





 下载:
下载:







-
随着全球气候变暖和江河、湖泊等水体富营养化程度加剧,蓝藻水华现象日益严重[1]。蓝藻暴发会导致水体溶解氧(DO)含量急剧降低、水生生物大量死亡,其在代谢过程中释放的具有极强肝毒性和遗传毒性的藻毒素(MCs)[2],也会对人类的生命健康造成严重威胁。根据理化性质及适用条件,传统蓝藻水华治理方法可分为物理法、化学法和生物法。物理法可在短期内大幅削减蓝藻密度,但存在二次污染、滤池堵塞及成本较高等不足[3];化学法灭藻效率高,但难以避免IOMs释放、DBPs产生及出水金属离子含量增加等环境风险[4-5];生物法操作简单、对环境影响较小,但见效周期长,且易受气候、温度等环境因素影响[6]。因此,如何高效、安全地治理蓝藻水华,已成为国内外学者关注和研究的热点。
AOPs是一种通过诱发链式反应产生·OH或
SO−4⋅ 等自由基[7],从而降解有机污染物的水处理技术,具有操作简便、反应条件温和等优点。但目前的研究主要集中于产生·OH的AOPs,有关产生硫酸根自由基(SO−4⋅ )的AOPs研究相对甚少。SO−4⋅ 氧化还原电位为2.50~3.10 V,高于·OH(2.80 V)和O3(2.07 V)[8],并且同·OH相比,SO−4⋅ 具有选择性强、受pH影响小以及半衰期更长等优点。基于AOPs产生的SO−4⋅ ,可以通过UV辐照、过渡金属离子和氧化剂协同等方式催化PMS或过硫酸盐(PS)来实现[9]。本研究以铜绿微囊藻为研究对象,探究了在FeSO4-PMS体系中FeSO4投加量、pH、HPO42−及NO−3 对藻细胞去除效果的影响,同时对藻液中溶解性有机碳(DOC)含量和UV254进行了测定;结合三维荧光光谱(EEM)、Zeta电位以及扫描电镜(SEM)等分析表征方法对藻细胞的去除机理进行了探究,以期为FeSO4-PMS治理蓝藻水华提供参考。 -
实验藻种为铜绿微囊藻,FACHB526,购自中国科学院水生生物研究所;培养基为BG11培养基[10];过硫酸氢钾(PMS) 为分析纯,麦克林生化科技有限公司;七水合硫酸亚铁(FeSO4·7H2O)、五水合硫代硫酸钠(Na2S2O3·5H2O)、硫酸(H2SO4)、氢氧化钠(NaOH)、磷酸氢二钠(Na2HPO4)、硝酸钠(NaNO3)、戊二醛、无水乙醇、叔丁醇、甲醇为分析纯,国药集团化学试剂有限公司;磷酸盐(PBS)缓冲液,北京索莱宝科技有限公司。实验用水均为超纯水。
-
AR124CN型电子天平,上海奥豪斯仪器有限公司;S400-K型多参数测定仪,上海梅特勒-托利多仪器有限公司;TDZ4-WS型台式低速离心机,湖南湘仪实验室仪器开发有限公司;BXM-75型立式压力蒸汽灭菌器,上海博迅医疗生物仪器股份有限公司;Smart2pure12型超纯水仪,美国赛默飞世尔公司;台式低速离心机,湖南湘仪实验室仪器开发有限公司;UV-8000型紫外可见分光光度计,上海元析仪器有限公司;六联数显磁力搅拌器,宁波市鄞州群安实验仪器有限公司;F-320型荧光分光光度计,天津港东科技股份有限公司;SCIENTZ-18N型冷冻干燥机,宁波新芝生物科技股份有限公司;TOC-VSH型有机碳测定仪,岛津国际贸易有限公司;Zeta电位分析仪,美国布鲁克海文仪器公司;S-3400N型扫描电子显微镜,日本日立公司。
-
取对数期的铜绿微囊藻液,利用超纯水依次稀释至原体积的1、1.33、2、4、6、8和10倍,采用血球计数板在400倍双目显微镜下进行藻细胞计数。藻细胞浓度与藻液在680 nm处的光吸收(OD680)有良好的线性关系[11],如图1所示。藻细胞浓度和藻细胞去除率分别采用式(1)和式(2)计算。
式中:y为藻液在680 nm处的吸光度值;x为藻细胞浓度,106 细胞数·mL−1。
式中:R为藻细胞去除率,%;x0为初始藻细胞浓度,106 细胞数·mL−1;xt为处理不同时刻的藻细胞浓度,106 细胞数·mL−1。
-
1)藻液的稀释和储备液的配置。取对数期的铜绿微囊藻液,使用超纯水稀释混匀,藻细胞终浓度为106 细胞数·mL−1,初始OD680为0.144,调节pH至7.5±0.1[12]。称取2.78 g FeSO4·7H2O、1.52 g PMS、2.48 g Na2S2O3·5H2O、1.419 g Na2HPO4和0.849 9 g NaNO3分别溶于超纯水并定容至100 mL,配制成浓度均为0.1 mol·L−1的相应储备液。
2)考察FeSO4投加量、pH、HPO42−和
NO−3 浓度对FeSO4-PMS除藻效果的影响。分别取1 L稀释后的藻液于6个烧杯中,PMS投加量均为0.1 mmol·L−1,FeSO4投加量分别为0、0.025、0.05、0.075、0.10和0.15 mmol·L−1,300 r·min−1下快速搅拌2 min后静置,在10、20、30、40、50、60、90和120 min处取样,并立即用Na2S2O3进行淬灭反应[13],依据式(2)计算藻细胞去除率。pH对FeSO4-PMS除藻效果的影响:分别取1 L稀释后的藻液于5个烧杯中,调节pH分别为6.0、7.0、8.0、9.0和10.0,FeSO4和PMS投加量均为0.1 mmol·L−1,后续操作同上。HPO42−和NO−3 浓度对FeSO4-PMS除藻效果的影响:分别取1 L稀释后的藻液于9个烧杯中,烧杯编号为1~9,1号为对照(不添加Na2HPO4和NaNO3),2~5号分别添加0.5、1.0、1.5和2.0 mmol·L−1 Na2HPO4,6~9号分别添加2.5、5.0、7.5和10 mmol·L−1 NaNO3,FeSO4和PMS投加量均为0.1 mmol·L−1,后续操作同上。 -
