

-
臭氧混凝耦合工艺(hybrid ozonation-coagulation,HOC)是基于传统污水深度处理工艺“混凝-沉淀-过滤”处理流程长、溶解性有机物去除效果差的问题提出并构建的,具有臭氧氧化与混凝同时进行、溶解性有机物去除率高的特点[1-3]。然而在进行高Br−浓度废水处理时,例如工业盐水、肥料废水等[4-5],HOC工艺中的臭氧和由于臭氧与混凝剂的互促增效反应产生的·OH[6]会将Br−氧化为潜在致癌物质溴酸盐(BrO3−)[7-9],而我国《生活饮用水卫生标准》(GB 5749-2022)规定,饮用水中BrO3−含量不能高于0.01 mg·L−1。
近年来,臭氧氧化过程的BrO3−消毒副产物的控制方法受到国内外相关研究领域学者的广泛重视[10-13]。在臭氧氧化体系中,BrO3−的生成途径主要分为2种[14-15]:臭氧直接氧化和·OH间接氧化,2种氧化途径均伴随着中间产物HBrO/BrO−的生成,因此,HBrO/BrO−的继续氧化是控制Br−转化为BrO3−的重要限制反应[16]。而有研究表明,H2O2能通过与HBrO/BrO−反应阻碍其继续氧化为BrO3−,从而有效地延缓溴酸盐消毒副产物的生成[13]。此外,H2O2能加速臭氧分解生成·OH,控制BrO3−生成的同时可提高有机物的去除效果,但·OH的间接氧化也会促进BrO3−的生成[11, 17]。目前HOC工艺中H2O2控制下的溴酸盐消毒副产物的抑制效能尚未探讨,抑制原理也有待进一步探究。
基于以上研究结果,本研究探究在不同臭氧投加量下HOC工艺的有机物处理效果和BrO3−生成情况,考察H2O2投加量对HOC工艺中BrO3−生成的抑制效能,通过对BrO3−生成贡献率和Br−消耗动力学的分析,明确HOC体系中BrO3−的主要生成途径和H2O2抑制原理。
-
实验用水为外加KBr(Br−质量浓度为500 μg·L−1)的二级出水,二级出水来自西安某城市污水处理厂的二沉池,主要水质指标参数如下:pH为7.56±0.23,TOC为(5.43±1.66) mg·L−1,Br−为 (117±8) μg·L−1。
-
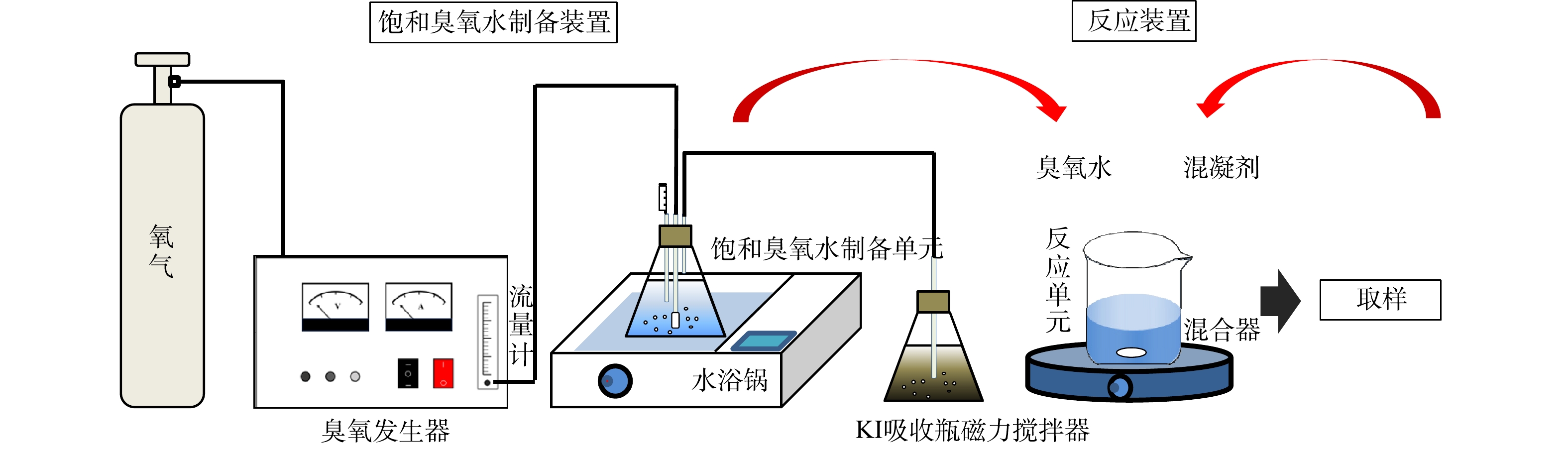
饱和臭氧水制备装置和HOC反应装置如图1所示,由气体流量计控制氧气源臭氧发生器(济南三康,SK-CFQ-3P)的出口流量为16 L·h−1,整个装置在0~4 ℃的水浴温度下持续曝气2 h后臭氧达饱和状态,本研究所投加的臭氧质量浓度为4.5、8.0、11.3 mg·L−1。HOC实验在外加Br−的100 mL二级出水中进行,混凝剂AlCl3·6H2O的投加量为15 mg·L−1。实验前使用0.2 mmol·L−1的磷酸缓冲溶液和1 mol·L−1的H2SO4溶液调节pH在7±0.2左右,然后加入混凝剂快搅1 min (转速300 r·min−1),饱和臭氧水和过氧化氢(n(H2O2):n(O3) = 0、0.5、1.0、1.5、2.0)在快搅结束后迅速加入体系,继而慢搅25 min (转速100 r·min−1),取不同反应时间的水样进行总有机碳(TOC)、溴离子(Br−)、溴酸根(BrO3−)、次溴酸(HBrO/BrO−)等指标的测定。
在探究HOC体系中BrO3−的生成贡献率时,取不同反应时间的水样经0.45 μm滤头过滤后快速测定臭氧质量浓度。由于对氯苯甲酸(pCBA)与·OH的反应速率k·OH,pCBA=5.0×109 L·(mol·s)−1远高于与O3的反应速率kO3,pCBA<0.15 L·(mol·s)−1[18],因此采用pCBA作为·OH的探针间接测定·OH的浓度。实验前向反应体系中加入0.5 μmol·L−1的pCBA,水样中pCBA的浓度测定前使用Na2S2O3淬灭·OH。
-
样品经0.45 μm滤头过滤、H2SO4酸化、N2吹脱3 min处理后,使用岛津TOC-VCPH分析仪测定TOC。饱和臭氧水质量浓度采用靛蓝比色法测定[19],气态臭氧采用碘量法测定[20]。Br−和BrO3−的质量浓度由热电阴离子色谱(ICS-1100 Dionex)测定,色谱柱为Dionex IonPacTM AS23(4×250 mm)。样品经固相萃取装置进行预处理:首先使用Bond Elut-C18小柱去除有机物;再使用Dionex OnGuardTM Ⅱ Ag/H小柱去除过渡金属离子和氯离子。HBrO/BrO−的质量浓度由苯酚衍生法测定[21],样品在pH为3、70 ℃的水浴条件下与苯酚反应1 h,反应后使用高效液相色谱法测定4-溴苯酚的质量浓度[22]。·OH浓度由pCBA探针法间接测定,pCBA浓度采用高效液相色谱法测定[23]。
-
Rct为某段时间内·OH暴露量与O3暴露量的比值(式(1))[24],Rct值通过式(2)计算。HOC体系中臭氧直接氧化和·OH间接氧化对BrO3−生成的贡献f(O3)和f(·OH)由式(3)~(5)[25-26]计算。
式中:C(·OH)为·OH的浓度,mol·L−1;C(O3)为O3的浓度,mol·L−1;C(pCBA)0和C(pCBA)分别为pCBA的初始浓度和反应后浓度,mol·L−1;k·OH,pCBA为pCBA与·OH的反应速率常数,5.0×109 L·(mol·s)−1。
式中:fBr−,·OH为·OH间接氧化的贡献率f(·OH);fBr−,O3为臭氧直接氧化的贡献率f(O3);C(Br−)为Br−的浓度,mol·L−1;C(·OH)为·OH的浓度,mol·L−1;kBr−,·OH为Br−与·OH的反应速率常数,1.1×109 L·(mol·s)−1[13];kBr−,O3为Br−与O3的反应速率常数,160 L·(mol·s)−1;C(O3)为O3的浓度,mol·L−1[13]。
-
图2为不同臭氧投加量下HOC体系对二级出水中有机物的处理效果和生成的BrO3−质量浓度的变化。当臭氧投加量为4.5、8.0、11.3 mg·L−1时,反应25 min后HOC体系对TOC的去除率分别达到36.29%、40.56%和42.41%,BrO3−的生成量分别为0.026、0.030和0.040 mg·L−1,有机物的去除率和BrO3−质量浓度均随着臭氧投加量的增大而增加。这是因为在HOC体系中混凝剂既可以作为混凝剂发挥混凝作用,又可以作为催化剂催化臭氧产生·OH[1-3, 6],随着臭氧投加量的增加,O3和·OH的含量增大,体系的氧化能力增强,强化了有机物去除的同时使更多的Br−被O3直接或·OH间接氧化为BrO3−[14-15]。同时,由图2(b)可以看到,在3种臭氧投加量下,反应8 min后HOC体系中的BrO3−质量浓度均已经超过0.01 mg·L−1,即国家饮用水标准规范(GB 5749-2022)的BrO3−质量浓度限值,因此需对HOC体系中溴酸盐的生成进行控制。
图3为不同H2O2投加量(n(H2O2):n(O3)=0.5、1、1.5和2)下HOC体系BrO3−质量浓度的变化。对于不同臭氧投加量的HOC体系,当n(H2O2):n(O3)>0.5时,H2O2对体系中BrO3−的生成具有明显的抑制作用,并且抑制作用随着H2O2投加量的增大而增强,其中当臭氧投加量为4.5 mg·L−1、n(H2O2):n(O3)=2时(图3(a)),H2O2对BrO3−的抑制率可达60%,BrO3−的生成量略低于0.01 mg·L−1。这与MIZUNO等[27]的研究结果一致,即n(H2O2):n(O3)超过1.25时能将BrO3−质量浓度控制在0.01 mg·L−1以下。这是因为过量的H2O2与生成BrO3−的重要中间产物HBrO/BrO−反应,将其还原回Br−,阻断了BrO3−的生成路径[28],同时H2O2的投加还会促进O3分解,降低了O3的暴露量,减少了HBrO/BrO−的生成,进而抑制了BrO3−的产生[11]。
由图3还可以看出,随着臭氧投加量的增加,H2O2对HOC体系中BrO3−生成的抑制效果变差,在臭氧投加量分别为4.5、8.0和11.3 mg·L−1时,当n(H2O2):n(O3)=2时对BrO3−的最大抑制率分别为60%、52%和42%,说明随着臭氧投加量的增大,O3暴露量升高,BrO3−生成量也随之增加[29]。因此,在较高臭氧投加量下投加相同摩尔比的H2O2难以达到低臭氧投加量时较好的抑制效果[30],但H2O2的投加延缓了HOC体系中BrO3−的生成。
-
