


 下载:
下载:


-
氯苯(CB)是一种重要的化工原料,常通过工业泄露以及废物、废气的不合理排放等途径进入土壤和地下水环境中。由于CB难以被生物降解并具有致癌、致畸、致突变效应[1],因此,CB成为多数污染场地地下水修复的主要目标污染物[2-4]。
高级氧化技术被广泛应用于污染场地下水修复[5]。高级氧化技术主要有芬顿(Fenton)法、过硫酸盐氧化法等[6]。芬顿(Fenton)法、过硫酸盐氧化法主要通过产生羟基自由基(·OH)或硫酸根自由基(
$ \cdot {\rm{SO}}_4^ - $ )去除CB。然而两者在修复污染场地地下水中CB的污染时,会存在CB氧化不彻底而产生有害稳定中间体的问题,从而造成二次污染[7]。目前,国内外有许多关于CB高效降解和其在氧化过程中有毒中间体种类的研究。JIANG等[8]通过改性的Fe2+/PS体系降解CB,发现在氧化CB的过程中生成了2-氯苯酚、3-氯苯酚、4-氯苯酚等中间体,他们认为是·OH攻击苯环,通过亲电加成反应生成的。OUYANG等[9]以氧还原产生的H2O2降解废水中CB,发现·OH通过亲电子取代、加成反应以及键断裂等一系列作用,将CB氧化成草酸、醇、乙酸以及其他小分子有机酸。ZHANG等[10]证明了Fe2+活化过碳酸钠(2Na2CO3·3H2O2,与水混合会释放H2O2)可有效去除CB,且在氧化过程中产生2-氯苯酚、乙酸酐、乙醛酸丁酯等中间体。由上述研究可发现在运用不同高级氧化体系降解CB时,氧化体系中均生成了氯代酚类中间体,但并未对CB氧化过程中氯代酚类中间体进行定量研究。氯代酚类化合物对水生生物具有毒性,在环境中能够稳定存在,且较CB毒性更高[11-13],对环境危害更大[14-17]。《污染地块地下水修复和风险管控技术导则》(HJ 25.6-2019)中明确指出修复后的地下水检测指标应关注二次污染物。在实际CB污染场地下水的修复时,即使CB已达到污染场地地下水的修复目标值,仍需对氯代酚类中间体浓度进行监测,防止氯代酚类中间体引发地下水的二次污染。因此,运用高级氧化技术进行CB污染物降解时,研究其氧化过程中稳定氯代酚类中间体的产生量对于污染场地地下水的修复效果评估具有重要意义。
基于此,本研究选用地下水修复项目中常用的芬顿(Fe2+/H2O2)、亚铁离子活化过硫酸钠(Fe2+/PS)和碱活化过硫酸钠(NaOH/PS) 3种高级氧化体系降解CB。在氧化过程中,发现了毒性强的稳定中间体对氯苯酚(4-CP)的产生,并使用GC-MS对其进行了定量分析,进而探究了3种氧化体系中氧化剂的用量对降解CB和去除4-CP的影响。此外,通过自由基捕获实验、氯离子(Cl−)浓度的变化、总有机碳(TOC)来分析3种氧化体系降解CB的机制,并对实际污染地下水修复工法的优化进行了验证实验。
