



-
氮氧化物(NOx)和挥发性有机化合物(volatile organic compounds,VOCs)等污染物是造成灰霾和近地面臭氧等大气污染问题的重要前体物。活性较强的VOCs在一定条件下与NOx发生光化学反应,造成O3体积增加,形成光化学烟雾,从而在更大范围内产生污染,对空气质量和人群健康造成威胁[1-2]。因此,开发高效、环保的催化剂以协同控制NO和甲苯的技术受到广泛重视。
目前,NOx控制技术主要有选择性催化还原、NOx储存还原和选择性催化氧化等。其中,NO催化氧化是反应过程中的关键步骤[3],同时催化氧化也是一种高效的甲苯控制技术,可将甲苯氧化成H2O和CO2 [4]。因此,可采用催化氧化法协同控制氮氧化物(NOx)和甲苯。然而,单独催化氧化NOx和甲苯的工艺会导致占地面积大、成本高[5]。因此,开发一种协同催化氧化NO和甲苯的催化剂具有重要意义。
贵金属催化剂是催化氧化技术中常用的催化剂,但其高成本和反应易团聚的缺点严重制约了其大规模应用[6]。钙钛矿催化剂由于成本低、环境友好及优异的催化活性等优点被认为是贵金属催化剂的替代品。CHEN等[7] 发现LaMe(Me=Mn, Fe, Co)钙钛矿催化剂具有良好的催化活性。其中,LaFeO3具有较好的热稳定性和较低的相形成温度,可应用较高温度条件下,但仍存在比表面积低、较少孔结构及B位Fe离子对甲苯和NO的氧化还原性能不够理想等缺点[8]。此外,工业尾气常含有的SO2会导致催化剂产生SO2中毒失活,从而严重抑制了其催化活性[9]。一般来说,钙钛矿催化剂的催化活性主要取决于氧迁移率、过渡金属的氧化还原特性及钙钛矿结构[10]。A或B位被不同价态的阳离子取代时,B位阳离子的氧数量会发生变化,钙钛矿结构中出现缺陷,诱导形成的氧空位可促进表面活性氧离子的迁移,从而提高其氧化还原能力[11]。一些过渡金属离子(如 Ce、Cu、Mn和 Co)具有 经济性和价态可变等优点[12],已被引入钙钛矿催化体系中以提高催化剂活性。如SHI等[10]将Ce部分取代LaMnO3钙钛矿复合氧化物的A位离子以提高催化性能和抗硫性能。WU等[12]发现LaM0.5Mn0.5O3 (M = Cu、Co、Fe、Ni、Cr)钙钛矿催化剂表现出优异的抗硫性能,主要原因是金属元素的引入提高了氧化还原能力和氧缺陷密度。ZHENG等 [13]发现Mn改性的LaFeO3催化剂具有较大表面积,因而表现出良好的活性。此外,ZHAO等[8]发现多孔 LaFeO3 钙钛矿催化剂具有更高比表面积,能提供更多表面活性位点。以上研究表明可通过过渡金属氧化物A位掺杂改性钙钛矿催化剂以提高其活性和抗硫性能。然而,不同过渡金属(X=Cu, Ce, Mn, Co)元素A位掺杂改性的多孔LaFeO3催化剂协同催化氧化甲苯和NO的研究较少,且SO2对其的影响机制尚不明确。
本研究通过溶胶-凝胶法制备一系列不同过渡金属X (X=Cu, Ce, Mn, Co)元素 A位取代的La0.65X0.35FeO3 (X=Cu, Ce, Mn, Co)钙钛矿催化剂,基于BET、SEM、XRD、XPS等表征结果探讨不同过渡金属元素A掺杂对La0.65X0.35FeO3协同催化氧化甲苯和NO的影响,并讨论SO2 对协同催化氧化反应的影响机理,以期为此类催化剂工业应用中的活性保持与再生提供参考。
-
不同过渡金属X (X=Cu, Ce, Mn, Co)元素A位掺杂改性的La0.65X0.35FeO3钙钛矿催化剂采用溶胶凝胶法制备。KIT-6硬模板基于文献[14]合成。首先,将一定量的硝酸镧(La(NO3)3·6H2O,分析纯,科密欧)、硝酸X(Co(NO3)3·6H2O、Ce(NO3)3· 6H2O、Cu(NO3)2· 3H2O、Mn(NO3)2·4H2O,均是分析纯,科密欧)、硝酸铁(Fe(NO3)3·9H2O,分析纯,国药集团)和柠檬酸 (分析纯,湖南汇虹) 按一定的物质的量之比(La:X:柠檬酸=4:4:8)加至20mL乙醇(分析纯,科密欧)水溶液中(V乙醇:V水=3),在室温下搅拌 6 h;再将1.0 g KIT-6加入上述混合溶液中,在 60 ℃搅拌2 h 后,于室温下超声搅拌至凝胶状态;然后将凝胶在 60 ℃ 下干燥 12 h后放入马弗炉中先在350 ℃煅烧2 h,后升温至750 ℃煅烧3 h,升温速率为 10 ℃·min−1;将所得样品用NaOH溶液(分析纯,天津大茂,浓度为2.0 mol·L−1)在75 ℃下处理1 h,并重复操作一次以去除SiO2模板,再用去离子水洗涤数次;最后将样品在 105 ℃ 下干燥过夜,研磨过筛(100~120目)得到催化剂。下标0.65表示La/(X+La)(物质的量之比)之值,0.35表示X/(X + La)(物质的量之比)之值。
-
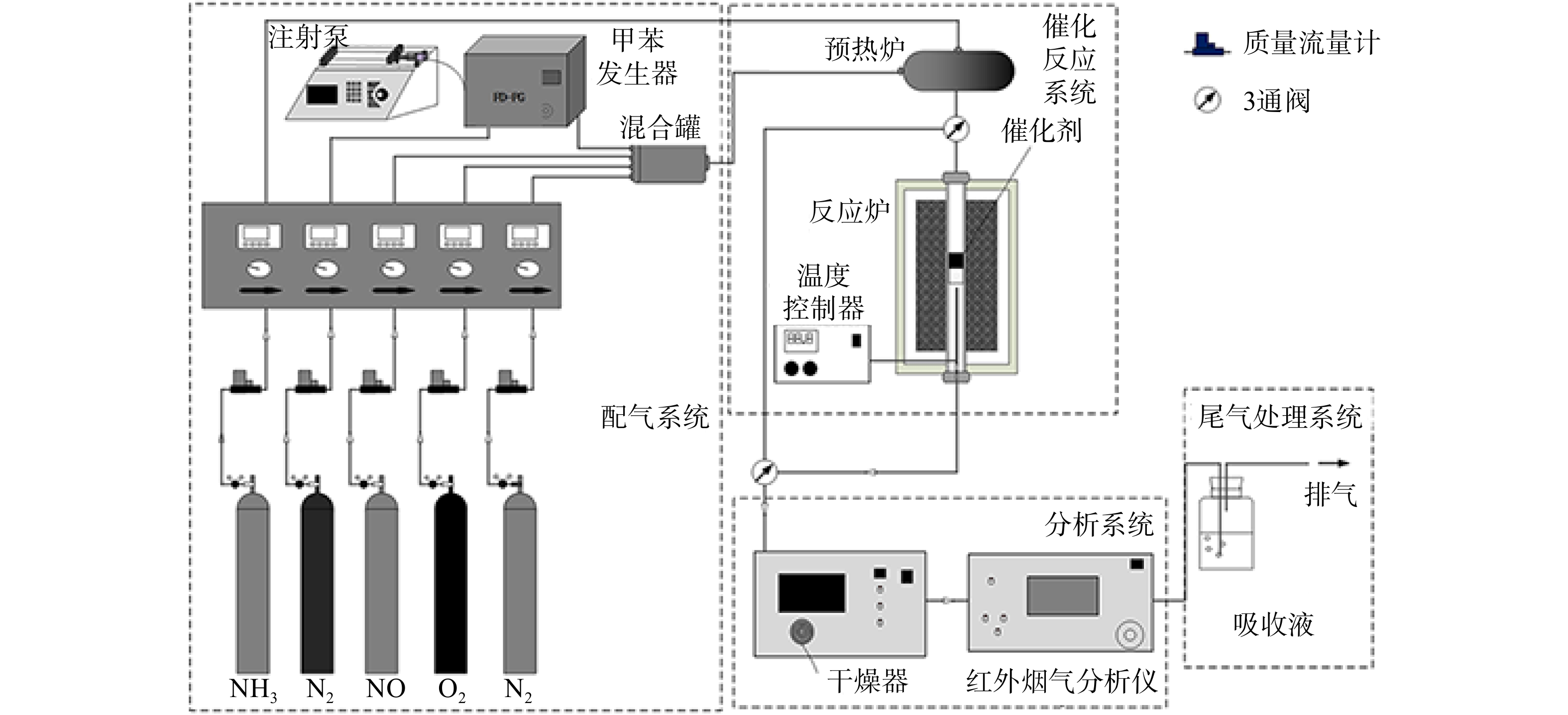
如图1 所示,La0.65X0.35FeO3(X=Cu、Ce、Mn、Co)催化剂协同催化氧化NO和甲苯的活性测试在固定床催化反应装置内进行(内径为 6 mm),测试温度为100~400 ℃。由质量浓度为804 mg·m−3的NO、1 645 mg·m−3的甲苯、0或571 mg·m−3 (使用时)的SO2、体积分数10% 的O2 ,以及平衡气体N2组成反应气体,总流量为300 mL·min−1。催化剂用量为0.2 g,空速(GHSV)为30 000 h−1。甲苯质量浓度由VOCs 发生装置(FD-PG,中国)精确控制。待反应稳定后,通过C600便携式NOx分析仪(Seitron,意大利)来检测进、出口气体中NO的质量浓度,采用Photon马杜红外烟气分析仪(深圳市昂为电子有限公司,奥地利)来检测进、出口气体中甲苯的质量浓度。NO和甲苯氧化效率按式(1)和(2)计算。
式中:
ηNO 为NO氧化效率,%;甲苯Toluene缩写为T,故η T为甲苯氧化效率,%;CNO,in/CNO,out和CT,out/CT,out分别表示NO、甲苯的进口和出口质量浓度,mg·m−3。在动力学状态下确定了同时氧化NO 和甲苯的反应速率,通过控制流量使不同温度下NO 和甲苯的转化率低于 20%,使得体系对传热几乎没有影响。 由质量浓度为402 mg·m−3 的NO、822 mg·m−3的甲苯、0或285 mg·m−3 的SO2(使用时)、 体积分数5%的 O2 和N2组成反应气体,总流量为 150 mL·min−1。 反应速率 (rT/rNO, μmol·(g·h)−1), BET 表面积归一化的比反应速率 (r*T/r*NO, μmol·(h·m2)−1),TOF (TOFT/TOFNO, S−1 ),表观活化能 (Ea, kJ·mol−1)根据式(3)~(6)计算。
式中:F是总流量,m3·h−1;mcat是催化剂的质量,g;SBET是Brunauer-Emmett-Teller表面积,m2·g−1;表观活化能Ea是采用阿伦尼乌斯定律(k = A exp(Ea/RT) )的线性拟合斜率计算,kJ·mol−1;R和T分别表示理想气体常数8.314 J·mol−1和反应温度(K);
SCO2 为CO2选择性,%。 -
采用X射线衍射(XRD)研究催化剂的物相组成和晶体结构。测试条件:在Cu-Kα辐射下,λ=0.154 18 nm,最大工作电压和电流为60 kV,最大工作电流为300 mA。使用Micrometeritics的TriStar II 3020分析仪(BET)对催化剂的比表面积和孔径分布进行表征。测试条件:取0.1 g样品预处理后,N2作为吸附质,在温度为77 K条件下进行测试。采用扫描电子显微镜(SEM)观察催化剂的形貌特征;通过氢气程序升温还原实验(H2-TPR)评价新鲜和反应后催化剂的氧化还原能力。反应后催化剂的反应条件为:在质量浓度为804 mg·m−3 的NO、1 645 mg·m−3的甲苯、571 mg·m−3的SO2,体积分数10%的O2,平衡气体为N2,总流量为300 mL·min−1,空速为30 000 h−1,处理时长为8 h。此时的催化剂表达式为S-La0.65X0.35FeO3。使用STA449 F5同步热分析仪测试热重分析(TGA)。测试条件:取10 mg催化剂, 在50 mL·min−1纯N2气流中以10 ℃·min−1的升温速率加热至1 000 ℃后进行测试。SO2-TPD的程序升温解吸实验使用AutoChem II 2920仪器进行,以研究SO2与催化剂样品表面的相互作用。测试条件:取100 mg催化剂预处理后,100 ℃下吸附SO2至饱和,在50 mL·min−1纯He气流中加热至800 ℃进行解析。采用X射线光电子能谱技术(XPS)分析催化剂掺杂前后和反应前后表面元素化学价态变化,基础压力为101.010 Pa。
-
图2展示了SO2对La0.65X0.35FeO3(X=Ce、Cu、Mn、Co)催化剂协同催化氧化NO和甲苯活性的影响。所有催化剂的NO转化率均随温度升高呈先升高后降低的趋势,甲苯转化率则随温度升高而逐渐升高。当通入SO2后,所有催化剂的NO和甲苯转化率均显著降低,表明SO2抑制了La0.65X0.35FeO3催化剂的催化性能。这归因于SO2在La0.65X0.35FeO3催化剂的表面产生了硫酸盐化,导致活性位点减少,从而催化剂活性降低。此外,不管有无SO2,不同过渡金属元素X掺杂后催化剂的活性均优于LaFeO3催化剂。这表明过渡金属元素X掺杂改性有效提升了催化活性。其中,Co元素A位掺杂的La0.65Co0.35FeO3催化剂具有最佳活性,在300 ℃时,NO转化率达到最高(60%),T90为330 ℃,这表明Co与Fe之间存在良好的协同效应。另外,无论是通入或不通入SO2,La0.65X0.35FeO3催化剂均显示出比LaFeO3更低的EaNO和EaT,其中La0.65Co0.35FeO3表现出最低Ea(表1)。
-
图3为La0.65X0.35FeO3催化剂的SEM图。LaFeO3催化剂表面粗糙,形成了一些 FeOx 团聚物,孔结构不显著(图3(a))。La0.65Ce0.35FeO3催化剂颗粒排列紧密、分布均匀,没有看到团聚现象,且形成较小的孔,这表明Ce掺杂促进了Fe的分散(图3(b))。La0.65Cu0.35FeO3和La0.65Mn0.35FeO3催化剂内部有不规则的较大孔道结构,但表面粗糙、颗粒较大(图3(c)和(d))。而La0.65Co0.35FeO3催化剂表面光滑、孔结构丰富,颗粒为纳米结构,颗粒直径为5~10 nm,分散性良好(图3(e))。结合XRD分析得出,过渡金属Ce和Co的掺杂促进了Fe活性组分的分散,而La0.65Co0.35FeO3中介孔和大孔的多孔结构明显增加了比表面积,有利于提升 La0.65Co0.35FeO3催化剂的性能。
-
研究了SO2对La0.65X0.35FeO3催化剂晶体结构的变化,如图4所示。新鲜LaFeO3催化剂的衍射峰为正交相钙钛矿结构的LaFeO3衍射峰(JCPDS PDF NO.88-0641)[4]。在过渡金属掺杂后,并未观察到新的衍射峰。这表明过渡金属元素X掺杂进了LaFeO3晶格中,且保持了正交相钙钛矿结构。此外,掺杂后衍射峰强度减弱、半峰宽变宽、晶胞参数变小(见表1)。这表明过渡金属元素X掺杂使得LaFeO3催化剂晶胞和晶粒尺寸减小,引起晶格畸变,提升了氧迁移率。另外,反应后的钙钛矿催化剂衍射峰强度变弱、晶粒尺寸减小。这可能是SO2与钙钛矿催化剂作用,促使钙钛矿结构遭到破坏的结果。
-
图5为La0.65X0.35FeO3催化剂的N2吸附-脱附等温线和孔径分布图,比表面积、孔容和平均孔径列于表2。所有催化剂的等温线均为IV型,在相对压力为0.4~1.0时存在H3型滞回环.。这表明所有催化剂均存在大孔和介孔结构。此外,LaFeO3催化剂比表面积为63.9 m2·g−1。Ce掺杂后,比表面积变化不明显。Cu和Mn掺杂后,比表面积降低。然而,Co掺杂明显增大了比表面积(74.6 m2·g−1),这是存在丰富的介孔和大孔结构所致。以上结果表明:不同过渡金属元素X掺杂对催化剂比表面积和孔结构影响不同;结合活性结果表明催化剂的性能除了与比表面积有关,还与孔结构有关;高比表面积可提供更多反应位点,而多孔结构有利于甲苯分子的吸附和扩散[15]。
-
采用H2-TPR方法考察SO2对La0.65X0.35FeO3催化剂氧化还原性能的影响,结果如图6所示。新鲜的LaFeO3催化剂在460 ℃处有一个归因于Fe的弱还原峰[16]。反应后LaFeO3催化剂则在500、600和800 ℃处出现3个还原峰。这归因于表面吸附的硫酸盐被还原为硫化氢[9]。新鲜的La0.65Ce0.35FeO3催化剂在570 ℃处有一个归因于Ce4+→Ce3+的宽还原峰[17],反应后催化剂的该峰变窄。这是因为部分CeO2物质与SO3生成了稳定的硫酸铈盐。新鲜的La0.65Cu0.35FeO3催化剂在低温310 ℃处有一个归因于CuO→Cu2O的还原峰[18],反应后催化剂的低温还原峰消失,这表明CuO→Cu2O的还原失效。在475 ℃、600 ℃处出现宽的强还原峰,这归因于吸附的硫酸盐被还原为硫化氢和高温下CuO→Cu的还原[18]。新鲜的La0.65Mn0.35FeO3催化剂在高温区域520~900 ℃出现的宽还原峰为Mn3+→Mn2+的还原[19]。反应后的催化剂在520 ℃处还原峰增强,这归因于金属和硫酸盐还原峰的叠加[20]。新鲜的La0.65Co0.35FeO3催化剂还原峰面积最大,这表明Co与Fe的相互协同作用增强了氧化还原能力[17]。而在570 ℃处的还原峰则归因于Co3+→Co2+→Co的还原。反应后La0.65Co0.35FeO3催化剂的还原峰强度增强,这同样归因于金属和硫酸盐还原峰的叠加。值得注意的是,反应后H2消耗量增加,但不代表催化剂氧化能力的提高[20];相反,硫酸盐的存在严重阻塞金属位点并影响了催化剂的氧化还原能力。