在HOC工艺中,BrO3−由Br−经臭氧直接氧化和·OH间接氧化2种途径生成[14-15],实验探究了不同臭氧投加量下投加H2O2时2种途径对BrO3−生成的贡献率。H2O2抑制条件下不同臭氧投加量的HOC体系中pCBA去除曲线(·OH的含量变化)如图4所示。随着臭氧投加量的增大,pCBA去除量增加,说明HOC体系中生成了更多的·OH。在同一臭氧投加量下,随着H2O2投加量的增大,pCBA去除量减少,说明HOC体系中的·OH浓度降低,这归因于低浓度的H2O2能促进O3分解产生·OH,而高浓度的H2O2不仅促进·OH的产生还会与·OH反应,消耗体系中的·OH[31-32]。
臭氧自分解反应一级动力学方程拟合曲线如图5所示,随着H2O2投加量的增大,HOC体系中臭氧分解前15 s的瞬时臭氧需求量(instantaneous O3 demand,IOD)增大,说明H2O2促进了臭氧的分解,降低了臭氧的质量浓度,进而抑制了BrO3−的臭氧直接氧化产生过程[33]。
H2O2抑制条件下的不同臭氧投加量的Rct值如图6所示,在HOC的反应初始阶段Rct值快速下降,中后期保持恒定。当臭氧投加量相同时,随着H2O2投加量的增大,Rct值增大,说明H2O2促进了O3的分解,使单位臭氧产生的·OH量增加;而随着臭氧投加量的增大,H2O2投加量相同时,Rct降低,说明O3的暴露量随着臭氧投加量的增加而增大。
HOC体系中·OH间接氧化和臭氧直接氧化对BrO3−生成的贡献f(·OH)和f(O3)如表1所示,结果表明,未投加H2O2时HOC体系中臭氧直接氧化对BrO3−的生成起主要作用,在不同臭氧投加量下,臭氧直接氧化的贡献率f(O3)均维持在80%以上。随着臭氧投加量的增加,HOC体系中O3的暴露量增大,臭氧直接氧化的贡献率增大,当臭氧投加量为11.3 mg·L−1时f(O3)可达到98%。
随着H2O2投加量的增大,·OH对BrO3−生成的贡献率f(·OH)增大,f(O3)相应降低,臭氧投加量为4.5 mg·L−1时,投加n(H2O2):n(O3)=2的H2O2相较于未投加H2O2使反应初始阶段f(·OH)由15.83%增加至34.44%,反应后期f(·OH)由7.90%增加至16.88%。在H2O2的抑制条件下,反应时间在0~5 min内f(·OH)逐渐减少,f(O3)逐渐增加且均在60%以上;在5~15 min的反应时间内臭氧与·OH的贡献率基本保持稳定,且f(O3)保持在80%以上。因此,在H2O2抑制条件下臭氧直接氧化仍是HOC体系中BrO3−生成的主要途径。
-
为探究HOC体系中H2O2对BrO3−生成的抑制原理,测定了臭氧投加量为4.5 mg·L−1时H2O2抑制条件下的Br−和HBrO/BrO−质量浓度变化如图7所示,BrO3−质量浓度变化如图3(a)所示。反应前期,由于H2O2的抑制,体系中HBrO/BrO−质量浓度保持在较低水平,Br−的消耗和BrO3−的生成都较为缓慢;在反应中后期,随着H2O2的消耗,其抑制作用减弱,难以继续抑制Br−的氧化,HBrO/BrO−开始积累,BrO3−生成速度加快。H2O2投加量越大,HBrO/BrO−维持低质量浓度的时间越长,BrO3−的最终生成量越低,当n(H2O2):n(O3)=2时,HBrO/BrO−在反应前10 min的质量浓度均在0.01 mg·L−1以下,BrO3−最终生成量也在0.01 mg·L−1以下。
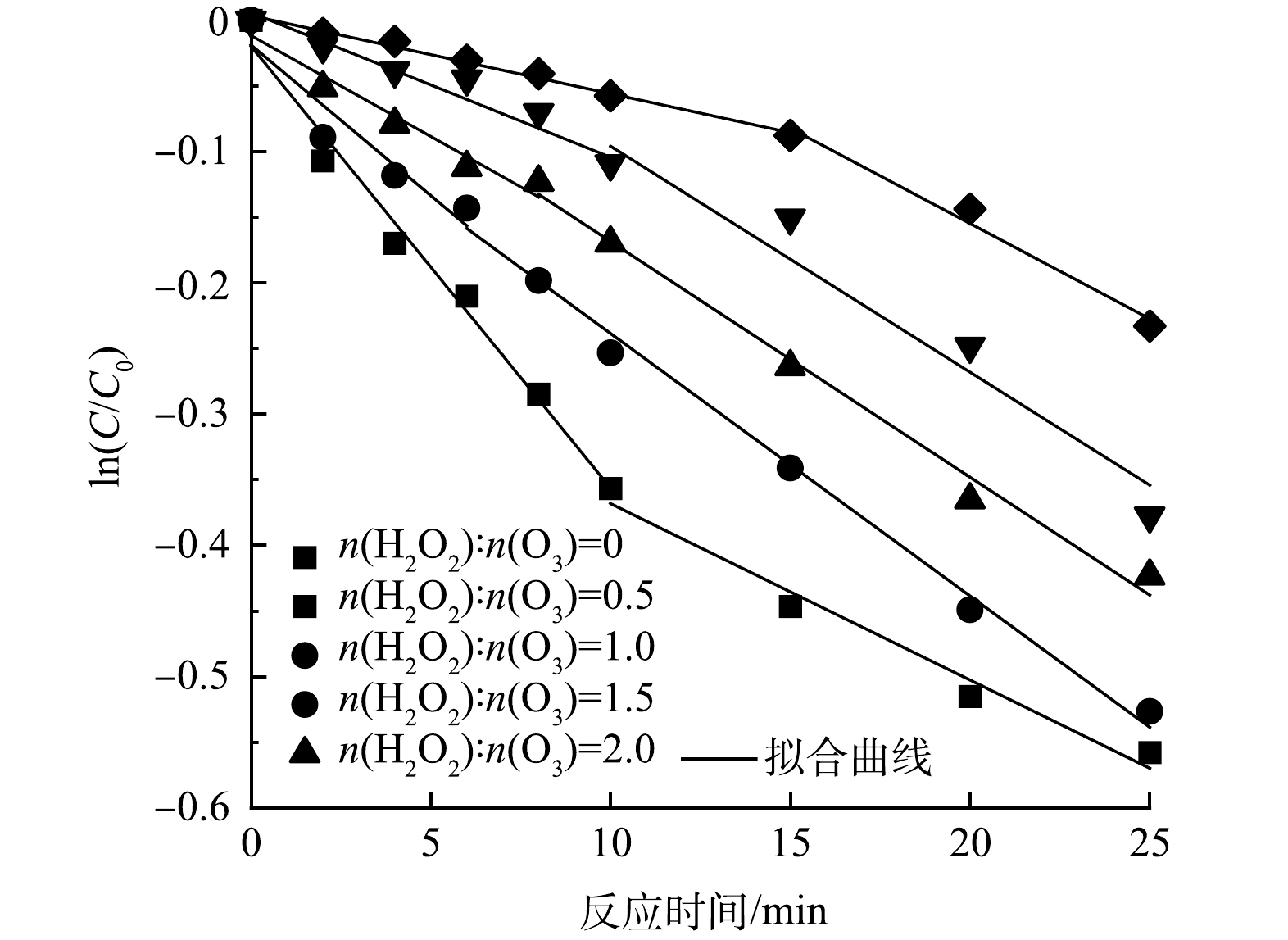
Br−的ln(C/C0)-t散点图如图8所示,可将Br−质量浓度的消耗过程分为快速阶段和慢速阶段,对两消耗阶段进行拟合,得到了不同H2O2投加量下的Br−拟一级反应速率常数kobs,具体拟合结果见表2。
根据表2,在H2O2抑制条件下的HOC体系中,当n(H2O2):n(O3)>0.5时,Br−的消耗由原本的先快速消耗后慢速消耗[34]变成了先慢速消耗后快速消耗。在反应前期,由于H2O2的抑制作用,Br−的消耗速率常数降低,且H2O2投加量越大,消耗速率常数越低,当n(H2O2):n(O3)=2时,Br−的消耗速率常数低至0.005 9 min−1。同时,H2O2投加量越大,Br−的慢速消耗时间越长,相同反应时间下BrO3−的生成量越低,说明H2O2的投加有效延缓了HOC体系中Br−的消耗和BrO3−的生成[11, 17]。在反应后期,H2O2被消耗,抑制能力减弱,Br−的消耗速率常数开始增大,BrO3−质量浓度逐渐增大。随着H2O2投加量的增大,反应后期剩余H2O2增多,促进了臭氧自分解以及·OH和H2O2的反应,快速消耗阶段的kobs减小,有效的降低了反应后期BrO3−的生成量。
因此,在HOC体系中,H2O2对BrO3−消毒副产物的抑制主要通过两个阶段进行:在反应前期,H2O2阻断了HBrO/BrO−被氧化为BrO3−的反应,有效抑制了Br−的消耗和BrO3−的生成;在反应后期,残留的H2O2加速了体系中臭氧的分解,当H2O2足够时还会与·OH反应,使反应后期体系中活性氧物质含量降低,有效减少了BrO3−的生成。在HOC体系中使用H2O2控制BrO3−的生成时,可以在保证有机物去除效果的同时通过控制反应时间处于慢速反应阶段控制BrO3−的生成。
-
在HOC体系中,H2O2对BrO3−消毒副产物的抑制主要通过慢速反应前期和快速反应后期进行,而慢速反应前期充分的H2O2能迅速地与HBrO/BrO−反应,减少BrO3−的生成,是抑制BrO3−的生成的主要阶段。
TOC去除结果表明,8.0 mg·L−1和11.3 mg·L−1的臭氧投加量对TOC的去除效果并未显著优于4.5 mg·L−1臭氧投加量,因此,控制臭氧投加量在4.5 mg·L−1,可在源头上降低BrO3−的生成量。同时,控制HOC工艺反应时间不超过15 min可以保证HOC工艺具有30%以上的有机物去除率;当n(H2O2):n(O3)=2时,可以将HOC工艺中的慢速反应前期的反应时间延长至15 min,进而控制BrO3−的生成量在0.01 mg·L−1以下。
-
1) HOC体系中随着臭氧投加量增大,TOC去除率升高, BrO3−质量浓度增大。当臭氧投加量为11.3 mg·L−1时,TOC去除率可达42.41%,但生成的BrO3−质量浓度达0.04 mg·L−1,远超国家饮用水标准规范(GB 5749-2022)。当n(H2O2):n(O3)>0.5时,H2O2能有效的抑制HOC体系中BrO3−的生成。
2) HOC体系中臭氧直接氧化是BrO3−生成的主要途径,f(O3)维持在80%以上,随着臭氧投加量增大,O3暴露量增大,f(O3)增大。在H2O2抑制条件下,反应前期f(·OH)增大,最大可达35.77%,而f(O3)均在60%以上,臭氧氧化仍是HOC体系中BrO3−生成的主要途径。
3)在H2O2抑制条件下的HOC体系中,Br−的消耗反应为先慢速反应后快速反应。反应前期H2O2抑制了Br−的氧化,且随着H2O2投加量增大,慢速反应时间增长,有效延缓了BrO3−的生成;反应后期H2O2被消耗,Br−消耗速率增大,BrO3−浓度增大,而随着H2O2投加量的增大,体系中活性氧物质减少,抑制了BrO3−的生成。
4)通过控制臭氧投加量为4.5 mg·L−1、反应时间不超过15 min以及n(H2O2):n(O3)=2可以保证HOC体系的有机物去除率在30%以上,同时控制BrO3−的生成量在0.01 mg·L−1以下。
臭氧混凝耦合体系中H2O2对溴酸盐生成的抑制效果及相关机理
Inhibitory effect and mechanism of bromate formation by H2O2 in the hybrid ozonation-coagulation process
-
摘要: 探究了臭氧混凝耦合体系(hybrid ozonation-coagulation,HOC)中H2O2对溴酸盐消毒副产物(BrO3−)的抑制效能,明确了BrO3−的主要生成途径和H2O2对BrO3−生成的抑制原理。结果表明,当n(H2O2):n(O3)>0.5时,H2O2能有效延缓BrO3−的生成。当臭氧投加量为4.5 mg·L−1、n(H2O2):n(O3)=2时,能有效的将BrO3−生成量控制在0.01 mg·L−1以下。通过对BrO3−生成贡献率和Br−消耗动力学的分析,臭氧直接氧化是HOC工艺中BrO3−生成的主要途径,贡献率维持在80%以上。在H2O2抑制条件下,•OH间接氧化贡献率有所提高,但臭氧直接氧化仍然是HOC工艺中BrO3−生成的主要途径,贡献率在60%以上。在H2O2抑制条件下,Br−的消耗反应为先慢速后快速,前期慢速反应阶段Br−的氧化过程被抑制,后期快速反应阶段中残余H2O2的存在减少了体系中活性氧物质的浓度,进而有效抑制了BrO3−的生成。
