





-
磷过量是水华爆发重要的诱因之一[1]。近年来,随着排污管控和面源污染治理,水体外源磷已得到逐步控制,内源成为水体中磷的主要来源,而底泥中磷向上覆水的释放是内源磷的重要成因之一[2-3]。
原位钝化技术具有操作简便、见效快等优势,可有效控制底泥中磷的释放,近年来得到了广泛的推广应用[4-5]。镧系钝化剂是目前使用最为广泛的底泥磷钝化剂,其效果远优于传统的铝盐和钙盐[6-8]。笔者在先前研究中,合成了具有强控磷能力的新型钝化剂——镧沸石,与市售锁磷剂Phoslock®相比具有一定的优势[9-12]。
钝化剂使用过程中,泥水界面有机质对其效果有显著影响[13-14]。从比重分组的角度,有机质可以分为轻组和重组有机质[15-18]。其中,轻组有机质由动植物残体、微生物等组成,具有周转时间短(最短为数周)和碳氮比高等特点,其含量呈现明显的季节变化[19-22]。将土壤或沉积物分散在密度大于轻组有机质的重液(如NaI、Na6(H2W12O40)溶液中,轻组有机质就会悬浮在溶液中,实现其与土壤或沉积物中其他组分的分离[23]。轻组有机质虽然占比不足有机质总量的10%,但其对底泥的磷释放行为有着重要影响[24]。有研究表明,去除轻组有机质后太湖贡湖湾底泥磷释放量增加了12.8倍,吸附磷的速率减少到原先的40%[25];底泥对磷的饱和吸附量也有了明显的降低[26]。但是,底泥轻组有机质对于钝化剂控磷效果的影响,目前仍然不清楚。
本研究以黑臭河道底泥为对象,考察了镧沸石对原底泥(raw sediment,简称R-S)和去除轻组有机质底泥(light fraction organics removal sediment,简称LFOR-S)磷吸附性能的影响,分析了镧沸石对R-S和LFOR-S磷释放的控制效果及其对温度变化的响应,并通过XPS和磷分级揭示了相关机理。研究结果将为原位钝化技术在河湖磷污染控制中的应用提供参考。
-
镧沸石的制备按照文献[9]进行。具体来说,25 g粉煤灰与150 mL NaOH溶液(浓度2 mol·L−1)充分混合,在水热反应釜中以95 ℃反应24 h。反应结束后,向得到的固液混合物中逐滴加入浓度为0.67 mol·L−1的LaCl3溶液150 mL,同时保持搅拌。之后离心分离,弃去上清液。用纯水和乙醇清洗固体2~3次,冻干研磨过80目筛备用。
-
底泥样品于2022年4月在新河西安鄠邑区黑臭段(34°13′34.95″N、108°39′57.672″E)使用重力取泥器采集。采集后尽快冻干,过80目筛获得原始底泥(R-S)。去除轻组有机质的方法参考文献[22],具体方法为:向5 g底泥中加入20 mL比重为1.7的NaI溶液,超声状态下混合10 min,之后在4 000 r·min−1条件下离心10 min,弃去上清液。将上述步骤重复2次后,使用去离子水将固体残渣清洗3次,冻干获得去除轻组有机质底泥(LFOR-S)。使用低温外热重铬酸钾氧化法测定R-S和LFOR-S的有机质含量,使用过硫酸钾-硫酸消解法测定R-S和LFOR-S的总磷含量,使用比表面与孔隙度分析仪(北京金埃谱公司F-sorb3400型)测定BET比表面积。
-
分别称取0.4 g R-S或LFOR-S于50 mL离心管中,添加质量分数为2%(0.008 0 g)或4%(0.016 0 g)的镧沸石。加入40 mL质量浓度为0~100 mg·L−1的KH2PO4溶液(包含0.01 M的NaCl离子强度)。在25 ℃下恒温振荡(180 r·min−1)24 h,取出后在3 000 r·min−1下离心15 min,使用钼锑抗分光光度法分析上清液正磷酸盐浓度,根据式(1)计算底泥对磷的吸附量;使用Langmuir(式(2))和Freundlich模型(式(3))拟合吸附等温线[27]。
式中:Qe为平衡吸附量,mg·kg−1;C0和Ce为反应前后的磷质量浓度,mg·L−1;V为液体体积,L;m为底泥或底泥/镧沸石混合物的重量,g。
式中:KL为Langmuir常数;Qmax为根据Langmuir模型计算出的最大吸附量,mg·kg−1;KF为Freundlich常数;其余符号意义同前。
为明确镧沸石对底泥吸附量增加的贡献,通过差减,得到投加镧沸石前后底泥磷吸附量的增加量,再除以投加的镧沸石质量,再根据式(4)计算镧沸石的吸附量。
式中:QL为镧沸石的吸附量,mg·g−1;QS+L为投加镧沸石后底泥的吸附量,mg·kg−1;QS为底泥自身的磷吸附量,mg·kg−1;m为底泥质量,g;mL为镧沸石投加量,g;1 000为单位转换系数。
-
分别称取0.4 g R-S或LFOR-S于50 mL离心管中,添加质量分数2%(0.008 0 g)或4%(0.016 0 g)的镧沸石。加入40 mL浓度为0.01 mmol·L−1的NaCl溶液,恒温振荡(180 r·min−1)一定时间。反应结束后,在3 000 r·min−1下离心15 min,测定上清液的正磷酸盐浓度。在磷释放动力学实验中,反应时间为0.25、0.5、1.5、3、5、8、12、24 h,反应温度为25 oC。在不同温度下的磷释放实验中,反应时间为24 h,反应温度为5、10、15和30 ℃。
通过实验后水中磷的浓度计算磷的释放量(式(5))。通过特定温度下,底泥在投加镧沸石前后磷释放量的变化,来计算镧沸石在该温度下的磷释放控制率(式(6))。
式中:R为磷释放量,mg·kg−1;C为释放实验结束后水中磷的质量浓度,mg·L−1。
式中:K为特定温度下的磷释放控制率,%;RS+L和RS分别为底泥在投加镧沸石后和投加前的磷释放量,mg·kg−1。
-
按照1.4中方法,不添加镧沸石的情况下获得R-S或LFOR-S磷释放上清液,反应时间为24 h。上述两种上清液中正磷酸盐质量浓度分别为1.79和2.89 mg·L−1。将2种上清液分别与0.008 0 g镧沸石混合,恒温振荡(180 r·min−1)24 h,离心分离得到反应后的镧沸石,45 oC下烘干。使用日本Ulvac-Phi公司PHI Quantera SXM 扫描成像X射线光电子能谱仪测定XPS图谱,按照C1s峰结合能为248.8 eV进行校正,并使用casaXPS进行处理。
-
分别称取1 g R-S或LFOR-S于50 mL离心管中,添加质量分数2%(0.020 0 g)或4%(0.040 0 g)的镧沸石。添加40 mL去离子水,在25 ℃下恒温振荡(180 r·min−1)24 h,取出后在3 000 r·min−1的条件下离心15 min,分析剩余底泥的磷形态,同时测定水相中磷的浓度。磷形态主要采用连续提取法测定[28],将底泥中的磷分为6种形态,其中NH4Cl-P和BD-P形态具有较强的释放潜力,NaOH-P和Org-P形态具有中等释放潜力,HCl-P和Res-P形态较为稳定一般不会释放。通过磷形态转化实验前后特定形态磷含量的变化计算转化率(式(7))。
式中:T为某一形态磷在转化实验前后的转化率,%;Qf和Qi分别为转化实验结束后和开始前该形态磷的含量,mg·kg−1。
-
去除轻组有机质前后底泥理化性质的变化如表1所示。可以看出,去除轻组有机质后,底泥中有机质含量下降了0.18%。此处下降的有机质属于轻组有机质。换算后发现,黑臭河道底泥中轻组有机质占总有机质的比例约为7%,远高于在西辽河[29]、西湖[15]、洱海[24]、太湖和鄱阳湖[19]等自然水体底泥中得到结果,而低于玄武湖、月湖[19]等城市内湖底泥中的水平。这也印证了前人有关重污染城市河湖底泥中轻组有机质含量较高的观点[19]。去除轻组有机质后,底泥中总磷从2 272.11下降到2 116.22 mg·kg−1,下降的部分应随轻组有机质的去除离开。底泥的比表面积在去除轻组有机质前后没有发生明显的变化。底泥的pH在去除轻组有机质后有了明显了上升。根据磷分级结果(见2.6),R-S和LFOR-S中BD-P和NaOH-P含量之和占到总磷的一半左右。研究表明,这些与底泥中铁、铝氧化物结合的磷,在碱性条件下会被水中OH−基团置换从而释放出来[26, 30-31]。因此,LFOR-S对磷的“束缚”能力相比R-S会出现明显的下降,进而产生较高的磷释放量。
-
为了解投加镧沸石对R-S和LFOR-S磷吸附能力的影响,测定了R-S和LFOR-S在投加镧沸石前后的吸附等温线,并使用Langmuir和Freundlich模型对其进行了拟合,结果如图1和表2所示。结果表明,R-S和LFOR-S在投加镧沸石前后的吸附等温线均可以被上述两个模型较好地拟合。去除轻组有机质后,底泥的最大磷吸附量从2 568下降到2 071 mg·kg−1。Freundlich模型拟合的结果表明,R-S和LFOR-S的Freundlich常数分别为68.13和13.57,同样说明去除轻组有机质后底泥对磷的吸附能力有所下降[32]。这与WANG等(2011)对太湖贡湖湾、梅梁湾和武汉月湖底泥进行类似研究的结果相近[26]。底泥去除轻组有机质后磷吸附量的下降主要是由于以下2个原因:1)磷酸盐可与底泥中铁铝氧化物表面的OH基团发生配体交换被吸附,同时释放出OH−,pH升高时水中OH−浓度增加会抑制这一反应;2)有机质对底泥中铁铝矿物表面吸附的磷起到一定的“保护”作用,轻组有机质的去除使得部分磷直接暴露于固液界面,其释放趋势因而增强[26]。
投加镧沸石后,无论是R-S还是LFOR-S的磷吸附量都有了显著的提升。对于R-S,投加质量百分数为2%和4%的镧沸石后,磷最大吸附量上升到3 055 mg·kg−1和4 054 mg·kg−1;对于LFOR-S,在相同的条件下,磷的最大吸附量则上升到3 004 mg·kg−1和4 014 mg·kg−1。镧沸石中的有效组分为水合氧化镧,其中La3+可以通过与PO43-形成沉淀的方式去除水中的磷,且沉淀产物LaPO4不会受到环境中pH和氧化还原电位变化的影响[9, 11]。在包括太湖、滇池、洱海等多个湖泊底泥中,镧沸石已经显示出较好的磷钝化能力[10-12]。
为明确镧沸石对底泥磷吸附量提升的贡献,根据式(4)计算了镧沸石在R-S和LFOR-S中的实际磷吸附量。结果表明,投加2%和4%镧沸石后,镧沸石在R-S中的实际磷吸附量分别为27.41 mg·g−1和41.20 mg·g−1,在LFOR-S中的实际磷吸附量为49.65 mg·g−1和52.59 mg·g−1。LFOR-S中镧沸石的实际吸附量大于R-S,说明镧沸石的吸附能力在LFOR-S中可得到更好地发挥。
-
R-S和LFOR-S的磷释放动力学过程如图2所示。可以看出,两种底泥磷释放量逐随时间逐渐上升,在12 h后逐渐平衡。R-S的磷释放量179.40 mg·kg−1,相比前人在我国浅水湖泊中得到的结果[19],本研究中底泥的磷释放量明显更高,这可能是与本研究采取的底泥中磷的本底值较高有关。去除轻组有机质后,底泥的磷释放量上升到289.03 mg·kg−1,相比R-S上升了1.61倍,这与易文利等在武汉月湖、太湖五里湖和贡湖开展的研究结果相近[25]。去除轻组有机质后,底泥磷释放量的增加主要与pH的升高有关,原因详见本文2.1节。
投加镧沸石可以有效控制底泥中磷的释放。如图2(a)所示,投加2%的镧沸石可以将磷释放量降低到9.97 mg·kg−1,而镧沸石投加量在4%时则可以完全控制R-S的磷释放。去除轻组有机质后,镧沸石对磷释放的控制效果明显变差,投加2%和4%的镧沸石分别仅可将释放量降低到94.68 mg·kg−1和59.80 mg·kg−1(图2(b))。如前所述,镧沸石在LFOR-S中有着更高的吸附量,因此LFOR-S磷释放量过高应该是控磷效果较差的原因。要实现对LFOR-S磷释放的有效控制,需增大镧沸石投量[33]。
-