-
采用TG方法进一步考察反应后S-La0.65X0.35FeO3催化剂上的表面硫物种,如图7(a)所示。所有催化剂在温度升至1 000 ℃之前一直处于失重状态。结合XPS结果,反应后S-LaFeO3在382 ℃处出现的失重峰归因于Fe2(SO4)3分解生成氧化铁并释放SO2。在温度为723 ℃、752 ℃、740 ℃和816 ℃处的失重峰分别对应于硫酸铜、硫酸铈、硫酸锰、硫酸钴等硫酸盐物质[20-21]。通常重量损失大小与硫酸盐生成量有关,重量损失大小排列为: S-La0.65Cu0.35FeO3> S-La0.65Ce0.35FeO3>S-LaFeO3>S-La0.65Mn0.35FeO3> S-La0.65Co0.35FeO3。以上结果表明,SO2在催化剂表面形成硫酸盐的含量越多,对催化活性的影响越大。
-
SO2-TPD用于探索SO2在La0.65X0.35FeO3催化剂表面上的吸附,如图7(b)所示。所有催化剂在90 ℃和300 ℃都存在低温峰,这归因于吸附在弱酸性中心的SO2。另外,LaFeO3催化剂在516 ℃和702 ℃出现的峰归因于FeSO4、Fe2(SO4)3的热分解及SO2的解吸[22]。过渡金属X掺杂后,100 ℃和300 ℃的峰强度降低,中心峰向高温移动。这是因为金属之间相互协同作用使得SO2在催化剂表面上的吸附变弱,生成较少的硫酸盐物质。La0.65Ce0.35FeO3催化剂没有新峰出现。而La0.65Co0.35FeO3、La0.65Cu0.35FeO3和La0.65Mn0.35FeO3催化剂则在790 ℃出现失重峰。这分别归因于CoSO4、CuSO4和MnSO4的硫酸盐物质分解[20, 23]。再结合反应后催化剂的H2-TPR和TGA结果分析表明,O2在催化剂上的表面硫酸盐化会导致金属位点减少,从而降低催化性能。另外,金属元素间的协同作用及氧物种数量亦会对金属的硫酸盐化产生影响。
-
La0.65X0.35FeO3催化剂反应前后O1s的XPS图谱如图8 (a)~(j)所示。在529.0、530.1和531.7 eV处的峰分别为晶格氧(Olatt,O2−)和化学吸附氧(Oads,如O2−、O22−)[24]。通常,用 Oads/Olatt来评估样品表面的氧空位浓度。反应前的LaFeO3催化剂Oads/Olat比例为2∶1,这表明主要氧物种为化学吸附氧。掺杂后,Oads/Olat比例增加且Co掺杂的Oads/Olat比例最高[25],这表明过渡金属元素A位掺杂增加了氧空位,气相氧吸附在氧空位上形成化学吸附氧,从而提高催化氧化反应。反应后所有催化剂表面Oads/Olatt的比例均下降,这表明反应过程中消耗了化学吸附氧,这是由于SO2会与催化剂表面的化学吸附氧结合生成SO3[26]。值得注意的是,在532 eV处峰值显著增加,这对应于硫酸盐形式的氧的特征电子状态,证明了这是来自于SO42−的氧,也进一步证明了在催化剂表面生成了硫酸盐。
图8(k)~(t)为Fe 2p的XPS图谱。所有催化剂均在710.1 eV和723.8 eV处有2个峰,分别对应于Fe 2p3/2和Fe 2p1/2轨道[27]。Fe 2p3/2轨道分裂的2个峰分别为Fe2 + (710.0 eV)和Fe3+ (712.1 eV)[28]。新鲜的LaFeO3催化剂主要以Fe2+为主要物种。在掺杂后,Fe3+/Fe2+比例增加,这表明过渡金属X元素A位掺杂导致了Fe2+→Fe3+的转化。此外,Fe 2p3/2的峰向低结合能方向移动,这表明掺杂金属与Fe之间存在一定的相互作用从而改变了Fe的配位环境。在反应后,所有催化剂表面Fe3+/Fe2+的比例增加,这是因为发生Fe2++Xn+1→Fe3++Xn氧化还原反应。
La0.65X0.35FeO3催化剂各掺杂元素X(Ce 3d、Cu 2p、Mn 2p和Co 2p)的 XPS能谱如图9。图9 (a)和(b)为Ce 3d的XPS能谱。标记为µ、µ2、µ3、v、v2和v3的谱带归属于Ce4+,而标记为µ1和v1的峰归属于Ce3+[28]。新鲜的La0.65Ce0.35FeO3催化剂主要以Ce4+存在。在反应后,Ce3+/Ce4+的比例从0.2增至0.3,发生了Ce4+到Ce3+的还原。图9 (e)和(f)为Cu 2p图谱,在932.9和952.7 eV左右的峰被分配给Cu+,在934.5和954.5 eV左右的峰归因于Cu2+,其余为卫星峰[1]。新鲜的La0.65Cu0.35FeO3催化剂表面Cu主要以Cu+形式存在。在反应后,Cu+/Cu2+的比率从1.3降至1.8,且Cu 2p3/2峰向高结合能方向移动,Fe 2p3/2的结合能向低结合能方向移动。这是因为发生了Fe2++Cu2+↔Fe3++Cu+反应[29]。图9 (g)和(h)为Mn 2p XPS图谱,结合能在642.7 eV和641.9 eV的峰分别为Mn4+和Mn3+物种,其余为卫星峰[30]。新鲜的La0.65Mn0.35FeO3催化剂表面Mn主要为Mn4+物种。在反应后,Mn4+/Mn3+的比例从1.5降至1.1。这表明发生了Mn4+到Mn3+的还原。图9(c)和(d)为Co 2p图谱,在783.6和797.4 eV处的峰归属于Co2+物种,而780.8和796.0 eV处的峰归属于Co3+物种[31]。新鲜的La0.65Co0.35FeO3催化剂表面以Co3+物种为主。反应后,Co3+/Co2+的比例从1.5降至0.7。这表明发生了Co3+向Co2+的还原反应(Fe2+ + Co3+ ↔ Fe3+ + Co2+)。根据文献[24] ,Co掺杂改性后存在的高价Co3+物种可充当活性位点,有利于催化氧化反应。
-
图10为S 2p的XPS图谱,结合能在168.2~168.5 eV和169.2~169.7 eV处的峰归属于S 2p3/2和S 2p1/2[32]。反应后的La0.65X0.35FeO3催化剂均在169.5eV和168.2 eV处存在2个峰,这归因于Fe2(SO4)3的沉积[29]。这表明SO2存在条件下,催化剂表面会沉积硫酸盐和亚硫酸盐,从而降低催化剂的活性。该结果与TGA和SO2-TPD分析结果一致。此外,进一步研究了SO2对La0.65Co0.35FeO3催化剂稳定性的影响(图11)。在添加SO2后,La0.65Co0.35FeO3催化剂活性降低,稳定性在4 h后略微下降,这表明SO2在催化剂表面形成的硫酸盐覆盖了活性位点并破坏了钙钛矿结构。值得注意的是,停止通入SO2后,LaFeO3催化剂的效率不能恢复,而La0.65Co0.35FeO3催化剂则能恢复。结合BET和XRD结果发现,Co掺杂的催化剂具有良好的比表面积、多孔结构,从而削弱了SO2的抑制作用。由此推断SO2的影响机制过程为:SO2被 La0.65X0.35FeO3催化剂表面化学吸附氧氧化成SO3,并与金属阳离子反应生成SO42-硫酸盐物种,覆盖在催化剂表面造成孔道堵塞、减少活性位点,影响反应气体的吸附扩散,从而导致催化活性下降。然而,过渡金属X掺杂提供了丰富氧空位,产生了更多的活性位点和多孔结构,故削弱了这种抑制作用。
-
不同过渡金属(Ce、Cu、Mn、Co)的掺杂使得催化剂拥有更大的表面积,丰富的孔结构,更好的氧化还原能力和更多的活性位点,可显著提高催化剂的催化性能和抗硫性能,并削弱了SO2对催化剂结构和硫酸盐化的影响。SO2对La0.65X0.35FeO3(X=Cu、Ce、Mn、Co)钙钛矿催化剂协同催化氧化NO和甲苯活性具有抑制作用。这是由于SO2竞争吸附活性氧生成的SO3与金属阳离子反应生成硫酸盐,并覆盖在催化剂表面造成孔道堵塞,减少活性位点和减弱催化剂氧化还原能力。
SO2对La0.65X0.35FeO3钙钛矿催化剂协同催化氧化NO和甲苯的影响
Effect of SO2 on the simultaneous oxidation of NO and toluene by La0.65X0.35FeO3(X= Cu,Ce,Mn,Co) perovskite catalysts
-
摘要: 采用KIT-6硬模板辅助溶胶凝胶法制备了不同过渡金属元素A位掺杂改性的La0.65X0.35FeO3(X=Cu、Ce、Mn、Co)钙钛矿催化剂,并利用XRD、BET、SEM、H2-TPR、热重(TGA)、SO2-TPD及XPS表征了SO2对La0.65X0.35FeO3钙钛矿催化剂协同催化氧化NO和甲苯的活性影响机制。结果表明,过渡金属的引入可提高催化剂催化性能,La0.65X0.35FeO3钙钛矿催化剂在温度为100~400 ℃时表现出比LaFeO3催化剂更高的活性。其中,La0.65Co0.35FeO3催化剂活性最佳,在300 ℃时其NO转化率为60%,甲苯的T90为330 ℃。另外,SO2对La0.65X0.35FeO3钙钛矿催化剂协同催化氧化NO和甲苯的活性均表现为抑制作用。这是由于SO2容易和催化剂的金属位点发生反应生成表面硫酸盐沉积物,导致活性位点失效并堵塞孔道结构,从而会抑制NO和甲苯的氧化反应。然而,过渡金属的A位掺杂可增大表面积、增强氧化还原性,以引起结构畸变而产生更多的氧空位,提供更多活性位点,从而减弱SO2的抑制作用。本研究可为开发高效协同催化氧化氮氧化物(NOx)和挥发性有机化合物(VOCs)的抗硫催化剂提供参考。Abstract: In this study, a series of A-site substituted La0.65X0.35FeO3 (X=Cu, Ce, Mn, Co) catalysts were synthesized via a sol-gel method using KIT-6 as the hard template. And the characterizations of XRD, BET, SEM, H2-TPR, thermogravimetry (TGA), SO2-TPD and XPS were used to investigate the effect mechanism of SO2 on the activity of simultaneous catalytic oxidation of NO and toluene. The results showed that the introduction of transition metals could improve the performance of the catalyst. The La0.65X0.35FeO3 perovskite catalyst exhibited higher activities than LaFeO3 catalyst in the temperature range of 100~400 ℃. The La0.65X0.35FeO3 catalyst performed the best activity. The NO conversion was 60% at 300 ℃, and the T90 of toluene was 330 ℃. Besides, SO2 had an inhibitory effect on the co-catalytic oxidation of NO and toluene by La0.65X0.35FeO3 (X=Cu, Ce, Mn, Co) catalysts, which was due to the fact that SO2 was easy to oxidize with the metal sites to form sulfates covering the surface of catalysts, resulting in the reduction of the active sites and blocking of the pore structure, thereby inhibiting the oxidation reaction of NO and toluene. However, the A-site doping of transition metals X (X=Cu, Ce, Mn, Co) could increase the surface area and enhance the redox property, thus causing structural distortion and generating more oxygen vacancies and providing more active sites, thereby weakening the inhibitory effect of SO2. This study can provide a reference for the development of sulfur-resistant catalysts for the efficient simultaneous catalytic oxidation of nitrogen oxides (NOx) and volatile organic compounds (VOCs).
