







-
磷(P)是所有动植物生长过程中必需的营养元素,在自然界中主要以结合态和游离态的形式存在[1]。当水生生态系统中存在过量的正磷酸盐时,就会引起水体富营养化而导致水环境质量恶化[2]。迄今为止,几乎所有用于农业生产的磷都来自于磷矿的开采。美国地质调查局(USGS)报告,以目前磷矿的开采速度计算,地球上的磷酸盐储量将在2051至2092年间耗尽[3],磷资源已成为人类发展的限制性因素之一。因而,对散失到水环境中的磷资源进行有效回收,控制磷污染的同时缓解磷资源危机,引起研究者的广泛关注。
目前去除水体中磷的方法主要有化学沉淀、生物处理、吸附技术、膜技术、离子交换、人工湿地和结晶法等[4-5]。这些处理过程中,沉淀法和生物处理(活性污泥法)会产生大量的污泥,从而对环境造成二次污染;此外,膜工艺经济成本较高[6]。近年来,利用结晶法回收水体中的磷引起研究人员的广泛兴趣,如磷酸钙结晶法、鸟粪石结晶法、蓝铁矿结晶法等[7]。其中磷酸钙具有多种存在形态,例如磷酸三钙(TCP)、羟基磷灰石(HAP)、磷酸氢钙(DCPA)等。HAP不仅是一种优质的肥料资源,而且是人体和动物骨骼的主要无机成分[8],可用作生物材料,具有良好的应用前景。DCPD(CaHPO4·2H2O)在一定条件下可以转化成HAP[9]。并且磷酸钙具有磷矿的有效成分,可在产磷工业体系中加工利用。因此,通过磷酸钙结晶回收散失到水体中的磷资源被广泛的研究[10]。然而,一般来说将含磷产物从水体中直接回收较为困难,通常需要通过离心、过滤等方式,一定程度上增加了磷回收的成本。如何实现水体中磷的高效回收已成为一项挑战。磁分离是一种通过借助外部磁场实现物质有效分离的技术,具有操作简单、反应条件温和、成本低廉等优势,被广泛应用于污水处理、酶反应工程以及生物医药等领域[11]。磁性复合材料的应用有助于实现废水中磷资源的结晶回收。
本研究采用微波冷却回流法首先制备多级花球状方解石和纳米四氧化三铁,进一步通过超声法将纳米磁铁矿与多级结构方解石结合,形成Ca-Fe基磁性纳米复合材料。通过调控初始pH、反应时间、磷的初始浓度、固液比、共存离子等单因素系统研究了Ca-Fe纳米复合材料对水体中磷的去除效率及影响因素,阐释Ca-Fe纳米复合材料对水体中磷的去除机制。
-
实验所使用的乙酸钙(Ca(CH3COO)2·H2O)、尿素(CO(NH2)2)、乙二醇((CH2OH2))、氯化铁(FeCl3·6H2O)、氯化亚铁(FeCl2·4H2O)、氢氧化钠(NaOH)、磷酸二氢钠(NaH2PO4)、乙醇(C2H5OH)均为分析纯,购置于国药集团化学试剂有限公司,实验用水为去离子水。
-
多级结构方解石的制备。采用前驱体煅烧法制备多级结构方解石[12-13]:将0.53 g (3 mmol) Ca(CH3COO)2·H2O和0.18 g (3 mmol) CO(NH2)2分别超声溶解于25 mL和5 mL乙二醇中,然后将上述溶液加入到100 mL圆底烧瓶中。溶液混合均匀后将圆底烧瓶置于微波化学反应器(额定输出功率为800 W)中,在50%的额定输出功率下反应10 min,反应结束后冷却至室温,将产物用乙醇洗涤3次,然后在40 ℃下进行真空干燥,即获得方解石前驱体。将干燥后的前驱体置于马弗炉中,在500 ℃空气气氛中煅烧2 h(升温速率为5 ℃·min−1),获得多级结构方解石。
纳米四氧化三铁的制备。采用微波辅助法制备纳米磁铁矿[14]:将0.54 g (2 mmol) FeCl3·6H2O和0.20 g (1 mmol) FeCl2·4H2O超声溶解于20 mL乙二醇,将0.32 g (8 mmol)氢氧化钠溶解于2 mL去离子水中。然后将上述溶液在100 mL圆底烧瓶中充分混匀,置于微波化学反应器中,在80%的额定输出功率下反应20 min。反应结束后,冷却至室温,用乙醇洗涤后将所得固体在60 ℃真空条件下干燥,获得纳米四氧化三铁。
Ca-Fe基磁性纳米复合材料的制备。采用超声法制备磁性复合材料[15]:称取0.12 g四氧化三铁和0.25 g方解石分别放入2个烧杯中,分别加入10 mL乙醇进行超声分散。然后将上述2种悬浊液混合、密封,并在室温下超声30 min。反应结束后通过离心分离,在60 ℃条件下进行真空干燥,获得Ca-Fe纳米磁性复合材料。
通过改变方解石的加入量,制备具有不同磁铁矿负载率的Ca-Fe基复合材料,将复合材料充分溶解于稀酸溶液后,利用电感耦合等离子体-原子发射光谱仪(ICP-AES)测定Fe和Ca的含量,通过计算获得复合材料中Fe3O4的实际负载率,具体结果见表1。在后续实验过程中,除特别说明外,所使用的材料均为C-2。
-
将磷酸二氢钠(NaH2PO4)溶解于去离子水配置模拟含磷废水贮备液,实验过程中所需的含磷废水均通过稀释该贮备液获得。在典型的实验过程中,取50 mL 1 mmol·L−1的含磷废水于烧杯中,使用1 mol·L−1的HCl和NaOH溶液调节含磷废水的初始pH后加入50 mg吸附剂,在室温下将烧杯置于恒温摇床中,以150 r·min−1振荡反应24 h,反应结束后取样,使用0.22 μm滤头过滤,并测定磷的残留浓度。同时对固体样品进行回收、干燥,以备分析。有关pH、接触时间、材料用量、不同磷的初始浓度、共存离子(K+、Mg2+、CO32-、SO42-)对复合材料去除磷的影响均采用相似步骤研究。通过钼酸盐分光光度法测定溶液中的磷含量,所有实验均进行3次,取其平均值。
-
本研究采用X射线衍射仪(XRD,D/MAX 2600)测定材料去除磷前后物质的组成;通过傅里叶红外光谱仪(FTIR,INVENIO)对材料表面官能团进行分析;采用光电子能谱仪(XPS,ESCALAB 250Xi)分析材料的元素及价态;利用场发射扫描电子显微镜(SEM,Zeiss Gemini 300)观察样品的形貌特征;通过比表面积测试仪(BET,ASAP2460)测定材料的比表面积和孔径分布;采用紫外分光光度仪(T2602S)测定磷的质量浓度。
-
图1(a)为碳酸钙负载Fe3O4前后的XRD谱图。可见,29.41°、39.40°、43.14°等位置出现的衍射峰为方解石(JCPDS 05-0586)的特征峰,峰型尖锐且强烈,表明实验所合成的CaCO3(方解石)具有良好的结晶性。经超声负载处理之后,除了方解石原有特征峰外,在37.07°(222)处出现了新的特征峰,为Fe3O4(JCPDS 85-1436)的特征衍射峰。此外,通过FTIR对产物进一步分析。如图1(b)所示,在713、876和1 429 cm−1处的吸收峰归因于方解石中碳酸盐的伸缩振动峰[16],1 795、2 513、2 870、2 978 cm−1处的吸收峰为碳酸盐的泛音或组合带[17]。红外光谱分析结果进一步证实了方解石的存在。负载Fe3O4之后的复合材料与方解石特征峰相比,主要区别在590 cm−1附近出现了1个较宽的吸收峰,该吸收峰是由于Fe3O4的四面体位和八面体位的Fe-O键的拉伸振动和扭转振动产生[18];634 cm−1附近出现的吸收峰归因于—OH 振动峰[19],在3 430 cm−1和1 080 cm−1等吸收峰为水分子[20]和—OH官能团的伸缩振动特征峰[21]。FTIR表征结果表明,通过超声处理后方解石与磁铁矿形成了Fe3O4- CaCO3复合材料。
进一步利用SEM对所制备的方解石和复合材料进行形貌和结构分析。由图1(c)可见,所制备的方解石CaCO3呈现出3~5 μm的均匀的花球状结构,并且微米结构花球由多孔纳米片相互交叉的纳米片形成。由图1(d)可见,在超声引入Fe3O4后,方解石的花球状结构未发生明显改变,然而在表面发现纳米颗粒的存在,SEM结果进一步表明通过超声法成功制备出Fe3O4-CaCO3复合材料。图1(e)中复合材料的Mapping表征结果表明,Ca、Fe、O、C等元素分布于材料表面,这进一步说明Fe3O4-CaCO3复合材料的生成。
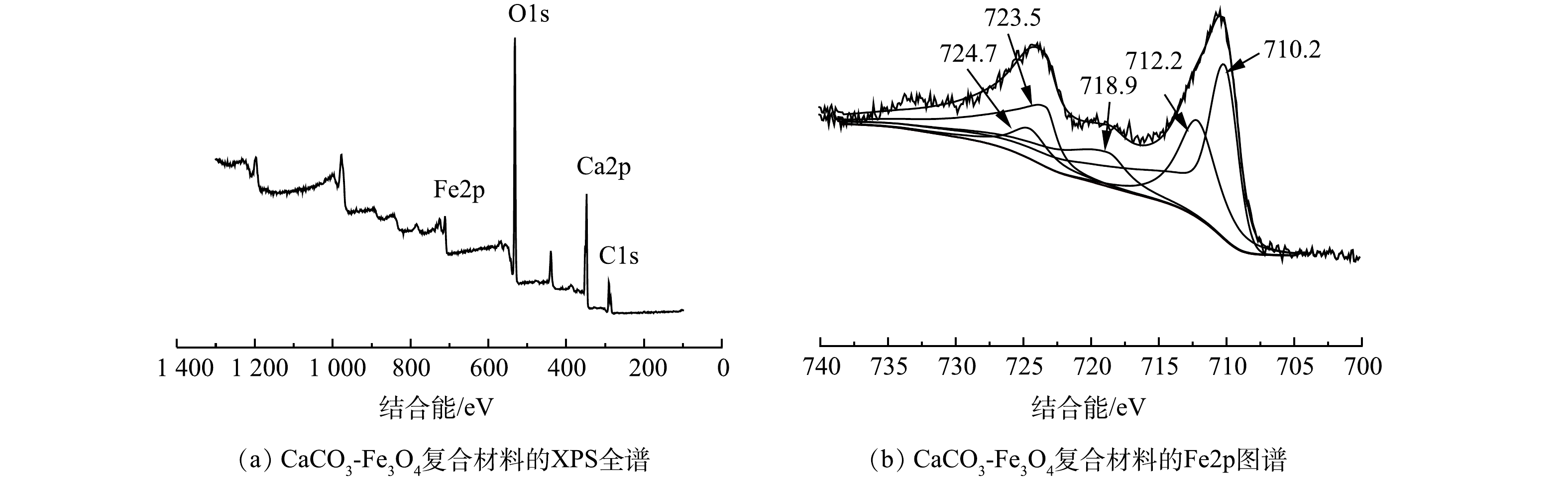
利用XPS分析复合材料元素组成和价态结构。图2(a)为Ca-Fe基复合材料的XPS全谱。由图2(a)可见,复合材料含有Ca、Fe、O、C元素。图3(b)中Fe2p的XPS分峰图谱表明,在710.2、712.2、723.5和724.7 eV处出现的峰分别与Fe2+的Fe2p3/2、Fe3+的Fe2p3/2、Fe2+的Fe2p1/2和 Fe3+的Fe2p1/2的峰值相对应[22],证明铁的氧化物以Fe2+和Fe3+混合组成[23]。Fe2p3/2的肩峰对Fe3O4的氧化态非常敏感[24-25],被用来定性地验证铁的存在形态。一般来说,Fe2p3/2卫星峰的位置比主峰数值高6 eV,则 Fe为+2价;差值为8 eV,则Fe为+3价[26-27]。所制备的复合材料中Fe2p3/2的卫星峰值(718.9 eV)与主峰值(712.2 eV)之间的差值为6.7 eV,表明同时存在Fe3+和Fe2+。因此,XPS分析结果进一步表明Fe3O4成功的负载于方解石,形成Ca-Fe基纳米复合材料,并且Fe3O4的形态在超声负载过程中未发显著变化。
通过比表面积测定仪对材料的比表面积和孔体积进行分析。如图3所示,多级方解石和CaCO3-Fe3O4复合材料的N2吸附-解吸等温线呈现IV型结构,复合材料的等温线中能够观测到较为明显的滞后回环,表明复合材料存在内部孔隙[16]。方解石花球和CaCO3-Fe3O4复合材料的比表面积和孔体积分别为7.17 m2·g−1、0.02 cm3·g−1和57.79 m2·g−1、0.35 cm3·g−1。由于纳米磁铁矿的负载,复合材料的比表面积和孔体积增加,可提供更多的活性位点,有利于污染物的去除。
