


 下载:
下载:

-
药品和个人护理品类污染物(如内分泌干扰物、抗生素等)导致的水体污染问题是生态安全和人类身体健康的一大威胁[1-3]。基于过硫酸盐的高级氧化技术可通过产生羟基自由基(OH•)、硫酸根自由基(SO4•−)、单线态氧(1O2)等活性氧物种降解水中多种类型的有机污染物,在药、护品类污染物处理方面受到了广泛关注[1]。与过一硫酸盐(peroxymonosulfate,PMS)相比,过二硫酸盐(peroxydisulfate,PDS)价格低廉、水溶性好、稳定性高便于储存和运输,已被广泛用于催化降解有机污染物[1]。PDS可通过热活化、碱活化、金属离子催化、金属氧化物催化等多种方法活化,其中基于纳米金属氧化物(如Co3O4等)的异相催化由于催化剂价格低廉、反应条件温和、催化活性高等优点,是目前研究的一个重要方向[2]。然而,纳米尺寸的金属氧化物分散于水中易团聚,导致催化剂活性位点暴露有限,并且分散于水中的纳米催化剂还存在难以分离和回收的问题,极大地限制了其应用。
将金属氧化物纳米催化剂负载于活性炭、泡沫金属、矿物颗粒以及陶瓷膜等三维载体的表面,不仅可以有效缓解氧化物纳米颗粒的团聚问题,而且便于分离和回收,对纳米金属氧化物活化过硫酸盐降解有机物具有重要意义[4]。现阶段,金属氧化物主要通过浆料涂覆的方式负载于载体表面,合成方法简单但稳定性较差[5]。添加高分子粘结剂可增强其稳定性,但催化体系中引入导电性较差的高分子不利于过硫酸活化过程中电子的转移,同时添加粘结剂也可能会带来二次污染[6]。因此,亟需发展一种可将金属氧化物原位负载于三维载体表面的绿色合成方法。植物多酚如单宁酸(tannic acid,TA)普遍存在于植物的根、茎、叶及果实中,是一种价格低廉的生物质[7]。TA分子富含邻二酚羟基,具有较强的金属离子络合能力和优异的表面粘附性能,可与多种金属离子(Co2+、Fe2+、Cu2+等)在聚乙烯球、陶瓷、玻璃等不同载体表面配位形成金属-多酚配合物[7]。作为由金属离子和有机配体组成的复合物,金属-多酚配合物可进一步热解制成不同种类的金属氧化物,并且研究表明有机配体TA分解还会产生多孔结构而形成多孔结构的金属氧化物[8]。
基于TA分子的金属离子络合能力和表面粘附性能,本研究以具有稳定物理化学性质且价格低廉的气泡石为载体,通过在其表面包覆Co2+与TA的配合物,进一步热解将纳米尺寸的Co3O4原位负载于气泡石表面,制备易分离回收的负载型Co3O4催化剂,通过SEM、XRD、ICP等详细分析了负载型Co3O4的结构和组成。利用负载型Co3O4催化剂活化PDS降解水中的双酚A(bis-phenol A,BPA)、磺胺甲恶唑(sulfamethoxazole,SMX)等药、护品类有机污染物,分析了溶液初始pH、PDS浓度、催化剂投加量、共存化学组分等对有机物降解性能的影响,并通过电子顺磁共振分析、化学淬灭实验、光谱分析等手段深入分析了PDS的活化机制和有机物的降解机理。
-
主要试剂包括六水合硝酸钴(Co(NO3)2·6H2O)、单宁酸(tannic acid)、双酚A(bisphenol A)、盐酸(HCl,1 mol·L−1)、过硫酸钠(sodium persulfate, Na2S2O8)、氢氧化钠(NaOH,1 mol·L−1)、叔丁醇(tert-butyl alcohol, TBA)、对苯醌(p-benzoquinone, BQ)、碳酸钠(Na2CO3)、氯化钠(NaCl)、硝酸钠(NaNO3)、硫酸钠(Na2SO4)、腐殖酸(HA)、甲醇、乙腈、无水乙醇等,所有试剂均为分析纯,实验用水为超纯水。
-
1)负载型Co3O4的制备:将尺寸0.5 cm×0.5 cm×0.5 cm的气泡石颗粒置于Co(NO3)2与TA的混合溶液中,利用TA分子的表面粘附特性及其与Co2+的配位反应,在气泡石颗粒表面形成Co-TA包覆层,进一步在空气气氛下焙烧制得负载于气泡石表面的Co3O4催化剂。具体步骤如下:将5 g Co(NO3)2和10 g TA依次溶解于250 mL纯水,然后加入30 g气泡石载体搅拌30 min,加入5 mL氨水后继续搅拌1 h,将修饰后的气泡石用纯水反复清洗。重复上述负载步骤4次后,制得负载有Co-TA配合物的气泡石颗粒,充分干燥后将其置于马弗炉中在400 °C下焙烧2 h (升温速率设置为2 ℃·min−1),即制得负载型Co3O4催化剂。
2)材料表征:通过场发射扫描电子显微镜(Hitachi, S-4800)分析负载型Co3O4的形貌与元素组成,利用Rigaku Ultimate IV型X射线衍射(XRD)表征其晶体结构,借助X射线光电子能谱(XPS)研究催化剂反应前后各元素的化学价态,采用电感耦合等离子体发射光谱仪(ICP, NexION 350D)测定浸出钴离子的浓度以及气泡石颗粒表面Co3O4的负载量。ATR-FTIR测试以超纯水为背景,将PDS与Co3O4纳米催化剂的混合物滴至ATR附件晶体表面,在400~3000 cm−1内至少扫描3次,扫描分辨率为4 cm−1。
3)BPA降解实验:本研究通过催化降解BPA评价负载型Co3O4活化PDS的性能。室温条件下,配制50 mL 0.04 mmol·L−1的BPA溶液,加入3.0 g负载型Co3O4催化剂和0.5 mL 40 mmol·L−1的PDS溶液,然后于特定时间取1 mL水样与0.5 mL甲醇混合以终止反应,进一步用0.22 μm微孔滤膜过滤后,通过高效液相色谱仪测定BPA的浓度,流动相为水和乙腈的混合溶液,两者的体积比为1:1,流速为1 mL·min−1。
-
图1为气泡石载体负载Co3O4前后的照片及其表面Co3O4纳米催化剂的微观结构。如图1(a)所示,气泡石表面粗糙呈白色,负载Co3O4后变为灰褐色(图1(b)),表明Co3O4纳米催化剂成功负载于其表面。通过SEM表征了气泡石表面Co3O4纳米催化剂的微观结构,如图1(c)所示,气泡石光滑的表面存在大量的纳米颗粒团簇,进一步表明Co3O4纳米催化剂的成功负载。纳米团簇的高倍SEM图表明Co3O4纳米催化剂呈多孔结构(图1(d)),这可能是Co-TA配合物中有机组分TA分解导致的。多孔结构利于催化位点的充分暴露,利于PDS的活化和有机污染物的降解[9]。
图2(a)为Co-TA焙烧衍生粉体Co3O4及气泡石负载Co3O4的XRD谱图。粉体催化剂在2θ为31.27°、36.85°、44.8°、59.35°、65.23°的衍射峰分别对应于立方相Co3O4的(220)、(311)、(400)、(511)和(440)晶面(JCPDS-no.42-1467)。气泡石的XRD图谱表明其主要成分为SiO2 (JCPDS-no.78-2315);SiO2化学性质稳定,便于在复杂环境中使用。气泡石表面负载催化剂后,在2θ为31.27°、44.81°检测到了属于Co3O4的衍射峰,分别对应于立方相Co3O4的(220)和(400)晶面,进一步表明Co3O4成功负载于气泡石表面。通过热重分析研究了Co-TA配合物的热分解过程。如图2(b)所示,Co-TA配合物的残余质量在20~400 °C内持续降低,这主要归咎配合物表面水分子的脱附以及有机配体TA的氧化分解[10]。DTA分析表明Co-TA分解过程中在380 °C出现了明显的吸热峰,表明Co-TA热解生成了新的固相。400 °C后配合物的质量损失保持不变,表明Co-TA中的有机配体在400 °C下完全分解,因此本研究将负载型Co3O4催化剂的合成温度设定为400 °C。通过ICP-MS分析了气泡石表面Co3O4的负载量,结果表明每克气泡石表面负载有1.25 mg的Co3O4纳米颗粒。
-
