

-
据《全国土壤污染状况调查公报》[1],我国土壤污染物总超标率为16.1%,其中,油类有机物是导致土壤污染的重要污染物之一。绝缘油是通过石油蒸馏、精炼得到的一种矿物油,广泛用在变压器、电容器等电力设备中[2-3]。矿物绝缘油主要由烷烃、环烷烃和芳香烃等化合物组成,具有良好的绝缘性、稳定性和冷却性,但难以在自然界中降解[4-5]。其中,绝缘油中含有的多环芳烃(PAHs)具有“三致”效应,可严重危害环境和人群健康[6-7]。当这些电力设备在检修或者发生故障时,通常会发生绝缘油泄露,因而可能会对周围的土壤、地下水等造成污染[8]。
污染土壤修复方法主要分为物理修复、化学修复和生物修复,其中,物理修复中热脱附技术是处理油类有机污染物有效的方式之一[9-12]。微波加热具有整体加热、升温速率快等优点,热脱附处理过程中采用微波辅助加热的方式能够使土壤中的污染物得以快速挥发、分解或固定,从而提高修复效率[13-16]。目前,国内外采用微波加热修复污染土壤的研究主要集中在污染土壤中挥发性和半挥发性有机物(如甲苯、PAHs、PCBs等)[17-19]、重金属[20-21]、有机氯农药类[22]等污染物的治理。ABRAMOVITCH等[23-24]将微波加热应用于PCBs污染土壤的修复,采用活性炭和铅笔芯作为微波吸收介质,在PCBs的初始质量分数为146 mg·kg−1的土壤中加入400 mg活性炭,微波功率750 W加热25 min后,PCBs的去除率达到87.8%;当土壤中加入长2.7 cm、直径2.0 mm的铅笔芯,微波功率1 000 W加热3 min后,PCBs的去除率达到100%。田勐等[25]采用微波修复六氯苯(HCB)污染土壤,以MnO2作为吸收介质,发现在微波功率750 W加热20 min,酸性条件下HCB的去除率可达到100%。孙磊等[26]对五氯酚污染土壤的热修复实验中发现,五氯酚的去除率随着含水率的增加先升高再降低。而另有研究表明,污染物的去除率随着含水率的增加先降低再趋于稳定[22]。可见,对于含水率的研究还存在分歧,因此,有待进一步研究含水率对污染物去除率的影响。周翠红等[27]采用微波热脱附技术对二甲苯污染土壤进行了工艺参数研究,研究结果表明,微波功率、含水率和辐射时间对二甲苯的去除率有显著影响。在石油烃污染土壤修复方面,LIU等[28]研究了微波修复油类污染土壤中PCBs的适用性,在添加微波吸收剂的情况下微波辐射10 min,PCBs的去除效果达到80%以上。PETARCA等[29]采用微波作为热源,考察了温度、含水率和处理时间对石油烃类污染土壤去除效果的影响,结果表明,高介电损耗因子的污染物更容易被去除,此外,土壤中水分的含量对污染物的去除起到关键作用。
绝缘油是石油烃类矿物油,是从石油中提炼出来的中性烃类分子的混合物,具有良好的化学稳定性,较难降解。实际绝缘油污染土壤中的成分复杂,微波修复绝缘油污染土壤修复的工艺参数和机理鲜有报道。本研究采用微波热脱附技术对绝缘油污染土壤进行修复,重点考察了温度、停留时间、含水率、初始浓度和微波功率5个因素对绝缘油去除效果的影响,并对土壤中绝缘油微波热脱附机理进行了分析,以期为微波热脱附技术应用于绝缘油污染土壤修复提供参考。
-
微波热脱附实验装置如图1所示,实验装置包括气体装置、热脱附系统和尾气处理系统组成。采用氮气作为吹扫气体。热脱附系统采用CY-TH1000C-S微波气氛热重炉,频率为2.45 GHz±25 MHz,功率0.2~1.40 kW连续可调,采用热电偶实时测定系统炉膛温度,控温精度±0.05。尾气处理系统包括冷凝管、收集瓶和洗气瓶,微波热脱附过程中产生的不凝气体通过装有乙醇水溶液的洗气瓶吸收,处理后的尾气排入近地面大气中。
实验仪器。SX2-10-12N箱式电阻炉;DHG-9145AD电热恒温鼓风干燥箱;ME203电子天平;JLBG-129U红外分光测油仪;JP-100S超声波清洗机;CY-TH1000C-S微波气氛热重炉;安捷伦7890B-5977B MSD气相色谱-质谱联用仪(GC-MS);QSC-12T氮吹仪。
实验材料。绝缘油取自变电站25号变压器油,密度883 kg·m−3,闪点158 ℃,初馏点>250 ℃。硅胶、氧化铝(Al2O3)、无水硫酸钠(Na2SO4)、四氯乙烯(C2Cl4)、正己烷(C6H14)、二氯甲烷(CH2Cl2)均为分析纯;正辛烷(C8H18,99.4%)、正葵烷(C10H22,99.8%)、正十二烷(C12H26,99.6%)、正十六烷(C16H34,99.5%)、正二十一烷(C21H44,99.6%)、正三十四烷(C34H70,99.4%)、甲苯(C7H8,99.9%)、1,2,3-三甲基苯(C9H12,93.3%)、萘(C10H8,99.6%)、苊(C12H10,99.9%)、芘(C16H10,98.9%)、苯并苝(C22H12,95.7%)。
-
1)污染土壤制备。由于从污染场地取来的绝缘油污染土壤中绝缘油含量不均匀,不利于比较微波热脱附修复效果,因此,本研究采用自配绝缘油污染土壤。土壤采集自变电站周围棕壤,土壤的孔隙度48.64%、含水率11.53%、pH值5.7、有机质质量分数为9.72 mg·g−1。为了实验样品的均一性,对土壤样品进行了预处理。在实验室将植物根茎、砂石等杂物去除,在通风橱中自然风干过28目筛搅拌均匀,将风干过28目筛的采集土壤在850 ℃下充分预处理6 h,以去除土壤中的有机污染物,待土壤冷却后放入棕色瓶中保存。
准确称取100 g经过预处理的土壤,并加入50 mL绝缘油四氯乙烯(10 g·L−1)溶液在恒温摇床振荡24 h,转速设置为150 r·min−1。在旋转蒸发器中蒸干溶液,放置在通风橱中风干12 h,陈化7 d,即可制得绝缘油质量分数约为5 000 mg·kg−1的土壤样品。通过改变绝缘油加入量制得其他质量分数的土壤样品。
2)实验方法。将绝缘油污染土壤放入微波热脱附反应系统中,以氮气为载气,设置不同微波条件进行实验;处理之后,继续用氮气氛围保护炉膛内温度冷却至室温,将处理后的土壤进行分析。
温度和停留时间:取含油率和含水率均为5%的土壤30 g,在功率1 000 W下,升温速率为30 ℃·min−1,进行微波加热,分别考察300、350、400、450 ℃和不同微波辐照停留时间对土壤中绝缘油去除率的影响。
含水率:取含油率5%,含水率分别为0、5%、10%和15%的土壤30 g,在微波功率1 000 W下进行加热,考察土壤含水率对土壤中绝缘油去除率的影响。
初始浓度:取含油率分别为0.5%、1.5%、3%和5%,含水率为5%的土壤30 g,在1 000 W功率下进行加热,考察绝缘油初始浓度对土壤中绝缘油去除率的影响。
微波功率:取含油率和含水率均为5%的土壤30 g,分别在400、600、800、1 000和1 200 W功率下进行加热,考察微波功率对绝缘油去除率和能耗的影响。
土壤中绝缘油微波热脱附机理:取含油率和含水率均为5%的土壤30 g,在微波功率1 000 W下,分别加热到300、350、400和450 ℃,保持恒温5 min,采用GC-MS对处理前后土壤中绝缘油进行分析。
-
1)土壤中绝缘油浓度分析。实验前后土壤样品中绝缘油浓度的测定参考《土壤石油类的测定 红外分光光度法》(HJ 1051-2019)[30]分析方法。实验土壤中绝缘油的去除率计算参考式(1)。
式中:ER为土壤中绝缘油去除率;
M 为土壤中初始绝缘油质量分数,mg·kg−1;m 为土壤中残留绝缘油质量分数,mg·kg−1。2)土壤中绝缘油成分分析。实验中采用气相色谱-质谱联用(GC-MS)方法对微波处理前后土壤中绝缘油进行分析检测,具体参考《土壤和沉积物 石油烃(C10-C40)的测定 气相色谱法》(HJ 1021-2019)[31]。
-
取含水率和含油率均为5%的土壤30 g,在功率1 000 W下,升温速率为30 ℃·min−1,进行微波加热,分别加热到300、350、400、450 ℃保持恒温,温度的波动范围为±10 ℃,考察温度和不同微波辐照停留时间对土壤中绝缘油去除率的影响,结果如图2所示。在同一热脱附温度下,随着停留时间的增加,土壤中绝缘油的去除率呈现先快速升高后趋于稳定的趋势。此结果表明,土壤中绝缘油的去除率基本在5 min时趋于平衡。当微波处理温度为300、350、400和450 ℃,停留时间5 min时,土壤中绝缘油的去除率分别为37.5%、68.5%、98.6%和99.9%;当停留时间达8 min时,土壤中绝缘油的去除率分别为38.3%、71.8%、99.6%和99.9%。由此可见,停留时间0~5 min是绝缘油的快速脱附阶段,在同一温度下,当微波热脱附平衡后,延长停留时间对土壤中绝缘油的去除影响较小。FALCIGLIA等[32]研究了在280 ℃下停留时间对土壤中柴油去除率的影响,结果与本实验相似。
-
取含油率5%,含水率分别为0、5%、10%和15%的土壤30 g,在1 000 W功率下进行加热,考察土壤含水率对土壤中绝缘油去除率的影响,结果如图3所示。在微波处理时间0~15 min,含水率从0增加到5%,土壤中绝缘油的去除率逐渐升高。当含水率为5%,微波处理20 min,绝缘油去除率达到99.9%。当含水率从5%增加到15%,微波处理0~15 min,土壤中绝缘油去除率则逐渐降低。当微波处理足够长时间,绝缘油去除效果基本一致。由此可知,含水率过多或过少都不利于土壤中绝缘油的去除。土壤含水率从0到5%时绝缘油去除率升高,这可能是因为:在含水率较低的情况下,含水率增加使强极性水分子占据了更多的土壤吸附位点[33],使得较多的绝缘油可以从土壤中被去除。土壤含水率从5%到15%时绝缘油的去除率反而降低,这可能是因为:在微波热脱附过程中,过多的水分会导致微波加热挥发水分消耗较多能量[34],使得土壤中绝缘油被脱附出来后得到的能量降低,造成绝缘油去除率降低。
-