由图4(a)可见,Ca-Fe基复合材料的磁化强度为18.50 emu·g−1,复合材料具有良好的顺磁性,能够满足材料使用后进行磁分离的需要。图4(b)直观的反映了材料的磁效应,经外加磁铁装置后,材料能够较好地被磁铁吸引。以上结果表明,所制备的Ca-Fe基磁性纳米复合材料具备良好的磁分离性能。
-
在反应体系中,不同pH下PO43-具有不同的存在形态,同时吸附材料的表面电荷也有所不同。因此,首先探究pH对材料去除磷的影响(磷溶液初始浓度为1 mmol·L−1,反应时间24 h)。如图5(a)所示,当pH为3~6时,复合材料对磷具有较高的去除率,并且随着初始pH的升高磷的去除率呈现缓慢降低的趋势;在初始pH为6~8时,磷的去除率从77.21%迅速下降至31.39%;当pH大于8时,下降趋势变缓。就反应后pH而言,在反应达到平衡后溶液的pH较初始pH有不同程度的增加,均为碱性。有研究[28]表明,方解石在不同的pH下具有不同的溶解度。在酸性条件下,方解石具有较大的溶解度,当pH小于6.5时,超过50%的方解石会发生溶解,同时溶液中的磷主要以H2PO4−为主,Ca2+与H2PO4−生成沉淀,从而实现对水体中磷的有效去除。当pH为6.5~7.5时,仅有10%~20%的方解石发生溶解,方解石主要通过吸附作用实现对对磷的去除。在碱性条件下方解石几乎不发生溶解,同时OH−还会与PO43-产生竞争,从而导致磷的去除率降低。综上所述,在磷去除过程中,方解石在溶解过程中释放出Ca2+,通过与磷酸根形成钙的磷酸盐矿物,进促进磷的去除;随着pH的增大,Ca2+的释放量减少,导致对体系中磷去除率的降低。
图5(b)为花球状方解石和Ca-Fe基复合材料对磷的去除率随反应时间的变化(磷溶液浓度为1 mmol·L−1,反应初始pH为5.0)。花球状方解石对磷的去除率随着时间的延长而升高,反应时间为1 h,磷的去除率为26.7%;在1~4 h内,去除率几乎保持不变,呈现出停滞状态;在4~24 h,方解石对磷的除率不断升高,反应12 h后去除容率达到80.7%。以上结果表明,方解石对磷的吸附主要发生在反应前1 h内,而后随着反应时间增加,方解石发生溶解释放的Ca2+与磷酸根发生反应,导致磷的去除率进一步升高。Ca-Fe基纳米复合材料对磷的去除趋势与花球状方解石规律几乎一致。然而,在反应进行到1 h,复合材料对水体中磷的去除效率明显高于方解石,12 h后去除率上升到86.7%。复合材料对磷的去除率高于方解石花球,是由于在Ca-Fe复合材料形成过程中,Fe的引入导致复合材料表面形成大量的氧的空位,从而提高了Ca-Fe复合材料表面的电子转移速率,减低吸附过程中的结合能障碍,加速了Ca-Fe复合材料与磷酸盐的配体交换反应动力学[29-30]。
吸附剂用量对水体中磷的去除的规律(磷溶液浓度为1 mmol·L−1,反应初始pH=5.0,反应时间24 h)见图5(c)。随着吸附剂用量的不断增大,复合材料对磷的去除率明显增大。当吸附剂由0.2 g·L−1增至0.6 g·L−1时,磷的去除率由67.4%增加90.7%。随着吸附剂浓度的持续增大,去除率逐渐达到平衡。这是由于随着吸附剂浓度增加,更多的方解石发生水解,提供了更多的活性位点,同时溶液的pH呈现出上升的趋势。为保证获得良好的去除率同时兼顾经济性,材料的使用剂量在接下来的实验中设定为1.0 g·L−1。
进一步在最佳初始pH为5.0条件下,探究了磷的初始浓度对复合材料去除磷的影响。图5(d)显示,在室温条件下,当初始浓度从0.1 mmol·L−1增大至25.0 mmol·L−1时,复合材料对磷的去除容量从2.01 mg·g−1迅速增大至189.21 mg·g−1。Ca-Fe基复合材料对磷的去除容量远高于天然方解石的去除容量(0.10 mg·g−1)[28],也高于文献所报道的Ca/Fe复合材料(161.40 mg·g−1)[29]、氧化镁负载硅藻土(160.94 mg·g−1)[31]、镁改性硅酸钙(71.05 mg·g−1)[32]、氧化镁改性磁性生物炭(149.25 mg·g−1)[33]、La-Fe氢氧化物(123.46 mg·g−1)[34] (表2)。
图5(e)为Ca-Fe基复合材料中Fe3O4负载率对磷去除容量的影响(磷溶液浓度为20 mmol·L−1,反应初始pH 5.0,反应时间24 h)。可以看出,未负载的花球状纳米方解石对磷表现出了良好的去除性能,其对磷的去除容量为218.43 mg·g−1。随着复合材料中Fe3O4负载率的不断增大,复合材料对水体中磷的去除容量呈现递减的趋势。这是由于随着Fe3O4负载率的增加,相同质量复合材料中的方解石的含量减少,导致复合材料对磷的去除容量降低。以上结果进一步说明方解石组分在复合材料中对磷的去除起到主导作用。
在实际应用中,水体中通常含有多种共存离子,因此探究了K+、Mg2+、CO32-和SO42-对复合材料去除磷的影响。由图5(f)可见,在相同反应条件下,共存离子种类和浓度的不同均会对复合材料除磷的性能产生一定的影响。当磷的初始浓度为20 mmol·L−1时,K+对磷的去除影响较为轻微,然而 Mg2+对磷的去除产生明显的抑制作用,Mg2+在2 mmol·L−1和10 mmol·L−1时,复合材料对磷的去除容量分别下降至154.31 mg·g−1和116.47 mg·g−1。这可能是由于,体系pH随着反应的进行不断升高,Mg2+发生水解产生氢氧化物吸附于复合材料表面,产生桥联作用[35],从而降低方解石的分散和水解能力,导致复合材料对磷的去除率降低[36]。同时发现CO32-和SO42-对体系中磷的去除也存在抑制作用,并且随着SO42-浓度的增大,对磷去除的抑制作用不断增强。Ca2+容易与CO32-和SO42-发生沉淀反应,CO32-和SO42-与HnPO4(3-n)-形成竞争关系,抑制了复合材料对HnPO4(3-n)-的有效去除。
-
为了进一步阐明Ca-Fe基磁性纳米复合材料对磷的去除机制,通过XRD、FTIR、SEM、XPS对复合材料除磷后的产物进行表征。如图6(a)所示,XRD结果表明在磷初始浓度为1 mmol·L−1时所获得的产物的物相与原材料相比未发生明显变化。这可能是由于复合材料对低浓度的磷主要以吸附的方式去除。此外,在反应过程中生成的钙的磷酸盐产物可能以非晶的形式存在,导致XRD无法精确检测。当磷的初始浓度为20 mmol·L−1时,产物在11.68°、17.97°、23.39°、29.25°等位置均出现了CaHPO4·2H2O(JCPDS 09-0077)的特征峰,表明在反应过程中体系中的HnPO4 (3-n)-与Ca2+发生化学反应生成CaHPO4·2H2O。此外,XRD图谱中依然可以观测到Fe3O4的特征峰存在,说明复合材料中的Fe3O4在磷的去除过程中未发生明显变化,并且反应后依然具有磁性,可通过磁分离技术对产物进行有效回收。结合图5中影响因素实验结果推测,Ca-Fe基复合材料在反应的初始阶段首先通过吸附作用对磷去除。随着反应的进行,由于复合材料中方解石的特殊花球纳米结构,Ca-Fe基复合材料中的方解石快速水解释放出大量的Ca2+,由图7(a)可见,反应体系中Ca2+的质量浓度在反应30 min后即可到达228.2 mg·L−1;与此同时,方解石的水解消耗了一定量的H+,导致反应体系pH逐步升高,图7(b)显示,反应体系pH从初始阶段的5.0左右逐步升高到8.2左右;在反应进行4 h后体系pH大于7.8,此时HnPO4(3-n)-主要以HPO42-的形式存在[5]。因此,反应体系中方解石所释放的Ca2+与HPO42-反应生成CaHPO4·2H2O (20 ℃时CaHPO4·2H2O的溶度积常数为4.3×10−3),复合材料通过结晶-吸附机制实现对磷的有效回收。图5(a)显示复合材料对磷的去除在反应1~4 h阶段出现停滞,该现象产生的原因可能是由于在反应4 h内体系的pH在7.3~7.5(图7(b)),此时溶液中的HnPO4(3-n)-主要以H2PO4−的形式存在[5],由于Ca(H2PO4)2溶解度较大(20 ℃时Ca(H2PO4)2的溶度积常数为1.8),不易产生结晶,导致反应停滞。值得注意的是,反应体系中虽然Ca2+释放量较高,然而Fe3+浓度低于0.01 5 mg·L−1(图8(b)),表明在反应过程中Fe3O4保持稳定,有利于反应后物质的回收。
在不同磷初始浓度下所得产物的FTIR图谱如图6(b)所示。在磷初始浓度为1 mmol·L−1时,所得产物在571 cm−1和1 037 cm−1附近出现了新的吸收峰,为PO43-的ν4和ν3反对称伸缩振动峰[37-38]。在磷初始浓度为20 mmol·L−1时,所得产物在876、1 425 cm−1处CO32-的吸收峰减弱,说明方解石在反应过程中发生溶解,CO32-含量降低。此外,FTIR谱图还显示出标准CaHPO4·2H2O的特征吸收峰[39-40]。在3 700~3 000 cm−1内出现了2个不同的双峰,分别位于3 543、3 489 cm−1和3 288、3 163 cm−1处,该吸收峰为2种结晶水分子的O—H 伸缩振动[41];同时在1 645 cm−1处的吸收峰也是由于水分子的 O—H 弯曲振动所产生[42]。PO43-的特征吸收峰位于1 150、900附近及650~520 cm−1[43]。此外,在1 134 cm−1和1 066 cm−1处出现了P=O的伸缩振动峰;在1 218、871和792 cm−1处的特征峰分别是由HPO42−的P—O—H和P—OH的振动引起的[44-45];576 cm−1和573 cm−1处为(H—O—)P=O的振动峰[46]。以上FTIR分析结果进一步说明CaHPO4·2H2O的生成。