全文HTML
-
气相色谱-质谱仪(GC-MS,8860-5977B,美国Agilent);总有机碳分析仪(TOC,Multi C/3100,德国Analytik Jena);离子色谱仪(ICS-600,美国DIONEX);pH计(PHS-3C,上海雷磁仪器有限公司)。
氯苯(C6H5Cl)、对氯苯酚(C6H4ClOH)、七水合硫酸亚铁(Fe2SO4·7H2O)、过硫酸钠(Na2S2O8)、双氧水(H2O2)、氢氧化钠(NaOH)、硫酸(H2SO4)、甲醇(CH3OH,MeOH)、叔丁醇(C4H9OH,TBA)和硫代硫酸钠(Na2S2O3)购买自国药集团化学试剂有限公司。药品均为分析纯,实验用水为超纯水。实验所用的CB溶液浓度为154.9 mg·L−1。污染场地地下水样品取自常州市某污染场地,CB为154.9 mg·L−1、pH为5.54、COD为700 mg·L−1。
-
1) Fe2+/H2O2、Fe2+/PS、NaOH/PS降解CB实验。向1 000 mL CB溶液中加入定量的活化剂(Fe2SO4·7H2O、NaOH)和氧化剂(H2O2、PS),在每个反应时间节点取样后立即加入Na2S2O3终止反应,置于4 ℃冰箱储存待测,降解实验设计见表1。
2)自由基捕获实验。分别使用TBA、MeOH作为自由基捕获剂进行实验,向反应溶液中添加一定量的TBA和MeOH来研究3种氧化体系中可能存在的自由基及其主要作用,其中溶液中TBA、MeOH与氧化剂的摩尔比均为1 000。
3)污染场地地下水降解实验。以A、B 2种方式向盛有1 000 mL污染场地地下水的棕色螺纹口瓶中添加NaOH和PS,其中NaOH与PS摩尔比为3,PS与CB的摩尔比为5、10、20、50、100。A方式为将全部的氧化药剂一次性添加至反应溶液中,B方式为分批次在8 h内将氧化药剂添加至反应溶液中,在反应0、1、2、4、6、8、10、12、14 d后取样并立即加入Na2S2O3终止反应,置于4 ℃冰箱储存待测。
以上所有实验每组设3个平行样,取平均值。
-
CB和CB中间体产物浓度使用配备有DB-5型色谱柱的Agilent 8860-5977B型GC-MS进行分析。仪器设置条件:进样口温度为220 ℃,进样方式为分流进样(分流比10∶1),程序升温为35 ℃恒温2 min,然后以15 ℃·min−1的升温速率升温至150 ℃,并恒温5 min,再以3 ℃·min−1升温至290 ℃,在290 ℃下保持2 min;载气为氦气,流量1.0 mL·min−1。质谱设置条件为:离子源温度为230 ℃、离子化能力为70 eV、接口温度为280 ℃、质量扫描范围为35~450 amu。
