAJ S, MILLOT Y, KRAFFT J-M, et al. Incorporation of silver atoms into the vacant T-atom sites of the framework of SiBEA zeolite as mononuclear Ag (I) evidenced by XRD, FTIR, NMR, DR UV–vis, XPS, and TPR[J]. The Journal of Physical Chemistry C, 2013, 117(24): 12552-12559. doi: 10.1021/jp401849e [13] SHI C, CHENG M J, QU Z P, et al. On the correlation between microstructural changes of Ag-H-ZSM-5 catalysts and their catalytic performances in the selective catalytic reduction of NOx by methane[J]. Journal of Molecular Catalysis A: Chemical, 2005, 235(1/2): 35-43. [14] KUNDAKOVIC L, FLYTZANI-STEPHANOPOULOS M. Deep oxidation of methane over zirconia supported Ag catalysts[J]. Applied Catalysis A: General, 1999, 183: 35-51. doi: 10.1016/S0926-860X(99)00043-5 [15] PALČIĆ A, VALTCHEV V. Analysis and control of acid sites in zeolites[J]. Applied Catalysis A: General, 2020, 606: 117795. doi: 10.1016/j.apcata.2020.117795 [16] BHARGAVI K, RAY D, CHAWDHURY P, et al. Oxidation of toluene by ozone over surface-modified γ-Al2O3: effect of Ag addition[J]. Catalysts, 2022, 12(4): 421. doi: 10.3390/catal12040421 [17] KRATZER J M M P. The role of the van der waals interactions in the adsorption of anthracene and pentacene on the Ag (111) surface[J]. The journal of chemical physics, 2017, 146(3): 4702. [18] XU X X, WANG P T, XU W C, et al. Plasma-catalysis of metal loaded SBA-15 for toluene removal: comparison of continuously introduced and adsorption-discharge plasma system[J]. Chemical Engineering Journal, 2016, 283: 276-284. doi: 10.1016/j.cej.2015.07.050 [19] AKIRA TAKAHASHI F H Y, AND RALPH T. YANG. Aromatics/Aliphatics separation by adsorption: new sorbents for selective aromatics adsorption by π-complexation[J]. Industrial & Engineering Chemistry Research, 2000, 39, (10): 3856-3867. [20] ELDRIDGE R B. Olefin/Paraffin separation technology: a review[J]. Industrial & Engineering Chemistry Research, 1993, 32(10): 2208-2212. [21] HOU X J, LI H, HE P, et al. Molecular-level investigation of the adsorption mechanisms of toluene and aniline on natural and organically modified montmorillonite[J]. The Journal of Chemical Physics, 2015, 119(45): 11199-11207. doi: 10.1021/acs.jpca.5b09475 [22] NETTO M S, OLIVEIRA J S, SALAU N P G, et al. Analysis of adsorption isotherms of Ag+, Co+2, and Cu+2 onto zeolites using computational intelligence models[J]. Journal of Environmental Chemical Engineering, 2021, 9(1): 104960. doi: 10.1016/j.jece.2020.104960 [23] HESSOU E P, BEDE L A, JABRAOUI H, et al. Adsorption of toluene and water over cationic-exchanged Y zeolites: a DFT exploration [J]. Molecules, 2021, 26 (18) . [24] ASPROMONTE S G, MIR E E, BOIX A V. FTIR studies of butane, toluene and nitric oxide adsorption on Ag exchanged NaMordenite[J]. Adsorption, 2011, 18(1): 1-12. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1684
- HTML全文浏览数: 1684
- PDF下载数: 43
- 施引文献: 0
