





-
氟是自然界的微量元素之一,氟在自然界中以萤石(CaF2)、冰晶石、氟磷灰石等形式存在,这些矿物埋藏在地下,随着地下水的侵蚀与沉积,F−被溶出并富集至地下水中,使得水中的F−浓度偏高。我国高氟水分布十分广泛,尤其集中在北方地区,比如辽宁省中部,山东省西南部,围绕渤海地区以及广袤的西北地区都有大面积的高氟地下水区域,这些含氟地下水不仅给当地居民的饮水带来诸多不便,而且会给当地的基础建设和经济发展带来阻碍,根据资料显示:氟与人体的身体健康密切相关,人体每天需要摄取的氟含量大约为0.1 mg,大约90%摄入的氟会随身体新陈代谢排除体外,留下的大部分氟会存在于身体的骨骼和牙齿之中,人体摄入氟的最主要途径是饮食,这也是地方性氟中毒的主要原因,因此需要确保饮用水氟安全,过量摄入氟化物会导致人体骨骼、牙齿、肝脏、肾脏、大脑等的多器官毒性[1],目前仍缺乏有效的治疗手段,除氟技术可以有效解决饮用水中氟含量超标的问题。
目前国内主要的除氟方法有吸附法[2]、混凝沉淀法[3]、离子交换法[4]、反渗透[5]等,吸附法由于其方法便捷、经济高效等原因是目前饮用水除氟的主要方式。目前主要的吸附剂有铝基金属材料、天然矿石材料等, SANINI等[6]利用CeO2改性活性氧化铝来去除水中氟离子; AYALEW 等[7]比较了高岭土和石灰石吸附剂对地下水中的除氟效果;LAONAPAKUL等[8]通过煅烧高岭土/羟基磷灰石复合材料来吸附除氟;THIRUNAVUKKARASU等[9]对生物吸附做了系统性探讨,阐述了生物吸附未来前景和发展潜力;AMIN等[10]利用白腐菌杏鲍菇来吸附水溶液中的氟;韩晓峰等[11]采用浸渍法制备了负载镧镁的活性氧化铝吸附剂,该吸附剂在3 h内可将10 mg·L−1的含氟溶液中的氟离子去除95%左右;BAKHTA等[12]利用通过金属浸渍改性活性炭可提高其吸附性能,是一种很有前途的水处理材料。目前研究报道的大多数吸附剂存在以下缺点:1)吸附量不足,吸附效率不能满足人们的需求。2)抗干扰离子能力弱,选择性差。3) pH适宜范围窄,尤其是生物吸附剂,水相pH对吸附剂吸附性能有很大影响,一般pH越低,除氟性能越好4)材料获取困难,制作过程复杂。5)铝基金属吸附材料会有Al3+溶出的问题,不仅会造成2次污染,而且长期饮用含高铝离子的水会给人体带来危害。因此选择一种高效、安全的饮用水除氟剂是吸附法的关键。
MgO作为我国储量很大的金属氧化物,具有高吸附能力、强亲和力、热稳定性,是一种十分安全并具有发展潜力的吸附除氟材料。刘理华等[13]研究了棒状改性氧化镁及其吸附性能,发现经过改性后的氧化镁有良好的吸附活性;王慧玲等[14]利用酸改性活性氧化镁,改性后吸附容量有所提高。本实验为了提高活性氧化镁的吸附容量和吸附效率,扩充活性氧化镁适宜pH的范围,增强活性氧化镁与其他离子共存时的吸附能力,提高其选择性和除氟稳定性,以活性氧化镁作为多孔性载体,通过铁盐,钙盐浸渍使其附着在多孔性载体上或者内表面,再通过焙烧制得改性吸附剂,探讨其最佳制备条件,并通过扫描电镜,X射线粉末衍射仪,傅里叶变换红外光谱仪对吸附剂进行表征。
-
氧化镁,柠檬酸三钠,冰乙酸,氯化钠,氯化铁,氯化钙,氢氧化钠,氟化钠,碳酸钠,硝酸钠,硫酸钠,磷酸钠,盐酸均为分析纯。
CJJ78-1磁力搅拌器;PF-2-01氟离子选择电极;232参比电极;Multi3620 pH计;PX223ZH/E电子分析天平;DHG-9013A恒温鼓风干燥箱;SX2-2.5-10箱式高温电阻炉;HWS-26恒温水浴锅;D8 advance X射线粉末衍射仪;MERLIN compact 扫描电子显微镜;is5 傅里叶变换红外光谱仪。
-
将氧化镁在420 ℃下灼烧1.5 h得到活性氧化镁,将活性氧化镁与金属盐浸渍液按照1 g:20 mL的比例混合,并置于磁力搅拌器上搅拌2 h,静置,待出现明显固液分离,用吸管将上清液吸出,将剩余含水的粘稠物质放入烘箱在105 ℃下烘干12 h,将干燥固体取出研磨成粉末,放入马弗炉内,在一定温度下高温焙烧2 h,制得改性活性氧化镁吸附剂。
对单一含氟水和模拟含氟饮用水用改性活性氧化镁吸附除氟,用盐酸和氢氧化钠溶液调节pH,单一含氟水中只含有氟离子,模拟含氟饮用水中杂质离子有Cl−、CO32−、SO42−、NO3−、HPO42−、Na+。在实验过程中,改变吸附时间、吸附剂投加量、pH等实验因素以进行不同条件下的吸附实验对比,吸附后,使用电极法测试滤液的氟离子浓度,计算除氟率和氟吸附量,每组实验重复3次,取平均值作为最终结果。
-
活性氧化镁、FeCl3-MgO、Fe2(SO4)3-MgO、CaCl2-MgO SEM表征结果如图1所示,活性氧化镁在电镜下结构松散,颗粒之间空隙较大,改性吸附剂表面变得粗糙,表面有凸起的颗粒,比表面积变得更大。BET比表面积测试结果表明,活性氧化镁、FeCl3-MgO、CaCl2-MgO的比表面积分别为17.724、47.423、56.642 m2·g−1,改性后的吸附剂比表面积均提升了2倍以上,特别是经过Fe2(SO4)3改性后的吸附剂,由于活性氧化镁在与水改性过程中生成了具有层状结构的氢氧化镁,而通过Fe2(SO4)3 改性过程中并没有破坏这一结构,而是在此结构上完成了负载,再经过高温焙烧后失去水时的层状结构被破坏转为交错的玫瑰状,使得比表面积进一步增大[15],比表面积由改性前的17.724 m2·g−1提高至62.492 m2·g−1。负载在氧化镁表面的铁盐和钙盐增加了吸附剂整体的正电性,F−本身带有负电,彼此之间的吸引有利于F−游离至改性吸附剂附近,改性吸附剂更大、更粗糙的比表面积有利于更多的与F−接触,使得吸附初期F−与吸附剂表面接触更加频繁,同时Fe与 F−直接的络合反应加快了反应速率。
-
活性氧化镁、FeCl3-MgO、Fe2(SO4)3-MgO、CaCl2-MgO的XRD表征结果如图2所示。由图2(b)可以看出,在衍射角为37.00、42.95、62.35、74.75、78.65°处出现MgO衍射峰,说明MgO仍然是改性吸附剂的主体成分。在FeCl3-MgO的图谱中出现了少量的Mg(OH)2 (在18.60、38.05、50.83、58.50、68.50°附近的衍射峰),但改性前后各组分峰形和峰位置并未发生明显的变化。说明改性过程并没有改变活性氧化镁的晶格结构。在图2(b)中未出现铁的吸收峰,说明铁可能以无定型的物质形式附着在氧化镁表面[11]。
Fe2(SO4)3-MgO的XRD图谱中只有氧化镁出现并没有出现Mg(OH)2,原因是在最佳焙烧温度(500 ℃)下Mg(OH)2重新分解为氧化镁,可见铁盐的改性并没有改变活性氧化镁的物质结构,铁盐并没有出现稳定的物质结构,可能是以无定型金属化合物的形式附着在活性氧化镁表面。
CaCl2-MgO的XRD图谱和FeCl3-MgO的 XRD图谱类似,图中的衍射峰依旧是以MgO为主体,强度较低的峰位置出现少量的Mg(OH)2。这是因为在400 ℃的焙烧温度下,钙盐的改性对于氧化镁晶体结构影响较小,钙类物质并没有出现稳定的物质结晶,可能是其以无定型的形式附着在氧化镁表面。
-
活性氧化镁、FeCl3-MgO、Fe2(SO4)3-MgO、CaCl2-MgO FTIR红外光谱如图3所示。改性对于活性氧化镁表面官能团或者化学键有一定影响。可见,吸附前的活性氧化镁在1 439 、1 633 、3 434 cm−1出现的吸收峰归属为CO32−非对称伸缩振动、吸附水O—H的弯曲振动和伸缩振动。FeCl3-MgO在1 409 、1 437 、3 404 cm−1处出现的吸收峰指示的官能团或共价键与活性氧化镁一致,在3 628 cm−1和3 697 cm−1处出现的吸收峰归属为O—H的伸缩振动。Fe2(SO4)3-MgO在1 477 、1 634、3 436 cm−1处出现的吸收峰对应的官能团与活性氧化镁一致,1 089 cm−1处出现SO42−中的S=O振动的吸收峰,其形成可能来自于高价位S与Fe,Mg氧化物的双配位吸附[16-17]。CaCl2-MgO在1 448、1 638、3 427、3 696 cm−1处出现的吸收峰指示与活性氧化镁一致,在2 939 cm−1处出现的吸收峰与钙,水汽和二氧化碳生成的化合物有关,可见CaCl2-MgO对二氧化碳等物质的吸收高于活性氧化镁。
由图3可以看出,吸附后的FeCl3-MgO在426 cm−1处出现新的吸收峰,其归属为Mg—F键和Fe—F键的出现,CO32−的非对称伸缩振动吸收峰和吸附水O—H的伸缩振动吸收峰均出现蓝移,说明FeCl3-MgO表面吸收水和CO32−结合更稳定。Fe2(SO4)3-MgO吸附后在439 cm−1和3 700 cm−1处出现新的吸收峰,分别归属为Mg—F键,Fe—F键和结构水O-H的伸缩振动,指示为SO42−中S=O振动的吸收峰和CO32−非对称伸缩振动的吸收峰均出现红移,说明Fe2(SO4)3-MgO表面的SO42−和CO32−结合更不稳定。CaCl2-MgO在443 cm−1处出现吸收峰,归属为Mg—F键和Ca2+的移动。吸收水O—H的弯曲振动峰和CO32−非对称伸缩振动峰均出现蓝移,说明CaCl2-MgO表面吸收水和CO32−的结合稳定性增强。活性氧化镁、FeCl3-MgO、Fe2(SO4)3-MgO、CaCl2-MgO在含氟废水中水解后,吸附剂表面附着大量O—H,因此活性氧化镁基吸附剂F−与吸附剂表面的羟基的离子互换为除氟的主要过程,Mg-F键的出现是因为水中F−离子与O—H发生离子交换吸附于吸附剂表面,其过程如式(1)所示。
同时MgO也可与F−发生选择性吸附,其可由共价键和络合反应实现,而水中的MgOH+和MgF+又可通过静电吸引进一步吸附F−[18],Fe—F键的生成说明改性活性氧化镁表面的无定型铁水解后生成Fe(OH)3,之后主要通过络合反应 (配位反应) 实现Fe与F的结合,同时释放OH−,络合反应吸附能量比离子反应更强,对F−的选择性更高。钙盐改性后的氧化镁虽然没有Ca—F键的出现,但钙盐是以无定型的形式附着在MgO表面,Ca2+本身可以与F−发生反应生成CaF2沉淀,CaF2比CaCO3,Ca(HCO3)2等其他阴离子结合的盐类更稳定,因此,F−会优先与Ca2+结合,使得钙盐改性后的吸附剂对氟离子选择性吸附增强,抵抗其他阴离子干扰的能力增强,并且铁盐和钙盐对氟离子的亲和力很高,氟离子被吸附后不易被替换。
-
在pH分别为2、4、6、8、10、12的条件下,活性氧化镁、FeCl3-MgO、Fe2(SO4)3-MgO、CaCl2-MgO的Zeta电位测试结果如图4所示。可见,随着pH的升高,3种改性吸附剂的Zeta电位在不断下降,由最初的22 mV降至−4.2 mV左右。这是因为在pH较低时溶液中含有较多的H+,H+与改性吸附剂结合,使得吸附剂表面电负性降低;当pH升高时,溶液中含有较多的OH−,OH−与改性吸附剂结合,使得表面电负性增加。3种改性吸附剂的零点电位在9.97左右,当pH<9.97时,改性吸附剂表面带有正电,有利于吸附带有负电的氟离子;当pH>9.97时,吸附剂表面带有负电会排斥带有负电的氟离子。改性后的吸附剂零电位点所对应的pH较高,在较宽的pH范围内都有利于吸附氟离子,活性氧化镁由于零点电位较低,吸附氟离子的有利pH范围较窄。
-
在温度为25 ℃,浸渍液中金属盐离子浓度分别为0.01、0.03、0.05、0.1 、0.15 mol·L−1,初始氟离子溶液质量浓度为20 mg·L−1,吸附时间为12 h,浸渍液中金属盐离子浓度对除氟率的影响如图5所示。可见,FeCl3-MgO会随着FeCl3浸渍液浓度增加除氟率先升高后降低。其原因是当FeCl3浓度较低时,浸渍改性会生成更多的氟离子吸附位点,当FeCl3浓度逐渐升高时,FeCl3会堵塞活性氧化镁表面的空隙,使得吸附位点与氟离子接触变少,导致除氟率下降,因此,FeCl3最佳浸渍浓度为0.05 mol·L−1。
随着CaCl2浸渍液离子浓度增加,CaCl2-MgO的除氟效果略微提升。这是因为少量CaCl2会提供更多的氟离子吸附位点,随着浓度的提高,过多的CaCl2会导致氧化镁表面吸附孔隙被堵塞,氟离子与活性氧化镁表面的吸附位点接触概率变低,导致除氟率下降。因此, CaCl2-MgO的最佳CaCl2浸渍液离子浓度为0.01 mol·L−1。
Fe2(SO4)3-MgO受Fe2(SO4)3浸渍液离子浓度变化影响较大。当Fe2(SO4)3浸渍液离子浓度浓度由0.01 mol·L−1升到0.03 mol·L−1时,Fe2(SO4)3-MgO的除氟率由80%迅速降至50%左右;当Fe2(SO4)3由0.05 mol·L−1增加到0.10 mol·L−1时,除氟率有一定上升,但除氟率仍低于Fe2(SO4)3为0.01 mol·L−1时制得的Fe2(SO4)3-MgO。这是因为Fe2(SO4)3有着较大的分子质量,当Fe2(SO4)3以无定型结构附着在氧化镁表面时,更容易堵塞氧化镁表面的吸附位点,使得氟离子难以被吸附,导致除氟率显著下降。因此,确定0.01 mol·L−1为Fe2(SO4)3-MgO的最佳浸渍液离子浓度。
