



-
氮氧化物(NOx)会导致光化学烟雾、酸雨、地面臭氧和细颗粒物等多种环境问题[1-2]。NOx的排放主要来自于固定源和移动源。在移动源排放中,柴油车对NOx排放贡献较大。传统NH3选择性催化还原(NH3-SCR)技术已被广泛用于NOx的去除,但其存在氨腐蚀、硫酸氢铵形成等诸多问题[3]。而柴油车尾气中除NOx外还含有大量碳氢化合物。与使用高成本NH3或尿素的NH3-SCR工艺相比,碳氢化合物选择性催化还原NOx(HC-SCR)工艺无需添加任何外源性还原剂,并且可同时去除尾气中的NOx和未燃烧的碳氢化合物[4-5]。因此,从经济、节能和安全的角度来看,HC-SCR是一种具有应用潜力的柴油车尾气控制技术[6-7]。
相较于CH4[8]、C3H8[9]和C2H6O[10],C3H6作为SCR还原剂的活性更高[11-13]。单一/复合金属氧化物及沸石基催化剂已受到广泛关注,如Sn[14-15]、In[16-17]、Co[18-19]、Cu[20-21]、Fe[22-23]等。过渡金属氧化物负载型催化剂具有较强的研究价值。然而,用于柴油车尾气的HC-SCR技术还存在诸多挑战,其中低温活性有待提高是其中之一。Cu基催化剂具有成本低和效率高的特点,其在SCR反应中具有明显低温优势[24-25]。Cu负载到Al2O3、Ti0.5Zr0.5O2-δ等载体表面后,在≤300 ℃条件下能实现NO向N2的转化[26-27]。Cu基催化剂的低温催化性能主要取决于催化剂表面Cu的价态和分散状态[28],而相比于低价Cu+和Cu0物质,Cu2+被证明是SCR的活性位点[29-31]。SHIMIZU等[32]发现Cu-铝酸盐催化剂表现出比Cu-ZSM-5更优的脱硝活性和水热稳定性,这是由于其中高度分散的Cu2+离子起到了作用。因此,通过调控催化剂表面Cu物质的分布以提高活性Cu含量是提升Cu基催化剂低温SCR性能的关键。助剂掺杂是一种催化剂改性的常用手段[33-34],In作为一种d10系主族金属元素,被广泛用作催化剂助剂,可调控活性位点的反应性能。KHARAT等[35]发现In的添加对三元CuO-ZnO-Al2O3催化剂结构和催化性能产生了影响,增加催化剂比表面积并减小CuOx晶粒尺寸可使催化剂在250 °C的活性得到显著提升。In3+离子具有较大离子半径,掺杂后可能导致Co3O4晶格结构变形,并形成氧空位,进而促进催化氧化反应进行[36];此外,掺杂In能改变载体[37]或负载物质的结构[38],使得其催化性能得以提升。尽管Cu基催化剂表现出一定的C3H6-SCR活性,但其低温NOx转化率还有待提升。而In2O3较弱的氧化性能可抑制碳氢化合物的过度燃烧,使更多碳氢化合物可用作还原剂,进而使得In/Al催化剂在高温区间表现出较高的NOx去除效率[39]。
基于此,本研究以γ-Al2O3为载体,采用浸渍法制备CuO-In2O3/γ-Al2O3催化剂用于C3H6-SCR反应,拟通过添加In来改善催化剂表面Cu物质的分布,以期提高Cu基催化剂的低温活性,再进一步通过X射线光电子谱(XPS)、氢气程序升温还原(H2-TPR)等表征方法,探明助剂In对Cu离子价态和反应中间产物的影响,以揭示C3H6-SCR的反应机理。本研究可为应用于柴油车尾气控制技术的低温SCR催化剂开发提供参考。
-
以γ-Al2O3为载体,通过浸渍法分别制备CuO-In2O3/γ-Al2O3、CuO/γ-Al2O3和In2O3/γ-Al2O3催化剂(记为Cu-In/Al、Cu/Al和In/Al)。采用硝酸铜(0.228 g,Cu(NO3)2·3H2O,西亚化学工业有限公司)和硝酸铟(0.081 g,In(NO3)3·4H2O,麦克林生化科技有限公司)水溶液浸渍γ-Al2O3粉末(2.88 g),制备CuO-In2O3/γ-Al2O3催化剂。将混合后的溶液在室温下搅拌2 h,然后慢慢加热至80 °C,搅拌至糊状。最后,在110 °C下,将样品干燥12 h,并在600 ℃温度下煅烧5 h(升温速率为1 ℃·min−1)。其他催化剂的制备方法也类似。本研究所用的Cu-In/Al催化剂已在前期经过比例优化,按Cu、In最佳配比计算得到其负载量分别为:CuO负载量 (质量分数) 为2.5%;In2O3负载量 (质量分数) 为1.25%。
-
1) XRD。在Cu Kα辐射(λ=1.5418 Å)、2θ为10°~90°、扫描速率为8°·min−1的条件下,使用X射线衍射仪(Bruker, D8 ADVANCE X)测试样品的X射线衍射谱图(XRD),以确定催化剂组成。2) BET。在77 K下,采用物理吸附仪(Micromeritics, ASAP2020-M)进行N2吸附-解吸分析,以测定催化剂的比表面积(BET)。3) ICP。采用电感耦合等离子体发射光谱仪(Agilent ICP-OES 725-ES)对制备的催化剂进行元素含量分析。4) H2-TPR。为测试催化剂氧化还原性能,利用化学吸附分析仪(Micromeritics, ChemiSorb 2720 TPx)进行氢气程序升温还原(H2-TPR)实验。样品在Ar中400 ℃预处理1 h,然后冷却至30 ℃并切换至10%H2/Ar的反应气氛,流量为30 mL·min−1,以10 ℃·min−1的升温速率从30 °C升至800 ℃。5) XPS。通过光谱仪(Thermo Escalab 250-XI)进行X射线光电子能谱(XPS),分析催化剂表面元素价态,以Al Kα X射线为单色光源,并根据C1s峰(284.8 eV)校正各个元素的结合能。6) NO+O2-TPD。采用气体分析仪(Thermo Scientific Antaris IGS)进行NO+O2程序升温脱附(NO+O2-TPD)实验,以测定催化剂对NOx的吸附性能。先将100 mg样品在180 mL·min−1的N2中400 ℃预处理1 h,然后冷却至室温,通入混合气体 (NO 500×10−6+O2 5%) 吸附1 h;用N2吹扫后,程序升温至600 ℃进行脱附实验。7) in situ DRIFTS。在红外光谱仪(Nicolet iS50)上进行程序升温C3H6氧化反应和瞬态原位漫反射红外光谱(in situ DRIFTS)实验,以探明催化剂表面的反应机制。样品在500 ℃条件下充以N2预处理1 h,然后冷却至所需温度。在每个温度下采集背景光谱,并在相同温度下采集样品光谱。
-
C3H6-SCR的活性通过固定石英床连续反应器(内部直径6 mm)来测试。测试中使用150 mg催化剂,其粒径为40~60目,并以N2为平衡气。通入的气体组分及其体积分数为:C3H6 1 000×10−6、NO 500×10−6、O2 2%。通入气体的总流速为200 mL·min−1,气体空速为38 000 h−1。C3H6、NO、NO2、N2O等气体进出口浓度通过气体分析仪(Thermo Scientific, Antaris IGS)检测。NOx、C3H6的转化效率和N2选择性计算参考式 (1)~(3) 进行。
式中:
ηNOx 和ηC3H6 分别表示NOx和C3H6的转化效率,%;ψN2 表示N2的选择性,%;方括号为气体体积分数,以下标“in”和“out”分别区分进口和出口气体。 -
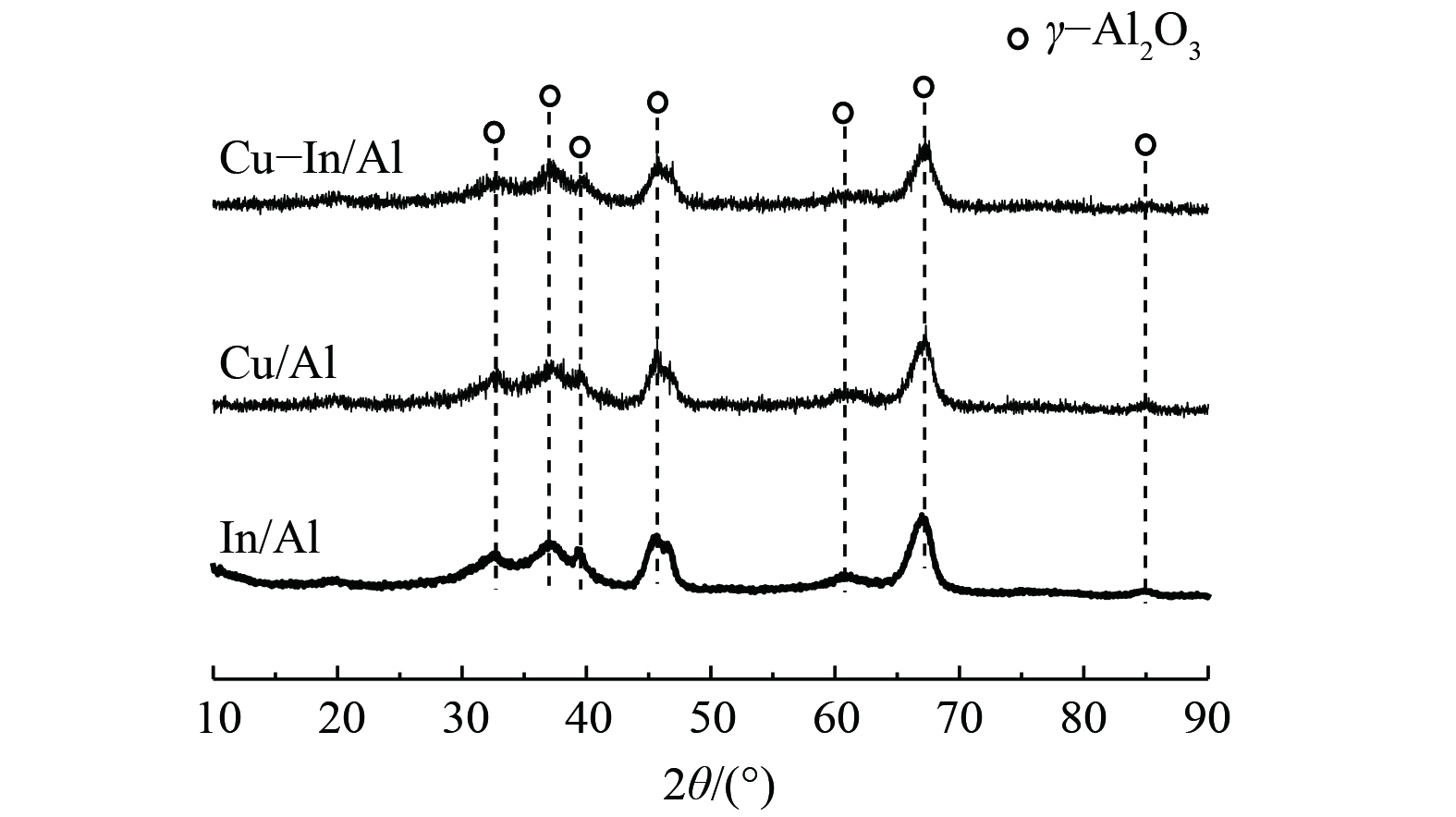
图1为Cu-In/Al、Cu/Al和In/Al催化剂的XRD测试结果。3种催化剂均显示出载体γ-Al2O3的衍射峰(PDF#79-1558),但未观察到CuO或In2O3的晶相。这表明Cu、In类物质的结晶度低或呈现无定形状态,并且高度分散在γ-Al2O3载体上。3种催化剂的BET测试结果较为相似,其比表面积分别为:In/Al 154 m2·g−1、Cu-In/Al 146 m2·g−1和Cu/Al 145 m2·g−1。与纯载体γ-Al2O3(比表面积为160 m2·g−1)相比,低结晶度或无定形的CuOx和InOx负载到γ-Al2O3表面后,并未显著改变载体的孔结构,对催化剂的比表面积影响亦较小,但Cu、In在表面的高度分散也将为催化反应提供更多反应位点。
