




-
四环素(tetracycline,TC)是最常见的一种广谱抗生素[1]。其化学结构稳定,不易被人体和动物消化吸收,大多通过粪便和尿液排出体外[2]。常规的污水处理方式对四环素的去除效果有限,因此,近年来在沉积物、地表水和地下水中经常检测到残留的四环素[3]。除了四环素本身的毒性,抗生素还会导致水体中产生抗性细菌和抗性基因,可危害生态系统和人体健康[4]。
近年来,芬顿氧化体系在水体污染物去除中受到广泛关注[5],其通过H2O2产生的羟基自由基(·OH)来降解有机污染物。但·OH的稳定性较差,从而限制对污染物的降解效果[6]。相比之下,半衰期更长、稳定性更好的硫酸根自由基(SO4·−)受到越来越多的探究。与液体H2O2相比,固体过硫酸盐在运输、使用和储存方面更安全。过一硫酸盐(PMS)是过硫酸盐的一种,在降解具有不饱和键和芳香族成分的有机污染物方面更具选择性[7]。PMS所产生的SO4·−在不同条件下被激发后还可以转化为·OH、SO5·−等多种活性物质[8]。目前较为常见的激活PMS的体系主要有2类,即均相反应体系和基于催化剂的非均相反应体系。其中,均相反应体系易受到水质等多方面因素影响,而基于催化剂的非均相体系催化效果相对更为稳定。因此,将PMS与基于催化剂的光催化技术相结合,具有更高的污染物去除效率和更广泛的应用前景。
目前,已有将PMS与TiO2[9]、ZnO[10]、g-C3N4[11]、CdS[12]等光催化剂相结合的研究。而在众多催化剂材料当中,含钴元素的材料被证明是激活PMS最有效的一类催化剂。JIANG等合成了多孔0D/3D NiCo2O4/g-C3N4 异质结,对卡马西平具有高效的去除效果[13]。JIN等合成了Z型异质结催化剂Co3O4/g-C3N4,在60 min内对四环素的去除率可以达到90.2%[14]。TIAN等制备出了蒲公英球状NiCo2O4催化剂,在120 min时可以去除90%的腐殖酸[15]。然而,钴元素的浸出可能会对水体环境造成二次污染。
铁酸镍(NiFe2O4)是一种常见的 p 型半导体,具有禁带宽度小、成本低、效益高、化学耐久性强和可磁性分离的优点[16]。然而,由于电荷载流子的快速复合和纳米粒子团聚效应引起的活性位点数量下降,导致NiFe2O4的催化活性较低[17]。钨酸铋(Bi2WO6)具有良好的可见光吸收能力和抗光腐蚀性[18]。将NiFe2O4和Bi2WO6复合构成p-n异质结结构,可促进电荷的分离且减弱NiFe2O4的团聚效应,提升污染物的去除率。
本研究利用静电自组装策略,在乙醇溶液中通过NiFe2O4和Bi2WO6合成得到复合催化剂NiFe-Bi-XY,并确定了NiFe2O4和Bi2WO6的最佳质量比,用于在太阳光/NiFe-Bi-XY/PMS体系中降解四环素。该催化剂NiFe-Bi-73含有双变价金属,可以很好地激发PMS,且所构建体系将光催化与非光催化体系相结合,大大提高了降解污染物的效率。此外,本研究将p-n异质结催化与PMS激活相结合,最大限度地发挥催化剂各组分的功能,进一步研究了在不同的反应条件下体系的降解效果,包括改变PMS浓度、溶液初始pH、催化剂剂量等影响因素。通过循环实验和一系列表征分析结果证明了所制备催化剂的稳定性。通过ESR、XPS价带谱和捕获实验等结果提出了多途径激活PMS以及基于自由基和非自由基的四环素降解途径。最后,通过HPLC-MS对降解产物进行检测,阐明了四环素可能的降解机制。
-
过一硫酸盐(K5H3S4O18,PMS)、四环素(tetracycline,TC)、五水合硝酸铋(Bi(NO3) 3·5H2O)、十六烷基三甲基溴化铵(CTAB)和二水合钨酸钠(Na2WO4·2H2O)购买于上海麦克林生化科技有限公司;六水合氯化镍(NiCl2·6H2O)购买于福晨(天津)化学试剂公司;六水合三氯化铁(FeCl3·6H2O)购买于阿拉丁试剂(上海)有限公司;氢氧化钠和硫酸购自国药集团化学试剂有限公司(上海)。所有化学品均未经进一步纯化,直接使用。
-
1) Bi2WO6的制备:以CTAB为模板,利用水热法制备Bi2WO6。称取165 mg Bi(NO3) 3·5H2O、485 mg Na2WO4·2H2O和25 mg CTAB加入35 mL去离子水中,剧烈搅拌30 min后,对混合溶液超声处理10 min。将溶液转移至100 mL反应釜中,置于120 ℃烘箱中加热24 h,自然冷却后通过离心获得沉淀,所得沉淀用去离子水洗涤两次,放入60 ℃烘箱中烘干待用。
2) NiFe2O4的制备:称取2 mmol FeCl3·6H2O和1 mmol NiCl2·6H2O加入100 mL去离子水中,搅拌30 min,然后逐滴加入5 mol·L−1的NaOH溶液直到pH调至13左右,搅拌1 h后通过离心获得沉淀,所得沉淀放入60℃烘箱中烘干,然后用马弗炉在500 ℃条件下烘干3 h,升温速度设置为5 ℃·min−1,自然冷却后待用。
3) NiFe-Bi-XY的制备:将Bi2WO6和NiFe2O4按照一定的质量比分别加入到100 mL无水乙醇中,两溶液均超声10 min后搅拌30 min,将Bi2WO6溶液逐滴加入NiFe2O4溶液中,搅拌2 h后离心得到沉淀,所得沉淀放入60℃烘箱中烘干待用。实验中Bi2WO6和NiFe2O4的质量比分别为5∶5、7∶3和9∶1,记为NiFe-Bi-55、NiFe-Bi-73和NiFe-Bi-91。
-
使用 Bruker D8衍射仪测试X射线衍射光谱(XRD)对晶体结构进行表征。为了测试样品的组成元素状态和价带谱,以Al-Ka射线作为激发源,使用EscaLab Xi+光谱仪测量X 射线光电子能谱(XPS)和价带位置。运用UV 3600 Plus分光光度计获得紫外-可见漫反射光谱(UV-Vis DRS)。以KBr为背景,运用ALPHA光谱仪(Bruker)对催化剂进行傅里叶变换红外光谱(FT-IR)检测。催化剂的形貌通过扫描电子显微镜(SEM, EM-30 Plus, COXEM)和透射电子显微镜(TEM, G2 F20, FEI)进行观察。用F7000光谱仪(HITACHI)测量光致发光光谱(PL)。用总有机碳(TOC)测定仪(multi N/C 3100, Analytik Jena AG)测定实验后溶液的矿化率。水样的化学需氧量(chemical oxygen demand,COD)通过分光光度计(DR3900, HACH)进行测定。利用电子顺磁共振仪(ESR, E500, BRUKER)对自由基和其他活性物质进行检测。以0.5 M的Na2SO4溶液为电解液,在配有碳棒对电极、饱和甘汞参比电极(SCE)和工作电极的标准三电极系统中测量光电流和电化学阻抗(EIS)。
-
光催化实验采用光化学反应器(CEL-PE300L-3A, 中教金源有限公司)作为反应装置来进行光催化反应。以300 W的氙灯作为模拟太阳光源,辐射通量确定为 30 mW·cm−2。在批量光降解实验中,将50 mg催化剂样品分散到100 mL浓度为20 mg·L−1的四环素溶液中,并在黑暗中搅拌溶液10 min以达到吸附和解吸平衡。PMS浓度为0.2 ~1.2 mmol·L−1,并且在辐照过程中每隔5 min取3 mL悬浮液。将样品离心并通过高效液相色谱(HPLC,LC-20A,岛津)测量四环素的浓度,根据式(1)计算光催化去除率。
式中:D为四环素去除率,%;A0为样品溶液初始吸光度;At为不同反应时间溶液的吸光度;C0为样品溶液初始浓度,mg·L−1;Ct为不同反应时间的浓度,mg·L−1。
此外,对NiFe-Bi-73的重复利用性能进行了测试。降解产物通过1290II-6460液相色谱-质谱联用(LC-MS,安捷伦)在正离子模式下鉴定。运用H2SO4和NaOH溶液(2 mol·L−1)调节四环素溶液的初始 pH。此外,将Cl−、HCO3−、NO3−和 H2PO4− 4种阴离子分别加入到四环素溶液中,对每种阴离子均设置1、5和10 mmol·L−1 3个浓度梯度,以测试NiFe-Bi-73的稳定性。本研究还测试了所合成催化剂对于二沉池出水和河水配制的四环素溶液中的污染物去除效率,以评估太阳光/NiFe-Bi-73/PMS体系的实际应用潜力。
-
为了判断在太阳光/NiFe-Bi-73 /PMS体系反应过程不同活性物种的作用,根据不同捕获剂与活性物种的反应速率,选用甲醇(MeOH,1.0 mmol·L−1)、叔丁醇(TBA,100.0 mmol·L−1)、对苯醌(BQ,1.0 mmol·L−1)、L-组氨酸(10.0 mmol·L−1)和甲酸(10.0 mmol·L−1)分别作为SO4·−和·OH、·OH、·O2−、1O2和h+的捕获剂。通过检测在加入捕获剂后对于四环素去除情况的影响,判断影响降解效果的主要活性物种。
-
通过X射线衍射仪对Bi2WO6,NiFe2O4 2种单体和3种NiFe-Bi-XY复合物的物相组成进行了测试,结果如图1(a)所示。由Bi2WO6的衍射图谱可见,衍射角位于28.28◦、32.71◦、47.05◦、55.84◦和58.57◦的特征峰,分别对应为(113)、(200)、(220)、(313)和(226)晶面(JCPDS 39-0256)[19]。NiFe2O4的特征峰位于30.29◦、35.66◦、37.35◦、43.31◦、53.80◦、57.36◦和62.98◦,分别对应于NiFe2O4图谱中的(220)、(311)、(222)、(400)、(442)、(511)和(440)晶面(JCPDS# 00-054-0964)[20]。此外,在NiFe-Bi-XY的XRD图谱清楚地可以看到Bi2WO6和NiFe2O4的出峰,说明催化剂在合成过程中保持着2种单体结构的完整性,并且不含有其它杂质。
Bi2WO6、NiFe2O4和NiFe-Bi-XY的FT-IR表征结果如图1(b)所示。在3种NiFe-Bi-XY中,均可以检测到2种单体的特征峰。位于3 404 cm−1和1 625 cm−1处的强宽峰分别源于水分子中羟基(—OH)基团的伸缩振动和弯曲振动[21]。Bi2WO6的吸收峰在400~1 000 cm−1,说明含有W−O、Bi−O键和W−O−W键[22]。对于NiFe2O4单体,在414.5~601 cm−1内观察到的2个特征峰分别对应于NiFe2O4的Ni—O和Fe—O键[23]。上述结果表明,合成的NiFe-Bi-XY有效地将Bi2WO6和NiFe2O4结合在一起。
XPS谱图用于研究所制备催化剂的元素组成和表面化学状态。由图2(a)可见,结合能为158.8 eV的峰对应于Bi4f7/2,164.1 eV处的峰对应于Bi4f5/2,其均为Bi3+的特征峰[24]。Fe2p的XPS谱图(图2(b))中有6个峰,分别是709.0、711.8、723.7和725.4 eV 4个主峰和2个卫星峰。其中,709.0 eV和711.8 eV处的峰对应Fe2p3/2,而725.4 eV和723.8 eV处的峰则对应Fe2p1/2[25]。由图2(c)可见,结合能为853.2 eV和872.4 eV的峰分别对应Ni2p3/2和Ni2p1/2[26]。此外,Ni2p3/2分别位于853.2 eV和855.8 eV,这说明同时存在Ni2+和Ni3+[27]。由图2(d)可以看出,O1s有3个峰,在529.4 eV处的峰为金属元素与氧原子成键,对应于Ni−Fe−O键[28];530.1 eV处的特征峰对应Ni−O−H键,532.4 eV处的峰则是水中氧元素的特征峰[29]。
在XPS图中可以看到,复合催化剂中各元素的出峰位置相较于单体的位置均有差异。对于异质结结构而言,两个半导体的费米能级平衡会产生内部电场,导致电荷向其中一侧扩散并改变相应半导体的能带位置[30]。由图2可以看出,相较于Bi2WO6,NiFe-Bi-73中Bi4f的数值整体向左移动0.1 eV,结合能减小。说明电子密度变大。铁、镍元素的结合能整体升高,证明电子密度变小[31]。由此推测,电子由NiFe2O4的导带流向了Bi2WO6的导带,形成了一个内部电场。
-
在图3(a)和图3(b)中呈现出NiFe2O4纳米粒子和片状的Bi2WO6,而由图3(c)可以观察到在复合催化剂中,NiFe2O4均匀分布在Bi2WO6表面。图3(d)则进一步证明2种单体之间产生了非常强的相互作用,交界面边界证明形成了异质结。在图3(e)~(f)中,高分辨率TEM图片中0.27 nm 的晶格条纹为Bi2WO6的(200)晶面[32],而0.25 nm和0.29 nm的间距分别对应了NiFe2O4中(311)和(220)的晶格平面[33]。
-
运用UV-Vis进一步检测了所合成催化剂的光学性质。如图4(a)所示,Bi2WO6在紫外波段具有很强的光吸收,光吸收波长接近464 nm;纯NiFe2O4在整个可见光和紫外光的范围内均有吸收。在复合材料中,随着NiFe2O4的增加,明显拓展了复合催化剂的光吸收范围。根据Kubelka-Munk(式(2)),如图4(b)和图4(c)所示,由于Bi2WO6和NiFe2O4是直接半导体,根据式(2)可计算得到禁带能量(Eg)分别为2.59 eV和2.63 eV。
一般来说,催化剂在PL谱图中的强度越弱,说明其抑制光生电子复合的能力越强。由图4(d)可以看出,3种NiFe-Bi-XY的强度均比Bi2WO6弱,说明光生电荷的分离效率更高且抑制光生载流子的能力更强。NiFe2O4的谱图强度在5个样品中是最弱的,是由于其团聚效应影响了对光的响应能力。光电流和阻抗被用来研究材料的光学和电化学性质。如图4(e)和图4(f)所示,在复合催化剂中,NiFe-Bi-73的光电流强度更高,阻抗也更小,表明光生电荷数量越多。相比较于NiFe2O4,复合物的光电流强度更高,这也间接说明异质结的形成改善了NiFe2O4的团聚效应。
-
如图5(a)所示,Bi2WO6在30 min内对四环素的去除率仅为70.0%,NiFe2O4对四环素的去除率为80.5%。将2种单体复合后,降解效果明显增强,其中NiFe-Bi-73的去除效果最好,最终对四环素的去除率可以达到91.1%。如图5(b)所示,各催化剂对应的一阶动力学常数顺序为NiFe-Bi-73>NiFe-Bi-91>NiFe-Bi-55> NiFe2O4 >Bi2WO6。由图5(c)可见,单纯的光照条件下无法去除四环素,单纯的PMS(42.9%)和催化剂(48.0%)对四环素去除效率较低。相比太阳光/PMS体系,太阳光/催化剂和催化剂/PMS体系去除四环素的效率更高,分别可以达到78%和83%。这说明催化剂可以很好地被光激发形成自由基,而太阳光对PMS的激发效果较差。显然,太阳光/催化剂/PMS体系的降解效果是最好的,对四环素的去除率可达91.1%。这说明有效去除四环素主要是多模式激发PMS和产生各种活性物质共同作用的结果。表1对单纯光催化、传统芬顿体系和光催化与PMS相结合3种体系降解四环素的性能进行了比较,可见,本研究所选用的光催化体系对四环素的降解效率较高。