OD680、UV254和叶绿素a(Chl-a)采用分光光度法测定;溶解性有机碳(DOC)采用有机碳测定仪测定;Zeta电位采用Zeta电位分析仪测定。利用荧光分光光度计对具有荧光特性的藻源性有机质(AOMs)进行表征,仪器参数设置:激发波长(Ex)为220~450 nm,发射波长(Em)为200~500 nm,激发间隔和发射间隔均为5 nm,扫描速度为2 400 nm·min−1,以超纯水为空白样,在数据处理中用以扣除拉曼散射。利用扫描电镜进行藻细胞形态观察,样品处理步骤为:以4 000 r·min−1离心10 min收集铜绿微囊藻细胞;在4 ℃下利用2.5%戊二醛固定藻细胞4 h;利用pH为7.0的PBS缓冲液反复清洗样品3次;分别采用30%、50%、70%、90%和100%的乙醇溶液依次对样品进行脱水处理;利用叔丁醇溶液置换乙醇溶液;在−40 ℃下冷冻干燥12 h后喷金;利用扫描电镜在15.0 kV下拍照。
-
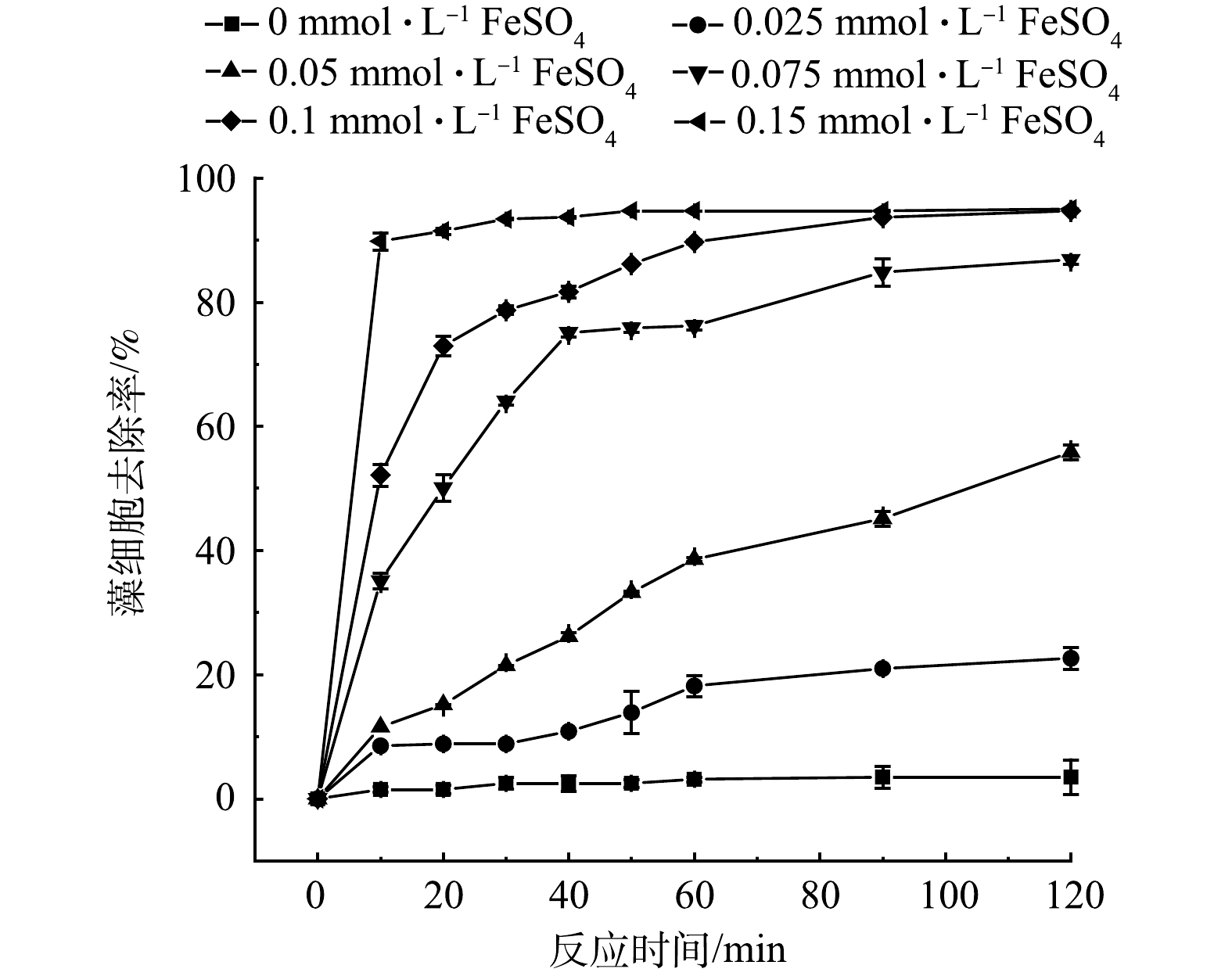
1) FeSO4投加量的影响。由图2可知,单独投加PMS时,藻细胞浓度无明显变化,藻细胞去除率仅为3.49%;而投加FeSO4后,随着反应进行,藻细胞去除率与反应时间的关系符合为二级反应动力学模型,藻细胞去除率在前30 min内急剧升高,而后趋于平稳。当FeSO4投加量依次增加为0.025、0.05、0.075、0.1和0.15 mmol·L−1时,藻细胞去除率分别提高至22.64%、55.81%、86.82%、94.66%和95.00%。
有研究[14]表明,带负电荷的AOMs可以提升藻细胞表面的负电性,藻细胞稳定性得以提高。常温下PMS极其稳定,对AOMs及藻细胞氧化效果较差,而投加FeSO4后,藻细胞去除率大幅度上升。这是因为Fe2+催化PMS分解产生了大量的
SO−4⋅ 和·OH用于氧化带负电的AOMs[15],藻细胞稳定性被破坏(式(3)和式(4))。此外,协同过程产生了大量的原位Fe3+对显负电性的藻细胞具有较好的絮凝效果,通过吸附架桥作用使藻细胞发生团聚而被去除[16]。FeSO4显著提高了PMS去除藻细胞的效果,而当FeSO4投加量增加至0.15 mmol·L−1时,相较FeSO4投加量为0.1 mmol·L−1,藻细胞去除率仅提高了0.34%。这是因为过量Fe2+对SO−4⋅ 和·OH的竞争反应导致自由基被清除(式(5))[17]。为分析藻细胞去除过程中氧化、絮凝阶段的贡献率,采用甲醇(MeOH)作为
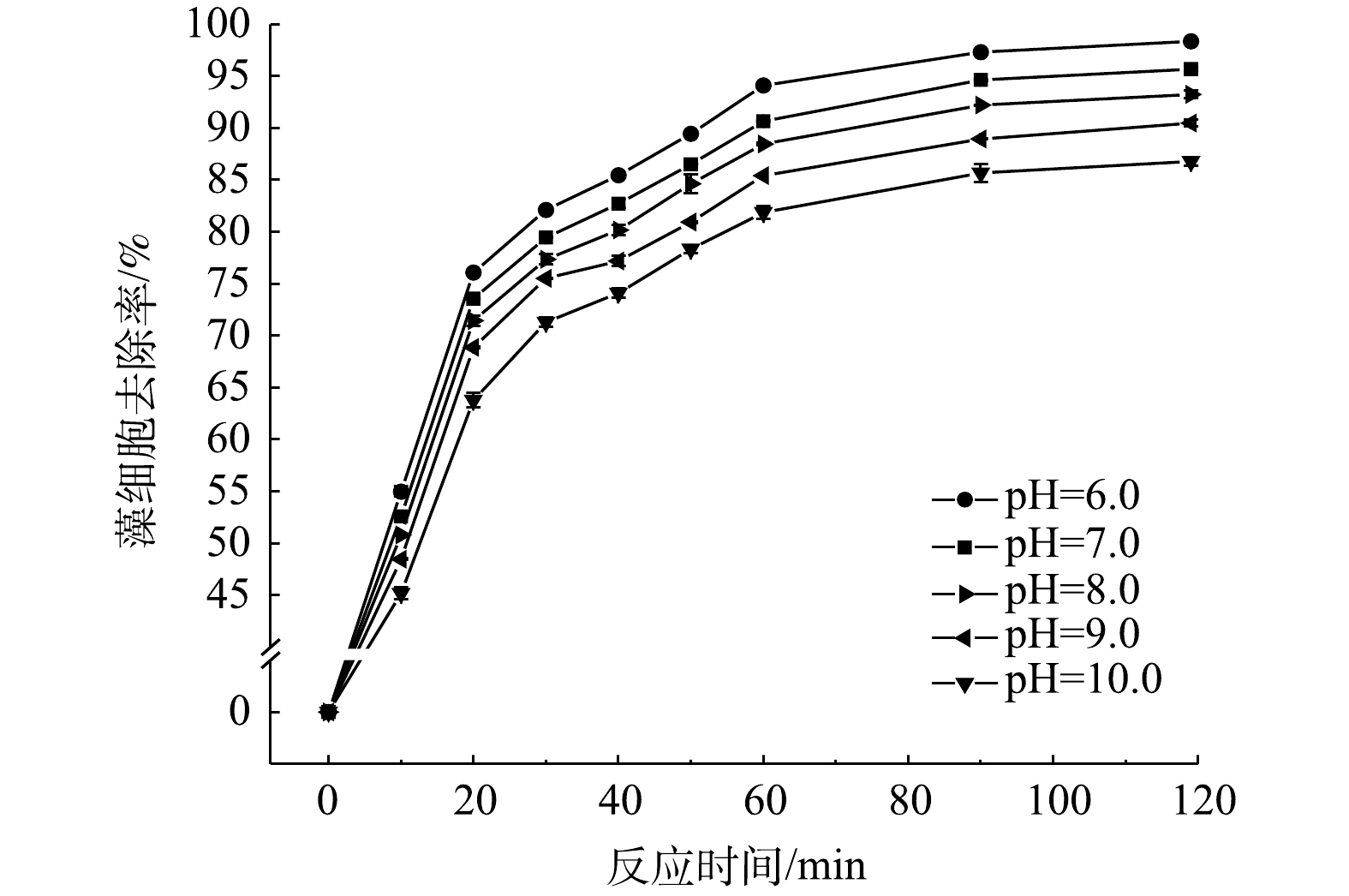
SO−4⋅ 和·OH的掩蔽剂。当未添加MeOH掩蔽剂时,在FeSO4和PMS投加量均为0.1 mmol·L−1时,由于氧化和絮凝作用,藻细胞去除率为94.66%;当添加10 mmol·L−1 MeOH掩蔽剂后,絮凝作用下,藻细胞的去除率为65.79%。通过计算分析可知,藻细胞去除的氧化、絮凝阶段的贡献率分别为30.50%和69.50%。2)初始pH的影响。当FeSO4和PMS投加量均为0.1 mmol·L−1时,不同pH条件下,藻细胞去除率随时间变化如图3所示。由图3可知,当pH为6.0~10.0时,藻细胞去除率均保持较高水平,随着pH升高,藻细胞去除率呈现缓慢降低的趋势。当pH分别为6.0、7.0、8.0、9.0和10.0时,在反应时间30 min时藻细胞去除率增幅最大,分别为81.12%、79.46%、78.37%、77.52%和74.32%;当反应时间为120 min时,藻细胞去除率趋于稳定,分别为97.37%、95.69%、94.25%、92.48%和89.79%。在碱性条件下,藻细胞去除率有所降低。这是因为:一方面Fe2+与OH−络合生成了Fe(OH)2沉淀,使参与协同反应的Fe2+减少;另一方面,FeSO4-PMS体系中自由基种类主要为
SO−4⋅ ,随着pH升高,·OH逐渐增多、SO−4⋅ 转化比例逐渐降低[18],由于·OH半衰期小于1 μs,远低于SO−4⋅ (4 s)[19],藻细胞与SO2−4⋅ 接触并发生反应的概率得以削弱。3)无机离子的影响。不同HPO42−浓度下,藻细胞去除率随时间的变化如图4所示。随着HPO42−浓度的升高,藻细胞去除率呈现出连续降低的趋势。当HPO42−浓度为0 mmol·L−1时,藻细胞去除率达到了94.20%;当HPO42−依次增加至0.5、1.0、1.5和2.0 mmol·L−1,藻细胞去除率分别降低至90.02%、85.45%、80.10%和74.64%。这是因为HPO42−是
SO−4⋅ 和·OH的清除剂[20],HPO42−通过竞争机制消耗了体系中产生的SO−4⋅ 和·OH(式(6)和式(7));此外,HPO42−会导致水溶液pH升高,碱性增强,从而阻碍SO−4⋅ 的形成,并且,HPO42−水解产生的H2PO4−,也会与溶液中游离的Fe2+和Fe3+发生络合形成难溶的磷酸盐复合物(式(8))[21],从而阻碍协同反应的进行。不同