不同温度下R-S和LFOR-S的磷释放情况如图3所示。对于R-S来说,温度在5 oC时磷释放量较大(218.24 mg·kg−1)。当温度上升到10 oC以上时,释放量降至119.04~145.49 mg·kg−1,并保持相对稳定。对LFOR-S来说,当温度由5 oC上升到30 oC的过程中,磷释放量持续上升。具体来说,5 oC时磷释放量为99.20 mg·kg−1,之后磷释放量稳步上升,到30 oC时达到322.96 mg·kg−1。底泥中磷的释放是底泥对磷的吸附和释放2个过程“净”的结果,即二者的差值[5]。从吸附来说,高温条件有利于底泥对磷的吸附[34];从释放来说,温度升高微生物活性增强会形成局部厌氧环境并生成CO2,导致Fe/Mn结合磷和Ca结合磷的释放[35, 36]。推测环境温度上升到10 oC以上时,R-S由于吸附作用增强磷释放量明显降低;而对LFOR-S,温度升高引起释放量的增加占主导作用。
投加镧沸石后,R-S和LFOR-S的磷释放均得到不同程度的控制。通过式(6)计算得到磷释放控制率(表3)。对R-S来说,温度从5 oC上升到30 oC,投加质量分数2%的镧沸石时,磷释放控制率从60.61%逐步上升到92.31%,这主要是由于镧沸石的吸附属于吸热反应,其性能会随着温度的升高而增加[37]。对LFOR-S来说,随温度升高磷释放控制率呈现先降低后增高的趋势,具体来说当镧沸石投加量为2%、温度由5 oC上升到10 oC时磷释放控制率由90%下降到62.67%,而温度继续升高控制率则逐步回升到75.26%。如前所述,随着温度升高,镧沸石的吸附性能会增加,但LFOR-S的磷释放量也会随之升高。因此,磷释放控制率随温度的变化,取决于以上2个因素中影响更大者。推测在5~10 oC,底泥磷释放量的增加影响更大,而在10 oC以上镧沸石的吸附性能的提升则占主导作用。
-
由于底泥成分复杂,很难直接使用光谱学手段表征镧沸石控制底泥磷释放的机理[38]。因此,使用R-S和LFOR-S释放24 h后得到的上清液与镧沸石反应,测定了反应前后镧沸石的XPS谱图,探讨实际环境中的控磷机理。
通过XPS宽谱获得的3种样品表面元素组成如表4所示。可以看出,镧沸石和R-S/LFOR-S释放上清液反应后,表面均有磷元素检出,说明镧沸石可有效吸附2种上清液中的磷。镧沸石反应后表面镧、氧原子摩尔比明显降低,侧面证实了具有较高氧原子含量的PO43-在镧沸石表面吸附[39]。镧沸石与R-S和LFOR-S释放上清液反应前后的La3d轨道XPS精细谱(图4)表明,镧沸石在反应后,La3d轨道结合能均有了明显的上升。有研究[40-42]表明,镧沸石中水合氧化镧组分表面的羟基,可以与PO43-之间发生配体交换形成内圈络合物。发生配体交换反应后,La原子的化学环境发生了明显的变化,其轨道结合能也会随之上升。因此,镧沸石与2种上清液中磷的作用机制均应为配体交换[40]。
-
投加镧沸石前后R-S和LFOR-S中磷形态分级情况如图5(a)所示。可以看出,无论对于R-S还是LFOR-S,投加镧沸石后非稳定态磷(LB-P、BD-P、NaOH-P、Org-P)的含量均有了明显的下降,而稳定形态磷(HCl-P和Res-P)的含量则有了明显的上升,说明镧沸石将底泥中非稳定态磷转化为了稳定态。为进一步比较镧沸石在R-S和LFOR-S中磷形态转化能力,根据式(7)计算了每一种磷形态的转化率[43],结果如图5(b)所示。从非稳定态磷来看,投加镧沸石后R-S和LFOR-S中的转化率基本上为正值且大小相近,说明两种底泥中非稳定态磷含量的下降幅度接近。从稳定形态磷来看,投加镧沸石后转化率均为负值,且LFOR-S中转化率的绝对值小于R-S。具体来说,投加2%的镧沸石时,R-S中HCl-P的转化率为-86.58%,LFOR-S中HCl-P的转化率仅为-75.92%;R-S中Res-P的转化率为-33.73%。LFOR-S中Res-P的转化率仅为-8.89%。这说明投加镧沸石后,LFOR-S中稳定形态磷含量的增幅小于R-S。衡算发现,投加2%镧沸石后R-S中非稳定态磷的含量降低了710.85 mg·kg−1,而稳定态磷的含量增加了755.48 mg·kg−1;在相同处理下,LFOR-S中非稳定态磷的含量降低了717.51 mg·kg−1,而稳定态磷的含量仅增加了632.52 mg·kg−1。也就是说,LFOR-S中减少的非稳定形态磷并未完全转化为稳定形态。
为明确LFOR-S中非稳定态磷的去向,测定了磷形态转化实验结束后水相中的磷含量。结果表明,实验后R-S中进入上清液的磷为79.91 mg·kg−1,投加2%镧沸石和4%镧沸石可以将水相中的磷分别降至22.64 mg·kg−1和25.30 mg·kg−1。而LFOR-S中进入水相的磷达到312.98 mg·kg−1,投加2%镧沸石和4%镧沸石仅能将磷降至106.55 mg·kg−1和74.58 mg·kg−1。因此,可以推断在磷形态转化过程中,LFOR-S中部分非稳定态磷直接进入了水相,由于其浓度较高无法被镧沸石有效控制,最终导致磷释放控制率有所下降。
-
本文的研究结果对于实际的原位钝化控磷工程具有一定的意义。根据谢锦升等[44]的研究,轻组有机质组分具有明显的季节变化特征,其含量在冬春季含量高于夏季58%~122%[44]。LI等[21]发现,在6~23 oC内,轻组有机质含量与环境温度呈反比关系。因此,春夏季气温逐渐升高时,底泥中轻组有机质含量会逐渐下降,磷的释放量则随之上升。在此条件下,镧沸石控磷效果相比升温前将有所下降。事实上,近期已有学者观测到夏季钝化控磷效果较差的现象[45]。根据2.3中的结果,对于LFOR-S,增大镧沸石投加量可以有效提高其磷释放控制率。因此,在底泥轻组有机质含量下降导致镧沸石控磷效果变差时,可以通过增大投量来保障钝化效果。总之,在使用镧沸石控制河湖内源磷的实践中,特别是在轻组有机质含量较高的重污染城镇河湖修复工程中,温度上升对钝化效果的抑制作用需要引起重视,并及时补充投加钝化剂。
-
1)投加镧沸石可以有效提高R-S和LFOR-S的磷吸附能力,且镧沸石在LFOR-S可以发挥出较好的固磷能力。LFOR-S的磷释放量为R-S的1.6倍,在相同投加量下控磷效果差于R-S。
2)无论在R-S还是LFOR-S中,镧沸石的控磷机制均为生成内圈络合物。镧沸石可以驱动底泥中非稳定形态的磷转化为稳定形态,但在LFOR-S中不能实现完全转化,这可能是镧沸石对LFOR-S磷释放控制效果不佳的原因。
3)在实际原位钝化工程中,如存在轻组有机质含量下降的情况,应考虑适当增加镧沸石的投量以实现较好的控磷效果。
底泥轻组有机质对镧沸石控磷效果的影响
Effect of light fraction organic matter in sediment on its phosphate releasing controlling by lanthanum modified zeolite
-
摘要: 底泥中轻组有机质对其中磷的吸附-释放行为影响显著。镧沸石可有效地控制底泥磷释放,但轻组有机质对控磷效果的影响尚不清楚。因此,考察了镧沸石对原底泥(raw sediment,简称R-S)和去除轻组有机质底泥(light fraction organic removal sediment,简称LFOR-S)中磷释放的控制效果及机理。R-S和LFOR-S的最大磷吸附量分别为2 568 mg·kg−1和2 071 mg·kg−1,投加镧沸石可将其提高到4 054 mg·kg−1和4 014 mg·kg−1。LFOR-S在24 h内磷释放量为R-S的1.61倍。温度从5 oC上升到30 oC时,镧沸石对R-S的磷释放控制率逐渐升高,而对LFOR-S的磷释放控制率由90%下降到76%后又回到原水平。XPS表征发现,配体交换是镧沸石控制2种底泥磷释放的主要机制。镧沸石可将底泥中非稳定态磷转化为稳定态,但LFOR-S中减少的非稳定磷的并未完全转化为稳定态,而是进入了水相中使其磷释放量高于R-S,导致控磷效果变差。在原位钝化实际工程中,温度升高引起底泥轻组有机质含量下降时,需提高镧沸石投量以获得稳定的控磷效果。Abstract: Light fraction organic matter in sediment has a significant impact on its phosphorus releasing behavior. Lanthanum Modified Zeolite (LMZ) could effectively control phosphorus release from sediment, but the effect of light fraction organic matter in sediment on its performance was still unclear. Therefore, control of phosphate release from raw sediment (R-S) and light fraction organic removal sediment (LFOR-S) by LMZ were investigated in the present study. The results showed that the maximum phosphate adsorption capacities of R-S and LFOR-S were 2568 and 2071 mg·kg−1, respectively, and they could increase to 4054 and 4014 mg·kg−1 after dosing LMZ, respectively. Phosphate release amount of LFOR-S was 1.61 times of that of R-S within 24 hours, leading to a lower controlling efficiency of LMZ in LFOR-S. Controlling performance of LMZ on phosphate release from R-S gradually became better when the temperature rose from 5 oC to 30 oC, while that of LFOR-S decreased from 90% to 76% and then returned to the original level under the same condition. XPS characterization showed that ligand exchange was the main mechanism of phosphate releasing inhibition by LMZ. LMZ could convert unstable phosphorus in R-S to stable forms, while a completely conversion of decreased liable phosphorus did not occur in LFOR-S, some unstable phosphorus entered water phase which led to a higher release than RS and worse phosphorus release control effect of LMZ. In engineering practice, it is necessary to increase the dosage of LMZ to maintain a stable controlling performance when the amount of light fraction organic matter in the sediment decreased under elevated temperature.