-
在温度为 25 ℃,初始氟离子质量浓度为20 mg·L−1,调节溶液pH至7,焙烧温度对吸附剂除氟效果的影响如图6所示。可见,随着焙烧温度增加,吸附容量随之增加,当达到一定温度后,吸附剂的吸附量基本维持不变, 400 ℃为FeCl3-MgO和CaCl2-MgO的最适焙烧温度,500 ℃为Fe2(SO4)3-MgO的最适焙烧温度。当焙烧温度过低时,吸附量较低,原因是吸附剂内结构水散失过少,不利于吸附位点的生成;焙烧温度过高,也会略微影响吸附量,原因是温度过高颗粒的过度结晶和团聚使吸附剂表面作用降低,吸附性能下降。焙烧使得铁盐和钙盐以无定型形态负载在活性氧化镁表面,当达到一定温度时,活性氧化镁表面负载量达到饱和,吸附剂的吸附量便基本维持不变。
-
在温度为25 ℃时,分别向实验水样中投加不同剂量的活性氧化镁、FeCl3-MgO、Fe2(SO4)3-MgO、CaCl2-MgO,初始氟离子溶液质量浓度为20 mg·L−1,吸附时间12 h, 吸附剂投加量对于吸附效果的影响如图7所示。4种吸附剂的除氟率均随其投加量的增加先快速升高然后趋于稳定,最终达到吸附平衡。由图7(a)、图7(b)、图7(d)可见,活性氧化镁、FeCl3-MgO、CaCl2-MgO3种吸附剂在投加量为2 g·L−1时除氟率均达到最大,吸附剂投加量继续增加,除氟率基本维持不变。由图7(c)可见,Fe2(SO4)3-MgO除氟率随着投加量的增加快速升高,但除氟率始终略低于活性氧化镁、FeCl3-MgO、CaCl2-MgO,当吸附剂用量达到4.5 g·L−1左右,除氟率趋于稳定并与活性氧化镁,FeCl3-MgO和CaCl2-MgO的除氟率相当,继续增加吸附剂的投加量,除氟率基本保持不变,达到吸附平衡。
由图7可见,在吸附剂用量未达到3 g·L−1时,随着吸附剂用量的增加,4种吸附剂的吸附容量均会快速降低,当吸附剂投加量大于3 g·L−1,吸附容量会缓慢下降。这是因为刚开始投加吸附剂的时候,吸附氟离子的活性位点较多,随着投加量的增加,吸附位点逐渐被占据,利用率下降,当吸附剂用量达到3 g·L−1左右时,基本达到吸附平衡,这时吸附剂会缓慢吸收溶液中的氟离子,吸附位点基本被占据,吸附容量会缓慢下降。
-
在温度为25 ℃,改性吸附剂投加量为2 g·L−1,初始氟离子溶液质量浓度为20 mg·L−1,吸附时间对于吸附效果的影响如图8所示。在吸附初期,4种吸附剂吸附速率均较快,随后,吸附速率减慢,直至达到吸附平衡,活性氧化镁吸附氟离子速率较慢,7 h达到吸附平衡,除氟效率低,CaCl2-MgO、FeCl3-MgO、Fe2(SO4)3-MgO除氟速率快,在2 h左右达到吸附平衡,除氟率均可达95%以上。
-
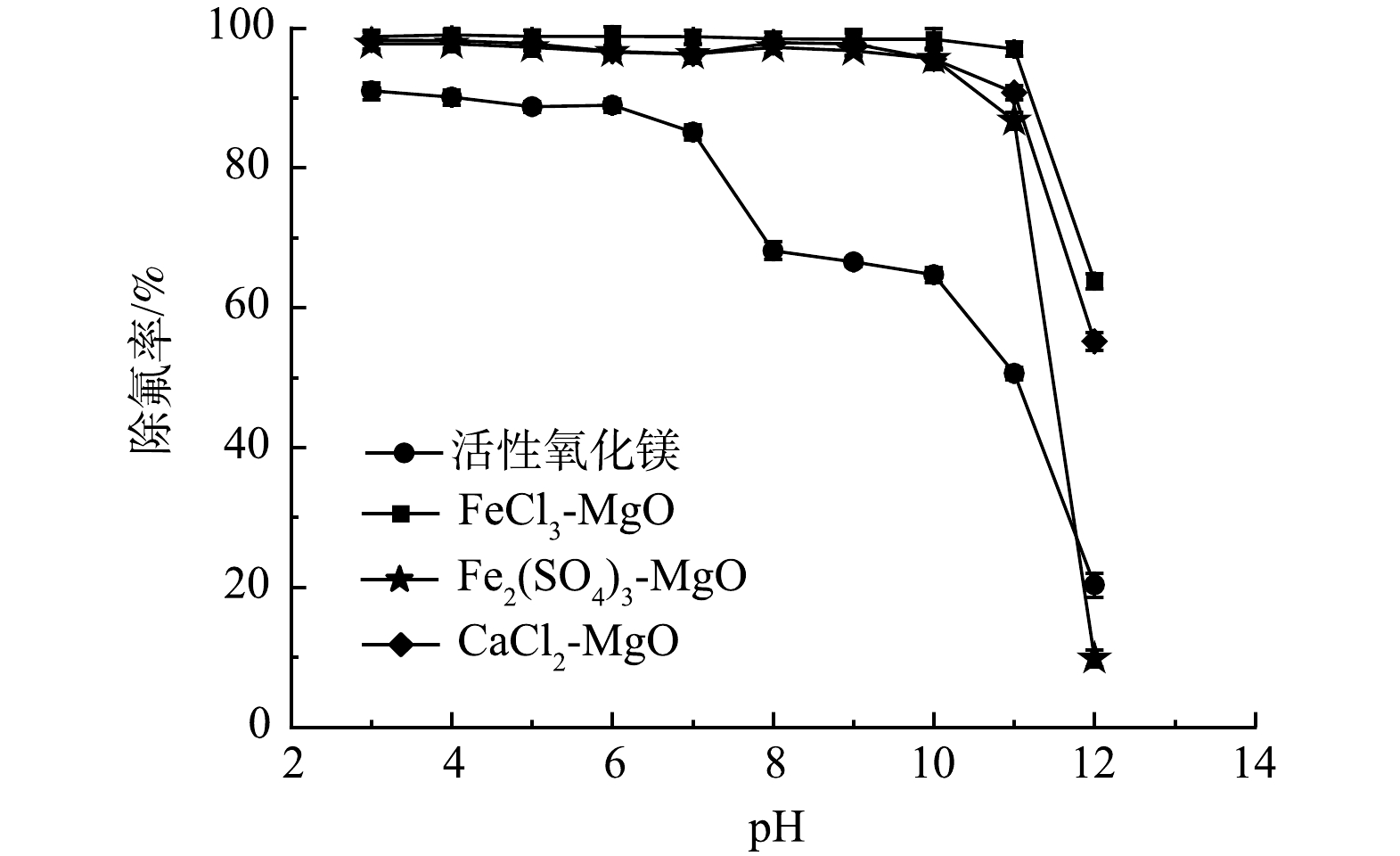
含氟溶液的pH会引起吸附剂表面电位的变化,影响吸附剂的吸附性能。在温度为25 ℃,改性吸附剂投加量为2 g·L−1,初始氟离子溶液质量浓度为20 mg·L−1,吸附时间为12 h,探究pH对去除率的影响。如图9所示, pH<7时活性氧化镁可以达到90%左右的去除率,当pH>7时活性氧化镁除氟率有明显的下降, pH>10时,活性氧化镁除氟率进一步下降。由此得出pH对于活性氧化镁除氟性能有很大的影响,活性氧化镁在酸性或者中性的环境下能有较好的除氟效果。随着pH升高,溶液中存在较多的OH−,OH−易于和金属吸附剂表面结合,使得活性氧化镁表面带负电,不利于吸附同为阴离子的氟离子。
改性后的3种吸附剂在pH>11后除氟率均有显著下降。这是因为改性后的吸附剂表面酸性位点增加,当pH升高时,溶液中的OH−会与吸附剂表面的酸性位点结合,从而提高吸附剂pH的缓冲能力,但吸附位点数量有限, OH−与吸附位点结合后使得F−不能与吸附位点结合,导致除氟率降低。由Zeta电位变化可以得出,改性后的吸附剂由于零电位点所对应pH较高,因此,在较宽的pH范围内均有良好的除氟效果。此外,有研究表明,SO42−与F−的交换可以在碱性条件下顺利进行,这也是经过Fe2(SO4)3改性后的活性氧化镁适宜pH扩大的原因之一。在pH<11的环境下,CaCl2-MgO、FeCl3-MgO、Fe2(SO4)3-MgO均能保持90%以上的去除率,改性后的吸附剂适应水溶液酸碱环境的能力显著增强。
-
天然的含氟水是一个复杂的水体环境,在进行除氟处理时,要考虑水体中其他离子对除氟的影响。为此,在25 ℃,吸附剂投加量为2 g·L−1,初始氟离子质量浓度为5mg·L−1,吸附时间12 h,共存离子(Cl−、SO42−、CO32−、NO3−、HPO42−、Na+)的质量浓度为5 mg·L−1的条件下,考察吸附剂在复杂水体环境中的除氟效果,结果如图10所示。
活性氧化镁在实际含氟饮用水环境中达到吸附平衡后对氟离子的去除率不足10%,除氟效果差, CaCl2-MgO、FeCl3-MgO和Fe2(SO4)3-MgO在实际含氟饮用水溶液中达到吸附平衡后依然保持了90%以上的氟离子去除率,剩余F−质量浓度均小于1 mg·L−1,满足国家饮用水含氟标准,可见改性后的吸附剂对氟离子的选择性显著增强,在实际饮用水除氟中有良好的效果。
-
取0.1 g活性氧化镁、FeCl3-MgO、Fe2(SO4)3-MgO、CaCl2-MgO分别置于100 mL初始氟离子溶液质量浓度为5、10、20、25、100、200、300 mg·L−1的模拟含氟水中,在室温25 ℃下测量吸附12 h后的氟离子质量浓度,分别对其进行Langmuir(式(2))和Freundlich(式(3))等温吸附模型拟合,拟合结果如表1所示。
式中:Ce为吸附后溶液的平衡质量浓度,mg∙L−1;Qe为平衡吸附量,mg∙g−1;Qmax为最大吸附量,mg∙g−1;KL为 Langmuir 平衡常数,L∙mg−1;KF为Freundlich 平衡常数,L∙mg−1
4种吸附剂的Langmuir吸附等温模型拟合相关性系数R2均大于Freundlich吸附等温模型拟合相关性系数,说明活性氧化镁、FeCl3-MgO 、Fe2(SO4)3-MgO、CaCl2-MgO,对F−的吸附更倾向于单分子层吸附[19],FeCl3-MgO、Fe2(SO4)3-MgO、 CaCl2-MgO的最大吸附容量均有显著增加,经过氯化铁改性后的吸附剂吸附容量大于经过硫酸铁改性的吸附容量,原因是经过硫酸铁改性后的吸附剂吸附达到短暂平衡后又有F−溶出。
在Freundlich吸附等温模型中,1/n可反映吸附的有利程度,0<1/n<1有利于吸附的发生。由表1可以看出,4种改性吸附剂1/n均为0<1/n<1,说明4种吸附剂均有较好的吸附强度,3种改性吸附剂的1/n均小于活性氧化镁1/n,说明FeCl3、Fe2(SO4)3和CaCl2的改性有利于活性氧化镁的吸附 [20]。
-
取0.1 g CaCl2-MgO于烧杯中,加入超纯水100 mL,于25 ℃下以150 r·min−1振荡120 min,利用离心机将振荡后的固液分离,采用镁试剂测试水样中的镁离子含量,结果未检出镁离子,另外对CaCl2-MgO除氟后的水样进行检测,同样未检测出镁离子;取0.1 g 铁盐改性吸附剂于烧杯中,在上述相同的条件下重复实验未见有沉淀析出。表明Mg2+不会随着反应渗入溶液中,不会造成二次污染。
取0.1 g铁盐改性吸附剂于烧杯中,在上述相同的条件下取上清液加入少量氢氧化钠溶液,未见有沉淀出现;取0.1 g CaCl2-MgO于烧杯中,在上述相同的条件下取上清液加入少量碳酸钠溶液,未见出现沉淀,表明浸渍盐的离子并不会随着反应渗到溶液中。这说明改性吸附剂稳定性良好,不会造成新的污染。
取0.1 g改性吸附剂与烧杯中,加入超纯水100 mL,于25 ℃下以150 r·min−1振荡120 min,采用镁试剂测试水样中的镁离子含量,结果未检出镁离子。另外对改性吸附剂除氟后的水样进行检测,同样未检测出镁离子,表明改性吸附剂稳定性好,除氟过程未有其他离子溶出,不会造成二次污染。
取0.1 g铁盐改性吸附剂与烧杯中,在上述相同的操作条件下,未见有沉淀出现;取0.1 g CaCl2-MgO于烧杯中,在同样的操作条件下,未出现沉淀,表明浸渍盐的离子并不会随着反应渗到溶液中。这说明改性吸附剂稳定性良好,不会造成新的污染。
-
1)改性后的氧化镁内部结构更加紧密,比表面积显著增加,与氟离子结合的活性位点增多,且活性位点更加稳定,从而提高了对氟离子的吸附效率,由铁盐和钙盐改性后的吸附剂仍以氧化镁为主体,钙盐和铁盐是以无定型的形态附着在活性氧化镁表面。
2)活性氧化镁通过F−与OH−的离子交换去除废水中的氟离子,铁盐改性的吸附剂由于Fe-F键的出现,吸附过程不仅有离子交换还有络合反应,络合反应的反应能量大于离子交换,络合反应可加强吸附剂对氟离子的选择性吸附;钙盐改性的吸附剂尽管没有形成Ca-F键,但Ca2+可与F−反应直接生成CaF2沉淀,CaF2比其他阴离子与F−结合的稳定性高,经过钙盐,铁盐改性后的吸附剂抵抗共存阴离子干扰的能力大大增强
3)改性吸附剂吸附容量显著增加,吸附速率显著提高。适宜的浸渍浓度和适宜的焙烧温度会增强改性吸附剂的除氟效果,FeCl3-MgO和CaCl2-MgO的最佳焙烧温度是400 ℃,Fe2(SO4)3-MgO的最佳焙烧温度是500 ℃。
4)改性后的吸附剂有较高的零电位点,适宜pH范围变宽,pH在3~10内均可维持较高的除氟率。改性吸附剂符合Langmuir等温模型,改性吸附剂除氟后水样未有镁离子和浸渍盐离子溶出,不会造成2次污染。
金属盐改性活性氧化镁对饮用水中氟离子的去除性能
Removal performance of metal salt modified active magnesium oxide on fluoride ions in drinking water
-
摘要: 饮用水中氟含量超标会损害人体的健康,本研究研制了一种改性吸附剂用于去除饮用水中过量的氟离子。本研究采用浸渍,焙烧的方法将金属盐负载到活性氧化镁上,制备改性活性氧化镁,考察了其除氟效果和吸附除氟的主要影响因素。结果表明,经过钙盐,铁盐改性后的活性氧化镁最大吸附容量显著增加,在复杂水体环境中仍有突出的除氟效果;改性吸附剂适宜pH为3~10;金属盐改性活性氧化镁处理水样不会有Mg2+和浸渍盐离子溶出,是一种安全可靠的吸附除氟材料。Abstract: Excessive fluoride content in drinking water can harm human health. In this study, a modified adsorbent was developed to remove excessive fluoride ions from drinking water. The impregnation and calcination methods were used to load metal salts onto activated magnesium oxide for the preparation of modified activated magnesium oxide. The fluoride removal effect and the main influencing factors were studied. The results showed that the maximum adsorption capacity of activated magnesium oxide modified with calcium and iron salts significantly increased, and it still had outstanding fluoride removal effects in complex water environments; The pH values of 3~10 were suitable for the modified adsorbent; Metal salt modified active magnesium oxide treatment of water samples did not release Mg2+ and immersion salt ions, it is a type of safe and reliable material for adsorption and fluoride removal.