产物的SEM和EDS表征结果如图8所示。可见,反应后方解石原有的形貌与初始形貌完全不同。在磷初始浓度为1 mmol·L−1条件下,方解石发生水解,原有的花球形貌发生变化,仍保留部分原有骨架。同时,由Mapping表征结果可见磷均匀分布于产物表面,CaPFeCO并且钙与磷元素的比值明显小于1(产物中钙的含量为10.05%,磷的含量为5.05%),说明在该条件下复合材料通过吸附对磷去除。当磷的初始浓度为20 mmol·L−1时,方解石的原有形貌被完全破坏,出现了由纳米颗粒状构成的块状物质;钙与磷元素的比值接近于1(产物中钙的含量为14.03%,磷的含量为15.49%),表明CaHPO4·2H2O矿物的生成,该结果与XRD分析结果一致。
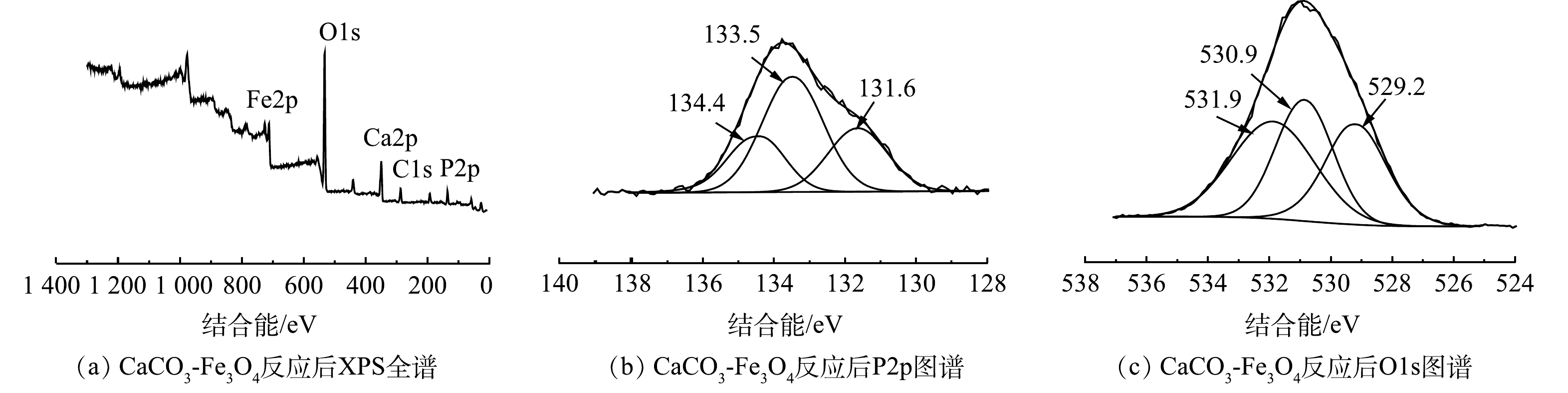
利用X射线光电子能谱仪对产物的化学成分以及所涉及元素的化学形貌进行表征分析。由图9(a)可见,XPS全谱中出现了明显的P2p和O1s的特征峰,证明产物中磷发生反应。由图9(b)可见,131.6、133.5和134.4 eV位置出现的3个特征峰分别对应于PO43-、HPO42-、H2PO4−[47-48]。其中,133.5 eV处的特征峰最强,表明在生成物中磷主要以HPO42-的形式存在。由图9(c)可见,在530.9 eV处出现的特征峰为P=O,531.9 eV处的特征峰为P-OH[49][和CaCO3中的C—O[50],529.2 eV处的吸收峰为表面羟基和结合水的O—H[51]。以上结果进一步表明生成物为CaHPO4·2H2O,与XRD和FTIR的分析结果一致。
所制备的Ca-Fe基磁性复合材料对磷的回收机制主要为吸附-结晶耦合。在结晶反应过程中,Ca-Fe基复合材料中方解石通过水解提供Ca2+,与磷酸根离子发生结晶反应生成CaHPO4·2H2O,从而实现磷资源的回收。所回收的磷矿物可作为肥料或生物材料被进一步利用。由于所制备的复合材料富有磁性,便于磁性回收,降低在实际应用中的回收成本,同时能够避免二次污染的产生。此外,Ca-Fe基磁性复合材料合成过程中所使用钙源和铁源易于获取,具有较好的经济性。
-
1)利用微波-冷却-回流和超声的方法制备Ca-Fe 基磁性纳米复合材料,系统研究了复合材料对水体中磷的回收性能,复合材料在pH=3.0~6.0内对水体中的磷表现出良好的回收效果,在最佳实验条件下复合材料对磷的最大去除容量为189.21 mg·g−1。
2)复合材料中的方解石组分在对磷的去除中起到主导作用。复合材料对低浓度的磷主要通过吸附作用实现有效去除;对于高浓度的磷,复合材料通过吸附-结晶耦合的机制实现对水体中磷的有效回收,产物中磷以CaHPO4·2H2O的形式存在。
3)由于复合材料具有良好的磁性,在处理含磷废水之后,可以通过磁分离技术将产物高效回收,避免产生二次污染,所制备的复合材料在磷回收领域具有潜在的应用价值。
Ca-Fe基磁性纳米复合材料对水体中磷的结晶回收
Crystallization recovery of phosphorus from water by Ca-Fe based magnetic nanocomposites
-
摘要: 人类活动导致大量的不可再生的磷资源流失到水环境中造成水体富营养化,磷的结晶回收对废水治理、地表水管理和可持续发展具有重要意义。采用微波-冷却-回流和超声的方法制备Ca-Fe 基磁性纳米复合材料(CaCO3-Fe3O4),通过批量吸附实验法系统探究了体系pH、接触时间、磷的初始浓度、共存离子等因素对复合材料去除水体中磷的影响规律。结果表明,CaCO3-Fe3O4纳米磁性复合材料在pH=3.0~6.0内对磷表现出良好的去除效果,对磷的最大去除容量为189.21 mg·g−1。复合材料对水体中的磷主要通过吸附-结晶耦合机制去除,在高浓度含磷废水中,磷以CaHPO4·2H2O的形式被回收。综合考虑磁分离的简易性、磷的去除容量和环境友好性,所制备的Ca-Fe基磁性复合材料在磷资源回收领域具有潜在的应用价值。Abstract: Human activities induced the loss of non-renewable phosphorus resources into aquatic environment, leading to eutrophication of waterbody. Crystallization recovery of phosphorus is crucial for wastewater treatment, surface water management and sustainable development. In this study, Ca-Fe based magnetic nanocomposites (CaCO3-Fe3O4) were synthesized by microwave-assisted reflux and ultrasound method. The effects of solution pH, contact time, initial phosphorus concentration, coexisting ions on phosphorus removal by magnetic nanocomposite were systematically examined by the batch adsorption method. The results demonstrated that the phosphate could be effectively recovered from water by CaCO3-Fe3O4 nanocomposites in the pH range of 3.0~6.0, and the maximum removal capacity of 189.21 mg·g−1(P). The phosphate was removed via sorption-crystallization coupling mechanism, and in high phosphate concentration effluent, P was harvested in a CaHPO4·2H2O form. Considering the easy magnetic separation, high phosphorus removal capacity and environmental friendliness, the prepared Ca-Fe based magnetic composites have potential applications in phosphorus resource recovery.
-
Key words:
- calcite /
- magnetic nanocomposites /
- phosphorus /
- sorption /
- crystallization recovery
-
水资源短缺与能源危机推动了水处理技术的革新。在“双碳”背景下,传统的以能耗换水质的水处理技术已无法满足可持续发展的要求[1]。开发以能源再生-资源回收为目的的新技术,降低水处理过程中的碳排放,对于实现我国的“双碳”目标有着重要意义[2]。与传统的好氧生物处理技术相比,厌氧生物处理技术能够在废水处理过程中实现资源回收和能源再生,在水处理中具有广阔的应用前景[3-4]。近年来厌氧生物处理技术开始逐步应用于低浓度废水(如市政污水)的处理,但有机物含量低时厌氧微生物生长缓慢,且低产气量较难实现泥水混合液的充分搅拌,导致有机物的去除转化效果不理想,从而限制了厌氧技术在低浓度废水处理中的应用[5]。厌氧膜生物反应器(anaerobic membrane bioreactors, AnMBR)采用厌氧处理与膜分离相结合的方式,将水力停留时间(hydraulic retention time, HRT)和污泥龄(sludge retention time, SRT)分开[6],有效弥补了传统厌氧生物处理技术存在的弊端,具有生物量大、出水水质高以及能耗低等优点[7],是实现低浓度废水厌氧生物处理的理想技术。