-
Key words:
- simultaneous catalytic oxidation /
- perovskite catalyst /
- transition metal /
- NO oxidation /
- toluene oxidation /
- SO2
-
厌氧氨氧化(anaerobic ammonium oxidation, anammox)工艺相较于传统硝化-反硝化脱氮工艺,具有曝气量少、不消耗有机物及污泥产率低等特点[1-3],并已成功应用于城市污水处理厂污泥水及与此类似的含有高浓度氨氮废水[4-6]。在厌氧氨氧化技术成功应用于处理高浓度含氮废水后,研究重点则从处理水量小、浓度高的污泥水(侧流)转变到处理水量大、浓度低的城镇污水(主流)处理[7-10]。
厌氧氨氧化反应的功能菌为厌氧氨氧化菌(AnAOB),而温度是AnAOB生长和代谢的一个重要参数,大多数厌氧氨氧化菌的最适生长温度为30~35 ℃[11]。而在实际污水处理厂主流的水温基本处于10~25 ℃。有研究[12-13]表明,温度每降低5 ℃,厌氧氨氧化菌的比生长速率下降30%~40%,从而影响反应器的脱氮效能。MA等[14]在利用厌氧氨氧化UASB反应器在中低温条件下处理低浓度废水时发现,当反应器温度由30 ℃降至16 ℃时,总氮去除率下降62%。因此,研究厌氧氨氧化菌活性随温度变化的规律对anammox技术应用于城市污水主流处理具有重要意义。
近年来,相比于传统活性污泥工艺,基于生物膜或颗粒污泥的主流工艺表现突出[15]。有研究表明,相较于活性污泥,在温度低于15 ℃的条件下,好氧颗粒污泥可长期稳定进行亚硝化过程并表现出0.63~0.7 kg·(m3·d)−1(以氮素计)的处理能力[16-17]。这说明不同形态的污泥在遭受温度变化时微生物响应特征存在差异。LOTTI等[18]在温度为10~30 ℃条件下,研究了以游离态和颗粒态存在的厌氧氨氧化菌的反应活化能,结果表明,以游离态存在的厌氧氨氧化菌对温度变化的敏感程度大于厌氧氨氧化颗粒污泥。这说明厌氧氨氧化菌的存在形态影响其对温度的适应性。随着以颗粒污泥和生物膜形式的厌氧氨氧化技术拟在城市污水处理厂主流工艺应用中的推进,厌氧氨氧化菌在不同温度和污泥形态条件下的反应活性和特性亟需了解和研究。
本研究以培养成熟的厌氧氨氧化颗粒污泥和生物膜为研究对象,通过测定anammox菌在不同温度(15~35 ℃)和不同形态下(游离态、颗粒污泥和生物膜)的活性,探讨了厌氧氨氧化反应的活化能和温度系数的变化,以期为厌氧氨氧化技术在主流系统脱氮的应用提供参考。
1. 材料与方法
1.1 实验装置及污泥
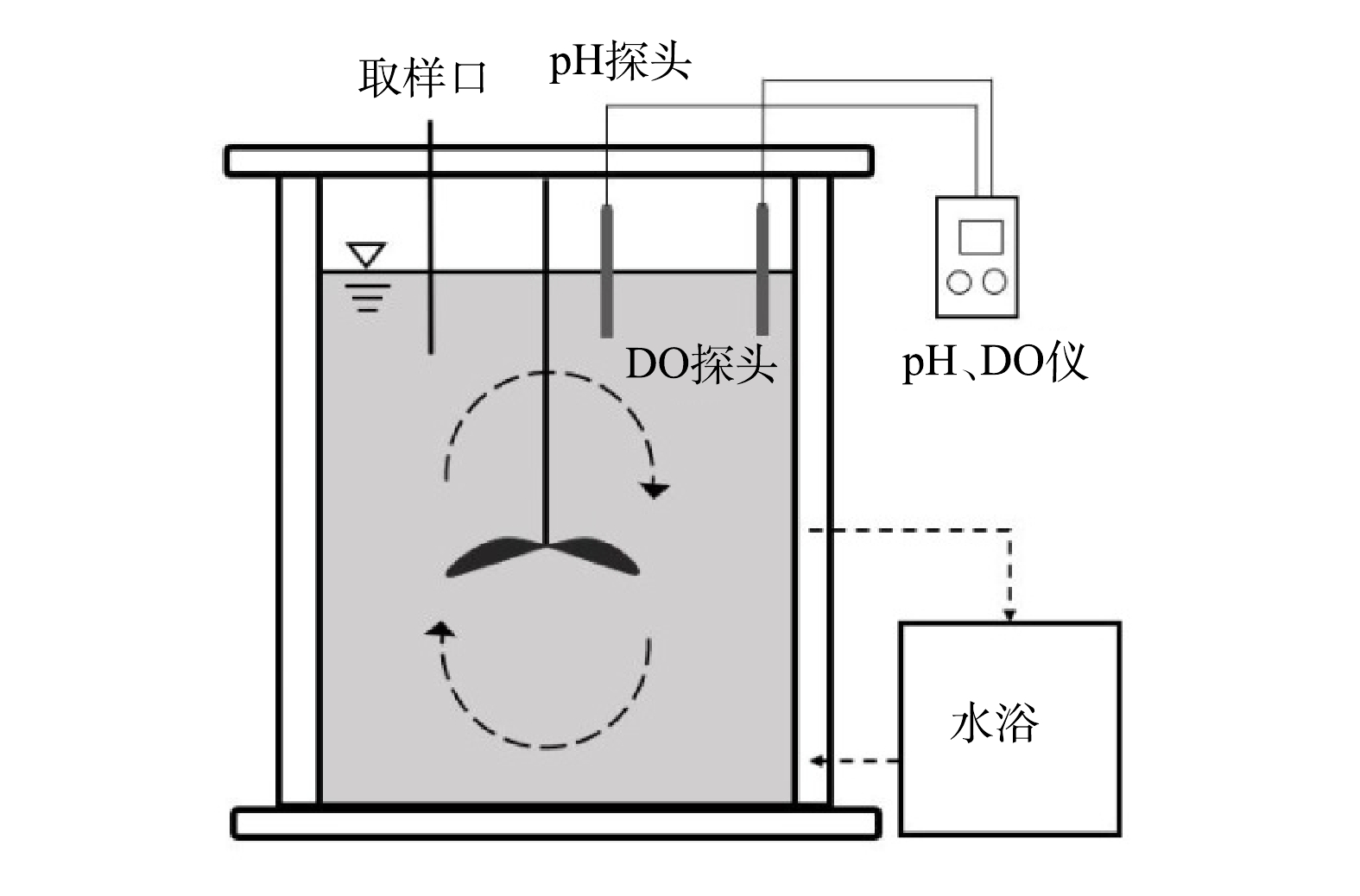
厌氧氨氧化活性(specific anammox activity, SAA)采用基质消耗速率法进行测定,测定装置如图1所示。反应器有效容积为7 L,内置推进器且在顶部设置进出水及取样口,温度由外部水浴控制,pH控制为7.5~8.1,DO控制在0.1 mg·L−1以下。活性测定过程中NH4+-N和NO2−-N由NH4Cl和NaNO2提供,质量浓度分别为30 mg·L−1和40 mg·L−1,每隔15~50 min(根据不同温度进行设定)取样并测定NH4+-N、NO2−-N和NO3−-N质量浓度,按式(1)计算厌氧氨氧化菌活性。
ki=dcidt⋅1X (1) 式中:ki为颗粒污泥、生物膜或游离状态厌氧氨氧化菌的厌氧氨氧化活性,g·(g·d)−1;ci为氮浓度变化(NH4+-N、NO2−-N及NO3−-N),mg·L−1;X为污泥浓度(mixed liquid volatile suspended solid,MLVSS),g·L−1。
厌氧氨氧化颗粒污泥取自实验室已稳定运行的厌氧氨氧化反应器,温度控制为35℃,进水负荷(nitrogen loading rate, NLR)为2.508 g·(L·d)−1,总氮(total nitrogen,TN)去除率为(87.77±1.59)%,MLVSS为(12.45±0.37) g·L−1,MLVSS/MLSS比值为0.67±0.04。厌氧氨氧化颗粒污泥呈红棕色且颗粒形态良好,直径为2 mm左右,SAA颗粒污泥为0.129 g·(g·d)−1。
厌氧氨氧化生物膜取自实验室已稳定运行的移动床生物膜反应器(moving bed biofilm reactor, MBBR),反应器温度控制为35 ℃,NLR为5.446 g·(L·d)−1,TN去除率为(73.15±5)%,填料型号为K3(比表面积为500 m2·m−3),生物膜厚度为2~3 mm,外观呈现红棕色,SAA生物膜为0.117 g·(g·d)−1 。
游离状态的厌氧氨氧化菌为实验所用颗粒污泥和生物膜通过磁力搅拌分散,以1 000 r·min−1搅拌时间3 min。由Microtrac Sync粒度仪(SYNC,美国)测定分散后的污泥粒径约为7.02~30.36 μm(≤30 μm),此时可认为污泥中的anammox处于游离态[19]。
1.2 水质指标测定
NH4+-N、NO2−-N和NO3−-N的测定参照标准方法[20]进行。NH4+-N使用纳氏试剂分光光度法测定;NO2−-N使用N-(1-萘基)-乙二胺光度法测定;NO3−-N使用紫外分光光度法测定。MLSS和MLVSS使用标准重量法测定;pH使用雷磁PH-3C pH计测定;DO使用便携式溶氧仪测定。
1.3 活化能及温度系数的计算
厌氧氨氧化菌的生物反应速率(活性)和温度的关系通过Arrhenius方程(式(2))表示,对式(2)进行积分可得式(3)。
dlnkdT=EaRT2 (2) 式中:k为厌氧氨氧化活性,g·(g·d)−1;Ea为反应所需活化能,J·mol−1;T为热力学温度,K;R为气体常数,8.314 J·(K·mol)−1。
lnk=−EaRT+lnA (3) 式中:A为Arrhenius常数。通过测定不同温度下的k,做lnk与1/T的关系图,直线的斜率为-Ea/R,由此可确定相应形态下的厌氧氨氧化活化能Ea。若温度分别为T1和T2时,反应速率分别为k1和k2,将其分别代入式(3),得式(4)和式(5)。将式(4)和式(5)相减可得式(6),再进一步换算可得式(7)。
lnk1=−EaRT1+lnA (4) lnk2=−EaRT2+lnA (5) lnk2k1=−EaR⋅T1−T2T1T2 (6) k2=k1⋅eEaRT1T2⋅(T2−T1) (7) 令Ea/(R·T1·T2)=KT (温度影响因子),e^(KT)=θ (温度系数),则(7)式可改为式(8)
k2=k1⋅θ⋅(T2−T1) (8) 根据不同温度下的Ea,计算温度影响因子KT,从而确定温度系数θ。
1.4 Anammox种群结构分析