-
关键词:
- 臭氧混凝耦合工艺(HOC) /
- 溴酸盐 /
- 消毒副产物 /
- 动力学分析
Abstract: In this study, the inhibitory effect of H2O2 on bromate disinfection byproducts (BrO3−) formation in the hybrid ozonation-coagulation (HOC) process was investigated, and the main generation pathway of BrO3− and the inhibitory mechanism of BrO3− generation by H2O2 addition were clarified. The results showed that when n(H2O2):n(O3)>0.5, BrO3− formation could be effectively mitigated. When the ozone dosage was 4.5 mg·L−1 and n(H2O2):n(O3)=2, BrO3− production could be effectively controlled below 0.01 mg·L−1. Through the analysis of BrO3− generation contribution rate and Br− consumption kinetics, direct ozone oxidation was the main pathway of BrO3− generation in the HOC process, and the contribution rate maintained above than 80%. With the addition of H2O2, the contribution rate of •OH indirect oxidation increased, while ozone direct oxidation was still the main pathway for BrO3− generation with the contribution rate above 60%. With the addition of H2O2, the consumption reaction of Br− was slow at first and then became fast. The oxidation process of Br− was inhibited during slow reaction at the early stage, and the presence of residual H2O2 during fast reaction at the later stage reduced the concentration of oxidizing substances in the process, thus effectively inhibited the formation of BrO3−. -
臭氧混凝耦合工艺(hybrid ozonation-coagulation,HOC)是基于传统污水深度处理工艺“混凝-沉淀-过滤”处理流程长、溶解性有机物去除效果差的问题提出并构建的,具有臭氧氧化与混凝同时进行、溶解性有机物去除率高的特点[1-3]。然而在进行高Br−浓度废水处理时,例如工业盐水、肥料废水等[4-5],HOC工艺中的臭氧和由于臭氧与混凝剂的互促增效反应产生的·OH[6]会将Br−氧化为潜在致癌物质溴酸盐(BrO3−)[7-9],而我国《生活饮用水卫生标准》(GB 5749-2022)规定,饮用水中BrO3−含量不能高于0.01 mg·L−1。
近年来,臭氧氧化过程的BrO3−消毒副产物的控制方法受到国内外相关研究领域学者的广泛重视[10-13]。在臭氧氧化体系中,BrO3−的生成途径主要分为2种[14-15]:臭氧直接氧化和·OH间接氧化,2种氧化途径均伴随着中间产物HBrO/BrO−的生成,因此,HBrO/BrO−的继续氧化是控制Br−转化为BrO3−的重要限制反应[16]。而有研究表明,H2O2能通过与HBrO/BrO−反应阻碍其继续氧化为BrO3−,从而有效地延缓溴酸盐消毒副产物的生成[13]。此外,H2O2能加速臭氧分解生成·OH,控制BrO3−生成的同时可提高有机物的去除效果,但·OH的间接氧化也会促进BrO3−的生成[11, 17]。目前HOC工艺中H2O2控制下的溴酸盐消毒副产物的抑制效能尚未探讨,抑制原理也有待进一步探究。
基于以上研究结果,本研究探究在不同臭氧投加量下HOC工艺的有机物处理效果和BrO3−生成情况,考察H2O2投加量对HOC工艺中BrO3−生成的抑制效能,通过对BrO3−生成贡献率和Br−消耗动力学的分析,明确HOC体系中BrO3−的主要生成途径和H2O2抑制原理。
1. 材料与方法
1.1 实验用水
实验用水为外加KBr(Br−质量浓度为500 μg·L−1)的二级出水,二级出水来自西安某城市污水处理厂的二沉池,主要水质指标参数如下:pH为7.56±0.23,TOC为(5.43±1.66) mg·L−1,Br−为 (117±8) μg·L−1。
1.2 实验方法与装置
饱和臭氧水制备装置和HOC反应装置如图1所示,由气体流量计控制氧气源臭氧发生器(济南三康,SK-CFQ-3P)的出口流量为16 L·h−1,整个装置在0~4 ℃的水浴温度下持续曝气2 h后臭氧达饱和状态,本研究所投加的臭氧质量浓度为4.5、8.0、11.3 mg·L−1。HOC实验在外加Br−的100 mL二级出水中进行,混凝剂AlCl3·6H2O的投加量为15 mg·L−1。实验前使用0.2 mmol·L−1的磷酸缓冲溶液和1 mol·L−1的H2SO4溶液调节pH在7±0.2左右,然后加入混凝剂快搅1 min (转速300 r·min−1),饱和臭氧水和过氧化氢(n(H2O2):n(O3) = 0、0.5、1.0、1.5、2.0)在快搅结束后迅速加入体系,继而慢搅25 min (转速100 r·min−1),取不同反应时间的水样进行总有机碳(TOC)、溴离子(Br−)、溴酸根(BrO3−)、次溴酸(HBrO/BrO−)等指标的测定。
在探究HOC体系中BrO3−的生成贡献率时,取不同反应时间的水样经0.45 μm滤头过滤后快速测定臭氧质量浓度。由于对氯苯甲酸(pCBA)与·OH的反应速率k·OH,pCBA=5.0×109 L·(mol·s)−1远高于与O3的反应速率kO3,pCBA<0.15 L·(mol·s)−1[18],因此采用pCBA作为·OH的探针间接测定·OH的浓度。实验前向反应体系中加入0.5 μmol·L−1的pCBA,水样中pCBA的浓度测定前使用Na2S2O3淬灭·OH。
1.3 分析测试方法
样品经0.45 μm滤头过滤、H2SO4酸化、N2吹脱3 min处理后,使用岛津TOC-VCPH分析仪测定TOC。饱和臭氧水质量浓度采用靛蓝比色法测定[19],气态臭氧采用碘量法测定[20]。Br−和BrO3−的质量浓度由热电阴离子色谱(ICS-1100 Dionex)测定,色谱柱为Dionex IonPacTM AS23(4×250 mm)。样品经固相萃取装置进行预处理:首先使用Bond Elut-C18小柱去除有机物;再使用Dionex OnGuardTM Ⅱ Ag/H小柱去除过渡金属离子和氯离子。HBrO/BrO−的质量浓度由苯酚衍生法测定[21],样品在pH为3、70 ℃的水浴条件下与苯酚反应1 h,反应后使用高效液相色谱法测定4-溴苯酚的质量浓度[22]。·OH浓度由pCBA探针法间接测定,pCBA浓度采用高效液相色谱法测定[23]。
1.4 数据分析方法
Rct为某段时间内·OH暴露量与O3暴露量的比值(式(1))[24],Rct值通过式(2)计算。HOC体系中臭氧直接氧化和·OH间接氧化对BrO3−生成的贡献f(O3)和f(·OH)由式(3)~(5)[25-26]计算。