对影响污染物去除的因素进行了探究,主要包括PMS浓度、初始溶液pH、催化剂用量和四环素初始浓度。由图6(a)可知,当加入PMS的初始浓度由0.3 mmol·L−1增加到1.2 mmol·L−1时,NiFe-Bi-73对四环素的去除率由74.2%迅速增加到91.1%,这是由于更多的PMS会产生更多SO4·−、·OH等活性成分,促进降解反应的进行。但是,当PMS的浓度继续增加到2.4 mmol·L−1时,催化剂对四环素的去除率反而降至84.1%。这可能是因为过多的SO4·−会自猝灭或者与PMS反应,生成氧化能力较弱的SO5·−自由基或者无氧化能力的离子基团,导致四环素降解效果降低(式(3)~式(4))[40]。
由图6(b)可见,该体系在所研究的pH范围内(3.01~11.13)均可保持较好的降解性能,中性条件对于四环素去除的抑制最为明显,但去除率依然可以达到77%。PMS的pKa1和pKa2分别是0和9.4,即当pH为3.0~9.0时,HSO5−是主要离子;当pH大于11时,PMS会形成SO52−。因此,当溶液是中性时,PMS产生的自由基数量有所下降,影响了降解效果[41]。此外,在pH为3、5、7、9时测得NiFe-Bi-73的Zeta电位分别−15.0、−23.4、−39.6和−36.3 mV,即NiFe-Bi-73在pH为7时,电位最负,四环素与催化剂之间产生静电排斥,不利于降解反应进行[42]。
如图6(c)所示,将催化剂的初始投加量从0.1 g·L−1增加到0.5 g·L−1,四环素的去除率由72%迅速增加到91%。这是因为催化剂提供的电子和空穴数量增加,使得更多的PMS被激活。当继续增加催化剂到0.7 g·L−1时,四环素的去除率反而降至86%。这可能是由于催化剂使得溶液对光的透射能力减弱,导致光利用率降低[43];也可能是过量催化剂之间发生团聚作用,导致催化剂的活性位点减少,进而降低催化效率[44]。如图6(d)所示,将四环素的初始质量浓度从10 mg·L−1逐渐增加到40 mg·L−1,去除率由91%降低到80%。这是因为当加入相同剂量的光催化剂和PMS时,活性基团和电子−空穴对的数量是固定的,四环素的浓度越高,降解效率越低。
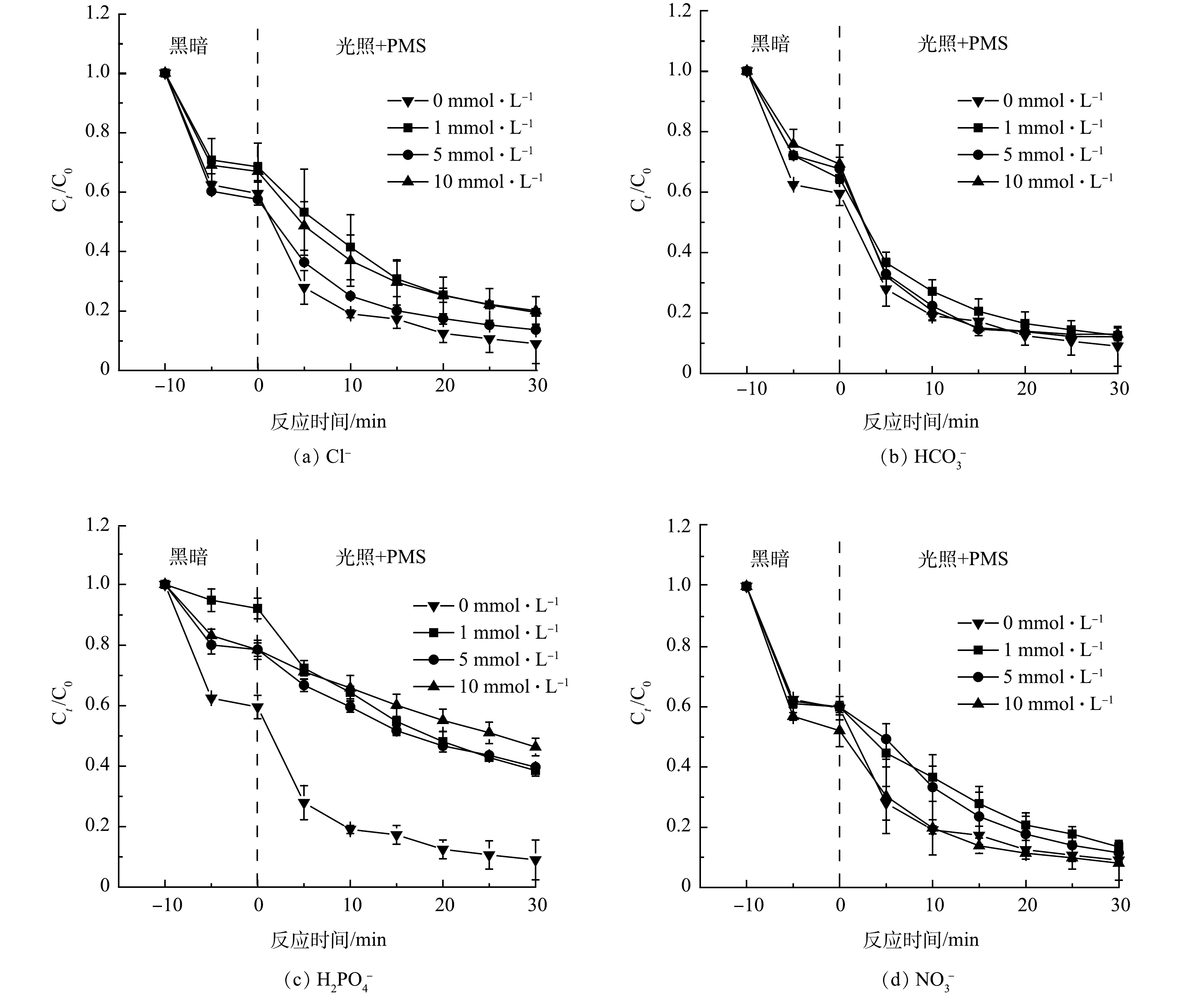
如图7(a)~(d)所示,进一步研究了实际水体中可能含有的阴离子对太阳光/NiFe-Bi-73/PMS体系降解四环素的影响。在Cl−、HCO3−、H2PO4− 和 NO3− 4种离子中,只有H2PO4−对降解表现出明显的抑制作用。当H2PO4−的浓度由1 mmol·L−1增加到10 mmol·L−1,四环素的去除率由61.5%降低至53.7%,这是由于H2PO4−可以与SO4·−和·OH反应生成氧化能力较低的HPO4·−,从而影响了降解效率[45]。高浓度的HCO3−会与SO4·−和·OH反应生成反应活性较低的CO3·−,从而导致四环素的去除效率下降[46]。高浓度的Cl−同样会影响降解效率,这可能是由于Cl−和四环素之间争夺催化剂表面的位点所致[47]。NO3−对降解效果并没有明显的影响。
为了检测太阳光/NiFe-Bi-73/PMS体系对实际水体中四环素的降解效果,采集了某河水和某污水处理厂二沉池出水的水样(水质参数见表2)配制污染物溶液。在这2种溶液中,所构建的反应体系对四环素的去除率均可以达到90%(图8(a)),且对COD的去除率分别可以达到100%和90.3%。如图8(b),将催化剂多次重复利用测试降解效果,发现催化剂四次重复后,反应体系对四环素的去除率率仍可以保持在77.6%。测得4次重复利用的矿化率分别为56.3%、50.3%、46.2%和43.1%。如图8(c)和8(d),对重复利用后的催化剂进行了XRD和XPS分析,发现与未重复利用前的催化剂进行对比,催化剂在重复利用后依然保持了原有的结构,结构和催化性能都比较稳定。图8(e)和8(f)为重复利用前后的Fe和Ni的高分辨率XPS图,从图中可以发现使用前后两种元素的出峰位置发生明显偏移,且二价离子与三价离子的相对峰强度变化表明二者的相对含量较使用之前发生变化,证明复合催化剂中的金属元素参与激活PMS。
-
根据对降解中间产物的质谱分析,本文推导出四环素可能的降解机理。如图9所示,四环素分子具有双键、胺基和酚羟基,这使其易于与多种活性物质反应。在过程A中,四环素首先脱去氨基和二甲氨基生成TC1(m/z = 389)。之后TC1通过开环反应生成TC2(m/z = 365),后者经历脱甲基、羟基氧化、开环等一系列反应生成TC3(m/z = 300)。此外,TC1也可以通过类似的一系列反应生成TC4(m/z = 337)。在过程B中,四环素分子首先与水分子通过加成反应生成TC5(m/z = 459),TC5再通过开环反应、脱水、脱甲基、羟基氧化等反应生成TC6(m/z = 389),最终经过一系列的反应过程生成TC7(m/z = 337)。TC3、TC4和TC7 最终可以被进一步降解成一系列小分子物质,包括TC8(m/z = 223)、TC9(m/z = 152)、TC10(m/z = 149)、TC11(m/z = 135)、TC12(m/z = 116)、TC13(m/z = 114)和TC14(m/z = 60)。结合测得的TOC去除率,可以证明部分有机分子被彻底矿化,转化成H2O和CO2。
复合催化剂的能带位置与反应过程中电荷的转移和活性物质的生成具有密切联系。由图10(a)和图10(b)可以看出,Bi2WO6和NiFe2O4的价带值分别为1.88 eV和0.86 eV。结合禁带宽度和式(5)可以进一步计算出Bi2WO6和NiFe2O4的导带值分别为−0.71 eV和−1.77 eV。
在太阳光/NiFe-Bi-73/PMS体系中,运用自由基淬灭实验来反推降解四环素过程中的主要活性物种。根据淬灭剂和相应活性物种的反应速率常数,选择甲醇、叔丁醇、对苯醌、L-组氨酸和甲酸分别作为 SO4·−和·OH、·OH、·O2−、1O2和h+的淬灭剂[48]。由图10(c)可以看出,加入对苯醌后抑制效果最为明显,四环素的去除率下降至61.3%,说明·O2−是降解四环素的主要活性物种。L-组氨酸也表现出了较为明显的抑制作用,四环素的去除率下降至76%,因此,1O2也是反应中重要的活性物种。加入叔丁醇和甲醇后的抑制效果并不明显,四环素的去除率仍然可以分别达到82.8%和86.1%,表明·OH和SO4·−在反应过程中并不发挥主要作用。这主要是因为这2种自由基在短时间内转化成其它活性物种。
为了进一步研究反应过程中自由基的生成和转化过程,进行了ESR测试。如图10(d)所示,在加入PMS的情况下,所处体系均可以产生·OH和SO4·−,证明光照和催化剂材料均可以激活PMS。此外,在催化剂光照的情况下,可以检测到明显的·OH四重峰,证明催化剂材料可以被光照激发。对于·O2−而言(图10(e))所示,光照情况下可以观察到明显的出峰信号,而在催化剂与PMS共存的情况下,由于催化剂中的变价金属可以与PMS产生中间体并进一步生成·O2− [49]。而体系中的1O2则是来源于SO4·−、·OH和·O2−的转化过程,使得其成为主要的活性物质之一。
为了证明NiFe-Bi-XY形成了p-n异质结,分别对Bi2WO6、NiFe2O4和NiFe-Bi-73进行了Mott-Schottky 测试,结果如图11所示。可以看出,Bi2WO6谱图斜率为正向,为n型半导体;NiFe2O4谱图斜率为负向,为p型半导体。此外,NiFe-Bi-73的谱图曲线呈现出倒V型趋势,证明复合催化剂形成了p-n异质结结构[50]。
基于上述结果,提出了基于PMS活化和光催化去除四环素的反应机制(图12)。光照条件下在Bi2WO6和NiFe2O4表面产生了光生电子和空穴(式(6))。在形成p-n异质结时,由于在Bi2WO6和NiFe2O4的界面处存在内部电场(internal electric field, IEF),n型半导体Bi2WO6的价导带位置下移,而p型半导体NiFe2O4的价导带位置上移[51]。NiFe2O4处的光生电子可以转移到Bi2WO6的导带处,而空穴被继续保留在NiFe2O4的价带里。电子可以与体系中的溶解氧反应生成·O2−,也可以和PMS反应生成SO4·−,SO4·−又可以进一步与水反应生成1O2(式(7)~式(9))。虽然光生空穴不具有足够的氧化能力直接与水反应生成·OH,但·OH的生成可以通过·O2−→H2O2→·OH这一途径实现(式(10)~式(11))[52]。一般来说,1O2并不能直接反应生成,但·O2−、·OH和SO4·−在一定条件下可以转化为1O2(式(12)~式(14))[53]。此外,过渡金属离子可以与HSO5−进行络合反应形成M-SO5中间体,所形成的中间体可再与HSO5−反应产生·O2−(式(15)~式(18))。这些活性物种将四环素分子降解成小分子物质,从而实现对污染物的高效去除(式(19))。
-
1)通过自组装法成功制备了复合催化剂NiFe-Bi-XY,当Bi2WO6和NiFe2O4的质量比为7∶3时,催化剂具有最佳的太阳光利用效率及最强的催化能力。在最佳反应条件下,在30 min时NiFe-Bi-73对20 mg·L−1四环素的去除率可达91%。
2)由于体系中同时存在自由基和非自由基降解途径,复合催化剂大大提升了太阳光/NiFe-Bi-73/PMS体系对四环素的去除率。
3)复合催化剂中NiFe2O4和Bi2WO6界面紧密结合,很好地形成了p-n异质结结构,促进了光生电荷的有效分离和自由基的高效产生。
4)在该体系中,Cl−、HCO3−和H2PO4− 会抑制四环素的降解, 而NO3−对降解效果没有明显的影响。
5)在本研究降解体系中主要活性物种为1O2、·O2−和·OH,同时也证实所合成的催化剂为p-n异质结。
太阳光/NiFe-Bi-XY异质结催化剂/过一硫酸盐体系对四环素的降解性能及机理
Performance and mechanism of tetracycline degradation by sunlight/NiFe-Bi-XY heterojunctions /permonosulfate system
-
摘要: 近年来,尖晶石型铁氧体在光催化领域展现出良好的应用前景,但其团聚作用会影响催化效果,构建异质结结构可以有效提高催化效率。通过自组装法合成了一系列Bi2WO6/NiFe2O4 p-n型异质结催化剂(NiFe-Bi-XY),并将其应用于去除水体中的四环素污染物。在太阳光/NiFe-Bi-73/过一硫酸盐(PMS)体系中,在反应30 min时对20 mg·L−1四环素溶液的去除率可以达到91.1%,矿化率可以达到56.3%,所构建的反应体系在碱性环境中依然保持着对四环素良好的去除效果。通过XPS价带谱、禁带宽度计算、Mott-Schottky和ESR测试证明NiFe-Bi-XY形成了p-n型异质结结构。在所构建的体系中,四环素的降解主要是通过光催化和非光催化降解2种途径共同实现的。淬灭实验结果表明,·O2−和1O2是降解四环素的主要活性物种。以上研究结果可为合成高效的二元异质结催化剂,并将其用于环境修复提供参考。Abstract: In recent years, spinel-type ferrites have promising application prospects in the field of photocatalysis. However, its agglomeration will affect the catalytic effect, and the construction of a heterojunction structure can effectively improve the catalytic efficiency. In this study, a series of Bi2WO6/NiFe2O4 p-n type heterojunctions (NiFe-Bi-XY) by self-assemble were prepared and used to remove tetracycline in water. In the sunlight/NiFe-Bi-73/permonosulfate (PMS) systems, yhe optimal removal efficiency towards 20 mg·L−1 tetracycline could reach 91.1% at 30 min, and the mineralization rate was 56.3%. The constructed reaction system still maintained a good removal effect of tetracycline in an alkaline environment. For NiFe-Bi-XY, the formation of p-n type heterojunctions was proved by XPS valence band spectra, Kubelka-Munk plots, Mott-Schottky test and ESR measurements. In this system, the degradation of tetracycline was mainly achieved by photocatalytic and non-photocatalytic pathways. Radical trapping experiments confirmed that ·O2− and 1O2 radicals were the most critical active species during the catalytic process. This study provides a feasible approach to synthesize efficient binary heterojunction catalysts for environmental remediation.