对实验污泥采用高通量测序,确定污泥中anammox菌的种群结构。取一定数量的生物膜及颗粒污泥,经去离子水淘洗离心后按照试剂盒E.Z.N.A.® soil DNA Kit (Omega Bio-tek,Norcross GA, U.S.)规定的方法对DNA进行提取。以提取的DNA为PCR模板,采用V3-V4引物(序列为338F:5'-ACTCCTACGGGAGGCAGCAG-3'和806R:5'-GGACTACHVGGGTWTATAAT-3'),在ABI Gene Amp® 9700 PCR thermo-cycler(ABI, CA,USA)上进行PCR反应。将同一样本的PCR产物混合后使用2%琼脂糖凝胶回收PCR产物,利用AxyPrep DNA Gel Extraction Kit (Axygen Biosciences,Union City,CA,USA)进行回收产物纯化,2%琼脂糖凝胶电泳检测,并用Quantus™ Fluorometer (Promega,USA)对回收产物进行检测定量。使用NEXTflexTM Rapid DNA-Seq Kit (Bioo Scientific,美国)进行建库,利用Illumina公司的Miseq PE300/NovaSeq PE250平台进行测序(上海美吉生物医药科技有限公司,www.majorbio.com)。
2. 结果与讨论
2.1 厌氧氨氧化颗粒污泥和生物膜微生物群落结构分析
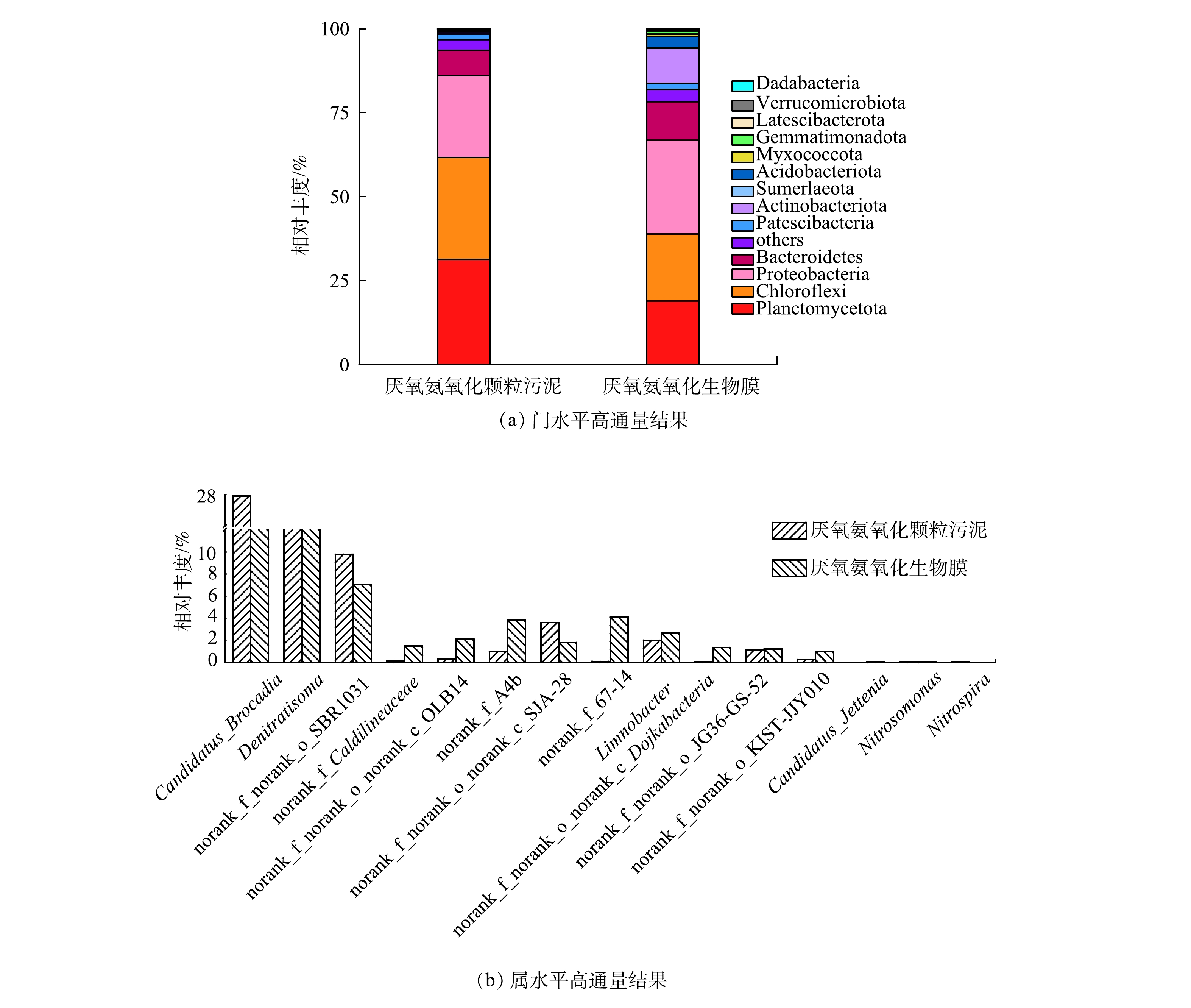
由图2可见,从门水平上来看,Planctomycetes门的微生物相对丰度分别为31.36%和18.86%,而Planctomycetes门中包含所有已知的anammox菌[21-22]。在属水平上,颗粒污泥和生物膜上探明的主要anammox菌属为Ca. Brocadia,此结果和主流系统内已探明的典型种属相同[23-24],相对丰度达到27.96%和17.52%。该结果表明anammox菌在颗粒污泥上和生物膜上均占据主导地位。除此之外,污泥中还含有Chloroflexi、Proteobacteria、Bacteroidetes。这些微生物的存在对颗粒污泥颗粒化及颗粒形态维持的过程中具有重要的作用[25]。因此,以上述2种污泥样品进行不同温度条件下的厌氧氨氧化活性特性研究是可行的。
2.2 颗粒污泥和生物膜的SAA
图3为不同温度条件下SAA的测定结果。SAA颗粒污泥随温度的下降而下降,当温度由35 ℃降至15 ℃时,SAA颗粒污泥由0.128 g·(g·d)−1下降至0.013 g·(g·d)−1,活性损失了89.89%。在对颗粒污泥进行分散处理后,SAA颗粒污泥(游离)由0.118 g·(g·d)−1(35 ℃)下降至0.008 g·(g·d)−1(15 ℃),活性损失增至93.58%。同样,当温度由35 ℃降至15 ℃时,SAA生物膜由0.117 g·(g·d)−1下降至0.016 g·(g·d)−1,活性损失为86.19%。在对生物膜进行分散处理后,SAA生物膜(游离)由0.106 g·(g·d)−1(35 ℃)下降至0.010 g·(g·d)−1(15 ℃),活性损失升至90.17%。由此可见,随着温度降低,SAA颗粒污泥和SAA生物膜均有不同程度的减小,但生物膜抵抗温度变化的能力较强。
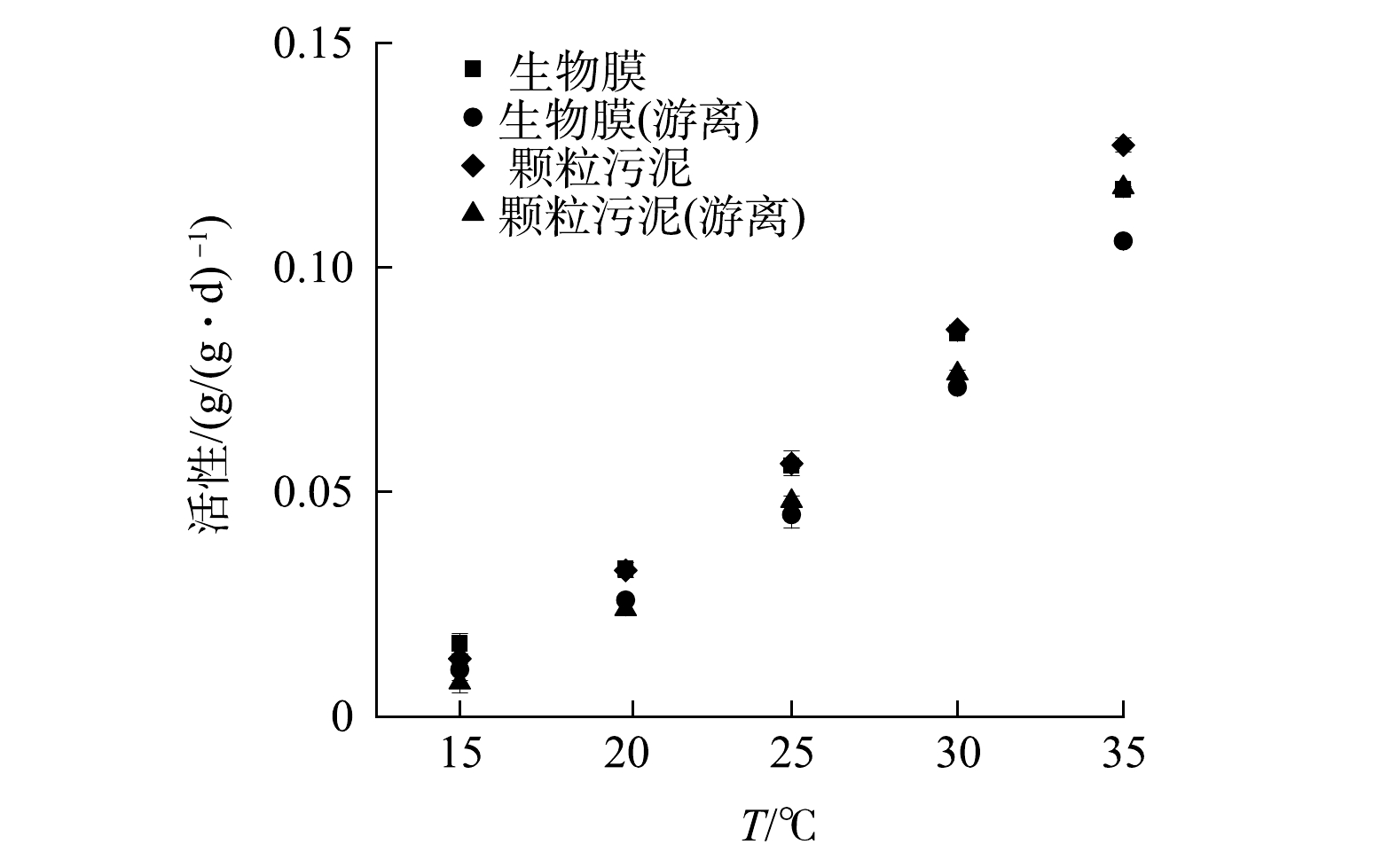 图 3 颗粒污泥和生物膜中厌氧氨氧化菌活性随温度的变化Figure 3. Variations of specific anammox activity in granule and biofilm under different temperature
图 3 颗粒污泥和生物膜中厌氧氨氧化菌活性随温度的变化Figure 3. Variations of specific anammox activity in granule and biofilm under different temperature2.3 厌氧氨氧化反应Ea
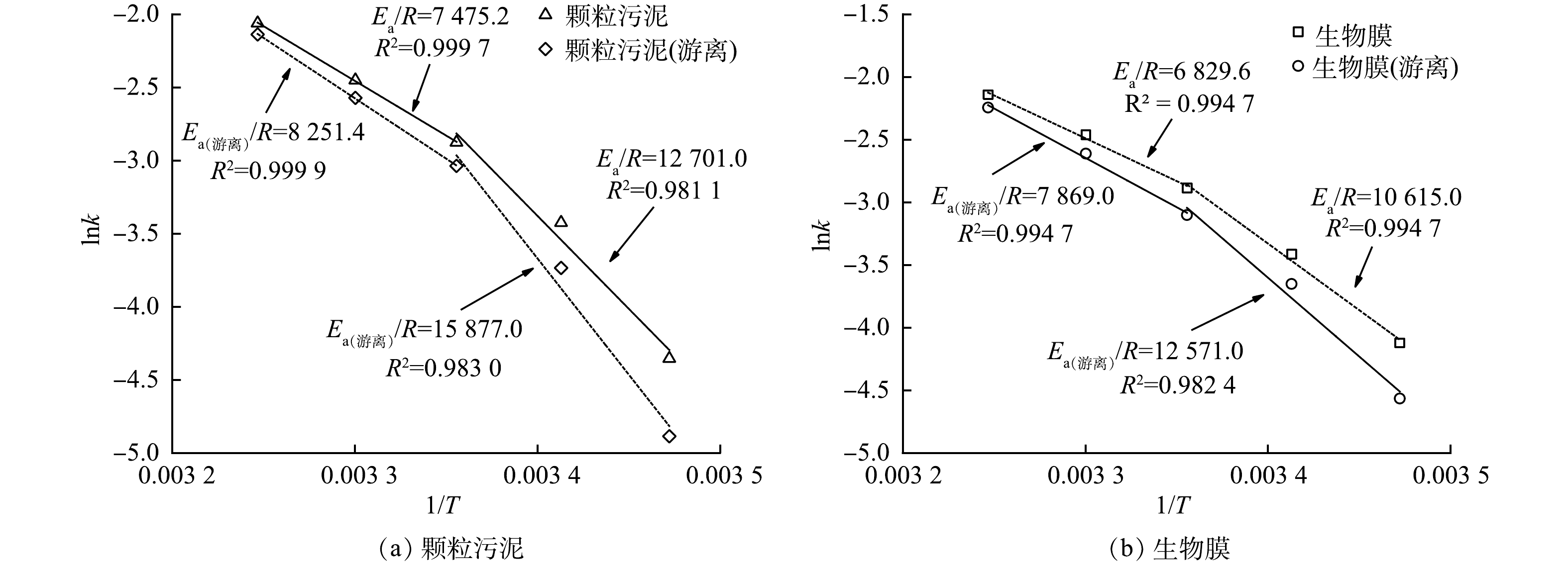
Ea值越大,说明反应过程中要跨越的能垒越大,该反应越难进行;反之,Ea值越小,则说明反应过程中要跨越的能垒越小,反应则越容易进行。由图4可见,不论颗粒污泥还是生物膜,若以15~35 ℃进行拟合,lnk和1/T线性关系较差,而若分别以15~25 ℃和25~35 ℃拟合,则能取得较好的线性关系。这表明颗粒污泥和生物膜的厌氧氨氧反应在不同温度范围内的活化能不同。谭锡诚等[26]在运行厌氧氨氧化反应器时也有相同发现。针对厌氧氨氧化活性存在拐点这一现象,ISAKA等[27]认为可能是由于厌氧氨氧化反应在不同温度区间酶活性存在差异所致。因此,将15~35 ℃分为15~25 ℃和25~35 ℃ 2个区间更符合anammox对温度的效应。由图4(a)可见,在15~25 ℃和25~35 ℃的活化能Ea-颗粒污泥分别为105.60 kJ·mol−1和62.15 kJ·mol−1;在15~25 ℃和25~35 ℃ Ea-颗粒污泥(游离)分别为132.00 kJ·mol−1和68.60 kJ·mol−1。由图4(b)可见,对生物膜而言,在15~25 ℃和25~35 ℃的活化能Ea-生物膜分别为88.25 kJ·mol−1和56.78 kJ·mol−1;在15~25 ℃和25~35 ℃ Ea-生物膜(游离)分别为104.52 kJ·mol−1和65.42 kJ·mol−1。以上数据说明不同形态的厌氧氨氧化菌活化能在不同温度区间有明显差异,温度较高时(25~35 ℃)的活化能明显小于温度较低时(15~25 ℃)的活化能。因此,厌氧氨氧化反应在温度较高时更容易进行。LOTTI等[18]的研究结果也证实了这一点。颗粒污泥和生物膜在进行分散处理后,Ea值均有不同程度的升高,说明Anammox菌的Ea与其存在状态有关。当Anammox菌以颗粒污泥和生物膜形态存在时,Anammox菌被胞外聚合物(EPS)包裹,这有助于抵抗外界温度变化[28];而当anammox菌以游离态存在时,由于缺少EPS的保护,anammox菌对温度变化就变得敏感。王淑莹等[29]研究温度对硝化反应的影响时也发现,同一温度范围内颗粒污泥的硝化反应Ea值低于絮状污泥。这说明颗粒污泥或生物膜空间结构的确有助于微生物抵抗外界的温度变化。除分散前后Ea发生变化外,生物膜所得Ea值与颗粒污泥所得Ea值两者存在差异,推测其原因可能是颗粒污泥与生物膜内EPS的含量不同。有研究表明,厌氧氨氧化颗粒污泥EPS含量为71.82~140.3 mg·g−1[30-34](以EPS计),而生物膜中EPS含量可高达300.84 mg·g−1[35]。此外,郭静[36]发现厌氧氨氧化生物膜中的EPS总量略高于颗粒污泥。这可能是造成生物膜Ea值与颗粒污泥Ea值存在差异的主要原因,但还需进一步研究。