已有很多研究报道了AnMBR用于处理低浓度废水时可以实现较好的有机物去除和产甲烷性能,但运行过程中存在着膜污染的问题,常需要通过沼气循环、混合液回流等方式增大体系混合程度来减轻膜污染。另一方面,AnMBR出水中含有较多的溶解性甲烷(dissolved methane, DCH4),甲烷是一种温室气体,DCH4解吸后[8](释放到大气)会造成能量流失和温室效应。现有研究[9-11]表明,AnMBR出水DCH4的质量浓度为8.8~19.1 mg·L–1,因此,而流失的甲烷占总甲烷产量的23%~88%。在较多研究中,测得DCH4的浓度经常高于亨利定律计算的理论值,即AnMBR出水的DCH4往往是过饱和的[12],其过饱和度即相比于理论DCH4的倍数为1.3~4.1倍[9,13]。已有部分研究通过外置气体分离器[14-17]对AnMBR出水DCH4进行回收并取得了较好的效果,但上述过程存在着能量消耗、占地增加和工艺流程长等问题。甲烷的液-气传质会显著改变甲烷的溶解程度,其传质系数[10,18]主要受反应器运行条件(如负荷、混合效率、温度)的影响,在不同工况下AnMBR出水DCH4的过饱和度具有较大的差异[19]。YEO等[10]在采用混合液循环的AnMBR中研究结果表明,随着负荷提升产气速率不断增大,有利于解除甲烷气-液传质的限制,从而降低了DCH4的过饱和度。研究不同工况对AnMBR中甲烷气-液分配特征的影响,有望通过反应器运行方式的调控,实现原位降低DCH4的浓度。为此,本研究针对AnMBR出水存在DCH4过饱和问题,基于AnMBR的低浓度废水处理过程,探究了不同负荷和混合方式对有机物去除和DCH4的影响。揭示上述过程甲烷气-液传质系数的变化规律并阐明AnMBR中甲烷的气-液分配特征,进一步用于指导反应系统运行,以期为降低AnMBR的碳排放量和能耗提供参考。
1. 材料与方法
1.1 实验装置
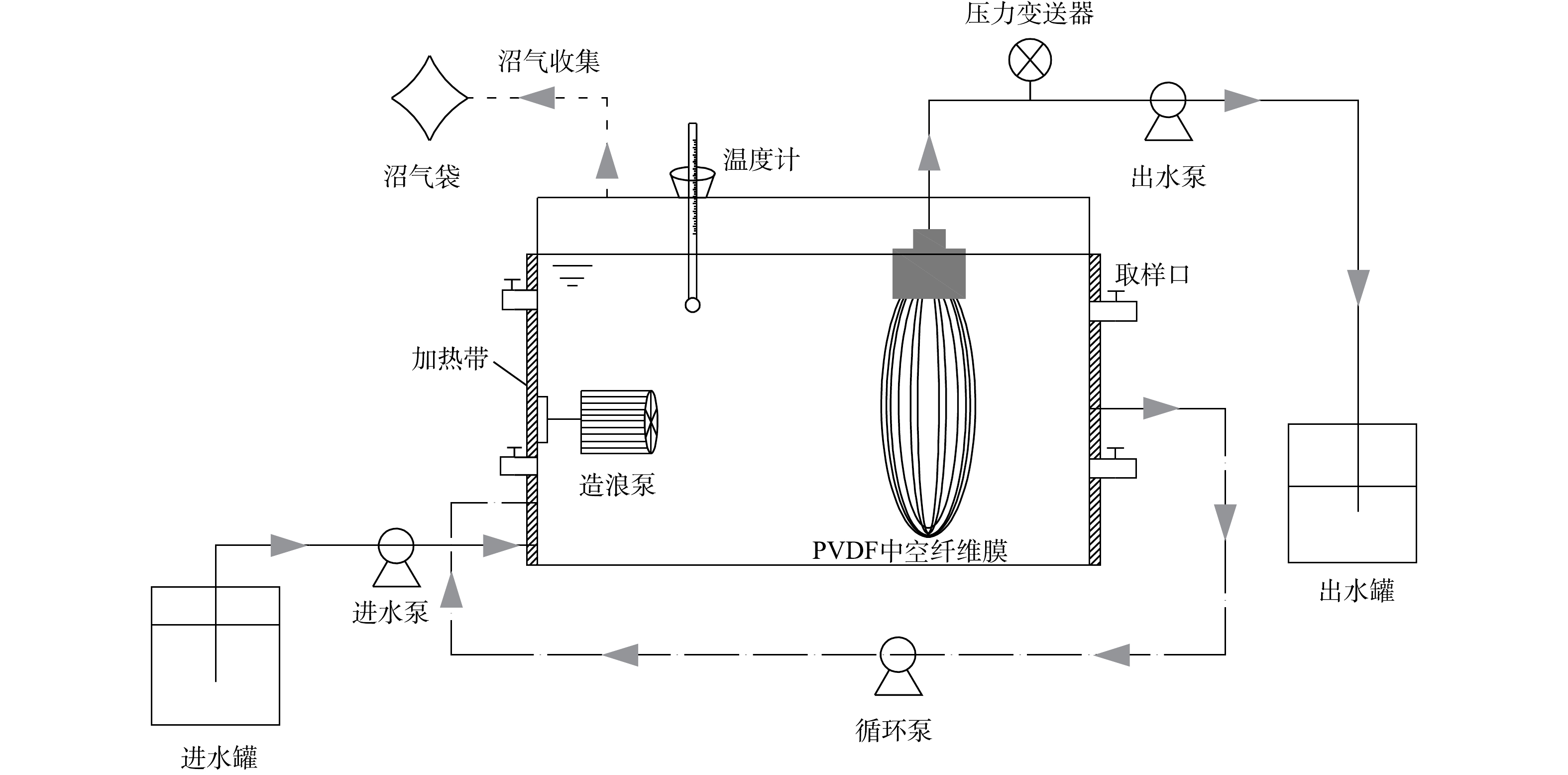
本实验所用AnMBR装置如图1所示,反应器主体为有机玻璃材质的长方体形容器(长×宽×高=30 cm×20 cm×25 cm),有效容积为11 L。反应器内设置聚偏氟乙烯(PVDF)材质的中空纤维膜(MBR-0.1F,海科滤膜),膜组件构型采用U型,膜丝数量共80根,单根膜丝的内、外径分别为0.8 mm和1.8 mm,膜孔径为0.1 μm,膜组件总有效面积0.19 m2。反应器进出水采用蠕动泵控制(BT100-2J,兰格恒流泵有限公司),出水泵与膜组件之间设有压力变送器(SIN-P300,中国杭州联测自动化技术有限公司),用于监测跨膜压差的变化。反应器外部覆盖有加热带(鸿泰电热配件),用于维持反应器内部的温度,内部温度由反应器顶部的温度计读取。反应器顶部设有阀门用于连接沼气袋和收集厌氧反应所产气体。反应器两侧均设有取样口用于采样分析。
AnMBR每个运行周期为10 min,其中出水8 min,松弛2 min。反应器设有2种不同的混合方式:从出水侧将泥水混合液泵回进水侧以进行循环混合;内部设置1个造浪泵(JVP-100AB)进行浪式脉冲混合。
1.2 接种污泥及实验用水
反应器接种污泥为黑色厌氧颗粒污泥,取自升流式厌氧污泥床反应器。实验进水采用由葡萄糖、尿素、磷酸二氢钾等人工配制而成的模拟废水,具体水质成分如下:0.23 g·L–1 尿素、0.11 g·L–1 KH2PO4、1 g·L–1 K2HPO4、0.05 g·L–1 Na2SO4、0.1 g·L–1 MgCl2·6H2O、0.05 g·L–1 CaCl2·2H2O、10 mL·L–1 微量元素、10 mL·L–1 维生素(注:微量元素和维生素具体组成参考文献[20])。缓冲物质为质量浓度为1 g·L–1的碳酸氢钠。葡萄糖的用量根据实验进行调整。
1.3 实验方法
控制AnMBR反应器温度为(35±1) oC,水力停留时间(HRT)为20 h,通过改变进水COD来改变反应器的负荷。反应器启动后共运行87 d,运行过程根据负荷和混合方式分为6个阶段,其中3个负荷水平(A、B、C)对应的进水COD分别为500、1 000、1 500 mg·L–1,每个负荷下混合方式分为循环混合与浪式脉冲混合,各运行工况条件见表1。
表 1 不同阶段的运行条件Table 1. Operation conditions of different phases阶段 负荷/(kg·(m3·d)–1) 进水COD/(mg·L–1) 混合方式 A1 0.6 500 循环 A2 0.6 500 浪式脉冲 B1 1.2 1 000 循环 B2 1.2 1 000 浪式脉冲 C1 1.8 1 500 循环 C2 1.8 1 500 浪式脉冲 | Show Table DownLoad:
CSV
DownLoad:
CSV
每天取进出水水样测定COD值(哈希(Hach)试剂盒,2125815-CN),取反应器内混合液样品用于DCH4的浓度测定(见1.4);沼气袋内收集气体的体积用注射器抽取测量,并用气相色谱(天美GC7900,中国)分析其组分及含量。各运行阶段结束时,取反应器内混合液用于生物量(MLSS和MLVSS)的测定(重量法)。
1.4 分析及测试方法
1)水中DCH4的测定。水中的DCH4采用顶空平衡法进行测定。在20 mL的密封小瓶中注入3.5 mol·L–1的盐酸1 mL,用于抑制产甲烷菌的生理活性,从反应器内采集9 mL混合液样品注入小瓶中。将小瓶在25 oC的振荡器中振荡5 min,并放置3 h使CH4达到气液平衡,平衡后取1 mL顶空气体注入气相色谱分析气体组成。反应器混合液中DCH4的浓度根据亨利定律计算(式(1))[21]所示。根据亨利定律推出式(2)。
CL=CG,eqVG+CL,eqVLVL (1) 式中:CL为液体中DCH4的质量浓度,mg·L–1;CG,eq是平衡后样品瓶顶空中CH4的质量浓度,mg·L–1;CL,eq是样品瓶液体中与顶空平衡的CH4质量浓度,mg·L–1;VG和VL分别为样品瓶中液体和气体的体积,L。
Hcp=CL,eqPP,eq⋅MCH4 (2) 式中:PP,eq是CH4在顶空中与液相平衡时的分压,Pa;
MCH4 2) COD质量平衡计算。在各工况运行稳定后,根据式(3)[18]进行COD的物料衡算,以分析有机物分布去向。
Cin=Ceff+CDCH4+CGCH4+Cother (3) 式中:Cin和Ceff分别为进出水COD,mg·L–1;
CDCH4 CGCH4 CH4+2O2=CO2+2H2O 3)CH4传质系数的计算。AnMBR中CH4由液相向气相的传质系数可由式(4)[10]进行计算。
KLa=(QgPCH4VRT)(1CL–Ceq)=QVKHRT(CLCeq–1) (4) 式中:KLa为由液相向气相的传质系数,h–1;CL为液体中DCH4的质量浓度,mg·L–1;Qg为甲烷产气速率,L·h–1;V是反应器的有效体积,L;PCH4为AnMBR顶空中CH4的分压,Pa;R是理想气体常数,8.31×103 L·Pa·(mol·K)–1;T是温度,K;QV (Qg/V)为反应器的CH4产气量,L·(L·h)–1;Ceq是与气相中CH4分压处于热力学平衡的DCH4的质量浓度,mg·L–1;KH是CH4的亨利常数,35 oC时为1.18×10–7 mol·(L·Pa)–1。
4)能量计算。CH4的热值约为37.8 MJ·m–3,按电能转换效率为35%计算CH4转换成的电能(式(5))[23]。反应器运行过程中混合所用设备的能量需求通过设备的功率和运行时间得出(式(6))。反应器运行过程中的净能量(E)由设备能源消耗与回收甲烷所得能源的差值计算(式(7))。
WCH4=37.8VCH4×35%3.6 (5) 式中:
WCH4 VCH4 W消耗=Pt1000 (6) 式中:W消耗为设备消耗的能量,kWh;P为设备的功率,浪式脉冲设备功率为3 W,混合液循环设备功率为20 W;t为设备运行时间,h。
E=W消耗−WCH4 (7) 2. 结果与讨论
2.1 有机物去除性能
本研究中AnMBR共运行87 d,固定反应器的HRT为20 h,通过增加进水COD来提升负荷,在此期间研究了反应器不同混合形式对有机物去除性能的影响,各运行工况下进出水的COD值及去除率变化如图2所示。结果表明,AnMBR可实现对有机物的高效去除,COD平均去除率超过90%,提升进水负荷AnMBR对有机物仍可保持着良好的去除效果,具有一定的抗冲击负荷性。如图2所示,各负荷下AnMBR浪式脉冲混合时对有机物的去除效果均好于循环混合,在进水COD为500 mg·L–1,负荷为0.6 kg·(m3·d)–1时,循环混合模式下反应器出水的COD值为(48±4.9) mg·L–1,对应COD去除率为(90.7±0.8)%;改用浪式脉冲混合模式后,COD去除率提升至(96.8±0.4)%。这是因为浪式脉冲混合的系统扰动强度更高,促进了微生物与底物的接触与利用,从而提高了有机物的去除效果[24]。
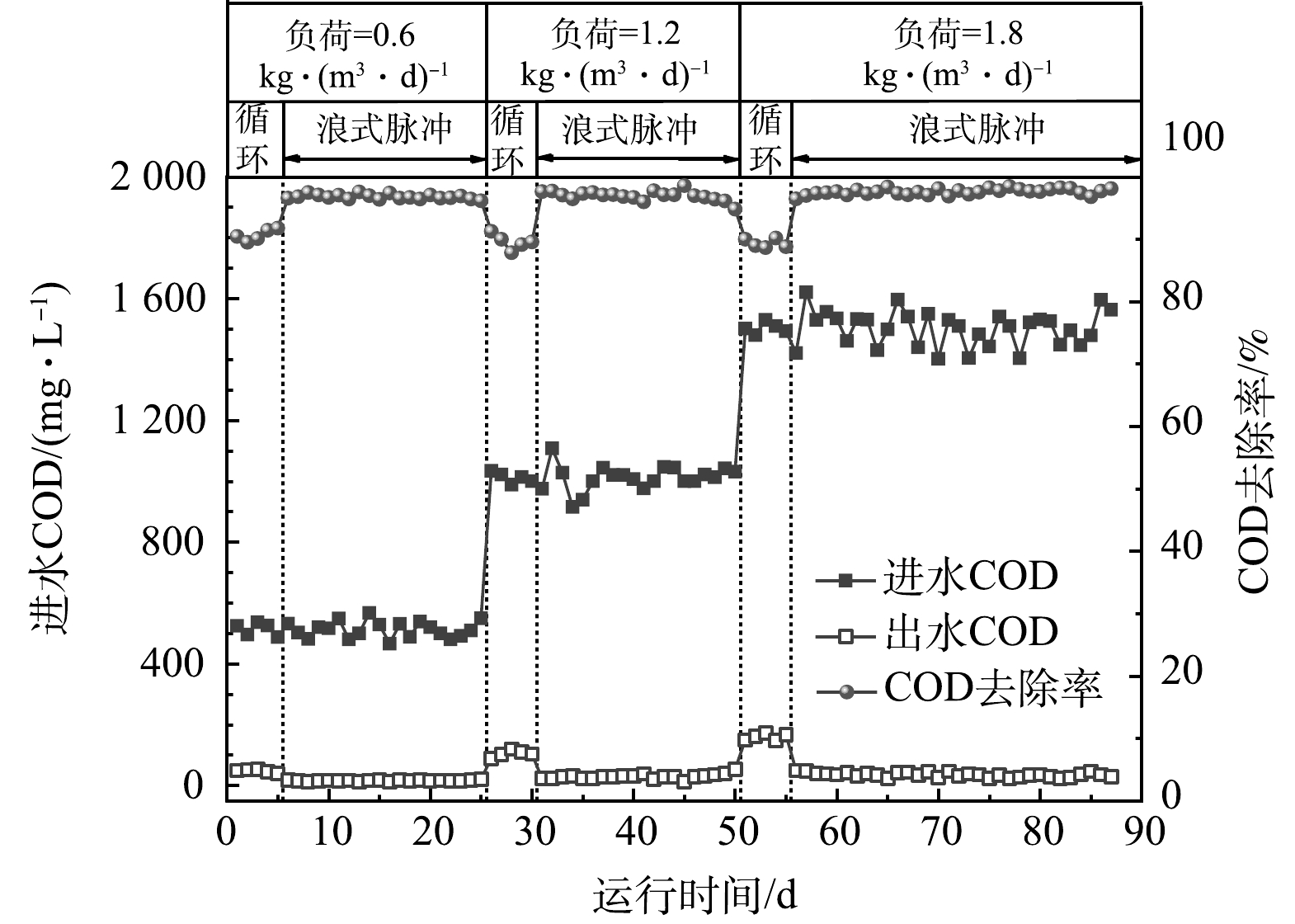 图 2 AnMBR各运行条件下有机物去除性能Figure 2. Removal performance of organic matter under different operating conditions of AnMBR
图 2 AnMBR各运行条件下有机物去除性能Figure 2. Removal performance of organic matter under different operating conditions of AnMBR2.2 甲烷气-液分配特征
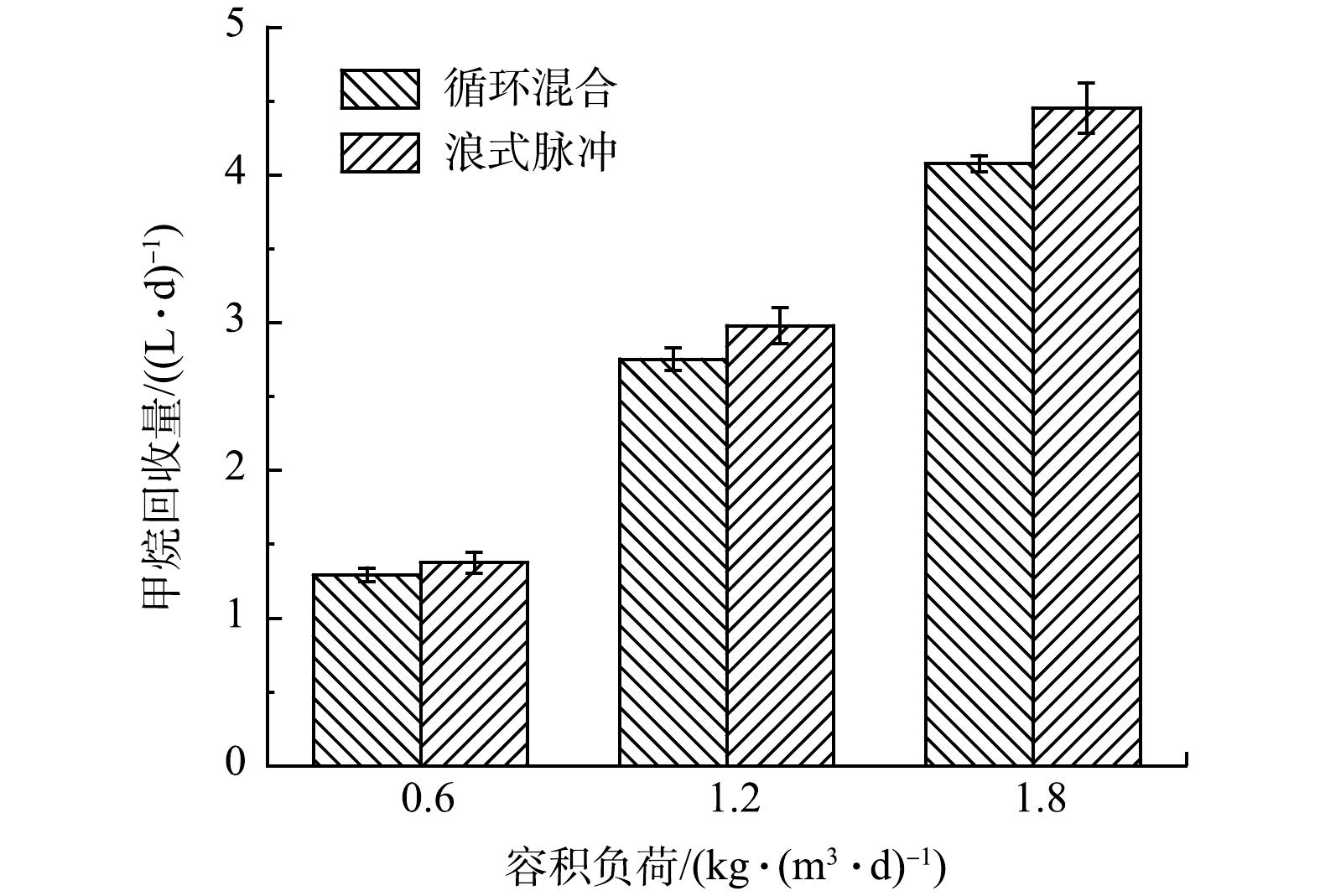
