


 下载:
下载:















-
氰化提金所带来的环境污染隐患是黄金冶炼行业面临的共性关键难题。据统计,我国每年产出的氰化尾渣数量约为2.45×107 t[1]。氰化尾渣中含有大量的氰化物和有价金属,如果仅仅进行堆存或填埋处理,不仅会污染环境,而且也浪费了资源。因此,氰化尾渣的无害化处理是黄金冶炼行业节能减排及可持续发展面临的关键问题。
氰化尾渣的综合利用主要包括预处理-二次提取金银,综合回收铜、铁、锌,以及无害化处理3大类。前2类侧重于有价资源的综合回收,无害化处理则侧重于氰化物的破坏及重金属离子的去除。综合回收一般流程长、工艺较为复杂,以浮选法为主;无害化处理工艺则比较简单,是解决氰化提金高污染问题最直接最有效的方法之一。目前,氰化尾渣的无害化处理主要包括化学氧化法、电解氧化法、微生物分解法及自然净化法。采用SO2-空气氧化法处理氰化尾渣,以铜为催化剂,SO2和空气的混合物在碱性条件下能将复合氰化物(CNWAD)氧化为氰酸盐(CNO−),同时沉淀除去金属和氰化铁[2-3]。MANORANJAN等[4]的研究结果表明,硫代硫酸盐和活性炭的混合物能够去除废水中的总氰化物和CNWAD。ADJEI等[5]发现,假单胞菌属、芽孢杆菌属等菌种都对氰化物有一定的降解能力。SAARELA等[2]发现,在电化学氧化过程中,氰化物和金属氰络合物首先在阳极被氧化成氰酸根离子,然后进一步分解成无毒的CO2和N2,释放出的金属阳离子在阴极处被还原成金属单质析出。