表1比较了本研究与文献报道的厌氧氨氧化反应Ea值。本研究颗粒污泥和生物膜中的厌氧氨氧化菌在25~35 ℃下的反应活化能分别为62.15 kJ·mol−1和56.78 kJ·mol−1,上述数值与STROUS等[13]报道的Ca. Brocadia在20~43 ℃下的Ea为70 kJ·mol−1 相近。此外,LOTTI等[18]报道,厌氧氨氧化菌活化能为68 kJ·mol−1(25~30 ℃),这也与本研究获得的Ea值相近。颗粒污泥中的厌氧氨氧化菌在15~25 ℃下的反应活化能为105.60 kJ·mol−1。PARK等[37]利用厌氧氨氧化颗粒污泥在13~23 ℃下获得Ea为89.6 kJ·mol−1,略低于本研究的结果。这可能是因为实验所用anammox菌不同而存在差异。本研究中Anammox菌为Ca. Brocadia菌,而PARK等[37]研究的anammox菌是Ca. Kuenenia菌。
表 1 不同实验厌氧氨氧化反应Ea值Table 1. Ea values for anaerobic ammonia oxidation reactions in different tests污泥形态 anammox种属 Ea/(kJ·mol−1) 参考文献 生物膜 Ca. Brocadia 88.25(15~25 ℃);56.78(25~35 ℃) 本研究 生物膜(游离) 104.52(15~25 ℃);65.42(25~35 ℃) 颗粒污泥 Ca. Brocadia 105.60(15~25 ℃); 62.15(25~35 ℃) 本研究 颗粒污泥(游离) 132.00(15~25 ℃);68.60(25~35 ℃) 颗粒污泥 Ca. KueneniaCa.Jettenia 93~94(6~28 ℃);33(28~37 ℃) [27] 颗粒污泥 Ca.Brocadia 230(10~15 ℃);105(15~20 ℃)68(20~25 ℃);46(25~30 ℃) [18] 颗粒污泥 Ca. Kuenenia 89(20~43 ℃) [37] 颗粒污泥 Ca. Kuenenia. 72.8(10~30 ℃) [37] 颗粒污泥 Ca.Brocadia 89.6(13~23 ℃);16.4(23~33 ℃) [37] 活性污泥 Ca. Brocadia 107.4(10~25 ℃) [37] 活性污泥 Ca. Brocadia 70(20~43 ℃) [13] 活性污泥 Ca.Brocadia 293(10~15 ℃);131(15~20 ℃)79(20~25 ℃);68(25~30 ℃) [18] | Show Table DownLoad:
CSV
DownLoad:
CSV
2.4 温度系数θ
依据各温度区间所得Ea,可得出不同形态下anammox污泥的温度系数θ。结果表明,颗粒污泥在15~25 ℃和25~35 ℃下的温度系数θ分别为1.14和1.09;生物膜在15~25 ℃和25~35 ℃的θ分别1.12和1.08。
在污水处理中,温度系数可以衡量系统温度对反应速率和净化能力的影响,θ值越大,表明温度变化对该微生物活性的影响越大。我国城市污水处理厂生物池中温度通常为15~25 ℃,且厌氧氨氧化过程必须与亚硝化或部分反硝化配合使用,因此,对比15~25 ℃厌氧氨氧化工艺中各功能微生物的KT和θ是必要的。表2为本研究和文献报道的厌氧氨氧化工艺中功能微生物的KT和θ值。可见,当温度为15~25 ℃时,各功能微生物的θ值大致为θ反硝化<θAOB<θanammox。这可能与3种功能菌的最适温度有关。Anammox菌、氨氧化菌(ammonium oxidizing bacteria, AOB)和反硝化菌的适宜温度依次为30~40、20~30和15~35 ℃,使得anammox菌的θ值较大,即anammox菌对低温环境的适应性弱于其他2种微生物。相比于反硝化菌,AOB温度系数θ值略小,这表明AOB抵抗温度变化的能力较强。因此,在主流系统内采用亚硝化厌氧氨氧化工艺比部分反硝化厌氧氨氧化工艺更有优势。
表 2 厌氧氨氧化工艺中功能微生物的KT和θTable 2. KT and θ of functional microorganisms in anammox process功能微生物 温度/℃ 污泥形态 KT/(℃-1) θ 参考文献 AMX 15~25 颗粒污泥 0.138 1.14 本研究 25~35 颗粒污泥 0.096 1.09 15~25 生物膜 0.116 1.12 25~35 生物膜 0.092 1.08 15~20 活性污泥 0.186 1.21 [18] 20~25 活性污泥 0.109 1.11 AOB 10~28 生物膜 0.058~0.095 1.06~1.10 [38] 17~28 颗粒污泥 0.039 1.04 [39] 13~25 活性污泥 0.048 1.05 [40] 20 活性污泥 0.095 1.10 [41] 反硝化菌 15~20 — 0.067 1.07 [42] 17~27 — 0.079 1.08 [43] | Show TableDownLoad:
CSV
3. 结论
1)当温度由35 ℃降至15 ℃时,以颗粒污泥形态存在的anammox菌活性由0.128 g·(g·d)−1下降至0.013 g·(g·d)−1,以生物膜形态存在的anammox菌活性由0.117 g·(g·d)−1下降至0.016 g·(g·d)−1。
2)以颗粒污泥形态存在的anammox菌在15~25 ℃和25~35 ℃的Ea分别为105.60 kJ·mol−1和62.15 kJ·mol−1;以生物膜形态存在的anammox菌在15~25 ℃和25~35 ℃的活化能分别为88.25 kJ·mol−1和56.78 kJ·mol−1。这表明以生物膜形态存在的anammox菌对于温度变化的抵抗能力较强。
3)以颗粒污泥形态存在的anammox菌在15~25 ℃和25~35 ℃的θ分别为1.14和1.09;以生物膜形态存在的anammox菌在15~25 ℃和25~35 ℃的θ分别为1.12和1.08。与硝化菌或反硝化菌相比,本实验所获得的厌氧氨氧化菌的温度系数θ偏大。这表明,厌氧氨氧化菌对温度的变化更为敏感,使得厌氧氨氧化在低温条件下首先将成为限制步。
-
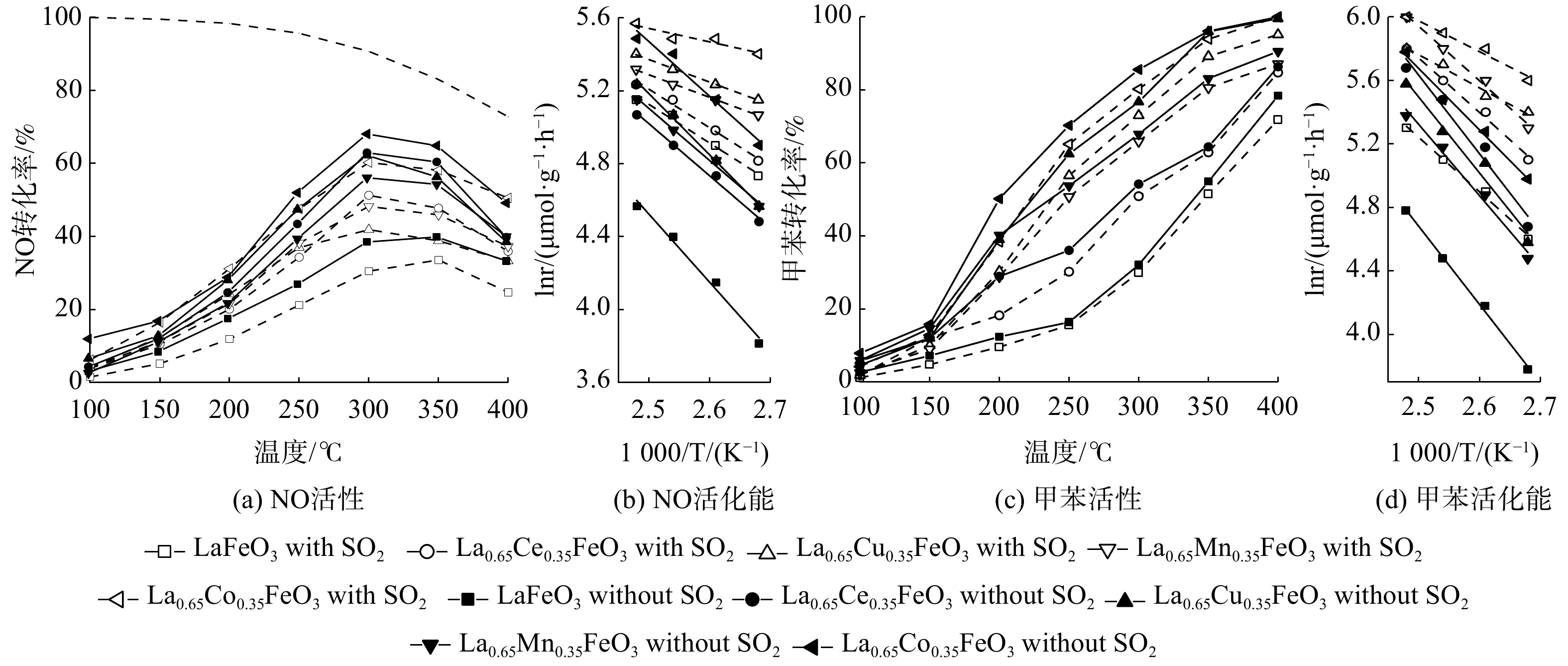
图 2 SO2对La0.65X0.35FeO3催化剂协同催化氧化NO和甲苯的影响
Figure 2. Effect of SO2 on the performance of La0.65X0.35FeO3 catalysts for simultaneous oxidation of NO and toluene
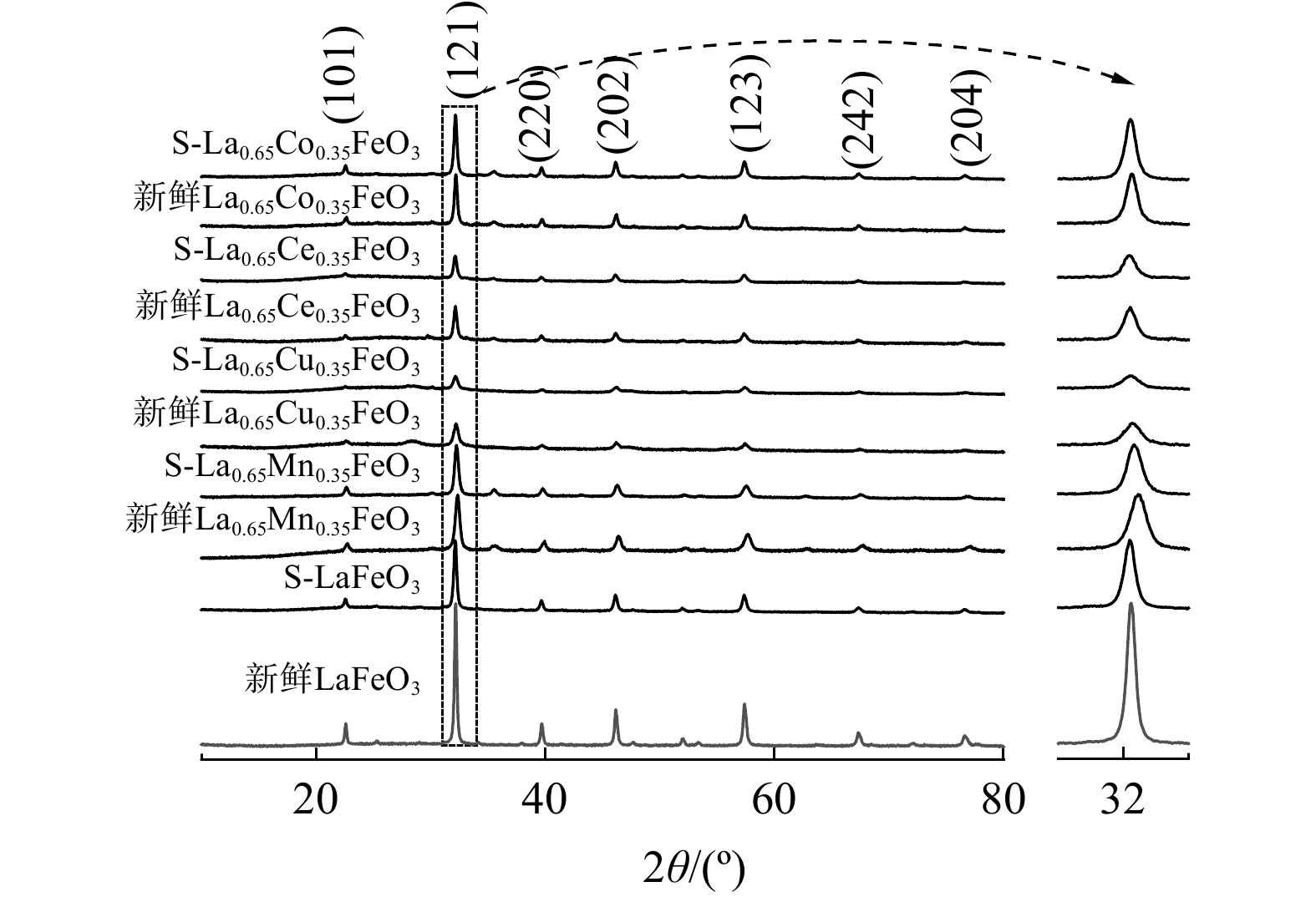
图 4 新鲜和反应后La0.65X0.35FeO3催化剂的XRD图谱
Figure 4. XRD profiles of the fresh and reacted La0.65X0.35FeO3 catalysts
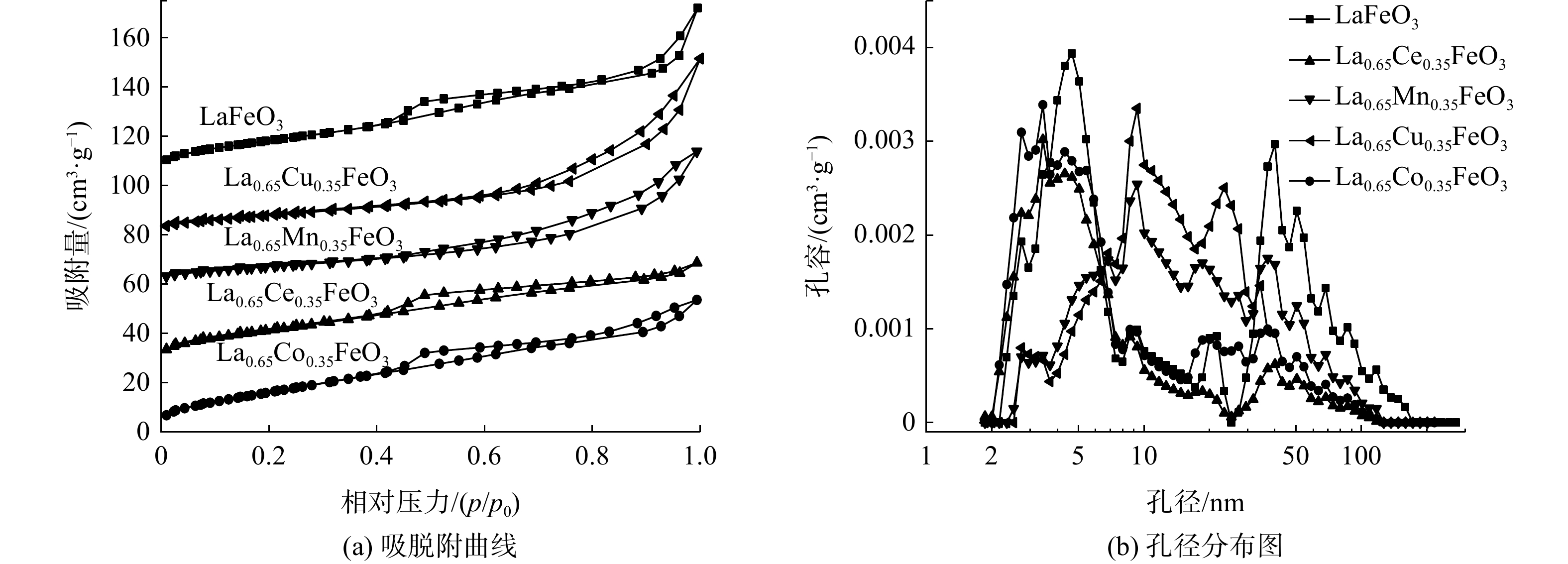