NO−3 浓度下,藻细胞去除率随时间变化如图5所示。由图5可知,藻细胞去除率随着NO−3 浓度的升高而降低。当NO−3 浓度依次为0、2.5、5.0、7.5和10.0 mmol·L−1时,藻细胞去除率分别为94.20%、88.53%、85.46%、80.39%和76.80%。其原因为:一方面NO−3 具有氧化性,Fe2+被氧化形成Fe3+;另一方面NO−3 消耗了SO−4⋅ ,被SO−4⋅ 氧化形成了活性较低的NO3·(式(9))[22]。而NO3·氧化能力偏弱[23],不足以降解AOMs。 -
UV254反映了AOMs中含不饱和碳键及芳香结构有机物的含量[24]。图6为在不同FeSO4投加量下反应120 min后DOC和UV254的去除效果。随着FeSO4投加量的增加,体系中DOC含量和UV254持续降低。当FeSO4投加量为0.025 mmol·L−1时,DOC含量为6.35 mg·L−1,DOC去除率为11.56%, UV254为0.167,UV254去除率仅为2.01%;继续增加FeSO4投加量分别至0.05、0.075、0.1和0.15 mmol·L−1时,DOC去除率分别为40.56%、47.36%、58.92%和61.00%,UV254去除率分别达到了13.01%、24.15%、40.83%和52.67%。
FeSO4-PMS产生的
SO−4⋅ 和·OH,对蛋白质类及含不饱和碳键的AOMs具有更好的降解效果,因此,随着FeSO4投加量的增加, DOC和UV254去除率持续升高[25]。此外,DOC含量下降表明藻细胞未出现大规模的裂解,因为藻细胞的破裂会导致大量IOMs释放、DOC浓度急剧上升[26]。 -
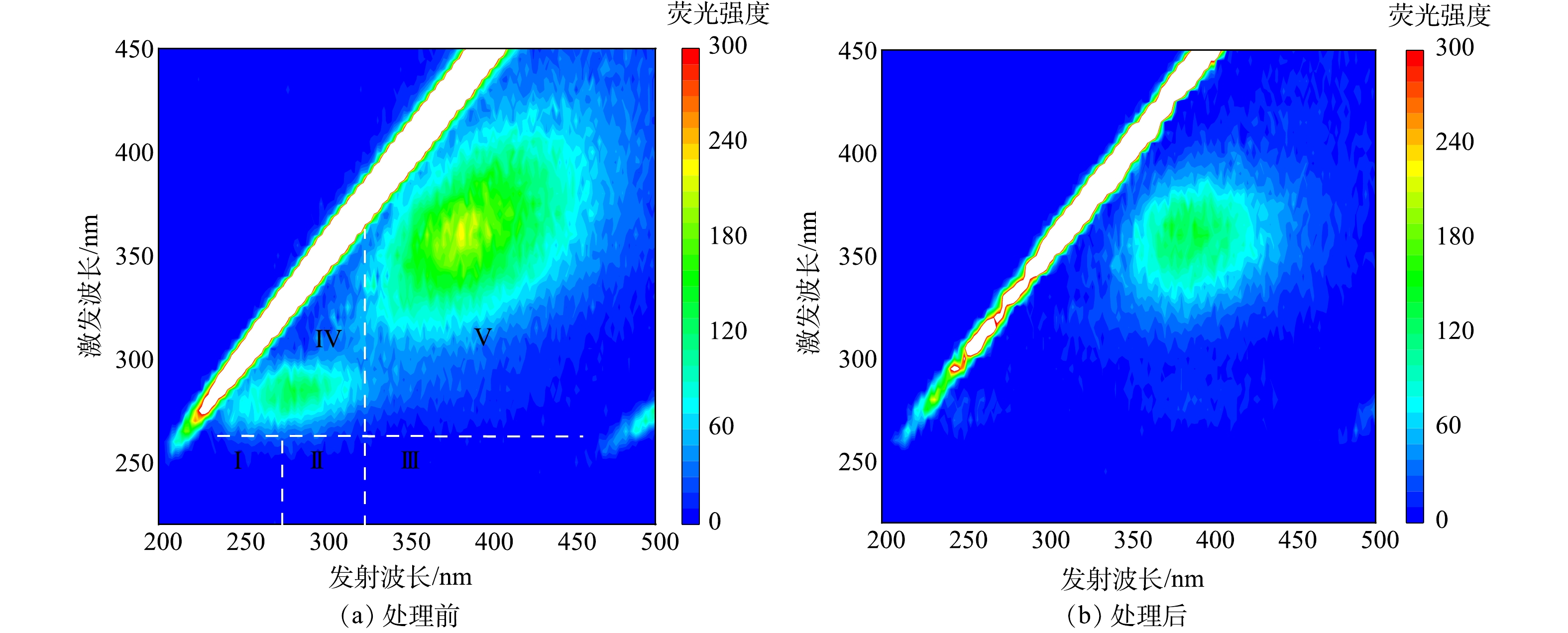
荧光光谱技术可以实时鉴别AOMs组成及变化[27],具有灵敏度高、选择性好等优点。CHEN等[28]将整个荧光光谱划分为5个区域:区域Ⅰ和Ⅱ代表芳香结构的蛋白质,区域Ⅲ代表富里酸类物质,区域Ⅳ代表溶解性微生物代谢类物质,区域Ⅴ代表腐殖酸类物质。
在FeSO4和PMS投加量均为0.1 mmol·L−1的条件下,水样经FeSO4-PMS处理前后的三维荧光光谱分别如图7(a)和图7(b)所示。原水以腐殖酸类物质和溶解性微生物代谢类物质为主,同时含有少量的芳香结构的蛋白质。经FeSO4-PMS协同氧化后,各区域荧光强度均出现不同程度降低,其中溶解性微生物代谢类物质和腐殖酸类代谢物质的荧光强度变化最为明显;区域Ⅰ、Ⅱ和Ⅳ荧光强度急剧减弱甚至消失,表明芳香结构的蛋白质类物质和微生物代谢类AOMs极易被去除,因为蛋白质类物质和微生物代谢类物质具有极强的亲水性以及较差的沉降性,极易被强氧化性的
SO−4⋅ 和·OH氧化[29] 。 -
Zeta电位反映藻细胞表面所带电荷大小,其净值越高,藻细胞间的静电斥力越大,藻细胞越稳定[30]。为进一步探究FeSO4-PMS去除藻细胞的机理,分析了在不同FeSO4投加量下,Zeta电位净值及藻细胞、Chl-a去除率的变化。由图8可知,原藻液含有大量带负电荷的AOMs[31],Zeta电位净值较高,为28.44 mV。当FeSO4浓度为0~0.1 mmol·L−1时,随着体系中FeSO4投加量的增加,Zeta电位净值持续降低,藻细胞和Chl-a去除率显著提高。当FeSO4投加量依次为0.025、0.05、0.075、0.10 mmol·L−1时,Zeta电位净值分别为22.01、10.57、7.97和0.33 mV,Chl-a去除率分别为29.42%、60.18%、89.05%和94.92%;当继续增加FeSO4投加量为0.15 mmol·L−1时,Zeta电位净值略有升高,为1.29 mV。这是因为:一方面,带负电荷的AOMs被
SO−4⋅ 和·OH有效降解;另一方面,经氧化形成的原位Fe3+可以有效中和藻细胞表面的负电荷,Zeta电位净值降低[32]。然而当FeSO4浓度高于0.1 mmol·L−1时,氧化过于剧烈,导致部分藻细胞裂解、带负电荷的IOMs释放,因此Zeta电位净值上升。FeSO4-PMS去除藻细胞的机理主要为氧化和絮凝。强氧化性的SO−4⋅ 和·OH可以有效降解水中的Chl-a和带负电荷的AOMs[33],使得藻细胞稳定性降低。此外,经氧化原位形成的Fe3+通过吸附架桥、电位中和作用与藻细胞结合,使其凝聚、沉降[34]。 -
使用高倍扫描电镜对铜绿微囊藻细胞的表面形态进行了分析。由图9(a)和图9(b)可知,正常的藻细胞为椭球形,形状饱满且通体光滑,尺寸为2.0~3.5 μm;而经FeSO4-PMS协同处理后,藻细胞被致密的絮体包裹、粘结,表面出现少量褶皱,整体结构依旧完整(图9(c)、图9(d))。其原因可能是:一方面
SO−4⋅ 和·OH可以有效穿过铜绿微囊藻细胞的细胞膜且主要作用于DNA,攻击嘌呤、嘧啶碱基,导致DNA链发生非选择性断裂[35],进而导致藻细胞失活;另一方面,经氧化原位形成的Fe3+对悬浮的藻细胞具有更好的絮凝效果[36],通过吸附架桥、电位中和作用,使藻细胞聚集形成块状絮体沉淀得以去除。 -
1)综合考虑藻细胞去除效果,FeSO4和PMS的最佳投加量均确定为0.1 mmol·L−1。在此条件下,反应120 min时的藻细胞、DOC和UV254的去除率分别为94.66%、58.92和41.52%,Zeta电位净值降至0.33 mV;藻细胞去除的氧化和絮凝的贡献率分别为30.50%和69.50%。