-
汞是一种具有较高毒性和生物累积性的重金属[1-2],其排放源通常分人为排放与自然排放2类。自然排放引起的汞污染约占汞排放总量的1/4,人为排放是汞污染的主要原因[3]。与燃煤电站烟气汞排放污染类似,冶炼行业烟气亦是汞人为排放的重要来源,其烟气中汞的质量浓度为9.8~30 mg·m−3,为普通燃煤电厂烟气中汞含量的近百倍。即使经过冷凝后回收烟气中部分高浓度液态Hg0,但剩余的Hg0含量依然较高,远超国家限制标准[4-5]。因此,亟需探寻清洁高效的冶炼行业烧结烟气Hg0治理方法。
光催化技术由于其绿色、高效和经济等优点已得到迅速发展。其中,TiO2是研究最广泛的光催化剂之一[6],具有高化学稳定性、无毒无害、较高的光电转换效率、低成本等优点。然而,TiO2基光催化剂不能利用资源丰富的太阳能,仅能在紫外线(ultraviolet, UV)照射下获得优异的脱汞性能。寻找高效、可见光驱动且低成本的光催化剂去除Hg0仍是当前一项极具挑战的工作。卤氧化铋BiOX(X=I、Br、Cl)具有成本低、无毒的优点,对有机污染物具有良好的光催化降解性能[7-9]。BiOX是层状结构材料,易在[Bi2O2]2+层和双[X]−层间形成内部静电场,能有效地分离和迁移光生电子-空穴(e−-h+)对[10-11]。三者之中,BiOCl较宽的禁带宽度限制了其可见光催化性能;BiOI则具有较窄的禁带宽度(1.77~1.92 eV)及较宽的可见光响应范围,致使其光生e−-h+对复合率较高,其光催化性能也不理想[12]。
有学者通过多种方法,如控制形貌[13]、引入氧空位[14]、离子掺杂[15]和构建异质结[16]等来提高BiOX的光催化能力。SUDHARANI[17]等采用简便水热法合成了BiOI和30%CuO/BiOI纳米复合材料,并发现与BiOI相比,纳米花状30%CuO/BiOI光催化剂在60 min内对MO的降解效率能提升40%。陈颖等[18]采用改进Hummers法制备出还原氧化石墨烯(reduced graphene oxide, RGO)并利用微波蚀刻法将其与BiOCl/Bi2WO6耦合,发现n(Bi2WO6):n(RGO-BiOCl/Bi2WO6)为50%时,降解率可达94.6%,远高于BiOCl。张群等[19]采用水热法制备出BiOI/BiOBr催化剂,发现荧光照射下的RhB脱色率高达100%,为BiOBr的1.5倍。复合光催化剂在降解有机污染物方面有优异表现,而卤氧化铋基复合光催化剂对气态单质汞降解的研究较少[20-21]。宽带隙能BiOCl和窄带隙能BiOI属于结构相似的层状结构BiOX材料。两者的复合可能综合各自优点,产生一种性能优异的光催化复合材料[22-23]。
本研究采用化学共沉淀法将BiOCl与BiOI耦合制备出复合光催化材料,在鼓泡式光催化测试平台上研究其在荧光灯辐照下对单质汞(Hg0)的脱除性能,通过N2吸附-脱附、扫描电子显微镜(SEM)、X射线衍射(XRD)、紫外-可见漫反射光谱(UV-vis DRS)、X射线光电子能谱(XPS)和电子自旋共振(ESR)等表征手段进行分析测试,以阐明复合光催化剂物理化学结构与脱汞性能间的关系。
1. 材料和方法
1.1 试剂与仪器
试剂:碘化钾(KI)和五水合硝酸铋(Bi(NO3)3·5H2O)购于天津市科密欧化学试剂有限公司;无水乙醇(C2H5OH)和冰乙酸(CH3COOH,HAc)购于山西同杰化学试剂有限公司;氯化钾(KCl)由天津市河东区红岩试剂厂提供。所有化学试剂为分析纯,用水为去离子水。
仪器:真空干燥箱(DZF-6050D,郑州南北仪器设备有限公司);磁力电动搅拌器(HJ-4AS,常州金坛良友仪器有限公司);电子天平(ESJ200-4B,沈阳龙腾电子有限公司);筛子(120目,浙江上虞五四筛具厂);比表面积及孔径分布测试仪(美国康塔仪器公司Autosorb-iQ型);场发射扫描电子显微镜(美国FEI公司FEI Quanta FEG 250);X射线衍射(XRD)仪(德国布鲁克D8 Advance型);紫外-可见分光光度计(日本日立U-4100型);X射线光电子能谱(XPS)分析仪(美国Thermo Fisher Scientific公司Escalab 250Xi型);电子自旋共振分析仪(德国Bruker公司 ER200-SRC型)。
1.2 光催化剂的制备
采用化学共沉淀法制备BiOI/BiOCl复合光催化剂,具体方法如下。在磁力搅拌作用下将0.03 mol的Bi(NO3)3·5H2O加至50 mL冰乙酸与150 mL去离子水混合液中,搅拌至完全溶解;然后加入0.027 mol的KCl和0.003 mol的KI,继续搅拌120 min;静置4 h,用去离子水和无水乙醇洗涤至中性,置于干燥箱中70 ℃干燥24 h,研磨、筛分至0.12 mm,记为1BiOI/BiOCl。其中,1表示n(I)/n(I+Cl)为10%。改变KI和KCl的浓度,制备不同n(I)/n(Cl)的光催化剂材料,分别命名为2BiOI/BiOCl和3BiOI/BiOCl。纯BiOCl和BiOI的制备方法同上,制备过程中不添加KI和KCl。
1.3 光催化剂活性评价
在鼓泡式可见光催化活性测试平台上完成光催化剂的性能评价。该平台的设备及实验流程见文献[24]。实验由N2(平衡气)、O2(体积占比约为6%)和CO2(体积占比约为12%)按一定配比组成模拟烟气。光催化反应器进口Hg0质量浓度(Cin)约为50 μg·m−3(在标准状况下)。可见光源选用商用11 W荧光灯(fluorescent lamp,FSL;佛山电器照明股份有限公司提供)。Hg0质量浓度由俄罗斯LUMEX汞仪器公司生产的RA-915M型在线测汞仪检测。模拟烟气携带汞渗透管挥发的气态Hg0进入鼓泡式光催化反应器,与反应器中光催化剂产生的活性物质反应,可达到Hg0光催化氧化为Hg2+的目的。由于反应器流出的模拟烟气中含有一定量的酸性气体和水蒸气,会影响测汞仪的测量精度,因此模拟烟气在进入测汞仪前需进行酸性气体的洗涤和干燥预处理。另外,为防止汞污染环境,采用活性炭吸附方法脱除尾气中未反应的Hg0。如无特殊说明,反应溶液温度为40 ℃,光催化剂剂量为0.5 g,反应器中溶液体积为1.0 L,模拟烟气总流量为1.5 L·min−1。光催化剂对Hg0的脱除效率按式(1)计算。
η=(1−Cout/Cin)×100% (1) 式中:Cin和Cout分别表示光催化反应器进口和出口的Hg0质量浓度,μg·m−3。
1.4 密度泛函理论计算