Rct=∫C(⋅OH)dt∫C(O3)dt (1) ln(C(pCBA)C(pCBA)0)=−Rctk⋅OH,pCBA∫C(O3)dt (2) 式中:C(·OH)为·OH的浓度,mol·L−1;C(O3)为O3的浓度,mol·L−1;C(pCBA)0和C(pCBA)分别为pCBA的初始浓度和反应后浓度,mol·L−1;k·OH,pCBA为pCBA与·OH的反应速率常数,5.0×109 L·(mol·s)−1。
fBr−,⋅OH=kBr−,⋅OHC(Br−)C(⋅OH)kBr - ,⋅OHC(Br−)C(⋅OH)+kBr−,O3C(Br−)C(O3) (3) fBr−,⋅OH=1.1×109Rct160+1.1×109Rct (4) fBr−,O3=1−fBr−,⋅OH (5) 式中:fBr−,·OH为·OH间接氧化的贡献率f(·OH);fBr−,O3为臭氧直接氧化的贡献率f(O3);C(Br−)为Br−的浓度,mol·L−1;C(·OH)为·OH的浓度,mol·L−1;kBr−,·OH为Br−与·OH的反应速率常数,1.1×109 L·(mol·s)−1[13];kBr−,O3为Br−与O3的反应速率常数,160 L·(mol·s)−1;C(O3)为O3的浓度,mol·L−1[13]。
2. 结果与讨论
2.1 有机物的去除及溴酸盐的产生和抑制
图2为不同臭氧投加量下HOC体系对二级出水中有机物的处理效果和生成的BrO3−质量浓度的变化。当臭氧投加量为4.5、8.0、11.3 mg·L−1时,反应25 min后HOC体系对TOC的去除率分别达到36.29%、40.56%和42.41%,BrO3−的生成量分别为0.026、0.030和0.040 mg·L−1,有机物的去除率和BrO3−质量浓度均随着臭氧投加量的增大而增加。这是因为在HOC体系中混凝剂既可以作为混凝剂发挥混凝作用,又可以作为催化剂催化臭氧产生·OH[1-3, 6],随着臭氧投加量的增加,O3和·OH的含量增大,体系的氧化能力增强,强化了有机物去除的同时使更多的Br−被O3直接或·OH间接氧化为BrO3−[14-15]。同时,由图2(b)可以看到,在3种臭氧投加量下,反应8 min后HOC体系中的BrO3−质量浓度均已经超过0.01 mg·L−1,即国家饮用水标准规范(GB 5749-2022)的BrO3−质量浓度限值,因此需对HOC体系中溴酸盐的生成进行控制。
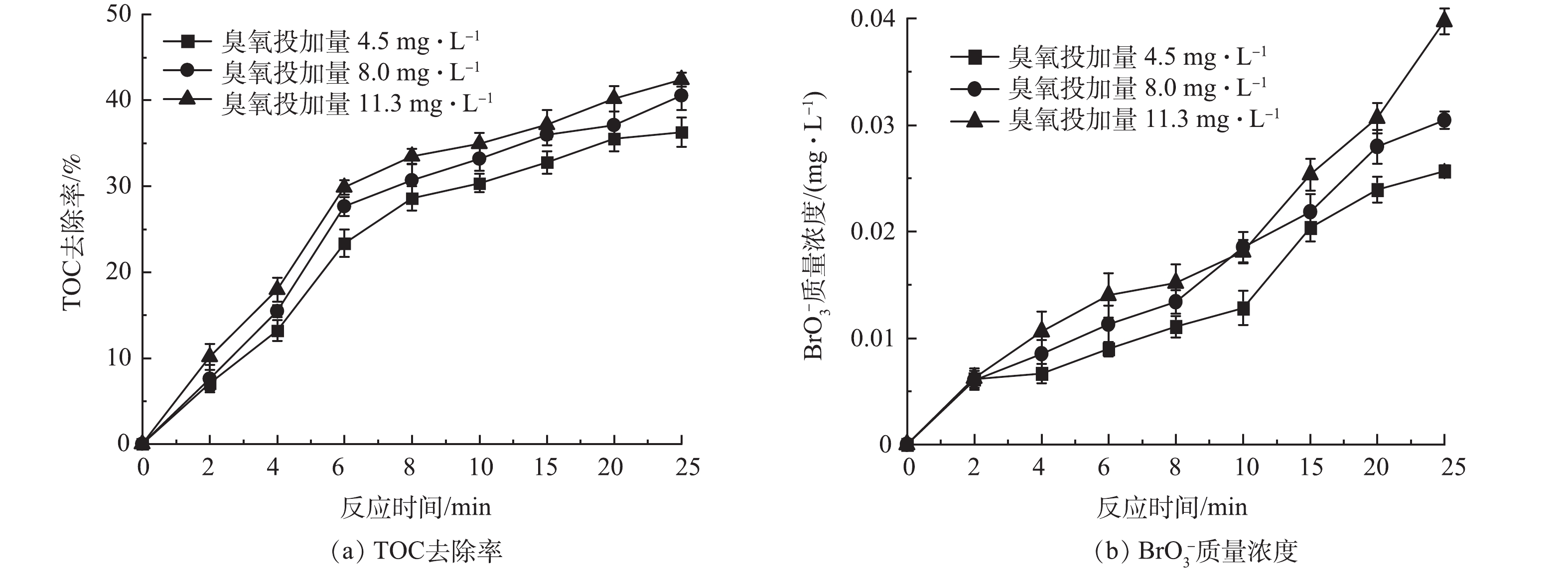 图 2 不同臭氧投加量下有机物的去除率和BrO3−质量浓度变化Figure 2. Variations of organic matter removal efficiency and BrO3− mass concentration at different ozone dosages
图 2 不同臭氧投加量下有机物的去除率和BrO3−质量浓度变化Figure 2. Variations of organic matter removal efficiency and BrO3− mass concentration at different ozone dosages图3为不同H2O2投加量(n(H2O2):n(O3)=0.5、1、1.5和2)下HOC体系BrO3−质量浓度的变化。对于不同臭氧投加量的HOC体系,当n(H2O2):n(O3)>0.5时,H2O2对体系中BrO3−的生成具有明显的抑制作用,并且抑制作用随着H2O2投加量的增大而增强,其中当臭氧投加量为4.5 mg·L−1、n(H2O2):n(O3)=2时(图3(a)),H2O2对BrO3−的抑制率可达60%,BrO3−的生成量略低于0.01 mg·L−1。这与MIZUNO等[27]的研究结果一致,即n(H2O2):n(O3)超过1.25时能将BrO3−质量浓度控制在0.01 mg·L−1以下。这是因为过量的H2O2与生成BrO3−的重要中间产物HBrO/BrO−反应,将其还原回Br−,阻断了BrO3−的生成路径[28],同时H2O2的投加还会促进O3分解,降低了O3的暴露量,减少了HBrO/BrO−的生成,进而抑制了BrO3−的产生[11]。
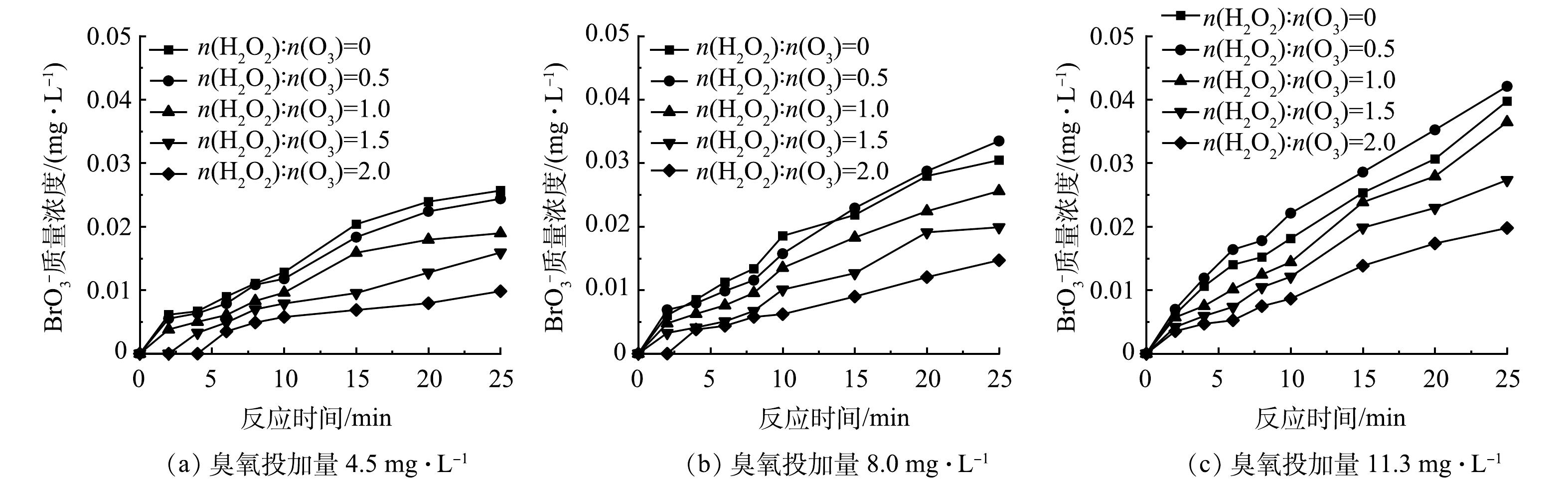 图 3 H2O2抑制条件下不同臭氧投加量下的BrO3-质量浓度变化Figure 3. Variations of BrO3- mass concentration at different ozone dosages with the addition of H2O2
图 3 H2O2抑制条件下不同臭氧投加量下的BrO3-质量浓度变化Figure 3. Variations of BrO3- mass concentration at different ozone dosages with the addition of H2O2由图3还可以看出,随着臭氧投加量的增加,H2O2对HOC体系中BrO3−生成的抑制效果变差,在臭氧投加量分别为4.5、8.0和11.3 mg·L−1时,当n(H2O2):n(O3)=2时对BrO3−的最大抑制率分别为60%、52%和42%,说明随着臭氧投加量的增大,O3暴露量升高,BrO3−生成量也随之增加[29]。因此,在较高臭氧投加量下投加相同摩尔比的H2O2难以达到低臭氧投加量时较好的抑制效果[30],但H2O2的投加延缓了HOC体系中BrO3−的生成。
2.2 H2O2抑制下的溴酸盐贡献率分析
在HOC工艺中,BrO3−由Br−经臭氧直接氧化和·OH间接氧化2种途径生成[14-15],实验探究了不同臭氧投加量下投加H2O2时2种途径对BrO3−生成的贡献率。H2O2抑制条件下不同臭氧投加量的HOC体系中pCBA去除曲线(·OH的含量变化)如图4所示。随着臭氧投加量的增大,pCBA去除量增加,说明HOC体系中生成了更多的·OH。在同一臭氧投加量下,随着H2O2投加量的增大,pCBA去除量减少,说明HOC体系中的·OH浓度降低,这归因于低浓度的H2O2能促进O3分解产生·OH,而高浓度的H2O2不仅促进·OH的产生还会与·OH反应,消耗体系中的·OH[31-32]。
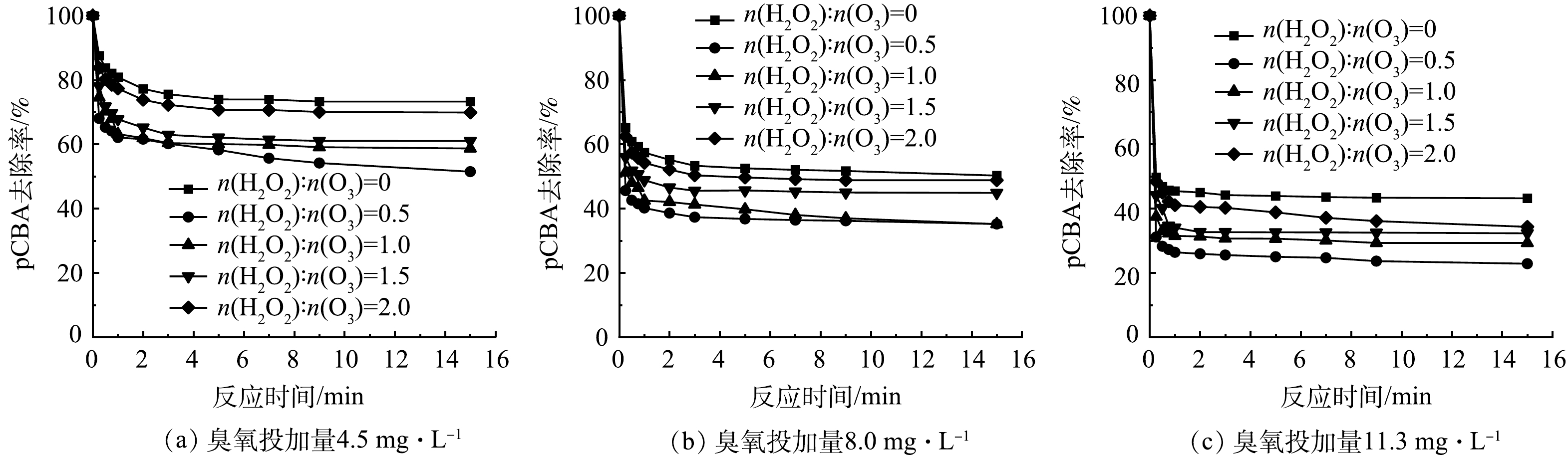 图 4 H2O2抑制条件下不同臭氧投加量的pCBA去除曲线Figure 4. pCBA removal curve at different ozone dosages with the addition of H2O2
图 4 H2O2抑制条件下不同臭氧投加量的pCBA去除曲线Figure 4. pCBA removal curve at different ozone dosages with the addition of H2O2臭氧自分解反应一级动力学方程拟合曲线如图5所示,随着H2O2投加量的增大,HOC体系中臭氧分解前15 s的瞬时臭氧需求量(instantaneous O3 demand,IOD)增大,说明H2O2促进了臭氧的分解,降低了臭氧的质量浓度,进而抑制了BrO3−的臭氧直接氧化产生过程[33]。
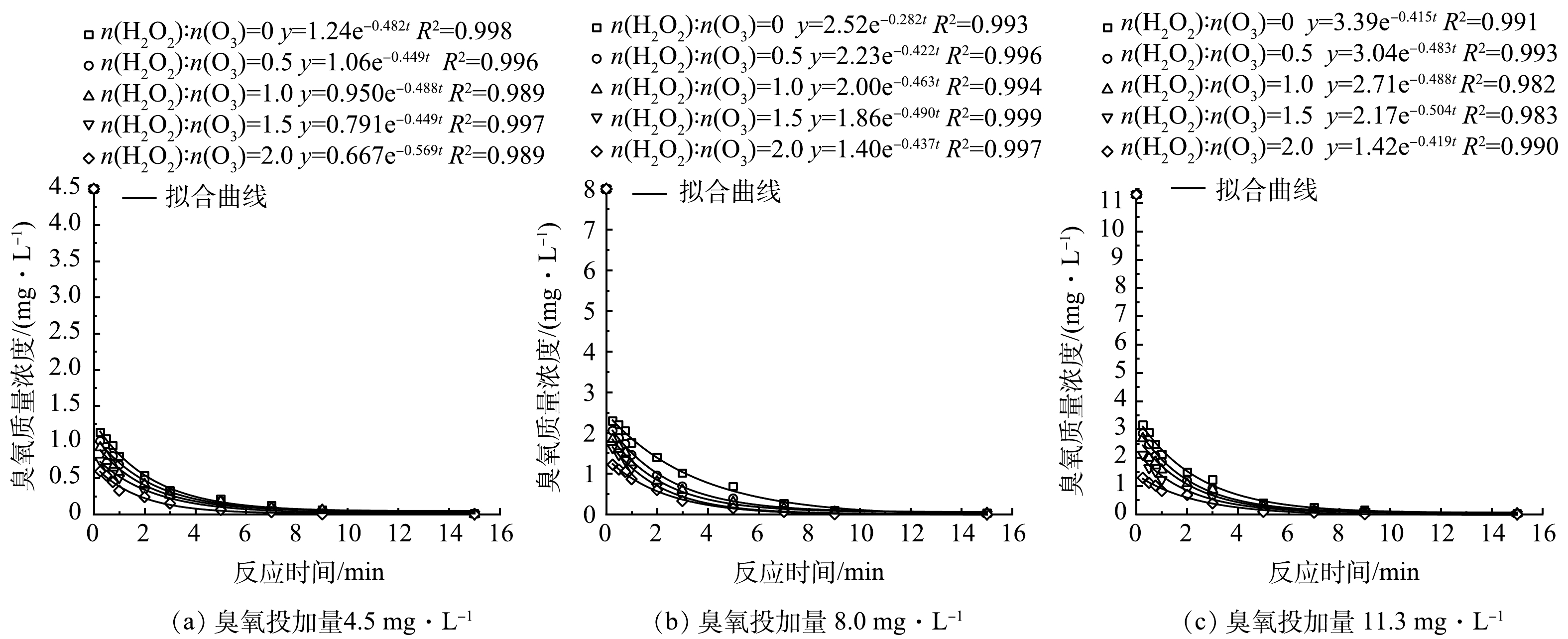 图 5 H2O2抑制条件下不同臭氧投加量的臭氧衰减曲线Figure 5. Ozone decay curve at different ozone dosages with the addition of H2O2
图 5 H2O2抑制条件下不同臭氧投加量的臭氧衰减曲线Figure 5. Ozone decay curve at different ozone dosages with the addition of H2O2H2O2抑制条件下的不同臭氧投加量的Rct值如图6所示,在HOC的反应初始阶段Rct值快速下降,中后期保持恒定。当臭氧投加量相同时,随着H2O2投加量的增大,Rct值增大,说明H2O2促进了O3的分解,使单位臭氧产生的·OH量增加;而随着臭氧投加量的增大,H2O2投加量相同时,Rct降低,说明O3的暴露量随着臭氧投加量的增加而增大。
 图 6 H2O2抑制条件下不同臭氧投加量的RctFigure 6. Rct at different ozone dosages with the addition of H2O2