一般情况下,氰化尾渣的无害化处理主要可分为矿浆直接氧化和矿渣洗涤后含氰废水氧化2种。前者氧化剂消耗量大,金属离子以沉淀形式进入渣中,氰化尾渣中的有价金属并没有得到有效的去除;而后者增加了洗涤、液固分离程序,处理过程比较繁杂,洗水量大,处理难度相对增加。因此,研究开发一种工艺简单、成本低、效果好的处理方法是黄金行业创新发展的迫切需要。
本研究中采用矿浆电解技术无害化处理氰化尾渣,将氰化尾渣的洗涤、氰化物的电解氧化、金属离子的电解沉积和矿物的氧化分解集成在同一反应器中进行,利用阳极反应生成的氯气/次氯酸根的强氧化性,破坏含氰离子,氧化包裹矿物,提高氰化尾渣中矿物的单体解离度,以期为后续有价金属的综合利用创造有利条件。
全文HTML
-
本研究中所用原料为陕西省太白金矿浮选-金精矿炭浆提金后的氰化尾渣,其主要组成为CNT 1 748.00 g·t−1、CN− 327.40 g·t−1、Cu 184.80 g·t−1、Fe 3 632.04 g·t−1、Zn 15.84 g·t−1,即该氰化尾渣中总氰与铁氰络合离子含量较高。
-
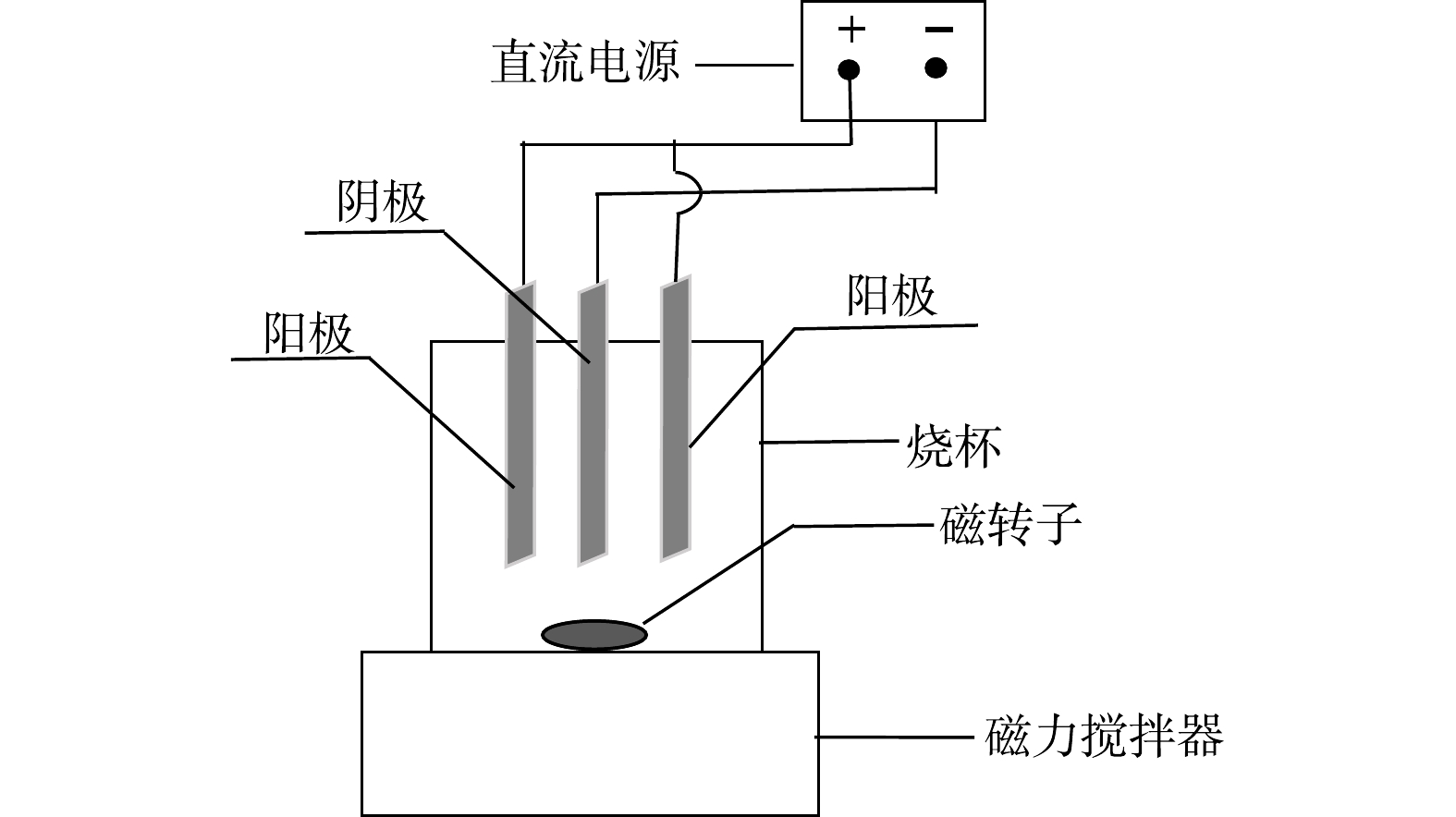
矿浆电解无害化处理氰化尾渣实验装置如图1所示。阴、阳极板均为TC4钛合金板,规格为20 mm×15 mm×2 mm,采用一阴两阳三极板体系。电压由直流稳压电源控制,型号为LP2002D型稳压电源,搅拌装置采用78-1型磁力搅拌器。
-