取含油率分别为0.5%、1.5%、3%和5%,含水率为5%的土壤30 g,在1 000 W功率下进行加热,考察绝缘油初始浓度对土壤中绝缘油去除率的影响,结果见图4。在处理15 min内,绝缘油初始浓度越低,去除效率越好。在微波处理5 min时,含油率为0.5%的污染土壤绝缘油去除率为83.4%,而含油率为1.5%、3%和5%的污染土壤绝缘油去除率分别为42.5%、38.1%和33.9%。在同一绝缘油浓度下,绝缘油去除效果随着处理时间的延长而增加,如从微波处理时间5 min增加到15 min,含油率为5%的土壤中绝缘油去除率从33.9%提高到99.8%。微波处理时间大于15 min后,不同绝缘油初始浓度下,土壤中绝缘油的去除率基本保持一致,且均大于99.5%。这可能是因为,当加热到5 min时,反应系统炉膛中的温度在200 ℃左右,从0~5 min这一加热阶段土壤中的水分大量蒸发,并会携带走绝缘油中易挥发的轻质油组分,该阶段绝缘油主要是通过挥发方式进行脱除。随着微波继续加热,系统温度不断升高,当温度达到绝缘油中各组分的沸点时,吸附于土壤颗粒表面和孔隙中的绝缘油,通过挥发和热解将土壤和绝缘油进行分离。对于污染土壤含水率相同但初始含油率不同的情形,在微波处理0~5 min阶段,通过挥发方式蒸发出去的挥发性物质相同,所以,绝缘油初始浓度高的土壤绝缘油去除效果较差[35]。在微波功率1 000 W条件下,微波辅助加热处理15 min后,土壤中绝缘油的含量远低于《土壤环境质量 建设用地土壤污染风险管控标准(试行)》(GB 36600-2018)[36]中第一类用地的石油烃类筛选值(826 mg·kg−1)。
-
取含油率和含水率均为5%的土壤30 g,在分别为400、600、800、1 000和1 200 W功率下进行加热,考察微波功率对绝缘油去除率和能耗的影响,结果见图5和表1。从图5可看出,在微波功率低于800 W时,绝缘油去除率较低。当微波功率增加到800 W以上,去除率显著提高。在同一功率下,土壤中绝缘油去除效果随着微波处理时间的延长而增加,达到平衡后,去除率趋于稳定。在1 000和1 200 W功率下,处理15 min,绝缘油去除率分别为99.8%和99.9%,效果相当。在800 W功率下,处理20 min,绝缘油去除率为91.8%。由表1可看出,微波功率1 000 W处理15 min时的能耗为0.250 kJ;微波功率1 200 W处理15 min时的能耗为0.300 kJ;微波功率800 W处理20 min时的能耗为0.267 kJ。由此可知,功率越小,提高绝缘油去除率需要延长加热时间,同样能耗也越高。因此,当功率为1 000 W时,去除效果高且能耗最小。
-
本实验对微波加热到不同温度处理前后的土壤中绝缘油进行了分析。通过与绝缘油原料GC-MS分析对比,其结果与绝缘油原成分基本相同。数次测试结果表明,回收率在85%~103%。微波处理前后土壤中绝缘油质量分数如表2和表3所示。由表2可看出,土壤中绝缘油脂肪烃的组分主要是C12~C34饱和烃类化合物,C12以下的低分子脂肪烃质量分数仅有5.7 mg·kg−1,在绝缘油中含量很少。由表3可看出,土壤中芳香烃组分主要以C15.5~C34.01为主,C11.7~C15.5质量分数仅有1.2 mg·kg−1。非烃类有机物在检测过程中未检出,因此认定其质量分数为0。由土壤中绝缘油成分分析可知,对于绝缘油污染土壤中脂肪烃组分,当微波加热到300 ℃恒温并停留5 min,处理后土壤中脂肪烃C10~C12组分去除率为100%。C10~C12组分沸点较低易挥发,这部分可以认为是通过蒸汽蒸馏去除。当微波加热到350 ℃后,土壤中C12~C16的去除率从72.2%升高到95.1%。350 ℃接近该组分沸点,因此,C12~C16组分可以认为是通过蒸汽蒸馏和蒸发方式脱除。对于脂肪烃C16~C21和C21~C34组分,300 ℃处理后其去除率在50%左右。经过400 ℃处理后,土壤中绝缘油脂肪烃各组分去除率均达到94%以上。这是因为,绝缘油各组分的沸点在260~380 ℃,这一部分可能是通过蒸发方式去除。对于C21~C34组分,在400 ℃处理后去除率为94.8%,450 ℃处理后去除率达到99.7%,约5%可能是通过热解方式脱除。对于高浓度绝缘油污染土壤,绝缘油各组分除了吸附在土壤颗粒表面外,还有一部分存在于土壤孔隙中,这部分污染物需要加热温度达到沸点后通过气化脱离土壤[37]。回收冷凝油通过GC-MS分析,脂肪烃组分的成分主要是C12~C34,没有发现较短碳链的脂肪烃。这表明,土壤中脂肪烃可能是通过热解吸方式脱除。由此可知,土壤中绝缘油脂肪烃中易挥发组分通过蒸汽蒸馏方式脱除,较难挥发的组分主要是通过蒸汽蒸馏和热解吸两种方式脱除。这一实验现象与李大伟[38]对于石油烃微波修复去除机制相似。
-
1)温度和停留时间显著影响土壤中绝缘油的去除率。随着温度及停留时间的增加,绝缘油去除率逐渐升高;当热脱附达到平衡后,绝缘油去除率趋于稳定。在400 ℃下停留5 min,绝缘油去除率高达98.6%。
2)在本实验条件下,随着土壤含水率的增加,土壤中绝缘油的去除率先增加再降低;土壤含水率为5%时,土壤中绝缘油去除率最佳。在微波处理15 min内,土壤中绝缘油去除率随着绝缘油初始浓度的升高逐渐降低。
3)微波功率为1 000和1 200 W,处理15 min时,绝缘油去除率分别为99.8%和99.9%,去除效果相当,此时能耗分别为0.250和0.300 kJ。微波功率为1 000 W时较佳。
4)土壤中绝缘油微波热脱附的机理为,脂肪烃易挥发组分通过蒸汽蒸馏方式得到去除,较难挥发组分主要通过蒸汽蒸馏和热解吸两种方式从土壤中脱除。
绝缘油污染土壤微波热脱附的影响因素
Influencing factors on the microwave thermal desorption of insulating oil contaminated soil
-
摘要: 为探究绝缘油污染土壤微波热脱附的影响条件,考察了温度、停留时间、土壤含水率、污染物初始浓度和微波功率对土壤中绝缘油去除效果的影响。结果表明,温度和停留时间显著影响土壤中绝缘油的去除率,在400 ℃、微波处理时间5 min的条件下,土壤中绝缘油的去除率为98.6%。当土壤含水率为5%时,土壤中绝缘油的去除率达到最佳。在微波处理15 min内,土壤中绝缘油的去除率随着绝缘油初始浓度的升高逐渐降低。微波功率越高,土壤中绝缘油的微波热脱附效率越高,综合考虑能耗和去除率,微波功率为1 000 W时较优。绝缘油污染土壤微波热脱附机理研究表明,脂肪烃主要通过蒸汽蒸馏和热解吸两种方式从土壤中脱除。本研究结果可为高浓度绝缘油污染土壤微波热脱附技术应用提供参考。Abstract: In order to explore the influencing conditions of microwave thermal desorption of insulating oil contaminated soil, the effects of temperature, residence time, soil moisture content, initial concentration of contaminate and microwave power on the removal of insulating oil in soil were investigated. The results showed that temperature and residence time had significant effects on the removal rate of insulating oil. The removal rate of insulating oil in the soil reached 98.6% when treated at 400 ℃ for 5 min. The removal rate of insulating oil reached the optimum when the moisture content was 5%. Within 15 minutes of microwave treatment, the removal rate of insulating oil gradually decreased with the increase of initial concentration. With the increase of microwave power, the thermal desorption efficiency of insulating oil showed an increasing trend. Considering the energy consumption and the removal rate, the microwave power of 1 000 W was optimal. The experimental results of the microwave thermal desorption mechanism of insulating oil contaminated soil showed that aliphatic hydrocarbons were mainly removed from the soil by steam distillation and thermal desorption. This study can provide a reference for the application of microwave thermal desorption technology in soil contaminated by high concentration insulating oil.