图 6 H2O2抑制条件下不同臭氧投加量的RctFigure 6. Rct at different ozone dosages with the addition of H2O2HOC体系中·OH间接氧化和臭氧直接氧化对BrO3−生成的贡献f(·OH)和f(O3)如表1所示,结果表明,未投加H2O2时HOC体系中臭氧直接氧化对BrO3−的生成起主要作用,在不同臭氧投加量下,臭氧直接氧化的贡献率f(O3)均维持在80%以上。随着臭氧投加量的增加,HOC体系中O3的暴露量增大,臭氧直接氧化的贡献率增大,当臭氧投加量为11.3 mg·L−1时f(O3)可达到98%。
表 1 H2O2抑制条件下不同臭氧投加量的溴酸盐生成贡献率Table 1. Contribution rate of Bromate formation at different ozone dosages with the addition of H2O2臭氧/(mg·L−1) n(H2O2):n(O3) 不同反应时间下的f(·OH)/f(O3) 0.5 min 0.75 min 1 min 2 min 3 min 5 min 7 min 9 min 15 min 4.5 0 0.16/0.84 0.13/0.87 0.11/0.89 0.10/0.90 0.09/0.91 0.08/0.92 0.08/0.92 0.08/0.92 0.08/0.92 0.5 0.17/0.83 0.13/0.87 0.14/0.86 0.09/0.91 0.08/0.92 0.08/0.92 0.10/0.90 0.11/0.89 0.13/0.87 1 0.25/0.75 0.21/0.79 0.26/0.74 0.17/0.83 0.15/0.85 0.13/0.87 0.13/0.87 0.13/0.87 0.13/0.87 1.5 0.36/0.64 0.28/0.72 0.26/0.74 0.19/0.81 0.18/0.82 0.15/0.85 0.15/0.85 0.15/0.85 0.15/0.85 2 0.34/0.66 0.27/0.73 0.23/0.77 0.20/0.80 0.18/0.82 0.17/0.83 0.16/0.84 0.17/0.83 0.17/0.83 8.0 0 0.12/0.88 0.09/0.91 0.08/0.92 0.05/0.95 0.05/0.95 0.04/0.96 0.03/0.97 0.03/0.97 0.03/0.97 0.5 0.13/0.87 0.10/0.90 0.09/0.91 0.07/0.93 0.06/0.94 0.05/0.95 0.05/0.95 0.05/0.95 0.06/0.94 1 0.15/0.85 0.12/0.88 0.16/0.84 0.09/0.91 0.08/0.92 0.08/0.92 0.08/0.92 0.09/0.91 0.10/0.90 1.5 0.19/0.81 0.14/0.86 0.13/0.87 0.10/0.90 0.09/0.91 0.07/0.93 0.07/0.93 0.07/0.93 0.07/0.93 2 0.24/0.76 0.18/0.82 0.16/0.84 0.11/0.89 0.11/0.89 0.09/0.91 0.09/0.91 0.09/0.91 0.09/0.91 11.3 0 0.08/0.92 0.06/0.94 0.05/0.95 0.03/0.97 0.03/0.97 0.02/0.98 0.02/0.98 0.02/0.98 0.02/0.98 0.5 0.14/0.86 0.11/0.89 0.10/0.90 0.06/0.94 0.05/0.95 0.05/0.95 0.05/0.95 0.05/0.95 0.06/0.94 1 0.20/0.80 0.13/0.87 0.11/0.89 0.07/0.93 0.06/0.94 0.05/0.95 0.05/0.95 0.05/0.95 0.05/0.95 1.5 0.18/0.82 0.24/0.76 0.19/0.81 0.13/0.87 0.10/0.90 0.09/0.91 0.08/0.92 0.08/0.92 0.08/0.92 2 0.21/0.79 0.21/0.79 0.18/0.82 0.11/0.89 0.09/0.91 0.08/0.92 0.09/0.91 0.10/0.90 0.11/0.89 | Show Table DownLoad:
CSV
DownLoad:
CSV
随着H2O2投加量的增大,·OH对BrO3−生成的贡献率f(·OH)增大,f(O3)相应降低,臭氧投加量为4.5 mg·L−1时,投加n(H2O2):n(O3)=2的H2O2相较于未投加H2O2使反应初始阶段f(·OH)由15.83%增加至34.44%,反应后期f(·OH)由7.90%增加至16.88%。在H2O2的抑制条件下,反应时间在0~5 min内f(·OH)逐渐减少,f(O3)逐渐增加且均在60%以上;在5~15 min的反应时间内臭氧与·OH的贡献率基本保持稳定,且f(O3)保持在80%以上。因此,在H2O2抑制条件下臭氧直接氧化仍是HOC体系中BrO3−生成的主要途径。
2.3 HOC体系中H2O2抑制原理
为探究HOC体系中H2O2对BrO3−生成的抑制原理,测定了臭氧投加量为4.5 mg·L−1时H2O2抑制条件下的Br−和HBrO/BrO−质量浓度变化如图7所示,BrO3−质量浓度变化如图3(a)所示。反应前期,由于H2O2的抑制,体系中HBrO/BrO−质量浓度保持在较低水平,Br−的消耗和BrO3−的生成都较为缓慢;在反应中后期,随着H2O2的消耗,其抑制作用减弱,难以继续抑制Br−的氧化,HBrO/BrO−开始积累,BrO3−生成速度加快。H2O2投加量越大,HBrO/BrO−维持低质量浓度的时间越长,BrO3−的最终生成量越低,当n(H2O2):n(O3)=2时,HBrO/BrO−在反应前10 min的质量浓度均在0.01 mg·L−1以下,BrO3−最终生成量也在0.01 mg·L−1以下。
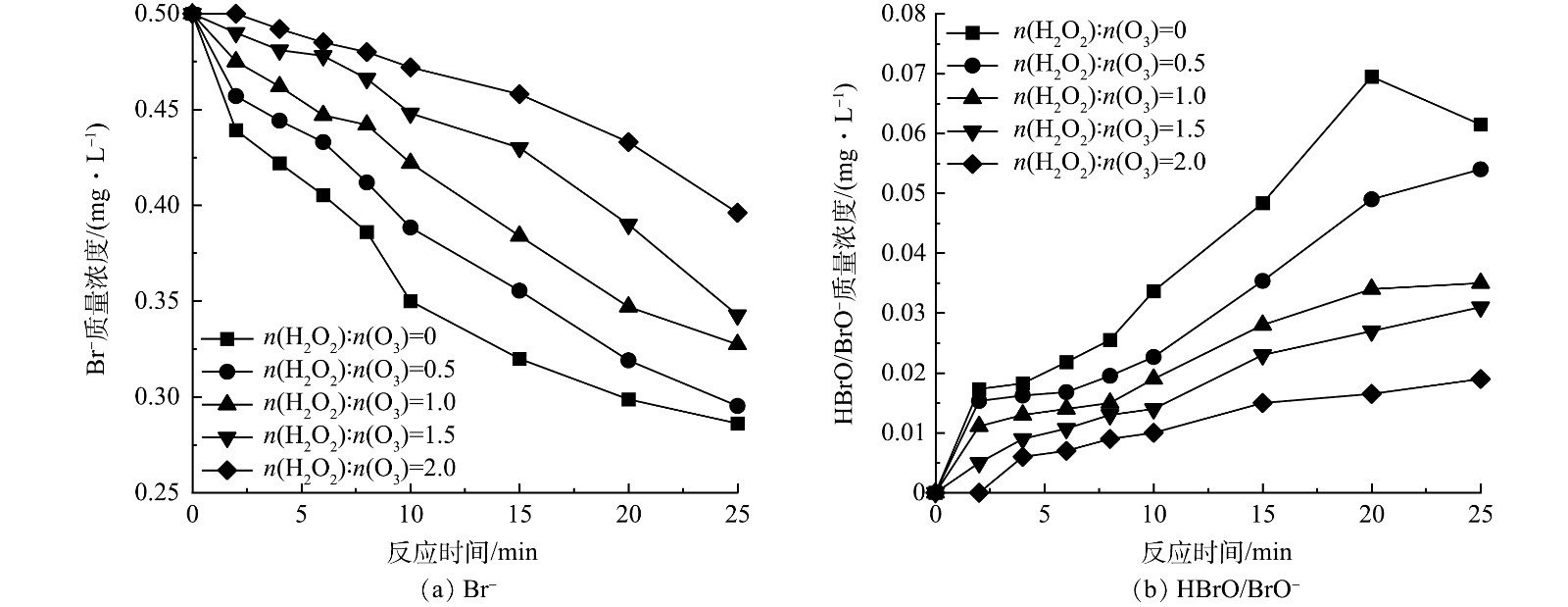 图 7 H2O2抑制条件下的溴类物质质量浓度变化Figure 7. Variation of Bromide species mass concentration with the addition of H2O2
图 7 H2O2抑制条件下的溴类物质质量浓度变化Figure 7. Variation of Bromide species mass concentration with the addition of H2O2Br−的ln(C/C0)-t散点图如图8所示,可将Br−质量浓度的消耗过程分为快速阶段和慢速阶段,对两消耗阶段进行拟合,得到了不同H2O2投加量下的Br−拟一级反应速率常数kobs,具体拟合结果见表2。
表 2 H2O2抑制条件下的Br-消耗的一级速率常数(kobs)Table 2. kobs of Br- consumption with the addition of H2O2n(H2O2):n(O3) 反应时间/min 拟合方程式 k/min-1 R2 0 0~10 y = −0.033 7x−0.019 70 0.033 7 0.985 10~25 y = −0.013 4x−0.234 00 0.013 4 0.976 0.5 0~6 y = −0.022 9x−0.018 86 0.022 9 0.950 6~25 y = −0.020 0x−0.038 59 0.020 0 0.990 1 0~8 y = −0.015 4x−0.011 67 0.015 4 0.955 8~25 y = −0.018 0x+0.011 12 0.018 0 0.990 1.5 0~10 y = −0.011 0x+0.005 83 0.011 0 0.971 10~25 y = −0.017 2x+0.076 27 0.017 2 0.983 2 0~15 y = −0.005 9x+0.003 51 0.005 9 0.991 15~25 y = -0.014 5x+0.135 60 0.014 5 0.983 | Show TableDownLoad:
CSV