图 5 La0.65X0.35FeO3催化剂的N2吸脱附等温线和孔径分布曲线
Figure 5. N2 adsorption/desorption isotherms and pore size distribution curves of La0.65X0.35FeO3 catalysts
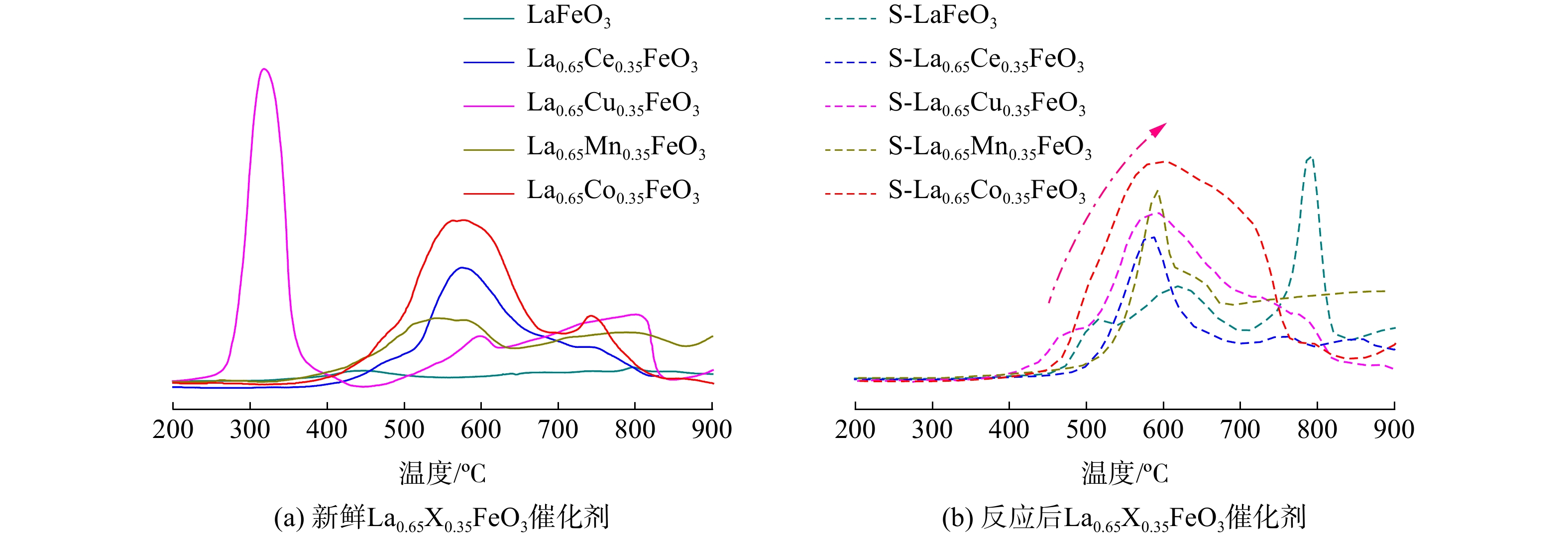
图 6 新鲜和反应后的La0.65X0.35FeO3催化剂H2-TPR图
Figure 6. H2-TPR profiles of fresh and used La0.65X0.35FeO3 catalysts
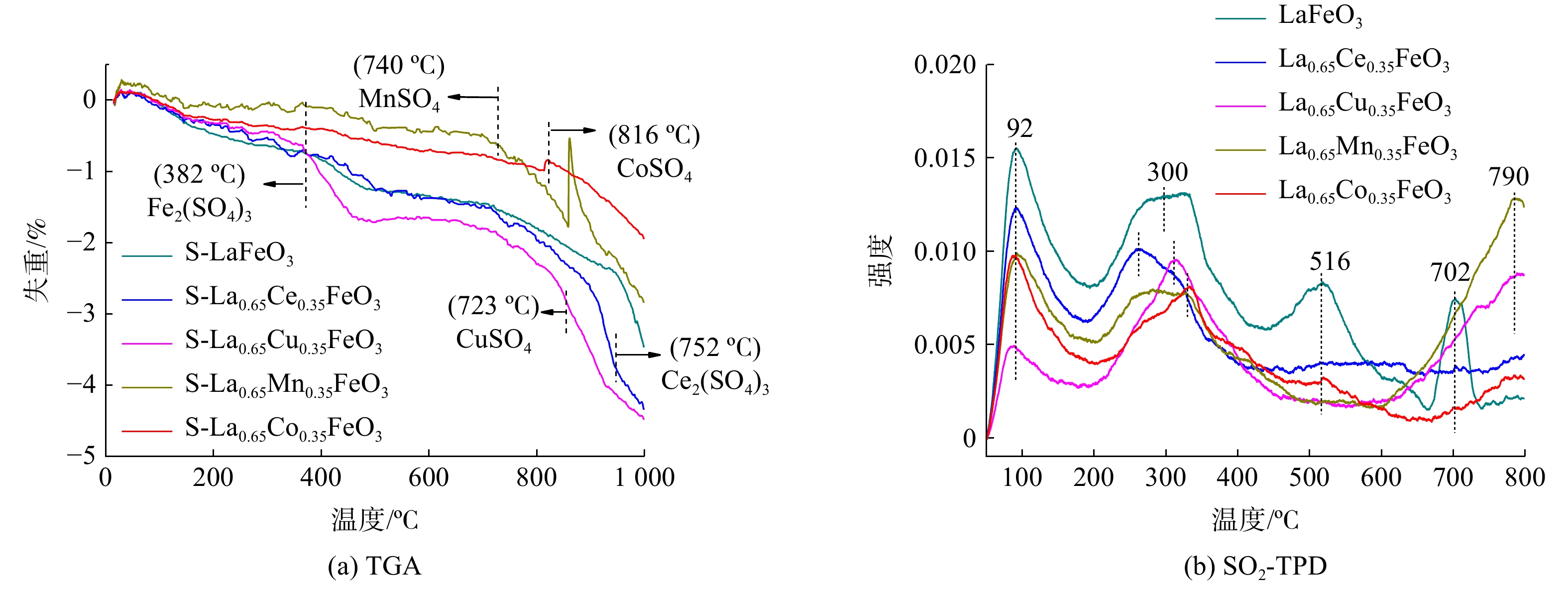
图 7 S-La0.65X0.35FeO3催化剂的TGA图和La0.65X0.35FeO3催化剂的SO2-TPD图
Figure 7. TGA diagrams of S-La0.65X0.35FeO3 catalysts and SO2-TPD profiles of La0.65X0.35FeO3 catalysts
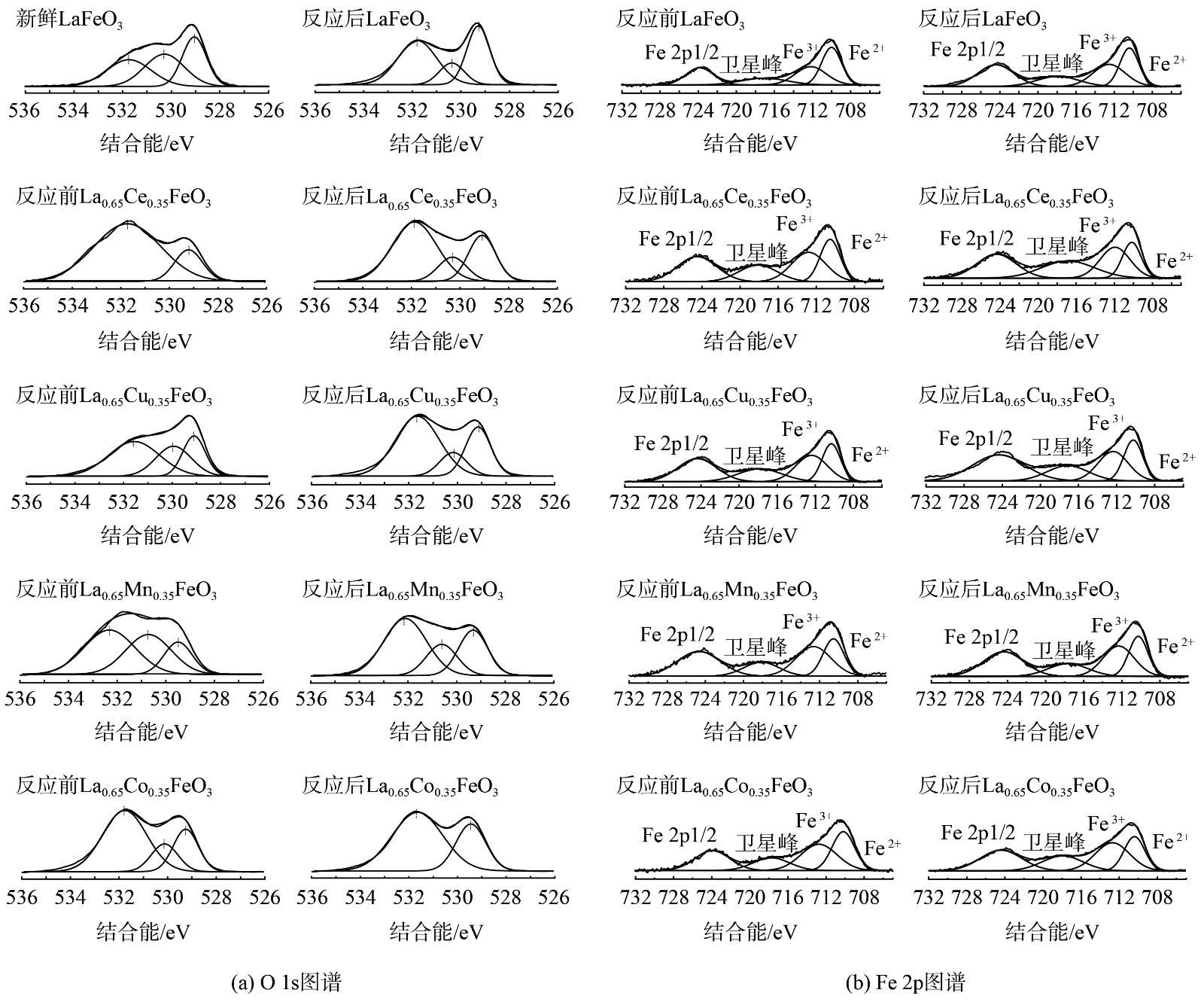
图 8 反应前后La0.65X0.35FeO3催化剂的O1s和Fe2p图谱
Figure 8. O1s (A) and Fe2p (B) spectra of the La0.65X0.35FeO3 catalysts before and after the test
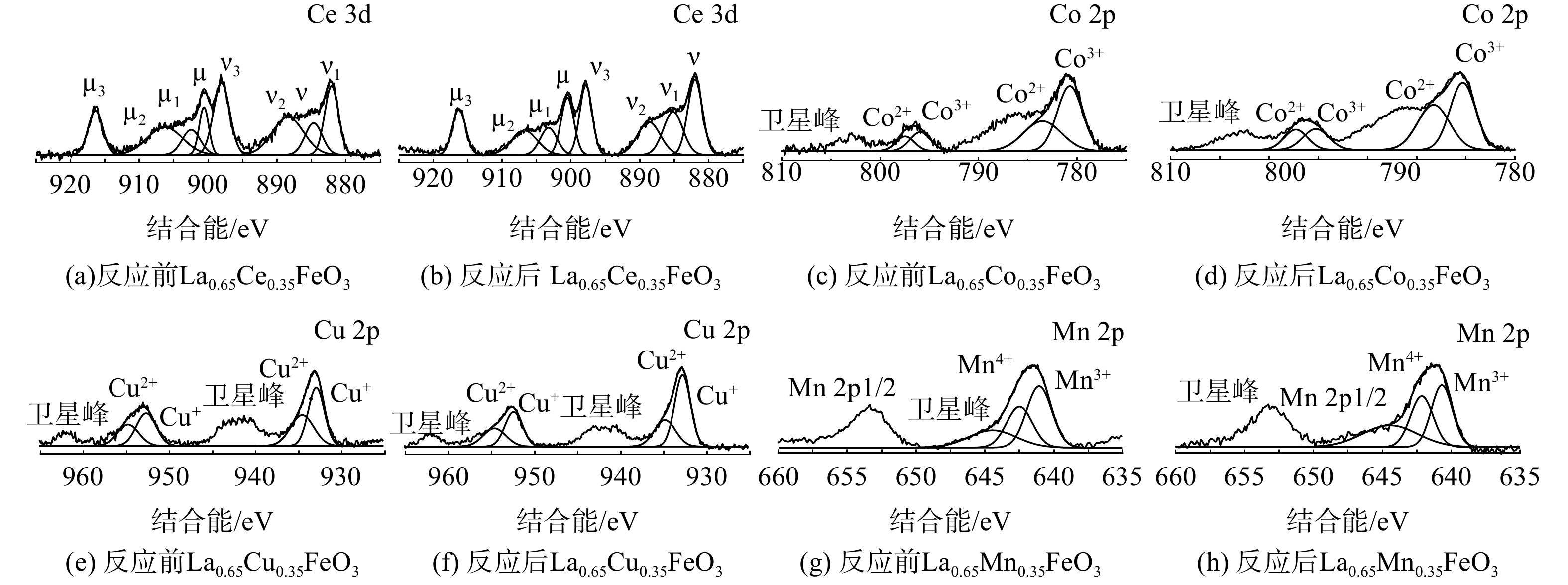
图 9 反应前后La0.65X0.35FeO3催化剂的Ce3d、Cu2p、Mn2p和Co2p图谱
Figure 9. Ce3d、Cu2p、Mn2p和Co2p spectra of the La0.65X0.35FeO3 catalysts before and after the test
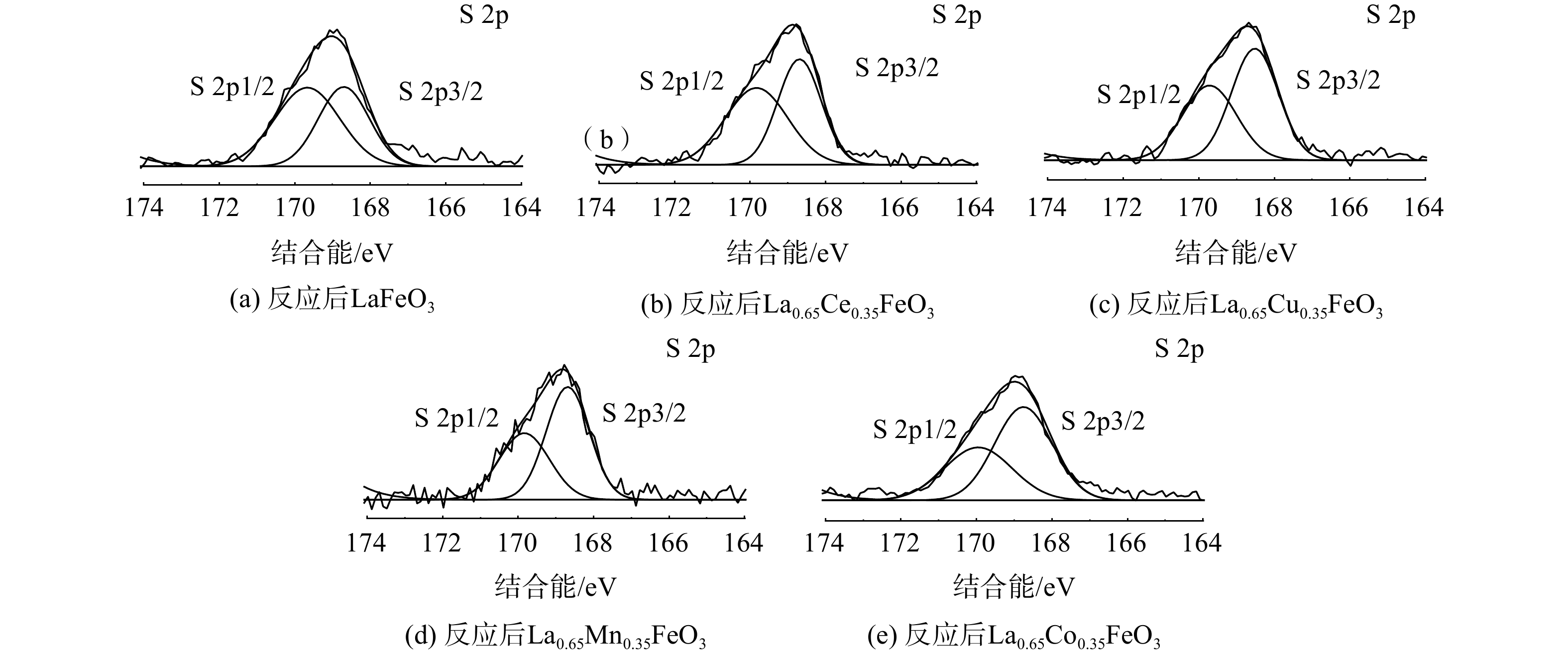
图 10 反应后La0.65X0.35FeO3催化剂的S2p图谱
Figure 10. S2p spectra of the La0.65X0.35FeO3 catalysts after the test
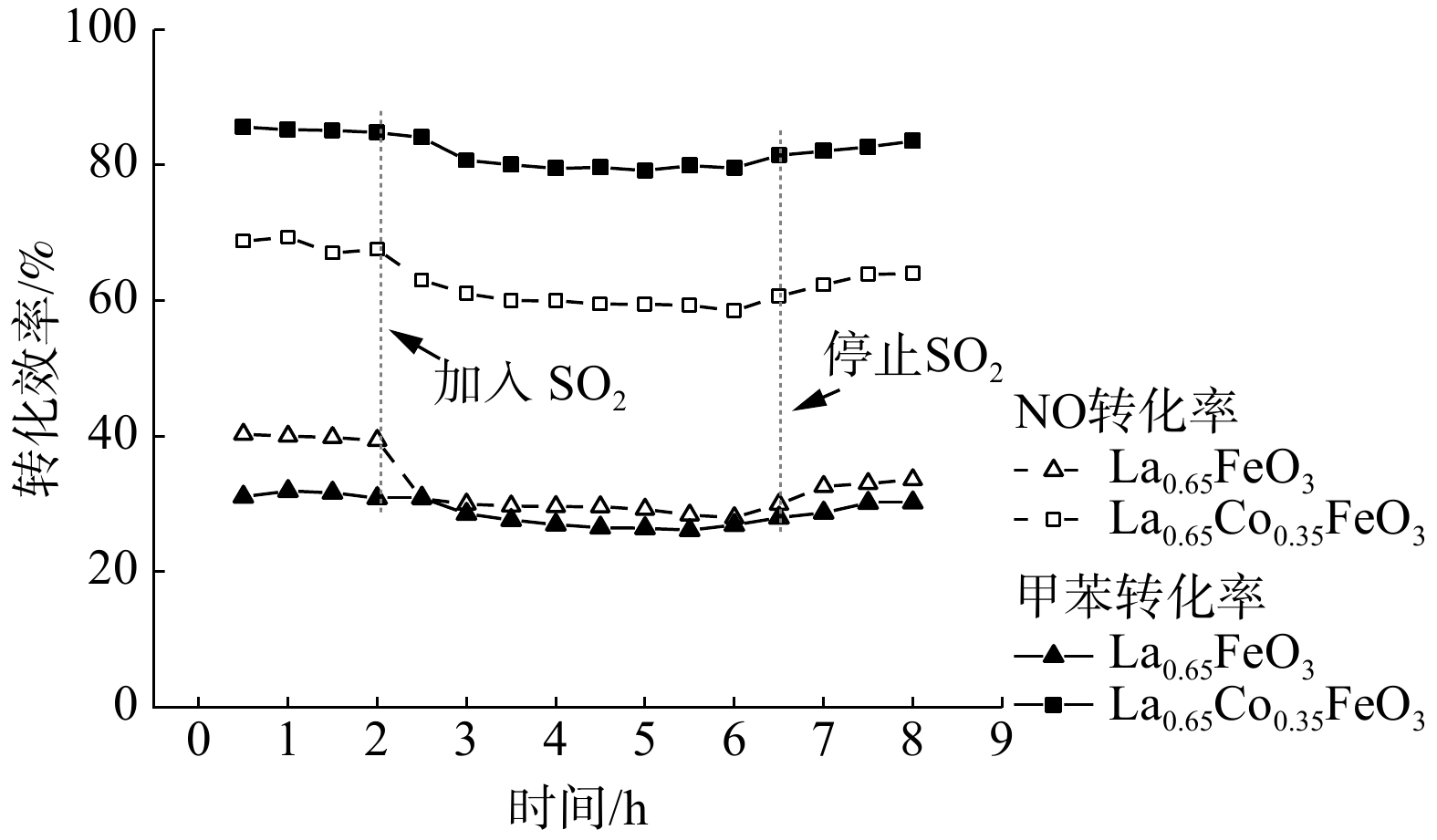
图 11 SO2对La0.65Co0.35FeO3催化剂稳定性的影响
Figure 11. The effect of SO2 on the stability on La0.65X0.35FeO3 catalysts
表 1 La0.65X0.35FeO3催化剂的XRD峰位、平均晶粒、晶格参数和活化能
Table 1. XRD peak position, crystal size, lattice parameter and apparent activation energies of La0.65X0.35FeO3 catalysts
催化剂种类 XRD峰位1)(o) 平均晶粒D2)/nm 晶格参数(Å)3) Ea-T/ (kJ·mol-1) Ea-NO/ (kJ·mol-1)hn a b c LaFeO3 32.16 35.98 5.920 7.855 5.556 28.6 21.2 La0.65Ce0.35FeO3 32.21 16.39 8.600 5.500 5.027 28.2 20.7 La0.65Cu0.35FeO3 32.14 26.77 5.662 7.852 5.475 17.4 12.3 La0.65Mn0.35FeO3 32.18 22.49 5.306 9.414 5.902 28.6 11.6 La0.65Co0.35FeO3 32.31 17.12 5.539 7.833 5.523 16.2 7.4 S-LaCeFeO3 32.14 26.32 5.571 7.857 8.579 41.0 37.33 S-La0.65Ce0.35FeO3 32.15 11.37 6.193 7.810 5.526 41.2 28.6 S-La0.65Cu0.35FeO3 32.15 21.50 5.566 7.846 5.577 39.7 33.6 S-La0.65Mn0.35FeO3 32.11 25.77 5.626 7.781 5.652 37.3 28.6 S-La0.65Co0.35FeO3 32.25 16.74 5.548 7.835 5.557 32.2 29.9 注:1)表示 La0.65X0.35FeO3催化剂121晶面的XRD峰位;2)基于(101、121、220、202、123、242和204)半峰宽的谢乐公式计算所得;3) 根据La0.65X0.35FeO3催化剂121晶面的XRD峰值。
下载: 导出CSV
表 2 La0.65X0.35FeO3催化剂的BET比表面积、孔容和平均孔径
Table 2. BET surface areas, pore volume and average pore sizes of La0.65X0.35FeO3 catalysts
催化剂种类 比表面积 /(m2·g-1) 孔容/ (cm3·g-1) 平均孔径 /nm LaFeO3 63.9 0.11 6.875 La0.65Ce0.35FeO3 63.5 0.069 4.339 La0.65Cu0.35FeO3 32.9 0.111 13.55 La0.65Mn0.35FeO3 31 0.085 10.96 La0.65Co0.35FeO3 74.6 0.088 4.738
下载: 导出CSV
-
[1] ZHAO L, HUANG Y, ZHANG J, et al. Al2O3-modified CuO-CeO2 catalyst for simultaneous removal of NO and toluene at wide temperature range[J]. Chemical Engineering Journal, 2020, 397: 125-419. [2] LU P, YE L, YAN X, et al. Impact of toluene poisoning on MnCe/HZSM-5 SCR catalyst[J]. Chemical Engineering Journal, 2021, 414: 128-838. [3] LI Z, DAI S, MA L, et al. Synergistic interaction and mechanistic evaluation of NO oxidation catalysis on Pt/Fe2O3 cubes[J]. Chemical Engineering Journal, 2021, 413: 127-447. [4] WU M, CHEN S, XIANG W, et