催化剂对NOx的吸附性能是SCR的重要参数。利用NO+O2-TPD考察了Cu-In/Al、Cu/Al和In/Al催化剂表面NOx的吸附性能,以探讨NOx在催化剂表面的吸附强度,结果如图2所示。在低于200 ℃条件下,吸附的物质多为NO,主要来自于弱结合的亚硝酸盐(ad-NO2−)分解;而在高于300 ℃条件下,吸附的物质多为NO2,主要来自于硝酸盐(ad-NO3−)的分解[39-40]。值得注意的是,在掺杂In后,Cu-In/Al催化剂上NO脱附峰强度明显增加,这说明In的掺杂有助于催化剂表面形成更多的亚硝酸盐。NOx总吸附量(表1)表明,Cu-In/Al催化剂表现出了最大NOx吸附量。同时,相比于In/Al,Cu/Al和Cu-In/Al表面硝酸盐的脱附温度较低。这可能是由于Cu位点上的硝酸盐结合能力不强,更容易分解,并易与C3H6活化后的中间产物反应,进而提升了SCR反应活性。
-
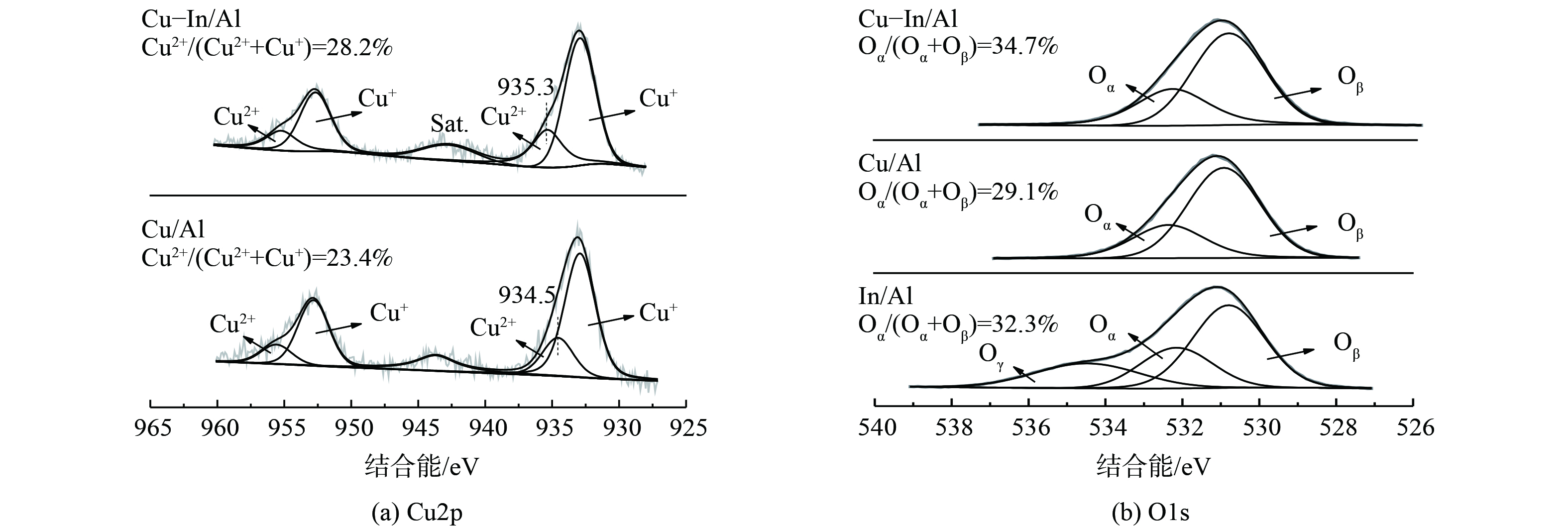
为进一步探讨In对催化剂表面组成和氧化状态的影响,Cu/Al、In/Al和Cu-In/Al催化剂的XPS结果如图3所示。Cu-In/Al和Cu/Al催化剂的Cu 2p光谱由2p1/2、2p3/2和卫星峰(943-945 eV)组成。分峰结果表明,催化剂表面同时存在Cu2+和Cu+类物质[27,41-42]。添加In后,Cu-In/Al催化剂表面上Cu2+增多。推测Cu2+可能是丙烯分子的主要吸附位点[43],表面更多的Cu2+有利于C—H键断裂,从而促进了C3H6活化氧化反应,最终使得C3H6-SCR的性能提升。此外,相比于Cu/Al催化剂,Cu-In/Al催化剂表面Cu2+的峰向更高结合能偏移 (由934.5 eV转移至935.3 eV) 。这表明催化剂上存在电子转移,导致Cu位点的电荷密度较低、存在高价态Cu。这可能是由于Cu2+与In有较强的相互作用,从而有利于气态反应物的活化氧化。催化剂的O1s谱图如图3(b)所示。经分峰后,2个主要O1s峰分别归属于催化剂的表面化学吸附氧(记为Oα)和晶格氧(记为Oβ)[44]。定量计算结果表明,Cu-In/Al催化剂的Oα/(Oα+Oβ)比例明显高于Cu/Al。这说明掺杂In会提高Cu-In/Al催化剂上的化学吸附氧物质的含量。
采用H2-TPR进一步研究催化剂的氧化还原性能,结果如图4所示。Cu-In/Al、Cu/Al和In/Al均显示出单一的H2还原峰,分别位于256、282和400 ℃温度下。Cu-In/Al和Cu/Al催化剂的峰主要归属于高度分散的Cu2+直接还原的金属Cu0[26,45],In/Al催化剂的宽峰主要归属于高度分散的In3+直接还原的金属In0[16,46]。相比Cu/Al和In/Al催化剂,Cu-In/Al催化剂的还原峰温度更低。这表明在CuIn位点的相互作用下,Cu2+表现出更强的氧化性。这与XPS中Cu-In/Al表面存在更高价态Cu类物质的结果相一致。在C3H6-SCR反应过程中,较强的氧化还原性能是催化剂展现出优异低温活性的原因之一。
-
Cu-In/Al、Cu/Al和In/Al催化剂在200~600 ℃条件下的C3H6-SCR活性如图5所示。In/Al催化剂在超过450 ℃的高温下的NOx去除效率较高,而Cu/Al催化剂在250 ℃开始起活,并表现出低温SCR活性。在In掺杂后,Cu-In/Al催化剂不但保持Cu/Al催化剂的低温活性优势,且NOx转化率显著提高,在350 ℃达到62%的NOx转化率。常见一元或二元非贵金属氧化物催化剂仅有高温SCR活性,在C3H6-SCR低温范围内 (< 400 ℃) 活性较低。In2O3/Al2O3[39,46]、CuO/Al2O3[47]、Co3O4/Al2O3[17]、Ga2O3-Al2O3[48]在350 ℃时的NOx转化率仅为约30%,且催化剂的起活温度较高。而掺杂In后的In2O3-Co3O4/Al2O3[17]、In2O3-SnO2/Al2O3[49]、In2O3-Ga2O3-Al2O3[48]催化剂在350 ℃的NOx转化率依然不高。相比之下,Cu-In/Al催化剂具有更好的低温活性潜力,在350 ℃的活性表现值得进一步研究,这可能与Cu和In之间的相互作用有关。
值得注意的是,图5 (b) 表明Cu-In/Al和Cu/Al催化剂上C3H6转化性能显著优于In/Al催化剂,而Cu/Al在掺杂In的基础上C3H6转化率又略有提高。在350 ℃时,催化剂的C3H6转化率按由大到小排列,依次为Cu-In/Al(93%)>Cu/Al(62%)>In/Al(6%)。此外,在图5(c)和(d)中,Cu-In/Al和Cu/Al催化剂表现出相似的N2选择性及产物分布。这表明C3H6还原NO后的产物主要是N2和CO2。因此,催化剂表面的CuOx可能是C3H6活化和NOx还原的活性位点,而In在其中是作为助剂促进了C3H6的活化转化,并提高了催化剂的NOx转化效率。
-
为阐明反应中间体和C3H6-SCR机理,采用原位DRIFTS方法测定不同反应中间产物的形成和转化情况。图6表明,在350 ℃条件下, (NO+O2) 在催化剂表面预吸附后,主要以单齿硝酸盐(峰值为1 258 cm−1)和双齿硝酸盐(峰值为1 300和1 554 cm−1)的形式存在[50]。反应气体切换为 (C3H6+O2) 后,Cu-In/Al和Cu/Al催化剂表面的硝酸盐快速消失,并出现乙酸盐υas(COO)、υs(COO)的振动峰[47,51](1 587和1 459 cm−1)和甲酸盐δ(—CH3)的振动峰(1 378 cm−1)[52-53]。这说明吸附态硝酸盐能与气态C3H6或表面活化CxHyOz快速反应。当硝酸盐被完全消耗后,C3H6吸附活化后形成的甲酸盐和乙酸盐在催化剂表面逐渐积累。但在In/Al催化剂上,随着气态C3H6的持续通入,吸附态硝酸盐仍稳定存在,几乎未被消耗减少,且未观察到深度氧化生成的甲酸盐,仅有少量乙酸盐吸附在表面。
在Cu-In/Al、Cu/Al和In/Al催化剂表面先进行C3H6的预吸附活化,然后再通入混合气体 (NO+O2) ,其原位红外结果如图7所示。在Cu-In/Al和Cu/Al催化剂表面,C3H6吸附活化后形成大量乙酸盐(1 587、1 580和1 459 cm−1)和甲酸盐(1 378 cm−1)。这说明C3H6在催化剂表面吸附,能被快速氧化活化生成CxHyOz类物质。另外,在Cu/Al催化剂上观察到明显的2 235 cm−1强峰。这个峰归属于L酸吸附的CO分子伸缩振动[54-55],表明了单独的CuOx位点会导致CxHyOz进一步氧化形成吸附的CO。这可能与其较低的NOx转化率相关。而在Cu-In/Al催化剂上2 235 cm−1处的峰较弱,可忽略,则表明掺杂In后,催化剂可抑制C3H6的过度氧化,更有利于C3H6活化形成乙酸盐或甲酸盐中间产物参与SCR反应。随着反应进一步进行,Cu基催化剂表面乙酸盐和甲酸盐逐渐被消耗,桥式硝酸盐(峰值为1 600 cm−1)和双齿硝酸盐(峰值为1 300和1 553 cm−1)出现并逐渐积累。这说明C3H6吸附活化后能与气态NO或吸附态硝酸盐反应。而对于In/Al催化剂 (图7(c)),C3H6在催化剂表面催化活化后,难以与气态NO反应,即使催化剂表面开始有硝酸盐形成,吸附态CxHyOz仍未减少。这表明2种吸附物质之间没有相互反应。这也是导致In/Al催化剂在350 ℃低温条件下C3H6-SCR性能差的主要原因。