2)碱性环境、HPO42−和
NO−3 对去除藻细胞具有抑制作用。当pH为6.0~10.0时,藻细胞去除率从97.37%降低至89.79%;HPO42−和NO−3 通过竞争机制消耗了SO−4⋅ ,当HPO42−浓度为0~2.0 mmol·L−1时,藻细胞去除率由94.20%降至74.64%;当NO−3 浓度为0~10 mmol·L−1时,藻细胞去除率由94.20%降至76.80%。3)当FeSO4浓度为0~0.1 mmol·L−1时,Zeta电位净值和藻细胞、Chl-a去除率呈负相关,藻细胞去除率随Zeta电位净值的降低持续升高。
4)藻细胞高效去除的原因是AOMs组分及藻细胞表面电荷的变化。带负电荷的AMOs被SO4−·和·OH有效降解,同时原位形成的Fe3+中和藻细胞表面的负电荷,导致Zeta电位净值降低,最后通过吸附、网捕作用,藻细胞聚集形成块状的絮体沉降得以去除。
硫酸亚铁协同过硫酸氢钾去除铜绿微囊藻
Removal of Microcystis aeruginosa by synergy of ferrous sulfate and potassium hydrogen persulfate
-
摘要: 避免消毒副产物(DBPs)形成和胞内有机物(IOMs)的释放,是解决传统工艺治理蓝藻水华潜在风险的关键。为此,开展了FeSO4协同过硫酸氢钾(PMS)高级氧化除藻研究,分别探讨了FeSO4投加量、pH及无机离子对铜绿微囊藻细胞去除效果的影响。结果表明:当FeSO4和PMS投加量均为0.1 mmol·L−1时,藻细胞、DOC和UV254的去除率分别为94.66%、58.92%和41.52%,藻细胞去除的氧化、絮凝的贡献率分别为30.50%和69.50%;当pH为6.0~10.0时,藻细胞去除率均保持较高水平,其中弱酸环境有利于提高藻细胞的去除;外加的HPO42−和
NO−3 对SO−4⋅ 的竞争抑制,可导致藻细胞去除率降低;FeSO4-PMS可以有效去除含有荧光特性的藻源性有机质(AOMs);当FeSO4投加量低于0.1 mmol·L−1时,藻细胞、叶绿素a去除率随着Zeta电位净值降低而升高。扫描电镜表征结果表明,协同氧化后大多数藻细胞形态结构完整,从而可有效避免IOMs释放。以上研究结果可为FeSO4-PMS治理富营养化水体中蓝藻水华提供参考。Abstract: Avoiding the formation of disinfection by-products (DBPs) and the release of intracellular organic matters (IOMs) are keys to resolve the potential risks of cyanobacterial blooms treated by traditional techniques. In this study, the advanced oxidation technology based on FeSO4 combined with potassium monopersulfate (PMS) for the treatment of Microcystis aeruginosa cells was investigated. The effects of FeSO4 dosage, pH and inorganic ions on the removal rates of Microcystis aeruginosa were studied. Results showed that when the doses of FeSO4 and PMS were controlled at 0.1 mmol·L−1, algal cell count, DOC concentration and UV254 measurements were reduced by 94.66%, 58.92% and 41.52%, respectively. The oxidation and flocculation process contributed to the algae cell removal rates were 30.50% and 69.50%, respectively. The removal rates of Microcystis aeruginosa could maintained at a high level when the pH was in the rage of 6.0~10.0, especial with mild acidic conditions. Due to competitive inhibition ofSO−4⋅ by extra added HPO42− andNO−3 , the removal efficiencies of algae cells began to decrease. FeSO4-PMS could effectively degrade organic matters (AOMs) with fluorescence characteristics. While