-
Key words:
- lanthanum modified zeolite /
- sediment /
- light fraction organic matter /
- phosphate
-
近年来,随着乡村经济的发展和农户生活水平的提高,农村生活污水对水环境的威胁日益严重[1]。为了有效整治农村人居环境,推进“美丽乡村”建设,以人工湿地(CW)主的污水生态处理工艺被广泛用于农村生活污水的处理[2-3]。CW种类多样,包括表面流人工湿地(SFCW)、水平潜流人工湿地(HFCW)、垂直人工湿地(VFCW)和潮汐流人工湿地 (TFCW)等[4]。其中HFCW对氮磷营养盐、有机物、总悬浮颗粒物(TSS)和病原体等污染物均有良好的去除效果,故成为农村生活污水处理中最常用的湿地类型[5-6]。通常认为,HFCW对TSS和有机物具有较理想的去除效果,但其对氮磷营养盐的去除效果却不甚理想[5-7]。究其原因,则应主要归因于进水中相对不足的有机碳源、HFCW较差的复氧能力及其填料有限的磷素吸附容量等[8-9]。鉴于此,亟需采用相应的技术手段或者调控措施对HFCW的脱氮除磷性能进行强化,以便使其能够高效稳定的处理农村生活污水。
利用电化学技术强化CW的脱氮除磷性能已成为当前CW强化脱氮的研究热点之一[4],先后有研究尝试将该技术与CW工艺耦合,以期提高CW系统的净化效能[10-11]。当利用电化学技术进行污水的处理时,污水中的N素可以通过电催化氧化法和电催化还原法来实现。前者可以直接或间接氧化污水中的氨氮,后者可以高效地去除污水中的硝酸盐和亚硝酸盐[12]。而基于牺牲阳极的电絮凝技术则可以利用铁或铝等阳极材料在电解时产生的金属阳离子生成高活性的多形态聚铁或聚铝絮凝剂,将水中磷酸盐予以去除[13]。
为了实现HFCW对农村生活污水的高效处理,本研究将电化学技术与前期构建而成的三级串联水平潜流人工湿地进行耦合,构建了E-HFCWs。然后考察了E-HFCWs的脱氮除磷性能和电解措施对其他水质指标的影响,分析了各级CW填料的全磷含量及其磷素形态和填料样品的微生物群落结构,以期为新型人工湿地工艺的研发和应用提供参考。
1. 材料与方法
1.1 实验装置
E-HFCWs实验装置如图1所示,装置(L×W×H=1 500 mm×540 mm×348 mm)由厚度为8 mm的钢化玻璃构建而成。该系统包含3个串联的湿地单元(L×W×H=1 264 mm×540 mm×348 mm)和1个出水池(L×W×H=237 mm×310 mm×150 mm),各湿地单元之间均使用管径为35 mm的PVC管进行连接。第1级湿地(H1)基质以沸石为主(粒径为3~8 mm,基质层厚度为220 mm),其主要成分是SiO2,表面粗糙,孔径均匀,吸附能力强,对COD 和氮素具有良好的去除效果。第2级(H2)和第3级(H3)湿地中的基质以红壤烧制而成的废砖块为主(粒径为10~50 mm,基质层厚度为220 mm),其铁、铝含量较高,具有较大的比表面积和多孔结构,有利于生物膜的生长。表层均用河砂(粒径为2 mm以下,厚度为30 mm)覆盖,基质层总厚度为250 mm。在H1和H2中安装电解装置,装置的电极材料均使用纯铁,阳极(L×W×T=250 mm×150 mm×0.3 mm、表面打孔、孔径为20 mm、孔距为20 mm)设置在湿地单元的中心,阴极均匀地设置在阳极的两侧,各极板相距为120 mm。使用铜线(直径为2 mm)将电极与直流稳压电源(eTM-305F、0~30 V、0~5 A)相连,为电解装置提供恒定电流。湿地植物选用生长正常、株型大小基本一致的芦苇和美人蕉,混种密度为76株·m−2。系统由蠕动泵、微电脑时控开关和液体流量计共同定时定量连续进水,以水平推流方式从另一端排出。植物移栽后,加入自来水至基质饱和。待植物成活、系统稳定后开始实验。
 图 1 电解强化三级串联人工湿地装置示意图Figure 1. Chart of electrolysis-three-stage constructed wetlands
图 1 电解强化三级串联人工湿地装置示意图Figure 1. Chart of electrolysis-three-stage constructed wetlands1.2 实验条件和模拟废水
该装置的HLR(水力负荷)为0.30 m3·(m2·d)−1,污水在系统中的水力停留时间(HRT)为3 d,即系统处理污水量为20 L·d−1。稳压直流电源每天以恒流模式输出8 h。本研究的实验时段为2018年4月至2019年9月,可分为3个阶段:第1阶段(2018年4—9月,电流强度为0 mA)、第2阶段(2018年10月—2019年2月,电流强度为100 mA)和第3阶段(2019年4—9月,电流强度为50 mA)。在实验开始前,装置先连续运行30 d,使湿地植物和基质微生物适应环境,并开始生长繁殖。接种污泥来自合肥市望塘城市污水处理厂。实验进水来自安徽农业大学园区内生活污水,原水经沉淀处理后,取上清液作为系统的进水。其污水水质如表1所示。
表 1 生活污水水质指标Table 1. Quality of domestic sewage阶段 TP/(mg·L−1)  -P/(mg·L−1)
-P/(mg·L−1)TN/(mg·L−1)  -N/(mg·L−1)
-N/(mg·L−1) -N/(mg·L−1)
-N/(mg·L−1) -N/(mg·L−1)
-N/(mg·L−1)温度/℃ DO/(mg·L−1) pH 氧化还原电位/mV 第1阶段 4.44±0.29 4.24±0.28 47.59±4.70 46.35±4.71 0.07±0.05 1.33±1.03 26.23±3.41 7.90±0.52 7.23±0.14 −13.06±18.74 第2阶段 5.33±0.60 5.19±0.79 68.87±10.68 67.43±9.95 0.02±0.02 1.44±1.59 12.51±8.21 8.47±0.48 7.21±0.81 −12.91±47.54 第3阶段 4.65±0.38 4.42±0.41 59.24±18.11 58.62±18.19 0.01±0.01 0.62±0.65 26.29±1.92 7.49±0.33 6.95±1.04 −0.46±62.31 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.3 采样点设置
实验装置的每个CW单元中用四周开孔的PVC管各设置1个固定的取水口(图1),进水的采样点设在污水桶,出水采样点设在出水池。每周二09:00用取样瓶从各采样点取水样并进行测定,测定取3次重复。各级湿地水质指标采样点的编号分别为H1、H2、H3。
1.4 水质测定与分析方法
水样的测定指标包括pH、温度、溶解氧(DO)、氧化还原电位(Eh)、TN、