采用密度泛函理论(density functional theory, DFT)了解2种材料的化学结构属性。采用Materials Studio软件中的CASTEP模块计算其功函数。利用P4/nmm空间群和晶格参数(a = b = 4.028 305 Å,c = 9.759 282 Å)建立BiOI模型。对于BiOCl,以a = b = 3.913872 Å、c = 7.827791 Å的晶格参数构建其模型[25-26]。为避免周期单元之间的干涉,真空层设为15 Å,截止能量为490 eV。使用3 × 3 × 1的伽玛中心k点网格对倒数空间中的布里渊区进行采样。能量收敛容差为2.0 × 10−5 eV和力收敛标准为0.01 eV·Å−1时,获得最优几何配置。
2. 结果与讨论
2.1 表征分析结果
2.1.1 孔隙结构分析结果
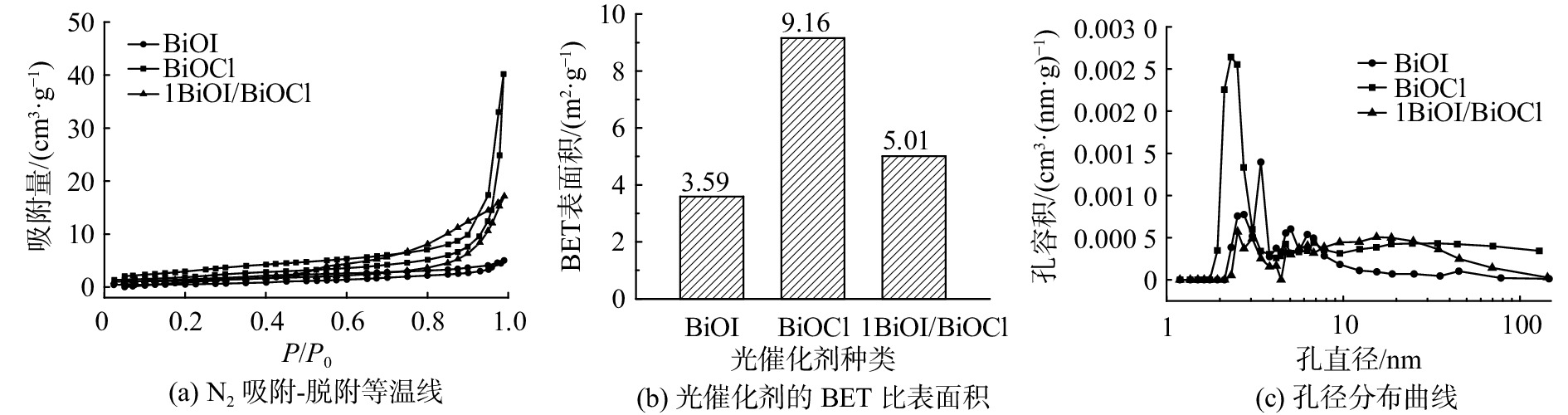
用Brunauer-Emmett-Teller(BET)方法测试得到BiOI、BiOCl和1BiOI/BiOCl样品的等温线、比表面积和孔径分布曲线,结果见图1。根据IUPAC分类可见,3种光催化剂均属于H3型滞后环的Ⅳ型等温线[13];1BiOI/BiOCl的比表面积为5.01 m2·g−1,介于BiOCl和BiOI之间。BiOI的平均孔径约为3.42 nm,多数分布于3~10 nm。BiOCl样品存在较宽的孔径分布。由于综合了BiOI和BiOCl的结构属性,1BiOI/BiOCl的平均孔径明显分为2部分,分别为3~6 nm和8~100 nm。这表明孔径分布不均,与BiOI和BiOCl不同的形貌结构有关。
2.1.2 SEM和HRTEM分析
图2为BiOI、BiOCl和1BiOI/BiOCl的SEM图。BiOI呈现多层薄片堆簇而成的花瓣结构,表面较光滑(图2(a))[27]。BiOCl样品为直径为5~15 μm的三维球状颗粒(图2(b)),由层状物质紧密构筑而成(图2(c)),BiOCl较大的比表面积应与此有关[28]。1BiOI/BiOCl耦合后的SEM与BiOCl颇为相似,为直径3~15 μm的微球结构,但在其表面上可明显观察到BiOI片状或微颗粒状物质,表明BiOI与BiOCl可良好耦合,紧密地接触将有利于异质结的形成,并促进光生e−-h+对的分离[29]。另外,其独特的微球状结构可能有利于可见光的进入和多次反射[30],进而提高光催化剂的活性。图3为1BiOI/BiOCl复合材料的HRTEM图像,呈现出0.19和0.28 nm 2种晶格条纹,分别对应BiOCl(200)晶面和BiOI(110)晶面。图4中EDS表明在1BiOI/BiOCl复合材料中存在Bi、Cl、I和O 4种元素,且n(I)/n(Cl)近似为1:9。EDS面扫描后发现,Bi、I、Cl和O元素分布颇为均匀,这表明已成功合成了高纯度BiOI/BiOCl复合光催化剂。
2.1.3 XRD分析
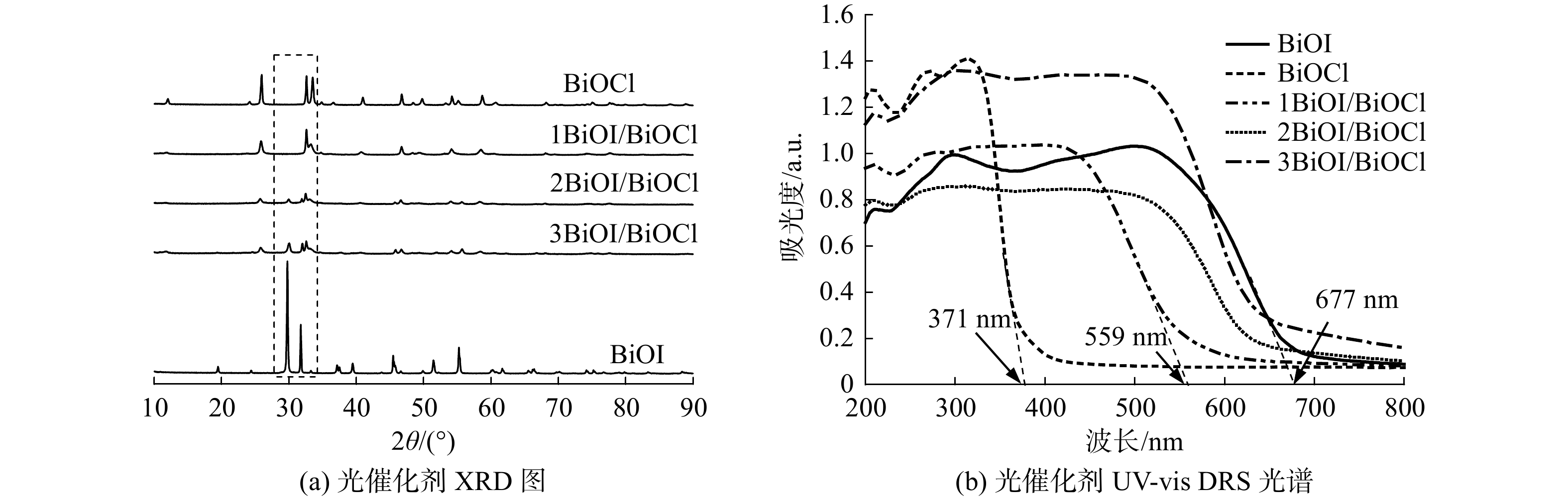
为了解复合光催化材料的晶体结构特征,采用XRD表征BiOI、BiOCl和BiOI/BiOCl光催化剂(图5(a))。BiOCl样品的XRD图谱表明,在2θ为 25.9°、32.4°、33.4°、40.8°、46.6°、49.6°、54.1°和58.7°时,分别获得较强衍射峰,对应BiOCl标准谱图(JCPDS No.06-0249)的(101)、(110)、(102)、(112)、(200)、(113)、(211)和(212)晶面[31]。BiOI样品在2θ为19.1°、29.6°、31.7°、37.1°、39.5°、45.4°、51.3°、55.3°和60°处观察到的衍射峰分别对应BiOI的(002)、(102)、(110)、(103)、(004)、(200)、(114)、(212)和(006)晶面(JCPDS No.10-0445)[32]。这意味着共沉淀法可制备纯BiOCl和BiOI。与BiOCl相比,1BiOI/BiOCl中BiOCl的特征衍射峰明显降低,未观察到明显的BiOI特征衍射峰。这说明BiOI在BiOCl表面负载较均匀。随着BiOI含量的增加,复合光催化材料中BiOI位于29.6°、31.7°和55.3°处的衍射峰开始增强,且BiOCl位于25.9°和32.4°的衍射峰大幅降低。这可能与BiOI/BiOCl异质结的形成有关,同时也验证了SEM结果。
2.1.4 UV-vis DRS分析
采用UV-vis DRS对样品的吸光特性进行对比研究。如图5(b)所示,在可见光区域(420 nm以上),1BiOI/BiOCl复合材料的吸收光谱强度明显强于BiOCl,表明BiOI掺杂改善了其可见光响应性能。通过UV-vis DRS观察到,BiOCl的吸收边缘波长为371 nm,BiOI的吸收边缘波长为677 nm,而1BiOI/BiOCl的吸收边缘波长则介于两者之间(559 nm)。且随着I−含量增加,吸收边缘的波长逐渐增加。因此,与BiOCl相比,复合材料均发生了红移,即提高了对可见光的吸收能力,这将有利于其活性的增强。样品的带隙能(Eg)可由经验公式(式(2))估算。其中,Eg和λ分别为带隙能和吸收边缘波长。因此,计算出BiOI和BiOCl的Eg分别约1.83和3.34 eV,这与文献[18,27]基本一致;而1BiOI/BiOCl的Eg计算值为2.22 eV。很明显,BiOI的掺杂降低了BiOCl的带隙值。根据Mulliken电负性理论,BiOCl和BiOI的导带电位(CB)和价带电位(VB)可由式(3)~(4)计算得到[33]。
Eg=1240/λ (2) ECB=χ−EC−0.5Eg (3) EVB=ECB+Eg (4) 式中:ECB和EVB分别为样品的CB和VB值;EC为自由电子在氢标准电势下的能量(4.5 eV);χ是半导体的绝对电负性(BiOI的χ值5.94 eV;BiOCl的χ值为6.36 eV)[16,34];Eg为禁带宽度。经计算,BiOCl和BiOI样品的CB分别为0.19和0.53 eV,VB分别为3.53和2.36 eV。
2.1.5 XPS分析
XPS可定性或定量分析样品的化学成分及其存在价态,已被广泛应用于物质表面成分研究。图6给出了3种光催化剂中Bi 4f、I 3d、Cl 2p和O 1s的高分辨XPS光谱。如图6(a)所示,光催化剂显示出2个位于159.40和164.70 eV附近的Bi 4f峰,分别对应Bi元素的Bi 4f7/2和Bi 4f5/2,这表明光催化剂中存在氧化物形式的Bi3+[35]。图6(b)中位于630.90和619.50 eV处I 3d峰,分别对应I 3d3/2和I 3d5/2,这表明复合材料中以I−形式存在[36]。在198.1 eV和199.4 eV附近,峰分别对应Cl 2p3/2和Cl 2p1/2[37]。在BiOCl和1BiOI/BiOCl光催化剂中,O 1s信号峰位于530.0 eV,该峰为Bi—O晶格氧中的O。与BiOCl相比,1BiOI/BiOCl的Bi 4f和Cl 2p峰均轻微的向高结合能方向移动,这表明BiOI和BiOCl之间存在电子转移,且形成了异质结结构[29]。
