




-
酸法地浸采铀即通过钻孔直接将酸性溶浸剂注入地下含矿水层使得其与矿物反应以获取铀的一种工艺。由于硫酸价格低廉、浸出能力强,酸法地浸采铀溶剂一般使用硫酸[1]。由于酸法地浸采铀人为改变了地下含水层的水文化学环境,酸法地浸采铀矿山退役后,矿层地下水中的硫酸、铀及重金属离子可能会污染矿区及周边的地下水环境[2-5]。还由于铀具有放射性与化学毒性,故对生态环境及人体健康存在潜在危害[6-7]。因此,酸法地浸采铀退役采区地下水的修复是亟待研究解决的问题。
近年来,部分研究学者开始研究酸法地浸采铀退役采区地下水的修复方法。目前,修复的主要方法有自然净化法、抽出处理法、微生物处理法、离子交换处理法及碱处理法等[8]。自然净化法[9-11]旨在利用天然环境条件实现水体净化。自然净化法具有环境友好的优点,但存在修复周期长及修复不彻底的缺点。抽出处理法[12-15]即将地下水抽出对其进行地表处理(如电渗析、反渗透法等),修复后的水再注入地下含水层。抽出处理的修复效果好,但存在修复成本高、修复周期长的缺点。微生物处理法[16-18]主要利用微生物及其代谢产物以实现铀的原位还原/矿化/吸附[19-21]。微生物修复无二次污染,但存在微生物适应性差及修复周期长的缺点。离子交换处理法[22-23]主要利用离子交换树脂等材料实现地下水中铀的分离,该方法修复效率高,但存在运行成本高的缺点。碱处理法[24-27]通过向地下水中注入碱性物质实现铀的沉淀/还原,此处理法具有修复效率高的优点,但存在易产生二次污染及处理成本高的缺点。因此,需研究修复成本低且无二次污染的碱性物质对酸性铀污染地下水进行修复。
硅酸盐、磷酸盐是一种绿色修复剂,其一般通过络合沉淀及共沉淀作用与地下水中污染物反应以达到修复的目的,其作用机制不会影响地下水的理化性质和结构,也不会产生二次污染。有研究表明,向退役铀矿采区地下含水层注入硅酸盐/磷酸盐可以固定地下水中的铀[28-31]。WEN等[32]研究了在Ca2+存在的条件下磷酸盐的加入对地下水中铀的固定作用,结果表明,磷酸根可与钙和铀结合形成三元表面复合物,从而可实现对铀污染地下水的修复。MEHTA等[33]研究了Na+与Ca2+在溶液中共存的情况下磷酸盐与铀的络合产物,结果表明,磷酸盐可与铀形成(Na2(UO2)2(PO4)2)和(Ca(UO2)2(PO4)2)络合物。SODERHOLM等[31]在水热处理条件下,向浓度为50 mmol·L−1的U(VI)溶液中加入硅酸钠,结果表明,在水热条件下硅酸钠可与铀反应形成硅铀矿沉淀,说明硅酸盐的加入可有效修复酸法地浸采铀退役采区铀污染地下水。也有研究表明,硅酸盐和磷酸盐通过共同作用可修复铀矿山退役采区污染地下水。如MASAKAZU等[34]研究了在硅酸钠存在的情况下,磷酸盐对合成酸性铀废水中铀的固定作用及硅酸盐/磷酸盐与铀作用产生的沉淀的形态和物理化学特性,结果表明,磷酸盐在反应过程中对铀的固定起到了重要作用。以上研究结果表明,硅酸盐和磷酸盐对酸法地浸采铀退役采区地下水有良好的修复效果,但硅酸盐、磷酸盐在酸法地浸采铀退役采区地下水的修复过程中会产生哪些作用,其作用机制如何,目前尚不明确。
因此,本研究拟从西北某酸法地浸采铀退役采区采取地下岩心样,同时制备了酸法地浸采铀退役采区地下水的模拟水样,构建了模拟修复反应的微模型;向微模型中分别加入硅酸钠溶液、磷酸二氢钾溶液、硅酸钠-磷酸二氢钾混合溶液,分析了微模型中铀及金属阳离子浓度的变化;对反应结束后的沉淀产物进行了表征及机理分析。本研究结果对于深入了解硅酸盐和磷酸盐原位修复酸法地浸采铀退役采区地下水的机理具有参考意义。
-
从西北某酸法地浸采铀退役采区采取砂岩铀矿岩心样,岩心样在取出后立即用保鲜膜包裹以保持厌氧环境送回实验室,并放置于冰箱中低温保存。同时制备酸法地浸采铀退役采区地下水的模拟水样,其特征是低pH(调节模拟废水pH为3.00±0.01)、多金属阳离子共存。水样的主要成分为1.60 mg·L−1 Fe3+、581.70 mg·L−1 Ca2+、3.90 mg·L−1 Mn2+、244.90 mg·L−1 Mg2+、1.70 mg·L−1 Zn2+、1 094.80 mg·L−1 Na+、5.00 mg·L−1 UO22+,以此构建模拟修复反应的微模型。
-
1)实验分组。为研究实验去除可溶性U(Ⅵ)的效果,向微模型中分别加入硅酸钠溶液、磷酸二氢钾溶液、硅酸钠-磷酸二氢钾混合溶液。实验分别设置空白组、对照组、实验组,每组设3个平行样。各实验体系中地下水体积为200 mL,地下水初始铀质量浓度为5 mg·L−1,200 mL模拟地下水中加入4 g的岩心样。向各实验体系通入45 min氮气以模拟地下水的厌氧环境。岩心样的主要化学成分如表1所示。由表1可以看出,岩心样主要由石英、高岭土和长石组成,三者总含量高达96.30%。
2)实验方法。用pH计测量微模型中水样的初始pH。向聚四氟乙烯瓶中加入200 mL待处理水样,记为空白组(G);向空白组(G)中加入铀矿山退役采区采取的岩心样,记为对照组(G+R);向空白组与对照组通入氮气以保持微模型的厌氧环境。向通入氮气的G+R对照组中缓慢滴加配制好的硅酸钠溶液(10 g·L−1),搅拌并测量水样pH,待水样pH达到7.00±0.05时,停止滴加硅酸钠溶液,并记录已加入的硅酸钠溶液体积,记为地下水+岩心样+硅酸钠(G+R+S)实验组;向通入氮气的G+R对照组与G+R+S实验组中加入与硅酸钠相同浓度的磷酸二氢钾溶液,记为地下水+岩心样+磷酸二氢钾(G+R+P)实验组与地下水+岩心样+硅酸钠+磷酸二氢钾(G+R+S+P)实验组,并测定各实验组中溶液的初始pH。实验过程中监测微模型中的pH及硅酸根、磷酸根、金属离子、铀离子浓度的变化;采用XRD、SEM、XPS对反应结束后的沉淀产物进行表征;采用改良后的连续提取法分析沉淀产物中铀的化学形态和稳定性。
3)样品分析检测方法。采用pH计测定溶液的pH;ICP-MS和紫外分光光度计测定溶液中铀、硅酸根、磷酸根的浓度;火焰原子吸收分光光度法测定溶液中阳离子的浓度;改良后的连续提取法分析沉淀产物中铀的化学形态;XRF分析初始岩心样成分;XPS分析沉淀产物中铀的价态;SEM观察沉淀产物的形貌;XRD鉴定矿物成分。
采用Origin 2018绘图,采用Jade 6软件匹配分析XRD图谱,采用Avantage软件匹配拟合XPS峰,采用SPSS软件分析铀去除率显著性变化。
-
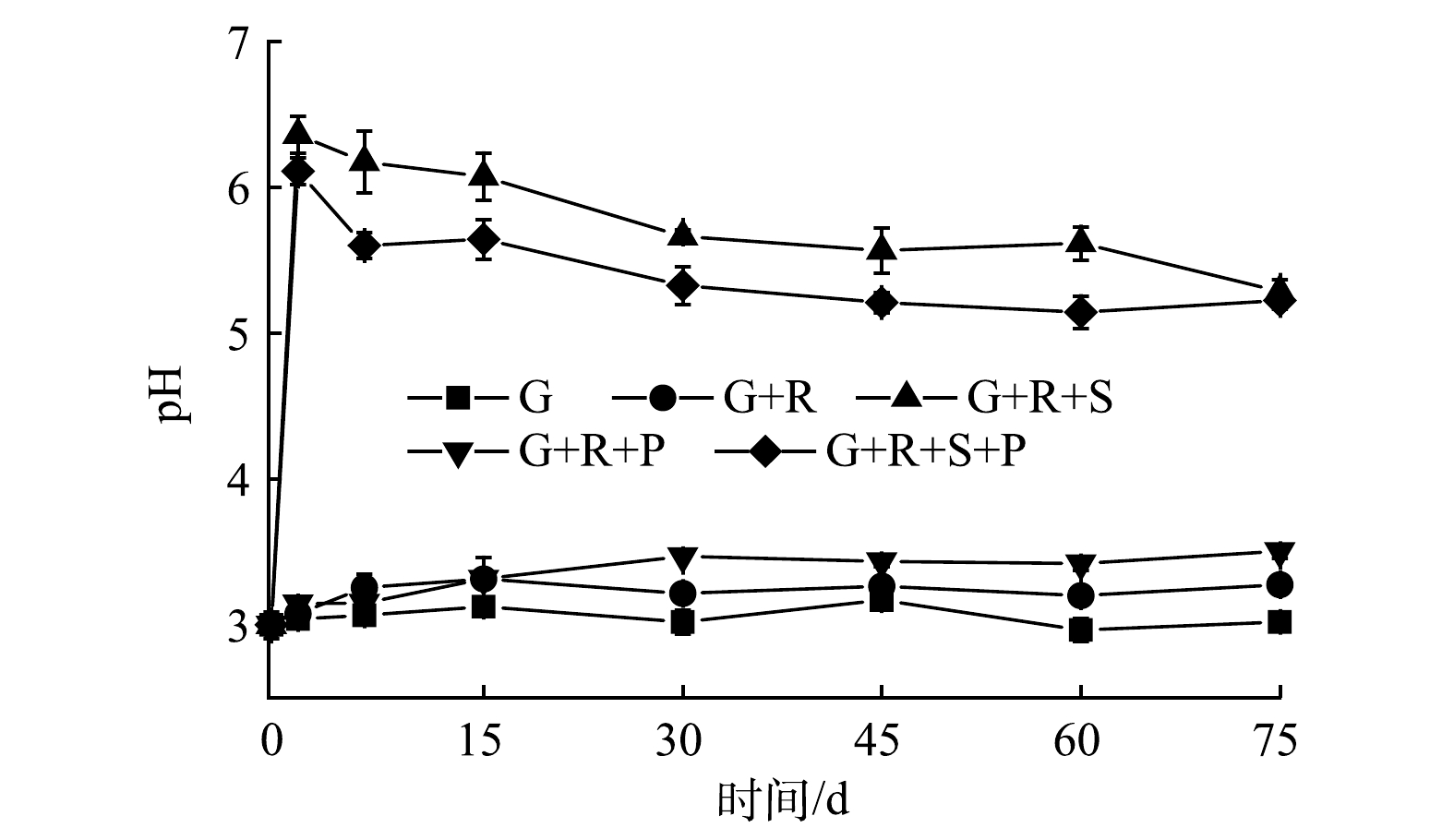