NH+4 -N、NO−2 -N、NO−3 -N、TP、PO3−4 -P、Fe2+和总溶解铁(TDFe)。上述部分水质指标测定时均参照《水与废水水质测定方法》[14],水中Fe3+的含量由TDFe含量减去Fe2+含量获得,pH、温度和Eh则分别采用便携式水质分析仪进行测定。在实验结束后,对各级湿地中填料样品的全磷含量及磷形态进行测定。填料中的全磷使用硫酸/高氯酸消解-钼锑抗分光光度法进行测定,有机磷的测定则采用马弗炉灼烧-钼锑抗分光光度法。填料中各种形态的无机磷首先利用不同化学浸提剂加以逐级分离,而后采用钼锑抗分光光度法测定,不同形态无机磷分离方法参照《土壤农化分析》[15]。直流稳压电源输出的电流、电压和电功率由其内置的4位数显电表测得。填料截留的磷素总量、TP去除量、TN去除量的计算如式(1)~式(3)所示。q=ΔCm (1) q2=(C0−C2)V (2) q3=(C1−C3)V (3) 式中:q为各级湿地的填料对溶液中P的吸附总量,mg;q2为各级湿地对溶液中P的去除量,mg;q3为各级湿地对溶液中P的吸附量,mg;ΔC为湿地填料的全磷变化量,mg·g−1;C0为初始TP浓度,mg·L−1;C2为经过电解处理后TP浓度,mg·L−1;C1为初始TN浓度,mg·L−1;C3经过电解处理后TN浓度,mg·L−1;V为装置中的污水体积,L;m为装置中填料的质量,g。
1.5 微生物群落分析
1)样品采集。装置稳定运行17个月后,采集了湿地单元的基质样品,用于高通量测序,分析其微生物群落结构。样品采集的具体过程如下:移除湿地表层凋落物和腐殖质后,使用梅花点阵法随机确定取样点。H1表层和底层极板间基质样品的编号分别为A1和a1,极板四周表层和底层的基质样品编号分别为A2和a2,H2表层和底层极板间基质样品的编号分别为B1和b1,极板四周表层和底层的基质样品编号分别为B2和b2,H3表层的基质样品编号为C1,底层基质样品编号为c2。共10个混合样品,各样品经充分混合后放入无菌袋中。
2) PCR扩增和测序。采用Illumina MiSeq技术平台进行测序,将湿地填料样品放至-80 ℃低温环境下,送至百迈客基因生物科技有限公司进行高通量测序。结果数据经过预处理和质量控制后,进行OTU聚类和注释,从而获得特定环境样品的细菌或古菌的物种组成及丰度信息;再通过多样性分析,从而寻找样品间的差异信息。分析湿地单元表层和底层以及电极之间的微生物群落结构和功能群的变化特征,以及其对污水的处理效果的影响。
1.6 电化学反应机理
本实验选择铁作为阳极材料,电催化和电絮凝反应过程中的化学方程式如下。其中电催化氧化分为直接氧化和间接氧化。直接氧化是指污染物在阳极表面通过电子传递被氧化(式(4))。间接氧化是指污染物被电解产生的活性物质如·OH等氧化(式(5)和式(6))。在电解过程中,系统会将
NO−3 和NO−2 电催化还原成NH+4 -N(式(7)),,然后通过电催化转化为N2(式(8))。P去除依赖于阳极产生的Fe2+,生成难溶于水的FePO4,沉淀下来。其反应过程如式(9)~式(11)所示。电催化脱氮反应见式(4)~式(8),电絮凝除磷反应见式(9)~式(11)。Fe−2e−→Fe2+ (4) H2O→⋅OH+H++e− (5) 6⋅OH+2NH+4→N2+6H2O+2H+ (6) Fe2++NO−3+9H2O+4H++13e−→Fe3++NH+412OH−+3H2 (7) NO−3+NH+4+H2O+2e−→N2+H2O+2OH− (8) Fe3++HnPO−3+n4→FePO4+nH+ (9) 4Fe2++10H2O+O2→Fe(OH)3+8H+ (10) Fe(OH)3+H2PO−4→FePO4+OH−+2H2O (11) 1.7 数据处理
实验数据的计算和处理使用Excel 2018完成,用Pearson检验方法和配对T检验来进行相关性分析和差异性分析,检验数据间相关水平的统计分析通过SPSS 20.0进行。利用GraphPad Prism 8软件作图,图中相关数据均为平均值±标准差。
2. 结果与分析
2.1 电解对脱氮除磷的影响
E-HFCWs在不同实验阶段对氮、磷的净化效果如图2所示。系统进水中N、P的浓度均有所波动。在第1阶段(即未电解阶段),系统出水中TN、
NH+4 -N、TP和PO3−4 -P的平均含量分别为(10.35±2.06)、(5.59±0.63)、(1.68±0.45)和(1.45±0.44) mg·L−1,平均去除率分别为(78.11±4.44)%、(87.81±1.99)%、(62.26±9.74)%和(65.89±10.15)%。而在第2、3阶段,电解强化人工湿地系统出水中TN、NH+4 -N、TP和PO3−4 -P的平均含量分别为(4.03±1.96)、(2.74±1.49)、(0.34±0.25)和(0.10±0.09) mg·L−1,平均去除率分别为(92.99±4.51)%、(95.22±3.04)%、(93.13±5.22)%和(97.84±1.96)% (如图2所示)。在第2阶段中,装置在秋冬季节运行,环境温度整体较低,人工湿地中植物和微生物作用较弱,因此,选择的电流强度为100 mA;出水中TN、
NH+4 -N、TP和PO3−4 -P含量分别为(2.60±1.42)、(1.83±0.97)、(0.19±0.14)和(0.11±0.09)mg·L−1,去除率分别为(96.19±1.99)%、(97.10±1.58)%、(96.66±2.23)%、(98.00±1.57)%。而在第3阶段,装置主要在春夏时节运行,环境温度较高,人工湿地中植物和微生物作用较强,因此,选择的电流强度为50 mA。出水中TN、NH+4 -N、TP和PO3−4 -P 的含量分别为(5.47±1.25)、(3.65±1.36)、(0.49±0.24)和(0.10±0.10) mg·L−1,去除率分别为(89.78±4.03)%、(93.28±2.90)%、(89.59±4.95)%、(97.69±2.30)%。第2阶段系统的氮磷去除能力要明显优于第3阶段。系统进出水中的NO−2 -N和NO−3 -N含量相对较少,但作为硝化和反硝化作用的中间产物,会在各级湿地中有不同程度的积累。而在电解阶段,NO−2 -N和NO−3 -N的积累量随电流强度越大而减少,且主要积累于未电解的H3中。由此可见,电化学技术与CW的结合大大提高了系统对氮磷营养盐的净化能力,且电解系统随着电流强度的增加,出水中氮磷含量持续下降,去除率显著升高。使出水达到了《城镇污水处理厂污染物排放标准》(GB 18918-2002)一级A标准。
2.2 电解对其他主要水质指标的影响