室温条件下(约25 ℃),取50 g氰化尾渣、一定量的NaCl以及200 mL的去离子水加入到500 mL烧杯中,用磁力搅拌器搅拌,同时控制极板间距。电解一定时间后固液分离,取滤液测定总氰、游离氰及各金属离子含量,并计算各离子去除率,尾渣用去离子水反复冲洗使pH稳定在7左右。
-
氰化尾渣中CNT、CN−的含量按照GB 5085.3-2007[6]所示硝酸银滴定法进行测定,金属离子采用化学滴定法测定,离子去除率按式(1)计算。
式中:Ca为氰化尾渣中各离子的浓度,mg∙L−1;Cb为处理后渣洗液中各离子的浓度,mg∙L−1。
实验中采用FEI MLA 250型高精度工艺矿物学参数自动测试系统(MLA)测定金属矿石的解离程度和连生关系。
1.1. 实验原料
1.2. 实验装置
1.3. 实验方法
1.4. 分析方法
-
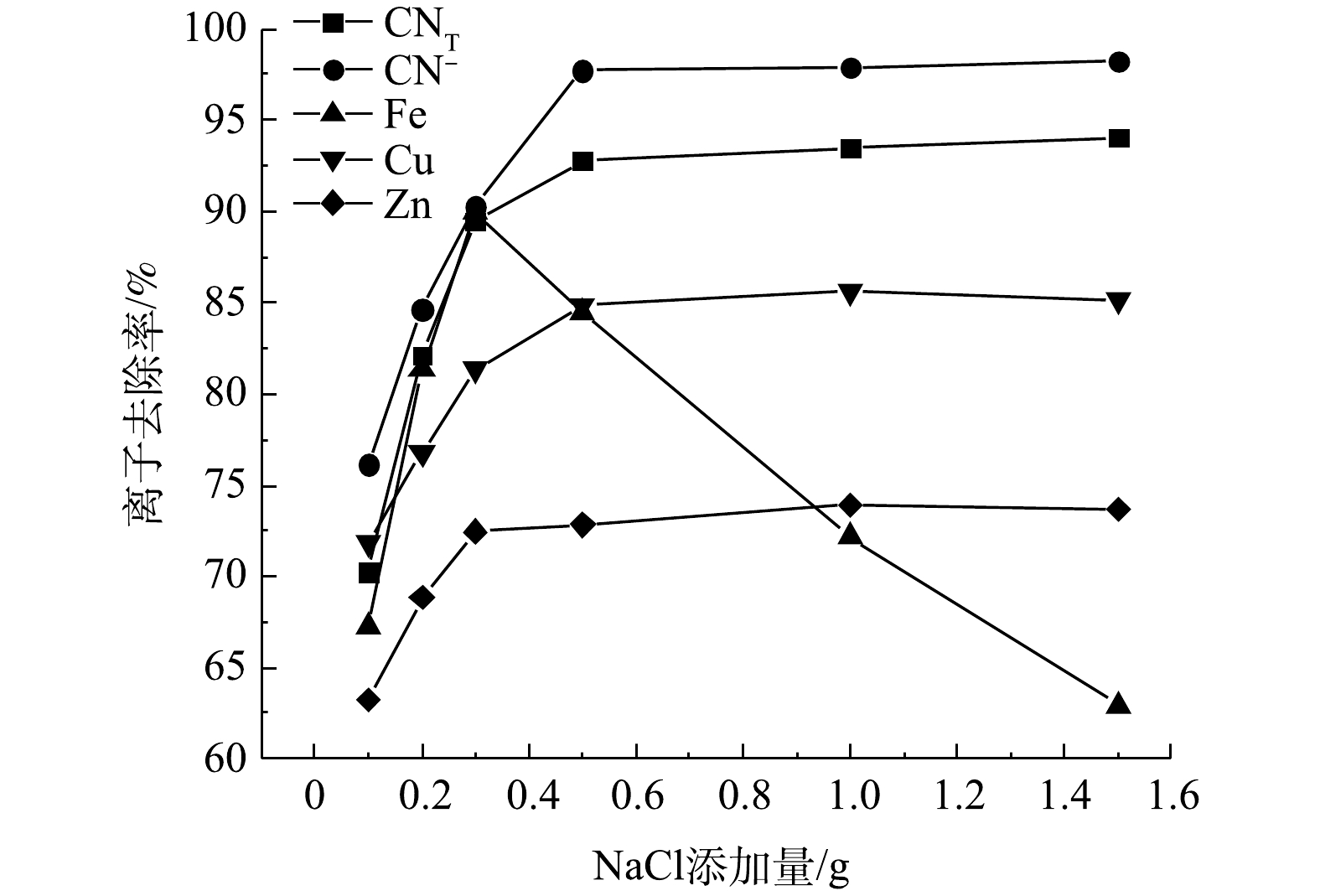
矿浆电解中,Cl−在阳极氧化生成的Cl2/ClO−是主要的氧化物,NaCl的添加量是影响氰化尾渣氧化效果的重要因素之一。在极板间距10 mm、电压8 V、电解时间4 h的条件下进行矿浆电解实验,结果如图2所示。随着NaCl添加量的增加,CNT、CN−及Cu、Zn离子的去除率逐渐增大,至NaCl添加量大于0.5 g后上述离子的去除率几乎不再增加,最大去除率分别为94.05%、98.25%、85.61%、73.92%。电解过程中,Cl−定向迁移至阳极表面,发生阳极氧化反应产生有效氯(ClO−和Cl2)(式(2)~式(4))。