在AnMBR的运行过程中,测定各运行工况下的CH4产气量(QV)和DCH4,结合亨利定律和式(4)计算出理论DCH4和甲烷传质系数(KLa)。如图3所示,各阶段反应器液相中DCH4均处于过饱和状态,DCH4浓度为理论浓度的1.3~2.1倍,该水平位于已报道的AnMBR中DCH4过饱和度范围之内[25-26]。在相同负荷下,浪式脉冲混合时DCH4的浓度和过饱和度均低于循环混合时(图3(c)~(d))。在负荷为0.6 kg·(m3·d)–1下,循环混合时DCH4为20.2 mg·L–1,对应过饱和度为2.1;而浪式脉冲混合时DCH4为12.6 mg·L–1,对应过饱和度为1.3。上述结果是由于不同条件下CH4的KLa不同导致的,KLa较大时,CH4能更好地从液相扩散至气相,从而有利于降低DCH4浓度及过饱和度。KLa通常与甲烷生成速率、混合效率、流体黏度和温度等因素有关[10,27]。在本研究中,反应器内流体黏度与温度基本保持不变,而浪式脉冲混合时CH4的KLa更大,这主要由2方面原因造成:一方面,浪式脉冲混合时体系的混匀度更高;另一方面,由上文可知,浪式脉冲混合时有机物的去除效果更好,对应CH4的Qv更大。在相同负荷水平,YEO等采用沼气循环混合时DCH4的过饱和度为2.2~2.5[10],对应KLa为(0.018±0.003)~(0.088±0.017) h–1,由此可见,浪式脉冲混合在降低DCH4浓度及过饱和度方面比液体循环或气体循环混合具有更好的效果。
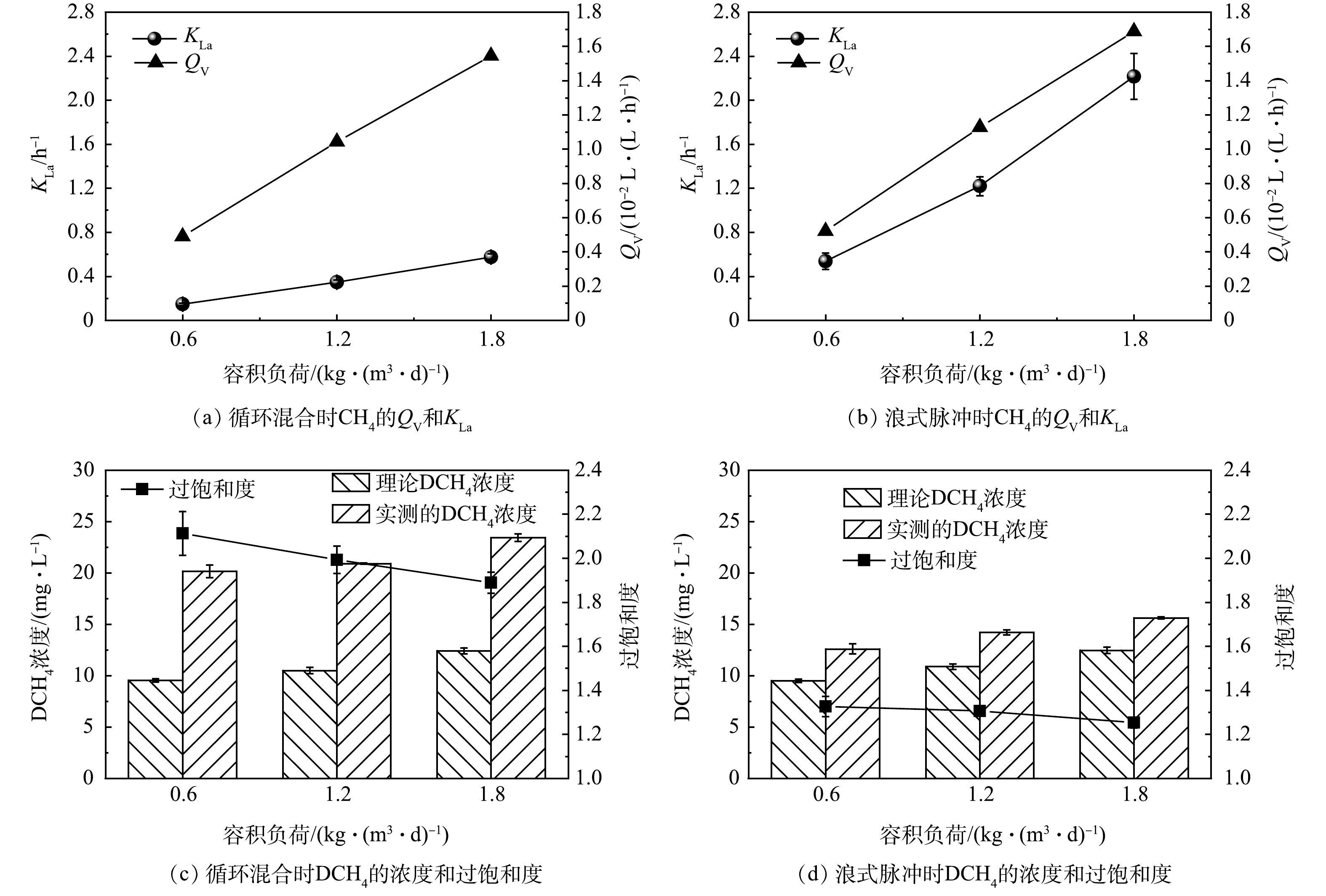 图 3 AnMBR运行过程中CH4的QV和KLa、DCH4的浓度和过饱和度Figure 3. QV and KLa of CH4, DCH4 concentration and supersaturation during AnMBR operation
图 3 AnMBR运行过程中CH4的QV和KLa、DCH4的浓度和过饱和度Figure 3. QV and KLa of CH4, DCH4 concentration and supersaturation during AnMBR operation如图3所示,随着进水负荷的提升,2种混合方式的DCH4浓度均有升高。其中,循环混合时的DCH4由20.2 mg·L–1上升至23.4 mg·L–1(图3(c)),对应DCH4的过饱和度由2.1降至1.9;浪式脉冲混合时的DCH4由12.6 mg·L–1升至15.6 mg·L–1(图3(d)),对应DCH4的过饱和度由1.3降至1.2。可以看出,随着负荷的提升,DCH4的过饱和度逐渐降低。这一结果与YEO等[10]的研究结果一致。CH4的QV随负荷的提升而增加(图3(a)~(b)),Qv与负荷呈现较好的正相关关系(R2=0.998),WIJEKOON等[28]的研究中也得出了相似的结论。因为CH4的KLa正比于QV(式(4)),所以KLa随负荷提升也不断增大,且与负荷也呈现出较好的正相关关系(R2=0.997)。高负荷(1.8 kg·(m3·d)–1)时CH4的KLa ((0.57±0.03) h–1)约为低负荷(0.6 kg·(m3·d)–1)时KLa ((0.15±0.01) h–1)的3~4倍。综上所述,CH4的Qv和KLa均与负荷显著相关,随着负荷提升DCH4不断升高,但DCH4的变化程度(增加倍数)小于Qv,这是因为随负荷提升KLa也同时增大,导致更多的DCH4扩散至气相,从而使DCH4在过饱和度上呈现下降趋势。
在AnMBR的运行过程中,计算不同运行工况下CH4在气/液相中的分布及在进水COD中的占比(见图4)。同负荷下,循环混合时气态CH4占总CH4及进水COD的比例均低于浪式脉冲混合时。如负荷为0.6 kg·(m3·d)–1,循环混合时气态CH4占总CH4及进水COD的比例分别为76.0%和50.2%;浪式脉冲混合时气态CH4占总CH4及进水COD的比例分别为84.5%和57.0%。这是因为浪式脉冲混合(KLa更大)可促进CH4从液相向气相的传质,这与上文的分析结果一致。
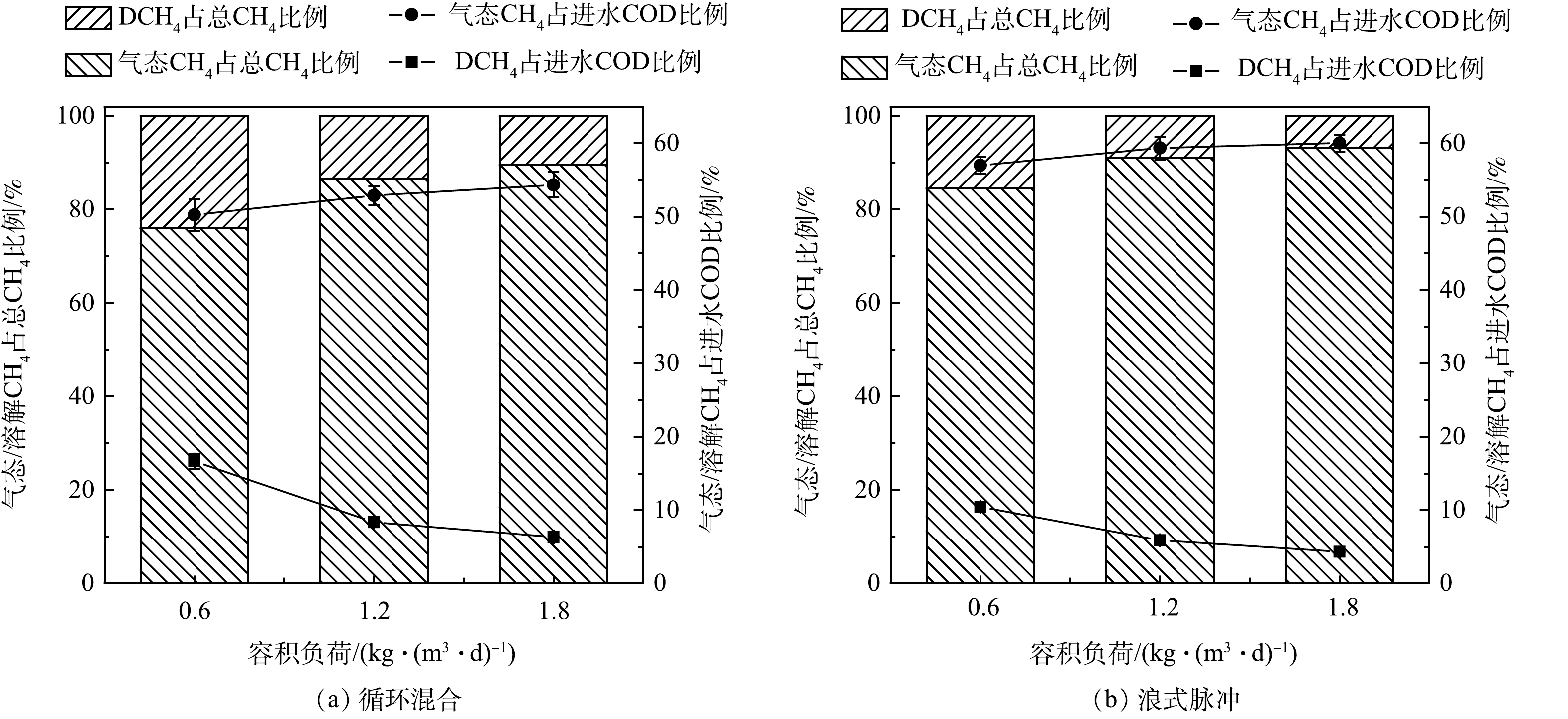 图 4 气态CH4和DCH4占总CH4和进水COD的比例Figure 4. The ratios of gaseous CH4 and DCH4 to total CH4 and influent COD
图 4 气态CH4和DCH4占总CH4和进水COD的比例Figure 4. The ratios of gaseous CH4 and DCH4 to total CH4 and influent COD如图4所示,随着进水负荷的提升,DCH4占总CH4及进水COD的比例逐渐下降,而气态CH4占总CH4及进水COD的比例不断升高,表明AnMBR在高负荷下更容易对有机物中的能量进行回收。此外,AnMBR中可回收的气态CH4占进水COD的比例(50.2%~60.0%)低于通常文献报道的CH4含量70%~85%[29-31],这是由于本研究关注的处理对象为低浓度废水,所用的进水COD和负荷水平要低于上述报道,根据上文分析此时的KLa较低从而导致了较多的进水COD以DCH4形式存在。
2.3 有机物转化
进水COD在AnMBR中的转化形式可能为气相CH4、DCH4、生物量增长和出水残留等,为了进一步阐明进水底物的电子流向,对各运行工况下AnMBR进行了COD质量衡算,结果见图5。由图5可知,总CH4 (DCH4+气态CH4)占进水COD的主要部分,为61.0%~67.4%,DCH4占比为4.3%~16.1%。以COD计的气态CH4、DCH4和出水COD加和后与进水COD的差值表示为Cother,在3种不同的负荷下的占比为24.4%~30.6%,这部分COD主要用于生物量合成、微生物代谢产物(SMP、EPS)的产生和微量的硫酸盐还原等,在其他AnMBR中也观察到了类似的COD转化[17,32]。整个运行过程没有对AnMBR进行排泥,但反应器内生物量(MLVSS)变化较小,从6 020.4 mg·L–1增加到6 820.7 mg·L–1。生物量(MLVSS)与COD的换算关系[33]约为1 g=1.4 g,计算后可知,用于生物增长的COD只占了Cother的3.1%~3.5%,以上结果表明大部分Cother可能以微生物代谢产物(SMP、EPS)的形式存在。微生物代谢产物主要为大分子的多糖和蛋白质[34-35],出水膜可能对这些化合物有很强的截留作用,进而导致了膜污染。关于不同负荷与混合方式对微生物代谢产物及膜污染的特征影响需要后续进一步的研究。
 图 5 不同工况下AnMBR进水COD质量平衡Figure 5. Influent COD mass balance of AnMBR under different operation conditions
图 5 不同工况下AnMBR进水COD质量平衡Figure 5. Influent COD mass balance of AnMBR under different operation conditions2.4 能量分析
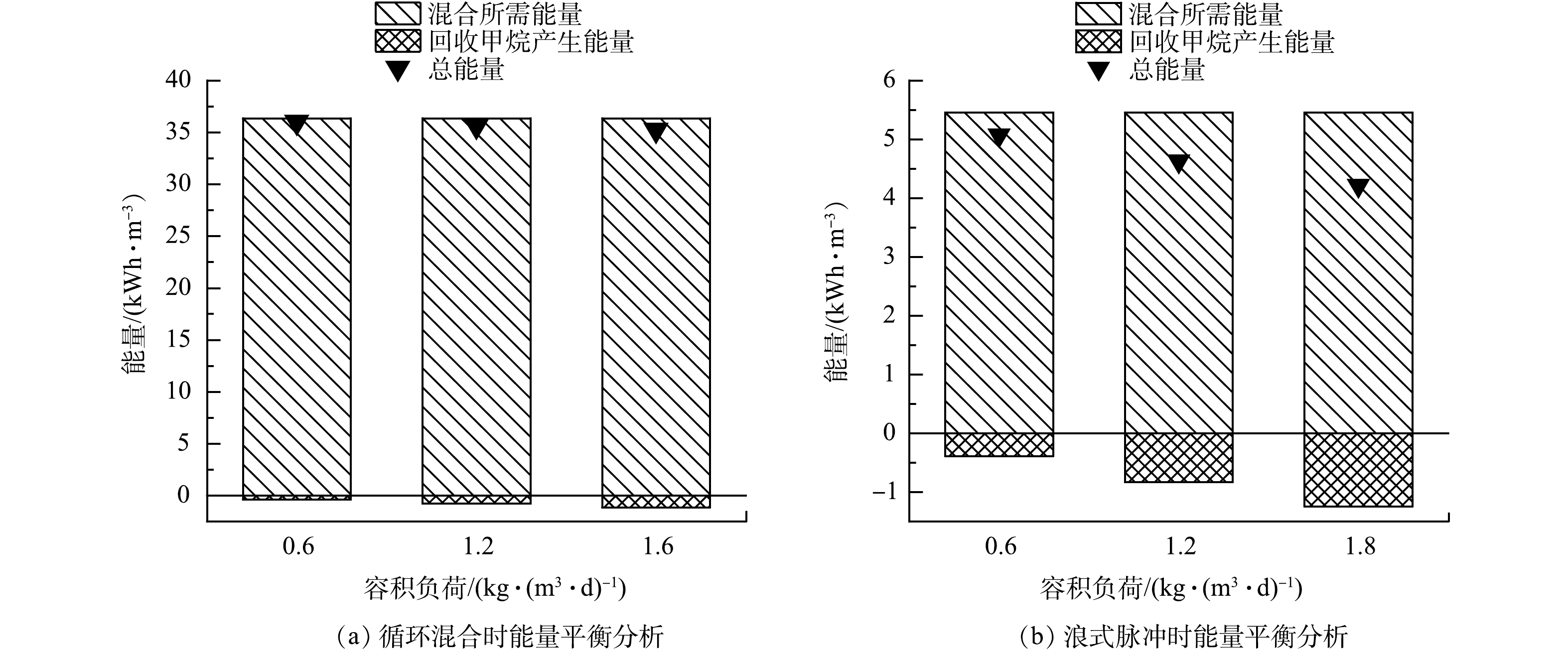
净能量平衡是评价系统应用时能量回收潜力的重要指标,通过能量需求与能量回收的差值来计算。本研究中,AnMBR运行过程保持HRT与温度恒定,各阶段的所需能量主要来自混合方式的电耗;各阶段的回收能量来自气相CH4。各运行条件下的CH4回收量和相关能量计算见图6和图7。
如图7所示,相比于循环混合,浪式脉冲混合时的CH4回收量更大且总能量消耗更低。浪式脉冲混合的CH4产量比循环混合条件下提高了6.5%~9.2%(图7)。由上文分析可知,这是由更多有机物转化和更高的气相CH4导致。另一方面,由图6和图7可知,浪式脉冲混合所需的能耗只有循环混合的15%,但甲烷回收率提升了6.5%~9.2%,因此,计算分析后,浪式脉冲混合时AnMBR总能量消耗比循环混合时降低了85.9%~88.0%。随着负荷的提升,AnMBR总能量消耗不断降低,高负荷(1.8 kg·(m3·d)–1)时总能耗比低负荷(0.6 kg·(m3·d)–1时下降了约16.9%。由此可知,通过调整运行参数如缩减HRT等进一步提升负荷有利于实现AnMBR处理废水过程的能量平衡甚至能量盈余。ASLAM等[36]在负荷为5.8 kg·(m3·d)–1的条件下,在流态化厌氧陶瓷膜生物反应器中实现了0.099 kWh·m–3的能量盈余,与本研究所得高负荷运行时AnMBR易实现能量平衡或盈余的结论一致。
3. 结论
1) HRT维持在20 h,进水负荷为0.6~1.8 kg·(m3·d)–1的运行条件下,AnMBR可实现高效稳定的有机物去除效果(COD去除率>90%),浪式脉冲混合时的有机物去除效果比循环混合时更好。
2) AnMBR在浪式脉冲混合时(KLa大),CH4能更好地从液相扩散到气相,其DCH4的浓度和过饱和度及占进水COD的比例均比循环混合时低,且随负荷提升,这些指标进一步降低。
3) AnMBR进水大部分COD (61.0%~67.4%)转化为了CH4,其中气相CH4占比为50.2%~60.0%,剩余进水COD主要转化为微生物代谢产物。
4) AnMBR浪式脉冲混合的总能耗比循环混合时低85.9%~88.0%,提高负荷有利于实现AnMBR处理污水过程的能量平衡甚至能量盈余。
-

图 1 CaCO3多级方解石和CaCO3-Fe3O4复合材料的组成和结构的表征分析
Figure 1. Characterizations of hierarchically structured CaCO3 and CaCO3-Fe3O4 composite,