-
Key words:
- microwave thermal desorption /
- insulating oil /
- soil contamination
-
贻贝是一种常见的海洋生物,它们可以强力附着在水体环境中有机或无机基底材料的表面[1]。研究发现,贻贝黏液中的黏附蛋白(mussel adhesive proteins,MAPs)是贻贝能够迅速在潮湿环境中附着在各种材料表面的主要成分[2]。而黏附蛋白中起到黏附作用的关键物质是3,4-二羟基苯丙氨酸(3,4-dihydroxylphenylalanine,L-多巴或L-DOPA)和含赖氨酸蛋白质[3]。2007年,Lee等的研究团队在表面化学的研究中发现,多巴胺(dopamine,DA)在含氧碱性水溶液中会发生自聚反应,并利用共价和非共价键作用在材料表面生成具有极强黏附性的聚多巴胺包覆层,该涂层拓宽了材料表面功能化的新途径[4]。聚多巴胺包覆层拥有大量的邻苯二酚和胺基等功能基团,为吸附结合目标物提供了大量活性位点。目前,聚多巴胺功能材料已广泛运用在生产生活的多个领域[5]。在环境护方面,聚多巴胺功能材料凭借着优异的吸附催化性能,成为净化去除水体中重金属离子和有机污染物的研究热点[6-7]。
1. 聚多巴胺的形成-附着机理(Formation-adhesion mechanism of polydopamine)
1.1 多巴胺的结构性质
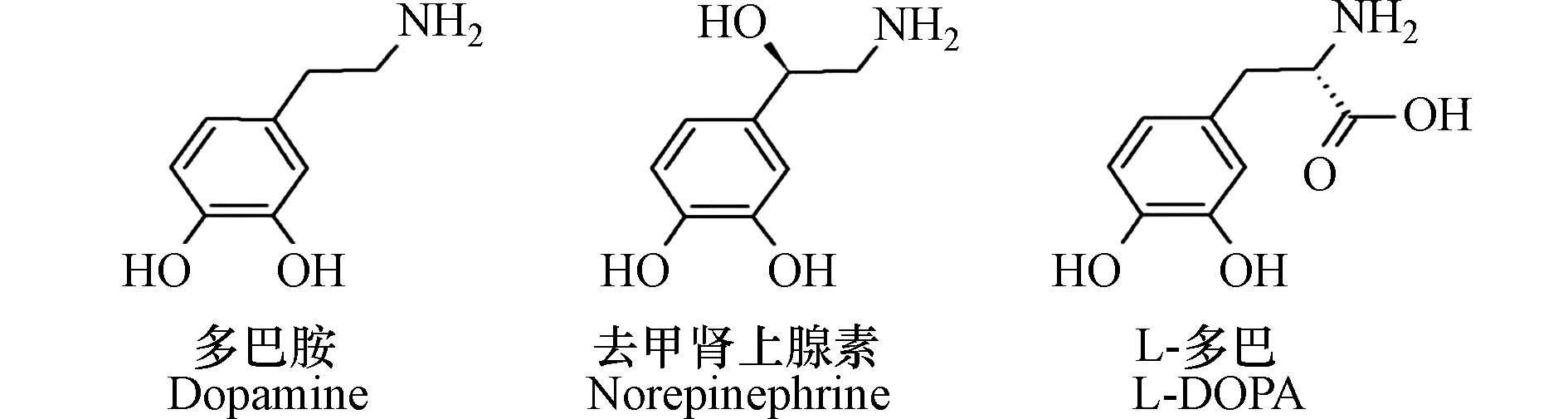
多巴胺是一种生物神经递质,它的化学名称为4-(2-乙氨基)-苯-1,2-二酚(4-(2-aminoethyl)benzene-1,2-diol),属于儿茶酚胺类物质[8]。在脱羧酶的作用下,L-多巴可转化形成多巴胺[9]。作为L-多巴的衍生物,多巴胺的化学结构中的邻苯二酚和氨基官能团为后续实现材料的修饰奠定了基础[10]。
儿茶酚胺类的不同结构(引自Barclay等[10])如下:
1.2 聚多巴胺的形成-附着机理
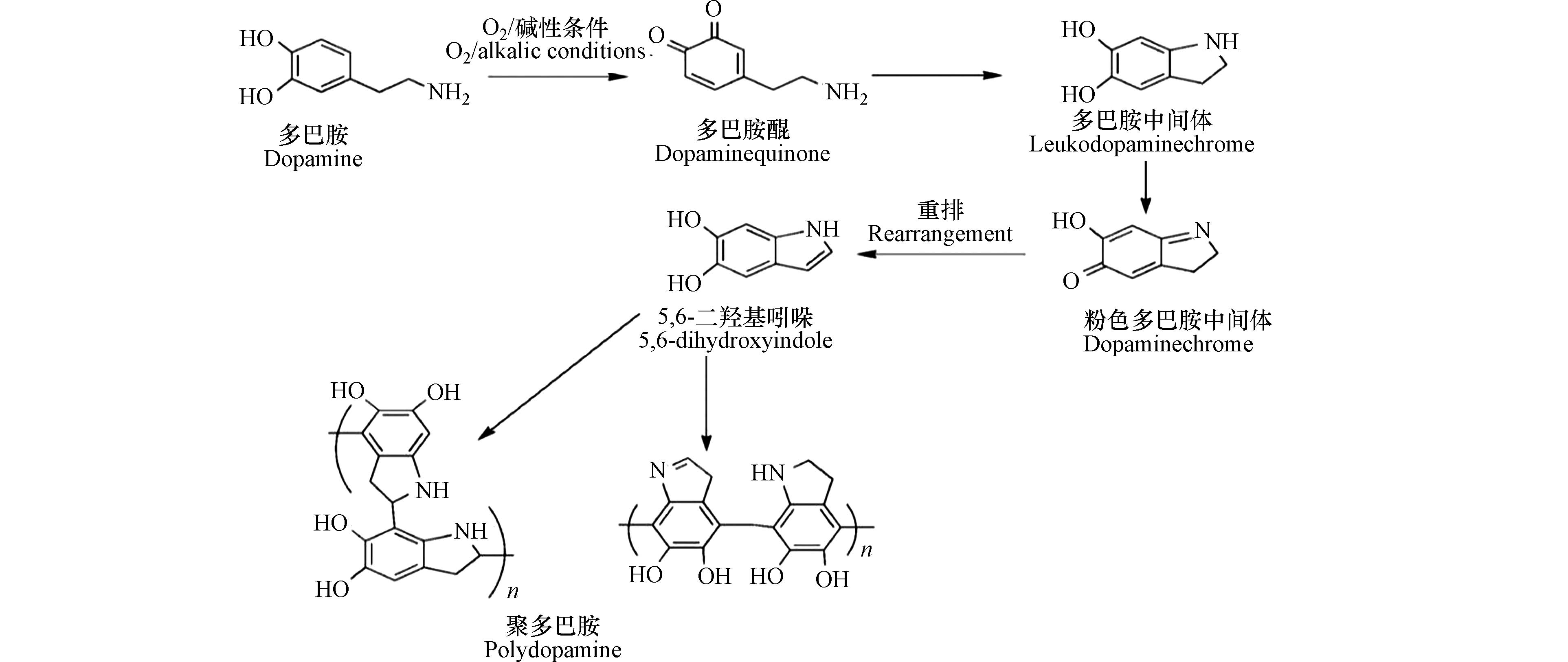
多巴胺在氧气的参与作用下,能够在弱碱性水溶液中自发反应生成聚合物—聚多巴胺(polydopamine,PDA)[11]。目前,关于聚多巴胺形成机理的研究众说纷纭,尚没有定论。主流的聚多巴胺形成理论包括 “氧化-聚合”机理、“真黑色素”形成理论和“共价-非共价”共同作用机理等[12-18]。
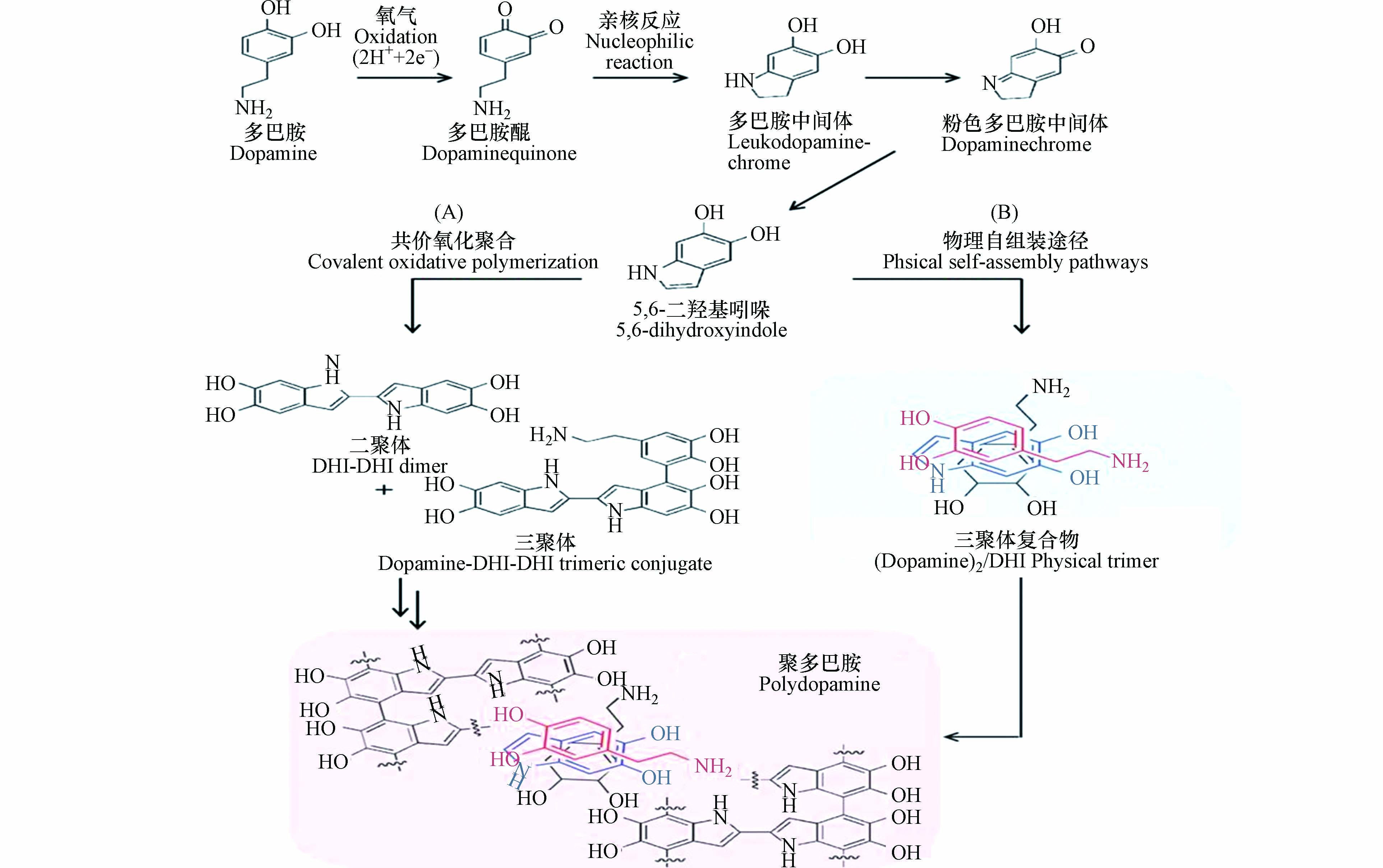
如图1所示,学者们提出了“氧化-聚合”机理:在反应过程中,多巴胺单体的邻苯二酚基团首先被氧化,生成结构性质不稳定的多巴胺醌(dopaminequinone)。多巴胺醌会发生内环化反应生成无色的多巴胺中间体(leukodopaminechrome)。该中间体发生氧化形成粉红色的多巴胺中间体(dopaminechrome),粉色中间体继续发生氧化重排生成5,6-二羟基吲哚(5,6-dihydroxyindole,DHI);5,6-二羟基吲哚与其产物5,6-醌再发生支化反应形成二聚体或其他低聚体,这些低聚体最后通过交联形成聚多巴胺包覆层[12-13]。多巴胺在溶液中的自聚反应伴随着颜色的变化,溶液颜色随着时间的延长由无色变为棕色,最终变为黑色[14]。针对该现象,科研人员通过模拟分析大量的分子数据,提出聚多巴胺的形成主要是依赖反应前期生成的低聚物,且这些受共价键束缚的低聚物以 π-π 键的相互作用堆积在一起,形成类似石墨结构的层状聚集体[15]。
Dreyer等也证明了聚多巴胺的形成与非共价键力(π-π 键、氢键和电荷转移)的作用密切相关,并且赋予了聚多巴胺在水溶液中较强的稳定性[16]。因此,大部分学者认为聚多巴胺的形成是非共价键和共价键共同作用的产物。如图2所示,Hong等[17]提出了“共价-非共价”共同作用机理:聚多巴胺是由共价聚合和非共价自组装共同作用形成的;反应前期生成的5,6-二羟基吲哚之间发生氧化聚合形成二聚体,二聚体再和一个多巴胺单体分子结合生成三聚体(DA-DHI-DHI);同时,两个多巴胺分子与5,6-二羟基吲哚通过自组装形成三聚体复合物(DA2/DHI)。DA2/DHI具有一定的生物毒性,但因其大部分被固定在聚多巴胺中,使得聚多巴胺具有较好的生物相容性。同时DA2/DHI也是聚多巴胺的形成过程中黑色沉淀产生的原因[18]。
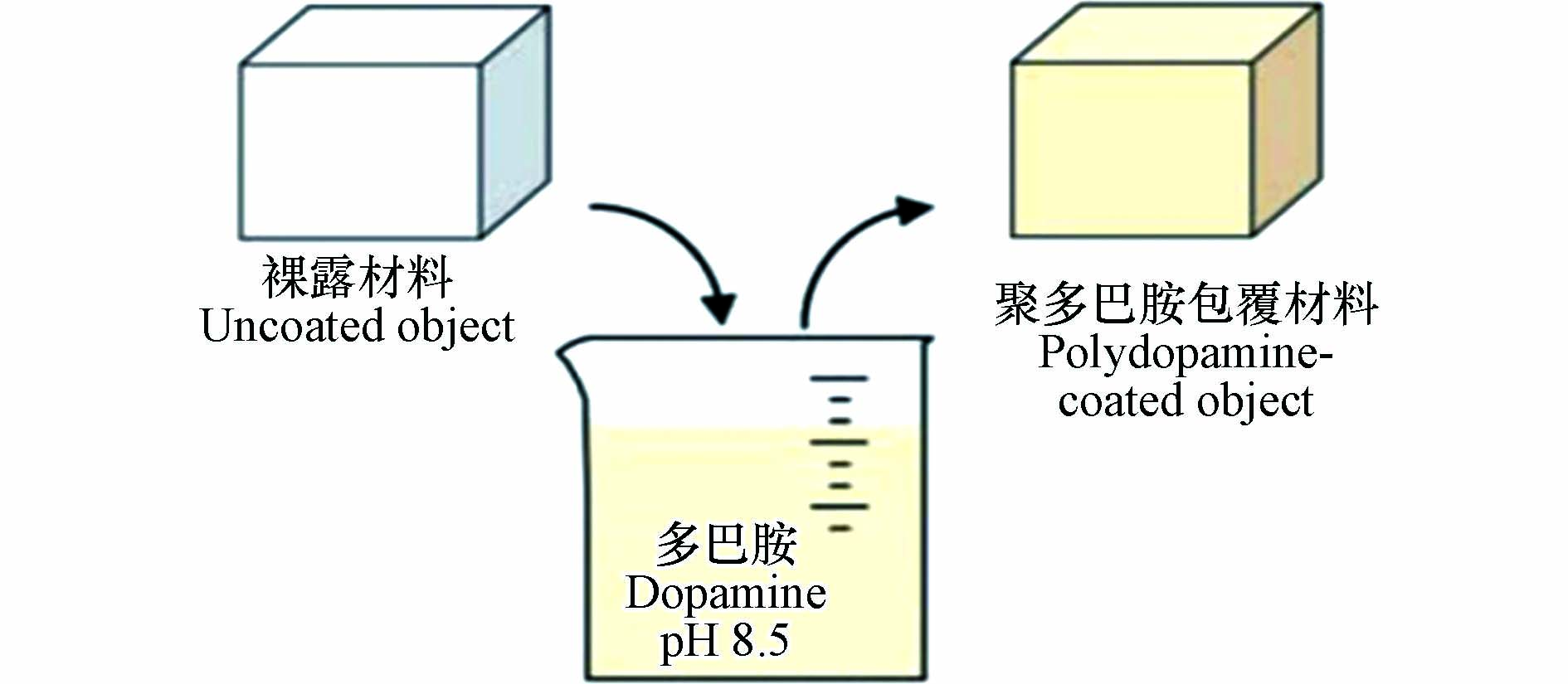
如图3所示,在避光有氧的环境条件下,基体材料置于三羟甲基氨基甲烷(Tris)的缓冲溶液(pH=8.5)中,多巴胺可以在其表面通过自聚反应生成聚多巴胺包覆层[4]。包覆层的厚度随反应时间的推移而增加。当反应时间为24 h时,厚度达到最大,为50 nm。因此在合适的反应条件下,多巴胺能够通过非共价和共价键力的相互作用,与不同的基底材料表面进行聚合附着来实现对材料的包覆[19-20]。在非共价作用方面,聚多巴胺利用非共价键力(π-π 堆积、氢键、金属离子螯合或配位等)在基底材料表面进行聚合包覆;而在共价作用方面,聚多巴胺不仅可以利用共价键的结合作用与一些表面含有硫醇基和胺基等官能团的材料发生迈克尔加成反应或碱性条件下的希夫反应,而且通过这些反应也可在聚多巴胺包覆层上接枝功能分子,实现复合材料进一步的功能化[21-22]。
1.3 影响聚多巴胺形成-附着的因素
影响聚多巴胺形成-附着过程的因素主要包括多巴胺单体浓度、溶液pH及沉积时间/温度等。多巴胺单体浓度会影响到聚多巴胺包覆层的厚度和表面粗糙度。一般包覆层的厚度和表面粗糙度会随着多巴胺浓度的提高而增加,但超出一定范围后,浓度基本不影响包覆层的沉积[23]。通常聚多巴胺的沉积发生在弱碱性溶液中,这是因为弱碱性环境有助于多巴胺自聚反应前驱体的形成,但Wei等研究发现,在反应溶液中加入一些氧化剂(Cu2+、过硫酸铵等)后也能实现酸性条件下的自聚反应[24]。此外,沉积时间/温度也是影响反应速率的重要因素,当沉积时间或温度适当增加,包覆层沉积的速率同样会提高[25]。