在反应温度为(25±1) °C、负载型Co3O4投加量60 g·L−1(即0.075 g·L−1 Co3O4纳米颗粒)、BPA初始浓度为0.04 mmol·L−1、PDS初始浓度为0.4 mmol·L−1和pH为7的条件下测试了负载型Co3O4活化PDS降解BPA的性能。如图3(a)所示,PDS氧化降解BPA的能力有限,120 min内BPA的去除率仅为9%,同时负载型Co3O4仅吸附了4.8%的BPA。负载型Co3O4活化PDS可有效降解BPA,120 min内BPA的去除率达到了100%。BPA的降解遵循拟一级反应动力学,反应动力学常数为0.04 min−1 (图3(b))。ICP分析表明催化反应结束后溶液中Co2+的质量浓度为0.55 mg·L−1,低于地表水环境质量标准(GB 3838-2002D)规定的标准(1.0 mg·L−1),表明负载型Co3O4具有良好的稳定性。溶出的Co2+在相同实验条件下催化PDS仅降解了11%的BPA,表明负载型Co3O4主要通过异相催化反应降解有机物[10]。图3(b)反映了负载型Co3O4活化PDS降解不同类型有机物的性能,苯酚(Phenol)、对乙酰氨基酚(AAP)、磺胺甲恶唑(SMX)均可被有效去除,相同反应条件下120 min内的去除率分别为78.2%、93.6%、67.3%,对应的反应动力学常数分别为0.01、0.02、0.01 min−1(图3(d))。以上结果表明负载型Co3O4活化PDS可催化降解不同类型的药、护品类有机污染物。
-
图4(a)反映了溶液初始pH分别为3、5、7、9、11时,负载型Co3O4活化PDS降解BPA的性能。在pH=3~9内,BPA均可得到有效去除,120 min内BPA的去除率均达到100%,同时反应速率也并未受到显著影响;然而,当pH提升至11时,BPA的降解受到抑制,120 min内BPA的去除率降低了24.4%,反应速率也由中性时的0.04 min−1降至0.01 min−1,这可能是由于碱性条件下OH•的氧化还原电位降低所致[11]。图4(b)为不同PDS投加量下,负载型Co3O4对BPA的催化降解性能。当PDS浓度由0.1 mmol·L−1增加到0.4 mmol·L−1时,BPA的去除率从20.9%增加到98.2%,反应速率也由0.002 min−1提升至0.03 min−1。增加PDS的浓度可增加活性氧物种的生成量,进而促进有机物的催化降解[12]。然而,进一步增加PDS的浓度至0.8 mmol·L−1和1.6 mmol·L−1时,BPA的降解性能并未得到进一步提升。这可能是由于负载型Co3O4的催化位点有限,不能完全活化反应体系中的PDS[13]。增加催化剂的投加量可提供更多的催化位点而充分活化PDS,进而产生更多的活性氧物种而显著提升有机物的降解效率[14]。如图4(c)所示,在BPA溶液体积为50 mL的情况下,当负载型Co3O4催化剂的投加量由1.5 g增加至3.0 g时,BPA的降解速率由0.02 min−1提高至0.037 min−1,当催化剂的投加量进一步增加至6.0 g时,BPA在60 min即可被完全去除,降解速率达0.039 min−1。这表明增加负载型Co3O4的剂量可显著提升PDS的有效利用率。
为考察水中常见无机阴离子对负载型Co3O4活化PDS降解有机物性能的影响,研究了Cl−、NO3−、CO32−对BPA去除效率和降解速率的影响。如图4(d)所示,在负载型Co3O4/PDS体系中加入10 mmol·L−1 Cl−并未降低BPA的去除效率,同样加入10 mmol·L−1的NO3−也未影响BPA的去除。Co3O4活化PDS降解有机物主要有2种途径:一种是基于SO4•−和OH•的自由基氧化;另一种是基于单线态氧(1O2)的非自由基氧化[15]。在自由基途径中,Cl−、NO3−等无机阴离子会竞争消耗强氧化性的SO4•−和OH•,进而抑制有机污染物的降解[16-18]。在本研究中,Cl−和NO3−对BPA的去除影响较弱,表明自由基氧化不是Co3O4活化PDS降解有机物的主要途径。然而,向反应体系中添加10 mmol·L−1的CO32−可显著抑制BPA的去除,同样反应条件下BPA仅去除了16.64%,同时反应速率由0.04 min−1降低至了0.001 min−1。CO32−是一种常用的1O2淬灭剂,其对BPA降解的显著抑制表明负载型Co3O4活化PDS降解有机物是1O2主导的非自由途径[19]。除了无机阴离子,天然有机物如腐殖酸(HA)也是一种常见的水体背景成分[6]。在10 mg·L−1 HA存在的情况下,负载型Co3O4催化降解有机物的效率也未受到显著影响,进一步说明本反应体系中有机物的降解为非自由基途径[20]。综上所述,负载型Co3O4/PDS体系不仅能够在较宽的pH范围内降解有机物,而且对无机阴离子和腐殖酸等常见的水体背景成分具有较强的抗干扰能力,因此,有着较强的应用潜力。
-
为明确负载型Co3O4活化PDS降解有机物的机理,通过EPR鉴定分析了该体系产生的活性氧的种类。如图5(a)所示,当仅有PDS时,以DMPO为自旋捕获剂未检测到任何信号,而当加入负载型Co3O4催化剂后,EPR谱图观测到了峰强度为1: 2 : 2 : 1的特征峰,这是催化产生的OH•被DMPO捕获所致,表明负载型Co3O4活化PDS产生了OH•[21]。此外,以TEMP为1O2捕获剂还检测到了TEMP-1O2的特征峰,因此,负载型Co3O4活化PDS也产生了1O2[22]。进一步通过化学淬灭实验判别了反应体系中OH•和1O2对BPA降解的贡献。图5(b)反映了不同浓度MeOH的淬灭效果,可以看出MeOH对BPA的降解影响有限,在1 000 mmol·L−1 MeOH存在的情况下,BPA的去除率仍高达94.7%。MeOH可与SO4•−和OH•快速反应,反应速率常数分别为k 分别为 2.5×107 (mol·s)−1和9.7×108 (mol·s)−1,是常用的自由基淬灭剂;MeOH对负载型Co3O4/PDS有限的淬灭能力表明该体系降解有机物的过程中,自由基的氧化作用有限[7]。然而,L-组氨酸可显著抑制BPA的降解,如图5(c)所示,1 mmol·L−1的组氨酸使得BPA的降解率由100%降低至89%;当L-组氨酸的浓度增加至5 mmol·L−1时,BPA的去除率进一步降低至21.98%,反应速率也由0.04 min−1降低至0.002 min−1。L-组氨酸具有一定的还原性,可能会消耗PDS而降低负载型Co3O4催化降解BPA的能力[23]。为排除L-组氨酸消耗PDS对淬灭实验的干扰,研究了不同浓度L-组氨酸对PDS的降解效率。如图5(d)所示,L-组氨酸分解PDS的能力有限,例如5 mmol·L−1的L-组氨酸在120 min内仅分解了5.8%的PDS。因此,L-组氨酸对负载型Co3O4/PDS降解BPA的抑制作用主要归咎于其对活性氧物种的淬灭,而L-组氨酸是常用的1O2淬灭剂,这表明负载型Co3O4活化PDS降解有机物是以1O2为主导的非自由基氧化途径。过渡金属氧化物活化PDS产生1O2已被广泛报道,有研究表明,O2•−是1O2生成的重要中间体(
2O2·−+2H2O→21O2+2OH−+H2O2 )[24]。本研究中,化学淬灭实验结果表明BQ(一种常用的O2•−淬灭剂)[25]可显著抑制BPA的降解。如图5(e)所示,加入5 mmol·L−1 BQ后BPA的去除率由100%降低至48.4%,同时速率常数由0.04 min−1降至0.008 min−1(图5(f)),表明O2•−的淬灭可显著抑制BPA的降解。O2•−的氧化还原电位较低,本身不能氧化分解有机物[24],而1O2是本研究中BPA降解的主要活性物种,这说明O2•−是1O2形成的中间体,也就是说负载型Co3O4活化PDS先生成了O2•−,O2•−再进一步反应生成1O2。 -