 图 6 BiOI、BiOCl和1BiOI/BiOCl样品的高分辨XPS能谱图Figure 6. High resolution XPS spectra of BiOI, BiOCl and 1BiOI/BiOCl samples
图 6 BiOI、BiOCl和1BiOI/BiOCl样品的高分辨XPS能谱图Figure 6. High resolution XPS spectra of BiOI, BiOCl and 1BiOI/BiOCl samples2.1.6 ESR分析
为探究BiOI/BiOCl光催化体系中活性物质的存在,采用ESR技术检测反应过程中活性自由基的生成情况。实验首先在黑暗条件下进行,然后使用可见光照射5和10 min后检测自由基的特征信号。图7表明,在可见光照射5 min后,可清晰检测出羟基自由基DMPO–·OH和阴离子超氧自由基DMPO–·O2–的信号。在辐射时间延长至10 min时,2种信号的强度进一步增强。HUANG等[38]发现自由基的含量与信号高度的平方成正比。因此,随着光照时间的增加,反应溶液中·OH和·O2–的含量大幅增加,将有利于反应溶液对污染物的高效去除。
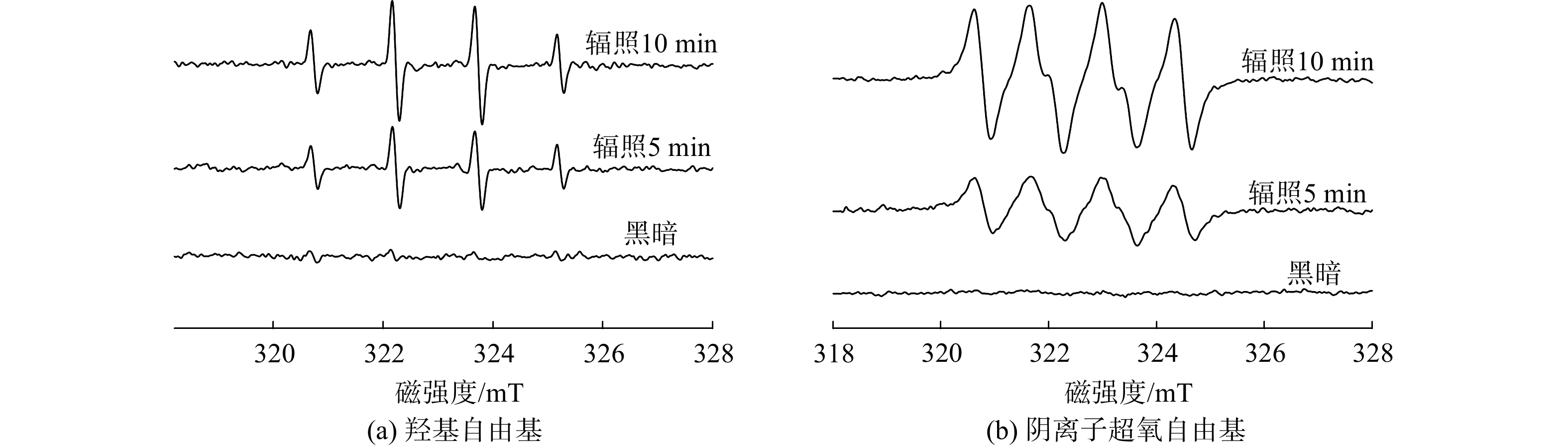 图 7 可见光照射下1BiOI/BiOCl光催化剂的ESR谱Figure 7. ESR spectra of 1BiOI/BiOCl photocatalyst under visible light irradiation
图 7 可见光照射下1BiOI/BiOCl光催化剂的ESR谱Figure 7. ESR spectra of 1BiOI/BiOCl photocatalyst under visible light irradiation2.2 光催化剂的活性
2.2.1 BiOI/BiOCl光催化剂的催化活性
图8为各光催化剂对Hg0的脱除效率。BiOCl的脱汞效率约为35%,BiOI的脱汞效率为68%。与BiOCl相比,BiOI具有更高的催化活性,推断其原因为BiOI的带隙能较窄,对可见光范围的响应强[18]。所制备的光催化剂脱汞性能顺序为1BiOI/BiOCl(96%) > 2BiOI/BiOCl (94%) > 3BiOI/BiOCl (93%) > BiOI (68%) > BiOCl (35%)。这表明BiOCl与BiOI之间存在良好的协同效应。基于式(5)可计算出脱汞过程初始反应阶段的反应速率常数(k)以了解样品活性[16]。
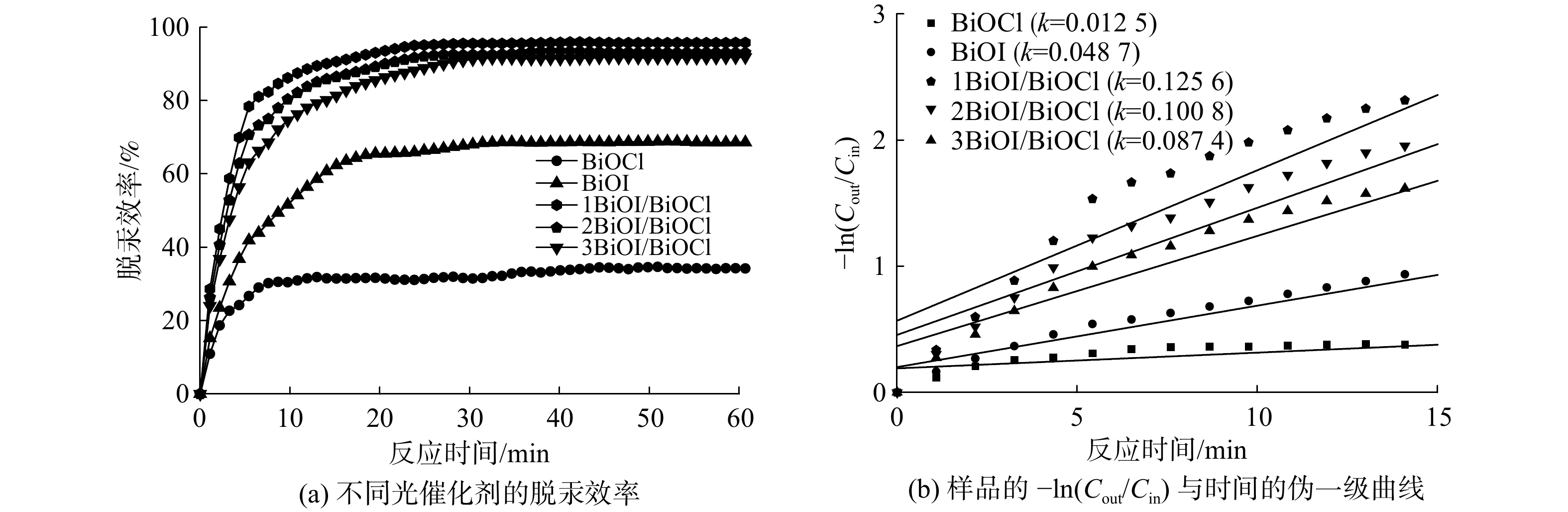 图 8 不同光催化剂的脱汞效率对比Figure 8. Comparison of mercury removal efficiencies of different photocatalysts
图 8 不同光催化剂的脱汞效率对比Figure 8. Comparison of mercury removal efficiencies of different photocatalysts−ln(Cout/Cin)=k×t (5) 式中:k是伪一级速率常数,t是反应时间。k值越大,光催化活性越高。经计算,反应15 min后,光催化剂对应的反应速率常数分别为0.012 5、0.004 8、0.125 6、0.100 8和0.087 4 min−1。这说明1BiOI/BiOCl光催化剂具有最高的催化活性,故接下来以该光催化剂为基础开展进一步研究。
2.2.2 光照-黑暗的影响
图9揭示了荧光灯辐照对1BiOI/BiOCl样品Hg0去除性能的影响。在反应最初的30 min内,1BiOI/BiOCl光催化剂在黑暗中的催化效率较低,光催化效率仅为36%。在30 min后,FSL开启,Hg0的去除效率急剧上升到93%。在60 min时刻关闭FSL,在60~90 min内样品的去汞性能出现略微下降。其原因可能是30~60 min FSL照射使光催化剂产生大量的活性物质,在30 min的光催化脱除Hg0过程中活性物质并未被完全消耗,所以仍能保持较高的效率。再次重复上述实验后,得到了预期结果。以上结果表明,光源照射对1BiOI/BiOCl样品稳定而优异的光催化性能是不可或缺的,可采用间歇性提供光源的方式保持样品良好的脱汞性能。
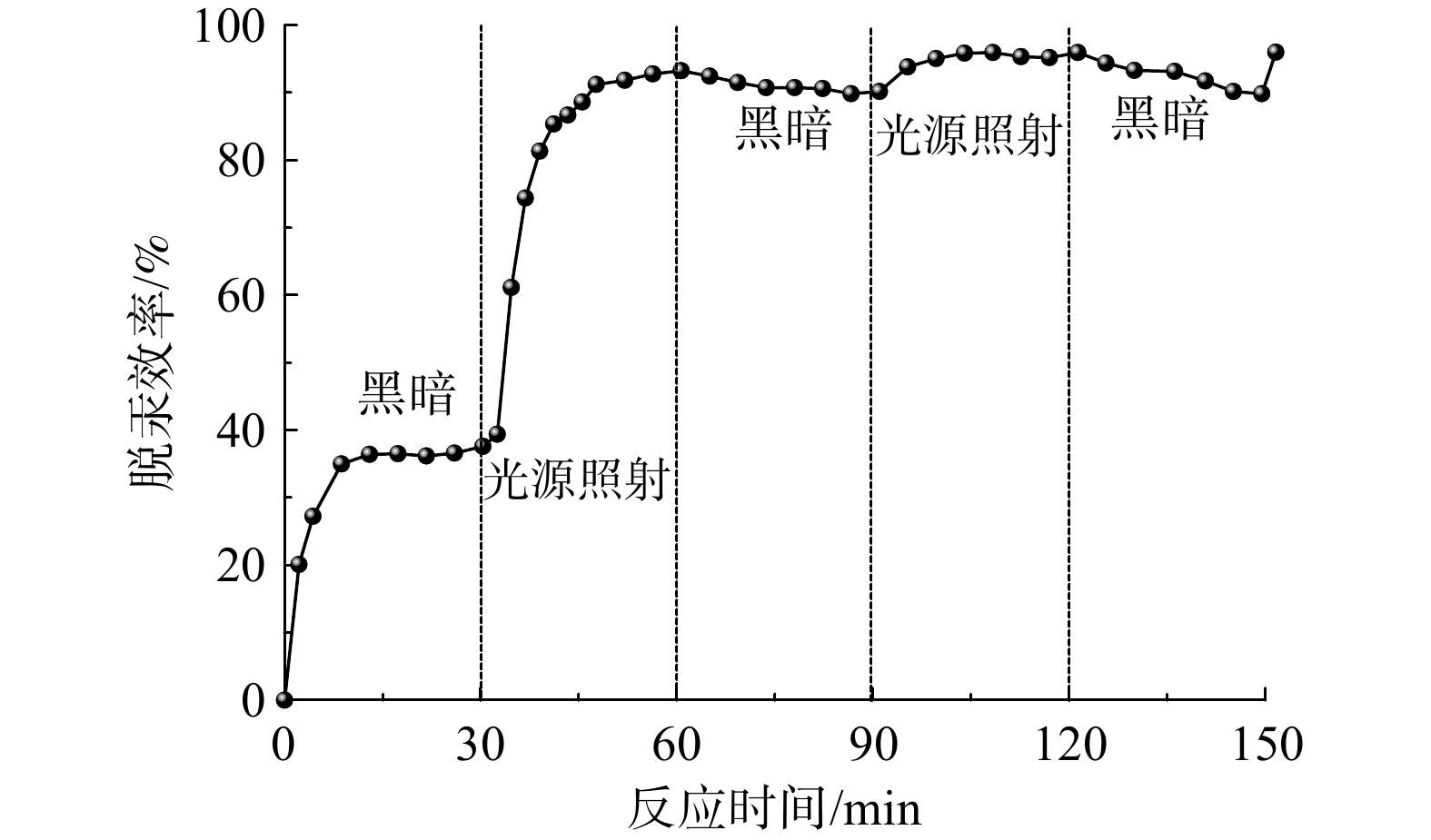 图 9 荧光灯照射对1BiOI/BiOCl光催化剂脱汞效率的影响Figure 9. Effect of fluorescent lamp irradiation on Hg0 removal using 1BiOI/BiOCl photocatalyst
图 9 荧光灯照射对1BiOI/BiOCl光催化剂脱汞效率的影响Figure 9. Effect of fluorescent lamp irradiation on Hg0 removal using 1BiOI/BiOCl photocatalyst2.2.3 光催化剂用量的影响
在环境污染治理的同时,为降低运行成本和增强经济效益,探究了光催化剂剂量对Hg0去除性能的影响。分别使用0.1、0.3、0.5和1.0 g·L−1光催化剂进行光催化脱汞实验(图10(a))。当1BiOI/BiOCl光催化剂剂量(质量浓度)为0.1和0.3 g·L−1时,Hg0的脱除效率分别为53%和82%。进一步提高光催化剂质量浓度至0.5和1.0 g·L−1时,催化效率的高达96%和98%。这意味着少量1BiOI/BiOCl光催化剂亦会产生大量活性物质,使反应溶液中具备较高的Hg0脱除条件。
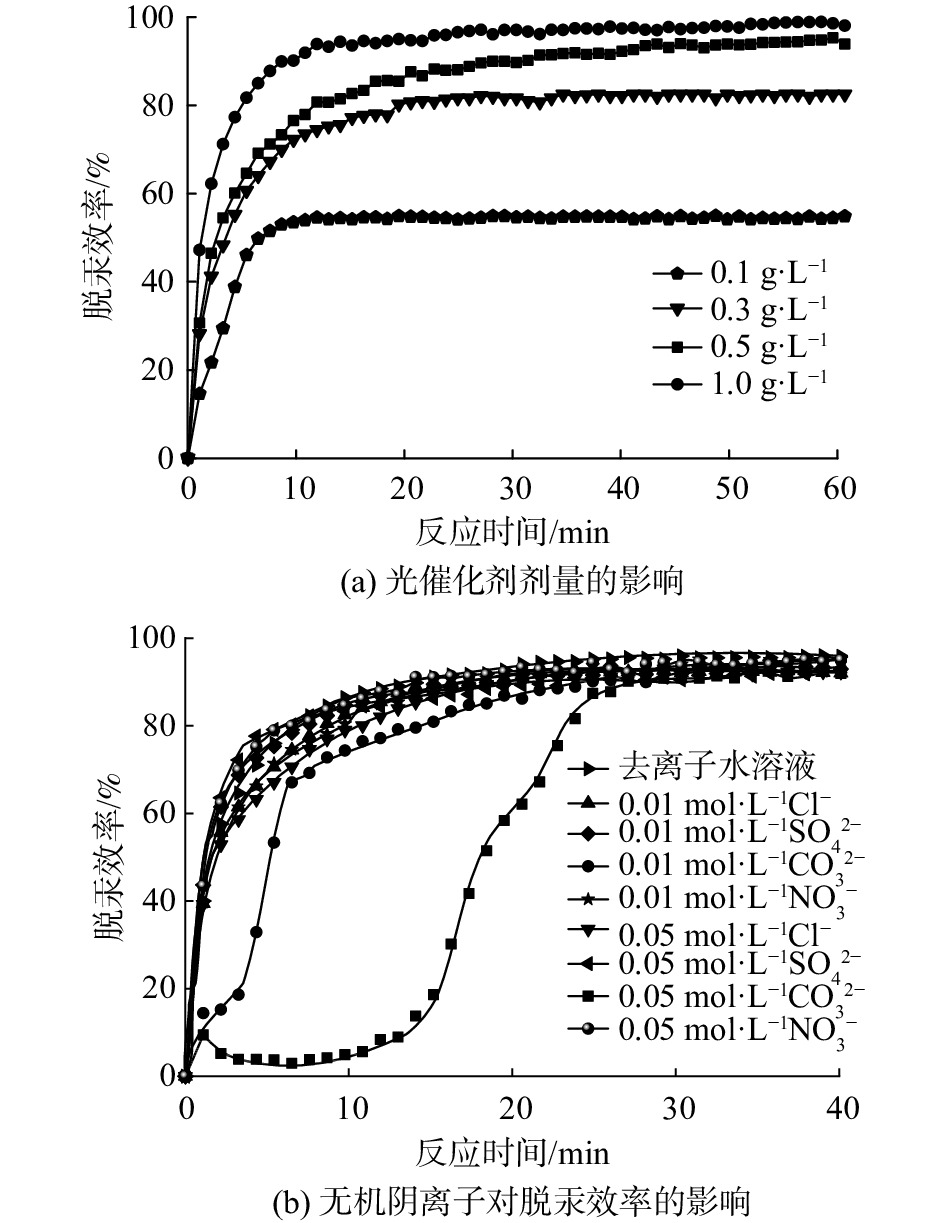 图 10 光催化剂剂量和无机阴离子对脱汞效率的影响Figure 10. Effects of photocatalyst dose and inorganic anions on mercury removal efficiency
图 10 光催化剂剂量和无机阴离子对脱汞效率的影响Figure 10. Effects of photocatalyst dose and inorganic anions on mercury removal efficiency2.2.4 无机阴离子的影响