2. 聚多巴胺功能材料在去除水中重金属和有机物污染物方面的应用(Applications ofpolydopamine-functional materials in the removal of heavy metals and organic pollutants in water)
随着当代社会的高速发展,人类的生产生活已严重影响到水体生态环境的稳定,水污染问题日益突出。水体污染物主要来源于工业废水、生活污水及农业污水,而污染物的种类一般分为有机物、无机物和微生物三大类[26]。目前,应用于水污染控制领域的处理技术有物理法、化学法、物理化学法及生物法。在水污染处理方面,聚多巴胺功能材料结合了基底材料的优良特性和聚多巴胺包覆层丰富的功能基团,通过吸附或催化等方法对水中重金属离子和有机污染物进行有效的去除[27]。
2.1 聚多巴胺功能材料在水中重金属去除方面的应用
作为一种利用吸附材料的高比表面积和特殊吸附位点等优势来去除水中重金属的方法,吸附法因材料廉价易得和操作简单的优势而被广泛使用[28]。聚多巴胺功能材料凭借着聚多巴胺层丰富的功能基团(邻苯二酚、胺基和亚胺基等),为与水中重金属离子结合提供了大量的吸附位点[29]。
目前,研究人员在努力开发新型的基底材料和聚多巴胺表面改性手段以提高复合材料吸附去除重金属离子的效率。马玉荣等利用共沉淀法合成的聚多巴胺包覆的Fe3O4(Fe3O4@PDA NPs),通过利用聚多巴胺具有丰富吸附位点和磁性材料易于分离回收的特点,用来吸附去除模拟废水中的Pb2+[30]。由电镜表征可以看出,聚多巴胺通过羟基-铁化学作用包覆在在Fe3O4 NPs表面,形成了具有核壳结构的Fe3O4@PDA NPs(图4C)。实验表明,在最佳条件下,Fe3O4@PDA NPs对Pb2+的最大吸附量约为20.68 mg∙g−1。作为石墨烯的衍生物,氧化石墨烯(GO)具有高比表面积、丰富功能基团和优秀力学性能等特点,因此可对GO进行表面改性来调控表面性质,以提升其吸附性能[31-32]。Dong等通过控制聚多巴胺的质量分数,合成了一系列亚纳米级厚的聚多巴胺包覆的GO复合材料(PDA/GO)[33]。PDA/GO对Pb2+、Cu2+、Cd2+及Hg2+等重金属离子的最大吸附量分别为53.6、24.4、33.3、15.2 mg∙g−1,吸附性能均优于单纯的GO和PDA,体现了复合材料中PDA和GO的协同作用。Ali等同样利用共沉淀法合成了磁性氧化石墨烯(GO/Fe3O4),然后通过聚多巴胺包覆GO/Fe3O4制备出 rGO/Fe3O4@PDA,作为吸附水中Pb2+的新型磁性吸附剂[34]。由于吸附材料存在丰富的胺基和羟基等官能团,rGO/Fe3O4@PDA对Pb2+的吸附能力可达35.2 mg∙g−1,吸附性能明显优于未修饰的GO/Fe3O4。碳纳米管也是一类具有较高比表面积的碳基材料,但因表面带有的官能团较少、水分散性较差等缺点导致其在实际运用过程中存在短板[35]。刘杏红等通过将多巴胺氧化自聚到磁性碳纳米管(mMWNTs)上,合成了聚多巴胺包覆的磁性碳纳米管材料(mMWNTs@PDA)[36]。实验结果证明,mMWNTs@PDA对水中Ni2+的吸附过程符合Freundlich等温吸附和准二级动力学模型,且在最优条件下,mMWNTs@PDA对Ni2+最大吸附量可达27.9788 mg∙g−1。针对一些特殊重金属离子污染水体的治理,Yang等通过采用多巴胺聚合沉积工艺和温和水热法合成了层状双氢氧化物(LDH)原位生长修饰的Fe3O4@PDA(Fe3O4@PDA@LDH)微球,对核工业产生的含U(Ⅵ)的废水进行吸附净化[37]。在实验过程中,通过控制PDA和LDH的用量制备了不同PDA厚度和LDH含量的复合材料微球(MP2L1、MP2L2、MP2L3、ML2及MP2等)。在pH=5.0和T=298.15 K的条件下,材料对U(Ⅵ)的最大吸附容量分别为MP2L2(344 mg∙g−1)> MP2L3(291 mg∙g−1)> MP3L2(245 mg∙g−1)> MP2L1(211 mg∙g−1)> ML2(142 mg∙g−1)> MP1L2(141 mg∙g−1)> MP2(71 mg∙g−1)> Fe3O4(34 mg∙g−1),其中MP2L2具有较高的吸附容量,处理效果在含铀废水的净化方面是较为可观的。

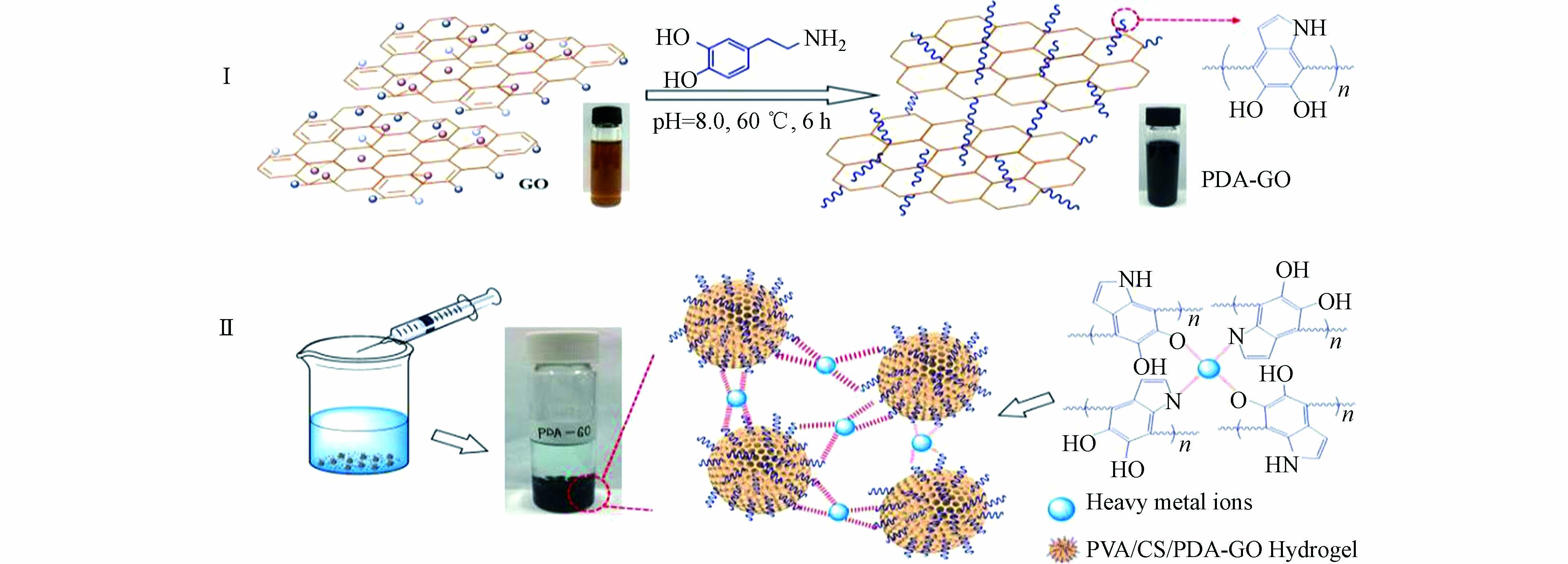
Zeng等通过将聚多巴胺掺入到支链淀粉水凝胶基质中,合成了具有高机械强度和可生物降解特性的支链淀粉/聚多巴胺(Pu/PDA)生物水凝胶吸附剂[38]。实验表明,通过调节预凝胶溶液中的聚多巴胺的浓度,可很好地控制该吸附剂的吸水率、机械强度及孔径。Pu/PDA对重金属离子具有较高的吸附能力,对Cu2+的吸附容量最高可达到100 mg∙g−1 。而Li等首先将聚多巴胺引入氧化石墨烯中合成了PDA-GO(图5-Ⅰ),然后通过瞬时凝胶法制备出聚乙烯醇/壳聚糖功能化的PDA-GO(PVA/CS/PDA-GO)水凝胶珠,用于吸附水中的Pb2+、Cu2+及Cd2+[39]。PDA-GO的存在不仅增强了水凝胶珠体系的稳定性,而且大量的活性基团的引入提高了吸附性能(图5-Ⅱ)。在最优条件下,通过Langmuir等温线对实验数据的拟合可知,PVA/CS/PDA-GO对Pb2+、Cu2+及Cd2+的最大吸附量分别为236.20、210.94、214.98 mg∙g−1。为解决实际水体环境中重金属离子共存污染的问题,Wang等采用水溶法快速制备了氨基水杨酸/聚多巴胺改性的PP无纺布吸附剂(PP/PDA/ASA)[40]。交联在聚合物末端的水杨酸分子增加了官能团的类型,有效地解决了因多组分体系中重金属离子的竞争吸附效应引起去除效率低的问题。在含有5 mg∙L−1的Cu(Ⅱ)/Cd(Ⅱ)/Pb(Ⅱ) 混合体系溶液中,由于表面氨基、羟基及羧基等官能团参与了重金属离子的吸附,PP/PDA/ASA可去除90%的Cu2+和Pb2+及80%的Cd2+。