由图1可以看出,空白组(G)的pH几乎未产生变化。与空白组相比,G+R对照组的pH略有上升。可能是由于地下水与岩心样混合体系需要一段时间才能达到平衡,平衡过程中由于岩心样内矿物成分的作用导致溶液pH略有上升。单独添加磷酸盐的G+R+P实验组溶液的pH随着反应的进行有小幅度上升,在75 d内,pH由初始的3.00升到3.50。其原因可能是:反应过程中磷酸二氢根在强酸条件下的电离受到了抑制,从而导致pH缓慢上升。添加硅酸盐的G+R+S组和G+R+S+P组的pH在1 d内急剧上升,由3.00上升到了6.00。这主要因为硅酸根水解产生氢氧根使得pH上升。随着反应的进行,2个实验组的pH存在一定幅度的下降,且在反应过程中G+R+S+P组的pH均低于G+R+S组。在反应75 d后,G+R+S和G+R+S+P实验组溶液的pH分别为5.29和5.23。G+R+S组溶液pH降低的主要原因可能是硅酸根水解产生氢氧根,而氢氧根与铀酰离子反应生成沉淀,导致pH降低。而在有磷酸盐存在的G+R+S+P组,硅酸根在中性条件下优先与铀酰离子发生络合反应形成沉淀,铀酰离子消耗后促进磷酸二氢根的电离产生H+,导致G+R+S+P组pH略低于G+R+S组。以上结果表明,添加磷酸盐的实验组对反应过程中pH的影响不大;添加硅酸盐的实验组使得pH急剧升高然后缓慢下降。
-
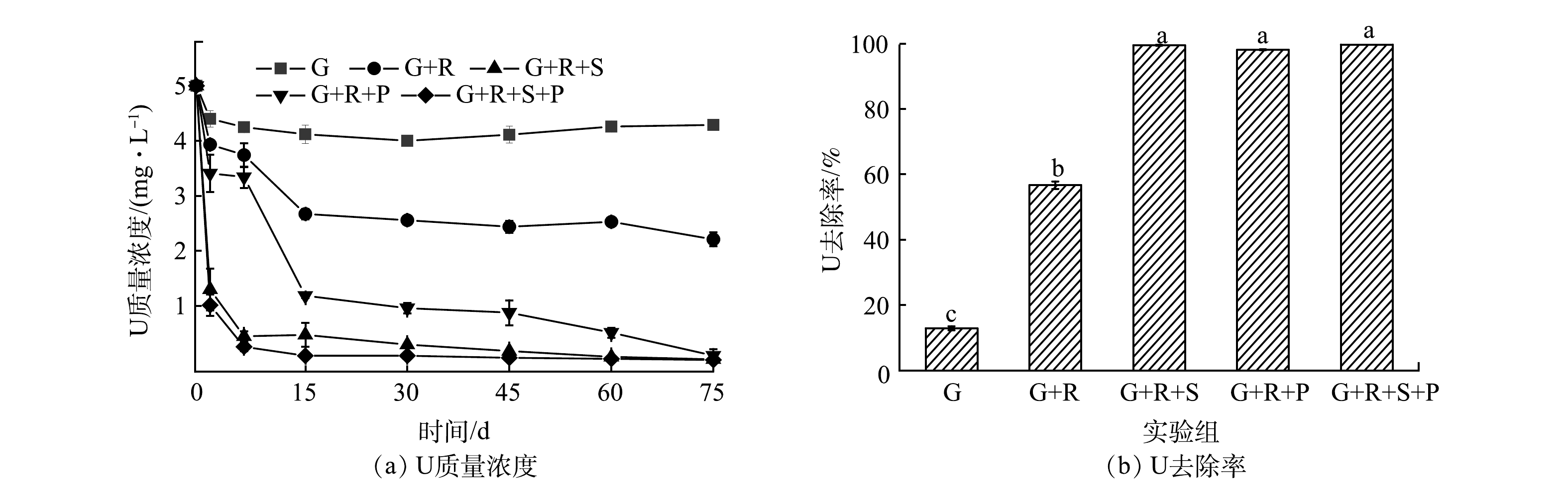
反应过程中溶液的铀质量浓度变化如图2所示。由图2(a)可以看出,空白组(G)与对照组(G+R)的铀质量浓度小幅下降;G+R+S组溶液的铀质量浓度在反应初始1 d内有明显的下降,由5 mg·L−1降至1.29 mg·L−1,并在反应45~75 d内趋于稳定,75 d时铀质量浓度为0.025 mg·L−1。铀质量浓度明显下降说明硅酸钠可以迅速与铀络合形成沉淀,从而降低地下水中的铀浓度。G+R+P组的铀质量浓度在反应进行至15 d时由原来的5 mg·L−1降至1.18 mg·L−1,75 d时铀质量浓度下降至0.092 mg·L−1;G+R+S+P组的铀质量浓度随着反应的进行大幅降低,在15 d后降到了0.09 mg·L−1,75 d后趋于平衡,最终质量浓度为0.014 mg·L−1。反应75 d后溶液中铀的去除率如图2(b)所示。由图2(b)可以看出,G+R+S+P组中溶液铀的去除率达到了99.7%,优于G+R+S组(99.5%)和G+R+P组(98.1%),且他们之间的差异性显著(P<0.01)。以上结果表明,G+R对照组相比于空白组G溶液中铀浓度减小,这可能是由于岩心样中的石英、高岭土、长石对铀有一定的吸附作用[35-36];相同条件下G+R+P组对铀的去除率不如G+R+S组,可能在中性条件下硅酸钠可以充分水解产生氢氧根,氢氧根可以更好地与铀酰离子络合形成沉淀;G+R+S+P组修复效率最高,说明在硅酸钠充分水解的条件下磷酸盐与铀也可以络合形成沉淀,从而提高修复效率。以上结果表明,硅酸钠-磷酸二氢钾混合溶液可有效去除酸性铀污染地下水中的铀。
-
由图3可以看出,钙、镁、铁、锰、锌离子的质量浓度随着反应的进行有所下降。由图3(a)可以看出,有磷酸二氢钾存在的条件下,钙离子浓度下降最快。这可能是由于钙离子和磷酸盐反应可形成磷酸钙固体,而磷酸钙可与铀络合形成钙铀云母所致。由图3(b)可以看出,在第75天时联合修复体系镁离子浓度下降最多。这可能是由于镁离子和硅酸盐、磷酸盐反应形成了沉淀。由图3(c)可以看出,在实验初始阶段,铁离子浓度呈快速下降趋势。这可能是由于三价铁离子与配体形成了含铁的不溶性沉淀。由图3(d)可以看出,在只添加磷酸二氢钾的实验组锰离子浓度下降最快。其原因可能是锰离子与磷酸根反应生成了难溶化合物。由图3(e)可以看出,在单独加入硅酸钠的实验组锌离子浓度下降很快,表明锌离子可与硅酸盐更好地反应。反应75 d后溶液中金属阳离子的去除率变化如图3(f)所示。可见,磷酸二氢钾对于镁、铁、锰的去除效果最好;硅酸钠-磷酸二氢钾对钙、锌的去除效果较好。以上结果说明硅酸钠、磷酸二氢钾、硅酸钠-磷酸二氢钾可有效去除溶液中重金属离子。
-
由图4(a)可以看出,G+R+P组磷酸盐质量浓度在反应前期(30 d内)急剧降低,并在反应时长为75 d时达到91.295 mg·L−1;G+R+S+P组中磷酸盐质量浓度随着反应的进行呈缓慢降低趋势,在反应75 d后下降至84.388 mg·L−1。以上结果表明,当溶液仅存高浓度磷酸盐时,反应前期磷酸二氢钾会与六价铀快速反应形成沉淀[37]。G+R+S+P组在反应初期磷酸盐的浓度下降趋势较慢,但在75 d时磷酸盐的浓度低于G+R+P组,说明在硅酸钠充分水解的条件下磷酸盐可以更好的与铀络合形成沉淀,提高修复效率。
由图4(b)可以看出,G+R+S组、G+R+S+P组硅酸盐质量浓度在反应前期(15 d)有较大幅度的降低,随后趋于稳定,在实验进行到第75天时分别降至为82.56 mg·L−1和114.98 mg·L−1。硅酸钠质量浓度在反应初期迅速下降,说明硅酸钠在反应初期与铀发生络合反应形成了沉淀,硅酸盐浓度变化趋势和KANEMATSU等[34]的研究结果一致。反应进行至第75天时,G+R+S+P组中硅酸钠的浓度高于G+R+S组,这说明在硅酸钠充分水解的条件下磷酸盐可以更好的与铀络合形成沉淀,从而提高修复效率。
-
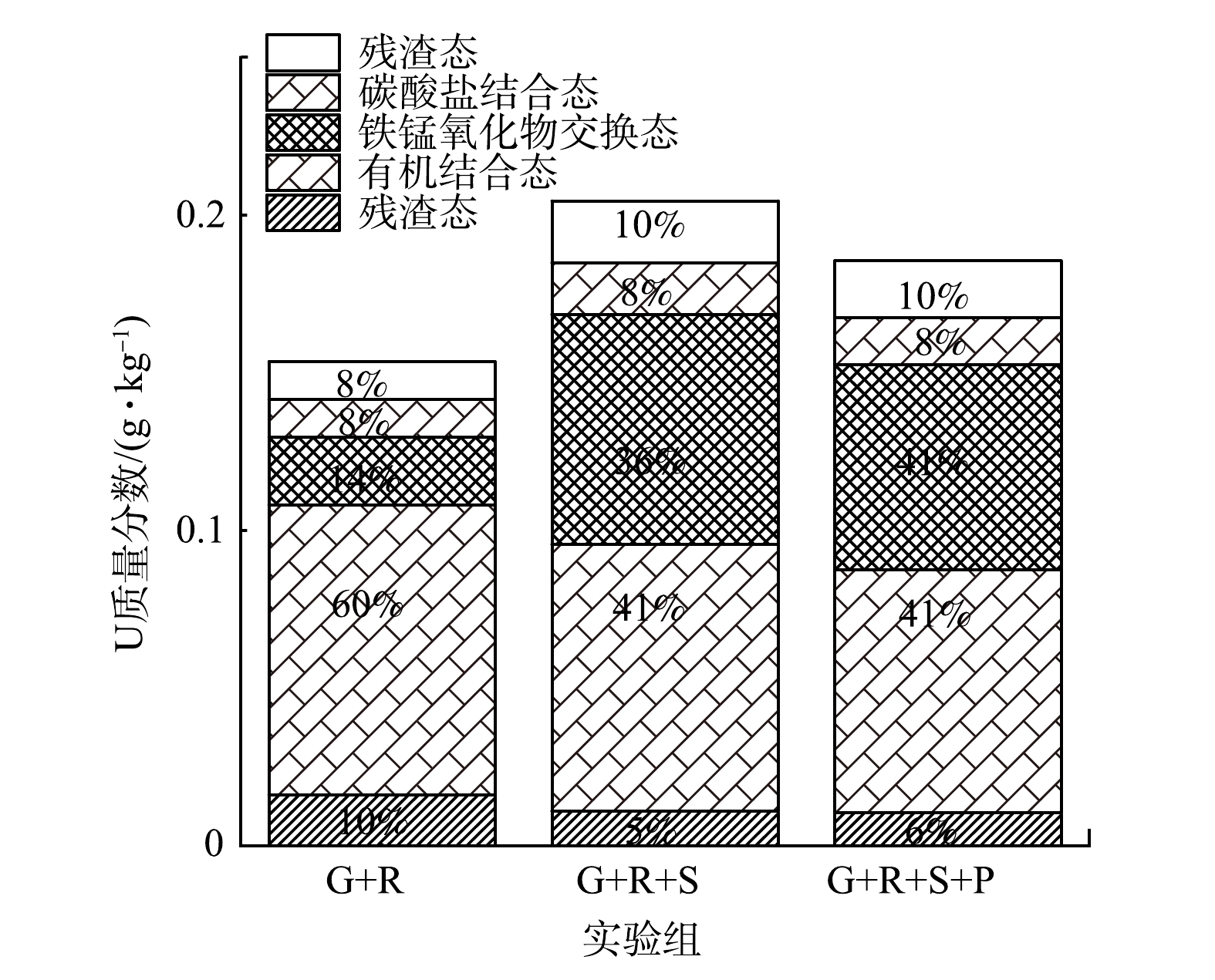
对铀浓度下降至排放标准的实验组的沉淀物进行连续提取。由图5可以看出,G+R对照组中可交换态铀、碳酸盐结合态铀、铁锰氧化物结合态铀、有机结合态铀、残渣态铀的含量分别为16.0、92.0、21.5、12.0、12.0 mg·kg−1,岩心样中铀的总含量为153.5 mg·kg−1;G+R+S实验组在反应至75 d后可交换态铀、碳酸盐结合态铀、铁锰氧化物结合态铀、有机结合态铀、残渣态铀的含量分别为11.0、84.5、73.0、65.0、19.5 mg·kg−1,铀的总含量为253 mg·kg−1;G+R+S+P实验组反应至75 d后可交换态铀、碳酸盐结合态铀、铁锰氧化物结合态铀、有机结合态铀、残渣态铀含量分别为10.5、77.0、62.5、15.0、18.0 mg·kg−1,铀总含量为183 mg·kg−1。与G+R对照组相比,G+R+S组、G+R+S+P组沉淀产物中可交换态铀、碳酸盐结合态铀的含量百分比有所下降,铁锰氧化物、有机结合态铀和残渣态铀的含量百分比有所增加。可交换态铀、碳酸盐结合态铀在环境中不稳定,易受到外界因素的影响,是环境中最具流动性和潜在可利用性的铀形态[38];铁锰氧化物结合态铀在氧化条件下比较稳定;有机结合态铀在自然条件下不易释放,但易受到碱性环境的影响;残渣态铀能够长期稳定地存在于地下岩心样中,对环境基本无有害影响[39]。以上结果说明硅酸钠、硅酸钠-磷酸二氢钾的加入可使沉淀物中不稳定态铀转变为稳定性较高的铀形态。
-
1)低浓度沉淀产物光谱分析。对反应结束后的沉淀产物进行了扫描电镜分析,结果见图6。由图6(a)~(c)可以看出,沉淀产物呈片状分布。与G+R对照组和G+R+S组相比,G+R+S+P组沉淀产物中的片状产物尺寸较大。以上结果说明,硅酸盐与磷酸盐共同作用可高效去除地下水中的铀,并形成较大的块状沉淀物。