其中,有效氯将游离氰化物、铜氰及锌氰络合离子氧化为氰酸盐及金属离子(式5~式10),氰酸盐进一步被氧化为无毒的N2和碳酸盐。同时,金属离子定向迁移至阴极附近,在阴极板上发生金属离子的沉积反应(式(11)~式(14))。随着NaCl用量的增大,各离子的氧化去除效果不断增强的原因是矿浆中的Cl−含量增加,使得电极的电流效率和有效氯浓度增大[7],能够不断氧化去除氰离子和金属氰络离子。NaCl添加量大于0.5 g后,离子去除率几乎不再变化的原因是,NaCl添加量的增大造成溶液中离子浓度增加,离子间距减小,传质作用降低[8],导致有效氯的生成量不会有大幅度提高,离子去除率便不再增加。
值得注意的是,Fe的去除率在NaCl添加量大于0.3 g时,随着NaCl添加量的增大而大幅度降低,最大仅为84.46%。这是因为有效氯的含量随着Cl−的增加而增加,ClO−会氧化部分黄铁矿和磁黄铁矿(式(15)~式(16)),使得矿石中的铁以离子形态再次回到矿浆液中,导致Fe3+去除率降低。
-
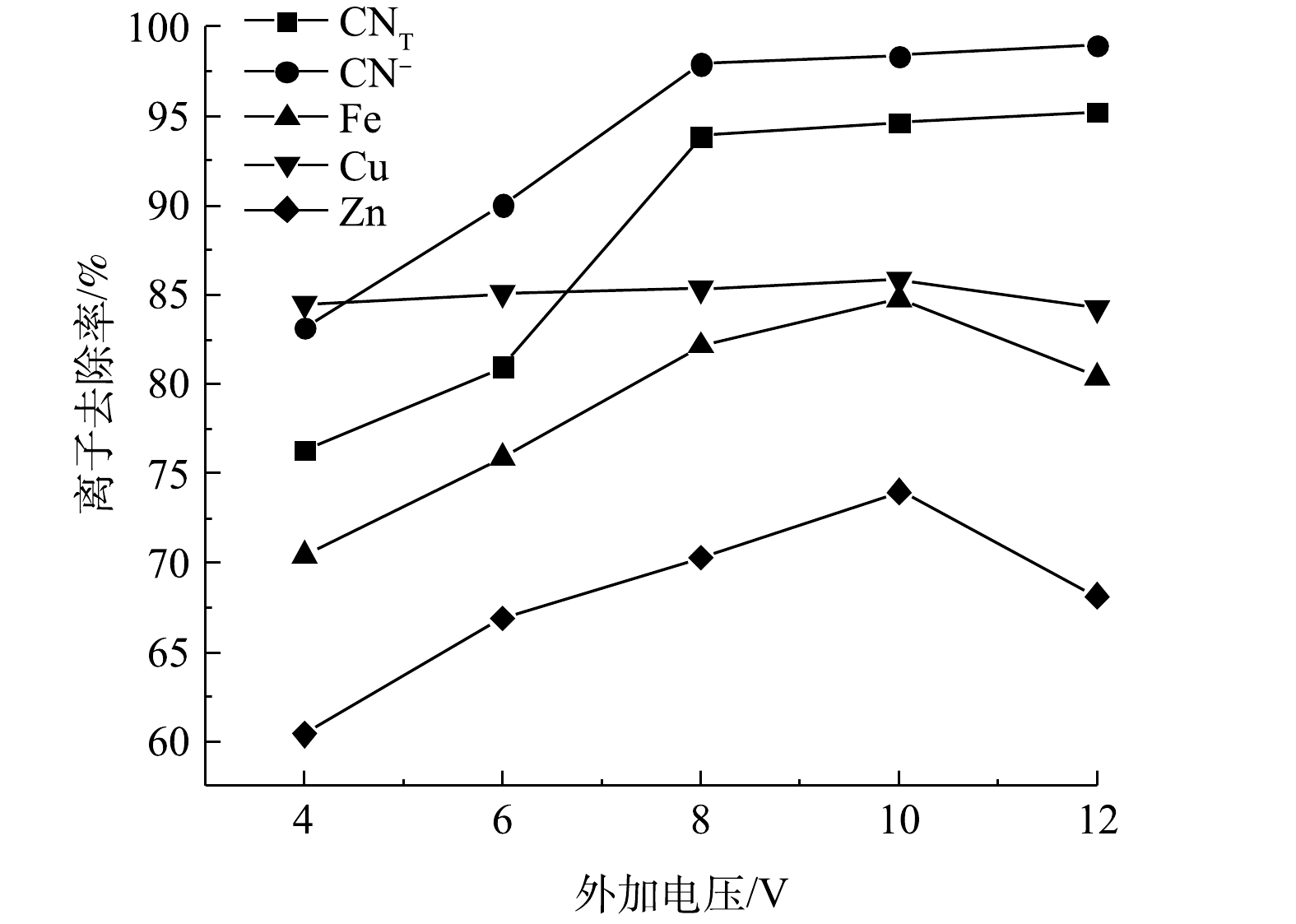
电压的大小直接影响NaCl的电解反应速率以及矿浆中CN−的迁移过程,进而影响氰化物的去除效果。在极板间距10 mm、NaCl添加量0.5 g、电解时间4 h的条件下进行矿浆电解实验,结果如图3所示。随着电压的升高,CNT、CN−的去除率先增大,在8 V之后趋于平缓,去除率最大为95.25%、98.97%;Cu、Fe、Zn离子的去除率随电压增大而增大,在10 V时达到最大值85.86%、84.76%和73.99%,随后有所下降。