the dosage of FeSO4 was below 0.1 mmol·L−1, the removal rate of algae cells and Chl-a increased with the decrease of the absolute Zeta potential. Scanning electron microscopy analysis also showed that most of algae cells had non-destructive morphology and structure after coordinated oxidation, which indicated this process effectively avoiding the release of IOMs. The above results can provide theoretical foundation for FeSO4-PMS treating cyanobacteria blooms in eutrophic water bodies. -
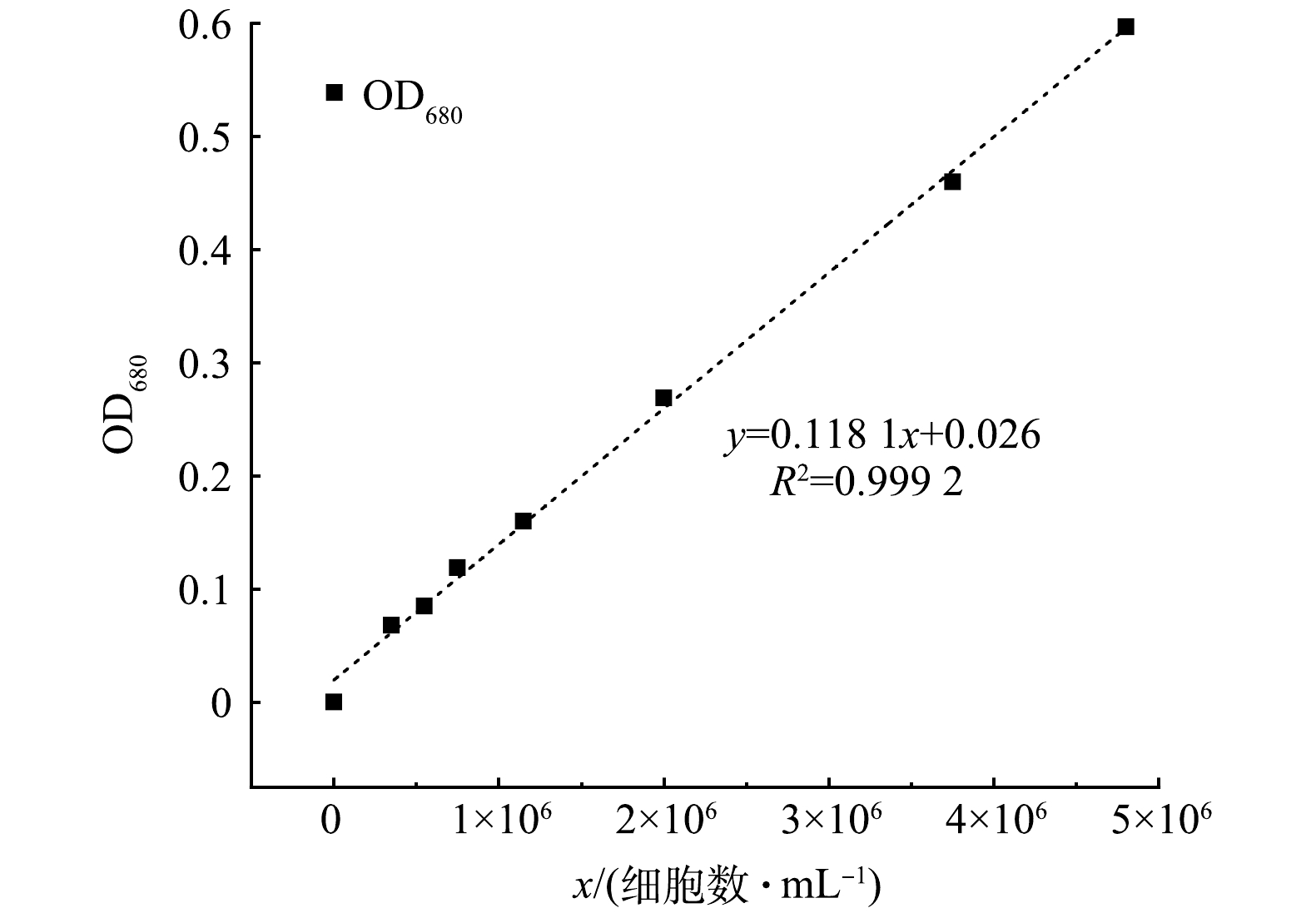
图 1 铜绿微囊藻细胞浓度与OD680线性关系
Figure 1. Linear relationship between the cell density of Microcystis aeruginosa and OD680
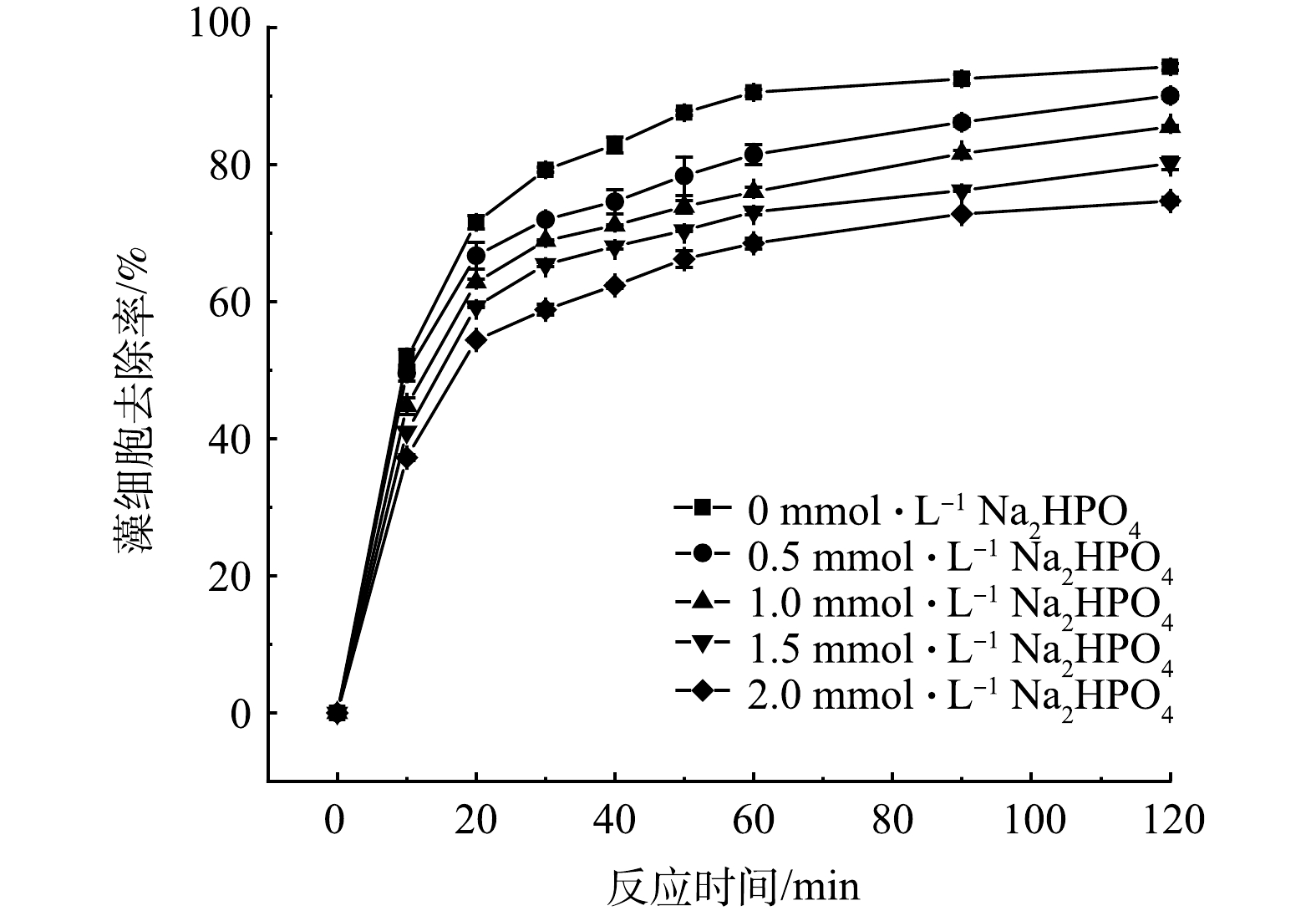
图 4 不同HPO42−浓度下藻细胞去除率
Figure 4. Effect of the concentration of HPO42− on the removal rate of algae cells
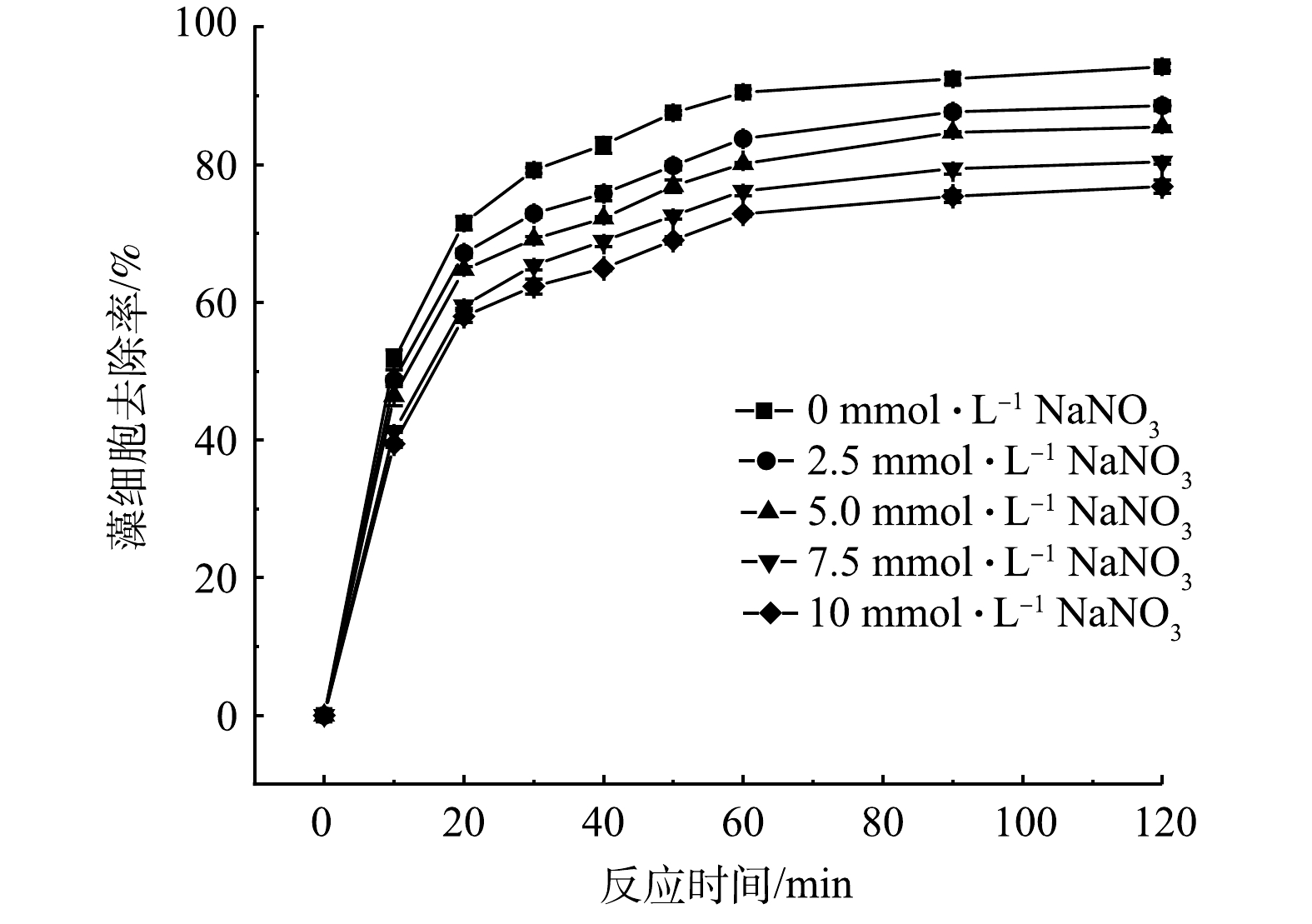
图 5 不同
NO−3 浓度下藻细胞去除率Figure 5. Effect of the concentration of
NO−3 on the removal rate of algae cells
图 6 不同FeSO4投加量下DOC和UV254的去除效果
Figure 6. DOC and UV254 removal effects at different FeSO4 dosages
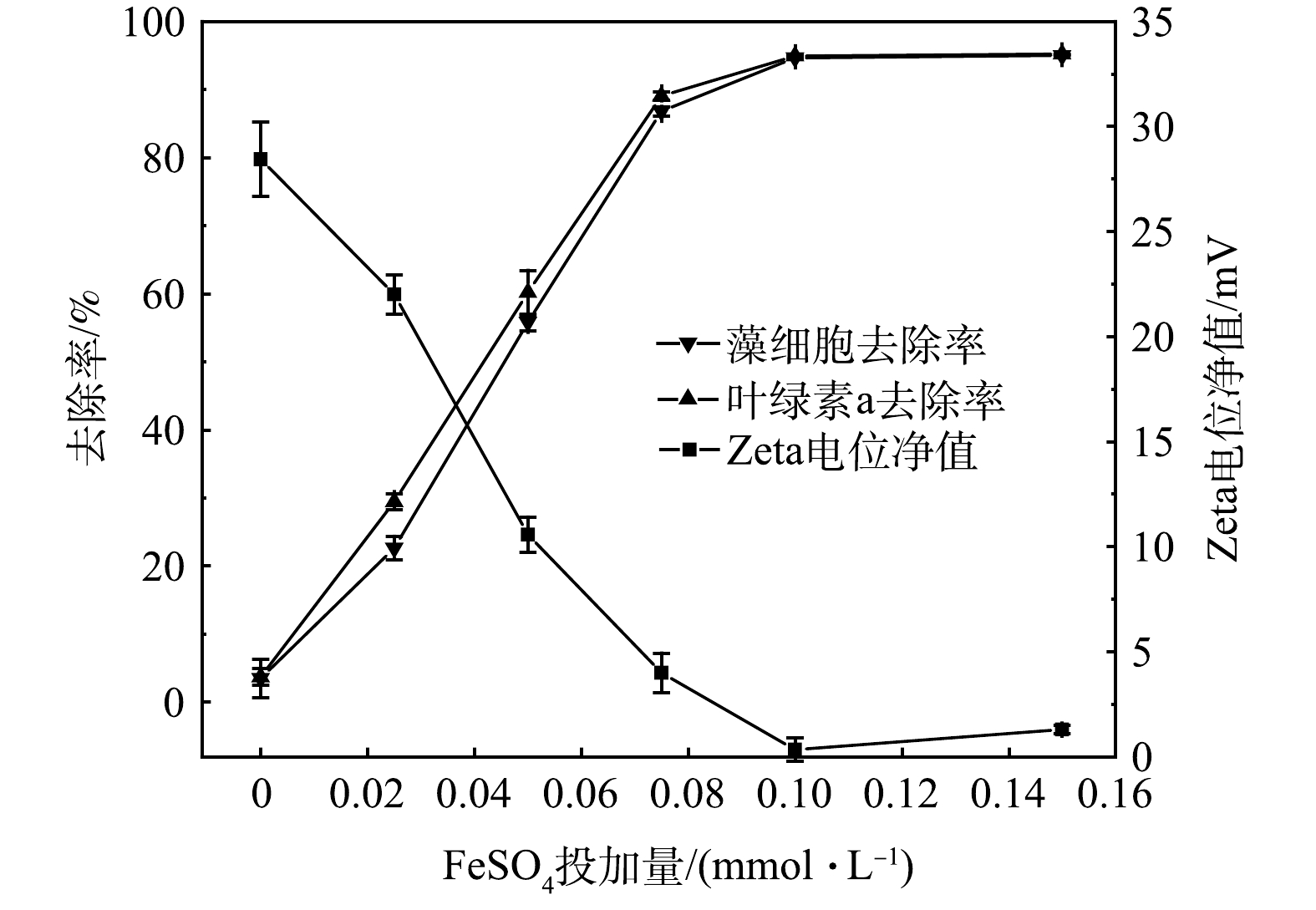
图 8 Zeta电位净值、藻细胞、叶绿素a去除率变化
Figure 8. Changes of the absolute value of zeta potentials and the removal rate of algae cells and Chl-a
-
[1] LI X, GUO M, DUAN X, et al. Distribution of organic phosphorus species in sediment profiles of shallow lakes and its effect on photo-release of phosphate during sediment resuspension[J]. Environment International, 2019, 130: 104916-104925. doi: 10.1016/j.envint.2019.104916 [2] 魏群, 王磊, 马湘蒙, 等. 淡水湖库蓝藻水华治理对策研究与展望[J]. 华北水利水电大学学报(自然科学版), 2021, 42(1): 22-30. [3] ZHAO F, CHU H, YU Z, et al. The filtration and fouling performance of membranes with different pore sizes in algae harvesting[J]. Science of the Total Environment, 2017, 587: 87-93. [4] PAERL H W, OTTEN T G. Harmful cyanobacterial blooms: Causes, consequences, and controls[J]. Microbial Ecology, 2013, 65(4): 995-1010. doi: 10.1007/s00248-012-0159-y [5] MOUSAVI S M S, DEHGHANZADEH R, EBRAHIMI S M. Comparative analysis of ozonation (O3) and activated carbon catalyzed ozonation (ACCO) for destroying chlorophyll a and reducing dissolved organic carbon from a eutrophic water reservoir[J]. Chemical Engineering Journal, 2017, 314: 396-405. doi: 10.1016/j.cej.2016.11.159 [6] DITTMANN E, WIEGAND C. Cyanobacterial toxins-occurrence, biosynthesis and impact on human affairs[J]. Molecular Nutrition & Food Research, 2010, 50(1): 7-17. [7] 刘宇程, 杨冰, 李沁蔓, 等. Cl-和pH对高级氧化工艺去除含盐废水中有机物的影响及机理[J]. 