过渡金属氧化物主要通过金属离子的价态循环活化过硫酸盐[26]。为解析负载型多孔Co3O4活化PDS的机理,通过XPS分析了Co3O4中Co元素的价态及其反应前后含量的变化。如图6(a)所示,Co2p在794.9 eV和779.7 eV的特征峰分别对应于Co2p1/2和Co2p3/2,而Co2p3/2的高分辨XPS谱图拟合结果表明,781.2 eV和779.7 eV处的峰分别对应于Co2+和Co3+,所占比例分别为34.2%和54.5%。反应后,Co3O4表面Co2+、Co3+比例变化表明Co3+/Co2+的氧化还原参与了PDS的活化[27]。进一步通过原位ATR-FTIR分析研究了PDS分子在负载型Co3O4表面的反应机理。如图6(b)所示,在1 050 cm−1和1 272 cm−1处检测到PDS振动峰,加入Co3O4催化剂后在其表面观测到了PDS的特征峰的位置并且未发生偏移,表明PDS吸附于Co3O4催化剂的表面。此外,在Co3O4/PDS体系中还观测到了SO42‒的特征峰(1 107 cm−1),表明Co3O4活化PDS产生了SO42‒;随着反应时间的增加PDS特征峰的强度逐渐降低,而SO42‒特征峰的强度逐渐增加,表明PDS在Co3O4催化剂表面持续分解[1]。
金属氧化物(MOx)活化过硫酸盐时,吸附于其表面的水分子会解离形成MOx-OH[19]。因此,结合XPS和ATR-FTIR分析,提出了负载型Co3O4活化PDS降解BPA的可能反应机理(图7)。首先,PDS分子通过氢键与[≡Co3+—OH]2+结合形成[≡Co3+—O—O—SO3]+络合物(式(1)),该络合物具有较高的氧化电势,还原PDS分子生成O2•−,同时Co3+被还原为Co2+形成中间态[≡Co2+—OH] (式(2)),随之生成的O2•−进一步反应生成1O2 (式(3))。中间态[≡Co2+—O—O—SO3]通过内电子转移被氧化为≡Co3+—OH,同时生成少量的自由基(式(4))。如上所述,负载型Co3O4/PDS体系中自由基对有机物的降解贡献有限,因此1O2进一步氧化有机物将其分解为中间产物或者H2O和CO2 (式(5))。本研究中,负载型Co3O4有效活化PDS主要有2点原因,一是负载型Co3O4避免了纳米催化剂的团聚,可暴露更多的活性位点而提升催化效率;另一方面,Co-TA配合物热解会产生富含氧缺陷的Co3O4[28],而氧缺陷可有效提升PDS分子在Co3O4表面的吸附,也会提升内电子催化PDS的效能。
催化剂的稳定性是衡量其实际应用潜力的一个重要因素,本研究通过循环降解实验研究了负载型Co3O4的稳定性。如图8(a)所示,3次循环后负载型Co3O4对BPA的去除率仍保持在89%,表明负载型Co3O4具有良好的稳定性。循环降解过程中催化活性的轻微降低可能是BPA的降解产物堵塞活性位点导致的[10]。此外,XRD分析表明循环使用后负载型Co3O4的晶相没有明显变化(图2(a)),进一步说明负载型Co3O4具有良好的稳定性。负载型Co3O4可有效矿化BPA(图8(b)),TOC去除率高达73%,而且3次循环后TOC的去除率仍高达63%,表明负载型Co3O4可持续稳定地处理水中酚类有机污染物。
-
1)基于植物多酚的界面配位原理,通过在三维矿物载体表面原位负载Co3O4纳米颗粒制得了易分离回收的负载型Co3O4催化剂,比Co3O4粉体催化剂展现出更优异的催化活性和良好的稳定性。
2)负载型Co3O4催化剂活化PDS可高效降解BPA、SMX、AAP等多种类型的药、护品类有机污染物,中性条件下,负载型Co3O4投加量为60 g·L−1(即0.075 g·L−1 Co3O4纳米颗粒),在BPA/PDS摩尔比为1:10的情况下可在120 min内将BPA完全降解。
3)负载型Co3O4主要通过内电子转移活化PDS,降解有机物时自由基氧化的作用有限,主要是1O2主导的非自由基氧化途径,对无机阴离子和腐殖酸等水体背景成分有着较强的抗干扰能力,在实际水体处理方面展现出广阔的应用前景。
负载型Co3O4活化过二硫酸盐非自由基降解有机污染物
Nonradical oxidation of bis-phenol A by peroxydisulfate activated over immobilized Co3O4 nanoparticles
-
摘要: 通过在三维矿物载体表面包覆金属-多酚配合物并煅烧实现了Co3O4纳米颗粒的原位负载,制得了易分离回收的负载型Co3O4催化剂,利用SEM、XRD以及XPS表征分析其形貌和微观结构,采用活化过二硫酸盐(PDS)降解药、护品类有机污染物以评价其催化性能。以双酚A为目标污染物,考察了初始pH、PDS浓度、催化剂投加量、共存阴离子(CO32−、SO42−、NO3−、Cl−)以及腐殖酸(HA)对BPA降解效率的影响。结果表明,负载型Co3O4能有效活化PDS降解有机物,在Co3O4投加量0.075 g·L−1,BPA初始浓度0.04 mmol·L−1,PDS初始浓度0.4 mmol·L−1以及初始pH=7的最优条件下,120 min内BPA可被完全去除。淬灭实验与EPR实验结果表明,负载型Co3O4活化PDS通过单线态氧(1O2)主导的非自由基途径氧化分解有机物。Abstract: Nanosized Co3O4 particles were in situ grown on the surface of three-dimensional mineral substrates by calcining surface-coated metal-phenolic coordination polymer. Immobilizing Co3O4 nanoparticles on the mineral substrate allows facile separation and recovery. SEM, XRD and XPS were performed to characterize the morphology and microstructure of the immobilized Co3O4 nanoparticles. The immobilized Co3O4 was applied to active peroxydisulfate (PDS) to degrade pharmaceutical and personal care products in water. The influence of initial pH, PDS concentration, catalyst dosage, inorganic ions (CO32−, SO42−, NO3−, Cl−) and humic acid (HA) on organics degradation was systematically investigated by using bis-phenol A(BPA) as a probe contaminant. The results showed that the immobilized Co3O4 could effectively active PDS to degrade BPA. Under the optimized conditions with a catalyst dosage of 0.075 g·L−1, PDS concentration of 0.4 mM, BPA with initial concentration of 0.04 mM and pH7 was completely removed within 120 min. Chemical scavenging experiments and EPR tests revealed the degradation of BPA followed a nonradical oxidation pathway based on singe oxygen (1O2).