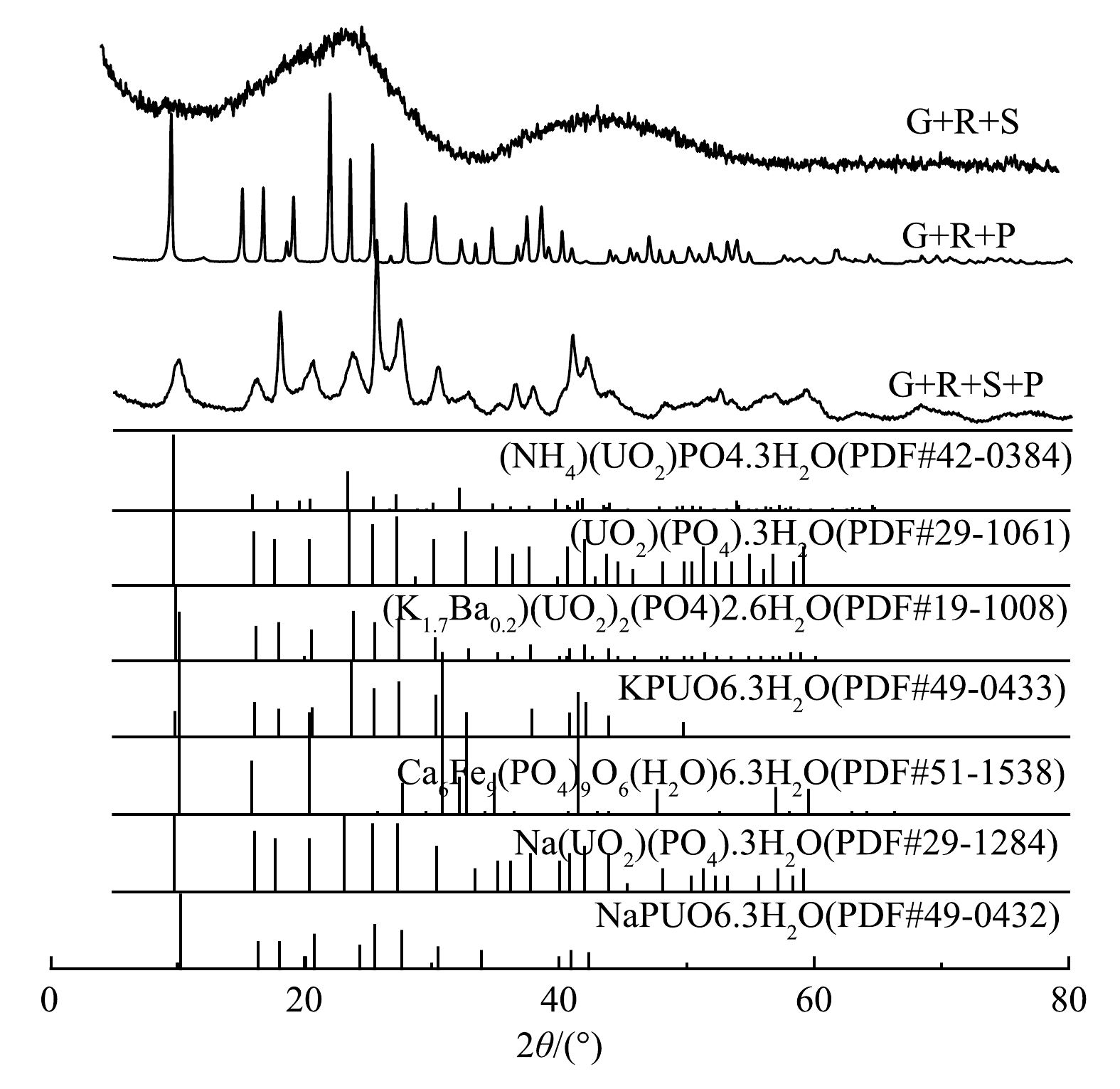
2)高浓度沉淀产物光谱分析。为了进一步分析铀的去除机理,调节模拟地下水中初始铀质量浓度为150 mg·L−1,其他条件不变。利用XRD和XPS分析了沉淀产物的主要成分及铀的价态。高浓度实验组中沉淀产物的XRD图谱如图7所示。图7(a)中有2个大的衍射宽峰,表明硅酸钠和铀形成的沉淀是非晶型结构。KANEMATSU等[34]的研究也表明,在常温条件下铀与硅形成的黄硅钾铀矿的结晶度较差。由图7(b)可以看出,G+R+P组生成的沉淀晶型较好。与XRD的PDF标准卡片图谱进行对比可发现,G+R+P组中的沉淀含有((NH4)(UO2)PO4.3H2O)、((UO2)(PO4).3H2O)、((K1.7Ba0.2)(UO2)2(PO4)2.6H2O)和(NaPUO6.3H2O)4种矿物。由图7(c)可以看出,G+R+S+P组反应生成的沉淀晶型较好,与XRD的PDF标准卡片图谱进行对比可发现,G+R+S+P组中的沉淀含有(Ca6Fe9(PO4)9O6(H2O)6.3H2O)、(Na(UO2)(PO4).3H2O)2和(NaPUO6.3H2O)3种矿物。
由图8(a)可以看出,各实验组中沉淀产物主要含有碳、氧、磷、硅元素,且在结合能为382 eV附近有明显的U4f特征峰,说明各实验组沉淀产物中均含有铀。由于铀的自旋轨道相互作用,U4f峰被分离为U4f5/2和U4f7/2峰[40]。文献报道[41]及XPS标准图谱库表明,U(VI)结合能为381.1~382.6 eV。由图8(b)可以看出,G+R+S组中U4f5/2和U4f7/2峰的结合能分离了10.7 eV[42],即381.79 eV处为U4f7/2峰, 392.49 eV处为U4f5/2峰;G+R+P组中结合能在381.77 eV处为U4f7/2峰,结合能在392.58 eV处为U4f5/2峰;在G+R+S+P组中结合能在381.72 eV处为U4f7/2峰,392.53 eV处为U4f5/2峰。上述结果说明,铀在实验过程中并未发生氧化还原反应,铀的特征峰均为六价铀的特征峰。
-
1) G+R+S组中铀的去除率为99.5%,铀与硅形成了非晶型沉淀。
2) G+R+P组中铀的去除率为98.1%,铀与磷酸盐形成了((NH4)(UO2)PO4.3H2O)、((UO2)(PO4).3H2O)、((K1.7Ba0.2)(UO2)2(PO4)2.6H2O)和(NaPUO6.3H2O) 4种矿物。
3) G+R+S+P组中铀的去除率为99.72%,铀与两者形成了(Ca6Fe9(PO4)9O6(H2O)6.3H2O)、(Na(UO2)(PO4).3H2O)2和(NaPUO6.3H2O) 3种矿物。
4)向构建模拟修复反应的微模型中加入硅酸钠-磷酸二氢钾混合溶液,可以大幅度降低模拟地下水中铀的浓度。这一结果表明,硅酸钠-磷酸二氢钾混合溶液是酸法地浸采铀退役采区地下水的高效修复剂。
硅酸盐-磷酸盐对酸法地浸采铀退役采区模拟地下水的修复
Remediation effect of silicate-phosphate on simulated groundwater in the decommissioned mining area of acid in situ leach uranium mining
-
摘要: 为探索酸法地浸采铀退役采区地下水的原位修复技术,本研究先从西北某酸法地浸采铀退役采区采取地下岩心样,同时制备了酸法地浸采铀退役采区地下水模拟水样,构建模拟修复反应的微模型;再向微模型中分别加入硅酸钠溶液、磷酸二氢钾溶液、硅酸钠-磷酸二氢钾混合溶液以考察其对地下水的修复效果。结果表明:加入的硅酸钠-磷酸二氢钾与铀反应形成了胶磷钙铁矿(Ca6Fe9(PO4)9O6(H2O)6.3H2O)、偏钠铀矿物(Na(UO2)(PO4).3H2O)和(NaPUO6.3H2O)三种矿物;沉淀物中铁锰氧化物交换态铀的含量增加到了41%;铀的去除率达到了99.72%。Abstract: In order to explore the in-situ remediation technique for groundwater in the decommissioned mining area of acid in situ leaching uranium, silicate and phosphate, which are green remediation agents with low cost and no secondary pollution, was used to repair it. The sandstone uranium ore samples were firstly taken from a decommissioned area of acid in situ leach uranium mining in northwest China, the simulated water samples were prepared based on the groundwater in the decommissioned field, and the microcosm for simulating the remediation reaction in the groundwater was constructed. Then, sodium silicate solution, potassium dihydrogen phosphate solution and the mixed solution including both of them were added into the microcosms, respectively, and their remediation effects on groundwater were monitored. The results show that sodium silicate-potassium dihydrogen phosphate could react with uranium, and three minerals of mitridatite [Ca6Fe9(PO4)9O6(H2O)6.3H2O], metanatroautunite (Na(UO2)(PO4).3H2O) and (NaPUO6.3H2O) occurred. The content of Fe-Mn oxides-bound uranium increased to 41%, and the removal rate of uranium reached 99.72%.