各级湿地出水中的N、P、Fe的含量及pH如表2所示。阳极铁片电解产生Fe2+,Fe2+在CW表面和植物根附近的有氧环境中易被氧化成Fe3+,而Fe3+在CW中下层的厌氧环境中被还原为Fe2+[16]。虽然Fe离子的引入能够强化N和P的去除[17],但是如果Fe离子含量较多,同样会污染水体。第2、3阶段系统出水的TDFe含量分别为(2.91±1.75) mg·L−1和(2.55±1.89) mg·L−1,均以Fe3+为主,且前后Fe离子含量没有显著性差异(n=15,P>0.05)。在第2阶段中,H1的TDFe的平均含量为(5.32±3.89) mg·L−1,且随着实验的进行缓慢地升高。而H2中的TDFe含量为(10.54±3.99) mg·L−1,显著高于H1(n=15,P<0.05),且H2中铁离子含量的上升速度明显快于H1。H1和H2具有相同的电解条件,但由于H1中的填料为沸石,而沸石独特的分子筛结构,对铁离子具有较强的吸附能力,故将电解产生的Fe2+直接吸附在填料的表面和孔隙中。H3中未添加电解装置,因此,TDFe含量较低,为(4.17±3.37) mg·L−1。在第3阶段电流强度降低,各级湿地的TF含量分别为(5.65±3.40)、(7.15±5.25)、(3.92±3.90) mg·L−1。除H1外,H2和H3中的Fe离子含量均显著降低(n=15,P <0.05)。除第2阶段H2中的Fe2+含量高于Fe3+铁以外,其他各级湿地均是Fe3+含量高于Fe2+。
表 2 湿地中N、P、Fe的含量和pHTable 2. Contents of N, P, Fe and pH in wetlands阶段 采样点 TN/(mg·L−1)  -N/(mg·L−1)
-N/(mg·L−1)TP/(mg·L−1)  -P/(mg·L−1)
-P/(mg·L−1)Fe2+/(mg·L−1) Fe3+/(mg·L−1) TDFe/(mg·L−1) pH 第1阶段 H1 28.89±3.63 24.76±5.53 3.89±0.41 3.91±0.45 — — — 7.25±0.29 H2 21.06±5.05 11.96±1.92 2.62±0.60 2.46±0.59 — — — 7.45±0.19 H3 12.90±4.54 6.73±1.04 1.57±0.49 1.33±0.49 — — — 7.64±0.14 出水 10.35±2.06 5.59±0.63 1.68±0.45 1.47±0.44 — — — 7.75±0.13 第2阶段 H1 18.84±7.92 17.71±8.04 2.65±1.82 2.40±1.84 2.32±1.60 3.00±2.35 5.32±3.89 6.90±0.56 H2 8.91±6.34 7.68±5.89 1.51±1.52 1.33±1.52 6.64±2.66 3.89±1.92 10.54±3.90 7.32±0.40 H3 3.83±2.17 2.82±1.41 0.24±0.14 0.17±0.14 1.84±1.28 2.32±2.18 4.17±3.29 7.53±0.30 出水 2.60±1.42 1.83±0.97 0.19±0.14 0.11±0.09 1.06±0.58 1.59±1.05 2.65±1.57 7.65±0.33 第3阶段 H1 13.53±3.47 12.77±3.24 2.83±1.64 1.46±1.34 3.04±2.79 2.62±1.46 5.65±3.32 7.35±0.36 H2 10.18±1.72 9.62±1.43 3.76±5.44 0.17±0.16 2.54±2.18 4.61±4.38 7.15±5.13 7.59±0.32 H3 7.84±1.88 5.00±1.91 2.30±2.69 0.15±0.14 0.91±0.39 3.01±3.77 3.92±3.81 7.74±0.31 出水 5.47±1.25 3.65±1.36 0.49±0.24 0.10±0.10 0.83±0.32 1.72±1.79 2.55±1.83 7.83±0.34 | Show TableDownLoad:
CSV
E-HFCWs中出水相较于进水,出水的温度和pH均在上升,DO、Eh均在下降(表2和表3)。第1阶段进水的pH为6.18~7.51,出水为7.62~8.03。第2阶段进水pH为4.59~7.64,出水为7.06~8.17。第3阶段进水pH为4.65~7.71,出水为7.26~8.41。H1在第2阶段中的平均pH(6.90±0.57)要低于进水(7.21±0.81),然而在第1和第3阶段,H1的碱度在增加(表2)。这可能是因为第2阶段环境温度较低,电流强度较大,产生的铁盐含量较多,其水解引起了碱度消耗所致。3个阶段进水的DO分别为6.67~7.77、7.82~9.15、6.85~7.94 mg·L−1,出水分别为0.61~0.91、0.43-0.71、0.60~0.83 mg·L−1。与未电解HFCWs相比,电解的效果可以诱导更碱性和更缺氧的环境。各阶段进水的Eh分别为−29.70~45.75、−45.15~140.22、−45.35~136.66 mV,出水分别为−64.16~−35.05、−72.67~−6.73、−86.74~−18.61 mV。
表 3 出水的主要水质指标Table 3. Quality of Effluent Water阶段 温度/℃ DO/(mg·L−1) 氧化还原电位/mV 第1阶段 28.51±1.83 0.77±0.73 −47.60±7.83 第2阶段 12.39±8.38 0.59±0.09 −41.61±19.81 第3阶段 26.83±1.90 0.74±0.06 −51.99±20.37 | Show TableDownLoad:
CSV
2.3 各级湿地中填料的TP含量及其P的形态
P在CW中的去除依赖于填料的吸附和沉淀、微生物的转化与吸收、植物的吸收与同化[6]。其中填料的吸附和沉淀作用被证明是最主要的去除方式[18]。为了探究填料对系统中P的去除贡献率和去除机理,测定了本实验前后各级CW中填料的全P含量及其P形态的情况,如表4所示。整个实验过程中,系统共截留P素23 609.82 mg,填料的吸附和沉淀作用占P去除贡献率的70%以上。H1、H2和H3的填料各截留了3 758.18、12 644.35、7 207.28 mg。H2中的全P含量最高,其次是H3和H1,全P的变化量分别为0.211、0.126、0.047 mg·g−1,分别占各自全P含量的67.32%、55.28%和43.85%。因此,废砖块作为E-HFCWs中的填料,对P有更好的吸附和沉淀能力。