-
药品和个人护理品类污染物(如内分泌干扰物、抗生素等)导致的水体污染问题是生态安全和人类身体健康的一大威胁[1-3]。基于过硫酸盐的高级氧化技术可通过产生羟基自由基(OH•)、硫酸根自由基(SO4•−)、单线态氧(1O2)等活性氧物种降解水中多种类型的有机污染物,在药、护品类污染物处理方面受到了广泛关注[1]。与过一硫酸盐(peroxymonosulfate,PMS)相比,过二硫酸盐(peroxydisulfate,PDS)价格低廉、水溶性好、稳定性高便于储存和运输,已被广泛用于催化降解有机污染物[1]。PDS可通过热活化、碱活化、金属离子催化、金属氧化物催化等多种方法活化,其中基于纳米金属氧化物(如Co3O4等)的异相催化由于催化剂价格低廉、反应条件温和、催化活性高等优点,是目前研究的一个重要方向[2]。然而,纳米尺寸的金属氧化物分散于水中易团聚,导致催化剂活性位点暴露有限,并且分散于水中的纳米催化剂还存在难以分离和回收的问题,极大地限制了其应用。
将金属氧化物纳米催化剂负载于活性炭、泡沫金属、矿物颗粒以及陶瓷膜等三维载体的表面,不仅可以有效缓解氧化物纳米颗粒的团聚问题,而且便于分离和回收,对纳米金属氧化物活化过硫酸盐降解有机物具有重要意义[4]。现阶段,金属氧化物主要通过浆料涂覆的方式负载于载体表面,合成方法简单但稳定性较差[5]。添加高分子粘结剂可增强其稳定性,但催化体系中引入导电性较差的高分子不利于过硫酸活化过程中电子的转移,同时添加粘结剂也可能会带来二次污染[6]。因此,亟需发展一种可将金属氧化物原位负载于三维载体表面的绿色合成方法。植物多酚如单宁酸(tannic acid,TA)普遍存在于植物的根、茎、叶及果实中,是一种价格低廉的生物质[7]。TA分子富含邻二酚羟基,具有较强的金属离子络合能力和优异的表面粘附性能,可与多种金属离子(Co2+、Fe2+、Cu2+等)在聚乙烯球、陶瓷、玻璃等不同载体表面配位形成金属-多酚配合物[7]。作为由金属离子和有机配体组成的复合物,金属-多酚配合物可进一步热解制成不同种类的金属氧化物,并且研究表明有机配体TA分解还会产生多孔结构而形成多孔结构的金属氧化物[8]。
基于TA分子的金属离子络合能力和表面粘附性能,本研究以具有稳定物理化学性质且价格低廉的气泡石为载体,通过在其表面包覆Co2+与TA的配合物,进一步热解将纳米尺寸的Co3O4原位负载于气泡石表面,制备易分离回收的负载型Co3O4催化剂,通过SEM、XRD、ICP等详细分析了负载型Co3O4的结构和组成。利用负载型Co3O4催化剂活化PDS降解水中的双酚A(bis-phenol A,BPA)、磺胺甲恶唑(sulfamethoxazole,SMX)等药、护品类有机污染物,分析了溶液初始pH、PDS浓度、催化剂投加量、共存化学组分等对有机物降解性能的影响,并通过电子顺磁共振分析、化学淬灭实验、光谱分析等手段深入分析了PDS的活化机制和有机物的降解机理。
1. 材料与方法
1.1 实验试剂
主要试剂包括六水合硝酸钴(Co(NO3)2·6H2O)、单宁酸(tannic acid)、双酚A(bisphenol A)、盐酸(HCl,1 mol·L−1)、过硫酸钠(sodium persulfate, Na2S2O8)、氢氧化钠(NaOH,1 mol·L−1)、叔丁醇(tert-butyl alcohol, TBA)、对苯醌(p-benzoquinone, BQ)、碳酸钠(Na2CO3)、氯化钠(NaCl)、硝酸钠(NaNO3)、硫酸钠(Na2SO4)、腐殖酸(HA)、甲醇、乙腈、无水乙醇等,所有试剂均为分析纯,实验用水为超纯水。
1.2 实验方法
1)负载型Co3O4的制备:将尺寸0.5 cm×0.5 cm×0.5 cm的气泡石颗粒置于Co(NO3)2与TA的混合溶液中,利用TA分子的表面粘附特性及其与Co2+的配位反应,在气泡石颗粒表面形成Co-TA包覆层,进一步在空气气氛下焙烧制得负载于气泡石表面的Co3O4催化剂。具体步骤如下:将5 g Co(NO3)2和10 g TA依次溶解于250 mL纯水,然后加入30 g气泡石载体搅拌30 min,加入5 mL氨水后继续搅拌1 h,将修饰后的气泡石用纯水反复清洗。重复上述负载步骤4次后,制得负载有Co-TA配合物的气泡石颗粒,充分干燥后将其置于马弗炉中在400 °C下焙烧2 h (升温速率设置为2 ℃·min−1),即制得负载型Co3O4催化剂。
2)材料表征:通过场发射扫描电子显微镜(Hitachi, S-4800)分析负载型Co3O4的形貌与元素组成,利用Rigaku Ultimate IV型X射线衍射(XRD)表征其晶体结构,借助X射线光电子能谱(XPS)研究催化剂反应前后各元素的化学价态,采用电感耦合等离子体发射光谱仪(ICP, NexION 350D)测定浸出钴离子的浓度以及气泡石颗粒表面Co3O4的负载量。ATR-FTIR测试以超纯水为背景,将PDS与Co3O4纳米催化剂的混合物滴至ATR附件晶体表面,在400~3000 cm−1内至少扫描3次,扫描分辨率为4 cm−1。
3)BPA降解实验:本研究通过催化降解BPA评价负载型Co3O4活化PDS的性能。室温条件下,配制50 mL 0.04 mmol·L−1的BPA溶液,加入3.0 g负载型Co3O4催化剂和0.5 mL 40 mmol·L−1的PDS溶液,然后于特定时间取1 mL水样与0.5 mL甲醇混合以终止反应,进一步用0.22 μm微孔滤膜过滤后,通过高效液相色谱仪测定BPA的浓度,流动相为水和乙腈的混合溶液,两者的体积比为1:1,流速为1 mL·min−1。
2. 结果与讨论
2.1 负载型Co3O4催化剂的表征
图1为气泡石载体负载Co3O4前后的照片及其表面Co3O4纳米催化剂的微观结构。如图1(a)所示,气泡石表面粗糙呈白色,负载Co3O4后变为灰褐色(图1(b)),表明Co3O4纳米催化剂成功负载于其表面。通过SEM表征了气泡石表面Co3O4纳米催化剂的微观结构,如图1(c)所示,气泡石光滑的表面存在大量的纳米颗粒团簇,进一步表明Co3O4纳米催化剂的成功负载。纳米团簇的高倍SEM图表明Co3O4纳米催化剂呈多孔结构(图1(d)),这可能是Co-TA配合物中有机组分TA分解导致的。多孔结构利于催化位点的充分暴露,利于PDS的活化和有机污染物的降解[9]。
 图 1 气泡石与气泡石负载Co3O4的电子照片与SEM分析Figure 1. Digital images of airstone with and without Co3O4 and their SEM analysis
图 1 气泡石与气泡石负载Co3O4的电子照片与SEM分析Figure 1. Digital images of airstone with and without Co3O4 and their SEM analysis图2(a)为Co-TA焙烧衍生粉体Co3O4及气泡石负载Co3O4的XRD谱图。粉体催化剂在2θ为31.27°、36.85°、44.8°、59.35°、65.23°的衍射峰分别对应于立方相Co3O4的(220)、(311)、(400)、(511)和(440)晶面(JCPDS-no.42-1467)。气泡石的XRD图谱表明其主要成分为SiO2 (JCPDS-no.78-2315);SiO2化学性质稳定,便于在复杂环境中使用。气泡石表面负载催化剂后,在2θ为31.27°、44.81°检测到了属于Co3O4的衍射峰,分别对应于立方相Co3O4的(220)和(400)晶面,进一步表明Co3O4成功负载于气泡石表面。通过热重分析研究了Co-TA配合物的热分解过程。如图2(b)所示,Co-TA配合物的残余质量在20~400 °C内持续降低,这主要归咎配合物表面水分子的脱附以及有机配体TA的氧化分解[10]。DTA分析表明Co-TA分解过程中在380 °C出现了明显的吸热峰,表明Co-TA热解生成了新的固相。400 °C后配合物的质量损失保持不变,表明Co-TA中的有机配体在400 °C下完全分解,因此本研究将负载型Co3O4催化剂的合成温度设定为400 °C。通过ICP-MS分析了气泡石表面Co3O4的负载量,结果表明每克气泡石表面负载有1.25 mg的Co3O4纳米颗粒。