根据Faraday第一定律,当电流通过电解质溶液时,在电极界面上发生化学反应的物质量与通过电极的电量呈正比,这说明电压是电化学反应过程的一种驱动力[9],随着电压的增大,电流密度逐渐增大,阳极析氯量增加,氧化反应加剧,各离子的去除率均不断上升。电压大于8 V以后,随着电压的增大,各离子去除几乎不再增加的原因:一方面,是由于电压过大会导致电极表面极化现象过大[10-11],造成电流密度减小,不利于氧化反应的发生;另一方面,是难降解的铁氰络离子会与其他金属离子结合转化成更加稳定的金属铁氰络合物沉淀(式(17)~式(18)),不易被氧化去除。此外,金属离子的去除率在大于10 V时呈现降低趋势,其原因可能与尾渣中矿物的氧化溶解(式(15)、式(19)、式(20))有关。
-
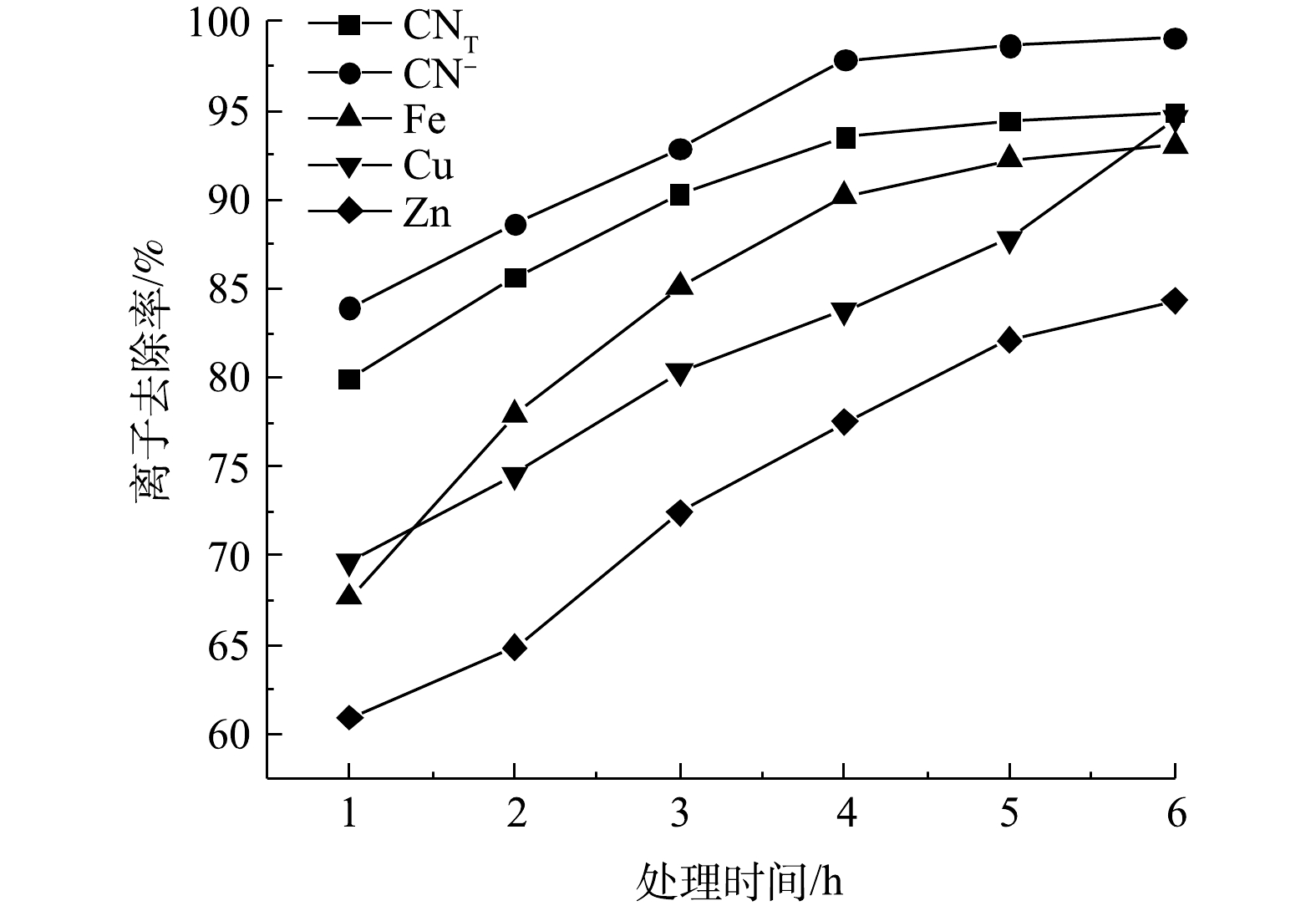
电解时间是影响氰化尾渣中离子去除效果的一个重要因素。在极板间距10 mm、NaCl添加量0.5 g、电压8 V的条件下,改变电解时间进行矿浆电解实验,结果如图4所示。随着电解时间的增长,CNT、CN−及金属离子的去除率不断增大,CNT与CN−的去除率在4 h之后趋于平缓。随着电解时间的增长,溶液中不断生成有效氯用于氧化氰化物和金属氰化物;电解4 h时,CNT与CN−去除率已经达到93.48%、97.84%,而Cu、Zn、Fe离子去除率分别为83.74%、77.52%和90.21%;随后金属离子去除率的增幅开始变小。考虑到能耗的影响,选择4 h为最佳的电解时间。