-
Key words:
- tetracycline /
- wastewater treatment /
- photocatalysis /
- peroxymonosulfate /
- heterojunction
-
塑料自出现以来极大地方便了人们的生活,其需求量和使用量不断增加,因此大量塑料垃圾进入并积累于环境中,在光照、热解、机械磨损以及生物作用下破碎形成粒径更小的塑料颗粒,其中直径小于5 mm的被定义为微塑料(microplastics,MPs)[1-2]. 微塑料作为一种新型污染物,广泛分布于水体、土壤、大气等环境介质中,在太平洋、大西洋、印度洋沿岸和深海地区,土壤及生物体内都能够检测到大量的微塑料[3-4],对生态环境安全及生物生存造成了严重的威胁[1]. 微塑料具有较大的比表面积,能够吸附大量的环境污染物,因此能够成为重金属、有机污染物等环境污染物质的运输载体,进一步增加其他环境污染物的风险[5]. 近期,重金属与微塑料之间的相互作用已成为环境领域关注的热点. 由于工业开采、金属冶炼、污泥回用等导致大量的重金属镉(Cd)进入土壤,造成严重的镉污染[6]. 据统计,我国Cd污染耕地土壤面积达2×105 km2,约占全国耕地总量的1/6,严重影响农业生产和粮食安全[7]. 重金属Cd具有高稳定性、不易降解、且易在生物体内累积的特点[8-9],严重威胁着人体健康和环境安全[10]. 同时,农业生产使用的大量农膜被遗留在环境中能够逐渐破碎形成微塑料,与重金属Cd形成复合污染[11]. 已有研究表明微塑料对水体环境中的重金属具有一定的富集作用[12],如海滩上微塑料表面镉、铬、铜、铁、锰、镍、铅、锌等重金属含量比周边海域中的重金属浓度高[13-14]. 然而,关于微塑料对Cd环境行为影响的研究仍存在不足.
微塑料吸附重金属还能影响其迁移特性与生物毒性,Boucher等[15]研究发现在潮汐带沉积物中,微塑料能够吸附重金属,而生物摄入微塑料后,重金属也随之进入食物链. 研究表明,微塑料吸附的重金属可能会在水体和肠道中被重新释放,从而使其生物毒性增加[16]. 镉与微塑料的复合污染对鲤鱼血浆中的酶活性、生化指标及免疫等具有明显的生物毒性效应,且二者表现出明显的协同作用[17]. 微塑料与重金属联合作用可导致海水青鳉肠道污染负荷增加,特定肠道微生物及肠道功能发生变化,免疫系统抵抗力受到明显抑制[18]. 目前,尚未有统一的方法对重金属与微塑料联合毒性进行测定. 研究发现,发光细菌毒性测定法具有高灵敏度、强相关性、快速、高自动化程度等特点,在重金属、有机物污染环境中的综合毒性分析中已得到广泛的应用[19]. 利用发光菌对取代苯酚和镉的混合物进行生物毒性测定,结果表明二者之间存在微弱的加成作用或近似加成作用的弱协同效应[19].
此外,环境中的微塑料在多种因素作用下逐渐发生老化,导致微塑料表面的氧化结合位点增多,对重金属及其他污染物的吸附能力增强[20],因此老化微塑料的吸附能力通常高于原始微塑料. 研究自然环境中老化微塑料对其他污染物环境行为的影响具有现实意义. 尽管已有国内外学者对实验室条件下微塑料与其他环境污染物的吸附进行了研究,但对于老化微塑料的研究仍不充分,且尚无一致定论.
微塑料高级氧化过程与自然老化过程中的氧化途径及老化产物具有极高的相似性[21],可克服自然环境中微塑料的老化速率极低的问题. 因此本研究采用热活化K2S2O8高级氧化法加速微塑料老化以获取不同老化程度的微塑料. 通过室内模拟实验,研究不同老化程度的微塑料对重金属的吸附动力学和吸附等温线的影响,以及在不同pH、微塑料浓度下对重金属的吸附,进一步探讨不同老化程度微塑料吸附行为的差异和作用机理. 采用发光菌毒性测试法对微塑料与重金属的复合污染的生物毒性进行测定,为复合污染的生物毒性评价提供科学依据.
1. 材料与方法 (Materials and methods)
1.1 实验材料
微塑料聚乙烯(PE)、聚丙烯(PP)购于东莞市特塑朗化工原料,平均粒径约100 µm;镉标准溶液(1 mg·mL−1)购于北京北方伟业计量技术研究院;过硫酸钾(K2S2O8)购于天津市风船化学试剂科技有限公司,纯度为99%;明亮发光杆菌T3(Photobacterium phosphoreum T3)由中国科学院南京土壤研究所提供.
1.2 微塑料老化与表征
将微塑料用甲醇超声3次,每次20 min,再以超纯水洗涤3次于40 ℃干燥,密封保存、备用. 采用热活化K2S2O8高级氧化法进行微塑料老化[21],将1.0 g微塑料(PE、PP)加入到40 mL 100 mmol·L−1新鲜制备的pH 7.0的K2S2O8溶液中,于70 ℃恒温搅拌. 每12 h添加等量K2S2O8溶液,每2 天更换新鲜溶液以避免K2SO4积累. 分别于5、10、20 d取出老化的微塑料样品,清洗后于45 ℃烘干避光保存. 将老化微塑料分别记为PE5、PE10、PE20和PP5、PP10、PP20,初始微塑料分别记为PE0和PP0.
利用傅里叶红外光谱仪(FTIR)在分辨率4 cm−1条件下检测500—4000 cm−1区域老化前后微塑料的表面官能团变化,并利用扫描电镜(SEM)表征微塑料的表面形貌变化.
1.3 镉吸附动力学
将镉标准溶液稀释至10 mg·L−1作为镉贮存液,并于4 ℃避光保存. 随后,进一步稀释成1 mg·L−1的镉吸附液. 在摇瓶中加入100 mL的镉吸附液,再分别加入0.2 g初始微塑料及不同老化程度的PE、PP微塑料,置于恒温摇床((25±2)°C、180 r·min−1)避光振荡72 h. 每处理重复3次,同时设置不添加微塑料的空白对照. 分别于0.5、1、3、6、12、24、36、48、60 h进行取样,过0.45 µm滤膜并稀释,采用火焰原子吸收分光光度计测定镉含量.
1.4 镉的吸附等温线
将镉贮存液准确地稀释至0.1、0.5、1、3、5、10 mg·L−1作为镉吸附液进行吸附等温线实验. 每100 mL镉吸附液添加0.2 g初始微塑料和老化20 d的微塑料,每处理重复3次,同时设置不添加微塑料的空白对照,于25 ℃,180 r·min−1振荡培养,并于24、48、72 h时取样. 将样品过0.45 µm滤膜并稀释,采用火焰原子吸收分光光度计对镉含量进行测定.
1.5 不同因素对镉吸附的影响
准确将镉贮存液稀释至1 mg·L−1,并用0.01 mol·L−1的NaOH和HNO3调节该溶液的pH,在不同pH(4、5、6、7、8)环境条件下进行微塑料对Cd的吸附,实验方法同1.4节.
将加入的微塑料浓度设为0.1、0.2、0.4、0.6 g·(100 mL) -1 以探究不同微塑料浓度对Cd吸附的影响,其实验方法同1.4节.
1.6 明亮发光杆菌毒性测定
将达到吸附平衡的微塑料于40 ℃干燥,准确称取0.2 g吸附镉的微塑料,并向其中加入100 mL 3% 的NaCl溶液,并超声使微塑料均匀分散,于摇床中恒温振荡24 h过滤后以浸出液作为待测液,用于发光菌毒性测定. 将明亮发光杆菌T3接种于高温灭菌的专用培养基,其中每100 mL水中酵母浸出粉0.5 g,胰蛋白胨0.5 g,NaCl 3 g,Na2HPO4 0.5 g,KH2PO4 0.1 g,甘油0.3 g,pH值6.5,25 ℃避光培养20 h,于10000 r·min−1离心收集菌体并重悬于3% NaCl溶液以获得合适的菌体浓度. 取待测液180 μL于96孔板,随后加入20 µL重悬后的菌液,每样品重复3次. 同时,按照不同的微塑料吸附量,将镉贮存液稀释至与其相当,测定镉的生物毒性;按照0.2 g·100 mL−1测定微塑料的生物毒性,在微孔板上添加200 μL 3% 的NaCl溶液作为空白对照,于25 ℃下静置15 min,在450—490 nm波长下测定明亮发光杆菌的发光强度,并计算不同处理的发光强度抑制率.
1.7 数据处理与分析
数据统计和分析采用 Excel 2010,图表使用 Origin 2018;用 SPSS进行统计分析.
2. 结果与讨论 (Results and discussion)
2.1 老化前后的微塑料特征
由图1可见,初始微塑料 PE、PP表面较光滑,而老化后微塑料表面的粗糙程度增加,并产生裂纹和孔隙,表面形貌更为破碎. 这一现象与以往的研究结果相似,即在老化过程中,微塑料表面的裂纹数目会增多,且裂缝深度会逐步扩展,会呈现碎片化现象[20]. 在老化作用下,微塑料的表面形态会发生变化,其比表面积、孔隙率可能增加,因此可能对其吸附性能造成一定的影响.
 图 1 微塑料PE(a)、老化20 d的微塑料PE(b)以及微塑料PP(c)、老化20 d的微塑料PP(d)的SEM图Figure 1. SEM images of initial microplastic PE (a), PE aged for 20 d (b), and initial microplastics PP (c), PP aged for 20 d (d)
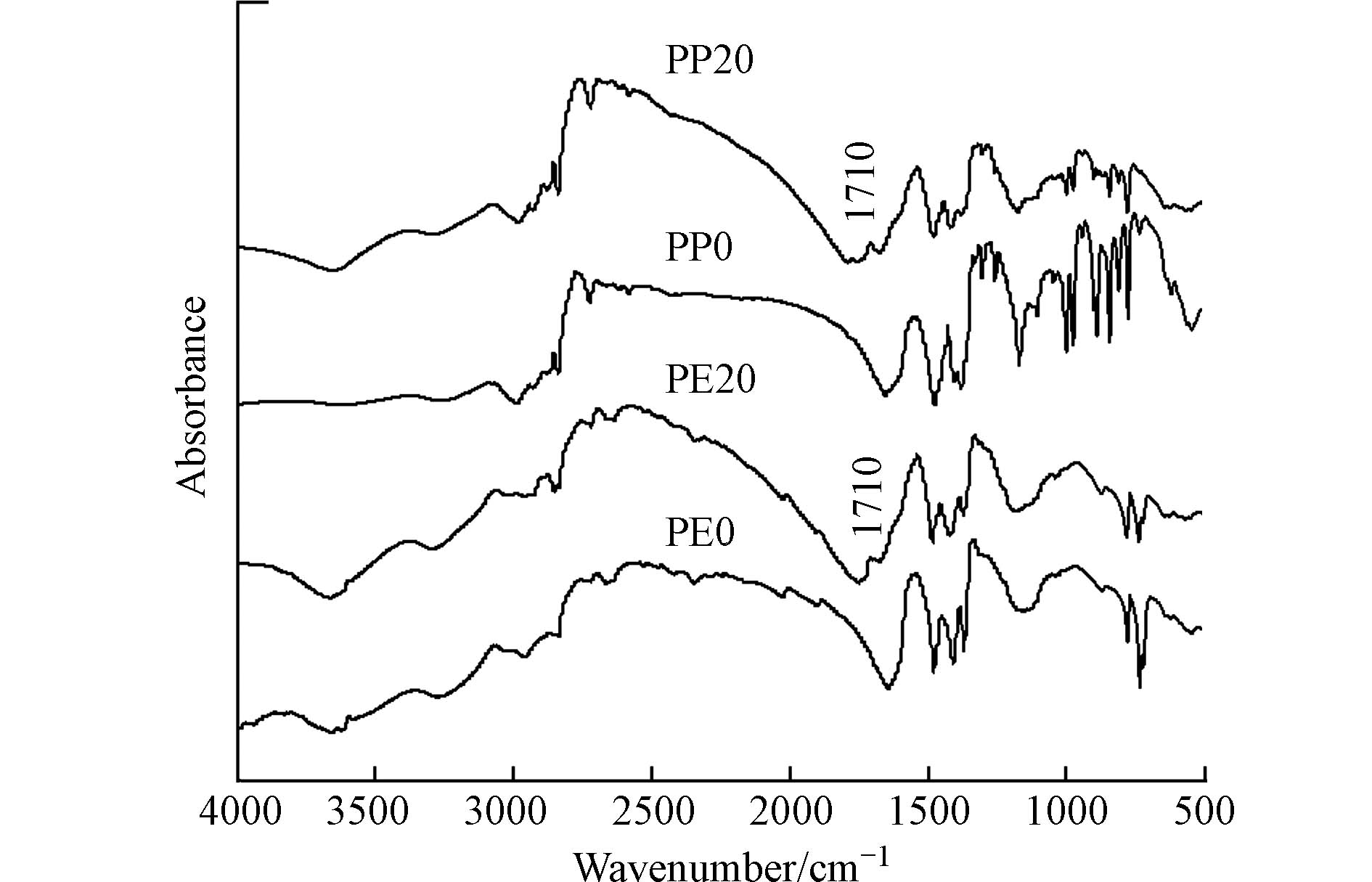
图 1 微塑料PE(a)、老化20 d的微塑料PE(b)以及微塑料PP(c)、老化20 d的微塑料PP(d)的SEM图Figure 1. SEM images of initial microplastic PE (a), PE aged for 20 d (b), and initial microplastics PP (c), PP aged for 20 d (d)采用FTIR对老化前后的微塑料PE和PP表面官能进行表征. 由图2可见,老化后微塑料PE和PP在1710 cm−1出现新的红外吸收峰,其特征峰为甲酸/乙酸等羧酸基团(−CH2COOH),这表明老化能够使微塑料PE、PP表面发生官能团的氧化. 这一结果与Liu等[20]的研究结果一致,微塑料表面的含氧官能团的出现可能会使微塑料的亲水性得到改善[21],进而影响微塑料对重金属的吸附性能.