-
Key words:
- acid in situ leaching uranium /
- groundwater /
- in situ remediation /
- silicate /
- phosphate
-
随着我国矿业得到更多重视和发展,采矿业发展必然带来大量的尾矿产生[1]。2019年我国铁尾矿产生量 5.36×108 t,综合利用量 1.16×108 t,铁尾矿综合利用率不足30%造成其堆积[2],而尾矿堆积引起的环境问题如累积潜在有毒元素等已成为全球性问题[3-4]。铁尾矿的铅、锌、镉、铜、镍、铬和锰等重金属,受到风化和沥滤等自然环境作用时,会产生具有毒性的酸性重金属废水污染地表水和地下水,而产生不可忽视地经济损失[5]。随着国家人民健康发展需求的日益增长,铁尾矿安全处置已引起广大关注。
目前,学者对铁尾矿资源化利用研究已有报道,如烧结固化技术[6-7]、制备改性材料[8]和磁化回收铁资源等。在烧结固化技术中,WANG等[5]在铁尾矿中添加高岭土和飞灰制备烧结砖,满足重金属浸出和抗压标准;在制备改性材料中,LI等[8]以铁尾矿和粉煤灰制备高比表面积(1.185 m2·g−1)和高孔隙率(62%)的多孔人工陶粒滤料。但以上两种途径对铁尾矿资源化回收方式没利用铁尾矿中赋存价值高的矿物,或是存在高能耗低价值等缺点[5, 8]。因而高效利用铁尾矿中赋存价值较高的铁元素显得格外重要。
中国因高品质铁矿石产量少,而成为高度依赖高品质铁矿石进口大国[1]。我国政协十三届全国委员会第四次会议也将铁矿列为战略性矿产,并大力加强铁矿石理论研究及其创新。可见,通过回收国内铁尾矿的铁以补充国内高品质铁矿石需求符合当代提倡的内循环模式。目前,学者通过磁化焙烧,对铁矿中的铁进行还原回收。按照还原剂不同,LI等[9]采用50% H2磁化焙烧铁尾矿获品位65.30%,回收率39.79%的铁精矿;YUAN等[10]采用20% CO磁化焙烧铁尾矿获品位68.31%,回收率96.34%的铁精矿;HUANG等[11]采用15%木屑磁化焙烧铁尾矿获品位62.84%,回收率94.58%的铁精矿。按照焙烧方式不同,其中YUAN等[10]采用悬浮磁化焙烧铁尾矿;HUANG等[11]采用固定床磁化焙烧铁尾矿。新颖的悬浮磁化焙烧法具有传热传质效率高等优点[12],但目前使用的还原剂多为单一还原剂或为理想性比例混气为主[9-10]。若采用还原性废气如高炉尾气和生物质造气等,按其主要成分为CO、H2、CO2和N2进行模拟还原混气研究[13-14],可寻找到一种低成本、节能、环保的工艺解决铁尾矿堆存资源浪费问题。
本研究以CO、H2、CO2和N2混气作为还原混气,研究不同温度、时间、混气H2和CO占比对铁尾矿磁化焙烧后铁品位和回收率的影响。利用X射线衍射(XRD)和扫描电子显微镜(SEM)研究焙烧前后铁尾矿基本特性和晶相结构,利用振动样品磁强计(VSM)测试样品磁性变化,利用光电子能谱仪(XPS)测试元素价态变化,利用N2吸脱附等温仪(BET)测试样品孔隙变化。本研究结果可为铁尾矿的资源化利用提供参考。
1. 材料与方法
1.1 供试材料
供试样品为广东省韶关市大宝山早期铁尾矿。铁尾矿元素含量分析如表1所示。可见,铁尾矿中铁为主要金属元素,品位为43.71%。二氧化硅及其氧化铝为主要杂质,并含有重金属。铁主要以赤铁矿、褐铁矿形式存在,占97.92%。本实验使用一氧化碳(CO, 99.95%);二氧化碳(CO2, 99.9%);氮气(N2, 99.9%)和由氢气发生装置(SPH – 300A,北京中惠普)制备的H2。
表 1 铁尾矿元素质量分数Table 1. Element content of iron tailings %Fe2O3 SiO2 Al2O3 SO3 K2O ZnO CuO 其他 46.78 27.90 19.59 4.11 0.54 0.20 0.17 0.71 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 实验方法
实验系统如图1所示。铁尾矿风干干燥,研磨至40%过200目,混合均匀储存密封袋中。待立式悬浮焙烧炉(SK–G03123K–D,天津中环)达到规定温度时,将10 g铁尾矿加入悬浮管,并组装悬浮焙烧炉。通N2(0.5 L·min−1)使物料保持悬浮,排出管中空气。通过4路混气系统(GSL–4Z,合肥科晶)将实验所配比的还原性混气(CO、H2、CO2、N2)通入悬浮管后反应一定时间。焙烧完成后,通入N2来及时排走过多还原混气,并对焙烧矿进行冷却至常温。随后将焙烧矿研磨过200目,依次使用磁场强度为120、80和60 mT的无极调节磁选管(XCGS–50,永盛选矿设备)湿法磁选获得铁精矿和磁选渣。最后在60 ℃烘箱处理12 h获得铁精矿和磁选渣固体,以待后续实验使用。
 图 1 悬浮磁化焙烧磁选装置图Figure 1. Suspension magnetization roasting magnetic separation device diagram
图 1 悬浮磁化焙烧磁选装置图Figure 1. Suspension magnetization roasting magnetic separation device diagram1.3 分析方法
铁矿石品位测定依据《铁矿石全铁含量的测定三氯化钛还原重铬酸钾滴定法(常规方法)》(GB/T 6730.65-2009)标准[15],回收率计算公式如式(1)所示。
R=cMc0M0 (1) 式中:R是精矿铁回收率; c0是磁化焙烧前原铁尾矿样品铁品位;M0是磁化焙烧前铁尾矿样品质量,g;c是铁精矿样品铁品位;M是铁精矿总质量,g。
采用XRD分析铁尾矿悬浮磁化焙烧和磁选工艺前后物相结构变化;采用XPS分析悬浮磁化焙烧和磁选前后元素含量和价态变化;采用VSM对铁尾矿悬浮磁化焙烧和磁选前后磁性变化进行分析;采用比表面积分析仪测定悬浮磁化焙烧前后铁尾矿比表面积和孔径分布;采用SEM – EDS观察悬浮磁化焙烧前后铁尾矿的表面结构、形态特征和元素含量。
2. 结果与讨论
2.1 悬浮磁化焙烧磁选
1)焙烧温度。在焙烧10 min、混气体积比H2∶CO∶CO2∶N2为10∶20∶15∶55条件下探究温度对铁精矿铁品位和回收率影响如图2所示。从图2可知,温度对铁精矿铁品位和回收率影响较大,随着温度上升,铁品位上升,而回收率稳定后急速下降。450 ℃时,铁尾矿磁化焙烧还原反应已能进行,获得铁品位和回收率分别为58.78%和95.73%的铁精矿,但此时较低的铁品位将导致利用率低下而增加炼铁厂成本[16];从450 ℃到600 ℃的过程中,铁品位提高至60.13%,而回收率仅下降5.83%,可知样品中更多的Fe以磁性Fe3O4的形式回收到铁精矿中;从600 ℃提升至700 ℃过程中,铁精矿回收率急速降至5.61%,即当前磁场无法回收铁精矿,而对于铁品位则提升至62.90%。600 ℃后,过高温度会让磁化焙烧副反应快速进行,导致铁尾矿原有的Fe2O3生成磁性Fe3O4后迅速生成弱磁性FeO等导致无法磁选收集[12,17];而铁品位的增加可能因为高温导致过还原的焙烧矿经过弱磁场磁选后,有大部分Fe以弱磁性铁相流失,表现为回收率低下,但弱磁场保留铁精矿虽质量少,但物质较纯,以强磁Fe3O4形式存在,所以铁品位有所上升。此时物质较纯的原因可能是:1)降低了精矿质量回收,从而降低了硅酸盐夹带的概率[18];2)降低了精矿质量回收,从而削弱了磁团聚现象[19]。综合考虑铁精矿产品质量和成本要求,选择600 ℃为最佳温度,此时铁品位和回收率分别为60.13%和89.90%。
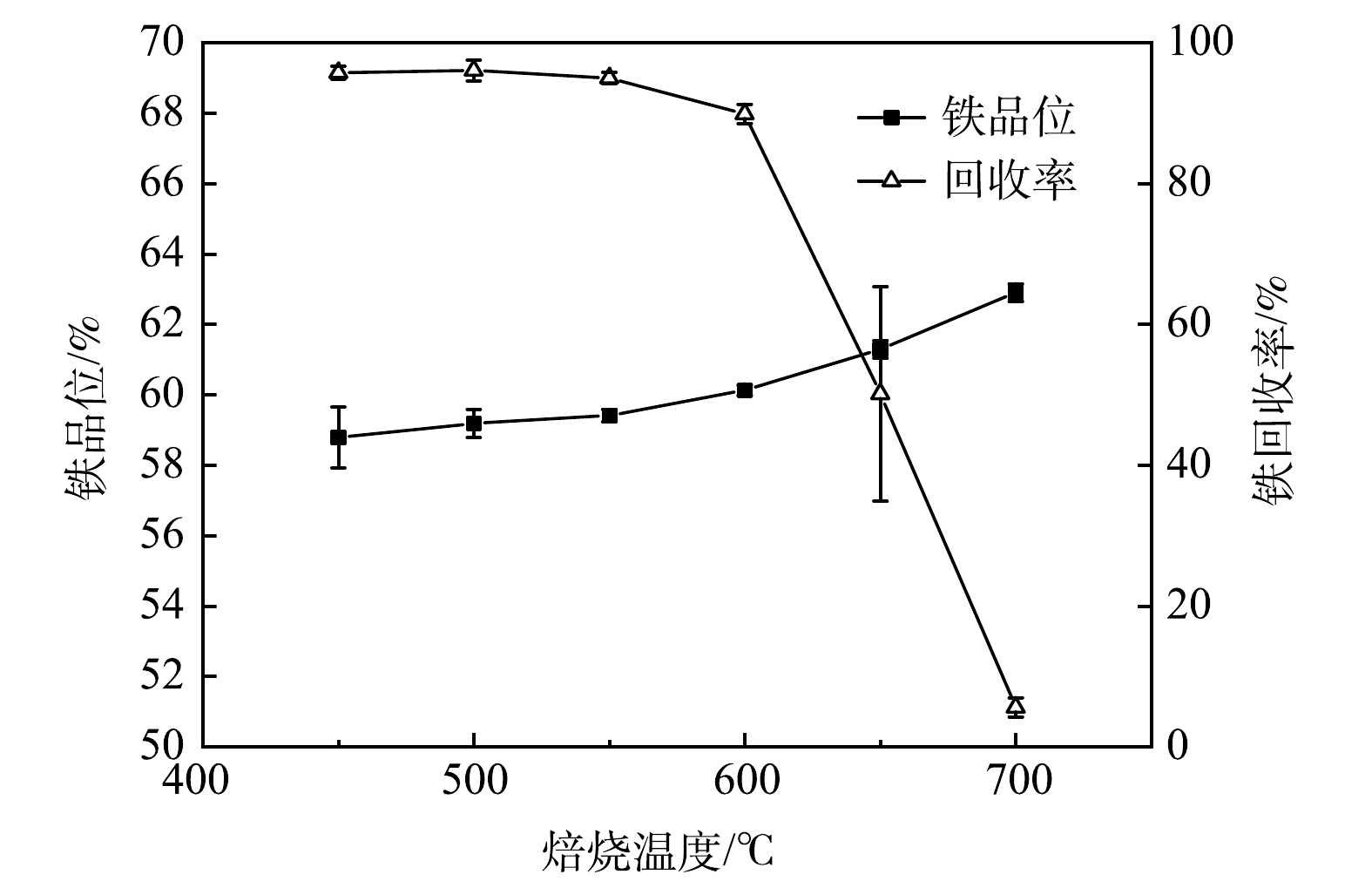 图 2 温度对铁精矿品位和回收率的影响Figure 2. Effect of temperature on grade and recovery of iron concentrate