表 4 湿地中填料样品的全磷含量及其磷形态Table 4. Contents of total phosphorus and phosphorus fractions in wetlands fillers mediamg·g−1 填料样品 全P 有机P 无机P 水溶性P AL-P Fe-P O-P Ca-P 沸石 0.060 0.001 0.059 0.001 0.045 0.010 0.001 0.002 废砖块 0.102 0.000 0.102 0.001 0.060 0.031 0.003 0.007 H1 0.107 0.001 0.106 0.003 0.061 0.034 0.004 0.004 H2 0.313 0.001 0.312 0.015 0.118 0.148 0.011 0.021 H3 0.229 0.001 0.228 0.012 0.111 0.080 0.008 0.017 | Show TableDownLoad:
CSV
在各基质样品中,无机P含量最高,其中Al-P和Fe-P为填料沉淀磷素的主要途径,分别占无机磷总量的90.48%、85.24%和83.80%,水溶性P、O-P和Ca-P占比较少。H1和H2的填料样品均是Fe-P的截留量高于Al-P,而H3则完全相反。这是由于H1和H2中的铁片电极在电解过程中产生的Fe2+,而其能与水中O2或电解过程中产生的·OH和H2O2形成Fe3+,生成高活性的絮凝基团,具有极强的吸附能力[19],可将废水中的磷酸盐吸附共沉,生成难溶性的FePO4进而固定下来[20],在湿地中形成黄褐色的沉淀物。而H3中没有电解装置且平均pH=7.62,容易与废砖块在水体中释放的Al3+,生成AlPO4沉淀下来[21]。
2.4 各湿地单元的多样性分析
本项目共完成10个样品的测序,共获得 1 115 342对序列,为了研究样品的物种组成及多样性信息,将相似性达到97%的序列聚类成一个OTU,各样品的OTUs数量和α多样性指数统计结果如表5所示。H3的OTUs数目>H2>H1,且各样品间共有597个相同的OTUs,并没有特有的OTUs。α多样性指数主要反映单个样品的物种丰度及其多样性,衡量指标包括:Chao1、Ace、Shannon、Simpson。通过表我们可以看出,样品的覆盖率最低为99.1%,说明此次测序的结果能够准确且完整地反映E-HFCWs中微生物样品的真实状况。在这10个样品中,C1的ACE、Chao1和Shannon指数最大,而Simpson指数最低,说明C1样品群落中物种丰度和多样性最高。而A1的ACE、Chao1和Shannon指数最低,而Simpson指数最高,即说明A1样品群落中物种丰度和多样性最低。H3的微生物物种丰度和多样性最高,其次是H2和H1。而在H1和H2的各样品中,除了b1和b2外,其余样品均是电极之间的物种丰富度和多样性要低于电极四周。这可能是由于电解作用降低了微生物群落中的物种丰富度和多样性。
表 5 α多样性分析结果Table 5. Result of Alpha diversity analysis基质微生物样品 OTUs ACE Chao1 Simpson Shannon 覆盖率/% A1 911 1 063.362 1 076.555 0.029 9 4.855 99.2 A2 1 034 1 141.203 1 166.759 0.012 2 5.537 99.1 B1 1 184 1 260.709 1 267.889 0.008 8 5.808 99.4 B2 1 225 1 306.517 1 311.628 0.006 1 5.964 99.3 C1 1 330 1 337.480 1 346.714 0.003 8 6.312 99.9 a1 938 1 095.810 1 098.308 0.026 4 4.929 99.1 a2 1 050 1 150.895 1 155.008 0.013 1 5.470 99.3 b1 1 245 1 298.371 1 302.904 0.007 1 5.940 99.5 b2 1 230 1 298.811 1 322.974 0.013 6 5.533 99.5 c2 1 297 1 317.396 1 324.500 0.004 4 6.224 99.8 | Show TableDownLoad:
CSV
为了进一步分析样品间物种多样性差异,依据不同微生物样品的OTUs 组成,对样品进行了非度量多维标定法(NMDS)分析。如图3所示,胁强系数远小于0.2,说明NMDS分析可以准确反映样本间的差异程度。A1、A2、a1、a2的差异程度较大,其余样品间的差异程度较小且聚集在一个共同的区域。这说明在H1中,无论在表层还是底层,极之间与电极四周的菌群组成有较大的差异,且在表层更为显著。
2.5 湿地单元基质微生物的群落结构
通过对OTU进行去低含量筛除(物种丰度小于0.005%),10个样品中共统计出2界、40门、91纲、192目、313科、501属、524属。为了方便分析,筛选出相对丰度前十的菌门,如图4所示,分别是变形菌门、拟杆菌门、Patescibacteria、绿弯菌门、厚壁菌门、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、螺旋体门(Spirochaetae)、嗜热菌门(Caldiserica)、硝化螺旋菌门(Nitrospirae)。前5个菌门是系统中的优势菌门,在各级CW中的相对丰度超过80%。其中变形菌门、拟杆菌门和Patescibacteria在前两级湿地中的相对丰度超过60%,在H3中超过56%。在各样品中,变形菌门的相对丰度最高,且CW表层相对丰度高于底层。在H1、H2表层电极之间的变形菌的相对丰度要低于四周,而在底层分布规律完全相反。有研究[22-23]表明,变形菌在人工湿地微生物氮磷去除中起到主要作用。其次是拟杆菌门,底层相对丰度大于表层,电极四周高于电极之间。Patescibacteria在前两级湿地广泛分布,平均相对丰度分别为16.42%和11.19%,而在H3中则分布较少(4.65%),且其在电极之间的相对丰度较高,因此,该类微生物的富集可能与电解作用有关。绿弯菌门主要分布在后两级湿地,能利用有机物进行光合作用,还可利用
NH+4 -N和有机N作为N源生长[24]。厚壁菌门在H2中分布较多(13.20%),且底层(16.03%)大于表层(10.37%),其次H1(10.52%)和H3(9.67%)。厚壁菌门中部分菌属具有反硝化的作用,对N和有机物具有良好的去除作用[25]。2.6 电解阶段水质指标间的相关性分析
为了进一步的分析电解对CW的影响,对数据进行了相关分析性和差异性分析。我们发现在第2、3阶段中,N和