根据表2,在H2O2抑制条件下的HOC体系中,当n(H2O2):n(O3)>0.5时,Br−的消耗由原本的先快速消耗后慢速消耗[34]变成了先慢速消耗后快速消耗。在反应前期,由于H2O2的抑制作用,Br−的消耗速率常数降低,且H2O2投加量越大,消耗速率常数越低,当n(H2O2):n(O3)=2时,Br−的消耗速率常数低至0.005 9 min−1。同时,H2O2投加量越大,Br−的慢速消耗时间越长,相同反应时间下BrO3−的生成量越低,说明H2O2的投加有效延缓了HOC体系中Br−的消耗和BrO3−的生成[11, 17]。在反应后期,H2O2被消耗,抑制能力减弱,Br−的消耗速率常数开始增大,BrO3−质量浓度逐渐增大。随着H2O2投加量的增大,反应后期剩余H2O2增多,促进了臭氧自分解以及·OH和H2O2的反应,快速消耗阶段的kobs减小,有效的降低了反应后期BrO3−的生成量。
因此,在HOC体系中,H2O2对BrO3−消毒副产物的抑制主要通过两个阶段进行:在反应前期,H2O2阻断了HBrO/BrO−被氧化为BrO3−的反应,有效抑制了Br−的消耗和BrO3−的生成;在反应后期,残留的H2O2加速了体系中臭氧的分解,当H2O2足够时还会与·OH反应,使反应后期体系中活性氧物质含量降低,有效减少了BrO3−的生成。在HOC体系中使用H2O2控制BrO3−的生成时,可以在保证有机物去除效果的同时通过控制反应时间处于慢速反应阶段控制BrO3−的生成。
2.4 HOC工艺运行条件的优化
在HOC体系中,H2O2对BrO3−消毒副产物的抑制主要通过慢速反应前期和快速反应后期进行,而慢速反应前期充分的H2O2能迅速地与HBrO/BrO−反应,减少BrO3−的生成,是抑制BrO3−的生成的主要阶段。
TOC去除结果表明,8.0 mg·L−1和11.3 mg·L−1的臭氧投加量对TOC的去除效果并未显著优于4.5 mg·L−1臭氧投加量,因此,控制臭氧投加量在4.5 mg·L−1,可在源头上降低BrO3−的生成量。同时,控制HOC工艺反应时间不超过15 min可以保证HOC工艺具有30%以上的有机物去除率;当n(H2O2):n(O3)=2时,可以将HOC工艺中的慢速反应前期的反应时间延长至15 min,进而控制BrO3−的生成量在0.01 mg·L−1以下。
3. 结论
1) HOC体系中随着臭氧投加量增大,TOC去除率升高, BrO3−质量浓度增大。当臭氧投加量为11.3 mg·L−1时,TOC去除率可达42.41%,但生成的BrO3−质量浓度达0.04 mg·L−1,远超国家饮用水标准规范(GB 5749-2022)。当n(H2O2):n(O3)>0.5时,H2O2能有效的抑制HOC体系中BrO3−的生成。
2) HOC体系中臭氧直接氧化是BrO3−生成的主要途径,f(O3)维持在80%以上,随着臭氧投加量增大,O3暴露量增大,f(O3)增大。在H2O2抑制条件下,反应前期f(·OH)增大,最大可达35.77%,而f(O3)均在60%以上,臭氧氧化仍是HOC体系中BrO3−生成的主要途径。
3)在H2O2抑制条件下的HOC体系中,Br−的消耗反应为先慢速反应后快速反应。反应前期H2O2抑制了Br−的氧化,且随着H2O2投加量增大,慢速反应时间增长,有效延缓了BrO3−的生成;反应后期H2O2被消耗,Br−消耗速率增大,BrO3−浓度增大,而随着H2O2投加量的增大,体系中活性氧物质减少,抑制了BrO3−的生成。
4)通过控制臭氧投加量为4.5 mg·L−1、反应时间不超过15 min以及n(H2O2):n(O3)=2可以保证HOC体系的有机物去除率在30%以上,同时控制BrO3−的生成量在0.01 mg·L−1以下。
-
图 2 不同臭氧投加量下有机物的去除率和BrO3−质量浓度变化
Figure 2. Variations of organic matter removal efficiency and BrO3− mass concentration at different ozone dosages
图 3 H2O2抑制条件下不同臭氧投加量下的BrO3-质量浓度变化
Figure 3. Variations of BrO3- mass concentration at different ozone dosages with the addition of H2O2
图 4 H2O2抑制条件下不同臭氧投加量的pCBA去除曲线
Figure 4. pCBA removal curve at different ozone dosages with the addition of H2O2
图 5 H2O2抑制条件下不同臭氧投加量的臭氧衰减曲线
Figure 5. Ozone decay curve at different ozone dosages with the addition of H2O2
图 6 H2O2抑制条件下不同臭氧投加量的Rct
Figure 6. Rct at different ozone dosages with the addition of H2O2
图 7 H2O2抑制条件下的溴类物质质量浓度变化
Figure 7. Variation of Bromide species mass concentration with the addition of H2O2
表 1 H2O2抑制条件下不同臭氧投加量的溴酸盐生成贡献率
Table 1. Contribution rate of Bromate formation at different ozone dosages with the addition of H2O2
臭氧/(mg·L−1) n(H2O2):n(O3) 不同反应时间下的f(·OH)/f(O3) 0.5 min 0.75 min 1 min 2 min 3 min 5 min 7 min 9 min 15 min 4.5 0 0.16/0.84 0.13/0.87 0.11/0.89 0.10/0.90 0.09/0.91 0.08/0.92 0.08/0.92 0.08/0.92 0.08/0.92 0.5 0.17/0.83 0.13/0.87 0.14/0.86 0.09/0.91 0.08/0.92 0.08/0.92 0.10/0.90 0.11/0.89 0.13/0.87 1 0.25/0.75 0.21/0.79 0.26/0.74 0.17/0.83 0.15/0.85 0.13/0.87 0.13/0.87 0.13/0.87 0.13/0.87 1.5 0.36/0.64 0.28/0.72 0.26/0.74 0.19/0.81 0.18/0.82 0.15/0.85 0.15/0.85 0.15/0.85 0.15/0.85 2 0.34/0.66 0.27/0.73 0.23/0.77 0.20/0.80 0.18/0.82 0.17/0.83 0.16/0.84 0.17/0.83 0.17/0.83 8.0 0 0.12/0.88 0.09/0.91 0.08/0.92 0.05/0.95 0.05/0.95 0.04/0.96 0.03/0.97 0.03/0.97 0.03/0.97 0.5 0.13/0.87 0.10/0.90 0.09/0.91 0.07/0.93 0.06/0.94 0.05/0.95 0.05/0.95 0.05/0.95 0.06/0.94 1 0.15/0.85 0.12/0.88 0.16/0.84 0.09/0.91 0.08/0.92 0.08/0.92 0.08/0.92 0.09/0.91 0.10/0.90 1.5 0.19/0.81 0.14/0.86 0.13/0.87 0.10/0.90 0.09/0.91 0.07/0.93 0.07/0.93 0.07/0.93 0.07/0.93 2 0.24/0.76 0.18/0.82 0.16/0.84 0.11/0.89 0.11/0.89 0.09/0.91 0.09/0.91 0.09/0.91 0.09/0.91 11.3 0 0.08/0.92 0.06/0.94 0.05/0.95 0.03/0.97 0.03/0.97 0.02/0.98 0.02/0.98 0.02/0.98 0.02/0.98 0.5 0.14/0.86 0.11/0.89 0.10/0.90 0.06/0.94 0.05/0.95 0.05/0.95 0.05/0.95 0.05/0.95 0.06/0.94 1 0.20/0.80 0.13/0.87 0.11/0.89 0.07/0.93 0.06/0.94 0.05/0.95 0.05/0.95 0.05/0.95 0.05/0.95 1.5 0.18/0.82 0.24/0.76 0.19/0.81 0.13/0.87 0.10/0.90 0.09/0.91 0.08/0.92 0.08/0.92 0.08/0.92 2 0.21/0.79 0.21/0.79 0.18/0.82 0.11/0.89 0.09/0.91 0.08/0.92 0.09/0.91 0.10/0.90 0.11/0.89