环境工程学报, 2021, 15(5): 1487-1499. doi: 10.12030/j.cjee.202009046 [8] FENG M, CIZMAS L, WANG Z, et al. Synergistic effect of aqueous removal of fluoroquinolones by a combined use of peroxymonosulfate and ferrate(VI)[J]. Chemosphere Environmental Toxicology & Risk Assessment, 2017, 177: 144-148. [9] MATILAINEN A, SILLANPAEAE M. Removal of natural organic matter from drinking water by advanced oxidation processes[J]. Chemosphere, 2010, 80(4): 351-365. doi: 10.1016/j.chemosphere.2010.04.067 [10] MA X, YAN X, YAO J, et al. Feasibility and comparative analysis of cadmium biosorption by living Scenedesmus obliquus FACHB-12 biofilms[J]. Chemosphere, 2021, 275: 130125-130133. doi: 10.1016/j.chemosphere.2021.130125 [11] MA X, CHEN Y, LIU F, et al. Enhanced tolerance and resistance characteristics of Scenedesmus obliquus FACHB-12 with K3 carrier in cadmium polluted water[J]. Algal Research, 2021, 55: 102267-102276. doi: 10.1016/j.algal.2021.102267 [12] GU N, WU Y, GAO J, et al. Microcystis aeruginosa removal by in situ chemical oxidation using persulfate activated by Fe2+ ions[J]. Ecological Engineering, 2017, 99: 290-297. doi: 10.1016/j.ecoleng.2016.11.048 [13] HAN D, JIANGYONG H. The optimal method for peroxydisulfate quenching: A comparison of commonly used reductants[J]. Chemosphere, 2021, 262: 128000-128005. doi: 10.1016/j.chemosphere.2020.128000 [14] MARTIN P, IVANA K, LENKA C, et al. Current knowledge in the field of algal organic matter adsorption onto activated carbon in drinking water treatment[J]. Science of the Total Environment, 2021, 799: 149455-149473. doi: 10.1016/j.scitotenv.2021.149455 [15] CHANIKYA P, NIDHEESH P V, BABU D S, et al. Treatment of dyeing wastewater by combined sulfate radical based electrochemical advanced oxidation and electrocoagulation processes[J]. Separation and Purification Technology, 2021, 254: 117570-117580. doi: 10.1016/j.seppur.2020.117570 [16] WANG Z, CHEN Y, XIE P, et al. Removal of Microcystis aeruginosa by UV-activated persulfate: Performance and characteristics[J]. Chemical Engineering Journal, 2016, 