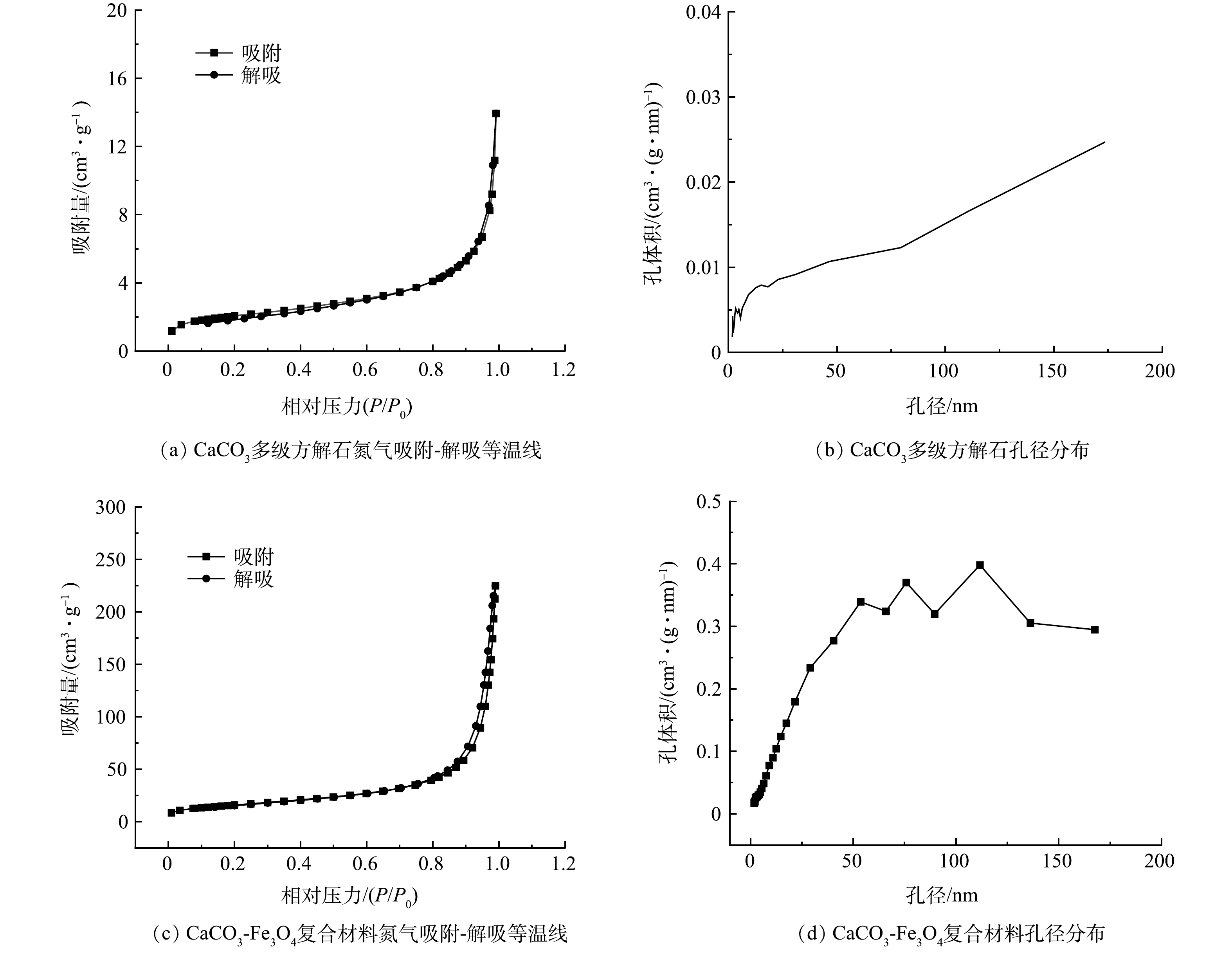
图 3 CaCO3和CaCO3-Fe3O4复合材料的氮气吸附-解吸等温线和孔径分布曲线
Figure 3. Nitrogen adsorption–desorption isotherms and pore size distribution of CaCO3 and CaCO3-Fe3O4 composite

图 4 CaCO3-Fe3O4复合材料磁滞回线图谱和磁效应图片
Figure 4. Magnetic hysteresis loop and magnetic effect of the CaCO3-Fe3O4 nanocomposites.

图 5 不同影响因子对复合材料去除磷的影响
Figure 5. Effects of different factors on the phosphorus removal of nanocomposites
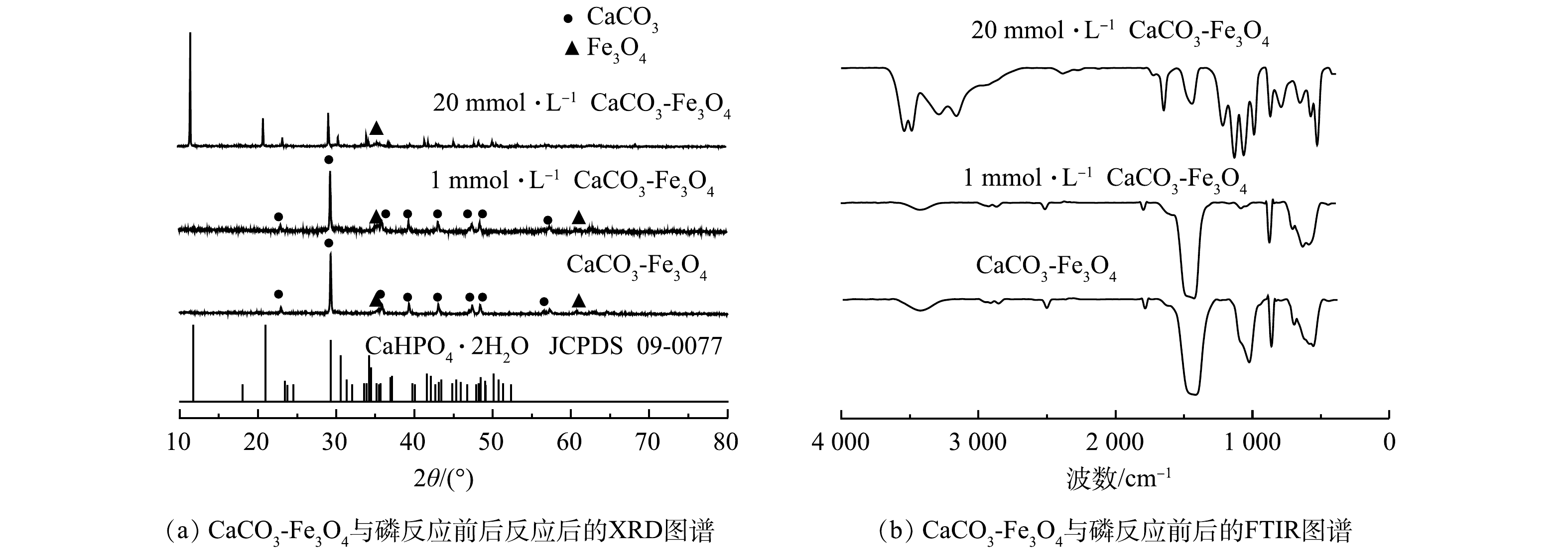
图 6 CaCO3-Fe3O4在不同磷初始浓度下产物的XRD 和FTIR图谱
Figure 6. Typical XRD patterns and FTIR spectra of CaCO3-Fe3O4 nanocomposites after the removal of phosphorus at different levels
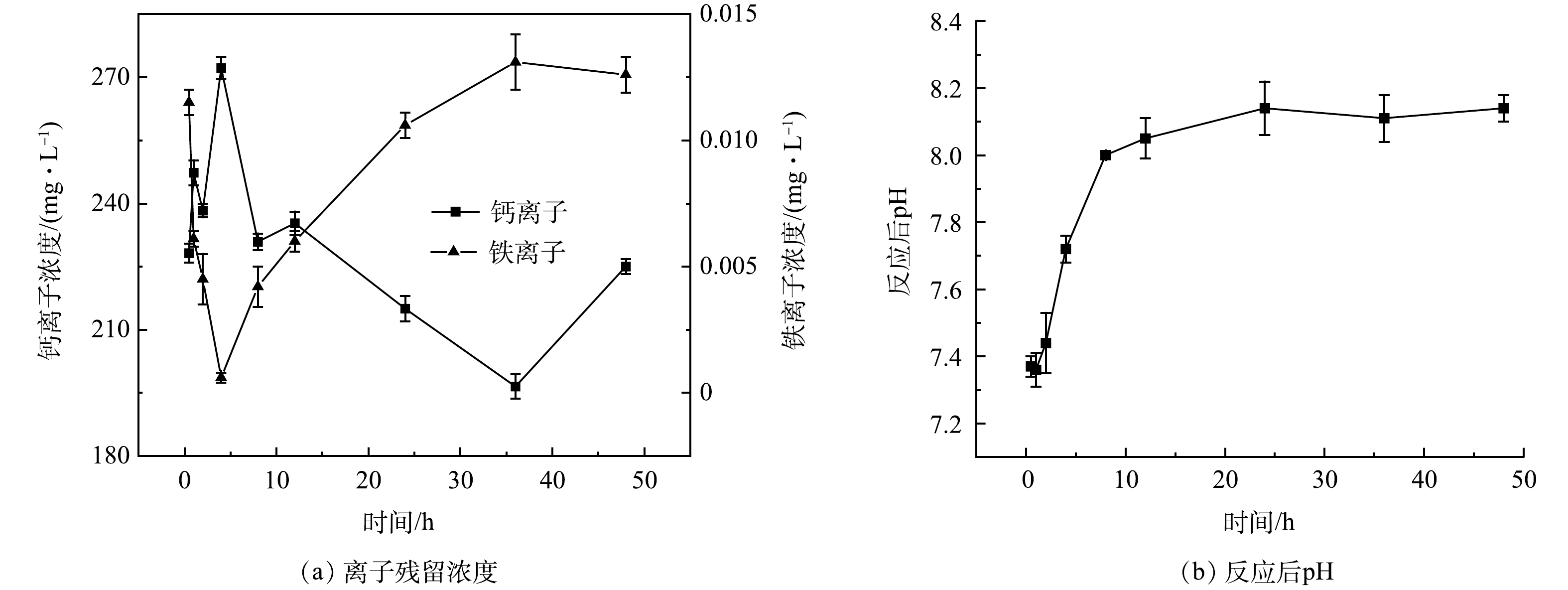
图 7 不同时间下反应体系Ca与Fe离子残留浓度和反应后pH
Figure 7. Ca and Fe concentration and pH at different time

图 8 在不同磷初始浓度下产物的SEM图片和Mapping分析图
Figure 8. SEM images and mapping analyses of CaCO3-Fe3O4 nanocomposites after the removal of phosphorus at different phosphorus concentrations
表 1 不同材料负载率
Table 1. Different loading rates of synthesized materials
样品编号 方解石/g Fe3O4/g 负载率(Fe3O4)/% C-1 0.125 0.12 41.7 C-2 0.250 0.12 22.4 C-3 0.500 0.12 11.9
下载: 导出CSV
表 2 不同吸附剂对磷去除性能的比较
Table 2. The phosphorus removal performance of different Mg-based sorbents
材料 初始pH 去除容量/(mg·g−1) 参考文献 天然方解石 6.5 0.10 29 Ca/Fe复合材料 5.4 161.40 30 氧化镁负载硅藻土 7.0 160.94 32 镁改性硅酸钙 7.0 71.05 33 氧化镁改性磁性生物炭 3.0 149.25 34 La-Fe氢氧化物 7.0 123.46 35 CaCO3-Fe3O4 5.0 189.21 本研究
下载: 导出CSV
-
[1] MAYER B K, BAKER L A, BOYER T H, et al. Total value of phosphorus recovery[J]. Environmental Science and Technology, 2016, 50(13): 6606-6620. doi: 10.1021/acs.est.6b01239 [2] MITROGIANNIS D, PSYCHOYOU M, BAZIOTIS I, et al. Removal of phosphate from aqueous solutions by adsorption onto Ca(OH)2 treated natural clinoptilolite[J]. Chemical Engineering Journal, 2017, 320: 510-522. doi: 10.1016/j.cej.2017.03.063 [3] FORREST A L, FATTAH K P, MAVINIC D S, et al. Optimizing struvite production for phosphate recovery in WWTP[J]. Journal of Environmental Engineering, 2008, 134(5): 395-402. doi: 10.1061/(ASCE)0733-9372(2008)134:5(395) [4] WANG S, WU Y, AN J, et al. Geobacter autogenically secretes fulvic acid to facilitate the dissimilated iron reduction and vivianite recovery[J]. Environmental Science & Technology, 2020, 54(17): 10850-10858. [5] TAO W, FATTAH K P, HUCHZERMEIER M P. Struvite recovery from anaerobically digested dairy manure: A review of application potential and hindrances[J]. Journal of Environmental Management, 2016, 169: 46-57. [6] FURUYA K, HAFUKA A, KUROIWA M, et al. Development of novel polysulfone membranes with embedded zirconium sulfate-surfactant micelle mesostructure for phosphate recovery from water through membrane filtration[J]. Water Research, 2017, 124: 521-526. doi: 10.1016/j.watres.2017.08.005 [7] HUANG H, ZHANG D D, LI J, et al. Phosphate recovery from swine wastewater using plant ash in chemical crystallization[J]. Journal of Cleaner Production, 2017, 168: 338-345. doi: 10.1016/j.jclepro.2017.09.042 [8] OBADA D O, OSSENI S A, SINA H, et al. Fabrication of novel kaolin-reinforced hydroxyapatite scaffolds with robust compressive strengths for bone regeneration[J]. Applied Clay Science, 2021, 215: 106298. doi: 10.1016/j.clay.2021.106298 [9] SHIH W J, CHEN Y F, WANG M C, et al. Crystal growth and morphology of the nano-sized hydroxyapatite powders synthesized from CaHPO4·2H2O and CaCO3 by hydrolysis method[J]. Journal of Crystal Growth, 2004, 270(1-2): 211-218. doi: 10.1016/j.jcrysgro.2004.06.023 [10] HERMASSI M, VALDERRAMA C, DOSTA J, et al. Evaluation of hydroxyapatite crystallization in a batch reactor for the valorization of alkaline phosphate concentrates from wastewater treatment plants using calcium chloride[J]. Chemical Engineering Journal, 2015, 267: 142-152. doi: 10.1016/j.cej.2014.12.079 [11] 彭诗珍, 黄启同, 甘滔等. 四氧化三铁基复合材料在生物磁分离中的应用[J]. 