 图 2 负载型Co3O4和粉体Co3O4的XRD以及Co-TA配合物的热解分析Figure 2. XRD patterns for powdery Co3O4 and supported Co3O4 and TG curve for Co-TA polymer
图 2 负载型Co3O4和粉体Co3O4的XRD以及Co-TA配合物的热解分析Figure 2. XRD patterns for powdery Co3O4 and supported Co3O4 and TG curve for Co-TA polymer2.2 负载型Co3O4的催化性能研究
在反应温度为(25±1) °C、负载型Co3O4投加量60 g·L−1(即0.075 g·L−1 Co3O4纳米颗粒)、BPA初始浓度为0.04 mmol·L−1、PDS初始浓度为0.4 mmol·L−1和pH为7的条件下测试了负载型Co3O4活化PDS降解BPA的性能。如图3(a)所示,PDS氧化降解BPA的能力有限,120 min内BPA的去除率仅为9%,同时负载型Co3O4仅吸附了4.8%的BPA。负载型Co3O4活化PDS可有效降解BPA,120 min内BPA的去除率达到了100%。BPA的降解遵循拟一级反应动力学,反应动力学常数为0.04 min−1 (图3(b))。ICP分析表明催化反应结束后溶液中Co2+的质量浓度为0.55 mg·L−1,低于地表水环境质量标准(GB 3838-2002D)规定的标准(1.0 mg·L−1),表明负载型Co3O4具有良好的稳定性。溶出的Co2+在相同实验条件下催化PDS仅降解了11%的BPA,表明负载型Co3O4主要通过异相催化反应降解有机物[10]。图3(b)反映了负载型Co3O4活化PDS降解不同类型有机物的性能,苯酚(Phenol)、对乙酰氨基酚(AAP)、磺胺甲恶唑(SMX)均可被有效去除,相同反应条件下120 min内的去除率分别为78.2%、93.6%、67.3%,对应的反应动力学常数分别为0.01、0.02、0.01 min−1(图3(d))。以上结果表明负载型Co3O4活化PDS可催化降解不同类型的药、护品类有机污染物。
 图 3 负载型Co3O4活化PDS降解不同有机物的性能Figure 3. Degradation of different organic pollutants by PDS activated over supported Co3O4
图 3 负载型Co3O4活化PDS降解不同有机物的性能Figure 3. Degradation of different organic pollutants by PDS activated over supported Co3O42.3 实验因素的影响
图4(a)反映了溶液初始pH分别为3、5、7、9、11时,负载型Co3O4活化PDS降解BPA的性能。在pH=3~9内,BPA均可得到有效去除,120 min内BPA的去除率均达到100%,同时反应速率也并未受到显著影响;然而,当pH提升至11时,BPA的降解受到抑制,120 min内BPA的去除率降低了24.4%,反应速率也由中性时的0.04 min−1降至0.01 min−1,这可能是由于碱性条件下OH•的氧化还原电位降低所致[11]。图4(b)为不同PDS投加量下,负载型Co3O4对BPA的催化降解性能。当PDS浓度由0.1 mmol·L−1增加到0.4 mmol·L−1时,BPA的去除率从20.9%增加到98.2%,反应速率也由0.002 min−1提升至0.03 min−1。增加PDS的浓度可增加活性氧物种的生成量,进而促进有机物的催化降解[12]。然而,进一步增加PDS的浓度至0.8 mmol·L−1和1.6 mmol·L−1时,BPA的降解性能并未得到进一步提升。这可能是由于负载型Co3O4的催化位点有限,不能完全活化反应体系中的PDS[13]。增加催化剂的投加量可提供更多的催化位点而充分活化PDS,进而产生更多的活性氧物种而显著提升有机物的降解效率[14]。如图4(c)所示,在BPA溶液体积为50 mL的情况下,当负载型Co3O4催化剂的投加量由1.5 g增加至3.0 g时,BPA的降解速率由0.02 min−1提高至0.037 min−1,当催化剂的投加量进一步增加至6.0 g时,BPA在60 min即可被完全去除,降解速率达0.039 min−1。这表明增加负载型Co3O4的剂量可显著提升PDS的有效利用率。
 图 4 负载型Co3O4活化PDS降解BPA的影响因素Figure 4. Effect of operation parameters on BPA removal in the supported Co3O4/PDS system
图 4 负载型Co3O4活化PDS降解BPA的影响因素Figure 4. Effect of operation parameters on BPA removal in the supported Co3O4/PDS system为考察水中常见无机阴离子对负载型Co3O4活化PDS降解有机物性能的影响,研究了Cl−、NO3−、CO32−对BPA去除效率和降解速率的影响。如图4(d)所示,在负载型Co3O4/PDS体系中加入10 mmol·L−1 Cl−并未降低BPA的去除效率,同样加入10 mmol·L−1的NO3−也未影响BPA的去除。Co3O4活化PDS降解有机物主要有2种途径:一种是基于SO4•−和OH•的自由基氧化;另一种是基于单线态氧(1O2)的非自由基氧化[15]。在自由基途径中,Cl−、NO3−等无机阴离子会竞争消耗强氧化性的SO4•−和OH•,进而抑制有机污染物的降解[16-18]。在本研究中,Cl−和NO3−对BPA的去除影响较弱,表明自由基氧化不是Co3O4活化PDS降解有机物的主要途径。然而,向反应体系中添加10 mmol·L−1的CO32−可显著抑制BPA的去除,同样反应条件下BPA仅去除了16.64%,同时反应速率由0.04 min−1降低至了0.001 min−1。CO32−是一种常用的1O2淬灭剂,其对BPA降解的显著抑制表明负载型Co3O4活化PDS降解有机物是1O2主导的非自由途径[19]。除了无机阴离子,天然有机物如腐殖酸(HA)也是一种常见的水体背景成分[6]。在10 mg·L−1 HA存在的情况下,负载型Co3O4催化降解有机物的效率也未受到显著影响,进一步说明本反应体系中有机物的降解为非自由基途径[20]。综上所述,负载型Co3O4/PDS体系不仅能够在较宽的pH范围内降解有机物,而且对无机阴离子和腐殖酸等常见的水体背景成分具有较强的抗干扰能力,因此,有着较强的应用潜力。
2.4 活性氧物种鉴定
为明确负载型Co3O4活化PDS降解有机物的机理,通过EPR鉴定分析了该体系产生的活性氧的种类。如图5(a)所示,当仅有PDS时,以DMPO为自旋捕获剂未检测到任何信号,而当加入负载型Co3O4催化剂后,EPR谱图观测到了峰强度为1: 2 : 2 : 1的特征峰,这是催化产生的OH•被DMPO捕获所致,表明负载型Co3O4活化PDS产生了OH•[21]。此外,以TEMP为1O2捕获剂还检测到了TEMP-1O2的特征峰,因此,负载型Co3O4活化PDS也产生了1O2[22]。进一步通过化学淬灭实验判别了反应体系中OH•和1O2对BPA降解的贡献。图5(b)反映了不同浓度MeOH的淬灭效果,可以看出MeOH对BPA的降解影响有限,在1 000 mmol·L−1 MeOH存在的情况下,BPA的去除率仍高达94.7%。MeOH可与SO4•−和OH•快速反应,反应速率常数分别为k 分别为 2.5×107 (mol·s)−1和9.7×108 (mol·s)−1,是常用的自由基淬灭剂;MeOH对负载型Co3O4/PDS有限的淬灭能力表明该体系降解有机物的过程中,自由基的氧化作用有限[7]。然而,L-组氨酸可显著抑制BPA的降解,如图5(c)所示,1 mmol·L−1的组氨酸使得BPA的降解率由100%降低至89%;当L-组氨酸的浓度增加至5 mmol·L−1时,BPA的去除率进一步降低至21.98%,反应速率也由0.04 min−1降低至0.002 min−1。L-组氨酸具有一定的还原性,可能会消耗PDS而降低负载型Co3O4催化降解BPA的能力[23]。为排除L-组氨酸消耗PDS对淬灭实验的干扰,研究了不同浓度L-组氨酸对PDS的降解效率。如图5(d)所示,L-组氨酸分解PDS的能力有限,例如5 mmol·L−1的L-组氨酸在120 min内仅分解了5.8%的PDS。因此,L-组氨酸对负载型Co3O4/PDS降解BPA的抑制作用主要归咎于其对活性氧物种的淬灭,而L-组氨酸是常用的1O2淬灭剂,这表明负载型Co3O4活化PDS降解有机物是以1O2为主导的非自由基氧化途径。过渡金属氧化物活化PDS产生1O2已被广泛报道,有研究表明,O2•−是1O2生成的重要中间体(