-
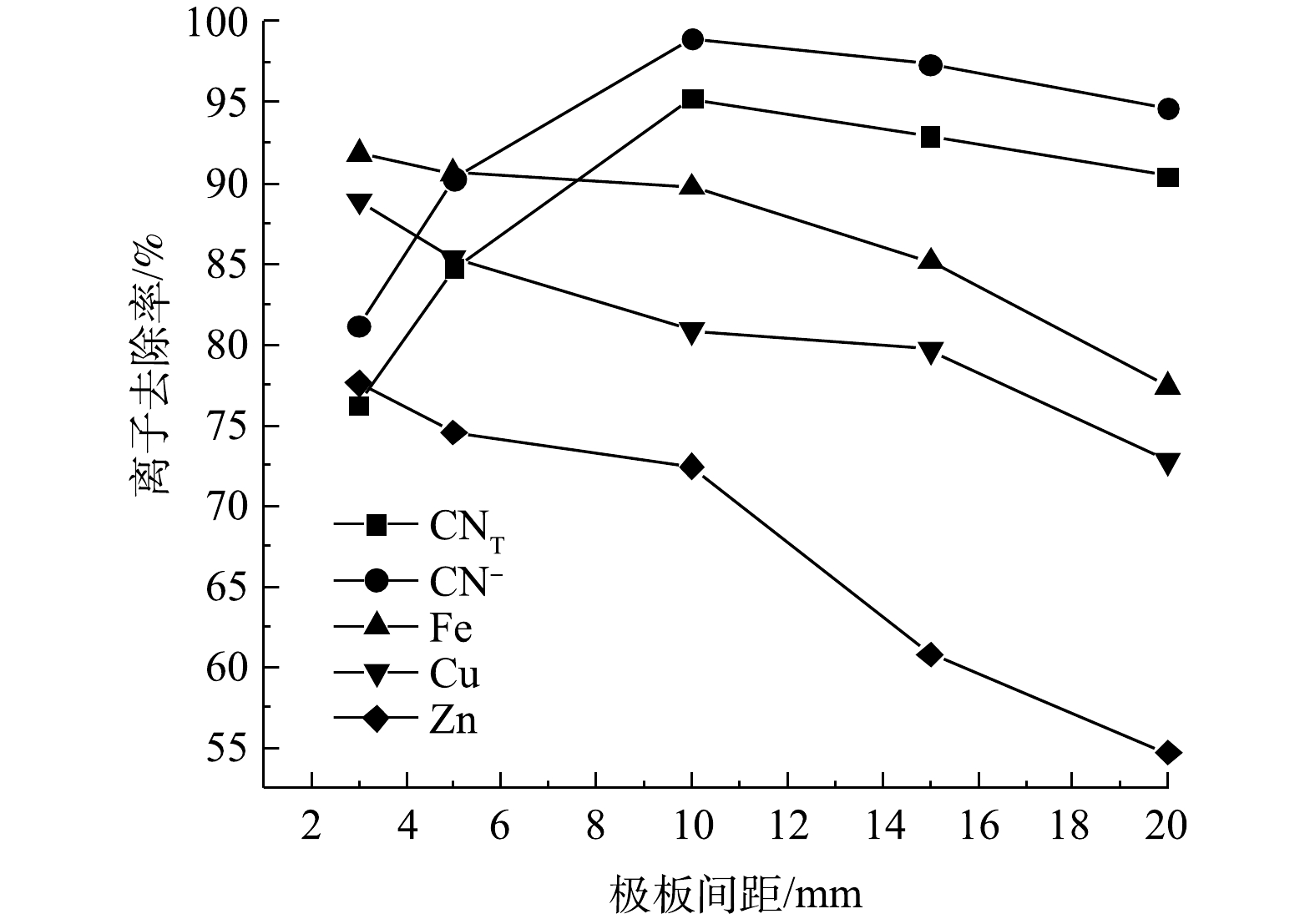
极板间距会影响电路电流大小、电解反应速率和传质效率。在NaCl添加量0.5 g、电压8 V、电解时间4 h的条件下,改变极板间距进行矿浆电解实验,结果如图5所示。随着极板间距增大,CNT与CN−的去除率先增大后减小,在极板间距为10 mm时达到最大,分别为95.25%和98.95%;Cu、Zn、Fe的离子去除率随极板间距增大而减小。极板间距过小时,极板间的电压差增大,易形成瞬时强大的电流,从而造成短路现象。随着极板间距的逐渐增大,极板上的析气反应逐渐正常,电解液流速提升,传质作用不断增强,氧化效果和离子去除率随之增大。但有研究者报道,当极板间距过大时,会导致槽电压和电能消耗不断增加[12],电解反应速率降低;随着反应的进行,离子浓度下降,导致浓差极化作用增强,亦造成金属单质在阴极表面沉积厚度增加,使得阴极电位降低[13],H+在金属单质表面的析出电位增高,最终氧化还原效果降低,从而导致氰化物及金属离子的去除率不断降低。
-
根据MLA分析结果统计所得的电解氯氧化处理前后氰化尾渣主要矿物的解离度,如图6所示。黄铁矿、磁黄铁矿与黄铜矿、闪锌矿的连生比例如表1所示。当NaCl添加量为0.1 g时,尾渣中与黄铜矿、闪锌矿连生的黄铁矿、磁黄铁矿的比例存在小幅度的下降,表明电解产生的有效氯能在一定程度上氧化矿石,添加量较少时氧化效果不太明显。