2.2 镉的吸附动力学
由图3可见,在不同的吸附时间下,微塑料PE、PP对重金属镉离子的吸附出现类似的变化,即随吸附时间延长,微塑料PE、PP对镉离子的吸附量都是增加的,而吸附速率均呈现先增加后减小的趋势. 由图3可见,吸附过程大致可分为3个阶段:0—10 h为快速吸附阶段,此时镉离子在微塑料上的吸附量约为其总量的1/2;10—30 h为第二阶段,微塑料PE、PP对镉离子的吸附速率逐步下降,此时微塑料上仍然有镉离子的吸附,但吸附速率逐渐降低;第三个阶段是30 h之后,吸附达到平衡. 这说明镉离子在微塑料上的吸附并不是瞬间完成的,而是一个由快速到缓慢的平衡过程.
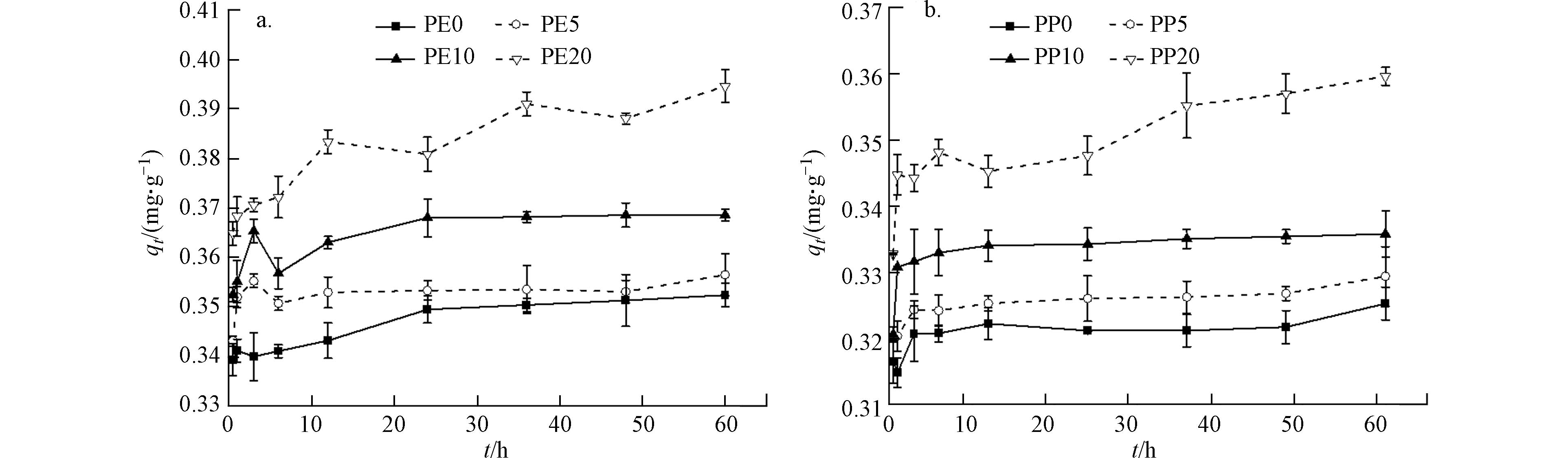 图 3 不同老化程度微塑料PE(a)和PP(b)对镉离子的吸附动力学曲线Figure 3. Adsorption kinetics curves of Cd on microplastics PE (a) and PP (b) with different ageing degrees
图 3 不同老化程度微塑料PE(a)和PP(b)对镉离子的吸附动力学曲线Figure 3. Adsorption kinetics curves of Cd on microplastics PE (a) and PP (b) with different ageing degrees通过微塑料的吸附实验,发现镉离子在微塑料上的吸附量依次为:PE20>PE10>PE5>PE0,PP20 >PP10 >PP5 >PP0. 由此可见,镉离子在老化微塑料的吸附量比初始微塑料的吸附量大,且随老化时间延长其吸附量增加,即微塑料吸附能力与老化时间具有正相关性. 这一发现与已有的研究结论一致,随着老化时间的推移,微塑料表面变得粗糙,其比表面积会增大,同时微塑料表面氧化结合位点、含氧官能团增多,这些都能导致微塑料表面负电性增大,与镉离子的络合作用增加,从而提高微塑料吸附镉离子的能力[22-23].
分别按照准一级动力学方程表达式(1)和准二级动力学方程表达式(2)对吸附动力学进行线性拟合,结果如图4所示. 由图4可见,按照准二级动力学方程表达式进行拟合时线性较好.
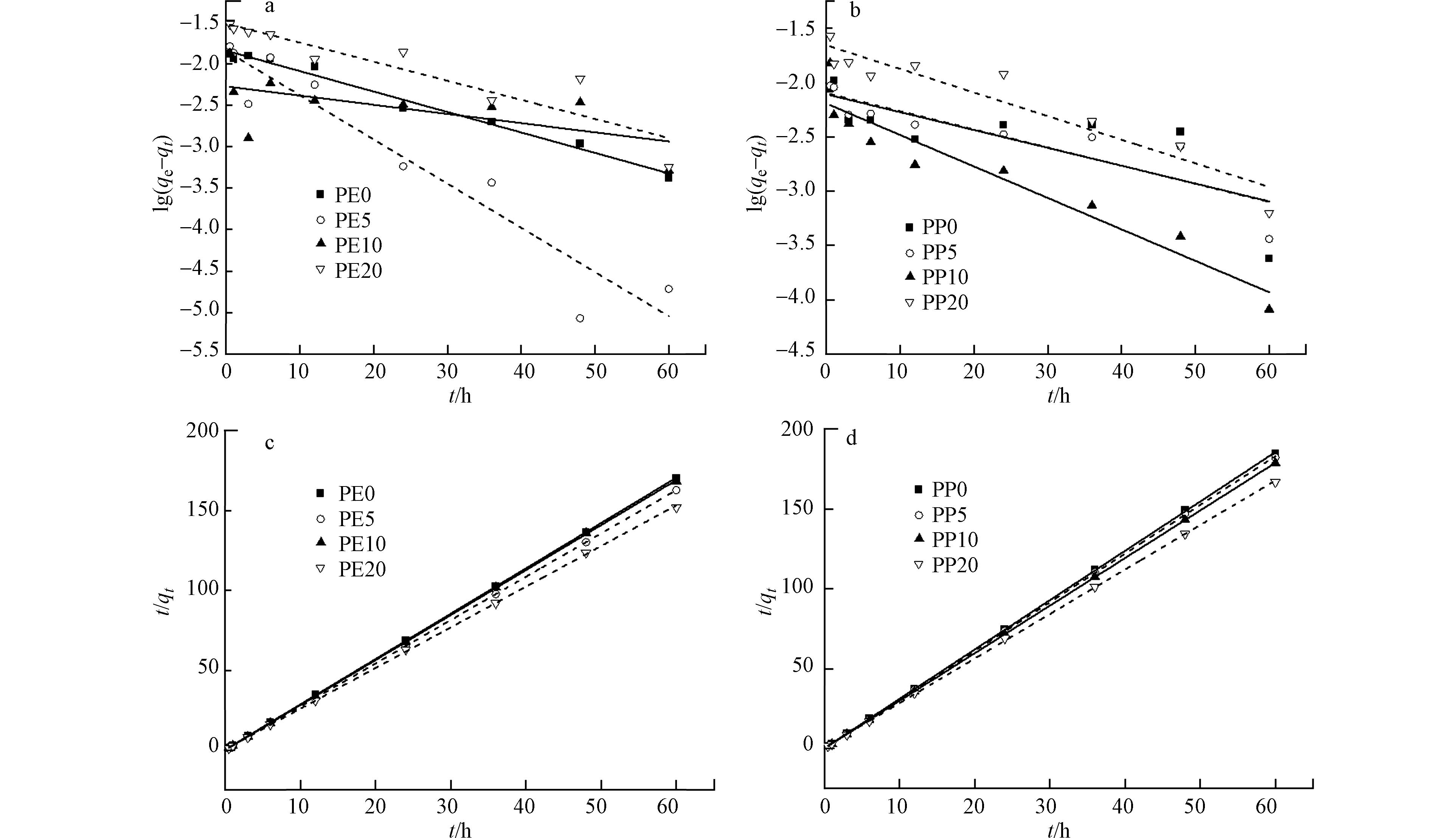 图 4 基于准一级模型(a、b)和准二级模型(c、d)绘制不同微塑料PE、PP吸附动力学线性图Figure 4. Linear diagram of adsorption kinetics based on quasi-first-order model (a, b)and quasi-second-order model (c, d)for different microplastics PE and PP
图 4 基于准一级模型(a、b)和准二级模型(c、d)绘制不同微塑料PE、PP吸附动力学线性图Figure 4. Linear diagram of adsorption kinetics based on quasi-first-order model (a, b)and quasi-second-order model (c, d)for different microplastics PE and PP准一级动力学方程表达式:
lg(qe−qt)=lgqe−k12.303t (1) 其中,k1(h−1)一级吸附速率常数,qt(mg·g−1)为t时刻吸附量;qe(mg·g−1)为平衡时吸附量;t(h)为时间.
准二级动力学方程表达式为:
tqt=1k2qe2+1qet (2) 其中,k2(g·mg−1·h−1)为二级吸附速率常数.
同时,表1列出了相关的动力学模型参数,其中按照准二级动力学方程表达式计算获得的相关系数R2在0.9994—0.9999之间,而按照准一级动力学方程表达拟合获得的相关系数R2在0.3788—0.9971之间. 由此可见,准二级模型对于微塑料对镉离子的吸附动态特性拟合效果更佳,而准一级模型则不能很好地拟合. 这表明镉离子在微塑料上的吸附行为符合准二级动力学方程,微塑料对镉离子的吸附存在多个吸附阶段,物理吸附和化学吸附同时存在,其中以化学吸附为主.