2.2 聚多巴胺功能材料在水中有机污染物去除方面的应用
有机污染是造成水污染的重要原因之一。水中有机污染物的来源主要有生活污水、工业废水及大气污染,污染水体一旦不进行净化处理,便会对自然环境造成严重破坏并威胁到人体生命健康[41]。聚多巴胺功能材料因聚多巴胺包覆层上邻苯二酚、胺基及芳香族基团的存在,为有机污染物的去除提供了大量的活性位点。聚多巴胺功能材料可通过静电相互作用、配位或螯合作用、氢键或 π-π 键堆积相互作用对有机染料和硝基苯酚等进行吸附或催化降解,因此在水中有机污染物的净化方面具有广阔前景[42-43]。
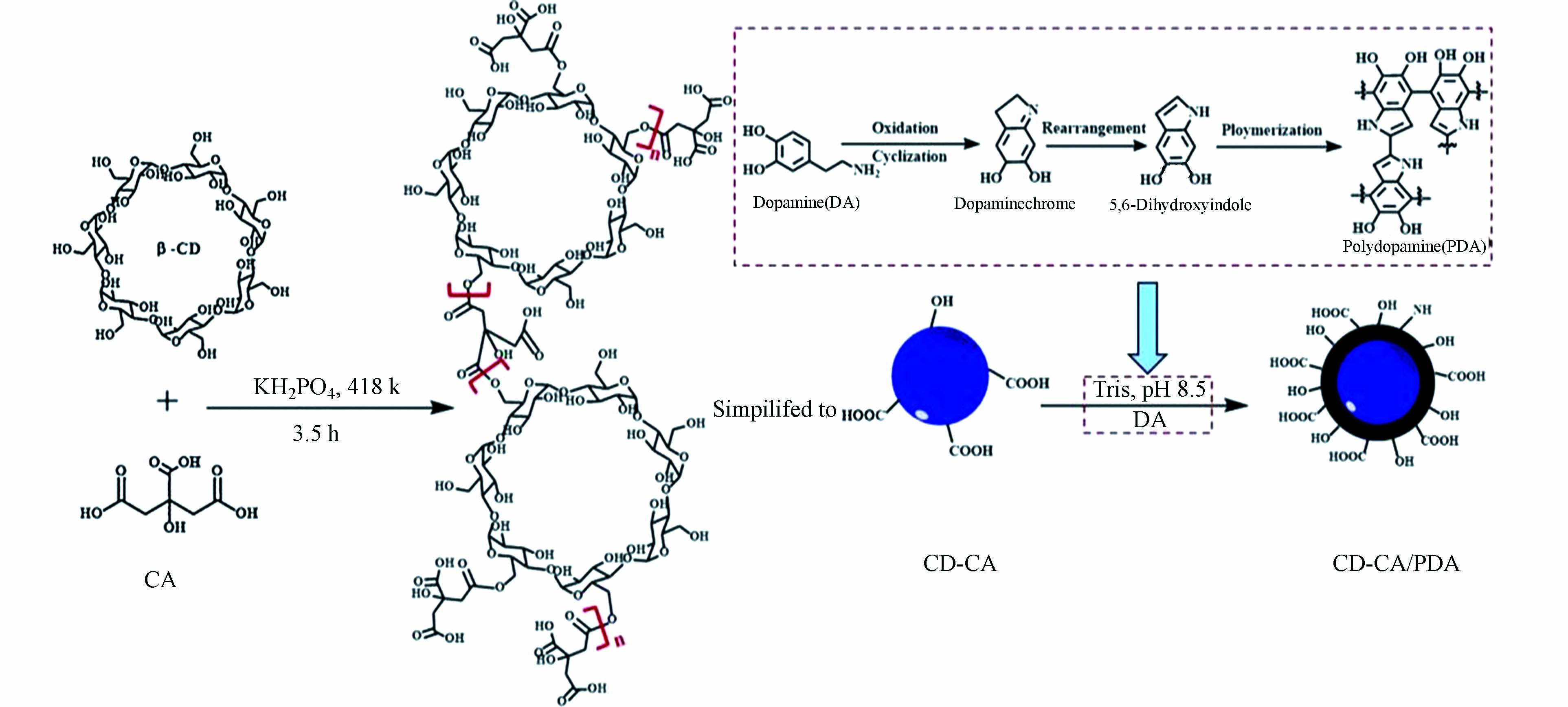
在污水中有机染料的吸附去除方面,Li等利用聚多巴胺为原料,通过将聚多巴胺包覆的CoFe2O4亚微球包裹在海藻酸钠微球中,合成了具有多孔结构和大量官能团的复合微球材料(SA@CoFe2O4-PDA),用来吸附去除亚甲基蓝(MB)、孔雀石绿(MG)及晶体紫(CV)等有机染料[44]。实验表明,SA@CoFe2O4-PDA对MB、CV及MG的最大吸附容量分别为466.60、456.52、248.78 mg∙g−1。Chen等首先将β-环糊精(β-CD)和柠檬酸(CA)酯化交联生成CD-CA,然后CD-CA与聚多巴胺结合,制备出聚多巴胺改性的环糊精聚合物(CD-CA/PDA)(如图6所示),用于MB、MG及CV等染料的吸附去除[45]。CD-CA/PDA对染料的吸附呈现出从单层吸附向多层吸附过渡的趋势,且对MB、MG及CV的吸附容量分别为582.95、1174.67、473.01 mg∙g−1。刘怡虹等将多巴胺和氧化石墨烯(GO)混合,通过聚多巴胺提供的强大粘合力,辅助GO自组装形成具有三维多孔网状结构的GO水凝胶,经水合肼对其进行还原后,生成了聚多巴胺交联的还原性石墨烯气凝胶(DA-rGA),以对MG、藏红T(ST)和罗丹明B(RhB)等阳离子染料进行吸附研究[46]。实验结果表明,DA-rGA的三维孔隙利于吸附分子在内部的快速扩散,因此对有机染料具有优异的吸附性能。Fu等通过合成的聚多巴胺微球对亚甲基蓝的吸附研究进一步证实,聚多巴胺在反应体系中会释放质子而带有负电荷,因此聚多巴胺功能材料可通过静电作用对水中阳离子染料进行吸附结合,而对阴离子染料的吸附效果就不如人意[47]。而何雪梅等利用双醛壳聚糖作为交联剂,通过在聚多巴胺修饰的羊毛织物表面上接枝季铵盐阳离子进行二次功能化,巧妙地得到了PDA/季铵盐阳离子改性的羊毛织物,用于吸附去除阴离子染料酸性大红G[48]。相较于未经处理的羊毛,羊毛织物的表面经改性后拥有更多的活性基团,大大提高了染料扩散进入纤维内部的速率。在pH值为2、染色时间为60 min等最佳实验条件下,PDA/季铵盐阳离子改性的羊毛织物对酸性大红G具有较高的吸附率。Zhan等通过将多巴胺聚合到多孔柚皮表面,成功制备了一种环保的生物吸附剂(PP-PDA)[49]。PP-PDA对水中阳离子染料表现出较高吸附能力,MB、MG和中性红(NR)的最大吸附容量分别为434.78、143.88、208.33 mg∙g−1。且PP-PDA的再生能力较强,经过20次循环后也可保持较高的吸附能力。
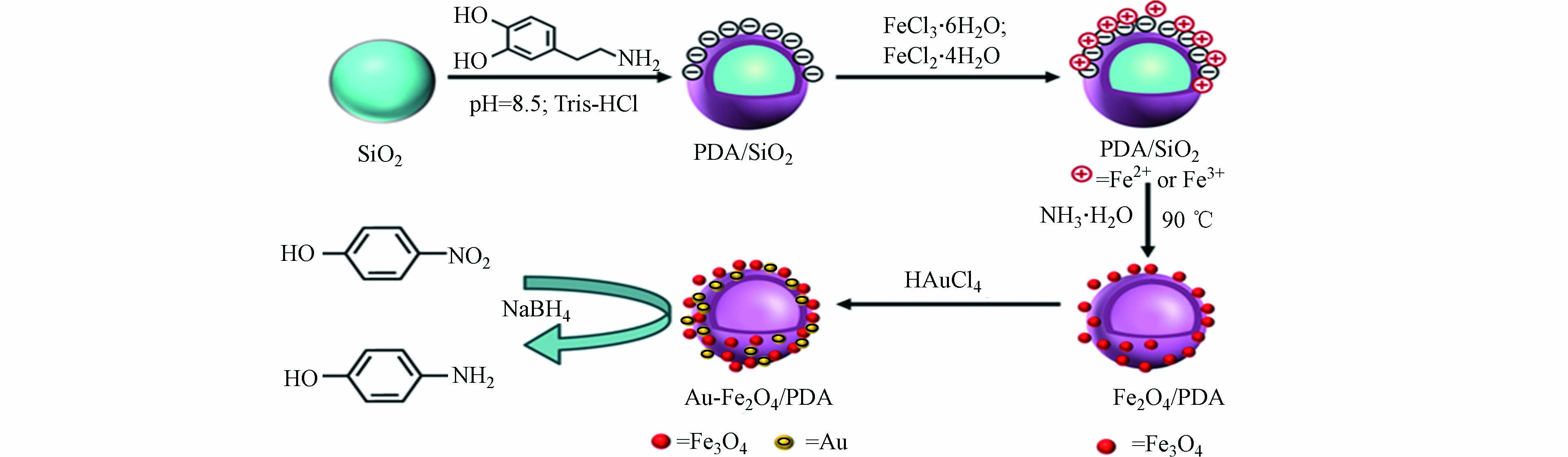
聚多巴胺功能材料也可利用催化法对水中有机染料和芳香族化合物进行去除。Ma等首次通过原位自聚反应合成了具有核壳结构的聚多巴胺包覆的CuFe2O4磁性纳米粒子(CuFe2O4@PDA MNPs),且该产品的PDA涂层厚度可通过调节制备过程中多巴胺的浓度来控制[7]。在H2O2的存在下,CuFe2O4@PDA MNPs表面吸附MB,H2O2分子被CuFe2O4@PDA激活产生的·OH可有效降解MB,最优条件下的催化降解效率可达97%以上。如图7所示,Niu等首先制备出PDA包覆的SiO2颗粒,接着利用PDA涂层作为交联剂和还原剂,在其表面接枝Fe3O4 NPs和Au NPs,合成了具有中空结构的Au-Fe3O4/PDA纳米颗粒[50]。在优化实验材料用量后,Au-Fe3O4/PDA利用金属纳米颗粒的催化性能,能够将溶液体系中的4-硝基苯酚完全还原为4-氨基苯酚,且复合材料具有较优异的可回收性。MASSARO等通过将铜离子螯合在PDA包覆的沙子表面,制备出低成本的砂载铜催化剂(Cu-PDA@Sand),对MB、CR及4-硝基苯酚进行催化还原[51]。实验表明,由于负载铜离子的存在,复合材料有效地对有机染料进行了催化降解。Cu-PDA@Sand不仅能够通过自沉淀法进行简单地回收,而且可重复使用来实现长期稳定的催化性能。而在聚多巴胺对膜材料表面的改性方面,张娇娇等利用DA作为涂层材料,聚丙烯腈(PAN)静电纺纳米纤维膜为基底材料,制备出PDA/PAN纳米纤维复合膜材料[52]。在保证较高纯水通量的前提下,经聚多巴胺涂覆后的PAN纤维膜对乳化油的截留率高达96.1%,大幅提升了处理含油废水的效果。
3. 结论与展望(Conclusions and prospects)