下载: 导出CSV
表 2 H2O2抑制条件下的Br-消耗的一级速率常数(kobs)
Table 2. kobs of Br- consumption with the addition of H2O2
n(H2O2):n(O3) 反应时间/min 拟合方程式 k/min-1 R2 0 0~10 y = −0.033 7x−0.019 70 0.033 7 0.985 10~25 y = −0.013 4x−0.234 00 0.013 4 0.976 0.5 0~6 y = −0.022 9x−0.018 86 0.022 9 0.950 6~25 y = −0.020 0x−0.038 59 0.020 0 0.990 1 0~8 y = −0.015 4x−0.011 67 0.015 4 0.955 8~25 y = −0.018 0x+0.011 12 0.018 0 0.990 1.5 0~10 y = −0.011 0x+0.005 83 0.011 0 0.971 10~25 y = −0.017 2x+0.076 27 0.017 2 0.983 2 0~15 y = −0.005 9x+0.003 51 0.005 9 0.991 15~25 y = -0.014 5x+0.135 60 0.014 5 0.983
下载: 导出CSV
-
[1] JIN X, JIN P, HOU R, et al. Enhanced WWTP effluent organic matter removal in hybrid ozonation-coagulation (HOC) process catalyzed by Al-based coagulant[J]. Journal of Hazardous Materials, 2017, 327: 216-224. doi: 10.1016/j.jhazmat.2016.12.043 [2] JIN X, SHI Y, HOU R, et al. Role of Al-based coagulants on a hybrid ozonation–coagulation (HOC) process for WWTP effluent organic matter and ibuprofen removal[J]. Environmental Science:Water Research & Technology, 2019, 5(3): 599-608. [3] JIN X, WANG Y, ZHANG W, et al. Mechanism of the hybrid ozonation-coagulation (HOC) process: Comparison of preformed Al13 polymer and in situ formed Al species[J]. Chemosphere, 2019, 229: 262-272. doi: 10.1016/j.chemosphere.2019.04.225 [4] 刘芳蕾, 张冬, 吕锡武. 臭氧生物活性炭工艺运行中产生溴酸盐的可能性分析[J]. 中国环境科学, 2014, 34(9): 2299-2305. [5] RUFFINO B, KORSHIN G V, ZANETTI M. Use of spectroscopic indicators for the monitoring of bromate generation in ozonated wastewater containing variable concentrations of bromide[J]. Water Research, 2020, 182: 116009. doi: 10.1016/j.watres.2020.116009 [6] JIN X, XIE X, LIU Y, et al. The role of synergistic effects between ozone and coagulants (SOC) in the electro-hybrid ozonation-coagulation process[J]. Water Research, 2020, 177: 115800. doi: 10.1016/j.watres.2020.115800 [7] 郝晓地, 甘微, 李季, 等. 臭氧降解污水厂二级出水有机物作用与效果分析[J]. 中国给水排水, 2021, 37(10): 1-7. [8] HOIGNé J. Chemistry of aqueous ozone and transformation of pollutants by ozonation and advanced oxidation processes[J]. Chemistry quality and treatment of drinking water, 1998, 5(2): 83-141. [9] QI Y. Ozonation of water and waste water: A practical guide to understanding ozone and its applications[J]. International Journal of Environmental Studies, 2010, 67(5): 795-796. doi: 10.1080/00207233.2010.517628 [10] 胡航恺, 徐浩丹, 卢晓辉, 等. 水中有机物对紫外光催化还原溴酸盐的影响[J]. 中国环境科学, 2022, 42(3): 1164-1172. [11] 吴纯德, 陈芳, 李商国, 等. NH4+和H2O2抑制臭氧氧化过程中溴酸盐形成的效能研究[J]. 环境工程学报, 2011, 5(10): 2233-2236. [12] 周娟, 陈欢, 李晓璐, 等. Pd/CeO2催化水中溴酸盐的加氢还原研究[J]. 中国环境科学, 2011, 31(8): 1274-1279. [13] VON GUNTEN U, OLIVERAS Y. Kinetics of the reaction between hydrogen peroxide and hypobromous acid: Implication on water treatment and natural systems[J]. Water Research, 1997, 31(4): 900-906. doi: 10.1016/S0043-1354(96)00368-5 [14] ULRICH P, URS V G. Bromate minimization during ozonation: Mechanistic considerations[J]. Environmental Science & Technology, 2001, 35(12): 2525-2531. [15] HOFMANN R, ANDREWS R C. Ammoniacal bromamines: a review of their influence on bromate formation during ozonation[J]. Water Research, 2001, 35(3): 599-604. doi: 10.1016/S0043-1354(00)00319-5 [16] VON GUNTEN U. Ozonation of drinking water: Part II. Disinfection and by-product formation in presence of bromide, iodide or chlorine[J]. Water Research, 2003, 37(7): 1469-1487. doi: 10.1016/S0043-1354(02)00458-X [17] 王永京, 杜旭, 金萌, 等. 