图 2 温度对铁精矿品位和回收率的影响Figure 2. Effect of temperature on grade and recovery of iron concentrate2)反应时间。在焙烧温度600 ℃、混气体积比H2∶CO∶CO2∶N2为10∶20∶15∶55条件下探究磁化焙烧时间对铁精矿铁品位和回收率的影响。从图3可知,随着时间从5 min提升到30 min,铁精矿品位维持在60.00%~61.30%,而回收率总体随时间呈现先上升后下降趋势。当时间从5 min增加到10 min时,回收率从79.36%提升到92.57%,提高了13.21%,而铁精矿的品位几乎不变。而从10 min继续增加焙烧时间时,回收率总体呈现下降趋势至69.35%。可知,焙烧时间对铁精矿回收率影响较大,在5 min时,磁化焙烧进行未完全,仍然残留着Fe2O3未被还原,而10 min时,铁尾矿的Fe2O3几乎以强磁Fe3O4形式存在,能尽可能回收;而继续增加焙烧时间后,磁化焙烧的副反应导致生成的Fe3O4过还原,降低了磁性,导致回收率下降[12]。综合考虑铁精矿产品质量和成本要求,选择10 min为最佳时间,此时铁品位和回收率分别为61.21%和92.57%。
 图 3 时间对铁精矿品位和回收率的影响Figure 3. Effect of time on grade and recovery of iron concentrate
图 3 时间对铁精矿品位和回收率的影响Figure 3. Effect of time on grade and recovery of iron concentrate3)还原混气配比。在焙烧温度600 ℃、焙烧时间10 min条件下探究混气中2种还原性气体H2和CO占比对铁精矿铁品位和回收率的影响。从图4中可以看出,随着H2或CO占比的上升,铁精矿铁品位先下降后于61.00%~62.00%间波动;而回收率则先上升后稳定于95.00%以上。对比600 ℃下,H2∶CO为5∶0、10∶0、15∶0和20∶0时的回收率均比对应的0∶5、0∶10、0∶15和0∶20高,可见此温度下,单独还原气H2的还原性比单独还原气CO的强;同时,对比H2∶CO为10∶0、0∶10与5∶5的回收率可知,在还原性气体总占比一定时,H2与CO混合气的共同作用与H2单独作用对回收率影响不明显,而远远高于CO单独作用[20]。当总还原气体未过剩时,随着还原气体占比增大,铁品位从66.04%下降至60.00%~62.00%,而回收率从3.16%上升至95.00%。这是因为还原气体未过剩,随着其占比增加,更多Fe2O3被还原成Fe3O4,从而回收率上升,并随着铁精矿质量增加,物质间包夹作用和磁团聚增强[18, 19],铁精矿铁品位下降。当总还原性气体过剩且占比逐渐加大时,铁品位和回收率会趋于稳定区间,可见在最佳温度和时间下,过还原反应受还原性气体浓度小幅度过量影响较小。这是因为在适宜温度600 ℃下,过还原反应不发生或反应较慢[10]。从铁精矿产品最佳而言,最佳理论还原性混气比例应为H2∶CO∶CO2∶N2为20∶15∶15∶50,此时铁精矿的铁品位和回收率分别为62.06%和98.03%。
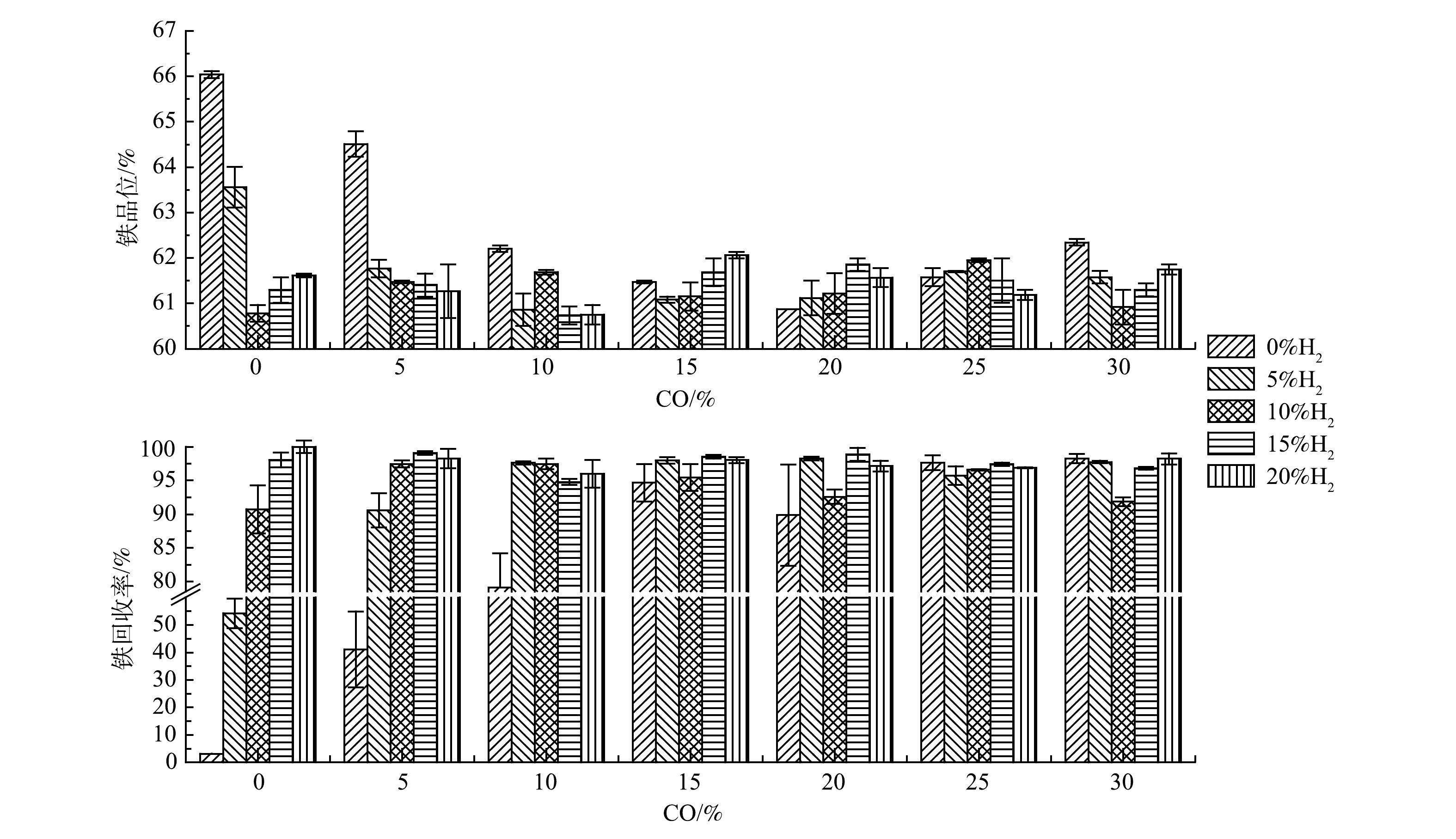 图 4 CO和H2占比对铁精矿品位和回收率的影响Figure 4. Effect of CO and H2 ratio on grade and recovery of iron concentrate
图 4 CO和H2占比对铁精矿品位和回收率的影响Figure 4. Effect of CO and H2 ratio on grade and recovery of iron concentrate2.2 产物性质及机理探究
1)物相分析。悬浮磁化焙烧磁选工艺出现的各种矿物形式、不同温度下焙烧矿和不同时间下焙烧矿的x射线衍射谱图见图5。可知,铁尾矿通过悬浮磁化焙烧磁选后,铁相从赤铁矿和针铁矿的形式转换成磁铁矿的形式富集于铁精矿中。原矿中含有石英相,是导致铁尾矿铁品位较低的主要原因[21]。悬浮磁化焙烧后,从焙烧矿衍射图谱与原矿对比可知,赤铁矿针铁矿的铁相消失,取代其的是磁铁矿出现,完成铁相转变[22]。并通过磁选过后,从铁精矿和磁选渣谱图对比可以看出,几乎所有石英相均于磁选渣中,而铁精矿以主要相磁铁矿的形式存在,完成铁的富集[12]。综上, 悬浮磁化焙烧磁选工艺确实能对铁尾矿进行铁富集回收,有效去除石英,提高精矿质量。
从图5(b)可知,温度变化对铁尾矿磁化焙烧铁相存在形式影响较大。随着温度从500 ℃上升到600 ℃,赤铁矿针铁矿衍射峰强度逐渐降低至消失,取而代之的是磁铁矿衍射峰增强,可知磁化反应顺利进行;提高温度至600 ℃时,焙烧矿不再出现赤铁矿衍射峰,铁相几乎全以磁铁矿的形式存在;随温度的继续上升,焙烧矿中磁铁矿衍射峰强度减弱,而出现了浮氏体这一弱磁性铁相。这就表明,600 ℃后提高温度会让过还原反应加快进行,导致磁铁矿含量下降,出现弱磁性浮氏体,导致铁流失在磁选渣中[23]。这一现象也能说明最佳温度为600 ℃,如继续提高温度导致铁回收率急速下降。
从图5(c)可知,时间变化对铁尾矿磁化焙烧铁相的存在形式影响较小,这也与随着时间变化,铁品位变化不大的现象一致。在焙烧时间为10 min时,焙烧矿中赤铁矿针铁矿衍射峰消失,铁相主要以磁铁矿的形式存在,但出现了微弱的浮氏体衍射峰,表明焙烧已经进行完全。结合继续提高焙烧时间时焙烧矿的磁铁矿衍射峰无明显变化,表明10 min焙烧时间已经足够。此时若继续提高焙烧时间不仅会提高工艺成本,而且无利于提高铁精矿质量[24]。
2) 磁性分析VSM。样品磁滞回线如图6所示,磁性参数如表2所示。由图6和表2可知,经过悬浮磁化焙烧的焙烧矿相比原矿,磁性有较大提升。饱和磁化强度和剩磁强度分别由0.77和0.05 Am2·kg−1提升至52.31和10.82 Am2·kg−1,提升了51.54和10.77 Am2·kg−1。这就说明,悬浮磁化焙烧能较高提升矿物磁性能[10, 12]。通过磁选工艺,能将磁性差异较大的磁铁矿和石英等进行分离,从而得到饱和磁化强度更接近纯磁铁矿的铁精矿,其饱和磁化强度和剩磁强度达到59.43和13.72Am2·kg−1。这也能侧面印证出悬浮磁化焙烧磁选工艺能提高铁精矿铁品位,同时保证较高的铁回收率,从而满足低成本炼钢炼铁要求。
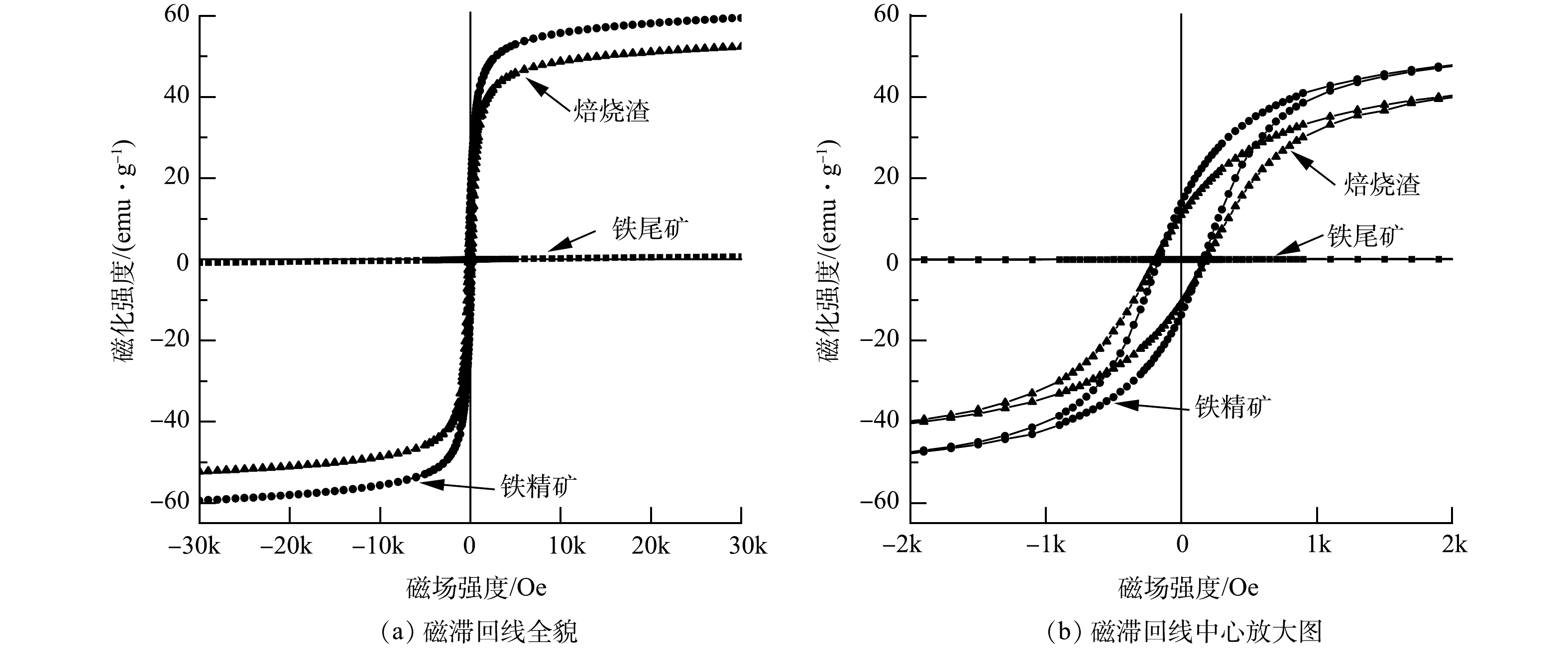 图 6 铁尾矿、焙烧矿和铁精矿的磁滞回线Figure 6. Hysteresis loop of iron tailing, roasted ore and iron concentrate表 2 样品及纯物质磁性参数Table 2. Magnetic parameters of samples and pure substances Am2·kg-1
图 6 铁尾矿、焙烧矿和铁精矿的磁滞回线Figure 6. Hysteresis loop of iron tailing, roasted ore and iron concentrate表 2 样品及纯物质磁性参数Table 2. Magnetic parameters of samples and pure substances Am2·kg-1样品 饱和磁化强度 剩磁 原矿 0.77 0.05 焙烧矿 52.31 10.82 铁精矿 59.43 13.72 纯赤铁矿 0.40 - 纯磁铁矿 92.00 - | Show TableDownLoad:
CSV
3) XPS分析。样品XPS分析总谱如图7所示,谱图均采用C1s的284.80 eV进行荷电校正。由总谱可知,元素Fe、O、Si、Al和C都有对应峰响应,且样品不同,Fe2p、O1s、Si2p和Al2p都有不同程度变化。Fe2p峰响应强度在焙烧矿中比原铁尾矿高,且结合O1s峰响应强度随着悬浮磁化焙烧磁选过程逐渐降低,可揭示过程中还原气CO和H2夺走铁尾矿部分O而生成Fe3O4[21]。Si2p和Al2p的峰响应强度在原铁尾矿和焙烧矿中变化不大,而铁精矿中几乎不存在Si2p和Al2p的峰响应,同时磁选渣中有较强的Si2p和Al2p的峰响应。可知,磁选这一过程能很好分离出Si和Al物质,以达到提高铁精矿品质[12]。这一结果与XRD分析结果保持一致。
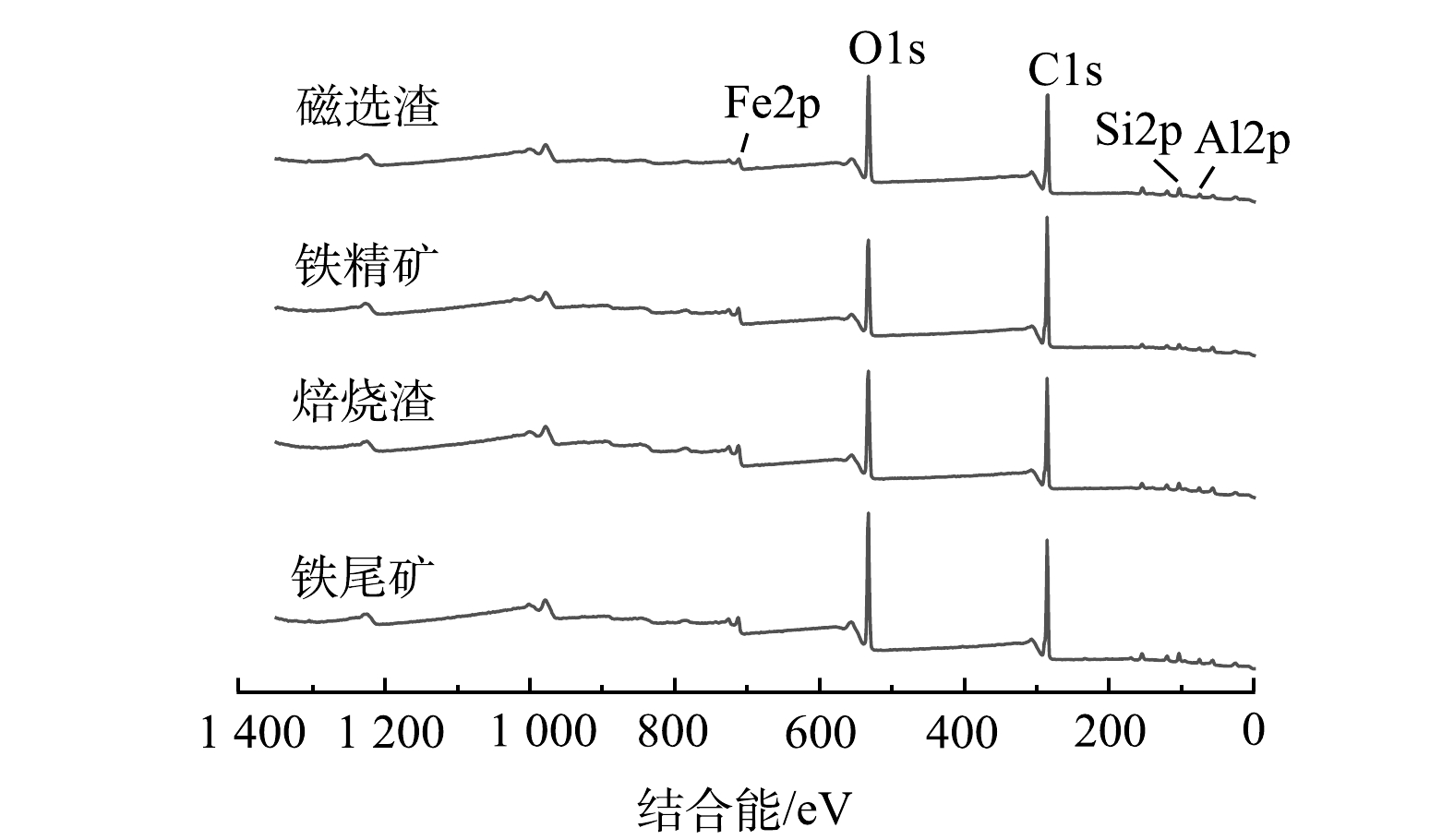 图 7 铁尾矿、焙烧矿、铁精矿和磁选渣的XPS分析谱图Figure 7. XPS analysis spectra of iron tailing, roasted ore, iron concentrate and magnetic separation slag
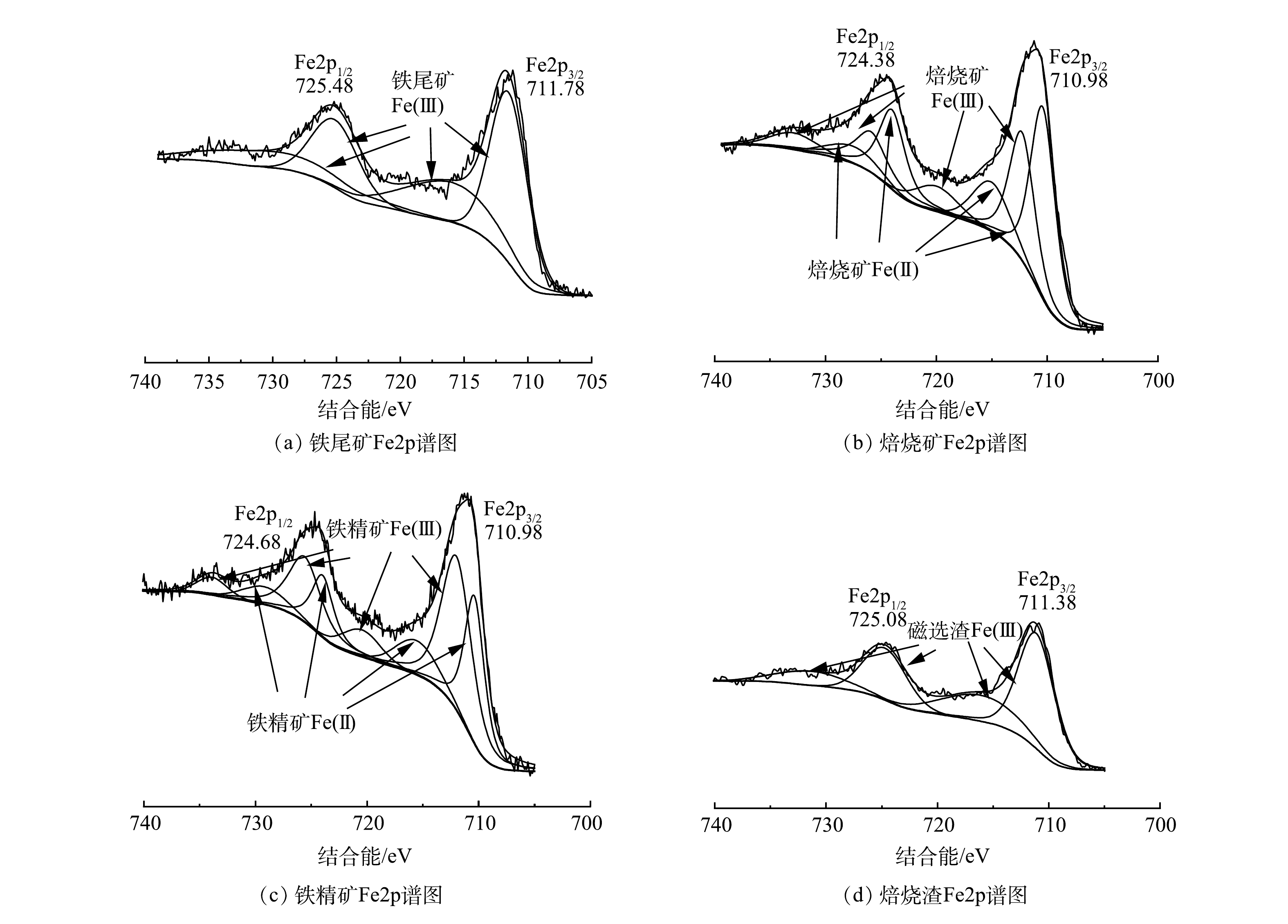