表 1 由准一级模型和准二级模型得到的吸附动力学参数Table 1. Adsorption kinetic parameters obtained from the quasi-first-order model and the quasi-second-order model微塑料Microplastics 准一级动力学方程Quasi-first-order model 准二级动力学方程Quasi-second-order model k1 qe/ (mg·g−1) R2 k2 qe/(mg·g−1) R2 PE0 14.4378 0.3496 0.9840 8.3893 0.3452 0.9999 PE5 6.5974 0.3652 0.9161 7.3099 0.3698 0.9999 PE10 6.8295 0.3547 0.3788 7.8927 0.3560 0.9996 PE20 6.1482 0.3813 0.8410 6.4735 0.3931 0.9998 PP0 8.5874 0.3207 0.6000 9.6525 0.3219 0.9994 PP5 10.1068 0.3250 0.7847 9.4340 0.3256 0.9997 PP10 7.6493 0.3337 0.9124 8.8810 0.3355 0.9996 PP20 5.9236 0.3504 0.9971 7.7520 0.3590 0.9998 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.3 镉的吸附等温线研究
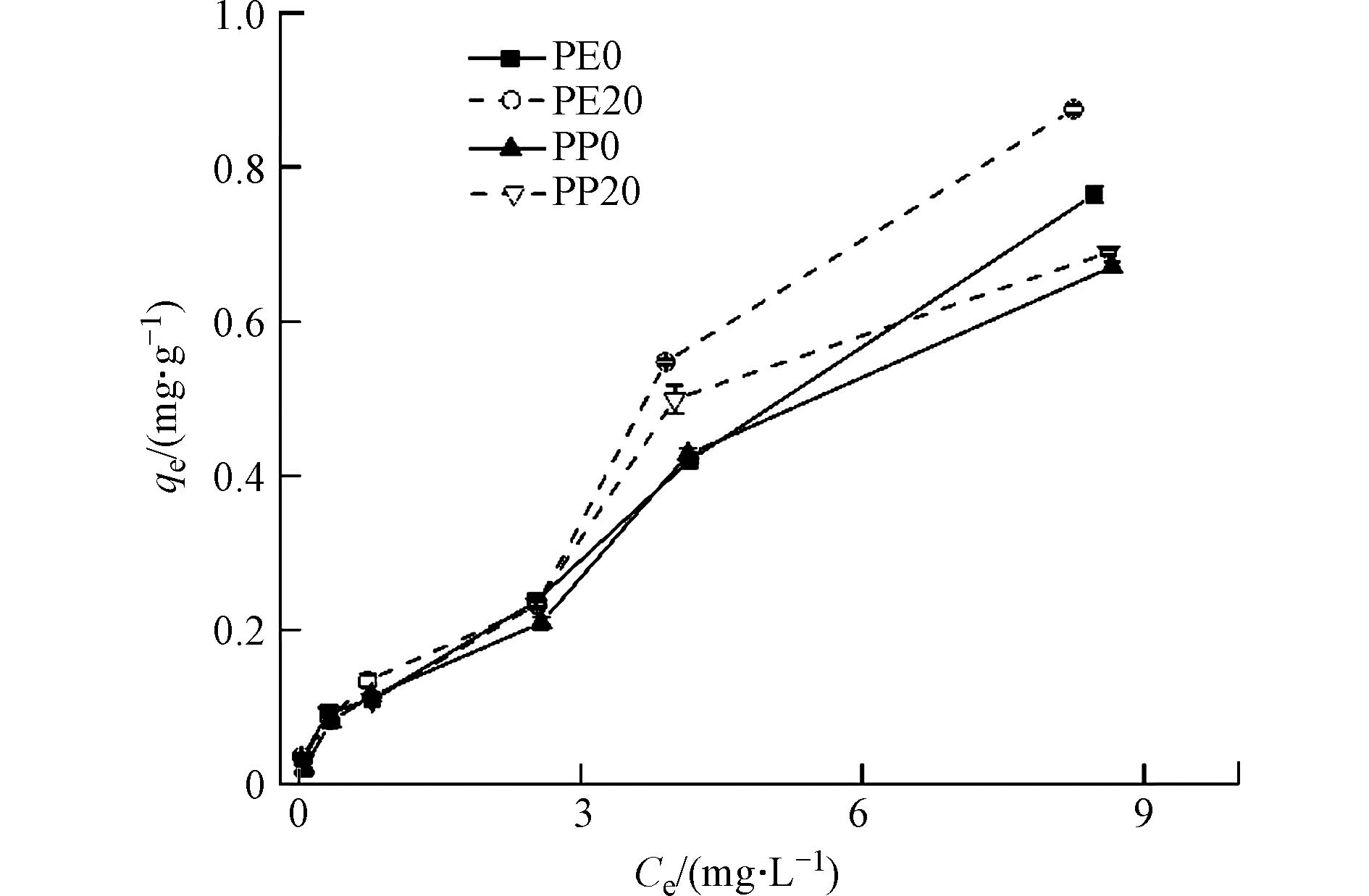
在25 ℃下,镉离子在微塑料PE、PP表面吸附达到平衡后,以平衡时溶液中镉离子浓度(Ce)为横坐标,以微塑料对镉离子的平衡吸附量为纵坐标绘图. 如图5所示,随着镉离子浓度增加,镉离子在微塑料PE、PP上的平衡吸附量也随之增加,但曲线的斜率却在不断降低. 这表明在镉离子浓度增加的同时微塑料表面镉的吸附量也逐渐增加,但其吸附速率却在逐渐降低. 这是由于微塑料与镉离子的吸附存在一定的定位性,即在较低浓度的镉离子溶液中,微塑料的表面存在大量的吸附位点,能够迅速吸附镉. 但随着镉离子浓度的升高,微塑料表面被占据的吸附点位也越来越多,吸附逐渐趋于饱和,进而影响吸附速率. 这表明镉离子的平衡吸附量与老化程度也存在一定的相关性,老化微塑料表面镉吸附量大于初始微塑料,即PE20 >PE0,PP20 >PP0. 由此可见,老化微塑料比初始微塑料具有更好的吸附性能,这也与老化微塑料表面形貌和官能团变化相关.
进一步利用Freundlich和Langmuir等温吸附模型对微塑料吸附等温线进行拟合,结果如图6和 表2所示. Freundlich和Langmuir模型拟合线性方程分别如公式(3)和(4)所示:
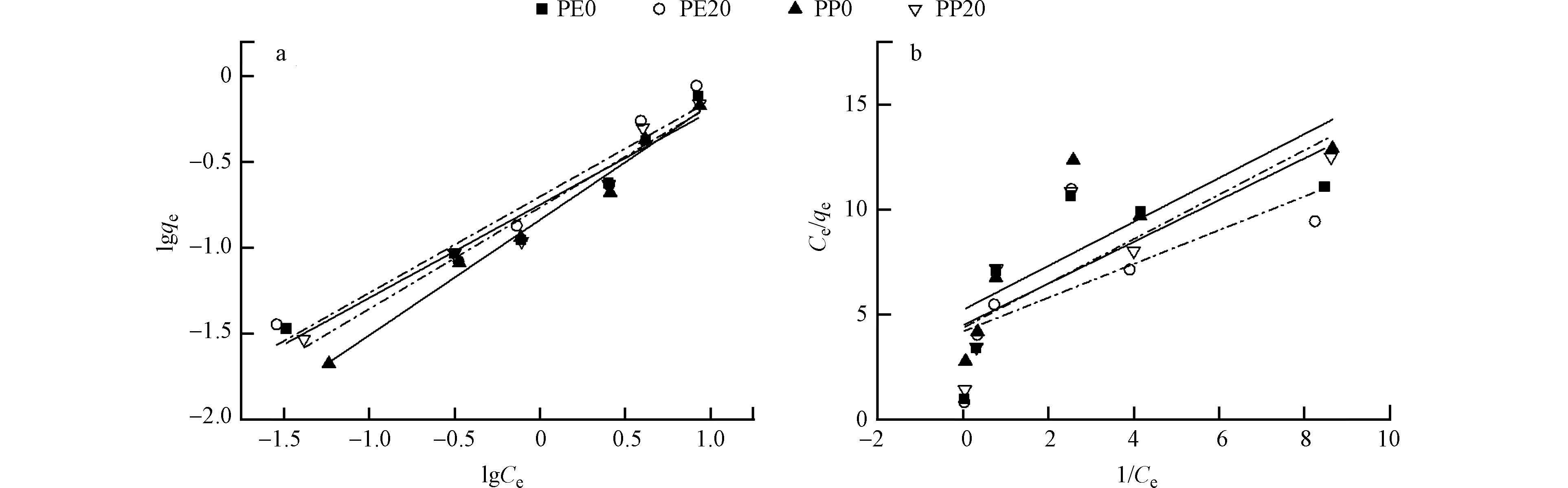 图 6 基于Freundlich模型(a)和Langmuir模型(b)绘制的吸附等温线线性图Figure 6. Linear adsorption isotherms based on Freundlich model (a) and Langmuir model (b)表 2 由Freundlich模型和Langmuir模型得到的镉离子在微塑料上的吸附等温线参数Table 2. Adsorption isotherm parameters of Cd on microplastics obtained by Freundlich model and Langmuir model
图 6 基于Freundlich模型(a)和Langmuir模型(b)绘制的吸附等温线线性图Figure 6. Linear adsorption isotherms based on Freundlich model (a) and Langmuir model (b)表 2 由Freundlich模型和Langmuir模型得到的镉离子在微塑料上的吸附等温线参数Table 2. Adsorption isotherm parameters of Cd on microplastics obtained by Freundlich model and Langmuir model微塑料Microplastics 温度/℃Temperature Freundlich Langmuir KF n R2 KL Qmax R2 PE0 25 0.0301 0.6621 0.9783 0.1562 1.178 0.5861 PE20 25 0.0300 0.6958 0.9694 0.1626 1.5196 0.4516 PP0 25 0.0216 0.7354 0.9886 0.1402 1.0987 0.6591 PP20 25 0.0275 0.6887 0.9791 0.1600 1.0959 0.6592 | Show TableDownLoad:
CSV
Freundlich:lgqe=lgKF+nlgCe (3) 其中,KF为吸附平衡常数,n为吸附过程中的支持力.
Langmuir:Ceqe=αKLCe+1KL (4) 其中,KL为平衡常数.
由表2中两种模型拟合的吸附等温线相关系数R2可以看出,Freundlich吸附模型R2的值在0.9694—0.9886之间,而Langmuir吸附模型R2的值在0.4516—0.6592之间,因此Freundlich等温模型能够较好地反映出镉离子在微塑料PE、PP上的吸附行为,即吸附等温线数据符合Freundlich吸附模型. 这一结果表明镉离子在微塑料 PE、PP上的吸附是非线性的,且其吸附过程具有不均匀性. 这与前人的研究结果相吻合[24],即镉离子先在微塑料表面的高能吸附点上进行吸附,再在低能吸附点上进行吸附. 这也进一步表明微塑料表面吸附镉的过程是由表面络合和静电引力共同控制的.
2.4 不同因素对镉在微塑料上吸附的影响
2.4.1 pH对微塑料吸附镉的影响
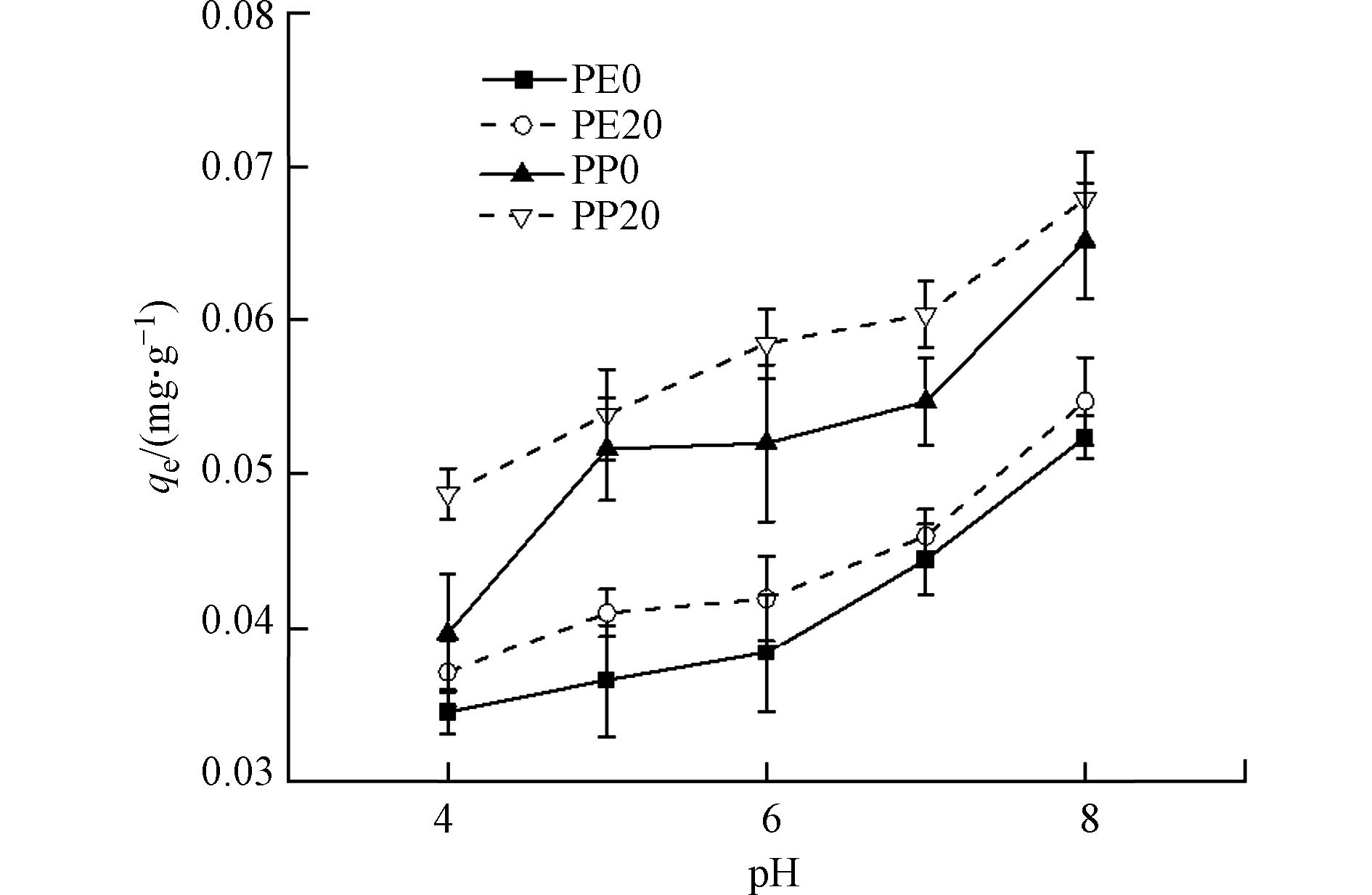
如图7所示,微塑料在不同pH值下对镉离子的吸附能力有所不同,随着 pH值的增加其吸附量增加,且老化微塑料对镉离子的吸附量大于初始微塑料. Brennecke等[25]研究发现,pH值对微塑料吸附镉离子的性能有很大影响,在低 pH值时,H+与镉离子能够竞争吸附点位导致微塑料对镉离子的吸附性能下降;而随着 pH值的增加,微塑料表面的官能团被去质子化,微塑料的负电性会增加,因此微塑料和镉离子间的静电作用会加强,微塑料吸附镉离子的能力则相应增加[25]. 这也进一步说明了静电作用对微塑料的吸附性能有很大的促进作用,微塑料吸附镉离子的主要机理可能是静电作用. 此外,受pH值的影响,老化及初始微塑料PP对镉离子的吸附量均高于PE,这可能与微塑料材质有关.
2.4.2 不同微塑料浓度对镉吸附的影响
图8比较了不同微塑料浓度对镉离子的吸附量的影响. 由图8可见,在保持其它条件不变的情况下,随着微塑料浓度增加,两种类型微塑料对镉离子的吸附量皆会相应地下降. 但老化微塑料对镉离子的吸附量仍比初始微塑料大,这一研究结果与以往的研究结果相符,即当微塑料浓度较低时,微塑料在水中的分布比较均匀,可以很好地与水中的镉离子相结合[9]. 当微塑料的浓度升高时,微塑料的吸附点位会增多,使得微塑料颗粒间发生相互竞争,并且微塑料之间会发生相互黏合,使得在水中分散不均匀,与水中的镉离子接触不足,导致单位吸附量下降[9].