由于烧结烟气可能含有少量SO2、NOx、HCl和较多的CO2气体,其存在会使反应溶液中含有少量的无机阴离子[39]。为考察常见无机阴离子对光催化活性的影响,研究了Cl−、NO3−、CO32−和SO42−对复合光催化剂脱汞性能的影响。本研究分别选取NaCl、NaNO3、Na2CO3和Na2SO4作为阴离子模型化合物,其结果见图10(b)。这表明Cl−、NO3−和SO42− 3种无机阴离子对Hg0的去除几乎没有影响;然而,在开始阶段0.01 mol·L−1的Na2CO3对光催化过程表现出明显的抑制作用。0.05 mol·L−1的Na2CO3加入反应溶液后,抑制效果颇为显著,10 min前Hg0去除效率在5%以下。随着实验的进行,CO32−的抑制作用逐渐消失,其原因可能是CO32−与活性物质发生了如下反应[40-41](式(6)~(10))。
CO2−3+⋅OH→⋅CO−3+OH− (6) CO2−3+h+→⋅CO−3 (7) CO2−3+H2O↔HCO−3+OH− (8) HCO−3+⋅OH→⋅CO−3+H2O (9) ⋅CO−3+e−→CO2−3 (10) 2.2.5 循环实验结果
为了解1BiOI/BiOCl复合光催化剂的稳定性,设计并开展了光催化剂循环实验。考虑在洗涤、干燥过程中,光催化剂会存在损失,故实验中1BiOI/BiOCl光催化剂剂量取0.8 g,并进行5次循环实验,单次实验时间为40 min,结果如图11所示。前4次实验光催化剂脱除Hg0的效率均在95%以上,第5次实验光催化剂脱除Hg0的效率略微下降,但仍能保持在约95%。分析其原因,可能是在回收、洗涤和干燥过程中光催化剂质量减少所致。1BiOI/BiOCl复合光催化剂稳定性较好,在多次循环实验后依旧能保持优异脱汞性能。
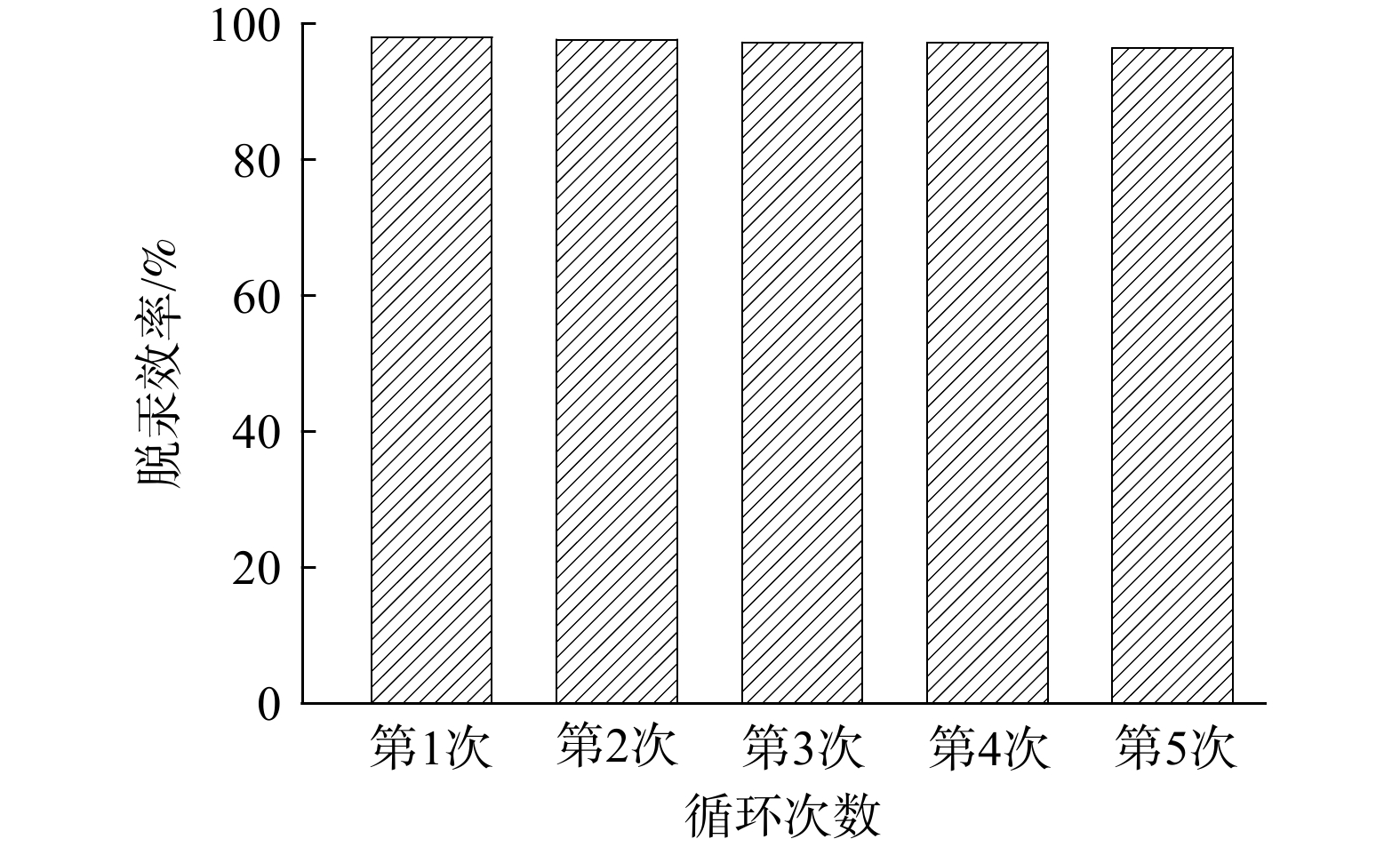 图 11 光催化剂的循环脱汞实验性能Figure 11. Experimental performance of cyclic Hg0 removal by photocatalyst
图 11 光催化剂的循环脱汞实验性能Figure 11. Experimental performance of cyclic Hg0 removal by photocatalyst2.3 反应机理
为进一步探明Hg0去除过程中活性物质的作用,将·O2−的消除剂苯醌(BQ,0.5 g)、·OH的消除剂异丙醇(IPA,15 mL)和h+的消除剂乙二胺四乙酸二钠盐(EDTA-2Na,0.5 g)分别与1BiOI/BiOCl光催化剂一起添加至反应溶液,记录40 min后光催化剂的脱汞效率(图12(a))。加入BQ或EDTA-2Na后,1BiOI/BiOCl的效果效率分别为10%和45%。与未添加消除剂相比,其脱汞性能均大幅降低。然而,在反应体系中加入IPA并未降低Hg0的去除效率。以上现象说明在光催化脱除Hg0的过程中,·O2−是最关键的活性物质,h+是次要活性物质,而·OH并非光催化反应过程中的主要活性物质,其主要原因可能是与·OH所处的溶液环境有关。
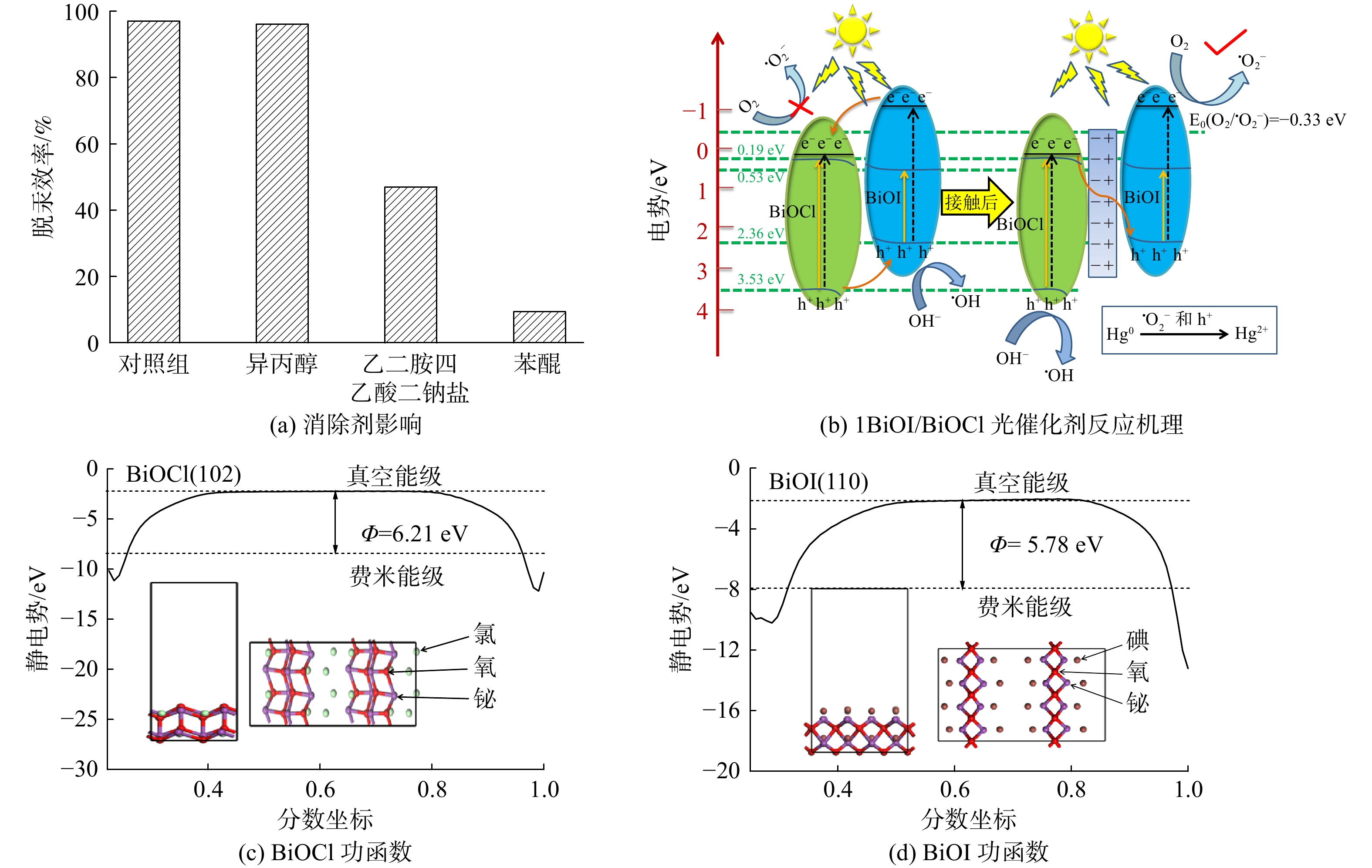 图 12 光催化剂高效脱除Hg0反应机理Figure 12. The reaction mechanism of efficient removal of Hg0 by photocatalyst
图 12 光催化剂高效脱除Hg0反应机理Figure 12. The reaction mechanism of efficient removal of Hg0 by photocatalyst基于上述实验和表征分析,推断出FSL辐照下1BiOI/BiOCl的高效脱汞机理(图12(b)所示)。UV-vis DRS分析表明:BiOI光催化剂的CB和VB分别为0.53 eV和2.36 eV,其带隙能为1.83 eV;BiOCl光催化剂的CB和VB分别为0.19 eV和3.53 eV,其带隙能为3.34 eV。由于BiOCl的带隙能较大,不能被可见光(2.95 eV)激发。然而,由于FSL中含有少量波长为366 nm的光,其携带能量约为3.39 eV,而BiOCl和BiOI的带隙能分别为3.34和1.83 eV,因此其均可被FSL激发产生电子-空穴(e−-h+)对。在FSL的照射下,BiOCl和BiOI导带上的电子会进一步跃迁至0.14和−1.03 eV[42]。图12(b)表明,BiOCl的CB更低,故e−会从BiOI的CB转移到BiOCl的CB上;同时,由于BiOCl的VB更高,所以h+从BiOCl的VB转移到BiOI的VB上。但由于O2的还原电位[E0(O2/·O2−)=−0.33 eV][34]比BiOCl的CB更低,因此BiOCl中CB上电子不能将O2还原为·O2−。但ESR测试证实存在·O2−,且消除剂实验表明,·O2−是光催化过程中最重要的活性自由基。因此,传统的交错间隙II型异质结电子转移方式不成立。
为了解BiOCl和BiOI之间的电子转移方式,采用DFT方法计算了2种物质的功函数,结果见图12(c)~(d)。BiOCl(102)的功函数为6.21 eV,BiOI(110)的功函数为5.78 eV。BiOCl的功函数高于BiOI,所以当2种材料接触时,而BiOCl是氧化光催化材料,BiOI将是还原光催化材料[43]。电子会自发地从BiOI流向BiOCl,直到新的费米能级平衡,且在界面形成1个从BiOI指向BiOCl的内电场。另外,由于BiOI失去电子带正电,其带边缘将向上弯曲;BiOCl得到电子带负电,其带边缘向下卷曲[43]。在内部电场、带边弯曲和库仑力相互作用下,BiOCl中CB上e−与BiOI中VB的h+复合。同时,内电场抑制了BiOI中CB中光生e−流入BiOCl的CB中,而BiOCl中VB上的h+转移到BiOI中VB的概率也显著降低。由于BiOI的CB能跃迁到−1.03 eV,低于O2的还原电位[E0(O2/·O2−)=−0.33 eV],因此,富集在BiOI导带上的e–能够与吸附在光催化剂表面的O2反应生成·O2−。此外,BiOCl中VB上的h+会与H2O和OH−反应生成·OH。这与ESR结果一致。消除剂结果表明,在光催化脱汞过程中,h+和·O2−是主要的活性自由基。以上电荷转移过程赋予了BiOI/BiOCl光催化剂最高的氧化还原能力,故1BiOI/BiOCl光催化剂的高效脱汞机理遵循Z型异质结电荷转移规律。
3. 结论