为进一步确认催化剂表面的C3H6-SCR反应路径,同时通入C3H6、NO和O2时的原位红外谱图如图8所示。在Cu-In/Al、Cu/Al和In/Al表面观察到桥式硝酸盐(峰值为1 600 cm−1)、双齿硝酸盐(峰值为1 300、1 550、1 555和1 568 cm−1)、硝基化合物(峰值为1 392 cm−1)、乙酸盐(峰值为1 587和1 459 cm−1)和甲酸盐(峰值为1 378 cm−1)等物质快速形成。结合图6和图7瞬态反应结果,C3H6和NO在催化剂表面会先形成吸附态中间产物,然后才能参与SCR反应,符合Langmuir-Hinshelwood (L-H)反应机制。在Cu-In/Al和Cu/Al催化剂上,吸附态硝酸盐仅存在于低温条件下,而In/Al催化剂上吸附态硝酸盐结合更强,这与NO-TPD结果一致。在350 ℃时,Cu-In/Al和Cu/Al表面上硝酸盐更活跃,更易于与吸附态CxHyOz反应,进而促进了SCR的低温活性。
-
NO+O2-TPD结果表明掺杂In提高了Cu-In/Al催化剂对亚硝酸盐的吸附量,但原位红外光谱中并未观察到明显的亚硝酸盐特征峰。这主要是由于在350 ℃时,亚硝酸盐已被氧化为硝酸盐。C3H6-SCR反应机理和原位实验结果表明,CuOx是Cu/Al和Cu-In/Al主要的活性位点,而L-H反应路径需要C3H6和NO同时在催化剂表面吸附活化。在350 ℃时,掺杂In对CuOx位点的NOx吸附及反应影响不大,NOx吸附性能的提高可能并不是In促进催化剂低温活性提升的主要原因。而In可能主要影响催化剂表面C3H6活化转化为中间产物甲/乙酸盐的过程。为了证明这一假设并深入探究In对C3H6-SCR过程的促进作用,采用了程序升温C3H6氧化与原位红外相结合的方法开展进一步研究。图9为混合气体 (1 000×10−6C3H6+2%O2+N2) 条件下,Cu-In/Al、Cu/Al和In/Al表面的程序升温原位红外光谱。在3种催化剂表面,C3H6在低温条件下能迅速活化氧化形成丙烯酸酯(峰值为1 643 cm−1)。随着温度的升高,丙烯酸酯开始减少,出现了更稳定的乙酸盐(峰值为1 459和1 587 cm−1)和甲酸盐(峰值为1 378 cm−1),并在400 ℃下仍稳定存在。相比于In/Al催化剂,Cu基催化剂能大量形成具有更高氧化态碳的甲酸盐,这与瞬态原位红外结果一致。在Cu-In/Al催化剂上,乙酸盐和甲酸盐的出现温度更低(图10)。这表明掺杂In后,Cu-In/Al催化剂更容易将气态C3H6氧化转化为乙酸盐和甲酸盐,从而有利于其作为吸附态中间产物参与C3H6-SCR反应。这与Cu-In/Al催化剂氧化还原性能较强有关。
综上所述,In掺杂改变了Cu-In/Al催化剂表面Cu2+的分布,提高了催化剂表面高度分散的Cu2+比例,进而导致催化剂的氧化还原性能增强,并通过C3H6实现更快速地活化氧化,在低温下即实现C3H6-SCR快速反应。通过揭示甲/乙酸盐的快速形成对C3H6-SCR低温活性提升的促进机制,强调了C3H6快速吸附活化是促进C3H6-SCR低温活性的关键步骤,可为新型低温催化剂的设计和开发提供参考。
-
1)同时负载Cu、In的催化剂表现出较好的C3H6-SCR活性,并在350 ℃达到最佳NOx和C3H6转化率。这主要是由于添加In使得Cu/Al催化剂的Cu2+和表面活性氧比例提高,从而增强其氧化还原性能和NOx吸附性能。
2)在Cu-In/Al催化剂上,C3H6能被快速活化形成乙酸盐和甲酸盐,且积累形成大量硝酸盐。两类吸附态中间产物的快速反应可能是C3H6-SCR反应活性提高的原因。
3) In掺杂可调控C3H6活化速率。Cu-In/Al催化剂的低温活性潜能与C3H6快速活化有关。C3H6活化机制是调控C3H6-SCR低温活性的一种有效方法。
铟掺杂促进铜铝催化剂低温C3H6-SCR反应的机理
Reaction mechanism of enhanced activity for C3H6-SCR at low temperature by indium-doped on CuO/Al2O3 catalyst
-
摘要: CuO/Al2O3催化剂为低温SCR催化剂,在其表面添加In组分,并用于丙烯选择性催化还原(C3H6-SCR)氮氧化物(NOx)的研究。结果表明,负载CuIn的催化剂表现出最好的反应活性,在350 °C时NOx转化率可达到62%。XPS表征结果显示,同时负载In改变了Cu的化合价态和表面氧的分布,提高了催化剂表面Cu2+和化学吸附氧的比例。H2-TPR和NO+O2-TPD结果表明,同时负载CuIn能提高催化剂氧化还原性,也促进了NOx的吸附,催化剂表面生成大量的亚硝酸盐/硝酸盐。反应机理研究表明,C3H6-SCR过程沿着L-H反应路径进行,同时负载CuIn能促进C3H6的快速氧化,并有助于催化剂表面甲酸盐和乙酸盐的形成。因此,Cu2+和化学吸附氧比例的提高,会增强催化剂的氧化还原性能,从而加速甲/乙酸盐的形成,这可能是促进C3H6-SCR低温活性得以提高的主要原因。本研究可为应用于柴油车尾气控制技术的低温SCR催化剂开发提供参考。Abstract: CuO/Al2O3 catalyst was a low-temperature SCR catalyst. In this paper, In was added to the surface of CuO/Al2O3 catalyst and used for selective catalytic reduction of nitrogen oxides (NOx) by propylene (C3H6-SCR). The results showed that the catalyst supported with CuIn exhibited the best activity, with NOx conversion up to 62% at 350 °C. XPS characterization results showed that the loading of In changed the valence state of Cu and the distribution of oxygen on the surface, and increased the ratio of Cu2+ and chemisorbed oxygen on the catalyst surface. The results of H2-TPR and NO+O2-TPD showed that loading CuIn could improve the reducibility of the catalyst and promote the adsorption of NOx, and a large number of nitrite/nitrate species were formed on the catalyst surface. Studies of reaction mechanism showed that C3H6-SCR process followed L-H reaction mechanism. Doping CuIn promoted the rapid oxidation of C3H6 and contributed to the formation of formate and acetate on the catalyst surface. Therefore, the increase of the ratio of Cu2+ and chemisorbed oxygen would enhance redox performance of the catalyst and accelerate the rapid formation of formate/acetate, which might be the main reasons for the improvement of activity of C3H6-SCR in low temperature range. This study can provide reference for the development of low temperature SCR catalyst applied in diesel vehicle exhaust control technology.