PO3−4 -P的去除率与进水的DO和Eh呈极显著负相关(n=15,P<0.01),与进水pH和温度呈极显著正相关(n=15,P<0.01)。第2阶段的环境温度较低,TP去除率与进水的DO成极显著负相关,与进水温度呈极显著正相关(n=15,P<0.01)。而在第3阶段,环境温度较高,TP去除率与进水的DO呈显著正相关,与进水温度无关(n=15,P<0.01)。在第2阶段,各级湿地水体中Fe2+和TDFe含量与其对应P的去除率呈极显著负相关(n=15,P<0.01)。除H2外,Fe3+含量与其对应的P去除率也呈极显著负相关(n=15,P<0.01)。H1和H2中Fe离子的含量均与其对应的TN去除率呈极显著负相关(n=15,P>0.05)。在第3阶段,各级湿地水体中Fe2+含量与其对应PO3−4 -P的去除率呈极显著负相关(n=15,P<0.01)。各级湿地水体中Fe3+和TDFe含量与其对应TP的去除率呈极显著负相关(n=15,P<0.01),Fe离子的含量与其对应N的去除率无关(n=15,P>0.05)。2、3阶段系统中出水的PO3−4 -P含量及其去除率均没有显著性差异(n=15,P>0.05),但在第2阶段,当电流强度为100 mA时,N和P的去除率均高于第3阶段。3. 结论
1)当水力负荷为0.30 m3·(m2·d)−1时,3个阶段中系统对TN和TP的平均去除率分别为(78.12±4.38)%、(96.19±2.03)%、(89.85±4.20) %,(62.27±9.86)%、(96.66±2.28)%、(89.59±5.06)%。电解强化了系统对N、P营养盐的净化能力。使2、3阶段的出水达到了《城镇污水处理厂污染物排放标准》(GB 18918-2002)一级A标准。
2)在整个实验过程中,系统共截留P素23 609.82mg,填料的吸附和沉淀作用占P去除贡献率的70%以上。各级湿地的填料中无机P含量最高,其中Al-P和Fe-P为填料沉淀磷素的主要途径。
3)各级湿地中的基质微生物种群和相对丰度均具有差异。共发现10个共同的优势菌门,分别是变形菌门、拟杆菌门、Patescibacteria、绿弯菌门、厚壁菌门、酸杆菌门、放线菌门、螺旋体门、嗜热菌门、硝化螺旋菌门。前5个菌门是系统中的优势菌门,在各级CW中的相对丰度均超过80%。其中,变形菌门、拟杆菌门和Patescibacteria在前两级湿地中的相对丰度超过60%,在H3中超过56%。在各样品中,变形菌门的相对丰度最高,对人工湿地N、P去除中起到主要作用。
-
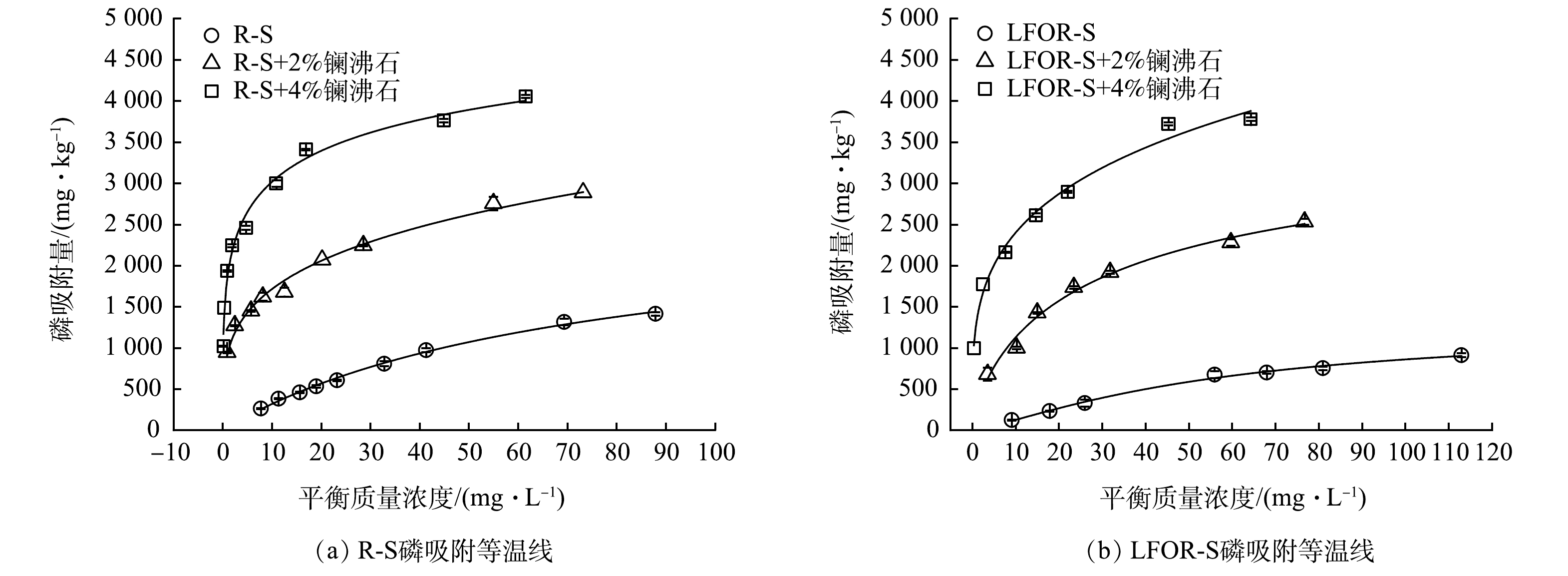
图 1 投加镧沸石前后R-S和LFOR-S对磷的吸附等温线
Figure 1. Phosphate adsorption isotherms of R-S and LFOR-S before and after LMZ dosing
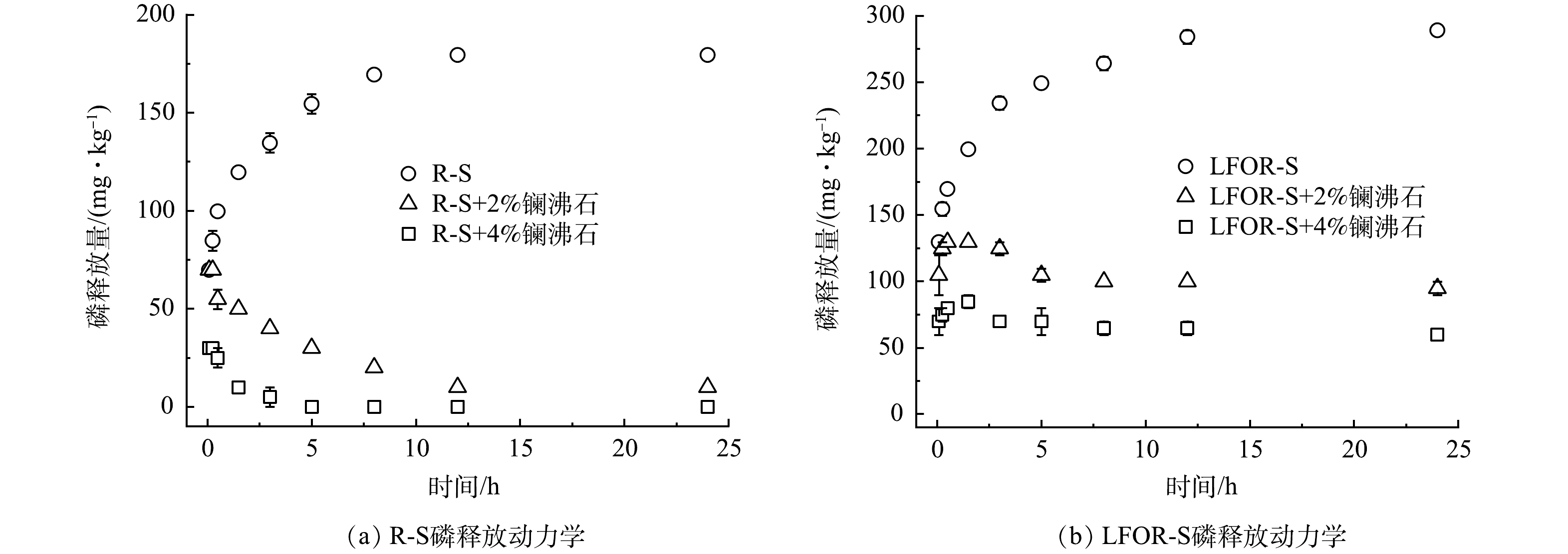
图 2 投加镧沸石前后R-S和LFOR-S的磷释放动力学
Figure 2. Phosphate releasing kinetics of R-S and LFOR-S before and after LMZ dosing
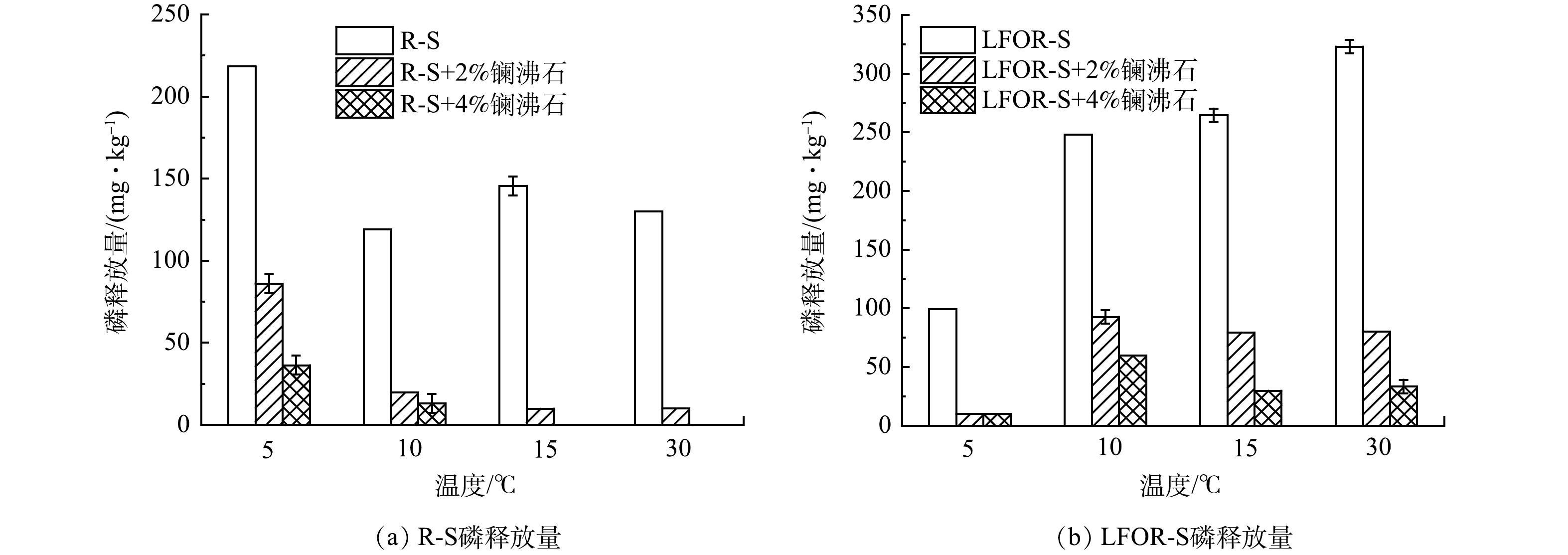
图 3 不同温度下R-S和LFOR-S在投加镧沸石前后磷释放量
Figure 3. Phosphate releasing amount of R-S and LFOR-S before and after LMZ dosing at different temperatures
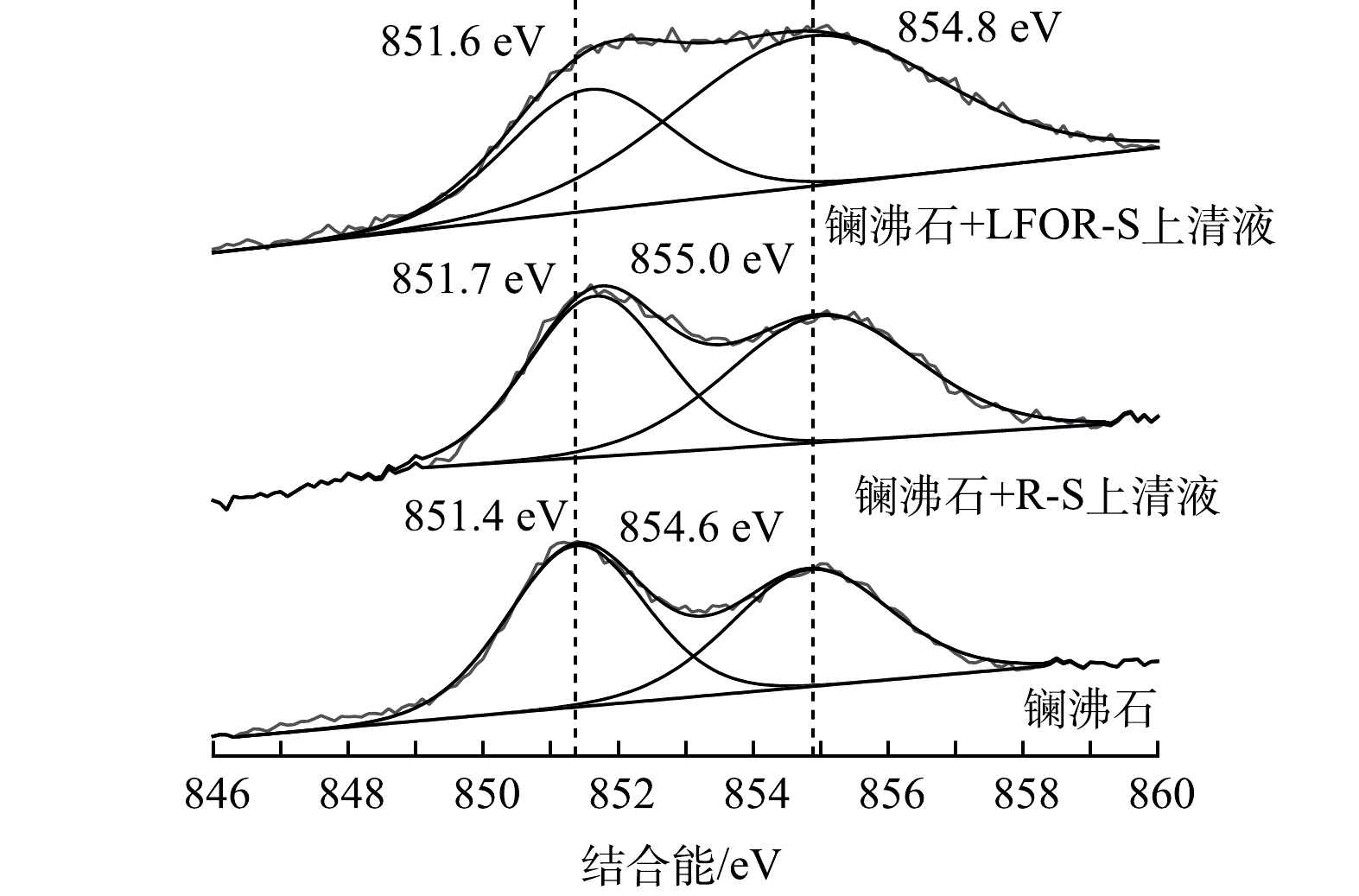
图 4 镧沸石与R-S和LFOR-S释放液反应前后的La 3d 5/2轨道精细谱
Figure 4. High resolution La3d5/2 XPS spectra of LMZ before and after reaction with releasing supernatant of R-S and LFOR-S
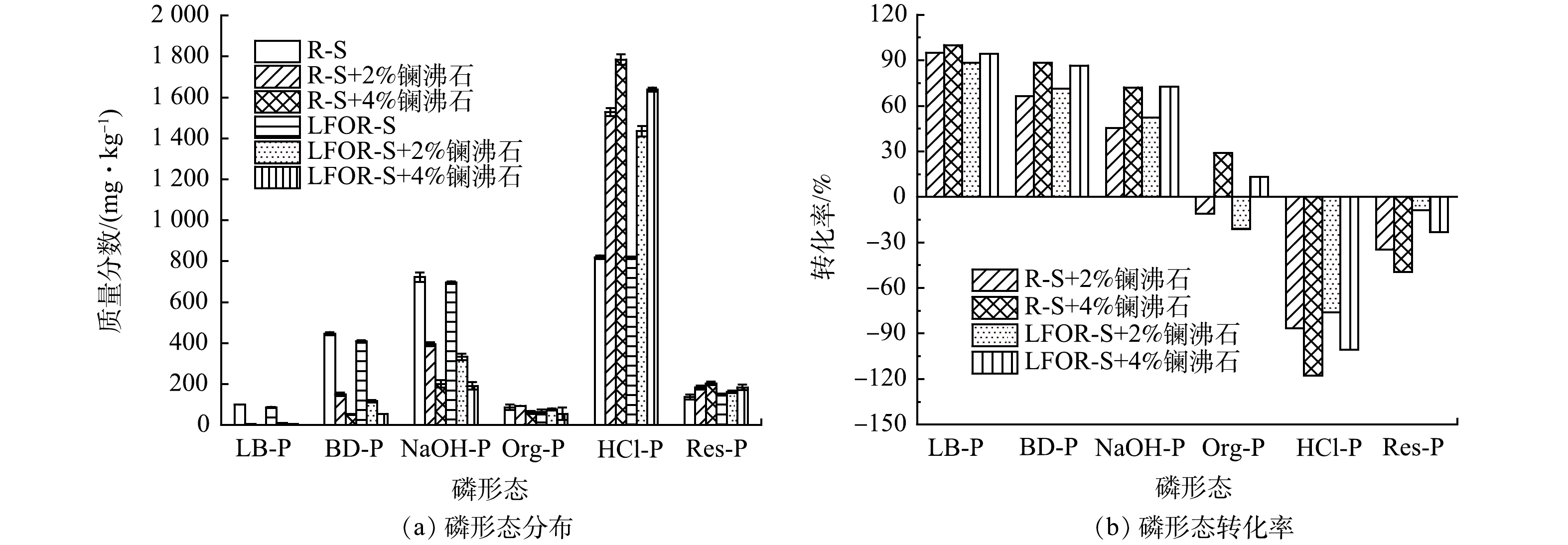
图 5 投加镧沸石前后R-S和LFOR-S中磷形态分布及磷形态转化率的变化
Figure 5. Phosphate fraction distribution and its transmission efficiency in R-S and LFOR-S before and after LMZ addition
表 1 去除轻组有机质前后底泥的基本理化性质
Table 1. Basic physical and chemical parameters of sediment before and after light fraction organic matter removal
底泥 有机质含量/% 总磷/(mg·kg−1) 比表面积/(m2·g−1) pH(固液比1:5) R-S 2.54 2 272.11 8.12 7.35 LFOR-S 2.36 2 116.22 8.15 10.21
下载: 导出CSV
表 2 投加镧沸石前后R-S和LFOR-S对磷的吸附等温线的拟合结果
Table 2. Fitting results of adsorption isotherms of R-S and LFOR-S before and after LMZ dosing
实验组合 Langmuir模型 Freundlich模型 Qmax/(mg·kg−1) KL/(L·mg−1) R2 KF/(mg·kg−1)·(mg·L−1)−1/n 1/n R2 R-S 2 568 0.014 0.984 68.13 0.696 0.992 R-S+2% 镧沸石 3 055 0.152 0.986 965.16 0.252 0.988 R-S+4% 镧沸石 4 054 0.532 0.996 1 805.09 0.209 0.966 LFOR-S 2 071 0.007 0.938 13.67 0.998 0.990 LFOR-S+2% 镧沸石 3 004 0.059 0.989 400.22 0.437 0.974 LFOR-S+4% 镧沸石 4 014 0.201 0.987 1 307.37 0.262 0.992
下载: 导出CSV
表 3 不同温度下镧沸石对R-S和LFOR-S磷释放控制率
Table 3. Phosphate releasing control efficiency of LMZ for R-S and LFOR-S at different temperatures %
温度/oC R-S LFOR-S 2%镧沸石 4%镧沸石 2%镧沸石 4%镧沸石 5 60.61 83.33 90.00 90.00 10 83.33 88.89 62.67 76.00 15 93.18 100.00 70.00 88.75 30 92.31 100.00 75.26 89.69
下载: 导出CSV
表 4 不同反应条件下镧沸石表面元素组成及摩尔比值
Table 4. Surface element composition and molar ratios of LMZ under different reaction conditions.