图 7 铁尾矿、焙烧矿、铁精矿和磁选渣的XPS分析谱图Figure 7. XPS analysis spectra of iron tailing, roasted ore, iron concentrate and magnetic separation slag样品XPS分析的Fe2p窄谱如图8所示,谱图均采用C1s的284.80 eV进行荷电校正。从图8(a)可知,原铁尾矿含有Fe2p3/2和Fe2p1/2结合能,分别在711.78和725.48 eV,两者间相差13.7(~13.6)eV。这些结合能位置信息暗示着原铁尾矿的Fe以Fe2O3和FeOOH的Fe+3存在[25],这也与XRD分析结果高度一致。对于图8(b)和8图(c)所示焙烧矿和铁精矿而言,Fe2p3/2和Fe2p1/2分别为710.98、724.38 eV和710.98、724.68 eV,其结合能对比原铁尾矿发生明显向右位移,即结合能下降,谱图出现了Fe2+。这是因为,在悬浮磁化焙烧过程中,一部分的Fe3+被还原成Fe2+,形成Fe3O4,所以导致Fe2p3/2和Fe2p1/2结合能右移[26-27]。8(d)图磁选渣中Fe2p响应相比较低,且不存在Fe2+。综上,XPS分析结果进一步证实了悬浮磁化焙烧Fe相从Fe2O3、FeOOH到Fe3O4转变的机理,并可明确通过悬浮磁化焙烧磁选工艺,获得高品质铁精矿是可行的。
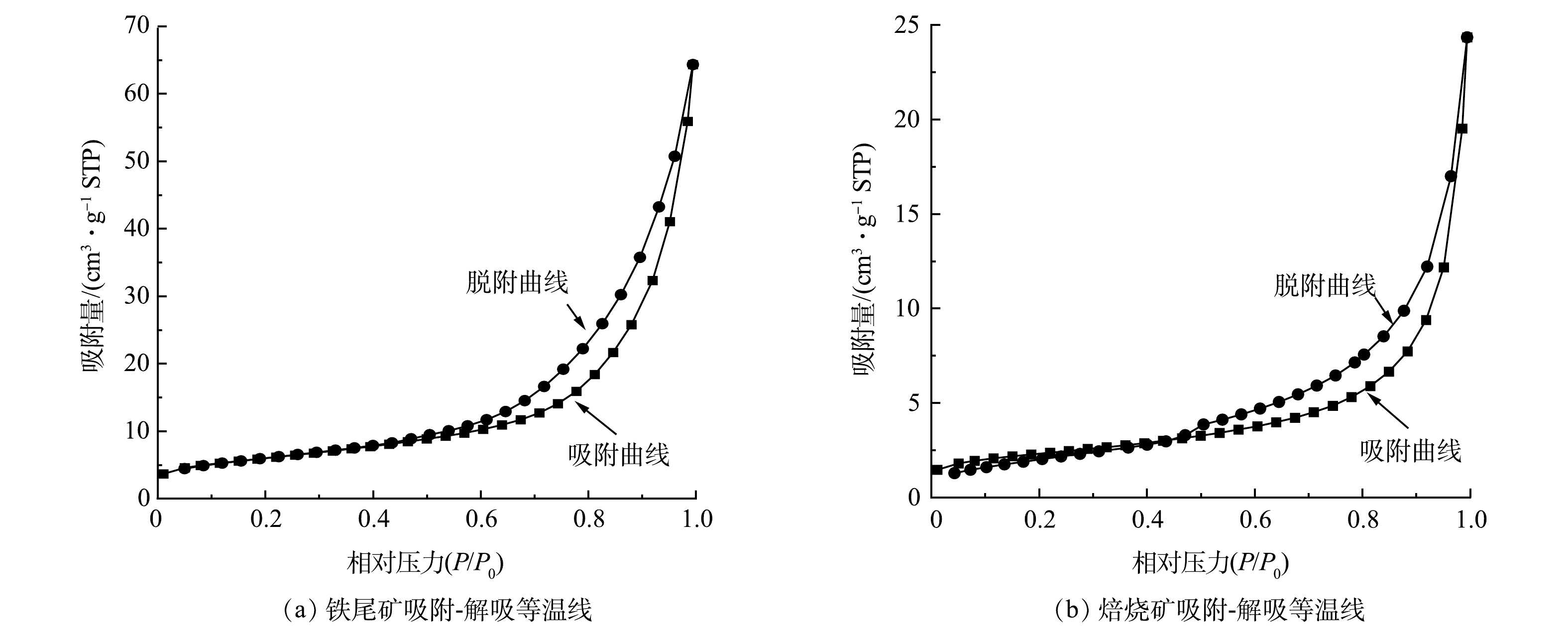
4)微观结构分析。原铁尾矿和焙烧矿的N2吸附-脱附等温曲线如图9所示。从图9可知,铁尾矿和焙烧矿的等温曲线类型是Ⅱ与Ⅳ型结合。在相对压力(P/P0)较低的情况下,原铁尾矿和焙烧矿的吸附容量均表现较低,微孔数量较少,这是Ⅱ型的表现;在相对压力(P/P0)较高的情况下,原铁尾矿和焙烧矿等温线出现H3型滞后环,表明产物存在狭缝状孔,这是Ⅳ型的表现[12]。由表3可知,铁尾矿经过悬浮磁化焙烧后BET表面积、朗缪尔表面积、孔隙体积和孔径分别提升了13.167 6 m2·g−1、274.842 2 m2·g−1、0.044 121 cm3·g−1和2.599 8 nm。较大孔隙能大大降低后续焙烧矿的研磨成本[23]。原铁尾矿和焙烧矿的BET表面积、朗缪尔表面积、孔隙体积和孔径如表4所示。综上,铁尾矿悬浮磁化焙烧过程,能较大程度提高铁尾矿的比表面积和孔隙等微观性能,可为后续焙烧矿研磨磁选提供便利。
表 4 原矿焙烧矿EDS分析Fe、O元素占比Table 4. EDS analysis of raw ore roasted ore Fe and O elements account wt%样品 Fe O 铁尾矿 61.98 33.71 焙烧矿 66.01 24.02 | Show TableDownLoad:
CSV
表 3 BET分析的相关参数Table 3. Relevant parameters of bet analysis供试样品 BET表面积/(m2·g−1) 朗缪尔表面积/(m2·g−1) 孔隙体积/(cm3·g−1) 孔径/nm 铁尾矿 8.122 6 81.491 1 0.018681 9.199 4 焙烧矿 21.290 2 356.333 3 0.0628 02 11.799 2 | Show TableDownLoad:
CSV
原铁尾矿和焙烧矿的SEM-EDS分析如图10所示。由图10(a)和(b)可知,原铁尾矿以有棱角的块状结构存在,质地紧密,表面吸附着片状块状且尺寸较小的物质;由图10(c)和(d)可知,焙烧矿以块状棒状团聚的形式存在,质地疏松。综上所述,经过悬浮磁化焙烧后,铁尾矿块状结构发生气固反应破裂,以尺寸更小的块状和棒状团聚形式存在,存在更多的孔隙[23]。这也和BET分析结果一致。从图10(e)原铁尾矿和图10(f)焙烧矿EDS对比可知,经过悬浮磁化焙烧后,Fe的相对含量上升,O相对含量下降。这个揭示了焙烧过程中Fe2O3经过CO和H2还原生成Fe3O4的反应机理,这也与XRD、XPS分析结果相一致。
3. 结论
1) 焙烧温度600 ℃、焙烧时间10 min、H2∶CO∶CO2∶N2体积比为20∶15∶15∶50时,铁精矿产品铁品位和回收率分别为62.06%和98.03%达到最优。
2) 悬浮磁化焙烧能有效将铁尾矿铁相转化成磁铁矿相,使饱和磁化强度由0.77 Am2·kg−1提升到59.43 Am2·kg−1,且磁选能有效将磁铁矿和石英分离。过高温度和过长时间会产生弱磁性浮氏体而阻碍铁的回收。
3) 经悬浮磁化焙烧后,焙烧矿颗粒出现裂缝,比表面积和孔隙率均有较大提升。
-
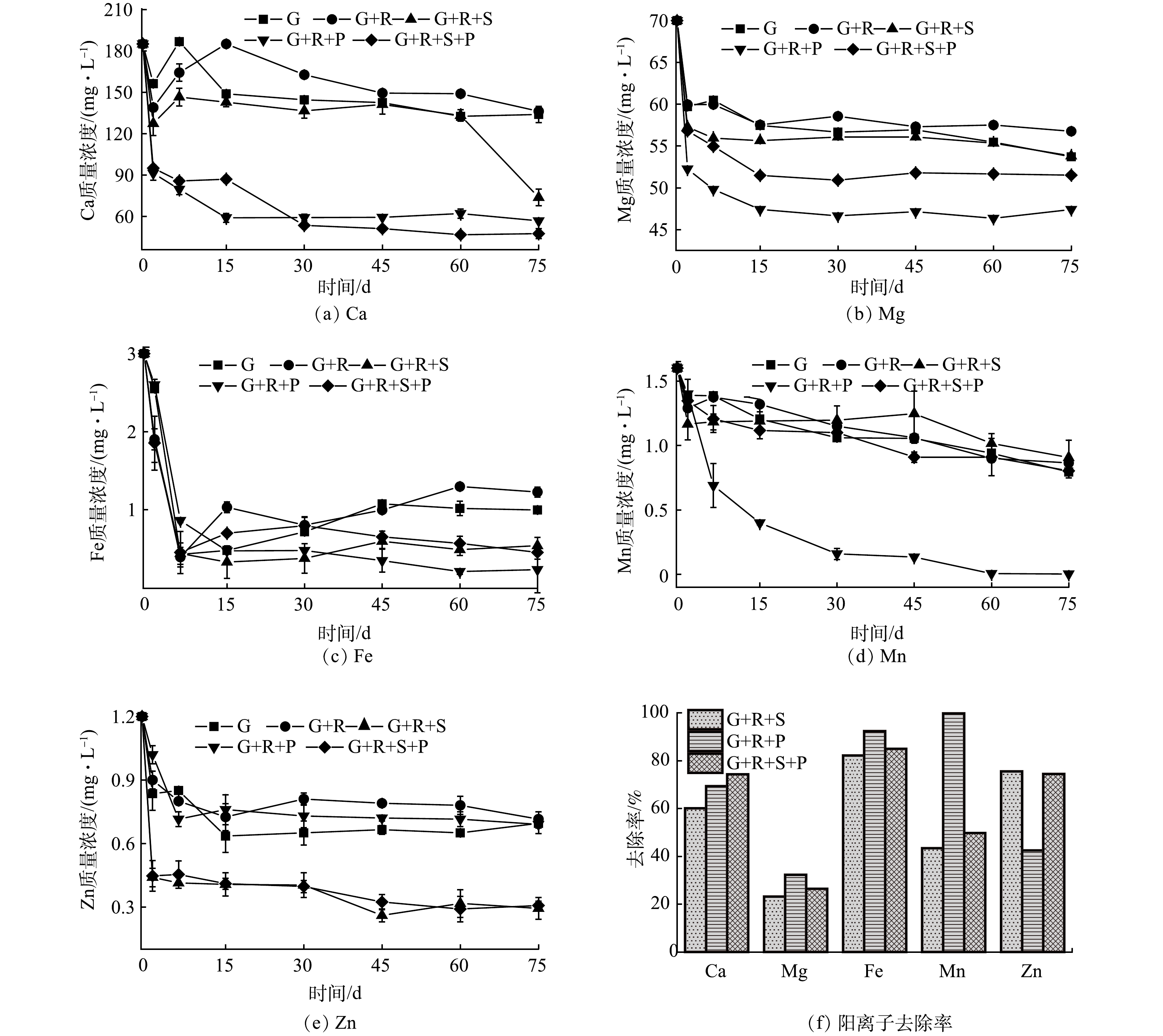
图 3 金属阳离子质量浓度变化及其去除率
Figure 3. Variations of metal cations mass concentration and their removal rate
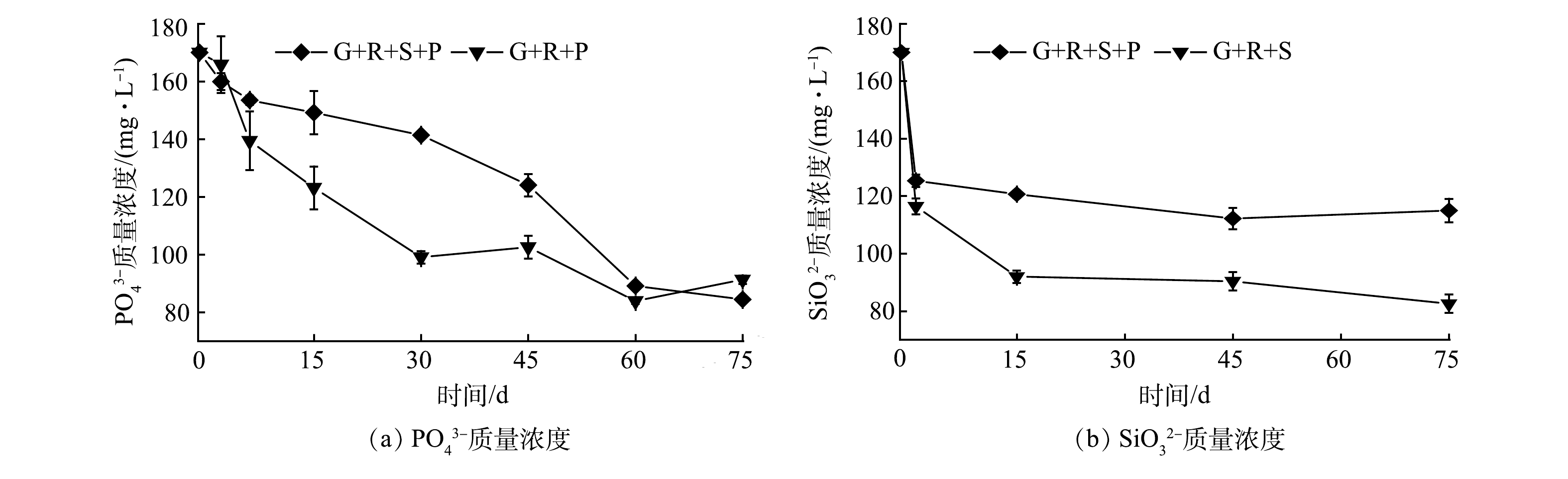
图 4 磷酸盐、硅酸盐质量浓度变化
Figure 4. Variations of mass concentration of phosphate and silicate
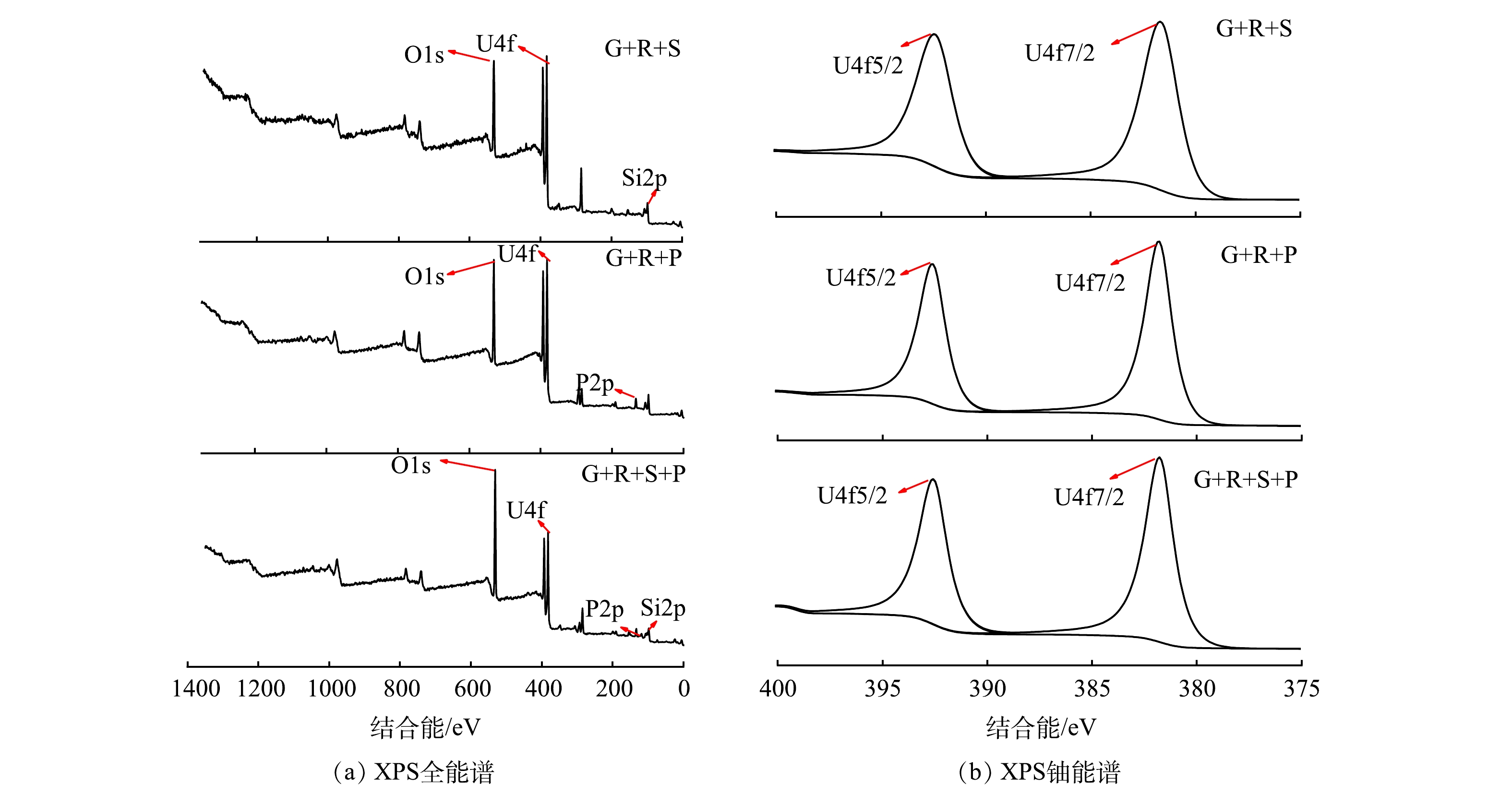
图 8 沉淀产物XPS全扫描图谱、窄扫描图谱
Figure 8. XPS full scanning spectra and narrow scanning spectra of precipitated products
表 1 初始岩心样的主要化学成分
Table 1. Main chemical components of initial core samples
组名 名称 含量/% 组名 名称 含量/% 1 SiO2 72.86 13 U3O8 0.0150 2 Al2O3 19.90 14 V2O5 0.0136 3 K2O 3.54 15 Cr2O3 0.0130 4 Fe2O3 0.728 16 MnO 0.0099 5 CaO 0.690 17 ZrO2 0.0079 6 SO3 0.673 18 Rb2O 0.0055 7 MgO 0.531 19 La2O3 0.0036 8 TiO2 0.446 20 CuO 0.0034 9 P2O5 0.364 21 SrO 0.0031 10 Na2O 0.134 22 NiO 0.0026 11 ZnO 0.0267 23 Co3O4 0.0022 12 Cl 0.0169 24 WO3 0.0021
下载: 导出CSV
-
[1] MA Q, FENG Z G, LIU P, et al. Uranium speciation and in situ leaching of a sandstone-type deposit from China[J]. Journal of Radioanalytical and Nuclear Chemistry, 2017, 311(3): 2129-2134. doi: 10.1007/s10967-016-5154-1 [2] PEREIRA R, BARBOSA S, CARVALHO F P. Uranium mining in Portugal: A review of the environmental legacies of the largest mines and environmental and human health impacts[J]. Environmental Geochemistry and Health, 2014, 36(2): 285-301. doi: 10.1007/s10653-013-9563-6 [3] MONTI M M, DAVID F, SHIN M, et al. Community drinking water data on the national environmental public health tracking network: a surveillance summary of data from 2000 to 2010[J]. Environmental Monitoring and Assessment, 2019, 191(9): 557. doi: 10.1007/s10661-019-7710-y [4] YIN M, SUN J, CHEN Y, et al. Mechanism of uranium release