2O2·−+2H2O→21O2+2OH−+H2O2 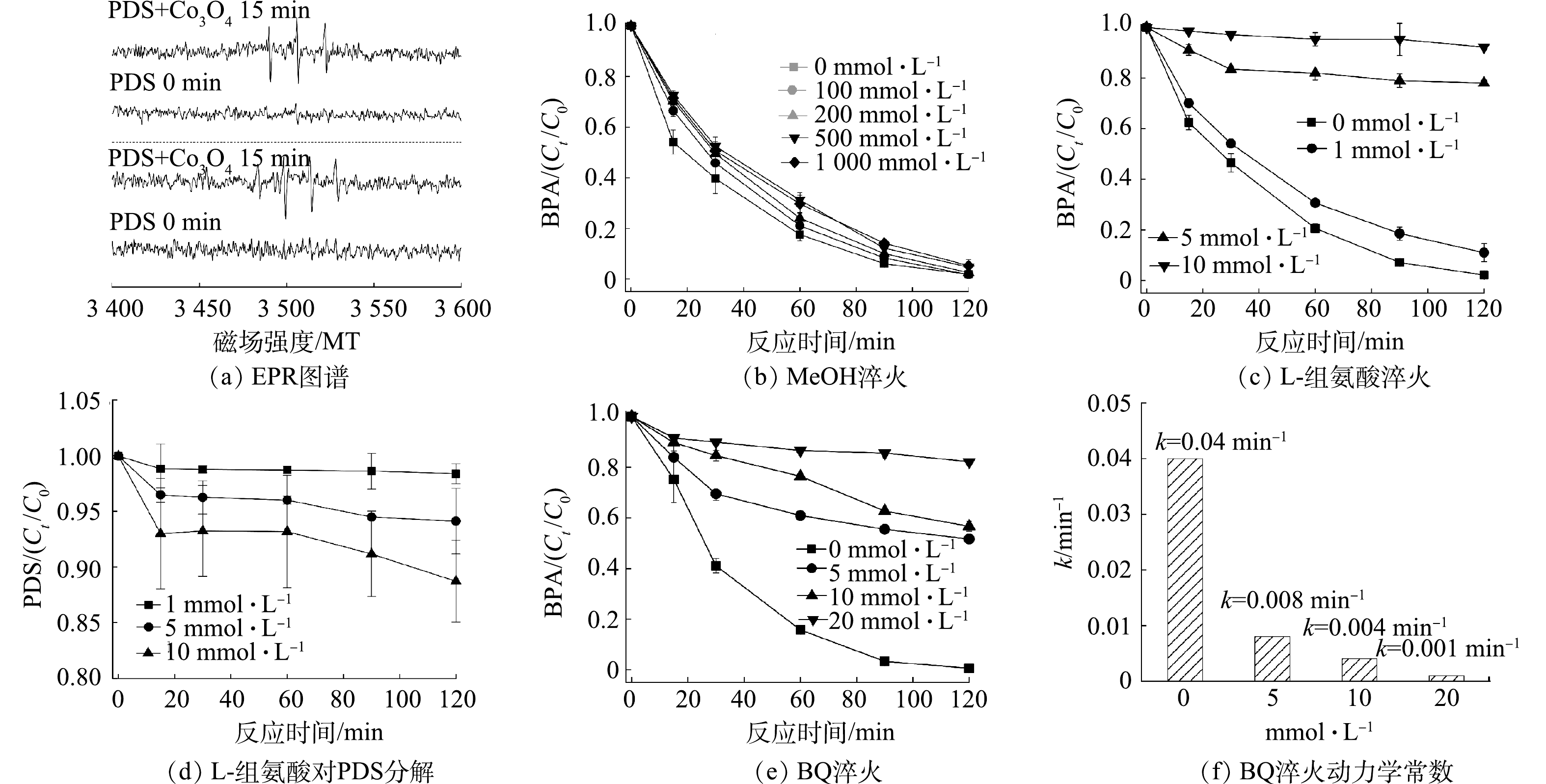 图 5 负载型Co3O4活化PDS的电子自旋共振测试和淬灭实验Figure 5. EPR spectra and quenching experiments on BPA removal
图 5 负载型Co3O4活化PDS的电子自旋共振测试和淬灭实验Figure 5. EPR spectra and quenching experiments on BPA removal2.5 PDS活化机理研究
过渡金属氧化物主要通过金属离子的价态循环活化过硫酸盐[26]。为解析负载型多孔Co3O4活化PDS的机理,通过XPS分析了Co3O4中Co元素的价态及其反应前后含量的变化。如图6(a)所示,Co2p在794.9 eV和779.7 eV的特征峰分别对应于Co2p1/2和Co2p3/2,而Co2p3/2的高分辨XPS谱图拟合结果表明,781.2 eV和779.7 eV处的峰分别对应于Co2+和Co3+,所占比例分别为34.2%和54.5%。反应后,Co3O4表面Co2+、Co3+比例变化表明Co3+/Co2+的氧化还原参与了PDS的活化[27]。进一步通过原位ATR-FTIR分析研究了PDS分子在负载型Co3O4表面的反应机理。如图6(b)所示,在1 050 cm−1和1 272 cm−1处检测到PDS振动峰,加入Co3O4催化剂后在其表面观测到了PDS的特征峰的位置并且未发生偏移,表明PDS吸附于Co3O4催化剂的表面。此外,在Co3O4/PDS体系中还观测到了SO42‒的特征峰(1 107 cm−1),表明Co3O4活化PDS产生了SO42‒;随着反应时间的增加PDS特征峰的强度逐渐降低,而SO42‒特征峰的强度逐渐增加,表明PDS在Co3O4催化剂表面持续分解[1]。
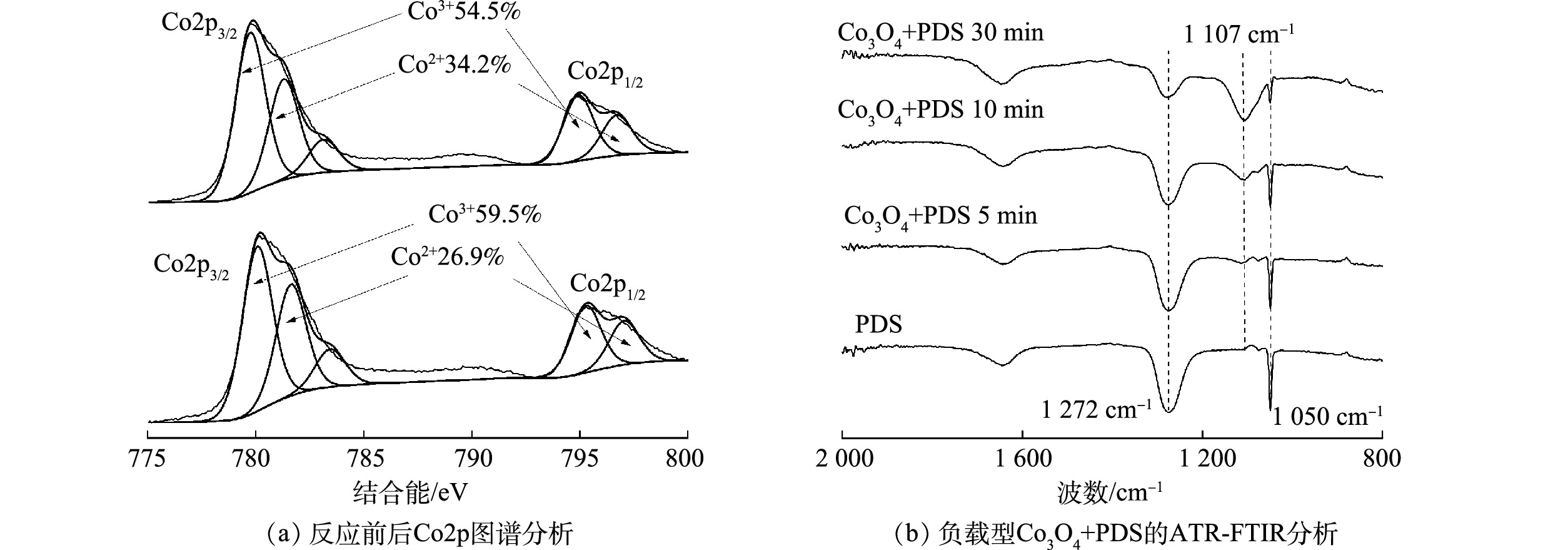 图 6 负载型Co3O4活化PDS的机理研究Figure 6. Mechanism of PDS activated over the supported Co3O4 catalyst
图 6 负载型Co3O4活化PDS的机理研究Figure 6. Mechanism of PDS activated over the supported Co3O4 catalyst金属氧化物(MOx)活化过硫酸盐时,吸附于其表面的水分子会解离形成MOx-OH[19]。因此,结合XPS和ATR-FTIR分析,提出了负载型Co3O4活化PDS降解BPA的可能反应机理(图7)。首先,PDS分子通过氢键与[≡Co3+—OH]2+结合形成[≡Co3+—O—O—SO3]+络合物(式(1)),该络合物具有较高的氧化电势,还原PDS分子生成O2•−,同时Co3+被还原为Co2+形成中间态[≡Co2+—OH] (式(2)),随之生成的O2•−进一步反应生成1O2 (式(3))。中间态[≡Co2+—O—O—SO3]通过内电子转移被氧化为≡Co3+—OH,同时生成少量的自由基(式(4))。如上所述,负载型Co3O4/PDS体系中自由基对有机物的降解贡献有限,因此1O2进一步氧化有机物将其分解为中间产物或者H2O和CO2 (式(5))。本研究中,负载型Co3O4有效活化PDS主要有2点原因,一是负载型Co3O4避免了纳米催化剂的团聚,可暴露更多的活性位点而提升催化效率;另一方面,Co-TA配合物热解会产生富含氧缺陷的Co3O4[28],而氧缺陷可有效提升PDS分子在Co3O4表面的吸附,也会提升内电子催化PDS的效能。
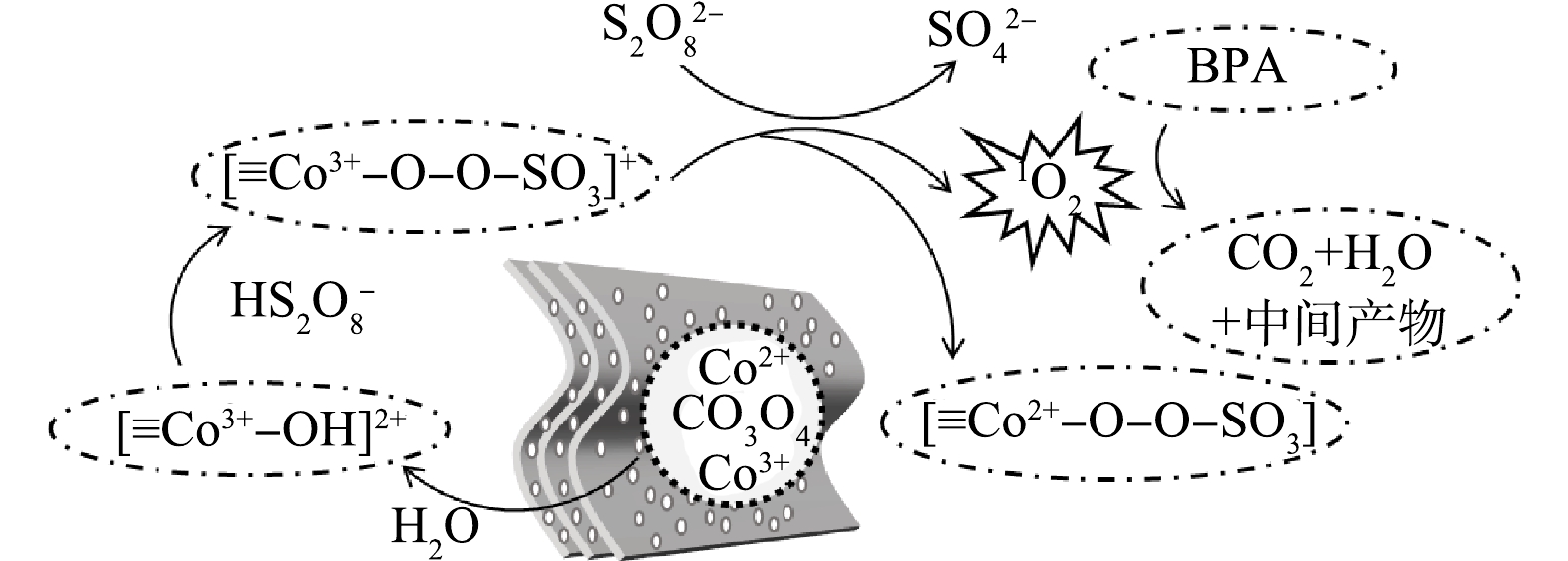 图 7 负载型Co3O4活化PDS降解BPA的机理示意图Figure 7. Schema for BPA degradation by PDS activated over the supported Co3O4 catalyst
图 7 负载型Co3O4活化PDS降解BPA的机理示意图Figure 7. Schema for BPA degradation by PDS activated over the supported Co3O4 catalyst2[≡Co3+−OH]2++HS2O8−→2[≡Co3+−O−O−SO3]++3H+ (1) 2[≡Co3+−O−O−SO3]++4H2O+S2O82−→2[≡Co2+−OH]+4SO42−+2O2·−+6H+ (2) 2O2·−+2H2O→21O2+2OH−+H2O2 (3) [≡Co2+−O−O−SO3]+H2O→≡Co3+−OH+SO4·−+OH· (4) 1O2+有机物→H2O+CO2+中间产物 (5) 催化剂的稳定性是衡量其实际应用潜力的一个重要因素,本研究通过循环降解实验研究了负载型Co3O4的稳定性。如图8(a)所示,3次循环后负载型Co3O4对BPA的去除率仍保持在89%,表明负载型Co3O4具有良好的稳定性。循环降解过程中催化活性的轻微降低可能是BPA的降解产物堵塞活性位点导致的[10]。此外,XRD分析表明循环使用后负载型Co3O4的晶相没有明显变化(图2(a)),进一步说明负载型Co3O4具有良好的稳定性。负载型Co3O4可有效矿化BPA(图8(b)),TOC去除率高达73%,而且3次循环后TOC的去除率仍高达63%,表明负载型Co3O4可持续稳定地处理水中酚类有机污染物。
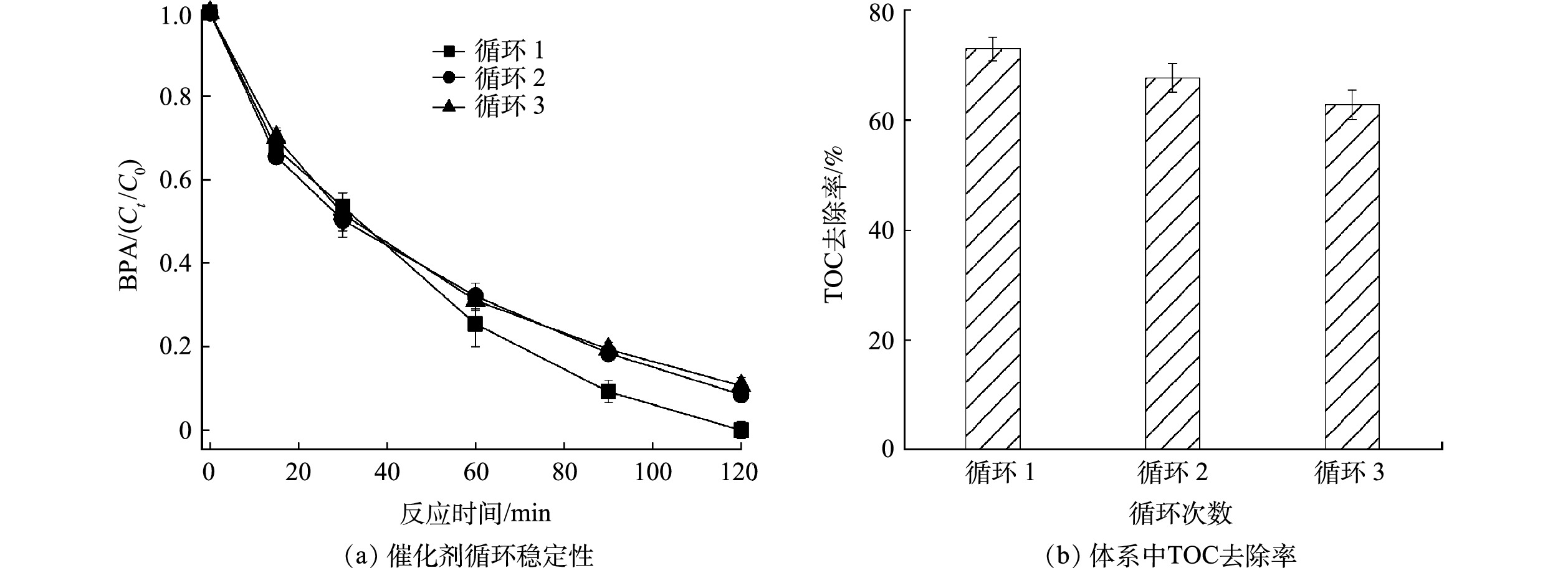 图 8 循环降解过程中BPA的去除性能和TOC的去除效率Figure 8. Removal of BPA and corresponding TOC during the consecutive runs over the supported Co3O4
图 8 循环降解过程中BPA的去除性能和TOC的去除效率Figure 8. Removal of BPA and corresponding TOC during the consecutive runs over the supported Co3O43. 结论
1)基于植物多酚的界面配位原理,通过在三维矿物载体表面原位负载Co3O4纳米颗粒制得了易分离回收的负载型Co3O4催化剂,比Co3O4粉体催化剂展现出更优异的催化活性和良好的稳定性。
2)负载型Co3O4催化剂活化PDS可高效降解BPA、SMX、AAP等多种类型的药、护品类有机污染物,中性条件下,负载型Co3O4投加量为60 g·L−1(即0.075 g·L−1 Co3O4纳米颗粒),在BPA/PDS摩尔比为1:10的情况下可在120 min内将BPA完全降解。
3)负载型Co3O4主要通过内电子转移活化PDS,降解有机物时自由基氧化的作用有限,主要是1O2主导的非自由基氧化途径,对无机阴离子和腐殖酸等水体背景成分有着较强的抗干扰能力,在实际水体处理方面展现出广阔的应用前景。
-
图 1 气泡石与气泡石负载Co3O4的电子照片与SEM分析
Figure 1. Digital images of airstone with and without Co3O4 and their SEM analysis
图 2 负载型Co3O4和粉体Co3O4的XRD以及Co-TA配合物的热解分析