1)与BiOCl相比,1BiOI/BiOCl中BiOCl的特征衍射峰明显降低,未观察到明显的BiOI特征衍射峰;1BiOI/BiOCl的SEM图像与BiOCl颇为相似,其表面上可明显地观察到BiOI片状或微颗粒状物质。这表明BiOI与BiOCl得到良好耦合,紧密地接触有利于异质结的形成和促进光生e−-h+对的分离。与BiOCl相比,复合材料均发生了红移,提高了对可见光的吸收能力,这将有利于其活性的增强。
2)所制备光催化剂的脱汞性能顺序为:1BiOI/BiOCl(96%) > 2BiOI/BiOCl (94%) > 3BiOI/BiOCl (93%) > BiOI (68%) > BiOCl (35%)。这说明BiOCl与BiOI存在良好的协同效应。Cl−、NO3−和SO42−这3种无机阴离子对Hg0的去除几乎没有影响。然而,CO32−在开始阶段对光催化过程表现出明显的抑制作用,随着实验的进行,CO32−的抑制作用逐渐消失。
3)由于不能产生强氧化性的·O2−,1BiOI/BiOCl光催化剂高效脱汞机理与传统的交错间隙II型异质结电子转移方式不符。消除剂实验和DFT计算表明,Z型异质结电荷转移规律符合BiOI/BiOCl复合材料的光催化氧化机理。
-

图 1 活性氧化镁改性前后SEM图像
Figure 1. SEM images of active magnesium oxide before and after modification
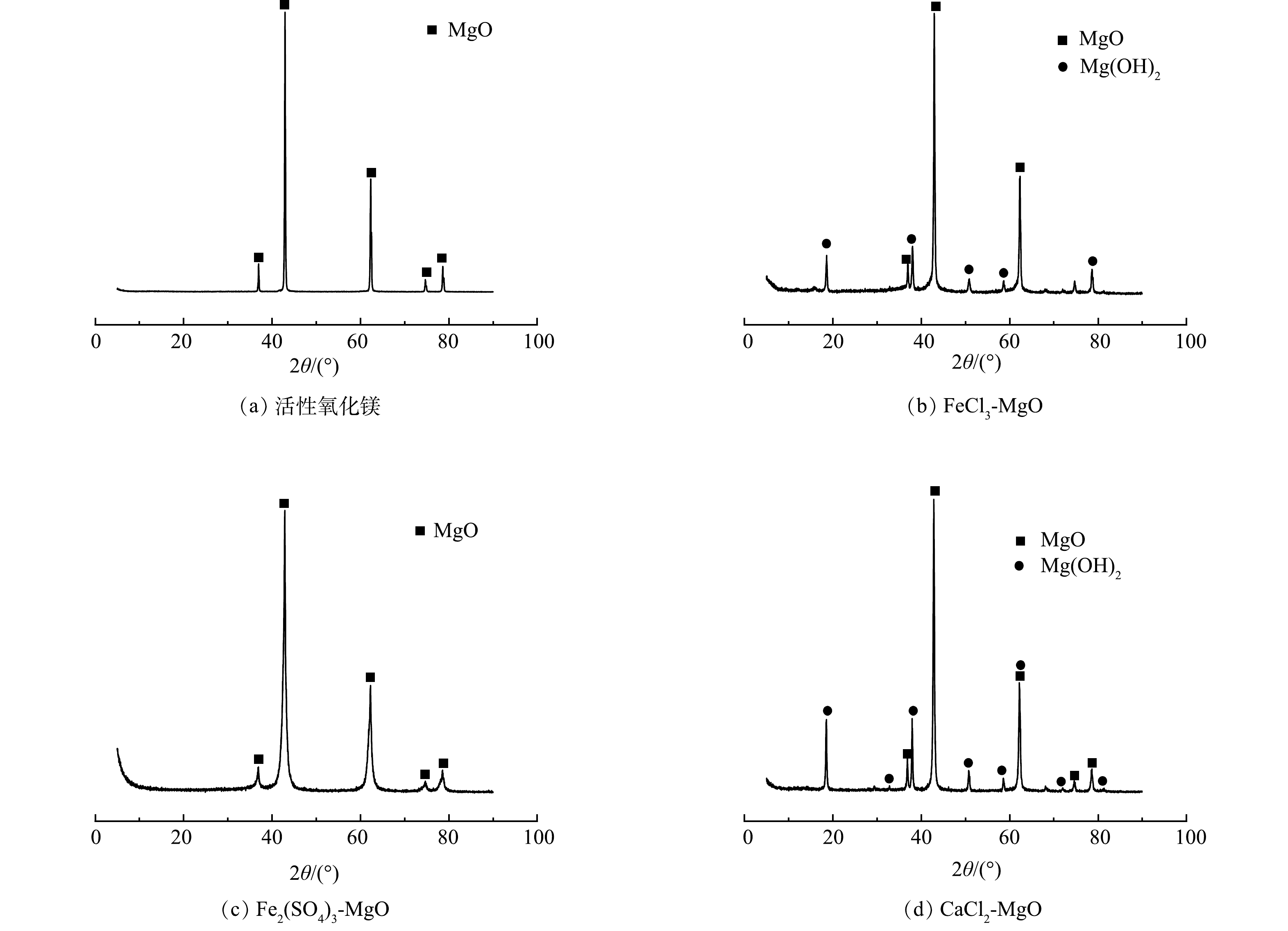
图 2 活性氧化镁改性前后的X射线衍射图谱
Figure 2. X-ray diffraction patterns of active magnesium oxide before and after modification

图 3 活性氧化镁改性前后的FTIR图谱
Figure 3. FTIR spectra of active magnesium oxide before and after modification
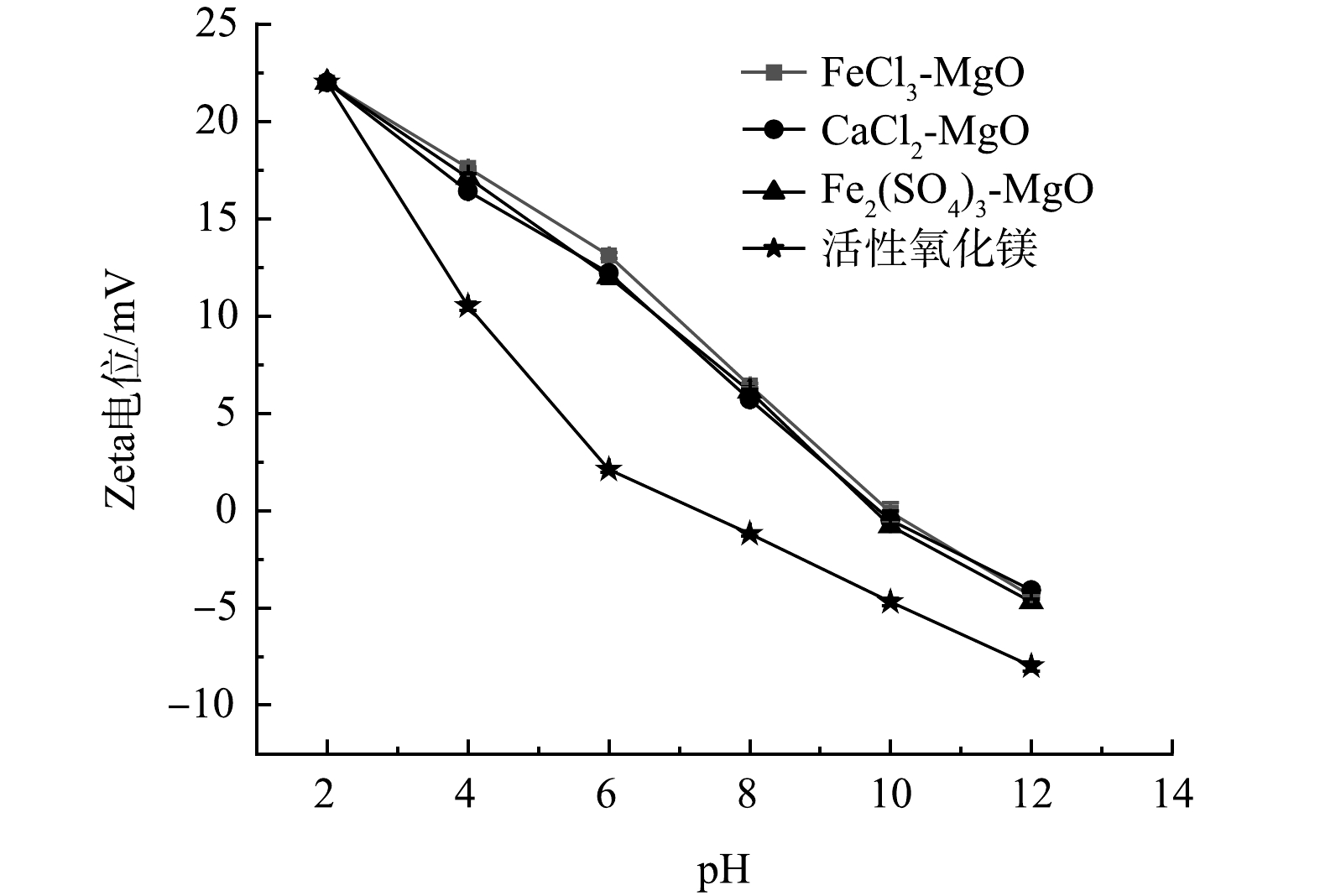
图 4 活性氧化镁改性前后Zeta电位图
Figure 4. Zeta potential of active magnesium oxide before and after modification
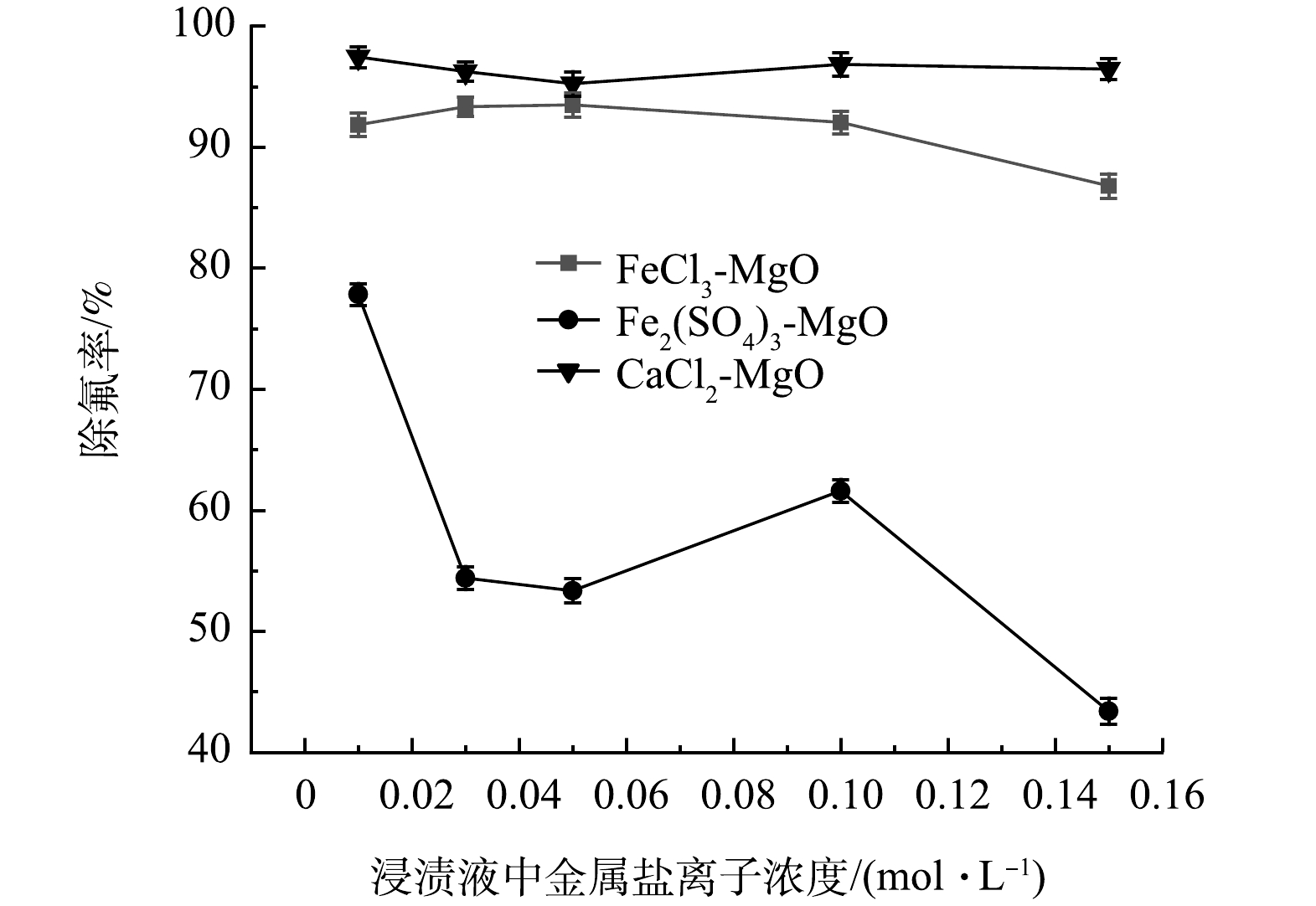
图 5 不同金属盐浸渍液离子浓度对改性吸附剂除氟率的影响
Figure 5. Effect of ion concentrations in different metal salt impregnations on the fluorine removal efficiency of modified adsorbents
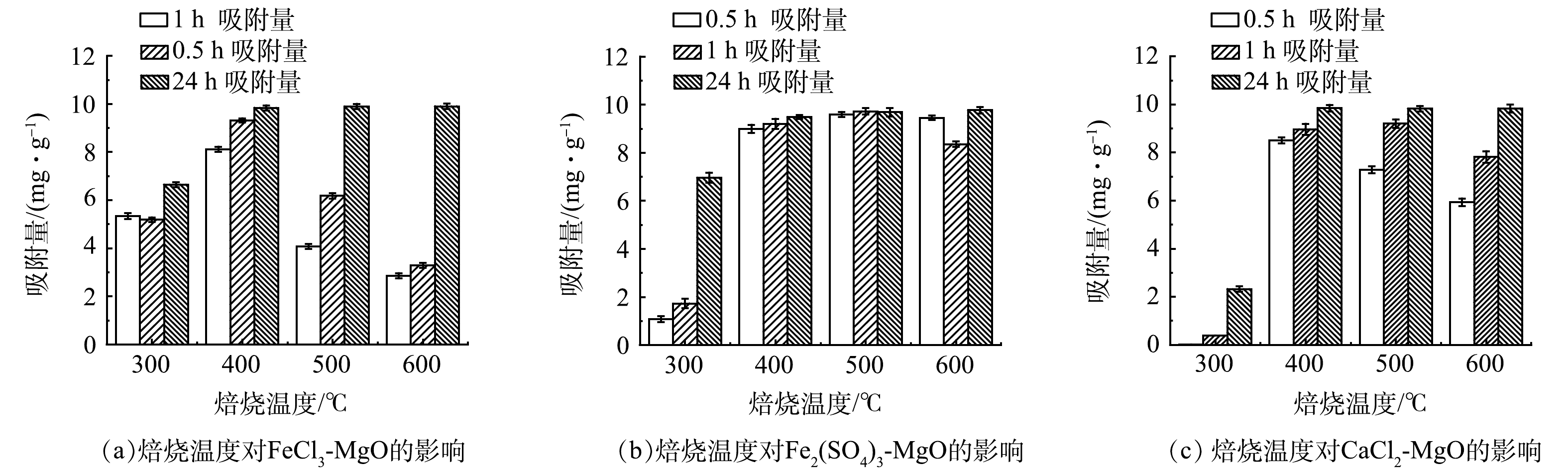
图 6 不同焙烧温度对改性活性氧化镁吸附量的影响
Figure 6. Effect of different calcination temperatures on the adsorption capacity of modified active magnesium oxide
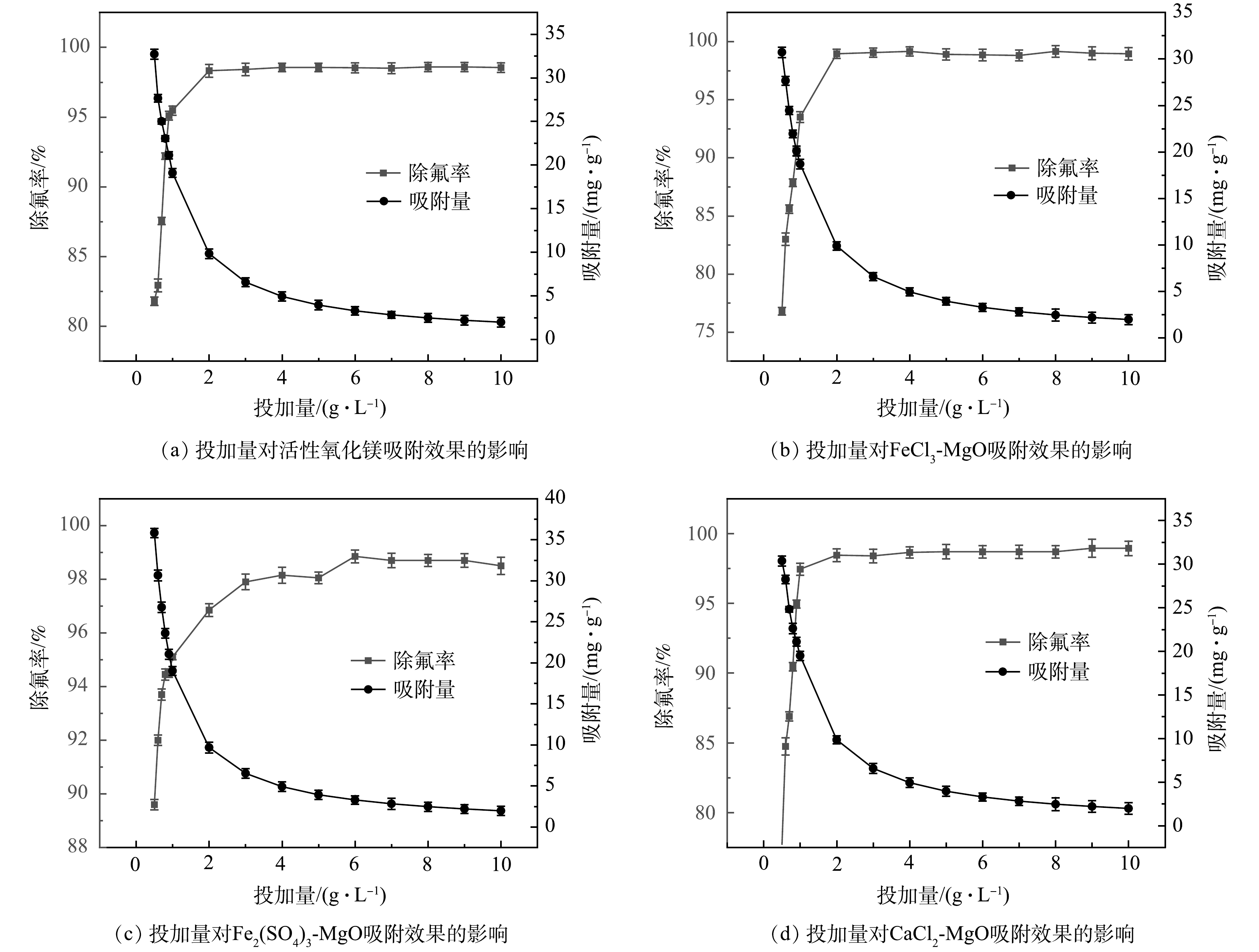
图 7 吸附剂投加量对于氟离子去除率和吸附容量的影响
Figure 7. Effect of adsorbent dosage on fluoride ion removal rate and adsorption capacity
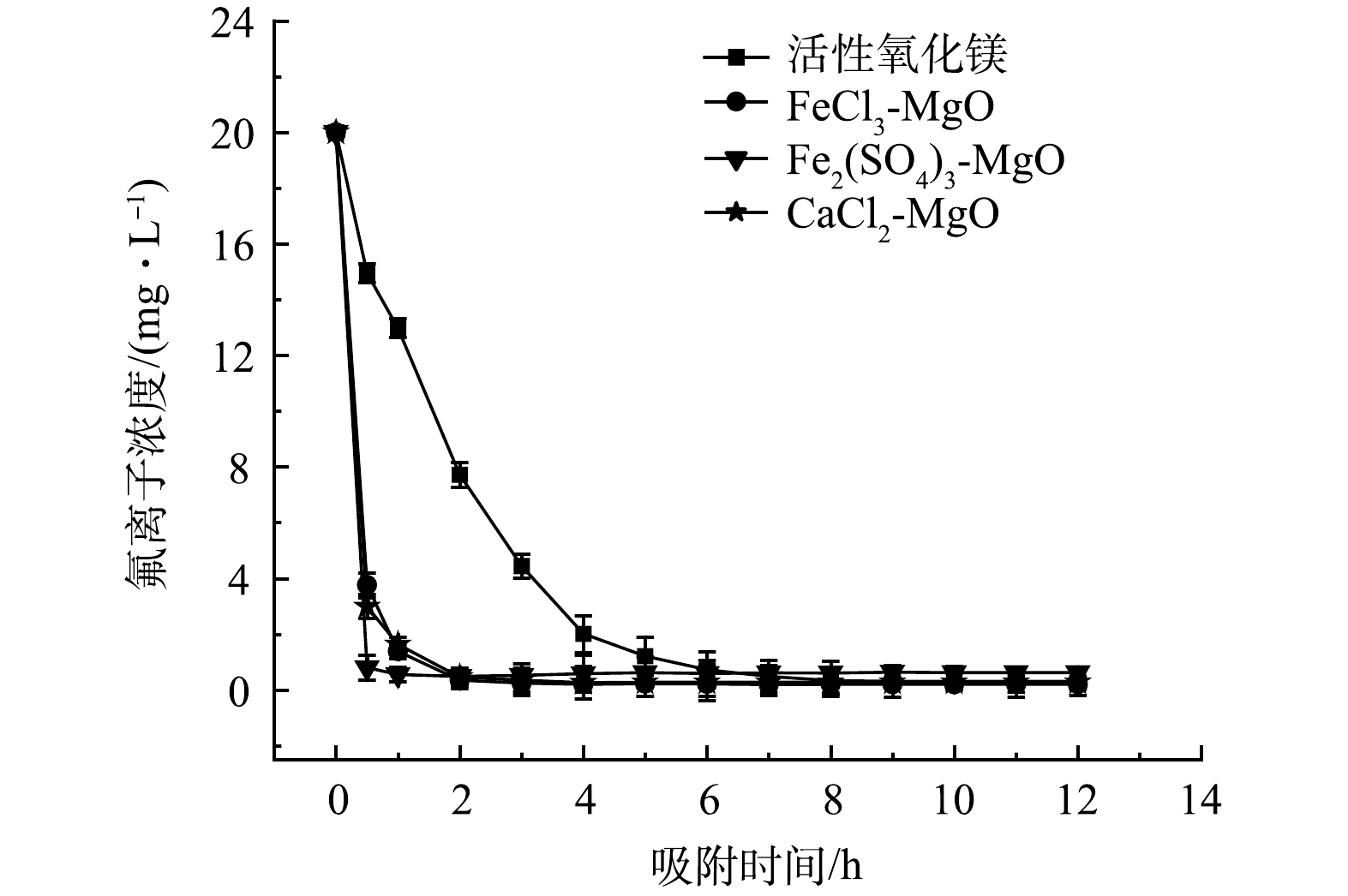
图 8 吸附时间对于吸附剂吸附氟离子的影响
Figure 8. Effect of adsorption time on fluorine ion adsorption by adsorbent
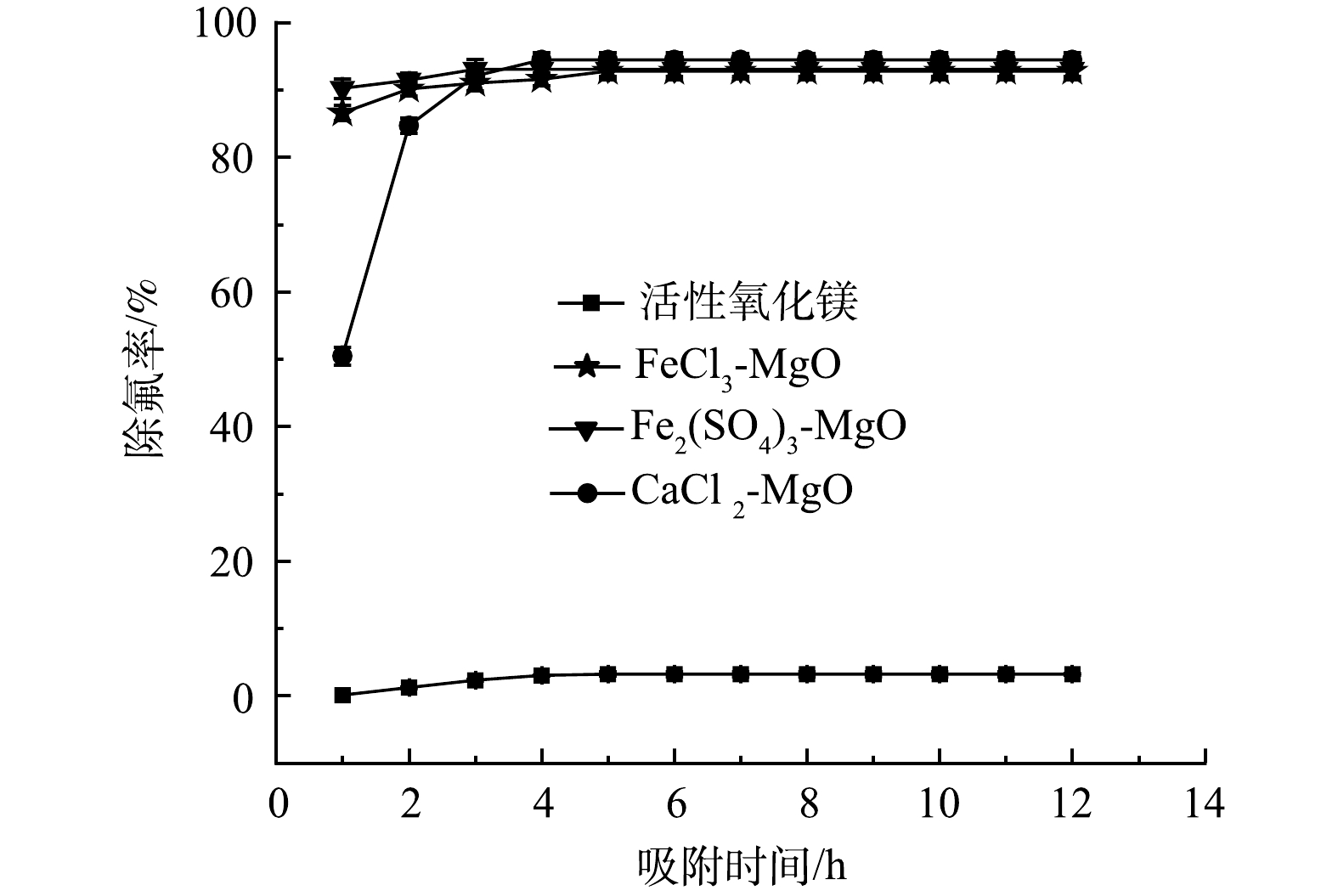
图 10 吸附剂在模拟5 mg·L−1实际含氟饮用水中的除氟率
Figure 10. The fluoride removal rate of adsorbent in simulated 5 mg·L−1 actual fluorinated drinking water
表 1 吸附剂的Langmuir和Freundlich等温吸附模型拟合参数
Table 1. Fitting parameters of Langmuir and Freundlich isothermal adsorption models for adsorbent
吸附剂 Langmuir Freundlich R2 Qmax/(mg∙g−1) KL/min R2 1/n KF/min−1 活性氧化镁 0.998 6 59.59 0.66 0.848 7 0.442 3 15.44 FeCl3-MgO 0.997 6 107.64 0.17 0.915 8 0.427 1 15.96 Fe2(SO4)3-MgO 0.997 2 89.29 0.15 0.886 4 0.414 9 13.12 CaCl2-MgO 0.994 6 108.34 0.22 0.875 4 0.398 4 18.71  下载: 导出CSV
下载: 导出CSV
-
[1] WANG H, YANG L, GAO P, et al. Fluoride exposure induces lysosomal dysfunction unveiled by an integrated transcriptomic and metabolomic study in bone marrow mesenchymal stem cells[J]. Ecotoxicology and Environmental Safety, 2022, 239: 113672. doi: 10.1016/j.ecoenv.2022.113672 [2] TANEJA L, KOCHAR C, KUMAR Y P, et al. Adsorption: A preferred technique for fluoride removal from water[J]. Materials Today:Proceedings, 2022, 71(2): 215-219. [3] LU N C, LIU J C. Removal of phosphate and fluoride from wastewater by a hybrid precipitation–microfiltration process[J]. Separation and Purification Technology, 2010, 74(3): 329-335. doi: 10.1016/j.seppur.2010.06.023 [4] 桑硕, 帖靖玺, 张南. 