赣南医学院学报, 2022, 42(04): 389-395. doi: 10.3969/j.issn.1001-5779.2022.04.012 [12] YU S H, LI H, YAO Q Z, et al. Preparation of mesoporous calcite with hierarchical architectures[J]. Materials Letters, 2015, 160: 167-170. doi: 10.1016/j.matlet.2015.07.116 [13] YIN W, LIU M, ZhAO T L, et al. Removal and recovery of silver nanoparticles by hierarchical mesoporous calcite: Performance, mechanism, and sustainable application[J]. Environmental Research, 2020, 187: 109699. doi: 10.1016/j.envres.2020.109699 [14] YU S H, LI H, YAO Q Z, et al. Microwave-assisted preparation of sepiolite-supported magnetite nanoparticles and their ability to remove low concentrations of Cr(VI)[J]. RSC Advances, 2015, 5: 84471. doi: 10.1039/C5RA14130C [15] 天津大学. 一种负载四氧化三铁的SiC泡沫陶瓷及其制备方法和应用: CN201810852660.0[P]. 2021-11-19. [16] ZHOU G T, GUAN Y B, YAO Q Z, et al. Biomimetic mineralization of prismatic calcite mesocrystals: Relevance to biomineralization[J]. Chemical Geology, 2010, 279(3/4): 63-72. doi: 10.1016/j.chemgeo.2010.08.020 [17] DLAPA P, BODI M B, MATAIX-SOLERA J, et al. FT-IR spectroscopy reveals that ash water repellency is highly dependent on ash chemical composition[J]. Catena, 2013, 108(108): 38-46. [18] NAMDURI H, NASRAZADANI S. Quantitative analysis of iron oxides using Fourier transform infrared spectrophotometry[J]. Corrosion Science, 2008, 50(9): 2493-2497. doi: 10.1016/j.corsci.2008.06.034 [19] MA M G, ZHU Y J, CHANG J. Monetite formed in mixed solvents of water and ethylene glycol and its transformation to hydroxyapatite[J]. Journal of Physical Chemistry B, 2006, 110(29): 14226-14230. doi: 10.1021/jp061738r [20] NEAGLE W, ROCHESTER C H. Infrared study of the adsorption of water and ammonia on calcium carbonate[J]. Journal of the Chemical Society Faraday Transactions, 1990, 86: 181-183. doi: 10.1039/ft9908600181 [21] 刘帅朋. 改进型共沉淀法制备四氧化三铁磁粉的工艺优化与表征[D]. 天津: 天津大学, 2018. [22] ZHAO K, XU T, CAO J, et al. Fabrication and adsorption properties of multiwall carbon nanotubes-coated/filled by various Fe3O4 nanoparticles[J]. Journal of Materials Science:Materials in Electronics, 2019, 30(20): 18802-18810. doi: 10.1007/s10854-019-02234-8 [23] ASUHA S, WAN H L, ZHAO S, et al. Water-soluble, mesoporous Fe3O4: Synthesis, characterization, and properties[J]. Ceramics International, 2012, 38(8): 6579-6584. doi: 10.1016/j.ceramint.2012.05.042 [24] LI B, CAO H, SHAO J, et al. Superparamagnetic Fe3O4 nanocrystals@graphene composites for energy storage devices[J]. Journal of Materials Chemistry, 2011, 21(13): 5069-5075. doi: 10.1039/c0jm03717f [25] CHASTAIN J, KING Jr R C. Handbook of X-ray photoelectron spectroscopy[J]. Perkin-Elmer Corporation, 1992, 40: 221. [26] MORDINA B, KUMAR R, TIWARI R K, et al. Fe3O4 nanoparticles embedded hollow mesoporous carbon nanofibers and polydimethylsiloxane-based nanocomposites as efficient microwave absorber[J]. The Journal of Physical Chemistry C, 2017, 121(14): 7810-7820. doi: 10.1021/acs.jpcc.6b12941 [27] MORDINA B, TIWARI R K, SETUA D K, et al. Impact of graphene oxide on the magnetorheological behaviour of BaFe12O19 nanoparticles filled polyacrylamide hydrogel[J]. Polymer, 2016, 97: 258-272. doi: 10.1016/j.polymer.2016.05.026 [28] 张小梅. 天然菱铁矿和方解石矿物及其改性产物除磷性能研究[D]. 南京: 南京大学, 2022. [29] WU D, TIAN S, LONG J, et al. Remarkable phosphate recovery from wastewater by a novel Ca/Fe composite: Synergistic effects of crystal structure and abundant oxygen-vacancies[J]. Chemosphere, 2021, 266: 129102. doi: 10.1016/j.chemosphere.2020.129102 [30] XIANG C, JI Q, ZHANG G, et al. In situ creation of oxygen vacancies in porous bimetallic La/Zr sorbent for aqueous phosphate: Hierarchical pores control mass transport and vacancy sites determine interaction[J]. Environmental Science & Technology, 2019, 54(1): 437-445. [31] XIA P, WANG X, WANG X, et al. Struvite crystallization combined adsorption of phosphate and ammonium from aqueous solutions by mesoporous MgO–loaded diatomite[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2016, 506: 220-227. doi: 10.1016/j.colsurfa.2016.05.101 [32] SI Q, ZHU Q, XING Z. Simultaneous removal of nitrogen and phosphorus by magnesium-modified calcium silicate core-shell material in water[J]. Ecotoxicology and Environmental Safety, 2018, 163: 656-664. doi: 10.1016/j.ecoenv.2018.07.120 [33] LIU J, JIANG J, AIHEMAITI A, et al. Removal of phosphate from aqueous solution using MgO-modified magnetic biochar derived from anaerobic digestion residue[J]. Journal of Environmental Management, 2019, 250: 109438. doi: 10.1016/j.jenvman.2019.109438 [34] YU J, XIANG C, ZHANG G, et al. Activation of lattice oxygen in LaFe (oxy) hydroxides for efficient phosphorus removal[J]. Environmental Science & Technology, 2019, 53(15): 9073-9080. [35] 方启学. 