多巴胺在弱碱条件下可通过自聚反应生成聚多巴胺,涂覆在各种基底材料表面形成聚多巴胺功能材料。目前,社会的高速发展已经对环境造成严重破坏,尤其在水体环境方面,水污染问题日益严峻。不同于一般的水污染修复材料,聚多巴胺功能材料不仅可利用邻苯二酚和胺基等丰富的功能基团,与水中重金属离子和有机污染物通过氢键、静电相互作用、π-π 键堆积及配位螯合作用进行结合,而且可利用共价和非共价键力在聚多巴胺包覆层接枝功能分子来提升复合材料的功能特性。聚多巴胺功能材料凭借着优异的吸附和催化还原性能、简单环保的制备方法、良好的生物相容性及可二次修饰等优势,对水中的重金属离子和有机污染物具有较高的去除效率且处理后不易对水体环境造成二次污染。本篇文章首先从多巴胺的结构性质入手,介绍了目前表面化学研究中主流的聚多巴胺形成和附着机理,然后从去除水中重金属离子和有机污染物这两个方面,总结了多种聚多巴胺功能材料在水污染处理方面的具体应用。
通过上述聚多巴胺功能材料在水体污染物去除方面的应用可以看出,基底材料经聚多巴胺修饰或在聚多巴胺涂层表面进行二次修饰后,吸附和催化还原性能均有大幅度的提升。但将这些复合材料从实验室的研究运用到实际生产生活中时,却存在一些关键问题需要解决。一方面,多巴胺的自聚过程、中间产物的组成及聚多巴胺的精确分子结构尚无定论,在缩短多巴胺聚合时间和调控聚多巴胺涂层的厚度等问题上仍需要摸索实验条件。另一方面,在实际污染水样的处理中,由于较难控制的水体含氧量和流速、复杂的污染物质及过酸或过碱的水体环境等多种因素的制约,导致聚多巴胺功能材料的使用场景受到严重限制且处理效率大幅降低。因此,需要针对聚多巴胺的形成机理做进一步的研究,探究限制多巴胺自聚反应的形成过程的限制因素(例如单次反应涂层的最大厚度不会超过50 nm、酸性条件下需要特定氧化剂的帮助等)[42]。此外,还需要通过优化聚多巴胺功能材料的制备过程来提高其在各种污染水体中的稳定性和高效性,并减少复合材料对水体的二次污染。目前,研究人员正努力开发制备简单、成本较低及环境友好的新型聚多巴胺功能材料,挖掘其在医学、光学及电化学等新领域的应用潜力。未来,相信聚多巴胺功能材料凭借其优异的性能会在水污染领域中具有更广阔的前景,也定会在更广泛的领域做出更大的贡献。
-
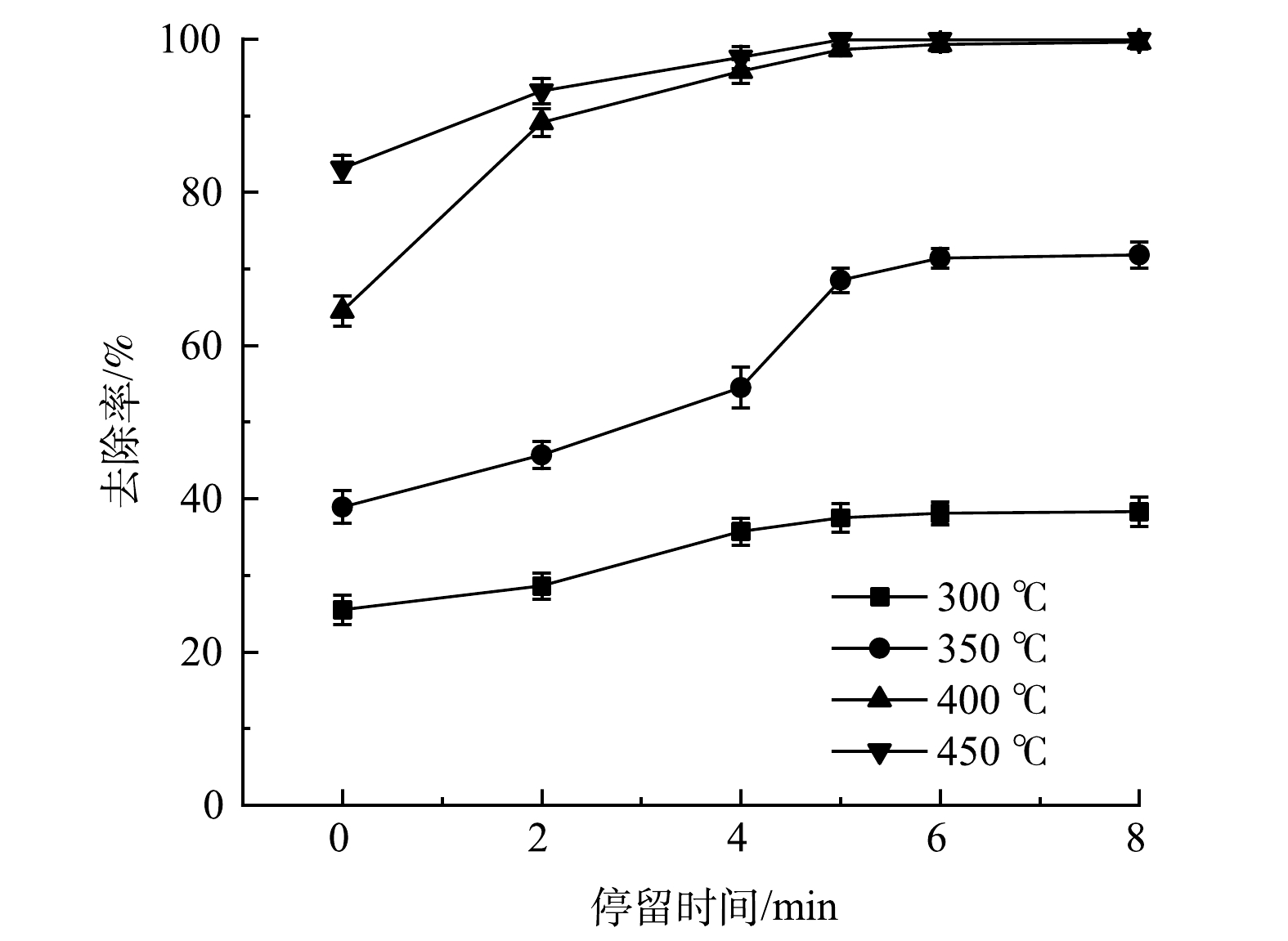
图 2 停留时间对绝缘油去除率的影响
Figure 2. Effect of residence time on the removal rate of insulating oil
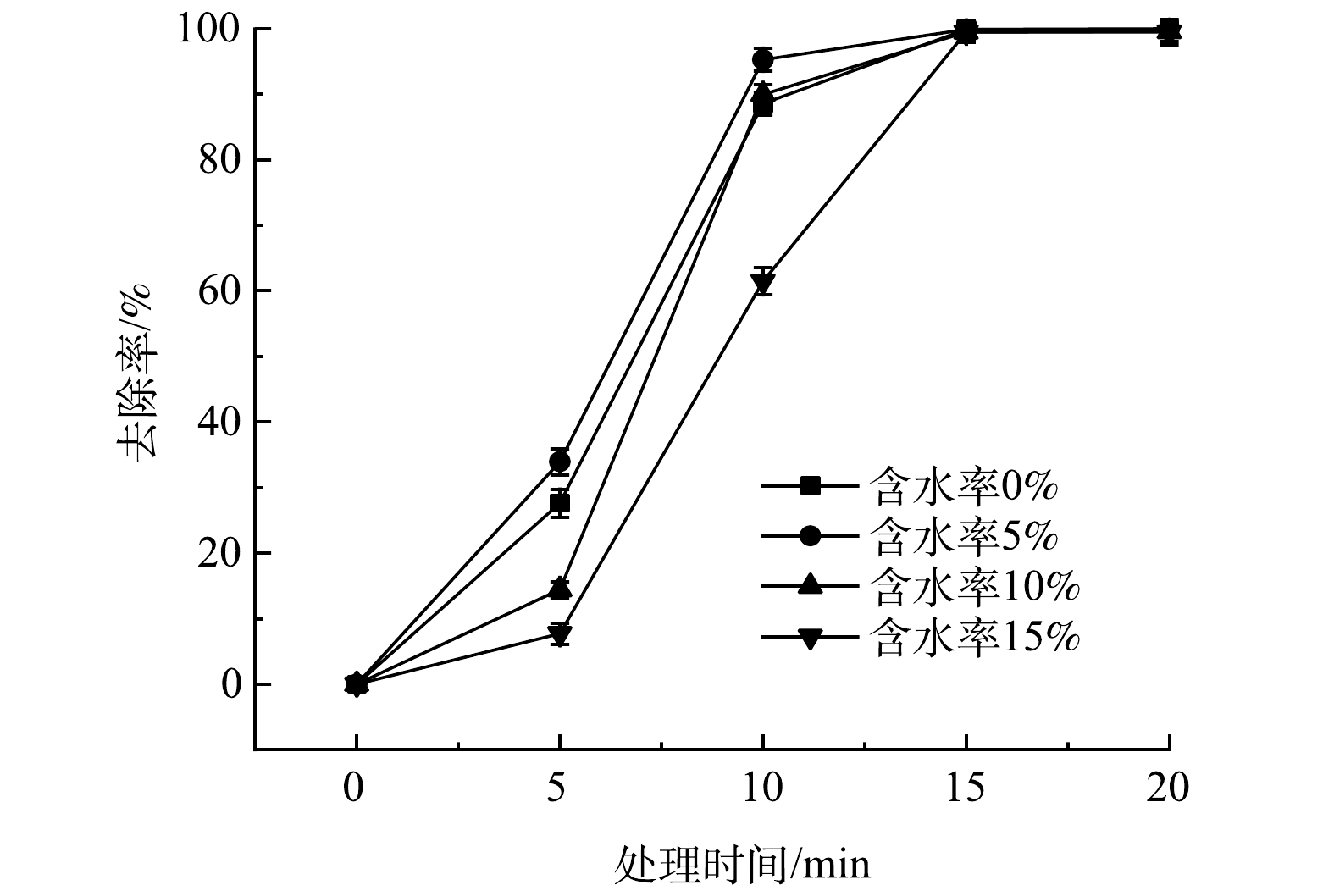
图 3 土壤含水率对绝缘油去除率的影响
Figure 3. Effect of soil moisture content on removal rate of insulating oil
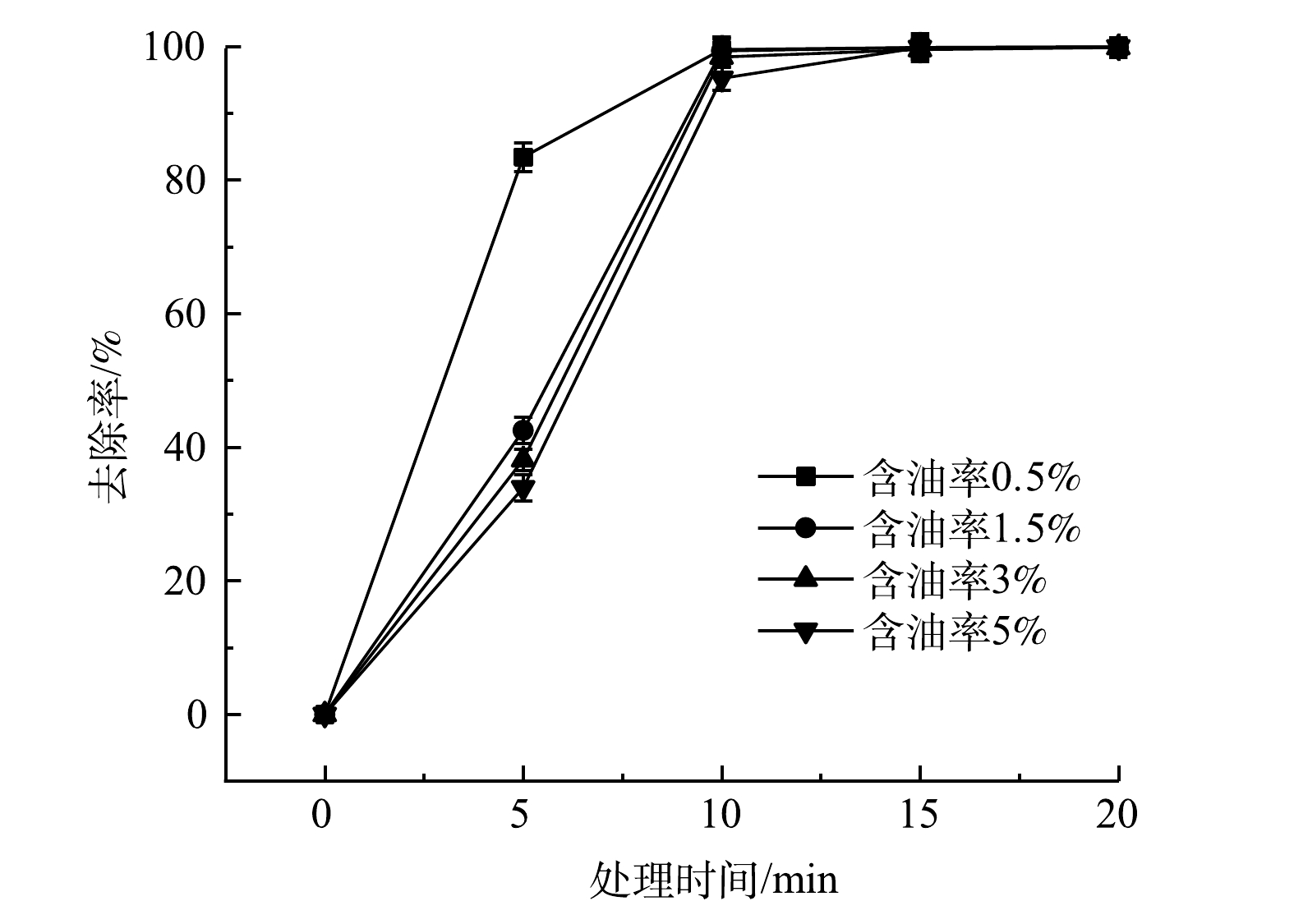
图 4 初始浓度对绝缘油去除率的影响
Figure 4. Effect of initial concentration on removal rate of insulating oil
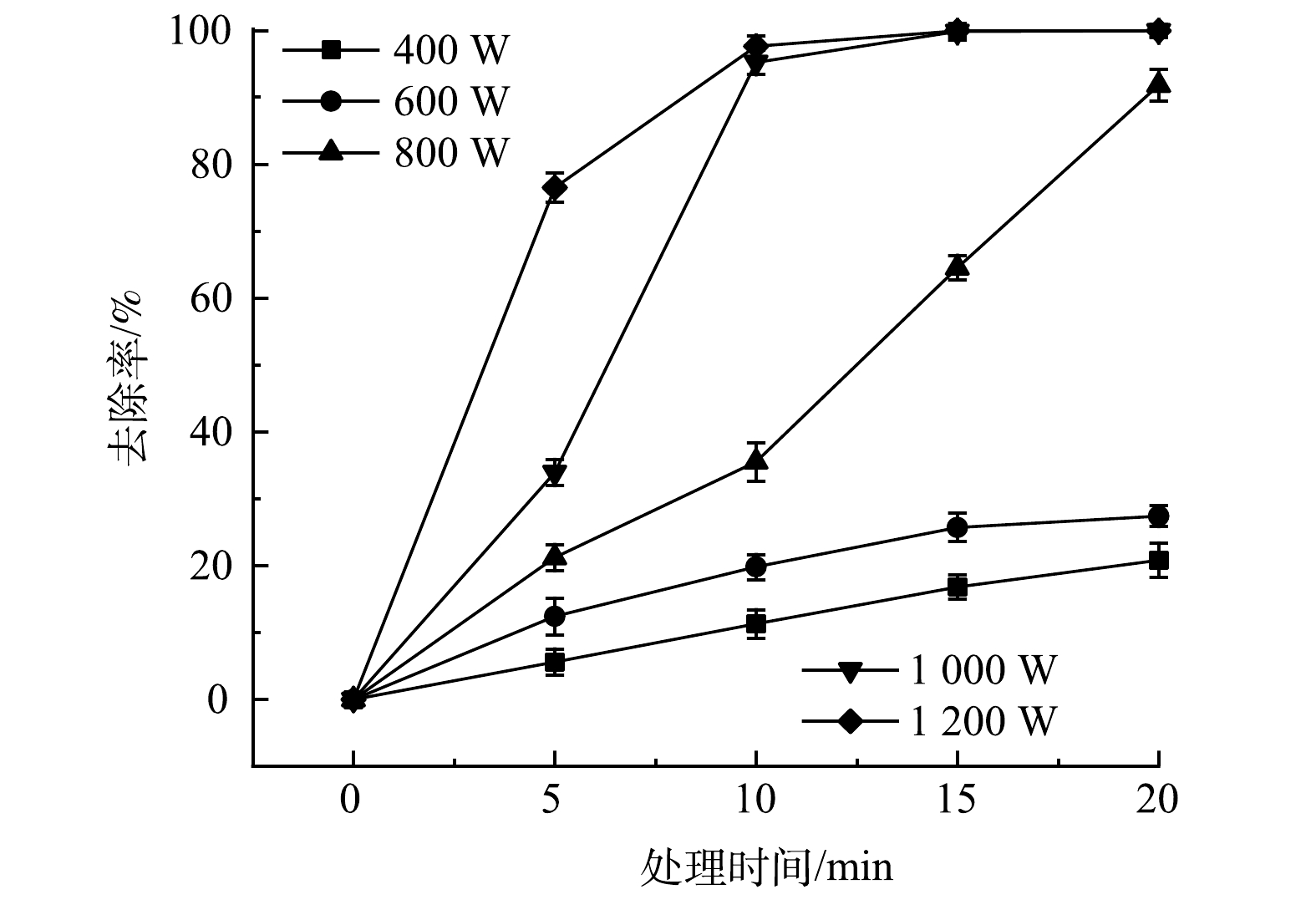
图 5 微波功率对绝缘油去除率的影响
Figure 5. Effect of microwave power on the removal rate of insulating oil
表 1 不同功率和微波时间下的能耗
Table 1. Energy consumption under different power and time
微波时间/min 能耗/kJ 400 W 600 W 800 W 1 000 W 1 200 W 5 0.033 0.050 0.067 0.083 0.100 10 0.067 0.100 0.133 0.167 0.200 15 0.100 0.150 0.200 0.250 0.300 20 0.133 0.200 0.267 0.333 0.400  下载: 导出CSV
下载: 导出CSV
表 2 微波处理前后土壤中脂肪烃质量分数
Table 2. Aliphatic content in soil before and after microwave treatment
mg·kg−1 脂肪烃类别 含油率 5% 土样 300 ℃ 处理后土样 350 ℃ 处理后土样 400 ℃ 处理后土样 450 ℃ 处理后土样 C8~C10 − − − − − C10~C12 5.7 − − − − C12~C16 5 823.3 1 620.9 284.9 1.1 0.9 C16~C21 41 596.9 20 882.8 9 187.2 251.1 96.8 C21~C34 6 075.1 2 984.9 1 641.6 317.9 18.3 注:−表示未检测出。
下载: 导出CSV
表 3 微波处理前后土壤中芳香烃质量分数
Table 3. Arene content in soil before and after microwave treatment
mg·kg−1 芳香烃类别 含油率 5% 土样 300 ℃ 处理后土样 350 ℃ 处理后土样 400 ℃ 处理后土样 450 ℃ 处理后土样 C7.6~C10.1 − − − − − C10.1~C11.7 − − − − − C11.7~C15.5 1.2 − − − − C15.5~C20.8 108.2 117.9 133.7 34.4 − C20.8~C34.01 32.9 61.8 68.3 22.3 − 注:−表示未检测出。
下载: 导出CSV
-