地下水除氟研究进展[J]. 科技创新与应用, 2022, 12(2): 78-82. [5] ANIS S F, HASHAIKEH R, HILAL N. Reverse osmosis pretreatment technologies and future trends: A comprehensive review[J]. Desalination, 2019, 452: 159-195. doi: 10.1016/j.desal.2018.11.006 [6] SANINI B, LEICHTWEIS J, SILVESTRI S, et al. Impregnation of activated alumina with CeO2 for water defluoridation[J]. Materials Chemistry and Physics, 2022, 291: 126648. doi: 10.1016/j.matchemphys.2022.126648 [7] AYALEW A A. Comparative adsorptive performance of adsorbents developed from kaolin clay and limestone for de-fluoridation of groundwater[J]. South African Journal of Chemical Engineering, 2023, 44: 1-13. doi: 10.1016/j.sajce.2022.11.002 [8] LAONAPAKUL T, SUTHI T, OTSUKA Y, et al. Fluoride adsorption enhancement of Calcined-Kaolin/Hydroxyapatite composite[J]. Arabian Journal of Chemistry, 2022, 15(11): 104220. doi: 10.1016/j.arabjc.2022.104220 [9] THIRUNAVUKKARASU A, NITHYA R, SIVASHANKAR R. Continuous fixed-bed biosorption process: A review[J]. Chemical Engineering Journal Advances, 2021, 8: 100188. doi: 10.1016/j.ceja.2021.100188 [10] AMIN F, TALPUR F N, BALOUCH A, et al. Biosorption of fluoride from aqueous solution by white—rot fungus Pleurotus eryngii ATCC 90888[J]. Environmental Nanotechnology, Monitoring & Management, 2015, 3: 30-37. [11] 张小磊, 李尚明, 李红艳, 等. 负载镧镁改性活性氧化铝的除氟性能[J]. 环境工程学报, 2016, 10(8): 4189-4195. [12] BAKHTA S, SADAOUI Z, LASSI U, et al. Performances of metals modified activated carbons for fluoride removal from aqueous solutions[J]. Chemical Physics Letters, 2020, 754: 137705. doi: 10.1016/j.cplett.2020.137705 [13] 刘理华, 车王燕, 刘书群. 棒状氧化镁的改性及其吸附性能研究[J]. 淮北师范大学学报(自然科学版), 2023, 44(1): 52-57. [14] 王慧玲, 张彦燕, 徐薇, 等. 改性活性氧化镁的除氟效能及机制[J]. 环境工程学报, 2015, 9(5): 2125-2130. [15] WANG X Y, WEI J J, PENG W C, et al. Evaluation and DFT analysis of 3D porous rhombohedral Fe-modified MgO for removing fluoride efficiently[J]. Applied Surface Science, 2021, 552: 149423. doi: 10.1016/j.apsusc.2021.149423 [16] 潘柳依, 孙晓红, 王军峰, 等. 固体酸催化剂SO42-/TiO2-SnO2的合成及其催化合成丙烯酸异冰片酯[J]. 精细化工, 2018, 35(5): 791-795. [17] 胡美秋, 袁红, 景艳红. SO42-/Al2O3催化剂的表征及其在环氧化反应中的催化活性[J]. 中国油脂, 2019, 44(12): 55-58. [18] LI L X, XU D, LI X Q, et al. Excellent fluoride removal properties of porous hollow MgO microspheres[J]. New Journal of Chemistry, 2014, 38(11): 5445-5452. doi: 10.1039/C4NJ01361A [19] LU M, GUAN X H, XU X H, et al. Characteristic and mechanism of Cr(VI) adsorption by ammonium sulfamate-bacterial cellulose in aqueous solutions[J]. Chinese Chemical Letters, 2013, 24: 253-256. doi: 10.1016/j.cclet.2013.01.034 [20] 付娆, 张文龙, 冯江涛, 等. 锐钛矿型二氧化钛的低温合成及其吸附除氟性能的研究[J]. 环境工程, 2020, 38(2): 70-76. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2743
- HTML全文浏览数: 2743
- PDF下载数: 99
- 施引文献: 0