Figure 2. XRD patterns for powdery Co3O4 and supported Co3O4 and TG curve for Co-TA polymer
图 3 负载型Co3O4活化PDS降解不同有机物的性能
Figure 3. Degradation of different organic pollutants by PDS activated over supported Co3O4
图 4 负载型Co3O4活化PDS降解BPA的影响因素
Figure 4. Effect of operation parameters on BPA removal in the supported Co3O4/PDS system
图 5 负载型Co3O4活化PDS的电子自旋共振测试和淬灭实验
Figure 5. EPR spectra and quenching experiments on BPA removal
图 6 负载型Co3O4活化PDS的机理研究
Figure 6. Mechanism of PDS activated over the supported Co3O4 catalyst
图 7 负载型Co3O4活化PDS降解BPA的机理示意图
Figure 7. Schema for BPA degradation by PDS activated over the supported Co3O4 catalyst
-
[1] WANG G, GE L, LIU Z, et al. Activation of peroxydisulfate by defect-rich CuO nanoparticles supported on layered MgO for organic pollutants degradation: An electron transfer mechanism[J]. Chemical Engineering Journal, 2022, 431: 134026. doi: 10.1016/j.cej.2021.134026 [2] NIKRAVESH B, SHOMALNASAB A, NAYYER A, et al. UV/Chlorine process for dye degradation in aqueous solution: Mechanism, affecting factors and toxicity evaluation for textile wastewater[J]. Journal of Environmental Chemical Engineering, 2020, 8(5): 104244. doi: 10.1016/j.jece.2020.104244 [3] 王丽娟, 路井义, 王汉铮, 等. 层状镍铁水滑石活化过二硫酸盐降解亚甲基蓝的研究[J]. 应用化工, 2021, 50(10): 2756-2760. doi: 10.3969/j.issn.1671-3206.2021.10.028 [4] DONG X, DUAN X, SUN Z, et al. Natural illite-based ultrafine cobalt oxide with abundant oxygen-vacancies for highly efficient Fenton-like catalysis[J]. Applied Catalysis B:Environmental, 2020, 261: 118214. doi: 10.1016/j.apcatb.2019.118214 [5] DONG X, REN B, ZGANG X, et al. Diatomite supported hierarchical 2D CoNi3O4 nanoribbons as highly efficient peroxymonosulfate catalyst for atrazine degradation[J]. Applied Catalysis B:Environmental, 2020, 272: 118971. doi: 10.1016/j.apcatb.2020.118971 [6] ZHONG Q Z, LI S, CHEN J, et al. Oxidation‐mediated kinetic strategies for engineering metal–phenolic networks[J]. Angewandte Chemie International Edition, 2019, 58(36): 12563-12568. doi: 10.1002/anie.201907666 [7] WANG G, QIN J, ZHOU X, et al. Self‐template synthesis of mesoporous metal oxide spheres with metal‐mediated inner architectures and superior sensing performance[J]. Advanced Functional Materials, 2018, 28(51): 1806144. doi: 10.1002/adfm.201806144 [8] WANG G, QIN J, FENG Y, et al. Sol–gel synthesis of spherical mesoporous high-entropy oxides[J]. ACS Applied Materials & Interfaces, 2020, 12(40): 45155-45164. [9] LI P, LIN Y, ZHAO S, et al. Defect-engineered Co3O4 with porous multishelled hollow architecture enables boosted advanced oxidation processes[J]. Applied Catalysis B:Environmental, 2021, 298: 120596. doi: 10.1016/j.apcatb.2021.120596 [10] WANG G, ZHANG Y, GE L, et al. Monodispersed CuO nanoparticles supported on mineral substrates for groundwater remediation via a nonradical pathway[J]. Journal of Hazardous Materials, 2022, 429: 128282. doi: 10.1016/j.jhazmat.2022.128282 [11] YANG S, XU S, TONG J, et al. Overlooked role of nitrogen dopant in carbon catalysts for peroxymonosulfate activation: Intrinsic defects or extrinsic defects[J]. Applied Catalysis B:Environmental, 2021, 295: 120291. doi: 10.1016/j.apcatb.2021.120291 [12] YANG Y, LI X, ZHOU C, et al. Recent advances in application of graphitic carbon nitride-based catalysts for degrading organic contaminants in water through advanced oxidation processes beyond photocatalysis: A critical review[J]. Water Research, 2020, 184: 116200. doi: 10.1016/j.watres.2020.116200 [13] WANG G, AN W, ZHANG Y, et al. Mesoporous carbon framework supported Cu-Fe oxides as efficient peroxymonosulfate catalyst for sustained water remediation[J]. Chemical Engineering Journal, 2022, 430: 133060. doi: 10.1016/j.cej.2021.133060 [14] 伍倩, 于芹芹, 戴友芝. CNQDs/Ag3PO4/g-C3N4复合光催化材料的制备及其对双酚A的降解性能[J]. 