300: 245-253. doi: 10.1016/j.cej.2016.04.125 [17] KAYLA P, LEI L, YOUCHUL J, et al. The application of potassium permanganate to treat cyanobacteria-laden water: A review[J]. Process Safety and Environmental Protection, 2021, 148: 400-414. doi: 10.1016/j.psep.2020.09.058 [18] FANG G, DIONYSIOU D D, WANG Y, et al. Sulfate radical-based degradation of polychlorinated biphenyls: Effects of chloride ion and reaction kinetics[J]. Journal of Hazardous Materials, 2012: 227-228. [19] BANERJEE M, KONAR R S. Comment on the paper "polymerization of acrylonitrile initiated by K2S2O8-Fe(II) redox system"[J]. Journal of Polymer ence Polymer Chemistry Edition, 2010, 22(5): 1193-1195. [20] LIPCZYNSKA-KOCHANY E, SPRAH G, HARMS S. Influence of some groundwater and surface waters constituents on the degradation of 4-chlorophenol by the Fenton reaction[J]. Chemosphere, 1995, 30(1): 9-20. doi: 10.1016/0045-6535(94)00371-Z [21] TAO Z, LILI D, HONGQIANG R, et al. Thermodynamic modeling of ferric phosphate precipitation for phosphorus removal and recovery from wastewater[J]. Journal of Hazardous Materials, 2010, 176: 444-450. doi: 10.1016/j.jhazmat.2009.11.049 [22] MPRA B, CSL A, UKA C, et al. Oxidative degradation of benzoic acid using Fe0- and sulfidized Fe0-activated persulfate: A comparative study[J]. Chemical Engineering Journal, 2017, 315: 426-436. doi: 10.1016/j.cej.2017.01.031 [23] 王庆良, 李倩倩, 童东革, 等. 光化学反应中自由基的作用及反应影响因素的研究进展[J]. 环境化学, 2020, 39(2): 301-316. doi: 10.7524/j.issn.0254-6108.2019061802 [24] ZHANG H, HUANG Q, KE Z, et al. Degradation of microcystin-LR in water by glow discharge plasma oxidation at the gas-solution interface and its safety evaluation[J]. Water Research, 2012, 46(19): 6554-6562. doi: 10.1016/j.watres.2012.09.041 [25] GUO T, YANG Y, LIU R, et al. Enhanced removal of intracellular organic matters (IOM) from Microcystic aeruginosa by aluminum coagulation[J]. Separation and Purification Technology, 2017, 189: 279-287. doi: 10.1016/j.seppur.2017.06.066 [26] YANG T, WANG L, LIU Y, et al. Removal of organoarsenic with ferrate and ferrate resultant nanoparticles: Oxidation and adsorption[J]. Environmental Science & Technology, 2018, 52(22): 13325-13335. [27] 黄芳, 温佳欣, 赵成, 等. 紫外光催化耦合化学絮凝工艺及其对腐殖酸抑制污泥发酵产酸的缓解效果[J]. 