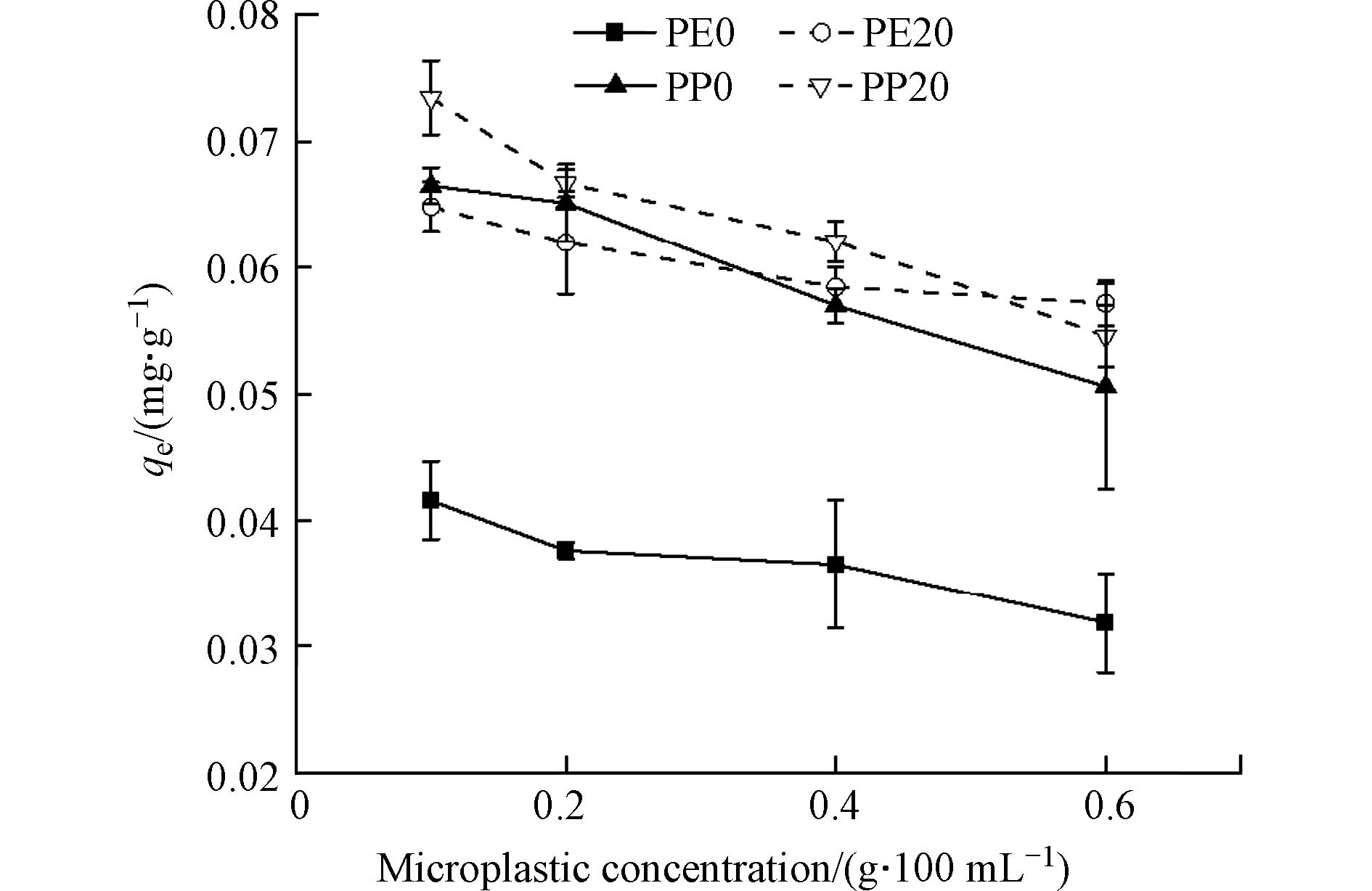 图 8 微塑料浓度对微塑料吸附镉的影响Figure 8. Influence of microplastic concentration on Cd adsorption
图 8 微塑料浓度对微塑料吸附镉的影响Figure 8. Influence of microplastic concentration on Cd adsorption2.5 微塑料与镉的联合生物毒性
利用明亮发光杆菌T3对镉、微塑料及其二者的联合生物毒性进行测定,结果如图9所示. 初始微塑料对T3的发光强度几乎没有影响,而老化微塑料对T3的发光强度有轻微抑制,其中PE20抑制了4.5%的发光强度,PP20对T3发光抑制率约为6.6%. 这可能是老化微塑料在溶液中释放出了一些有毒物质,对发光菌产生了生物毒性. 此外,不同镉离子浓度下,T3发光强度也受到了抑制,且随着镉离子浓度的升高发光强度逐渐降低,即发光强度抑制率随镉离子浓度升高而升高(图9a). 这是由于重金属可以改变细胞膜的渗透率,阻碍氧气的透过,从而能够影响到细胞的呼吸,影响到发光[26]. 另外,由于发光菌的呼吸链位于细胞膜中,所以能影响细胞膜的因素都会影响到细胞的正常代谢,从而影响到发光.
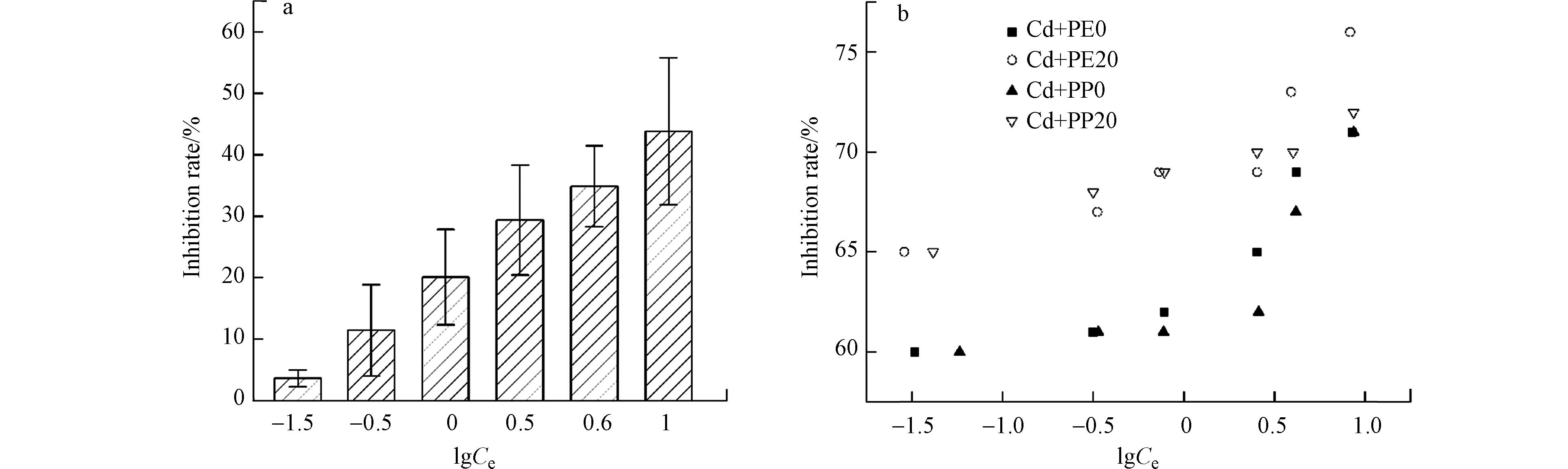 图 9 单独镉离子的生物毒性(a)及微塑料与镉的联合生物毒性(b)Figure 9. Biotoxicity of Cd (a) and combined biotoxicity of microplastics and Cd (b)
图 9 单独镉离子的生物毒性(a)及微塑料与镉的联合生物毒性(b)Figure 9. Biotoxicity of Cd (a) and combined biotoxicity of microplastics and Cd (b)由图9b可见,镉与微塑料的联合生物毒性大于二者单独作用时的生物毒性,对T3发光强度的抑制率均在60%以上. 随着微塑料吸附镉量的增加其联合生物毒性逐渐增强,这表明重金属镉与微塑料复合污染存在较强的生物毒性,其对发光强度的抑制程度与吸附的镉离子量之间存在着正相关关系. 由此可见,微塑料与镉对发光菌的生物毒性存在协同效应,且老化微塑料与镉的联合生物毒性较强. 这一结论与以往的研究相符合,即有毒物质如重金属、微塑料等,会抑制发光菌体内的核酸、蛋白质等的合成,从而抑制其新陈代谢,使得发光菌的正常代谢受到胁迫,而发光菌发光所依赖的荧光酶是其呼吸作用代谢过程中不可或缺的物质,因而镉离子与微塑料都会影响到发光菌的正常发光,而其复合形态的胁迫下,其对于发光强度抑制进一步加强,表现为协同作用[27].
3. 结论 (Conclusion)
(1)微塑料老化会使微塑料的表面结构发生变化,其孔隙率和比表面积增大并出现含氧官能团. 老化微塑料对重金属镉的吸附量大于初始微塑料,且与老化时间呈正相关.
(2)微塑料对重金属镉的吸附过程大致分成3个阶段:短时间内的快速吸附,较长时期的缓慢吸附,及达到吸附平衡后逐渐稳定. 吸附动力学数据符合准二级动力学模型,吸附等温线数据符合Freundlich模式,且吸附作用主要受表面络合和静电引力的影响.
(3)微塑料对镉离子的吸附量易受到外界环境因素影响. 随着pH值升高,由于微塑料表面的去质子化作用其对Cd的吸附量升高;随着微塑料浓度升高,由于微塑料表面结合位点的竞争作用其对Cd的吸附量下降.