环境工程学报, 2022, 16(1): 101-111. doi: 10.12030/j.cjee.202108170 [15] WANG C, JIA S, ZHANG Y, et al. Catalytic reactivity of Co3O4 with different facets in the hydrogen abstraction of phenol by persulfate[J]. Applied Catalysis B:Environmental, 2020, 270: 118819. doi: 10.1016/j.apcatb.2020.118819 [16] KARRER C, DE BOER W, DELMAAR C, et al. Linking probabilistic exposure and pharmacokinetic modeling to assess the cumulative risk from the bisphenols BPA, BPS, BPF, and BPAF for Europeans[J]. Environmental Science & Technology, 2019, 53(15): 9181-9191. [17] 安晓静, 刘东方, 梁啸夫, 等. 铁酸钴活化过一硫酸盐处理高盐废水中罗丹明B的研究[J]. 水处理技术, 2022, 48(06): 50-53. doi: 10.16796/j.cnki.1000-3770.2022.06.010 [18] GHANBARI F, MORADI M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants[J]. Chemical Engineering Journal, 2017, 310: 41-62. doi: 10.1016/j.cej.2016.10.064 [19] ZHU S, LI X, KANG J, et al. Persulfate activation on crystallographic manganese oxides: mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants[J]. Environmental Science & Technology, 2018, 53(1): 307-315. [20] Jawad A, Zhan K, Wang H, et al. Tuning of persulfate activation from a free radical to a nonradical pathway through the incorporation of non-redox magnesium oxide[J]. Environmental Science & Technology, 2020, 54(4): 2476-2488. [21] YANG S, QIU X, JIN P, et al. MOF-templated synthesis of CoFe2O4 nanocrystals and its coupling with peroxymonosulfate for degradation of bisphenol A[J]. Chemical Engineering Journal, 2018, 353: 329-339. doi: 10.1016/j.cej.2018.07.105 [22] ZHU X, ZHANG Y, YAN W, et al. Peroxymonosulfate activation by mesoporous CuO nanocage for organic pollutants degradation via a singlet oxygen-dominated pathway[J]. Journal of Environmental Chemical Engineering, 2021, 9(6): 106757. doi: 10.1016/j.jece.2021.106757 [23] ZHU X, GE L, YAN W, et al. Peroxymonosulfate activation by immobilized CoFe2O4 network for the degradation of sulfamethoxazole[J]. Journal of Environmental Chemical Engineering, 2022, 10(3): 107781. doi: 10.1016/j.jece.2022.107781 [24] LIU Y, LUO J, TANG L, et al. Origin of the enhanced reusability and electron transfer of the carbon-coated Mn3O4 nanocube for persulfate activation[J]. ACS Catalysis, 2020, 10(24): 14857-14870. doi: 10.1021/acscatal.0c04049 [25] LEE J, VON G U, KIM J H. Persulfate-based advanced oxidation: critical assessment of opportunities and roadblocks[J]. Environmental Science & Technology, 2020, 54(6): 3064-3081. [26] 许晟硕, 钱征, 王龄侦, 等. 氮掺杂碳催化剂活化过一硫酸盐的活性位点分析及其对双酚A的降解机制[J]. 环境工程学报, 2022, 16(02): 452-461. doi: 10.12030/j.cjee.202111044 [27] WANG G, ZHOU X, Qin J, et al. General synthesis of mixed semiconducting metal oxide hollow spheres with tunable compositions for low-temperature chemiresistive sensing[J]. ACS Applied Materials & Interfaces, 2019, 11(38): 35060-35067. [28] HU J, ZENG X, WANG G, et al. Modulating mesoporous Co3O4 hollow nanospheres with oxygen vacancies for highly efficient peroxymonosulfate activation[J]. Chemical Engineering Journal, 2020, 400: 125869. doi: 10.1016/j.cej.2020.125869 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4367
- HTML全文浏览数: 4367
- PDF下载数: 59
- 施引文献: 0