-
Key words:
- copper-based catalyst /
- selective catalytic reduction /
- C3H6 /
- NOx /
- Al2O3
-
室内空气污染对人类居住生活环境的影响日趋严重,据世界卫生组织估计,2016年全球有420万人因室内空气污染而过早死亡,相比之下,户外空气污染导致的过早死亡只有380万人[1]。糟糕的室内空气质量已成为全球第九大疾病风险负担[2]。室内空气的污染除室外污染空气的影响外,还与室内装修、人类活动以及室内家居用品等密切相关。随着人们健康意识的提高,空气净化器作为改善室内空气品质的一种有效途径,已逐渐成为室内空气净化的主要手段[3]。
目前市场上销售的空气净化器大都采用HEPA滤网过滤空气中的颗粒物,研究表明,HEPA滤网可以将颗粒物质量和颗粒物数浓度降低50%以上[4-7],技术相对成熟稳定。近年来,HEPA技术也一直占据着空气净化器市场的最主要份额[8-9]。相较于颗粒物的净化方式,对气态污染物的净化方式较多,但一直没有取得质的突破。较为主流的净化方式是应用改性活性炭制作成填充滤网或者夹炭复合布材料,整个过程中涉及到很多化学反应,如果选用的化学试剂不当,会将新的污染物引入到环境空气中,造成二次污染。近年来空气净化器在使用过程中出现酸臭异味的问题成为了消费者主要的投诉项目,这个酸臭异味也主要来源于活性炭滤网[10]。但由于各个厂商活性炭滤网的制作工艺及其中活性炭的改性配方各不相同[11-16],造成活性炭类滤网的性状差异较大,酸臭异味的成因来源一直没有明确的结论。
为了解决这一室内空气净化领域难题,本文通过感官评价与仪器分析相结合的方法对典型受试样本进行剖析[17-19],采用气相色谱-质谱、离子色谱及平板计数法测定了活性炭滤网中的挥发性有机物、有机酸、无机离子及菌落总数和霉菌等,并通过感官等级评价与不同成分的相关性分析,全面客观的对活性炭滤芯酸臭异味的成因及来源进行了探讨,为改善活性炭滤芯生产工艺与防治异味提供科学参考依据,以期促进空气净化器市场的持续健康发展。
1. 材料与方法(Materials and methods)
1.1 试验材料
试验选取的滤网分别采用不同的活性炭种类包括椰壳活性炭和煤质活性炭,改性与未经改性的活性炭,用胶粘炭和撒炭的形式,以及正常人居环境下使用过和未使用过的样品。本实验所用活性炭滤网样品由北京三五二环保科技有限公司提供。具体情况如表1所示。
表 1 不同类型活性炭滤网样品Table 1. Different types of activated carbon filter samples样品编号Sample number 活性炭种类Type of activated carbon 产品类型Product type 是否改性Modified or not 是否用胶Used glue or not 使用情况Usage condition 1 椰壳炭+催化剂 柱状碳 是 否 6个月 2 煤制碳 柱状碳 是 是 6个月 3 煤制碳+催化剂 柱状碳 是 是 6个月 4 煤制碳+催化剂 柱状碳 是 是 6个月 5 煤制碳 柱状碳 是 是 6个月 6 煤制碳 柱状碳 是 是 未使用 7 煤制碳 柱状碳 是 是 未使用 8 煤制碳 柱状碳 是 是 未使用 9 煤制碳 柱状碳 是 是 6个月 10 煤制碳 柱状碳 是 是 6个月 11 椰壳炭 夹炭布 是 是 6个月 12 椰壳炭 夹炭布 是 是 6个月 13 椰壳炭 夹炭布 是 是 6个月 14 椰壳炭 夹炭布 是 是 6个月 15 椰壳炭 夹炭布 是 是 6个月 16 椰壳炭 夹炭布 是 是 未使用 17 椰壳炭 夹炭布 是 是 未使用 18 椰壳炭 夹炭布 是 是 未使用 19 煤制碳 柱状碳 是 否 6个月 20 煤制碳 柱状碳 否 否 6个月 21 椰壳炭 柱状碳 是 否 6个月 22 椰壳炭 柱状碳 否 否 6个月 23 煤制碳 柱状碳 是 是 6个月 24 煤制碳 柱状碳 是 是 6个月 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 试验方法
1.2.1 气味感官评价
选取10位不吸烟,嗅觉器官灵敏且训练有素的普通人群作为感官评价人员(6名女性+4名男性),参照张晓等的研究方法[10]在室温下对活性炭滤网异味的强度进行了评估,强度分为0—5的6个等级,根据0表示无酸臭味,1表示似有似无,不敢确定,2表示微弱的酸臭味,3表示感觉到明显的酸臭味,4表示感觉到比较强烈的酸臭味,5表示感觉到非常强的酸臭味来打分。每位嗅闻员对同一样品嗅闻3次,3次数据中有2次重复时才被记录用来分析。
1.2.2 气相色谱-质谱分析条件
采用7890A/5975C气相色谱-质谱仪(美国Agilent公司)对滤网中的挥发性有机物进行了测定,为了让实验结果更贴近于实际应用情况,实验在容积14 m3大型环境舱(中国林业科学院木材工业研究所)中进行。将待测滤网放置于同一空气净化器中,以相同风量开启净化器一段时间后,在净化器出气口处架设恒流空气采样泵(美国Gilian IAQ—Pro—II),应用Tenax—TA吸附管(美国CAMSCO)采集出口处气体,采样流速200 mL·min−1,采样时间20 min。采集完毕后,应用ATD—35二级热脱附装置(美国Perkin Elmer公司)对吸附管进行加热脱附,然后进入气相色谱质谱仪分析。
热脱附条件:样品管除湿时间2 min;阀温225 ℃;传输线温度230 ℃;一级脱附温度80 ℃;一级脱附时间5 min;冷阱温度-30 ℃至290 ℃(升温速率40 ℃·s−1);二级脱附时间2 min;分流比10∶1。
气相色谱条件:色谱柱:HP—VOC(60 m×0.32 mm×1.8 μm);程序升温初始温度45 ℃,保留2 min,5 ℃·min−1升至250 ℃,保留5 min;载气为高纯He气,柱流量1 mL·min−1。
质谱条件:EI离子源,电子能量70 eV,离子源温度230 ℃,四极杆温度150 ℃,传输线温度280 ℃,扫描质量范围:45—450 amu。
1.2.3 离子色谱分析条件
采用ICS2000离子色谱仪(美国Dionex公司)对24个样品中的主要水溶性阴离子和有机酸进行了测定。准确称取柱状炭及颗粒状炭样品5 g,置于小烧杯中,加入50 mL去离子水后震荡萃取12 h,提取液经0.45 μm的水系滤膜过滤,待测。夹碳布样品,首先经炭粒与滤布分离后,称取2 g炭粒样品,加入20 mL去离子水,后续步骤同上。
阴离子测试条件:色谱柱型号:AS14A 4 mm×250 mm;淋洗液:30 mmol·L−1 KOH;流速:1.0 mL·min−1;抑制器电流:59 mA;进样体积:25 μL。
有机酸测试条件:色谱柱型号:AS11-HC 4 mm×250 mm;淋洗液:5 mmol·L−1 KOH;流速:1.2 mL·min−1;抑制器电流:223 mA;进样体积:25 μL。
分别配制5个不同浓度的阴离子(Cl−、NO2−、NO3−、SO42-和PO43-,国家有色金属及电子材料分析测试中心)和有机酸(甲酸、乙酸、丙酸、丁酸和乳酸,北京百灵威科技有限公司)混合标准品溶液系列,用标准溶液系列中各组分的峰面积与浓度数据作标准曲线,计算回归方程和相关系数,将低浓度的混合标准溶液不断稀释并进样分析,并以S/N=3计算出各阴离子和有机酸的检出限。无机阴离子相关系数均≥0.9995,有机酸均≥0.9993。5种阴离子的检出限为0.03—0.08 µg·mL−1,5种有机酸的检出限为0.02—0.07 µg·mL−1。
1.2.4 pH值分析
取5 g样品用50 mL去离子水浸泡,震荡12 h后经0.45 μm的水系滤膜过滤。滤液的pH值由上海雷磁PHS-3C pH计测定。
1.2.5 微生物分析
灭菌棉拭子于10 mL灭菌生理盐水内浸润(吸取约1 mL溶液),在净化器滤网的前后中央部位5 cm×5 cm面积上,用力均匀涂抹5次,再用灭菌剪刀剪去棉签手接触部分。棉拭子放入剩余9 mL生理盐水内,尽快测定。按照GB/T 18204.4—2013《公共场所微生物检验方法第4 部分:公共用品用具微生物》检测菌落总数和霉菌数量[20]。
2. 结果与讨论(Results and discussion)
2.1 气味感官分析
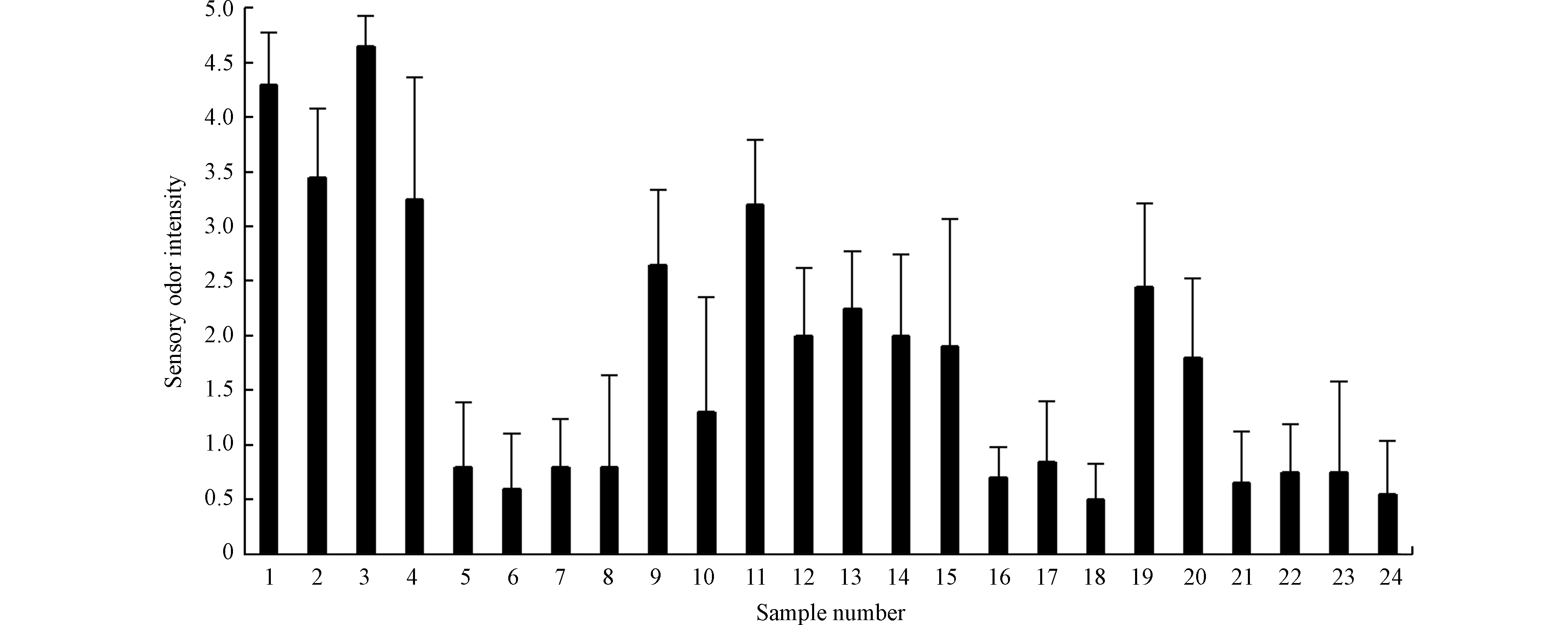
本试验首先对24种不同类型的活性炭滤网样品进行了感官评价,评分大于2的判定为存在异味,24个样品的感官气味评级如图1所示。由图1可以看出,所测样品中有8个明确存在异味。
同时,为了考察感官气味与滤网样品特征是否存在联系,对不同类型的活性炭样品进行赋值处理,运用SPSS软件对感官评分和滤网类型、是否添加催化剂、是否改性、是否用胶以及是否使用过总共8项因子进行了Spearman相关性分析,结果如表2所示。通过表2可以看出,酸臭味大小与活性炭滤网是否经过改性、活性炭种类、活性炭滤网的制备形式、是否在制作过程中使用有机胶粘剂无相关性,而与是否添加催化剂及是否使用过两项属性具有较高的相关性。由此可以初步判断,添加催化剂及长期使用的活性炭滤网更容易产生酸臭味。
表 2 滤网样品特征与感官评分的Spearman相关系数Table 2. Spearman’s correlation coefficients between filters sample characteristics and sensory scores样品特征Sample characteristics 感官评分Sensory evaluation 是否添加催化剂 0.558** 椰壳炭 −0.042 煤制炭 0.042 柱状炭 0.019 夹碳布 −0.019 是否改性 0.088 是否用胶 −0.060 是否使用过 0.559** **相关性在0.01置信水平下显著. **Correlation is significant at 0.01 level. | Show TableDownLoad:
CSV
2.2 气相色谱-质谱分析
考虑到这种难闻的酸臭味可能是由滤网释放的挥发性成分造成的,对感官评级结果中产生酸臭异味较明显的8块滤网使用吸附管采集-热脱附-气相色谱质谱进行测定。将检测的总离子流图中各峰一级质谱经计算机NIST08标准谱库检索鉴定,检测出挥发性成分匹配度大于80 的化合物。甲苯通常用于总挥发性有机化合物(TVOC)的测量,以及在参考化合物无法获得的情况下可用于单一VOC的定量[21],因此本实验采用甲苯对各VOCs的含量进行相对定量分析。在1.2.2节确定的实验条件下,甲苯的方法检出限为0.60 µg·m−3,实验结果如表3所示。