from uranium mill tailings under long-term exposure to simulated acid rain: geochemical evidence and environmental implication[J]. Environmental Pollution, 2019, 244: 174-181. doi: 10.1016/j.envpol.2018.10.018 [5] SAUNDERS J A, PIVETZ B E, VOORHIES N, et al. Potential aquifer vulnerability in regions down-gradient from uranium in situ recovery (ISR) sites[J]. Journal of Environmental Management, 2016, 183: 67-83. [6] BJORKLUND G, SEMENOVA Y, PIVINA L, et al. Uranium in drinking water: a public health threat[J]. Archives of Toxicology, 2020, 94(5): 1551-1560. doi: 10.1007/s00204-020-02676-8 [7] NOLAN J, WEBER K A. Natural uranium contamination in major US aquifers linked to nitrate[J]. Environmental Science & Technology Letters, 2015, 2(8): 215-220. [8] NEWSOME L, MORRIS K, LLOYD J R. The biogeochemistry and bioremediation of uranium and other priority radionuclides[J]. Chemical Geology, 2014, 363: 164-184. doi: 10.1016/j.chemgeo.2013.10.034 [9] JROUNDI F, DESCOSTES M, POVEDANO P C, et al. Profiling native aquifer bacteria in a uranium roll-front deposit and their role in biogeochemical cycle dynamics: insights regarding in situ recovery mining[J]. Science of the Total Environment, 2020,https://doi.org/10.1016/j.scitotenv.2020.137758. [10] CHANG Y J, PEACOCK A D, LONG P E, et al. Diversity and characterization of sulfate-reducing bacteria in groundwater at a uranium mill tailings site[J]. Applied and environmental microbiology, 2001, 67(7): 3149-3160. doi: 10.1128/AEM.67.7.3149-3160.2001 [11] REIMUS P W, DANGELMAYR M A, CLAY J T, et al. Uranium natural attenuation downgradient of an in situ recovery mine inferred from a cross-hole field test[J]. Environmental Science & Technology, 2019, 53(13): 7483-7493. [12] BORCH T, ROCHE N, JOHNSON T E. Determination of contaminant levels and remediation efficacy in groundwater at a former in situ recovery uranium mine[J]. Journal of Environmental Monitoring, 2012, 14(7): 1814-1823. doi: 10.1039/c2em30077j [13] 孙占学, 马文洁, 刘亚洁, 等. 地浸采铀矿山地下水环境修复研究进展[J]. 地学前缘, 2021, 28(5): 1-10. [14] 陈约余, 张辉, 胡南, 等. 地浸采铀地下水修复技术研究进展[J]. 矿业研究与开发, 2021, 41(2): 149-154. [15] 牛洁, 张学礼. 捷克Straz地浸铀矿山地下水恢复治理介绍[J]. 铀矿冶, 2016, 35(2): 110-117. [16] FINNERAN K T, ANDERSON R T, NEVIN K P, et al. Potential for bioremediation of uranium-contaminated aquifers with microbial U(VI) reduction[J]. Soil and Sediment Contamination:An International Journal, 2002, 11(3): 339-357. doi: 10.1080/20025891106781 [17] ANDERSON R T, VRIONIS H A, ORTIZ B I, et al. Stimulating the in situ activity of geobacter species to remove uranium from the groundwater of a uranium-contaminated aquifer[J]. Applied and environmental microbiology, 2003, 69(10): 5884-5891. doi: 10.1128/AEM.69.10.5884-5891.2003 [18] 张伟, 董发勤, 杨杰, 等. 三种非活性微生物对铀的吸附行为及其受γ辐照的动力学影响[J]. 核化学与放射化学, 2018, 40(4): 258-266. doi: 10.7538/hhx.2018.YX.2017038 [19] SIVASWAMY V, BOYANOV M I, PEYTON B M, et al. Multiple mechanisms of uranium immobilization by cellulomonas sp strain ES6[J]. Biotechnology and Bioengineering, 2011, 108(2): 264-276. doi: 10.1002/bit.22956 [20] 张健, 宋晗, 邓洪, 等. 铀与微生物相互作用研究进展[J]. 矿物岩石地球化学通报, 2018, 37(1): 55-62+158-159. [21] 张露, 刘峙嵘. 微生物法处理低浓度含铀废水研究[J]. 环境工程, 2017, 35(12): 36-40. [22] ZOU W, ZHAO L, HAN R P. Removal of uranium (VI) by fixed bed ion-exchange column using natural zeolite coated with manganese oxide[J]. Chinese Journal of Chemical Engineering, 2009, 17(4): 585-593. doi: 10.1016/S1004-9541(08)60248-7 [23] 许智慧, 李宏星, 胥国龙. 用纳滤膜从酸性溶液中富集铀的可行性研究[J]. 铀矿冶, 2018, 37(1): 37-41. [24] 张洪灿. 化学中和法和生物法联合治理酸法地浸采铀矿山污染地下水实验研究[D]. 