环境工程学报, 2021, 15(6): 2037-2045. doi: 10.12030/j.cjee.202102073 [28] CHEN W, WESTERHOFF P, LEENHEER J A, et al. Fluorescence excitation-emission matrix regional integration to quantify spectra for dissolved organic matter[J]. Environmental Science & Technology, 2015, 37(24): 5701-5710. [29] TIAN J, WU C, YU H, et al. Applying ultraviolet/persulfate (UV/PS) pre-oxidation for controlling ultrafiltration membrane fouling by natural organic matter (NOM) in surface water[J]. Water Research, 2018, 132: 190-199. doi: 10.1016/j.watres.2018.01.005 [30] RAUL E M, OLEG S P, JACQUES S, et al. Surface charge and zeta-potential of metabolically active and dead cyanobacteria[J]. Journal of Colloid and Interface Science, 2008, 323(2): 317-325. doi: 10.1016/j.jcis.2008.04.041 [31] ZHANG G, ZHANG P, FAN M. Ultrasound-enhanced coagulation for Microcystis aeruginosa removal[J]. Ultrasonics Sonochemistry, 2008, 16(3): 334-338. [32] ZHOU J, LIU J, ZHAO Z, et al. Microcystis aeruginosa-laden water treatment using peroxymonosulfate enhanced Fe(II) coagulation: Performance and the role of in situ formed Fe3O4[J]. Chemical Engineering Journal, 2020, 382: 123012-123023. doi: 10.1016/j.cej.2019.123012 [33] MATZEK L W, CARTER K E. Activated persulfate for organic chemical degradation: A review[J]. Chemosphere, 2016, 151: 178-188. doi: 10.1016/j.chemosphere.2016.02.055 [34] PIVOKONSKY M, SAFARIKOVA J, BUBAKOVA P, et al. Coagulation of peptides and proteins produced by Microcystis aeruginosa: Interaction mechanisms and the effect of Fe-peptide/protein complexes formation[J]. Water Research, 2012, 46(17): 5583-5590. doi: 10.1016/j.watres.2012.07.040 [35] LIN J, LI X, HAN L, et al. Folium Sennae protects against hydroxyl radical-induced DNA damage via antioxidant mechanism: An in vitro study[J]. Botanical Studies, 2014, 55(1): 1-8. doi: 10.1186/1999-3110-55-1 [36] LIU B, QU F, CHEN W, et al. Microcystis aeruginosa-laden water treatment using enhanced coagulation by persulfate/Fe(II), ozone and permanganate: Comparison of the simultaneous and successive oxidant dosing strategy[J]. Water Research, 2017, 125: 72-80. doi: 10.1016/j.watres.2017.08.035 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 7161
- HTML全文浏览数: 7161
- PDF下载数: 76
- 施引文献: 0


