表 3 挥发性有机化合物的测定结果(µg·m−3)Table 3. Determination results of the volatile organic compounds(µg·m−3)化合物Compound 1号Sample 1 2号Sample 2 3号Sample 3 4号Sample 4 9号Sample 9 11号Sample 11 19号Sample 19 20号Sample 20 环氧乙烷 3.21 10.18 5.28 7.76 1.55 2-甲基戊烷 5.62 3.03 5.16 1.65 4.97 3-甲基戊烷 12.22 9.02 11.39 6.35 2.11 12.72 正己烷 75.43 33.11 9.88 10.79 34.71 8.33 53.19 36.25 环己烷 10.68 5.66 8.84 3.86 6.24 4.40 庚烷 12.36 2.93 5.33 甲苯 17.58 11.41 8.55 24.37 6.06 19.15 30.38 乙酸丁酯 2.33 3.16 对、间二甲苯 9.32 6.76 3.15 2.10 8.31 4.88 1.55 环己酮 3.95 0.98 α-蒎烯 7.19 8.52 2.63 2.13 1.86 3-己醇 4.71 1.26 2.21 苯甲醛 6.10 3.24 1.75 癸烷 3.35 7.28 4.38 3.16 2.44 十六烷 9.26 11.33 5.66 6.12 3.43 | Show TableDownLoad:
CSV
由表3可以看出,样品中检出的挥发性有机物质含量均较低,且以烷烃类为主,这类物质的嗅阈值通常较高,故对整体酸臭味的贡献可能不大,由此可见,滤网吸附空气中的可检出高浓度挥发性有机污染物并不是滤网产生异味的主要来源。并由此可推断产生酸臭异味物质的嗅阈值较低,其挥发到空气中的含量较少,通过空气捕捉采样的方法很难被检测到。此外,值得指出的是,受限于检测方法,本实验中未检出挥发性脂肪酸,是否存在挥发性脂肪酸以及其可能的异味贡献,值得进一步研究确认。
2.3 离子色谱与pH分析
2.3.1 有机酸分析
考虑到一些小分子有机酸可能是导致酸臭味产生的一个重要因素,因此对24个实验样品中的甲酸、乙酸、丙酸、丁酸和乳酸5种有机酸含量进行了测定。实验结果如图2所示。
由图2可以看出,小分子有机酸在测试样品中普遍存在,其中乙酸在感官评分大于2的样品中含量最为丰富。研究表明[22],小分子有机酸嗅阈值很低,且都有强烈的不愉快的味道,例如,乙酸具有刺激性的醋酸味,丁酸具有腐臭味,初步判断这些酸类可能是滤网酸臭味的主要贡献者,因此进一步采用SPSS软件对感官气味与5种有机酸及总酸(5种有机酸之和)之间进行相关性分析(表4)。
表 4 感官气味与不同有机酸的相关性Table 4. Correlation between sensory odors and different organic acids感官气味Sensory odor 丙酸Propionic acid 甲酸Formic acid 丁酸Butyric acid 乙酸Acetic acid 乳酸Lactic acid 总酸Total acid 感官气味 1 丙酸 0.709** 1 甲酸 0.097 0.046 1 丁酸 0.167 −0.107 −0.134 1 乙酸 0.751** 0.888 0.126 −0.115 1 乳酸 −0.005 −0.063 −0.090 0.402 −0.011 1 总酸 0.712** 0.621** 0.115 0.610** 0.709** 0.285 1 **相关性在0.01置信水平下显著. **Correlation is significant at 0.01 level. | Show TableDownLoad:
CSV
由表4可以看出,滤网酸臭异味的产生和总酸、乙酸、丙酸之间具有良好的相关性,和乳酸的含量相关性较小。而总酸与乙酸、丙酸、丁酸的3种小分子酸显著相关,说明总酸主要是由上述3种物质构成。结合图2可以看出,以乙酸为主的小分子有机酸可能是活性炭滤网产生酸臭味的主要原因。但是人居环境的空气中含有小分子有机酸的量极少,因此由净化器吸收空气中有机酸而导致酸臭异味产生的可能性较小。众所周知,活性炭对有机化合物具有很大的吸附能力,这与它们发达的内部孔结构、比表面积和表面官能团的存在有关,因此,可以采用热处理或化学处理,以提高活性炭的特殊功能和表面极性[23]。大量研究表明,活性炭表面含氧官能团的多少对有机物吸附性能起着重要的作用,改变活性炭表面酸性含氧官能团的数量和极性已被证明是增强极性有机化合物吸附的一种有效手段[24-26]。以往研究发现,含氧官能团在改性活性炭中十分常见,而且很容易被硝酸、过氧化氢、高锰酸钾、过硫酸铵、臭氧等氧化而引入。研究普遍采用了Boehm滴定法对活性炭表面的含氧官能团进行定量,均发现氧化改性后的活性炭表面含有大量的羧基和羰基[27-29]。综合以上分析,可以初步判定小分子有机酸是由活性炭表面的羧基脱落产生,尽而导致滤网酸臭异味发生。值得指出的是,净化器滤网捕捉到的有机物经过长时间的氧化作用(臭氧或氧气),也可能形成羧酸类,Hyttinen等在对接触臭氧的通风过滤器的实验室测试中发现了包括有机酸在内的氧化产物[30-31],这也是使用一段时间的活性炭滤网产生酸臭异味的几率较大的原因。
另外,经调研,目前市内空气净化活性炭多会添加锰系催化剂对甲醛进行催化,从而加大甲醛清除效果。一方面,锰系催化剂在生成过程中,某些工艺会用到有机酸-锰化合物作为原料[32-33],这些原料在最终催化剂中的降解残留也可能是小分子有机酸的来源之一。另外在催化氧化有机污染物的过程中,有时也会有一些有毒的醛类、酮类以及有机酸等有机副产物的产生[34-35]。
2.3.2 阴离子、pH分析结果
为了考察无机酸根离子和酸碱性与滤网酸臭味的产生是否存在联系,利用离子色谱、酸度计对24个样品的阴离子及PH值进行了测定,并与感官等级评价进行相关性分析,结果如图3、表5和表6所示。
表 5 感官气味与不同阴离子的相关性Table 5. Correlation between sensory odors and different anion感官气味Sensory odor Cl− NO2− NO3− SO42- PO43- 总离子Total ion 感官气味 1 Cl− 0.376 1 NO2− −0.077 −0.072 1 NO3− 0.347 −0.017 −0.194 1 SO42- 0.037 −0.103 −0.230 0.059 1 PO43- −0.378 −0.301 −0.208 −0.220 −0.287 1 总离子 −0.315 −0.278 −0.316 −0.167 0.380 0.771** 1 **相关性在0.01置信水平下显著. **Correlation is significant at 0.01 level. | Show TableDownLoad:
CSV
由图3可以看出,感官气味强度并没有随无机离子含量的不同而出现规律性变化。相关性分析结果显示(表5和表6),感官气味与无机酸根离子及pH值无显著相关性,表明了酸臭异味不是由无机酸根离子产生,也不会随着活性炭自身pH值偏酸而加重。相比之下,总离子与PO43-离子及pH值之间呈现出了较高的相关性,说明活性炭上的无机阴离子以PO43-离子为主,这与图3离子色谱的测定结果相一致,另一方面也反映出活性炭pH值和活性炭水浸提液中的PO43-离子含量存在一定程度的联系。
表 6 感官气味与总酸、总离子、pH值的相关性Table 6. Correlation between sensory odour and total acid, total ion and pH value感官气味Sensory odor 总酸Total acid 总离子Total ion pH值pH value 感官气味 1 总酸 0.715** 1 总离子 −0.314 −0.017 1 pH值 −0.073 −0.151 −0.617** 1 **相关性在0.01置信水平下显著. **Correlation is significant at 0.01 level. | Show TableDownLoad:
CSV
2.4 微生物分析结果
人们通常认为空气净化器滤网产生酸臭异味可能是由于长期放置导致“发霉变质”产生的。为此本实验截取了不同种类的活性炭滤网材料,通过对滤网材料进行微生物培养,利用平板计数法进行统计分析,并与感官气味进行相关性分析,结果如表7所示。由表7可以看出,感官气味与菌落总数及霉菌数相关程度均较低,可以推断活性炭滤网的酸臭异味由细菌滋生的贡献较小。但滤网本身确有可能生长霉菌,建议使用者定期更换滤膜并进行清洁保养。
表 7 感官气味与菌落数及霉菌的相关性Table 7. Correlation between sensory odour and colony number and mold感官气味Sensory odor 菌落总数Aerobic bacterial count 霉菌Mould 感官气味 1 菌落总数 −0.234 1 霉菌 0.045 0.612** 1 **相关性在0.01置信水平下显著. **Correlation is significant at 0.01 level. | Show TableDownLoad:
CSV
3. 结论(Conclusion)
本文首先通过感官评价与滤网样品特征进行了Spearman相关性分析,发现使用过的滤网,以及添加催化剂的滤网比较容易产生酸臭异味。进一步通过气相色谱质谱、离子色谱等多种仪器从酸臭异味的角度对滤网样本进行剖析,通过与感官评分的相关性分析,判断出以乙酸为主的小分子有机酸可能是活性炭滤网酸臭异味的主要贡献者,并且很可能源于活性炭表面羧基基团脱落、常用催化剂的降解残留以及使用过程中的催化氧化。无机酸根离子、活性炭自身酸碱性及细菌滋生对滤网酸臭异味的贡献较小。值得指出的是,受限于检测方法,酸臭明显的气味与气相色谱鉴定出的主峰并不相关,这表明嗅阈值低的微量化合物可能是重要的气味贡献者之一。为了精确测定这些异味活性物质,需要更复杂和耗时的方法。目前这项研究的目标是对净化器滤网中不愉快的酸臭气味进行相关特性分析,以便了解潜在的异味来源和形成途径,以期为改善活性炭滤芯生产工艺与防治异味提供科学参考依据。