样品 元素原子百分比/% La:O La Si C O P 镧沸石 1.240 47.130 21.300 26.890 — 0.046 镧沸石+R-S释放上清液 1.120 42.920 24.130 28.460 0.870 0.039 镧沸石+LFOR-S释放上清液 0.830 36.580 29.530 28.730 0.650 0.029
下载: 导出CSV
-
[1] XIAO M, BURFORD M A, WOOD S A, et al. Schindler's legacy: From eutrophic lakes to the phosphorus utilization strategies of cyanobacteria[J]. FEMS Microbiology Reviews, 2022, 46(6): 1-24. [2] YIN H, ZHANG M, YIN P, et al. Characterization of internal phosphorus loading in the sediment of a large eutrophic lake (Lake Taihu, China)[J]. Water Research, 2022, 225: 119-125. [3] YAN Q, CHENG T, SONG J, et al. Internal nutrient loading is a potential source of eutrophication in Shenzhen Bay, China[J]. Ecological Indicators, 2021, 127: 107736. doi: 10.1016/j.ecolind.2021.107736 [4] 杨海全, 陈敬安, 刘文, 等. 草海底泥原位钝化工程示范及其生态环境效应[J]. 环境工程学报, 2017, 11(7): 4437-4444. [5] XUE W, LU S-Y. Effects of inactivation agents and temperature on phosphorus release from sediment in Dianchi Lake, China[J]. Environmental Earth Sciences, 2015, 74(5): 3857-3865. doi: 10.1007/s12665-014-3910-5 [6] 马鑫雨, 杨盼, 张曼, 等. 湖泊沉积物磷钝化材料的研究进展[J]. 湖泊科学, 2022, 34(1): 1-17. [7] MEIS S, SPEARS B M, MABERLY S C, et al. Sediment amendment with Phoslock® in Clatto Reservoir (Dundee, UK): Investigating changes in sediment elemental composition and phosphorus fractionation[J]. Journal of Environmental Management, 2012, 93(1): 185-193. doi: 10.1016/j.jenvman.2011.09.015 [8] FUNES A, ÁLVAREZ-MANZANEDA I, ARCO A D, et al. Evaluating the effect of CFH-12® and Phoslock® on phosphorus dynamics during anoxia and resuspension in shallow eutrophic lakes[J]. Environmental Pollution, 2021, 269: 116093. doi: 10.1016/j.envpol.2020.116093 [9] XIE J, WANG Z, FANG D, et al. Green synthesis of a novel hybrid sorbent of zeolite/lanthanum hydroxide and its application in the removal and recovery of phosphate from water[J]. Journal of Colloid and Interface Science, 2014, 423: 13-19. doi: 10.1016/j.jcis.2014.02.020 [10] WANG Z, FAN Y, LI Y, et al. Synthesis of zeolite/hydrous lanthanum oxide composite from coal fly ash for efficient phosphate removal from lake water[J]. Microporous and Mesoporous Materials, 2016, 222: 226-234. doi: 10.1016/j.micromeso.2015.10.028 [11] WANG Z, LU S, WU D, et al. Control of internal phosphorus loading in eutrophic lakes using lanthanum-modified zeolite[J]. Chemical Engineering Journal, 2017, 327: 505-513. doi: 10.1016/j.cej.2017.06.111 [12] 王哲, 朱俊, 李雯, 等. 镧沸石对磷和重金属的吸附与底泥钝化性能[J]. 环境科学, 2022, 43(11): 5106-5114. [13] LüRLING M, WAAJEN G, VAN OOSTERHOUT F. Humic substances interfere with phosphate removal by lanthanum modified clay in controlling eutrophication[J]. Water Research, 2014, 54: 78-88. doi: 10.1016/j.watres.2014.01.059 [14] DITHMER L, NIELSEN U G, LUNDBERG D, et al. Influence of dissolved organic carbon on the efficiency of P sequestration by a lanthanum modified clay[J]. Water Research, 2016, 97: 39-46. doi: 10.1016/j.watres.2015.07.003 [15] 李静, 朱广伟, 朱梦圆, 等. 杭州西湖“香灰土”沉积物轻、重有机质组成特征及其环境意义[J]. 环境科学, 2015, 36(6): 2038-2045. [16] 赵萱, 成杰民, 鲁成秀. 不同生态类型富营养化湖泊沉积物中有机质赋存形态[J]. 环境化学, 2012, 31(3): 302-307. [17] NDZELU B S, DOU S, ZHANG X, et al. Molecular composition and structure of organic matter in density fractions of soils amended with corn straw for five years[J/OL]. Pedosphere, 1-11,https://doi.org/10.1016/j.pedsph.2022.06.057, 2022-06-07. [18] 黄桥明, 吕茂奎, 聂阳意, 等. 武夷山不同海拔森林表层土壤轻组有机质特征[J]. 生态学报, 2020, 40(17): 6215-6222. [19] 易文利, 王圣瑞, 金相灿, 等. 长江中下游浅水湖沉积物中有机质及其组分的赋存特征[J]. 西北农林科技大学学报(自然科学版), 2008, 36(5): 141-148. [20] 赵海超, 王圣瑞, 焦立新, 等. 洱海沉积物有机质及其组分空间分布特征[J]. 环境科学研究, 2013, 26(3): 243-249. [21] LI X, YANG T, HICKS L C, et al. Latitudinal patterns of light and heavy organic matter fractions in arid and semi-arid soils[J]. CATENA, 2022, 215: 106293. doi: 10.1016/j.catena.2022.106293 [22] MAYER S, KöLBL A, VöLKEL J, et al. Organic matter in temperate cultivated floodplain soils: Light fractions highly contribute to subsoil organic carbon[J]. Geoderma, 2019, 337: 679-690. doi: 10.1016/j.geoderma.2018.10.014 [23] 张雨洁. 会稽山香榧林土壤有机碳特征研究[D]. 北京, 中国林业科学研究院, 2019. [24] 赵海超, 王圣瑞, 张莉, 等. 有机质含量及其组分对洱海沉积物磷吸附-释放影响[J]. 环境科学学报, 2014, 34(9): 2346-2354. [25] 易文利, 王圣瑞, 金相灿, 等. 去除轻组有机质对湖泊沉积物磷释放速率的影响研究[J]. 南开大学学报(自然科学版), 2008, 41(4): 1-7. [26] WANG S, YI W, YANG S, et al. Effects of light fraction organic matter removal on phosphate adsorption by lake sediments[J]. Applied Geochemistry, 2011, 26(3): 286-292. doi: 10.1016/j.apgeochem.2010.12.001 [27] 陈星, 陆莹, 黄威. 氧化-载钠改性黑臭河道底泥对磷削减的效果及机制[J]. 环境工程学报, 2017, 11(12): 6282-6289. [28] LIN J, ZHAO Y, ZHANG Z, et al. Immobilization of mobile and bioavailable phosphorus in sediments using lanthanum hydroxide and magnetite/lanthanum hydroxide composite as amendments[J]. Science of the Total Environment, 2019, 687: 232-243. doi: 10.1016/j.scitotenv.2019.06.042 [29] 王而力, 王嗣淇. 西辽河沉积物有机组分对磷的吸附影响[J]. 中国环境科学, 2012, 32(4): 687-694. [30] XU D, YAN P, LIU Z, et al. Spatial distribution of phosphorus forms and the release risk of sediments phosphorus in West Lake, Hangzhou, China[J]. Ecological Engineering, 2021, 173: 106421. doi: 10.1016/j.ecoleng.2021.106421 [31] 黄威, 靳郑海, 凃成琪, 等. 城市河网区河流沉积物磷形态分布特征及释放贡献[J]. 环境科学学报, 2022, 42(12): 171-185. [32] 金彦任, 黄振兴. 吸附与孔径分布[M]. 北京, 国防工业出版社, 2015. [33] VAN OOSTERHOUT F, LURLING M. The effect of phosphorus binding clay (Phoslock (R)) in mitigating cyanobacterial nuisance: A laboratory study on the effects on water quality variables and plankton[J]. Hydrobiologia, 2013, 710(1): 265-277. doi: 10.1007/s10750-012-1206-x [34] 王若凡, 田甜, 刘骅, 等. 黄河兰州段消落带表层沉积物对磷的吸附[J]. 环境工程学报, 2023, 17(1): 343-350. [35] 张义, 刘子森, 张垚磊, 等. 环境因子对杭州西湖沉积物各形态磷释放的影响[J]. 水生生物学报, 2017, 41(6): 1354-1361. [36] KONG M, HAN T, CHEN M, et al. High mobilization of phosphorus in black-odor river sediments with the increase of temperature[J]. Science of the Total Environment, 2021, 775: 145595. [37] XIE J, LAI L, LIN L, et al. Phosphate removal from water by a novel zeolite/lanthanum hydroxide hybrid material prepared from coal fly ash[J]. Journal of Environmental Science and Health, Part A:Toxic/Hazardous Substances and Environmental Engineering, 2015, 50(12): 1298-1305. [38] YIN H, ZHU J. In situ remediation of metal contaminated lake sediment using naturally occurring, calcium-rich clay mineral-based low-cost amendment[J]. Chemical Engineering Journal, 2016, 285: 112-120. doi: 10.1016/j.cej.2015.09.108 [39] MALLET M, BARTHELEMY K, RUBY C, et al. Investigation of phosphate adsorption onto ferrihydrite by X-ray Photoelectron Spectroscopy[J]. Journal of Colloid and Interface Science, 2013, 407: 95-101. doi: 10.1016/j.jcis.2013.06.049 [40] FANG L, LIU R, LI J, et al. Magnetite/Lanthanum hydroxide for phosphate sequestration and recovery from lake and the attenuation effects of sediment particles[J]. Water Research, 2018, 130: 243-254. doi: 10.1016/j.watres.2017.12.008 [41] WU Y, LI X, YANG Q, et al. Hydrated lanthanum oxide-modified diatomite as highly efficient adsorbent for low-concentration phosphate removal from secondary effluents[J]. Journal of Environmental Management, 2019, 231: 370-390. [42] WU B, FANG L, FORTNER J D, et al. Highly efficient and selective phosphate removal from wastewater by magnetically recoverable La(OH)(3)/Fe3O4 nanocomposites[J]. Water Research, 2017, 126: 179-188. doi: 10.1016/j.watres.2017.09.034 [43] XU Y, HU H, LIU J, et al. pH dependent phosphorus release from waste activated sludge: contributions of phosphorus speciation[J]. Chemical Engineering Journal, 2015, 267: 260-265. doi: 10.1016/j.cej.2015.01.037 [44] 谢锦升, 杨玉盛, 杨智杰, 等. 退化红壤植被恢复后土壤轻组有机质的季节动态[J]. 应用生态学报, 2008, 19(3): 557-563. [45] YANG C, YANG P, YIN H. In situ control of internal nutrient loading and fluxes in the confluence area of an eutrophic lake with combined P inactivation agents and modified zeolite[J]. Science of the Total Environment, 2021, 775: 145745. doi: 10.1016/j.scitotenv.2021.145745 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3216
- HTML全文浏览数: 3216
- PDF下载数: 117
- 施引文献: 0