随着NaCl添加量的增加,矿石的解离度不断增加,与黄铜矿、闪锌矿连生的黄铁矿、磁黄铁矿比例大幅下降,同时有部分矿物被氧化溶解,矿物自由面积百分比含量也存在一定程度的降低。NaCl添加量为1.0 g时,矿粒被氧化的程度继续加深,解离度进一步提高,但是,解离度过高使得更多的黄铁矿、磁黄铁矿、黄铜矿和闪锌矿暴露并被氧化,反而导致溶液中金属离子含量有所增加。
-
在极板间距10 mm、外加电压8 V、NaCl用量0.5 g、电解时间4 h的最佳工艺条件下进行了A、B、C 3组平行验证实验,结果如表2所示。由表2可以看出,最佳工艺条件下CNT、CN−以及Cu、Fe、Zn离子的去除率均值分别为94.83%、98.94%、85.65%、84.51%、73.85%,说明氰化尾渣中大部分氰化物及金属离子已被去除。氰化尾渣中游离氰含量达到了国家黄金行业氰渣污染控制技术规范(HJ 943-2018)的要求。
-
从热力学角度分析,氰化尾渣矿浆液中的还原性物质的氧化还原电极电位均小于氯的氧化还原电极电位,这些物质是可以被氯氧化的。各物质被氧化分解的顺序为:CN−>>
Zn(CN)2−4 >Cu(CN)2−3 >>Fe(CN)4−6 。在有NaCl存在的电解体系中,矿浆液中的CN−、Cu(CN)2−3 、Fe(CN)4−6 、Zn(CN)3−4 等阴离子在电场的作用下向阳极定向迁移,Cl−离子在阳极上反应析出的Cl2和进一步溶解生成的ClO−在阳极附近区域将CN−、Cu(CN)2−3 、Fe(CN)4−6 等阴离子氧化,铜离子由+1价被氧化至+2价,氰离子被氧化为无害的N2和碳酸盐,Fe(CN)4−6 被氧化为Fe(CN)3−6 。