-
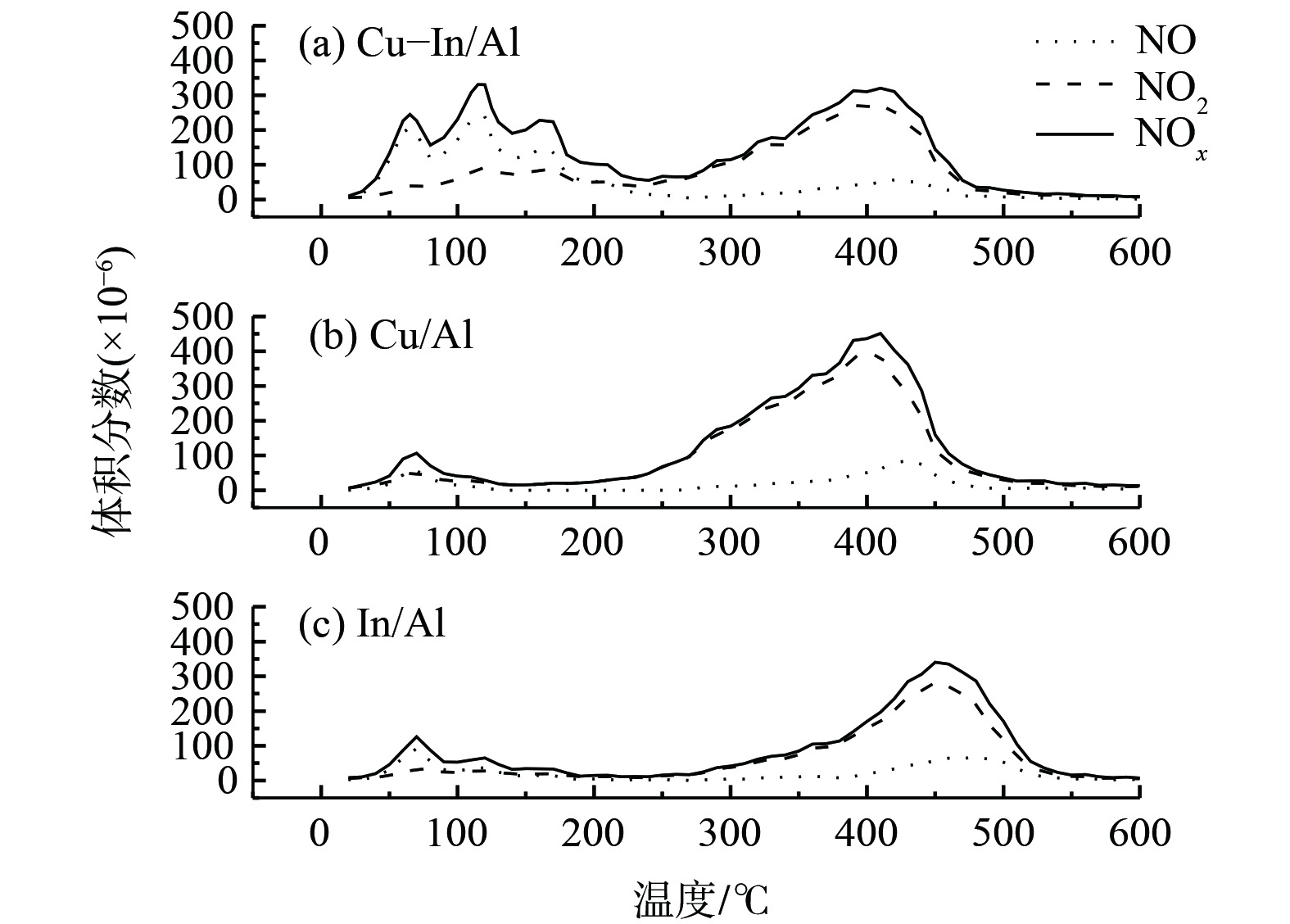
图 2 Cu-In/Al、Cu/Al和In/Al的NO+O2-TPD曲线图
Figure 2. NO+O2-TPD patterns of Cu-In/Al, Cu/Al and In/Al
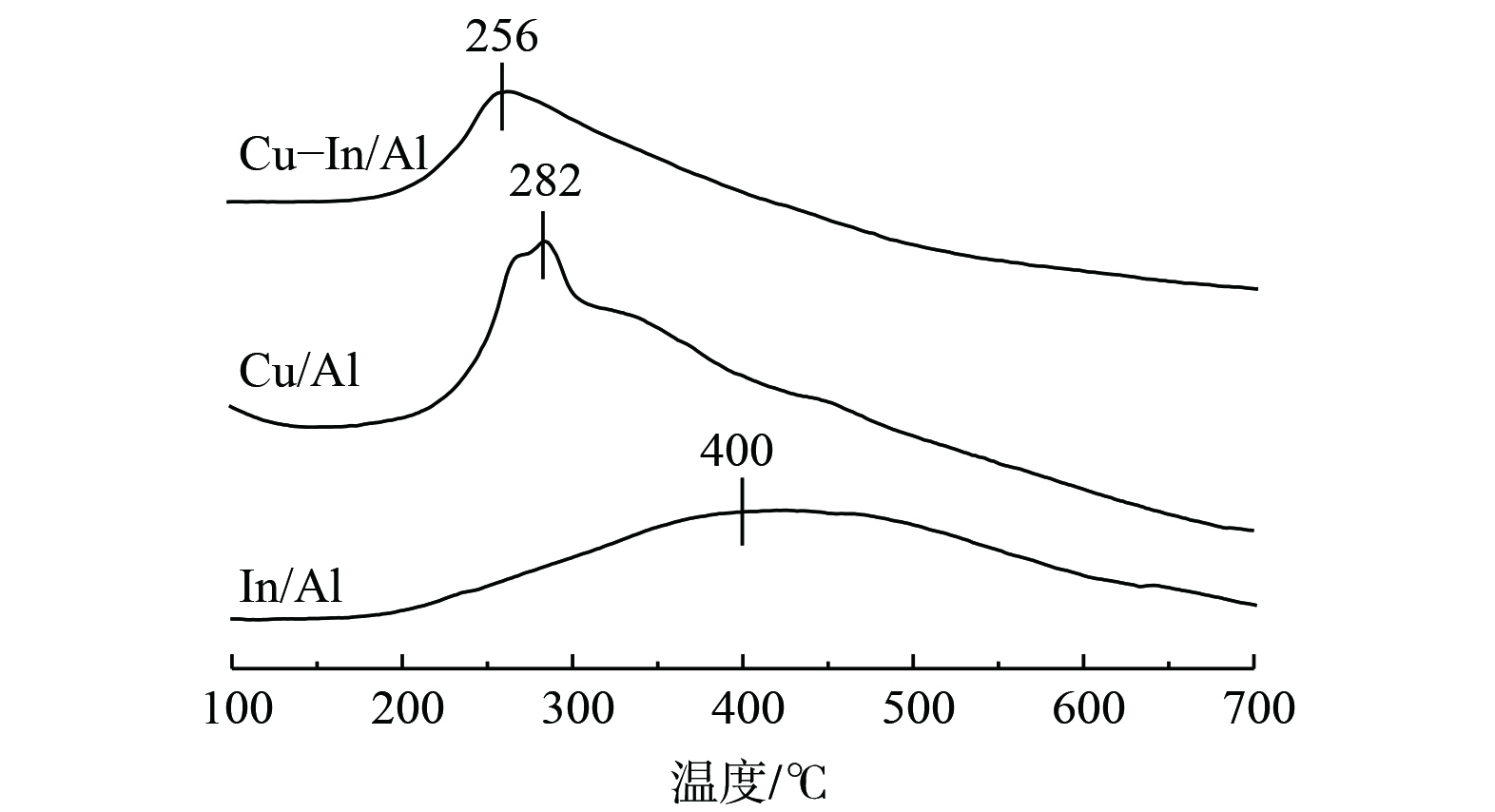
图 4 Cu-In/Al、Cu/Al和In/Al的H2-TPR谱图
Figure 4. H2-TPR spectra of Cu-In/Al, Cu/Al and In/Al

图 5 Cu-In/Al、Cu/Al和In/Al的C3H6-SCR活性测试结果
Figure 5. Catalytic performance of Cu-In/Al, Cu/Al and In/Al in C3H6-SCR
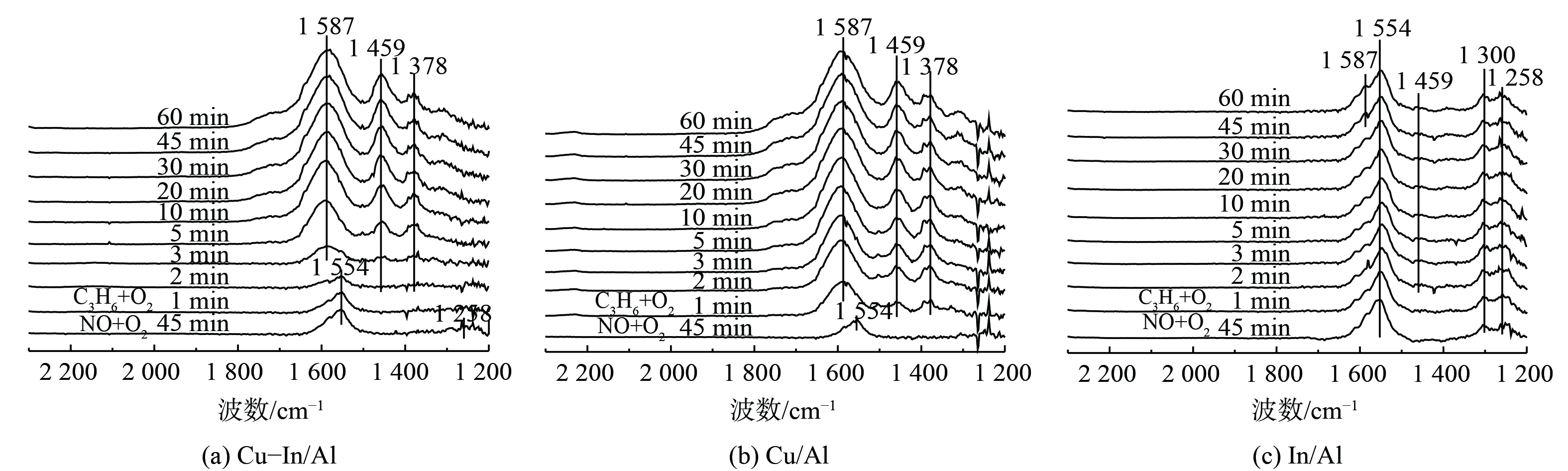
图 6 在350 ℃达到NO吸附饱和时,Cu-In/Al、Cu/Al和In/Al的in situ DRIFTS谱图
Figure 6. In situ DRIFTS spectra of Cu-In/Al, Cu/Al and In/Al at NO adsorption saturation at 350 °C
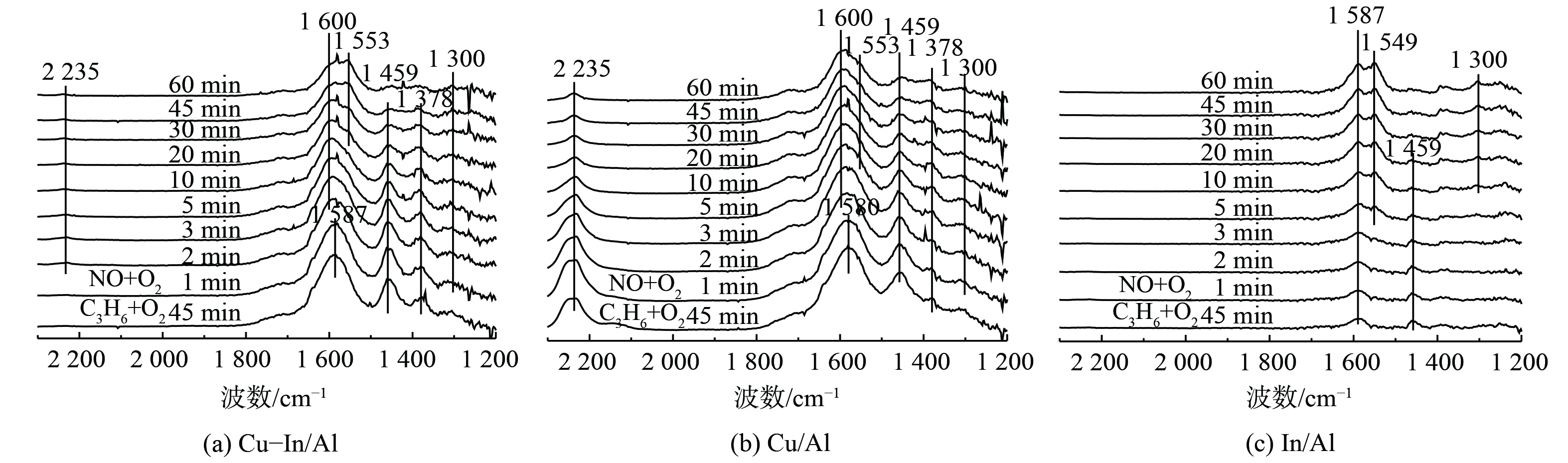
图 7 在350 ℃达到C3H6吸附饱和时,Cu-In/Al、Cu/Al和In/Al的in situ DRIFTS谱图
Figure 7. In situ DRIFTS spectra of Cu-In/Al, Cu/Al and In/Al at C3H6 adsorption saturation at 350 °C

图 8 温度为200~500 ℃时,Cu-In/Al、Cu/Al和In/Al的in situ DRIFTS谱图
Figure 8. In situ DRIFTS spectra of Cu-In/Al, Cu/Al and In/Al at a temperature ranging from 200 °C to 500 °C
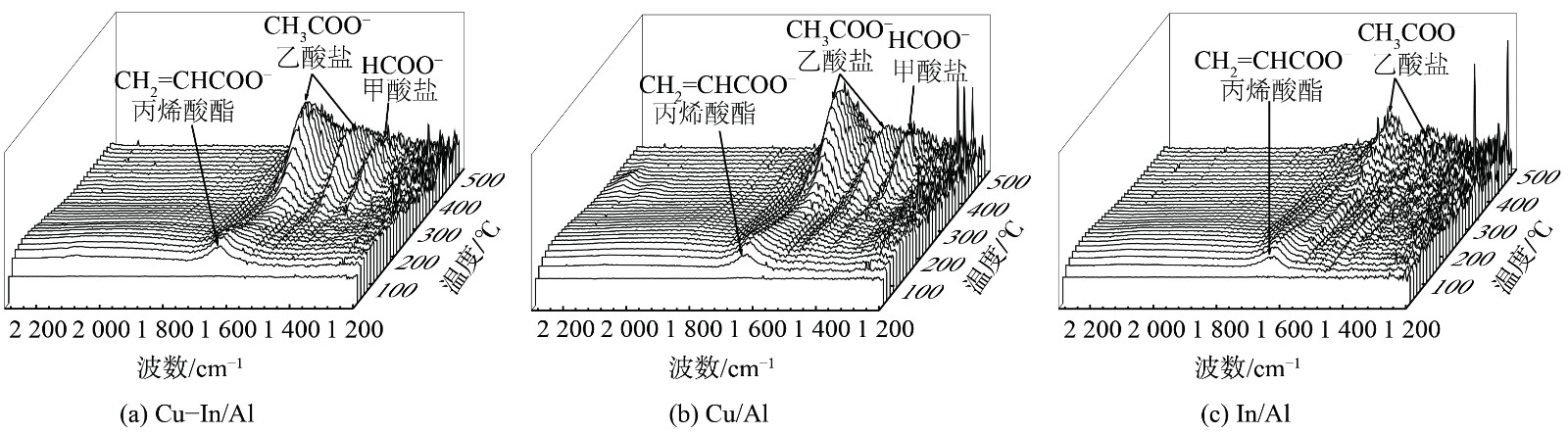
图 9 温度升高过程中Cu-In/Al、Cu/Al和In/AlC3H6氧化的in situ DRIFTS谱图
Figure 9. In situ DRIFTS spectra of Cu-In/Al, Cu/Al and In/Al for C3H6 oxidation during temperature increase

图 10 红外谱图表征的Cu-In/Al、Cu/Al和In/Al上中间产物的强度
Figure 10. Intensity of intermediates on Cu-In/Al, Cu/Al and In/Al as charaterized by IR spectra
表 1 Cu-In/Al、Cu/Al和In/Al催化剂的NOx吸附量
Table 1. The NOx adsorption capacity of Cu-In/Al, Cu/Al and In/Al catalysts
催化剂 NO吸附量/(μmol·g−1) NO2吸附量/(μmol·g−1) NOx吸附量/(μmol·g−1) Cu-In/Al 274.00 441.52 715.52 Cu/Al 86.74 528.61 615.35 In/Al 108.59 336.88 445.47
下载: 导出CSV
-
[1] IWAMOTO M, ZENYO T, HERNANDEZ A M, et al. Intermediate addition of reductant between an oxidation and a reduction catalyst for highly selective reduction of NO in excess oxygen[J]. Applied Catalysis B:Environmental, 1998, 17: 259-266. doi: 10.1016/S0926-3373(98)00018-6 [2] CHANG H Z, QIN X, MA L, et al. Cu/SAPO-34 prepared by a facile ball milling method for enhanced catalytic performance in the selective catalytic reduction of NOx with NH3[J]. Physical Chemistry Chemical Physics, 2019, 21(39): 22113-22120. doi: 10.1039/C9CP04519H [3] 荆国华, 李俊华, 杨栋, 等. 固体超强酸和金属氧化物类催化剂上CH4-SCR还原NOx研究进展[J]. 环境工程学报, 2010, 4(7): 1441-1447. [4] HALPOTO A, KASHIF M, Su Y X, et al. Preparations and characterization on Fe based catalyst supported on coconut shell activated carbon CS(AC) and SCR of NOx-HC[J]. Catalysis Surveys from Asia, 2020, 24(2): 123-133. doi: 10.1007/s10563-020-09293-6 [5] ZHAO L, ZHANG Y, BI S N, et al. Metal-organic framework-derived CeO2–ZnO catalysts for C3H6-SCR of NO: an in situ DRIFTS study[J]. RSC Advances, 2019, 9(33): 19236-19242. doi: 10.1039/C9RA03103K [6] PAN H, GUO Y H, Bi H T. NOx adsorption and reduction with C3H6 over Fe/zeolite catalysts: effect of catalyst support[J]. Chemical Engineering Journal, 2015, 280: 66-73. doi: 10.1016/j.cej.2015.05.093 [7] YANG W, ZHANG R D, CHEN B H, et al. New aspects on the mechanism of C3H6 selective catalytic reduction of NO in the presence of O2 over LaFe1-x(Cu, Pd)xO3-delta perovskites[J]. Environmental Science & Technology, 2012, 46(20): 11280-11288. [8] 林睿, 苏亚欣, 程江浩, 等. Fe/Ga2O3-Al2O3催化甲烷还原NO的性能[J]. 环境工程学报, 2020, 14(6): 1592-1604. doi: 10.12030/j.cjee.201908001 [9] ADAMOWSKA-TEYSSIER M, KRZTOŃ A, COSTA P D, et al. SCR NOx mechanistic study with a mixture of hydrocarbons representative of the exhaust gas from coal combustion over Rh/Ce0.62Zr0.38O2 catalyst[J]. Fuel, 2015, 150: 21-28. doi: 10.1016/j.fuel.2015.01.017 [10] WU Q, YU Y B, HE H. Mechanistic study of selective catalytic reduction of NOx with C2H5OH and CH3OCH3 over Ag/Al2O3 by in situ DRIFTS[J]. Chinese Journal of Catalysis, 2006, 27(11): 993-997. doi: 10.1016/S1872-2067(06)60052-1 [11] CAMPA M C, PIETROGIACOMI D, SCARFIELLO C, el al. CoOx and FeOx supported on ZrO2 for the simultaneous abatement of NOx and N2O with C3H6 in the presence of O2[J]. Applied Catalysis B:Environmental, 2019, 240: 367-372. doi: 10.1016/j.apcatb.2017.04.041 [12] 周皞, 苏亚欣, 邓文义, 等. 金属氧化物类催化剂上HC-SCR研究进展[J]. 