氨氮及H2O2对溴酸盐和消毒副产物控制的影响[J]. 环境科学, 2017, 38(2): 616-621. [18] QI S, MAO Y, LV M, et al. Pathway fraction of bromate formation during O3 and O3/H2O2 processes in drinking water treatment[J]. Chemosphere, 2016, 144: 2436-2442. doi: 10.1016/j.chemosphere.2015.11.022 [19] BADER H, HOIGNé J. Determination of ozone in water by the indigo method[J]. Water Research, 1981, 15(4): 449-456. doi: 10.1016/0043-1354(81)90054-3 [20] GALAPATE R P, BAES A U, OKADA M. Transformation of dissolved organic matter during ozonation effects on trihalomethane formation potential[J]. Water Research, 2001, 35(9): 2201-2206. doi: 10.1016/S0043-1354(00)00489-9 [21] ZHANG T, CHEN W, MA J, et al. Minimizing bromate formation with cerium dioxide during ozonation of bromide-containing water[J]. Water Research, 2008, 42(14): 3651-3658. doi: 10.1016/j.watres.2008.05.021 [22] WEN G, PAN Z-H, MA J, et al. Reuse of sewage sludge as a catalyst in ozonation: Efficiency for the removal of oxalic acid and the control of bromate formation[J]. Journal of Hazardous Materials, 2012, 239-240: 381-388. doi: 10.1016/j.jhazmat.2012.09.016 [23] PSALTOU S, KARAPATIS A, MITRAKAS M, et al. The role of metal ions on p-CBA degradation by catalytic ozonation[J]. Journal of Environmental Chemical Engineering, 2019, 7(5): 103324. doi: 10.1016/j.jece.2019.103324 [24] ELOVITZ M S, URS V G. Hydroxyl radical/ozone ratios during ozonation processes. I. The Rct concept[J]. Ozone:Science & Engineering, 1999, 21(3): 239-260. [25] WANG Y, MAN T, ZHANG R, et al. Effects of organic matter, ammonia, bromide, and hydrogen peroxide on bromate formation during water ozonation[J]. Chemosphere, 2021, 285: 131352. doi: 10.1016/j.chemosphere.2021.131352 [26] YU J, WANG Y, WANG Q, et al. Implications of bromate depression from H2O2 addition during ozonation of different bromide-bearing source waters[J]. Chemosphere, 2020, 252: 126596. doi: 10.1016/j.chemosphere.2020.126596 [27] MIZUNO T, OHARA S, NISHIMURA F, et al. O3/H2O2 process for both removal of odorous algal-derived compounds and control of bromate ion formation[J]. Ozone:Science & Engineering, 2011, 33(2): 121-135. [28] ACERO J L, HADERLEIN S B, SCHMIDT T C, et al. MTBE oxidation by conventional ozonation and the combination ozone/hydrogen peroxide: Efficiency of the processes and bromate formation[J]. Environmental Science & Technology, 2001, 35(21): 4252-4259. [29] LEGUBE B, PARINET B, GELINET K, et al. Modeling of bromate formation by ozonation of surface waters in drinking water treatment[J]. Water Research, 2004, 38(8): 2185-2195. doi: 10.1016/j.watres.2004.01.028 [30] 强晨. 臭氧/过一硫酸盐组合工艺氧化含溴水过程溴酸盐的形成规律与控制方法[D]. 西安: 西安建筑科技大学, 2019. [31] DAVID YAO C C, HAAG W R. Rate constants for direct reactions of ozone with several drinking water contaminants[J]. Water Research, 1991, 25(7): 761-773. doi: 10.1016/0043-1354(91)90155-J [32] JEKEL M, ZUCKER I, HüBNER U. Options and limitations of hydrogen peroxide addition to enhance radical formation during ozonation of secondary effluents[J]. Journal of Water Reuse and Desalination, 2015, 5(1): 8-16. doi: 10.2166/wrd.2014.036 [33] 韩帮军, 马军, 张涛, 等. 臭氧催化氧化抑制溴酸盐生成效能的研究[J]. 环境科学, 2008, 29(3): 665-670. doi: 10.3321/j.issn:0250-3301.2008.03.021 [34] PINKERNELL U, LüKE, HANS J, et al. Selective photometric determination of peroxycarboxylic acids in the presence of hydrogen peroxide[J]. Analyst, 1997, 122(6): 567-571. doi: 10.1039/a700509a -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2541
- HTML全文浏览数: 2541
- PDF下载数: 121
- 施引文献: 0