(4)微塑料复合镉污染对发光菌的生物毒性表现出协同效应,且随吸附镉离子量的增加其生物毒性增强,其中老化微塑料复合镉表现出更强的生物毒性.
-

图 1 NiFe2O4、Bi2WO6和NiFe-Bi-XY的晶相与成键表征
Figure 1. Characterization of crystal phase and bonding of NiFe2O4, Bi2WO6 and NiFe-Bi-XY
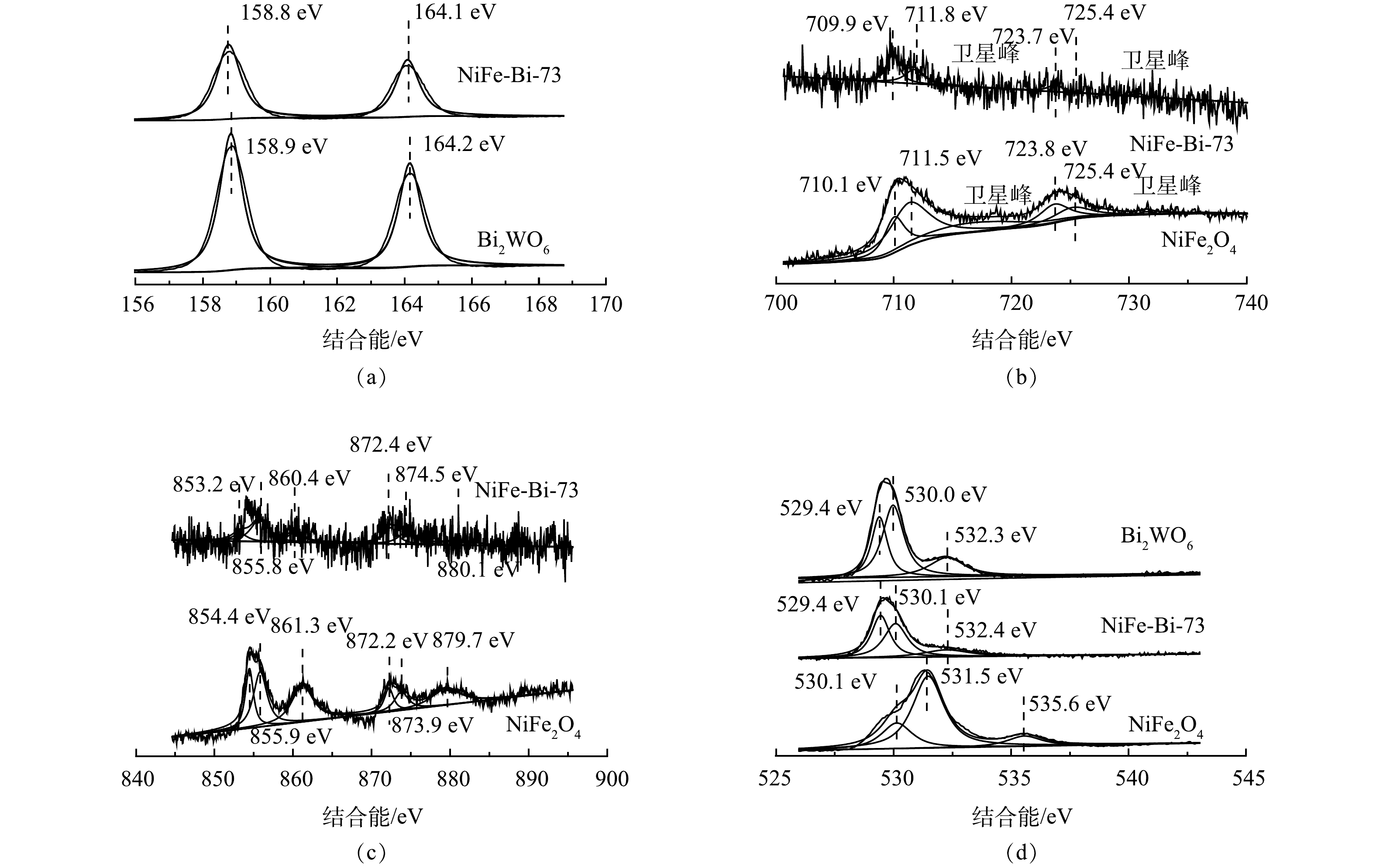
图 2 Bi2WO6,NiFe2O4和NiFe-Bi-73样品的XPS图谱
Figure 2. XPS spectra of Bi2WO6,NiFe2O4 and NiFe-Bi-73

图 4 催化剂材料的光化学与光电化学性质表征
Figure 4. Characterization of photochemical and photoelectrochemical properties of catalyst materials

图 6 太阳光/NiFe-Bi-73/PMS体系降解四环素的最佳反应条件确定
Figure 6. Optimal parameters for tetracycline degradation by NiFe-Bi-73/solar light/PMS system
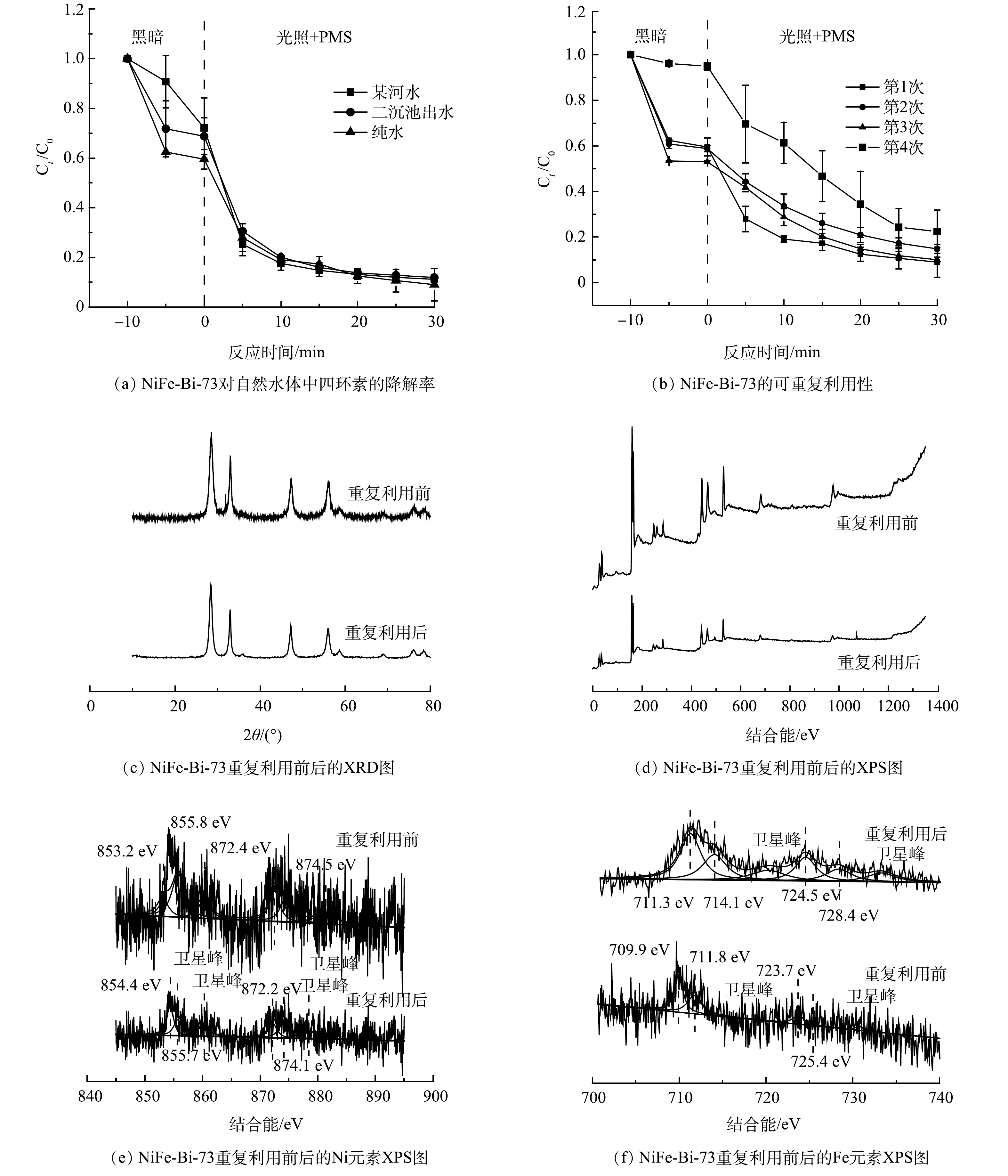
图 8 太阳光/NiFe-Bi-73催化剂/PMS对实际水体的适用性
Figure 8. Tetracycline removal in actual water by NiFe-Bi-73/solar light/PMS system
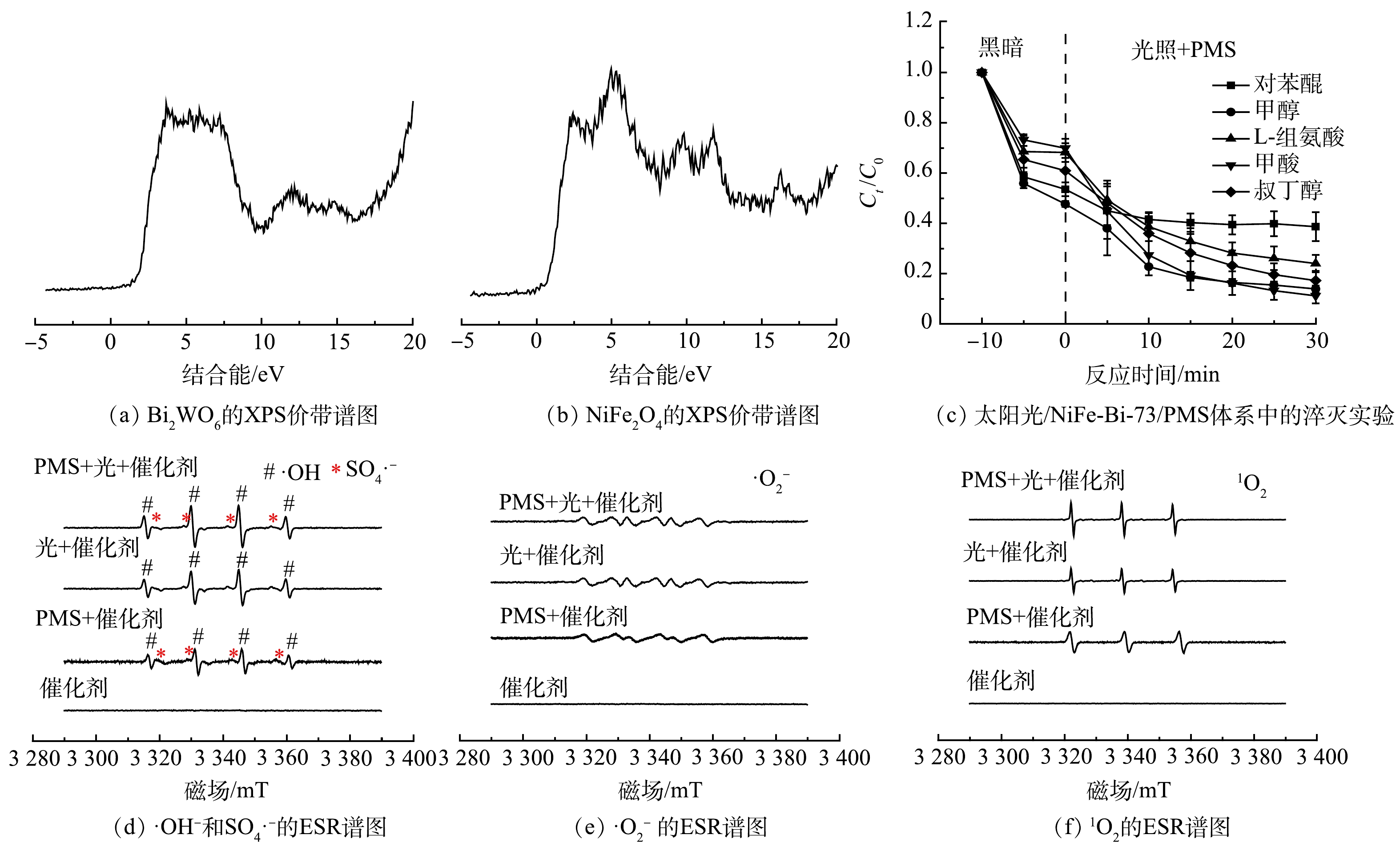
图 10 催化剂能带与活性物质的确定
Figure 10. Determination of catalyst energy band and active species
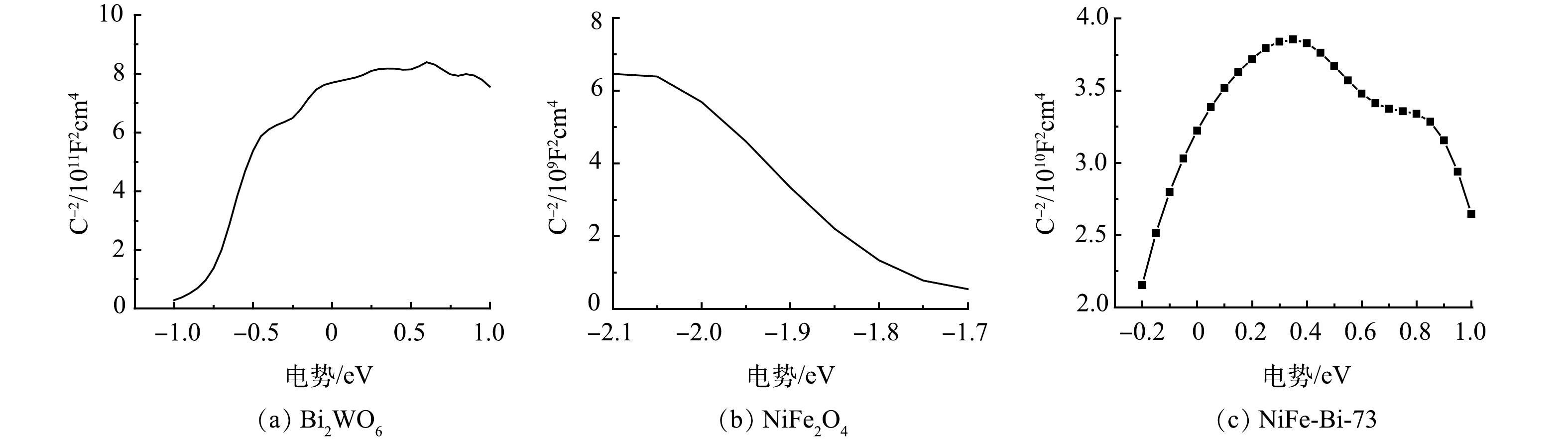
图 11 Bi2WO6、NiFe2O4和NiFe-Bi-73的Mott-Schottky测试
Figure 11. Mott-Schottky test of Bi2WO6、NiFe2O4 and NiFe-Bi-73
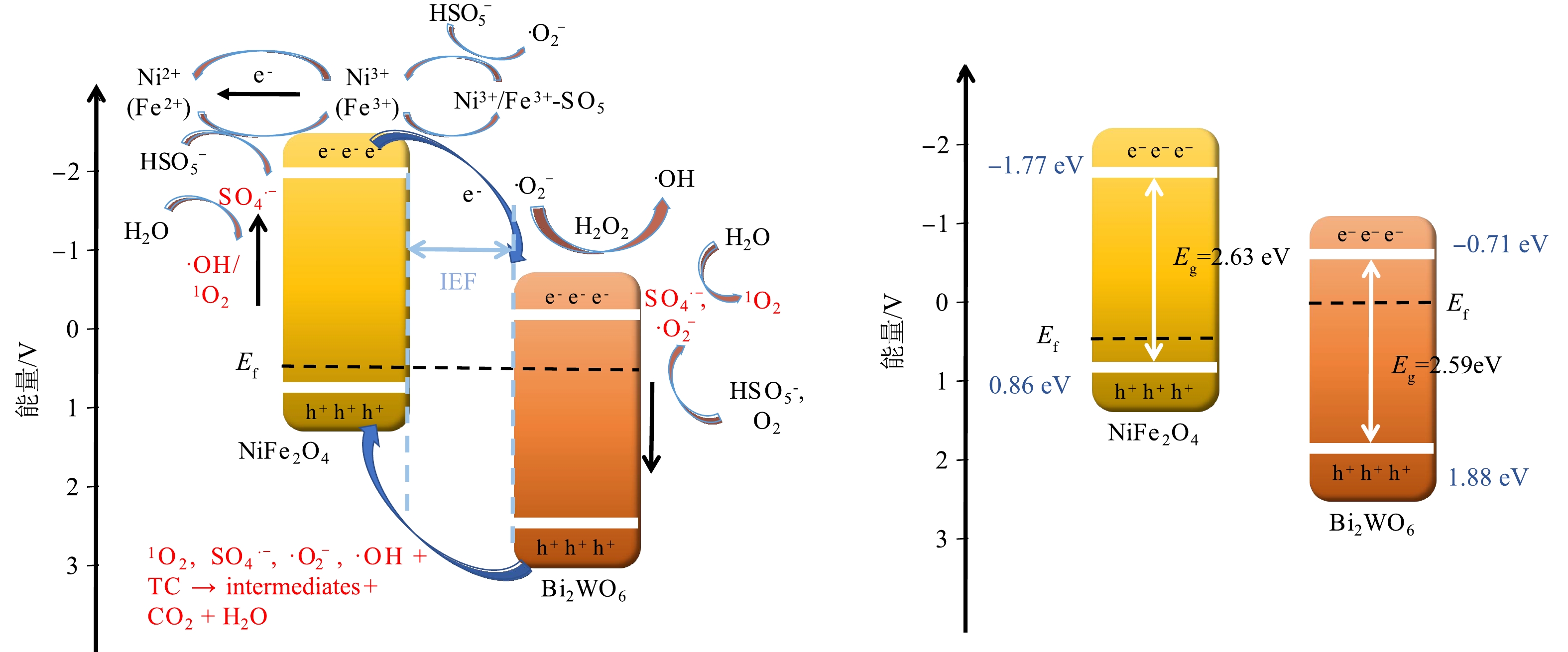
图 12 在太阳光/NiFe-Bi-73 /PMS体系中Bi2WO6/NiFe2O4异质结的催化反应机制
Figure 12. Catalytic charge transfer mechanism of Bi2WO6/NiFe2O4 heterojunction in NiFe-Bi-73/solar light/PMS system
表 1 不同催化剂材料对于四环素降解的效果对比
Table 1. Comparison of different catalyst materials for photocatalytic degradation of tetracycline
下载: 导出CSV
表 2 某污水处理厂二沉池出水和某地表河水的水质参数
Table 2. Water quality of a secondary sedimentation tank effluent and a surface river.