Cu(CN)2−3 和Zn(CN)3−4 的氧化在阳极附近释放出溶液中的金属离子大部分在阴极发生沉积反应,只有少量Cu2+及Zn2+会与溶液中的铁氰络合离子发生沉淀反应,生成亚铁氰化铜、亚铁氰化锌以及铁氰化亚铜[14-15],沉降到矿浆渣中。
2.1. NaCl用量对各离子去除效果的影响
2.2. 外加电压对各离子去除效果的影响
2.3. 电解时间对各离子去除效果的影响
2.4. 极板间距对各离子去除效果的影响
2.5. 氰化尾渣中矿物解离度分析
2.6. 矿浆电解氰化尾渣平行验证实验
2.7. 矿浆电解氧化氰化尾渣过程分析












-
1)采用一步矿浆电解技术可有效处理氰化尾渣中的氰化物,当极板间距10 mm、外加电压8 V、NaCl添加量0.5 g、电解时间4 h时,CNT、CN−、Cu、Fe以及Zn离子的去除率分别可达到94.83%、98.94%、85.65%、84.51%和73.85%。
2)矿浆电解处理氰化尾渣过程中阳极附近主要发生的是氰化物的直接氧化与间接氧化,阴极板主要是Cu、Fe、Zn等重金属离子的电解沉积。随着电解电压、NaCl添加量以及电解时间的增大,金属矿物的解离度不断增加,矿物之间的连生比例不断降低,尾渣中黄铁矿、磁黄铁矿的大颗粒连生体被分解为小颗粒的单体态并不断被电解氧化溶解。