钙镁对微细矿粒分散稳定性的影响及其机理研究[J]. 国外金属矿选矿, 1998(6): 42-45. [36] 刘忠义. 金属离子对菱锌矿和方解石分散行为的影响研究[D]. 北京: 中国矿业大学, 2019. [37] LI J, CAO X, PAN A, et al. Nanoflake-assembled three-dimensional Na3V2 (PO4)3/C cathode for high performance sodium ion batteries[J]. Chemical Engineering Journal, 2018, 335: 301-308. doi: 10.1016/j.cej.2017.10.164 [38] SHARIF F, TABASSUM S, MUSTAFA W, et al. Bioresorbable antibacterial PCL-PLA-nHA composite membranes for oral and maxillofacial defects[J]. Polymer Composites, 2019, 40(4): 1564-1575. doi: 10.1002/pc.24899 [39] HIRSCH A, AZURI I, ADDADI L, et al. Infrared absorption spectrum of brushite from first principles[J]. Chemistry of Materials, 2014, 26(9): 2934-2942. doi: 10.1021/cm500650t [40] TORTET L, GAVARRI J R, NIHOUL G, et al. Study of protonic mobility in CaHPO4· 2H2O (brushite) and CaHPO4 (monetite) by infrared spectroscopy and neutron scattering[J]. Journal of Solid State Chemistry, 1997, 132(1): 6-16. doi: 10.1006/jssc.1997.7383 [41] PETROV I, ŠOPTRAJANOV B, FUSON N, et al. Infra-red investigation of dicalcium phosphates[J]. Spectrochimica Acta Part A Molecular Spectroscopy, 1967, 23(10): 2637-2646. doi: 10.1016/0584-8539(67)80155-7 [42] JAYASREE R, KUMAR T S S, VENKATESWARI R, et al. Eggshell derived brushite bone cement with minimal inflammatory response and higher osteoconductive potential[J]. Journal of Materials Science:Materials in Medicine, 2019, 30(10): 1-14. [43] BERRY E E, BADDIEL C B. The infra-red spectrum of dicalcium phosphate dihydrate (brushite)[J]. Spectrochimica Acta Part A Molecular Spectroscopy, 1967, 23(7): 2089-2097. doi: 10.1016/0584-8539(67)80097-7 [44] CASCIANI F, CONDRATE SR R A. The vibrational spectra of brushite, CaHPO4·2H2O[J]. Spectroscopy Letters, 1979, 12(10): 699-713. doi: 10.1080/00387017908069196 [45] HAZZAT M E, HAMIDI A E, HALIM M, et al. Complex evolution of phase during the thermal investigation of Brushite-type calcium phosphate CaHPO4·2H2O[J]. Materialia, 2021, 16(4): 101055. [46] RAJENDRAN K, DALE K C. Growth and characterization of calcium hydrogen phosphate dihydrate crystals from single diffusion gel technique[J]. Crystal Research and Technology, 2010, 45(9): 939-945. doi: 10.1002/crat.200900700 [47] SONG Y W, SHAN D Y, CHEN R S, et al. A novel phosphate conversion film on Mg-8.8Li alloy[J]. Surface & Coatings Technology, 2009, 203(9): 1107-1113. [48] LI S, ZENG W, XU H, et al. Performance investigation of struvite high-efficiency precipitation from wastewater using silicon-doped magnesium oxide[J]. Environmental Science and Pollution Research, 2020, 27(13): 15463-15474. doi: 10.1007/s11356-019-07589-3 [49] WEI L, HONG T, LI X, et al. New insights into the adsorption behavior and mechanism of alginic acid onto struvite crystals[J]. Chemical Engineering Journal, 2019, 358: 1074-1082. doi: 10.1016/j.cej.2018.10.110 [50] LIU X, ZHONG H, YANG Y, et al. Phosphorus removal from wastewater by waste concrete: influence of P concentration and temperature on the product[J]. Environmental Science and Pollution Research, 2020, 27(10): 10766-10777. doi: 10.1007/s11356-019-07577-7 [51] ZHANG Y, SHI J, CHENG C, et al. Hydrothermal growth of Co3(OH)2(HPO4)2 nano-needles on LaTiO2N for enhanced water oxidation under visible-light irradiation[J]. Applied Catalysis B:Environmental, 2018, 232: 268-274. doi: 10.1016/j.apcatb.2018.03.067 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2793
- HTML全文浏览数: 2793
- PDF下载数: 75
- 施引文献: 0