衡阳: 南华大学, 2013. [25] DOUGLAS G, SHACKLETON M, WOODS P. Hydrotalcite formation facilitates effective contaminant and radionuclide removal from acidic uranium mine barren lixiviant[J]. Applied Geochemistry, 2014, 42: 27-37. doi: 10.1016/j.apgeochem.2013.12.018 [26] 魏榕, 黄健. 酸性矿山废水的污染与处理研究[J]. 能源与环境, 2006, 2: 31-33. [27] MERKEL B, SCHIPEK M. Neutralisation and trace element removal from beverley in-situ recovery uranium mine barren lixiviant via hydrotalcite formation[J]. The New Uranium Mining Boom, 2012, 12: 101-109. [28] SU M, TSANG D C, REN X, et al. Removal of U(VI) from nuclear mining effluent by porous hydroxyapatite: evaluation on characteristics, mechanisms and performance[J]. Environmental Pollution, 2019,https://doi.org/10.1016/j.envpol.2019.07.059. [29] LAMMERS L N, RASMUSSEN H, ADILMAN D, et al. Groundwater uranium stabilization by a metastable hydroxyapatite[J]. Applied Geochemistry, 2017, 84: 105-113. doi: 10.1016/j.apgeochem.2017.06.001 [30] RUIZ O, THOMSON B, CERRATO J M, et al. Groundwater restoration following in-situ recovery (ISR) mining of uranium[J]. Applied Geochemistry, 2019,https://doi.org/10.1016/j.apgeochem.2019.104418. [31] SODERHOLM L, SKANTHAKUMAR S, GORMAN L D, et al. Characterizing solution and solid-phase amorphous uranyl silicates[J]. Geochimica Et Cosmochimica Acta, 2008, 72(1): 140-150. doi: 10.1016/j.gca.2007.10.002 [32] WEN H, PAN Z, GIAMMAR D, et al. Enhanced uranium immobilization by phosphate amendment under variable geochemical and flow conditions: insights from reactive transport modeling[J]. Environmental Science & Technology, 2018, 52(10): 5841-5850. [33] MEHTA V S, MAILLOT F, WANG Z, et al. Effect of co-solutes on the products and solubility of uranium(VI) precipitated with phosphate[J]. Chemical Geology, 2014, 364: 66-75. doi: 10.1016/j.chemgeo.2013.12.002 [34] KANEMATSU M, PERDRIAL N, UM W Y, et al. Influence of phosphate and silica on U(VI) precipitation from acidic and neutralized wastewaters[J]. Environmental Science & Technology, 2014, 48(11): 6097-6106. [35] SEMIAO A J, ROSSITER H M, SCHAEFER A I. Impact of organic matter and speciation on the behaviour of uranium in submerged ultrafiltration[J]. Journal of Membrane Science, 2010, 348(1-2): 174-180. doi: 10.1016/j.memsci.2009.10.056 [36] ANTONINA P K, LYUDMILA Y Y, IRINA D A, et al. Ultrafiltration removal of U(VI) from contaminated water[J]. Desalination, 2004, 162(1): 229-236. [37] MEHTA V S, MAILLOT F, WANG Z, et al. Effect of reaction pathway on the extent and mechanism of uranium(VI) immobilization with calcium and phosphate[J]. Environmental Science & Technology, 2016, 50(6): 3128-3136. [38] ROUT S, KUMAR A, RAVI P M, et al. Understanding the solid phase chemical fractionation of uranium in soil and effect of ageing[J]. Journal of Hazardous Materials, 2016, 317: 457-465. doi: 10.1016/j.jhazmat.2016.05.082 [39] 雷鸣, 廖柏寒, 秦普丰. 土壤重金属化学形态的生物可利用性评价[J]. 生态环境, 2007, 5: 1551-1556. [40] LIU K, LUO L, LUO L, et al. Initial oxidation behaviors of nitride surfaces of uranium by XPS analysis[J]. Applied Surface Science, 2013, 280: 268-272. doi: 10.1016/j.apsusc.2013.04.147 [41] HAYCOCK D, URCH D S, GARNER C D. Electronic structure of the octachlorodimolybdenum(II) anion using X-ray emission and X-ray photoelectron spectroscopies[J]. Journal of Electron Spectroscopy & Related Phenomena, 1979, 17(5): 345-352. [42] EJIMA T, SATO S, SUZUKI S, et al. Line shapes of the XPS U 4f spectra in some uranium compounds[J]. Physical review. B, Condensed matter, 1996, 53(4): 1806-1813. doi: 10.1103/PhysRevB.53.1806 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5254
- HTML全文浏览数: 5254
- PDF下载数: 79
- 施引文献: 0