水源 电导率/(μs·cm−1) TOC/(mg·L−1) ORP/mV pH 其他无机离子的质量浓度/( mg·L−1) NO3− Cl− F− SO42− K+ Ca2+ Na+ Mg2+ Al3+ 二沉池 873 3.39 154.4 7.54 10.5 160.02 0.35 51.92 23.04 30.54 97 10.08 0.08 河水 325 3.95 138.3 7.73 6.49 15.9 0.39 31 9.21 38.47 16.89 3.68 0.06
下载: 导出CSV
-
[1] MANAGAKI S, MURATA A, TAKADA H, et al. Distribution of macrolides, sulfonamides, and trimethoprim in tropical waters: Ubiquitous occurrence of veterinary antibiotics in the Mekong Delta[J]. Environmental Science and Technology, 2007, 41(23): 8004-8010. doi: 10.1021/es0709021 [2] ZHU X D, WAND Y J, SUN R J, et al. Photocatalytic degradation of tetracycline in aqueous solution by nanosized TiO2[J]. Chemosphere, 2013, 92(8): 925-932. doi: 10.1016/j.chemosphere.2013.02.066 [3] HONG Y, LI C, YIN B, et al. Promoting visible-light-induced photocatalytic degradation of tetracycline by an efficient and stable beta-Bi2O3@g-C3N4 core/shell nanocomposite[J]. Chemical Engineering Journal, 2018, 338: 137-146. doi: 10.1016/j.cej.2017.12.108 [4] WERNER J J, ARNOLD W. A, MCNEILl K. Water hardness as a photochemical parameter: Tetracycline photolysis as a function of calcium concentration, magnesium concentration, and pH[J]. Environmental Science and Technology, 2006, 40: 7236-7241. doi: 10.1021/es060337m [5] CHEN X, ZHOU J b, CHEN Yi, et al. Degradation of tetracycline hydrochloride by coupling of photocatalysis and peroxymonosulfate oxidation processes using CuO-BiVO4 heterogeneous catalyst[J]. Process Safety and Environmental Protection, 2021, 145: 364-377. doi: 10.1016/j.psep.2020.08.016 [6] LU J, SUN J X, CHEN X X, et al. Efficient mineralization of aqueous antibiotics by simultaneous catalytic ozonation and photocatalysis using MgMnO3 as a bifunctional catalyst[J]. Chemical Engineering Journal, 2019, 358: 48-57. doi: 10.1016/j.cej.2018.08.198 [7] ZHANG Y, Zhou J B, Chen X, et al. Coupling of heterogeneous advanced oxidation processes and photocatalysis in efficient degradation of tetracycline hydrochloride by Fe-based MOFs: Synergistic effect and degradation pathway[J]. Chemical Engineering Journal, 2019, 369: 745-757. doi: 10.1016/j.cej.2019.03.108 [8] WANG, J, WANG S. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 2018, 334: 1502-1517. doi: 10.1016/j.cej.2017.11.059 [9] Xu B, Ahmed M. Zhou J, et al. Visible and UV photocatalysis of aqueous perfluorooctanoic acid by TiO2 and peroxymonosulfate: Process kinetics and mechanistic insights[J]. Chemosphere, 2020, 243: 25366. [10] TRUONG T, NGUYEN T, PHUONG La, et al. Insight into the degradation of p-nitrophenol by visible-light-induced activation of peroxymonosulfate over Ag/ZnO heterojunction[J]. Chemosphere, 2021, 268: 129291. doi: 10.1016/j.chemosphere.2020.129291 [11] QIU P. CHENG Z, XUE N, et al. The synergistic effect in metal-free graphene oxide coupled graphitic carbon nitride/light/peroxymonosulfate system: Photothermal effect and catalyst stability[J]. Carbon, 2021, 178: 81-91. doi: 10.1016/j.carbon.2021.02.088 [12] ZHU H, LI Z, YANG J. A novel composite hydrogel for adsorption and photocatalytic degradation of bisphenol A by visible light irradiation[J]. Chemical Engineering Journal, 2018, 334: 1679-1690. doi: 10.1016/j.cej.2017.11.148 [13] JIANG J J, WaANG X Y, ZHANG C J, et al. Porous 0D/ 3D NiCo2O4/g-C3N4 accelerate emerging pollutant degradation in PMS/vis system: Degradation mechanism, pathway and toxicity assessment[J]. Chemical Engineering Journal, 2020, 397: 125363. [14] JIN C Y, WANG M, LI Z L, et al. Two dimensional Co3O4/g-C3N4 Z-scheme heterojunction: Mechanism insight into enhanced peroxymonosulfate-mediated visible light photocatalytic performance[J]. Chemical Engineering Journal, 2020, 398: 125569. doi: 10.1016/j.cej.2020.125569 [15] TIAN X, TIAN C, NIE Y, et al. Controlled synthesis of dandelion-like NiCo2O4 microspheres and their catalytic performance for peroxymonosulfate activation in humic acid degradation[J]. Chemical Engineering Journal, 2018, 331: 144-151. doi: 10.1016/j.cej.2017.08.115 [16] LASSOUED A, LASSOUED M S, DKHIL B, et al. Photocatalytic degradation of methyl orange dye by NiFe2O4 nanoparticles under visible irradiation: Effect of varying the synthesis temperature[J]. Journal of Materials Science:Materials in Electronics, 2020, 31(13): 10943. doi: 10.1007/s10854-020-03457-w [17] GEBRESLASSIE G, BHARALI P, CHANDRA U, et al. Hydrothermal synthesis of g-C3N4/NiFe2O4 nanocomposite and its enhanced photocatalytic activity[J]. Applied Organometallic Chemistry, 2019, 33(8): e5002. [18] ZHANG L W, ZHU Y F. A Review of controllable synthesis and enhancement of performances of bismuth tungstate visible-light-driven photocatalysts[J]. Catalysis Science & Technology, 2012, 2(4): 694-76. [19] LI M, LAI C, YI H, et al. Chen. Multiple charge-carrier transfer channels of Z-scheme bismuth tungstate-based photocatalyst for tetracycline degradation: Transformation pathways and mechanism[J]. Journal of Colloid and Interface Science, 2019, 555: 770-782. doi: 10.1016/j.jcis.2019.08.035 [20] HE Z, XIA Y, TANG B, et al. Fabrication and photocatalytic property of magnetic NiFe2O4/Cu2O composites[J]. Materials Research Express, 2017, 4(9): 95501. doi: 10.1088/2053-1591/aa7cb8 [21] LU H, ZHU Z, ZHANG H, et al. Simultaneous removal of arsenate and antimonate in simulated and practical water samples by adsorption onto Zn/Fe layered double hydroxide[J]. Chemical Engineering Journal, 2015, 276: 365-375. doi: 10.1016/j.cej.2015.04.095 [22] YU J, XIONG J, CHENG B, et al. Hydrothermal preparation and visible-light photocatalytic activity of Bi2WO6 powders[J]. Journal of Solid State Chemistry, 2005, 178(6): 1968-1972. doi: 10.1016/j.jssc.2005.04.003 [23] BAIG M, PERVAIZ E, AZAD M, et al. NiFe2O4/SiO2 nanostructures as a potential electrode material for high rated supercapacitors[J]. Ceramics International, 2021, 47(9): 12557-12566. doi: 10.1016/j.ceramint.2021.01.113 [24] ISSARAPANACHEEWIN S, WETCHAKUN K, PHANICHPHANT S, et al. Efficient photocatalytic degradation of rhodamine by a novel CeO2/Bi2WO6 composite film[J]. Catalysis Today, 2016, 278: 280-290. doi: 10.1016/j.cattod.2015.12.028 [25] ZENG J, ZENG W, ZENG H. In situ plasmonic au nanoparticle anchored nickel ferrite: An efficient plasmonic photocatalyst for fluorescein-sensitized hydrogen evolution under visible light irradiation[J]. Journal of Solid State Chemistry, 2017, 253: 294-304. doi: 10.1016/j.jssc.2017.06.005 [26] BHUVANESWARI S, PRATHEEKSHA P M, ANANDAN S, et al. Efficient reduced graphene oxide grafted porous Fe3O4 composite as a high performance anode material for Li-ion batteries[J]. Physical Chemistry Chemical Physics, 2014, 16(11): 5284-5294. doi: 10.1039/c3cp54778g [27] LI X, XIN M, GUO S, et al. Yan. Insight of synergistic effect of different active metal ions in layered double hydroxides on their electrochemical behaviors[J]. Electrochimica Acta, 2017, 253: 302-310. doi: 10.1016/j.electacta.2017.09.075 [28] HE G, WANG Y, CHEN X, et al. Laser in situ synthesis of NiFe2O4 nanoparticle-anchored NiFe(OH)x nanosheets as advanced electrocatalysts for the oxygen evolution and urea oxidation reactions[J]. Electrochimica Acta, 2022, 411: 140074. doi: 10.1016/j.electacta.2022.140074 [29] CHENG M, FAN H, SONG Y, et al. Interconnected hierarchical NiCo2O4 microspheres as high-performance electrode materials for supercapacitors[J]. Dalton Transactions, 2017, 46(28): 9201-9209. doi: 10.1039/C7DT01289F [30] LI Y, SHEN J, QUAN W, et al. 2D/2D p-n heterojunctions of CaSb2O6/g-C3N4 for visible light-driven photocatalytic degradation of tetracycline[J]. European Journal of Inorganic Chemistry, 2020, 2020(40): 3852-3858. doi: 10.1002/ejic.202000635 [31] WANG Y H, DING L Z, LIU C, et al. 0D/2D/2D ZnFe2O4/Bi2O2CO3/BiOBr double Z-scheme heterojunctions for the removal of tetracycline antibiotics by permonosulfate activation: Photocatalytic and non-photocatalytic mechanisms, radical and non-radical pathways[J]. Separation and Purification Technology, 2022, 283: 120-164. [32] SHI Y L, FENG X J, GUAN H Y, et al. Porous sunflower plate-like NiFe2O4/CoNi-S heterostructure as efficient electrocatalyst for overall water splitting[J]. International Journal of Hydrogen Energy, 2021, 46(12): 8557-566. doi: 10.1016/j.ijhydene.2020.12.062 [33] WU S H, LI H R, LI X, et al. Performances and mechanisms of efficient degradation of atrazine using peroxymonosulfate and ferrate as oxidants[J]. Chemical Engineering Journal, 2018, 353: 533-541. doi: 10.1016/j.cej.2018.06.133 [34] NIU J, Ding S, Zhang L, et al. Visible-light-mediated Sr-Bi2O3 photocatalysis of tetracycline: Kinetics, mechanisms and toxicity assessment[J]. Chemosphere, 2013, 93(1): 1-8. doi: 10.1016/j.chemosphere.2013.04.043 [35] LIU H, Liang C, NIU C, et al. Facile assembly of g-C3N4/Ag2CO3/graphene oxide with a novel dual Z-scheme system for enhanced photocatalytic pollutant degradation[J]. Applied Surface Science, 2019, 475: 421-434. doi: 10.1016/j.apsusc.2019.01.018 [36] LIU W, Zhou J, LIU D, et al. Enhancing electronic transfer by magnetic iron materials and metal-organic framework via heterogeneous Fenton-like process and photocatalysis[J]. Materials Science in Semiconductor Processing, 2021, 135: 106096. doi: 10.1016/j.mssp.2021.106096 [37] ALPAY A, TUNA, SIMSEK E, et al. Deposition of perovskite-type LaFeO3 particles on spherical commercial polystyrene resin: A new platform for enhanced photo-Fenton-catalyzed degradation and simultaneous wastewater purification[J]. Environmental Technology & Innovation, 2020, 20: 101175. [38] JIN C, KANG J, LI Z, et al. Enhanced visible light photocatalytic degradation of tetracycline by MoS2/Ag/g-C3N4 Z-scheme composites with peroxymonosulfate[J]. Applied Surface Science, 2020, 514: 146076. doi: 10.1016/j.apsusc.2020.146076 [39] SHI Y, LI J, WAN D, et al. Peroxymonosulfate-enhanced photocatalysis by carbonyl-modified g-C3N4 for effective degradation of the tetracycline hydrochloride[J]. Science of the Total Environment, 2020, 749: 142313. doi: 10.1016/j.scitotenv.2020.142313 [40] YUE J, FAN A D, GUO J X, et al. Synthesis of an ultrathin MnO2 aanosheet-coated Bi2WO6 nanosheet as a heterojunction photocatalyst with enhanced photocatalytic activity[J]. Chemical Engineering Journal, 2022, 429: 132-193. [41] MAHDIAHMED M, CHIRON S. Ciprofloxacin oxidation by UV-C activated peroxymonosulfate in wastewater[J]. Journal of Hazardous Materials, 2014, 265: 41-46. doi: 10.1016/j.jhazmat.2013.11.034 [42] WU S, LI X, TIAN Y, et al. Excellent photocatalytic degradation of tetracycline over black anatase-TiO2 under visible light[J]. Chemical Engineering Journal, 2021, 406: 126747. doi: 10.1016/j.cej.2020.126747 [43] SUN H R, GUO F, PAN J J, et al. One-pot thermal polymerization route to prepare n-deficient modified G-C3N4 for the degradation of tetracycline by the synergistic effect of photocatalysis and persulfate-based advanced oxidation process[J]. Chemical Engineering Journal, 2021, 406: 126844. doi: 10.1016/j.cej.2020.126844 [44] 李立, 吴丽颖, 董正玉, 等. 高晶度Mn-Fe LDH催化剂活化过一硫酸盐降解偶氮染料RBK5[J]. 环境科学, 2020, 41(6): 2736-2745. [45] MA J, YANG Y Q, JIANG X C H, et al. Chen. Impacts of inorganic anions and natural organic matter on thermally activated persulfate oxidation of BTEX in water[J]. Chemosphere, 2018, 190: 296-306. doi: 10.1016/j.chemosphere.2017.09.148 [46] DEVI P, DAS U, DALAI A. K. In-situ chemical oxidation: Principle and applications of peroxide and persulfate treatments in wastewater systems[J]. The Science of the Total Environment, 2016, 571: 643-657. doi: 10.1016/j.scitotenv.2016.07.032 [47] JAAFARZADEH N, GHANBARI F, AHMADI M. Catalytic degradation of 2, 4-dichlorophenoxyacetic acid (2, 4-D) by nano-Fe2O3 activated peroxymonosulfate: Influential factors and mechanism determination[J]. Chemosphere, 2017, 169: 568-576. doi: 10.1016/j.chemosphere.2016.11.038 [48] FANG G D, GAO J, DIONYSIOU D. D, et al. Activation of persulfate by quinones: Free radical reactions and implication for the degradation of PCBs[J]. Environmental Science & Technology, 2013, 47: 4605-4611. [49] SHAHZAD A, ALI J, IFTHIKAR J, et al. Non-radical PMS activation by the nanohybrid material with periodic confinement of reduced graphene oxide (rGO) and Cu hydroxides[J]. Journal of Hazardous Materials, 2020, 392: 122316. doi: 10.1016/j.jhazmat.2020.122316 [50] SHI H, FAN J, ZHAO Y, et al. Visible light driven CuBi2O4/Bi2MoO6 p-n heterojunction with enhanced photocatalytic inactivation of E. coli and mechanism insight[J]. Journal of Hazardous Materials, 2020, 381: 121006. doi: 10.1016/j.jhazmat.2019.121006 [51] SARKAR D, GHOSH C. K, MUKHERJEE S, et al. Three dimensional Ag2O/TiO2 type-II (p-n) nanoheterojunctions for superior photocatalytic activity[J]. Applied Materials & Interfaces, 2013, 5(2): 331-337. [52] MA, J Z, WANG C X, HE H. Enhanced photocatalytic oxidation of NO over G-C3N4-TiO2 under UV and visible light[J]. Applied Catalysis. B, Environmental, 2016, 184: 28-34. doi: 10.1016/j.apcatb.2015.11.013 [53] WANG A, ZHENG Z, WANG H, et al. 3D hierarchical H2-reduced Mn-doped CeO2 microflowers assembled from nanotubes as a high-performance Fenton-like photocatalyst for tetracycline antibiotics degradation[J]. Applied Catalysis. B, Environmental, 2020, 277: 119171. doi: 10.1016/j.apcatb.2020.119171 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5878
- HTML全文浏览数: 5878
- PDF下载数: 79
- 施引文献: 0