[1] 全国土壤污染状况调查公报(2014年4月17日)[J]. 环境教育, 2014, (6): 8-10. [2] 韩超, 陈彬, 刘阁. 颗粒污染物对变压器油理化性能的影响[J]. 石油学报(石油化工), 2016, 32(6): 1156-1163. [3] 刘佳. 废变压器油中污染物及环境风险评价研究[D]. 保定: 华北电力大学, 2015. [4] 郭涛涛, 王达达, 高阔, 等. X射线对变压器油的影响研究[J]. 核电子学与探测技术, 2012, 32(12): 1437-1440. doi: 10.3969/j.issn.0258-0934.2012.12.023 [5] 侯朝鹏, 李永丹, 夏国富, 等. 典型单环和双环芳烃加氢热力学分析[J]. 石油化工, 2013, 42(6): 625-631. doi: 10.3969/j.issn.1000-8144.2013.06.007 [6] 孟祥帅, 陈鸿汉, 郑从奇, 等. 焦化厂不同污染源作用下土壤PAHs污染特征[J]. 中国环境科学, 2020, 40(11): 4857-4864. doi: 10.3969/j.issn.1000-6923.2020.11.026 [7] ENUNEKU A, OGBEIDE O, OKPARA B, et al. Ingestion and dermal cancer risk via exposure to polycyclic aromatic hydrocarbons (PAHs) contaminated soils in an oil producing community, Niger Delta, Nigeria[J]. Environmental Toxicology and Chemistry, 2020, 40(1): 261-271. [8] 王国秉. 关于俄罗斯萨扬·舒申斯克水电站事故的思考[J]. 山西水利科技, 2010(2): 1-5. doi: 10.3969/j.issn.1006-8139.2010.02.001 [9] 赵玉霞, 杨珂. 石油污染土壤修复技术研究综述[J]. 环境科技, 2009, 22(S1): 60-63. [10] 邢汉君, 蒋俊, 李晶, 等. 有机氯农药污染土壤异位热脱附修复研究[J]. 湖南农业科学, 2019(11): 62-64. [11] 赵中华, 李晓东, 陈彤, 等. 多氯联苯污染土壤热脱附研究综述[J]. 生态毒理学报, 2016, 11(2): 61-68. [12] 刘珑, 王殿生, 曾秋孙, 等. 微波修复石油污染土壤升温特性影响因素的实验研究[J]. 环境工程学报, 2011, 5(4): 898-902. [13] 桑义敏, 艾贤军, 马绍芳, 等. 基于超声波-微波耦合效应的石油烃类污染土壤的热脱附规律与参数优化[J]. 环境工程学报, 2019, 13(10): 2311-2319. doi: 10.12030/j.cjee.201905113 [14] 刘珑, 王殿生, 曾秋孙, 等. 微波修复原油污染土壤的均匀设计实验[J]. 环境工程学报, 2012, 6(6): 2034-2038. [15] LUO H, WANG H, KONG L, et al. Insights into oil recovery, soil rehabilitation and low temperature behaviors of microwave-assisted petroleum-contaminated soil remediation[J]. Journal of Hazardous Materials, 2019, 377: 341-348. doi: 10.1016/j.jhazmat.2019.05.092 [16] ELDOS H I, ASHFAQ M Y, AL-GHOUTI M A. Rapid assessment of the impact of microwave heating coupled with UV-C radiation on the degradation of PAHs from contaminated soil using FTIR and multivariate analysis[J]. Arabian Journal of Chemistry, 2020, 13(11): 7609-7625. doi: 10.1016/j.arabjc.2020.08.031 [17] DI P K, CHANG D P. Investigation of polychlorinated biphenyl removal from contaminated soil using microwave-generated steam[J]. Journal of the Air & Waste Management Association, 2001, 51(4): 482-488. [18] KAWALA Z A, ATAMANCZUK T. Microwave-enhanced thermal decontamination of soil[J]. Environmental Science & Technology, 1998, 32(17): 2602-2607. [19] 王贝贝, 朱湖地, 胡丽, 等. 硝基酚, 六氯苯污染土壤的微波修复[J]. 环境化学, 2013, 32(8): 1560-1565. doi: 10.7524/j.issn.0254-6108.2013.08.022 [20] TAI H S, JOU C J. Immobilization of chromium-contaminated soil by means of microwave energy[J]. Journal of Hazardous Materials, 1999, 65(3): 267-275. doi: 10.1016/S0304-3894(98)00274-X [21] 吴风光. 微波辐射修复Cr(Ⅵ)污染土壤的研究[D]. 长沙: 湖南大学, 2013. [22] 刘娜, 赵维, 赵浩, 等. 微波修复氯丹污染土壤中氯丹降解的影响因素研究[J]. 