al. Oxygen vacancy induced performance enhancement of toluene catalytic oxidation using LaFeO3 perovskite oxides[J]. Chemical Engineering Journal, 2020, 387: 124-101. [5] YE L, LU P, CHEN X, et al. The deactivation mechanism of applied surface science on MnOx-CeO2 SCRcatalyst[J]. Applied Catalysis B:Environmental, 2020, 277: 119-257. [6] LI X, GAO H, Influence of Ce doping on catalytic oxidation of NO on LaCoO3 (011) surface: A DFT study[J]. Applied Surface Science, 2020, 499: 143-866. [7] CHEN J, SHEN M, WANG X, et al. Catalytic performance of NO oxidation over LaMeO3 (Me=Mn, Fe, Co) perovskite prepared by the sol–gel method[J]. Catalysis Communications, 2013, 37: 105-108. doi: 10.1016/j.catcom.2013.03.039 [8] ZHAO S, WANG L, WANG Y, et al. Hierarchically porous LaFeO3 perovskite prepared from the pomelo peel bio-template for catalytic oxidation of NO[J]. Journal of Physics and Chemistry of Solids, 2018, 116: 43-49. doi: 10.1016/j.jpcs.2017.12.057 [9] MARTINOVIC F, TRAN Q N, DEORSOLA F A, et al. SO2 deactivation mechanism of NO oxidation and regeneration of the LaCoO3 perovskite[J]. Catalysis Science & Technology, 2020, 10: 2193-2202. [10] SHI X, GUO J, SHEN T, et al. Improvement of NH3-SCR activity and resistance to SO2 and H2O by Ce modified La-Mn perovskite catalyst[J]. Journal of the Taiwan Institute of Chemical Engineers, 2021, 126: 102-111. doi: 10.1016/j.jtice.2021.06.056 [11] SHI X, GUO J, SHEN T, et al. Enhancement of Ce doped La–Mn oxides for the selective catalytic reduction of NOx with NH3 and SO2 and/or H2O resistance[J]. Chemical Engineering Journal, 2021, 421: 129-995. [12] WU Y, LIU H, LI G, et al. Tuning composition on B sites of LaM0.5Mn0.5O3 (M = Cu, Co, Fe, Ni, Cr) perovskite catalysts in NOx efficient reduction[J]. Applied Surface Science, 2020, 508: 145-158. [13] ZHENG S, HUA Q, GU W, et al. Catalytic oxidation of CO on LaMn1−xFexO3 perovskites solid solution[J]. Journal of Molecular Catalysis A:Chemical, 2014, 391: 7-11. doi: 10.1016/j.molcata.2014.04.001 [14] PIREZ C, CADERON J M, DACQUIN J P, et al. Tunable KIT-6 Mesoporous Sulfonic Acid Catalysts for Fatty Acid Esterification[J]. ACS Catalysis, 2012, 2(8): 1607-1614. doi: 10.1021/cs300161a [15] 裴文波, 张兴, 侯志全, 等. 多孔复合金属氧化物的制备及其在消除挥发性有机物中的应用[J]. 工业催化, 2020, 28(4): 39-51. doi: 10.3969/j.issn.1008-1143.2020.04.004 [16] 李悦, 姜宏, 蔡思翔. 钒改性对铁基脱硝催化剂活性及抗碱性能的影响[J]. 人工晶体学报, 2021, 50(8): 1511-1517. doi: 10.3969/j.issn.1000-985X.2021.08.017 [17] SHEN B, ZHU S, ZHANG X, et al. Simultaneous removal of NO and Hg0 using Fe and Co co-doped Mn-Ce/TiO2 catalysts[J]. Fuel, 2018, 224: 241-249. doi: 10.1016/j.fuel.2018.03.080 [18] GLISENTI A, PACELLA M, ACELLA M, et al. Largely Cu-doped LaCo1−xCuxO3 perovskites for TWC: Toward new PGM-free catalysts[J]. Applied Catalysis B:Environmental, 2016, 180: 94-105. doi: 10.1016/j.apcatb.2015.06.017 [19] 魏永林, 陈红萍, 侯欣辛, 等. Fe-Mn/TiO2低温NH3-SCR脱硝催化剂的SO2中毒机理[J]. 