环境科学与技术, 2016, 39(1): 93-100. [13] BURCH R, BREEN J P, MEUNIER F C. A review of the selective reduction of NOx with hydrocarbons under lean-burn conditions with non-zeolitic oxide and platinum group metal catalysts[J]. Applied Catalysis B:Environmental, 2002, 39: 283-303. doi: 10.1016/S0926-3373(02)00118-2 [14] LIU Z M, LI J H, HAO J M. Selective catalytic reduction of NOx with propene over SnO2/Al2O3 catalyst[J]. Chemical Engineering Journal, 2010, 165(2): 420-425. doi: 10.1016/j.cej.2010.09.009 [15] LIU Y Q, LAI Q, SUN Y, el al. SnO2/Al2O3 catalysts for selective reduction of NOx by propylene: on the promotional effects of plasma treatment in air atmosphere[J]. Catalysis Today, 2019, 337: 171-181. doi: 10.1016/j.cattod.2019.04.013 [16] PERDIGON-MELON J A, GERVASINI A, AUROUX A. Study of the influence of the In2O3 loading on γ-alumina for the development of de-NOx catalysts[J]. Journal of Catalysis, 2005, 234(2): 421-430. doi: 10.1016/j.jcat.2005.07.001 [17] LIU Z M, HAO J M, FU L X, el al. Activity enhancement of bimetallic Co-In/Al2O3 catalyst for the selective reduction of NO by propene[J]. Applied Catalysis B:Environmental, 2004, 48(1): 37-48. doi: 10.1016/j.apcatb.2003.09.005 [18] HE C H, PAULUS M, CHU W, et al. Selective catalytic reduction of NO by C3H8 over CoOx/Al2O3: An investigation of structure–activity relationships[J]. Catalysis Today, 2008, 131(1/2/3/4): 305-313. [19] CARLO G D, LIOTTA L F, PANTALEO G, et al. Alumina and alumina-baria supported cobalt catalysts for deNOx: Influence of the support and cobalt content on the catalytic performance[J]. Topics in Catalysis, 2009, 52(13/14/15/16/17/18/19/20): 1826-1831. [20] 王琪莹, 刘自力, 邹汉波, 等. 焙烧温度对层柱粘土催化剂Cu/Ti-PILCs催化丙烯还原NO反应的影响[J]. 环境工程学报, 2015, 9(11): 5527-5530. doi: 10.12030/j.cjee.20151163 [21] AMIN N A S, TAN E F, MANAN Z A. SCR of NOx by C3H6: Comparison between Cu/Cr/CeO2 and Cu/Ag/CeO2 catalysts[J]. Journal of Catalysis, 2004, 222(1): 100-106. doi: 10.1016/j.jcat.2003.10.005 [22] ZHOU H, GE M Y, WU S G, et al. Iron based monolithic catalysts supported on Al2O3, SiO2, and TiO2: A comparison for NO reduction with propane[J]. Fuel, 2018, 220: 330-338. doi: 10.1016/j.fuel.2018.01.077 [23] 袁旻昊, 钱文燕, 邓文义, 等. 铁修饰铝柱撑黏土催化剂(Fe/Al-PILC)的制备及其对C3H6-SCR活性的影响[J]. 环境工程学报, 2020, 14(4): 1022-1032. doi: 10.12030/j.cjee.201911027 [24] ROY S, VISWANATH B, HEGDE M S, et al. Low-temperature selective catalytic reduction of NO with NH3 over Ti0.9M0.1O2-δ (M = Cr, Mn, Fe, Co, Cu)[J]. Journal of Physical Chemistry C, 2008, 112: 6002-6012. doi: 10.1021/jp7117086 [25] ZHANG R D, TEOH W Y, AMAL R, et al. Catalytic reduction of NO by CO over Cu/CexZr1−xO2 prepared by flame synt hesis[J]. Journal of Catalysis, 2010, 272(2): 210-219. doi: 10.1016/j.jcat.2010.04.001 [26] KUMAR P A, REDDY M P, JU L K, et al. Low temperature propylene SCR of NO by copper alumina catalyst[J]. Journal of Molecular Catalysis A:Chemical, 2008, 291(1/2): 66-74. [27] Liu J, Zhao Q D, LI X Y, et al. Structure sensitivity of selective catalytic reduction of NO with propylene over Cu-doped Ti0.5Zr0.5O2-catalysts[J]. Applied Catalysis B:Environmental, 2015, 165: 519-528. doi: 10.1016/j.apcatb.2014.10.038 [28] LU G, LI X Y, QU Z P, et al. Copper-ion exchanged Ti-pillared clays for selective catalytic reduction of NO by propylene[J]. Chemical Engineering Journal, 2011, 168(3): 1128-1133. doi: 10.1016/j.cej.2011.01.095 [29] VALVERD J L, DELUCAS A, SÁNCHEZ P, et al. Cation exchanged and impregnated Ti-pillared clays for selective catalytic reduction of NOx by propylene[J]. Applied Catalysis B:Environmental, 2003, 43(1): 43-56. doi: 10.1016/S0926-3373(02)00274-6 [30] VALVERDE J L, DELUCAS A, DORADO F, et al. Study by in situ FTIR of the SCR of NO by propene on Cu2+ ion-exchanged Ti-PILC[J]. Journal of Molecular Catalysis A:Chemical, 2005, 230(1/2): 23-28. [31] DORADO F, ROMERO R, CRUZ J, et al. Selective catalytic reduction of NO by propene in the presence of oxygen and water over catalysts prepared by the modified sol–gel method[J]. Catalysis Communications 2007, 8(4): 736-740. [32] SHIMIZU K, KAWABATA H, MAESHIMA H, et al. Intermediates in the selective reduction of NO by propene over Cu-Al2O3 catalysts: Transient in-Situ FTIR study[J]. Journal of Physical Chemistry B, 2000, 104: 2885-2893. doi: 10.1021/jp9930705 [33] YADAV D, KAVAIYA A R, MOHAN D, et al. Low temperature selective catalytic reduction (SCR) of NOx emissions by Mn-doped Cu/Al2O3 catalysts[J]. Bulletin of Chemical Reaction Engineering & Catalysis, 2017, 12(3): 415-429. [34] PANAHI P N, SALARI D, NIAEI A, et al. NO reduction over nanostructure M-Cu/ZSM-5 (M: Cr, Mn, Co and Fe) bimetallic catalysts and optimization of catalyst preparation by RSM[J]. Journal of Industrial and Engineering Chemistry, 2013, 19(6): 1793-1799. doi: 10.1016/j.jiec.2013.02.022 [35] SADEGHINIA M, REZAEI M, KHARAT A N, et al. Effect of In2O3 on the structural properties and catalytic performance of the CuO/ZnO/Al2O3 catalyst