环境污染与防治, 2012, 34(5): 43-47. doi: 10.3969/j.issn.1001-3865.2012.05.009 [23] ABRAMOVITCH R A, HUANG B Z, DAVIS M, et al. Decomposition of PCB's and other polychlorinated aromatics in soil using microwave energy[J]. Chemosphere, 1998, 37(8): 1427-1436. doi: 10.1016/S0045-6535(98)00133-7 [24] ABRAMOVITCH R A, HUANG B Z, ABRAMOVITCH D A, et al. Decomposition of PCBs in soil using microwave energy[J]. Chemosphere, 1999, 38(10): 2227-2236. doi: 10.1016/S0045-6535(98)00441-X [25] 田勐, 袁松虎, 陆晓华. 微波辐射在二氧化锰诱导下对六氯苯污染土壤的修复研究[J]. 环境保护科学, 2006, 32(2): 49-52. doi: 10.3969/j.issn.1004-6216.2006.02.016 [26] 孙磊, 蒋新, 周健民, 等. 五氯酚污染土壤的热修复初探[J]. 土壤学报, 2004, 41(3): 462-465. doi: 10.3321/j.issn:0564-3929.2004.03.021 [27] 周翠红, 王世晗, 周怡. 响应曲面法优化微波修复二甲苯污染土壤工艺参数[J]. 环境污染与防治, 2020, 42(3): 259-263. [28] LIU X T, ZHANG Q, ZHANG G X, et al. Application of microwave irradiation in the removal of polychlorinated biphenyls from soil contaminated by capacitor oil[J]. Chemosphere, 2008, 72(11): 1655-1658. doi: 10.1016/j.chemosphere.2008.05.030 [29] PETARCA L, CIONI B. Petroleum products removal from contaminated soils using microwave heating[J]. Chemical Engineering Transactions, 2011, 24: 1033-1038. [30] 中华人民共和国生态环境部. 土壤 石油类的测定 红外分光光度法: HJ 1051-2019[S]. 北京: 中国环境出版社, 2019. [31] 中华人民共和国生态环境部. 土壤和沉积物 石油烃(C10-C40)的测定 气相色谱法: HJ 1021-2019[S]. 北京: 中国环境出版社, 2019. [32] FALCIGLIA P P, GIUSTRA M G, VAGLIASINDI F G A. Low-temperature thermal desorption of diesel polluted soil: Influence of temperature and soil texture on contaminant removal kinetics[J]. Journal of Hazardous Materials, 2011, 185(1): 392-400. doi: 10.1016/j.jhazmat.2010.09.046 [33] GEORGE C E, LIGHTSEY G R, JUN I, et al. Soil decontamination via microwave and radio frequency co-volatilization[J]. Environmental Progress & Sustainable Energy, 2010, 11(3): 216-219. [34] LIU X T, YU G. Combined effect of microwave and activated carbon on the remediation of polychlorinated biphenyl-contaminated soil[J]. Chemosphere, 2006, 63(2): 228-235. doi: 10.1016/j.chemosphere.2005.08.030 [35] 齐红媛. 微波处理被原油和成品油污染土壤的规律研究[D]. 西安: 西安石油大学, 2013. [36] 中华人民共和国生态环境部. 土壤环境质量 建设用地土壤污染风险管控标准(试行): GB 36600-2018[S]. 北京: 中国环境科学出版社, 2018. [37] GILOT P, HOWARD J B, PETERS W A. Evaporation phenomena during thermal decontamination of soils[J]. Environmental Science and Technology, 1997, 31(2): 461-466. doi: 10.1021/es960293p [38] 李大伟. 石油污染土壤的碳材料增强微波热修复研究[D]. 大连: 大连理工大学, 2008. -

 点击查看大图
点击查看大图

计量
- 文章访问数: 4151
- HTML全文浏览数: 4151
- PDF下载数: 40
- 施引文献: 0