功能材料, 2021, 52(04): 4132-4139+4146. doi: 10.3969/j.issn.1001-9731.2021.04.020 [20] Y WANG Y, YI W, YU J, et al. Novel Methods for Assessing the SO2 Poisoning Effect and Thermal Regeneration Possibility of MOx-WO3/TiO2 (M = Fe, Mn, Cu, and V) Catalysts for NH3-SCR[J]. Environmental Science & Technology, 2020, 54: 12612-12620. [21] WANG F, SHEN B, ZHU S, et al. Promotion of Fe and Co doped Mn-Ce/TiO2 catalysts for low temperature NH3-SCR with SO2 tolerance[J]. Fuel, 2019, 249: 54-60. doi: 10.1016/j.fuel.2019.02.113 [22] FRANCE L J, YANG Q, LI W, et al. Ceria modified FeMnOx—Enhanced performance and sulphur resistance for low-temperature SCR of NOx[J]. Applied Catalysis B:Environmental, 2021, 206: 203-215. [23] GAO F, TANG X, YI H, et al. Promotional mechanisms of activity and SO2 tolerance of Co- or Ni-doped MnOx-CeO2 catalysts for SCR of NOx with NH3 at low temperature[J]. Chemical Engineering Journal, 2017, 317: 20-31. doi: 10.1016/j.cej.2017.02.042 [24] ZHAO M, DENG J, LIU J, et al. Roles of Surface-Active Oxygen Species on 3DOM cobalt-based spinel catalysts MxCo3–xO4 (M = Zn and Ni) for NOx-assisted soot oxidation[J]. ACS Catalysis, 2019, 9: 7548-7567. doi: 10.1021/acscatal.9b01995 [25] YI Y, LIU H, CHU B, et al. Catalytic removal NO by CO over LaNi0.5M0.5O3 (M = Co, Mn, Cu) perovskite oxide catalysts: Tune surface chemical composition to improve N2 selectivity[J]. Chemical Engineering Journal, 2019, 369: 511-521. doi: 10.1016/j.cej.2019.03.066 [26] WANG H, ZHNG C, MA Q, et al. The adsorption and oxidation of SO2 on MgO surface: experimental and DFT calculation studies[J]. Environmental Science:Nano, 2020, 7: 1092-1101. doi: 10.1039/C9EN01474H [27] KANG L, HAN L, HE J, et al. Improved NOx reduction in the presence of SO2 by using Fe2O3-promoted halloysite-supported CeO2-WO3 catalysts[J]. Environmental Science & Technology, 2019, 53: 938-945. [28] TANG X, SHI Y, GAO F, et al. Promotional Role of Mo on Ce0.3FeOx catalyst towards enhanced NH3-SCR catalytic performance and SO2 resistance[J]. Chemical Engineering Journal, 2020, 398: 125-619. [29] 房德仁, 李婉君, 刘中民, 等. Cu-Fe合成低碳醇催化剂性能研究[J]. 工业催化, 2013, 21(7): 39-44. doi: 10.3969/j.issn.1008-1143.2013.07.009 [30] ARANDIYAN H, DAI H, DENG J, et al. Three-dimensionally ordered macroporous La0.6Sr0.4MnO3 with high surface areas: Active catalysts for the combustion of methane[J]. Journal of Catalysis, 2013, 307: 327-339. doi: 10.1016/j.jcat.2013.07.013 [31] ZHU H, SONG X, HAN X, et al. Co3O4 Nanosheets preferentially growing (220) facet with a large amount of surface chemisorbed oxygen for efficient oxidation of elemental mercury from flue gas[J]. Environmental Science & Technology, 2020, 54: 8601-8611. [32] SMIRNOV M Y, KALINKIN A V, PASHIS A V, ea al. Interaction of Al2O3 and CeO2 Surfaces with SO2 and SO2 + O2 Studied by X-ray Photoelectron Spectroscopy[J]. The Journal of Physical Chemistry B, 2005, 109: 11712-11719. doi: 10.1021/jp0508249 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3673
- HTML全文浏览数: 3673
- PDF下载数: 79
- 施引文献: 0