in CO2 and CO hydrogenation to methanol[J]. Molecular Catalysis, 2020, 484: 110776. doi: 10.1016/j.mcat.2020.110776 [36] MA L, SEO C Y, CHEN X Y, et al. Indium-doped Co3O4 nanorods for catalytic oxidation of CO and C3H6 towards diesel exhaust[J]. Applied Catalysis B:Environmental, 2018, 222: 44-58. doi: 10.1016/j.apcatb.2017.10.001 [37] BERSANI D, LOTTICI P P, RANGEL G, et al. Micro-Raman study of indium doped zirconia obtained by sol–gel[J]. Journal of Non-Crystalline Solids, 2004, 345-346: 116-119. doi: 10.1016/j.jnoncrysol.2004.08.006 [38] SHENDE A G, GHUGAL S G, VIDYASAGAR D, et al. Magnetically separable indium doped ZnS NiFe2O4 heterostructure photocatalyst for mineralization of acid violet 7 dye[J]. Materials Chemistry and Physics, 2019, 221: 483-492. doi: 10.1016/j.matchemphys.2018.09.032 [39] LI J H, HAO J M, CUI X Y, et al. Influence of preparation methods of In2O3/Al2O3 catalyst on selective catalytic reduction of NO by propene in the presence of oxygen[J]. Catalysis Letters, 2005, 103(1/2): 75-82. [40] LI J H, HAO J M, FU L X, et al. Cooperation of Pt/Al2O3 and In/Al2O3 catalysts for NO reduction by propene in lean burn condition[J]. Applied Catalysis A:General, 2004, 265(1): 43-52. doi: 10.1016/j.apcata.2004.01.001 [41] HU S Y, XIAO W, YANG W W, et al. Molecular O2 activation over Cu(I)-mediated C identical with N bond for low-temperature CO oxidation[J]. ACS Applied Materials & Interfaces, 2018, 10(20): 17167-17174. [42] LUO Z, CETEGEN S A, MIAO R, et al. Structure–property relationships of copper modified mesoporous TiO2 materials on alkyne homocoupling reactions[J]. Journal of Catalysis, 2016, 338: 94-103. doi: 10.1016/j.jcat.2016.03.009 [43] FANG Y R, LI L, YANG J, et al. Engineering the nucleophilic active oxygen species in CuTiOx for efficient low-temperature propene combustion[J]. Environment Science & Technology, 2020, 54(23): 15476-15488. [44] CHANG H Z, LI M G, LI Z G, et al. Design strategies of surface basicity for NO oxidation over a novel Sn–Co–O catalyst in the presence of H2O[J]. Catalysis Science & Technology, 2017, 7(10): 2057-2064. [45] AMANO F, SUZUKI S, YAMAMOTO T, et al. One-electron reducibility of isolated copper oxide on alumina for selective NO–CO reaction[J]. Applied Catalysis B:Environmental, 2006, 64(3/4): 282-289. [46] PARK P W, RAGLE C S, BOYER C L, et al. In2O3/Al2O3 Catalysts for NOx Reduction in Lean Condition[J]. Journal of Catalysis, 2002, 210(1): 97-105. doi: 10.1006/jcat.2002.3667 [47] HE H, ZHANG C B, YU Y B. A comparative study of Ag/Al2O3 and Cu/Al2O3 catalysts for the selective catalytic reduction of NO by C3H6[J]. Catalysis Today, 2004, 90: 191-197. doi: 10.1016/j.cattod.2004.04.026 [48] HANEDA M, KINTAICHI Y, HAMADA H. Promotional effect of H2O on the activity of In2O3-doped Ga2O3–Al2O3 for the selective reduction of nitrogen monoxide[J]. Catalysis Letters, 1998, 55: 47-55. doi: 10.1023/A:1019002105560 [49] CUI X Y, LI J H, HAO J M, et al. Enhancement of activity of SnO2-doped In2O3/Al2O3 catalyst for NO reduction with propene in the presence of H2O and SO2[J]. Chinese Chemical Letters, 2005, 16(11): 1535-1538. [50] HADJIIVANOV K. Identification of neutral and charged NxOy surface species by IR spectroscopy[J]. Catalysis Reviews, 2000, 42(1/2): 71-144. [51] TAMM S, INGELSTEN H H, PALMQVIST A E C. On the different roles of isocyanate and cyanide species in propene-SCR over silver/alumina[J]. Journal of Catalysis, 2008, 255(2): 304-312. doi: 10.1016/j.jcat.2008.02.019 [52] LIU Z M, OH K S, WOO S I. Promoting Effect of CeO2 on NOx reduction with Propene over SnO2/Al2O3 catalyst studied with in situ FT-IR spectroscopy[J]. Catalysis Letters, 2007, 120(1/2): 143-147. [53] NGUYEN L Q, SALIM C, HINODE H. Roles of nano-sized Au in the reduction of NOx by propene over Au/TiO2: An in situ DRIFTS study[J]. Applied Catalysis B:Environmental, 2010, 96(3/4): 299-306. [54] PIETRZYK P, DUJARDIN C, GÓRA-MAREK K, et al. Spectroscopic IR, EPR, and operando DRIFT insights into surface reaction pathways of selective reduction of NO by propene over the Co-BEA zeolite[J]. Physical Chemistry Chemical Physics, 2012, 14(7): 2203-15. doi: 10.1039/C1CP23038G [55] GÓRA-MAREK K, GIL B, DATKA J. Quantitative IR studies of the concentration of Co2+ and Co3+ sites in zeolites CoZSM-5 and CoFER[J]. Applied Catalysis A:General, 2009, 353(1): 117-122. doi: 10.1016/j.apcata.2008.10.034 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4018
- HTML全文浏览数: 4018
- PDF下载数: 44
- 施引文献: 0