采用液液萃取法[18]对反应液中的CB及CB中间体产物进行萃取。用量筒移取20 mL反应溶液,加入少量Na2S2O3,用1∶20的稀H2SO4调节溶液呈酸性,后加入20 mLCH2Cl2,300 r·min−1水平恒温振荡30 min进行萃取。萃取完成后,其萃取液经有机滤膜过滤后上机检测。
配制CB与4-CP浓度分别为0~180.0 mg·L−1和0~100.0 mg·L−1的混标标准溶液,使用GCMS检测分析,以响应值为纵坐标,CB、4-CP浓度为横坐标分别绘制标准曲线。CB在0~180.0 mg·L−1内线性关系良好,回归方程为y =11 512.774 2x,R2 = 0.999;4-CP在0~100.0 mg·L−1内线性关系良好,回归方程为 y =7 991.708 1x,R2 = 0.993。
采用德国耶拿Multi N/C 3100总有机碳分析仪对反应液进行分析,以此来表征CB降解过程中的矿化效果。具体分析方法参考《水质总有机碳的测定 燃烧氧化-非分散红外吸收法》(HJ 501-2009)。
通过ICS型离子色谱仪(戴安中国有限公司)对反应液中的氯离子含量[19]进行测定。
-
CB的降解率按式(1)计算,CB的矿化率按式(2)计算,CB的氯离子产率按式(3)计算。
式中:ηCB为CB的降解率;
$ {C}_{{\rm{B}}t} $ 为反应t时间后CB浓度,mg·L−1;$ {C}_{\rm{B0}} $ 为反应初始时的CB浓度,mg·L−1。式中:ηTOC为CB的矿化率;Ct为反应t时间后TOC的浓度,mg·L−1;C0为CB初始时的TOC浓度,mg·L−1。
式中:
${\eta _{{\rm{C}}{{\rm{l}}^ - }}} $ 为Cl−的产率;Ct为反应t时间后Cl−浓度,mg·L−1;C0为1 000 mL浓度为154.86 mg·L−1 CB溶液中CB中Cl元素完全电离时的Cl−浓度,mg·L−1。
1.1. 仪器与试剂
1.2. 降解实验
1.3. 检测方法及CB去除率、矿化率和氯离子产率计算
1.4. CB降解率、矿化率和氯离子产率的计算
-
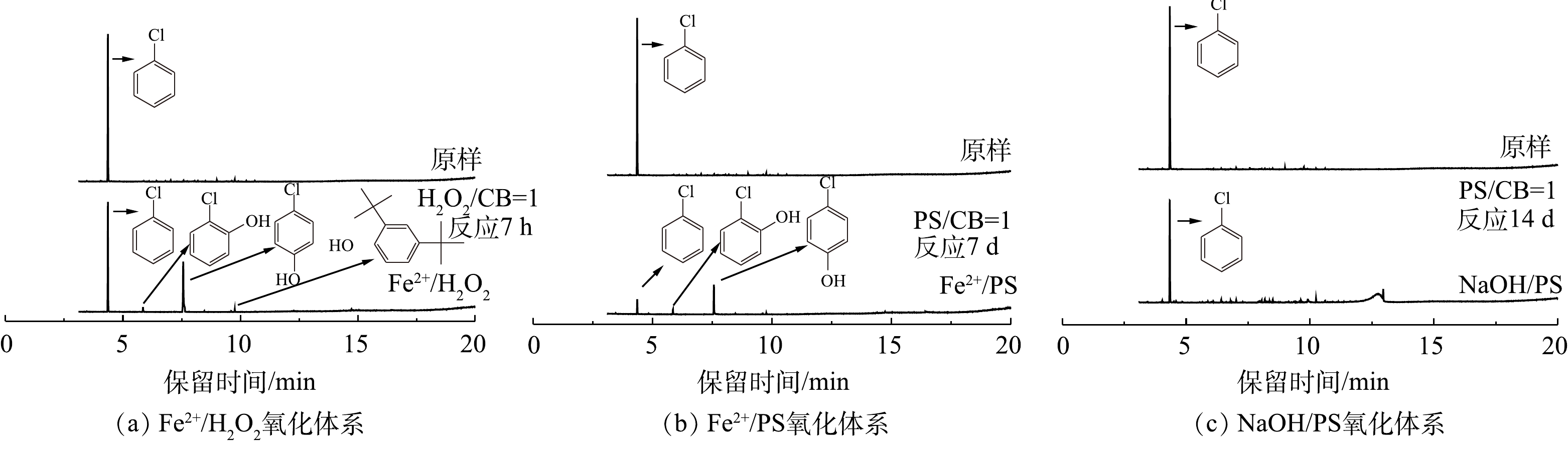
使用GC-MS对Fe2+/H2O2、Fe2+/PS和NaOH/PS 3种氧化体系降解CB前后中间体产物进行定性分析,结果如图1所示。对比CB降解前后GC-MS谱图(图1(a)~图1(c))可知,Fe2+/H2O2和Fe2+/PS体系在氧化CB的过程中主要生成4-CP、2-CP等中间体,且4-CP含量最高,故以4-CP作为代表性中间体开展研究。NaOH/PS体系在氧化CB的过程中不产生氯代酚类中间体,这可能是NaOH/PS体系为强碱性,氯代酚类中间体在碱性条件下会形成酚钠盐,可进一步被迅速氧化。
-
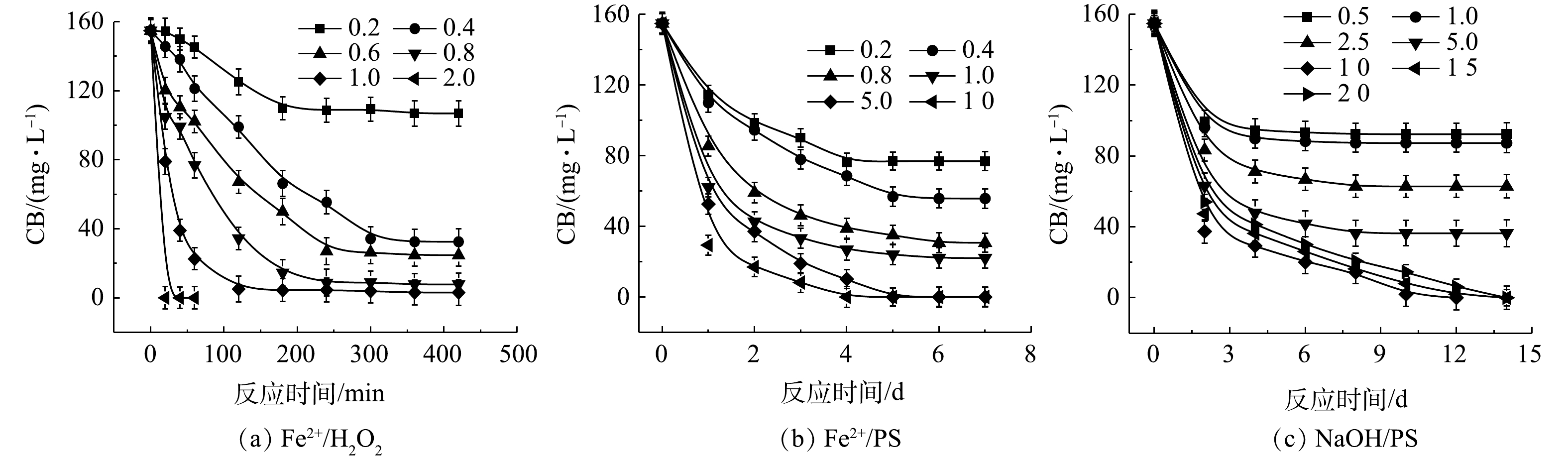
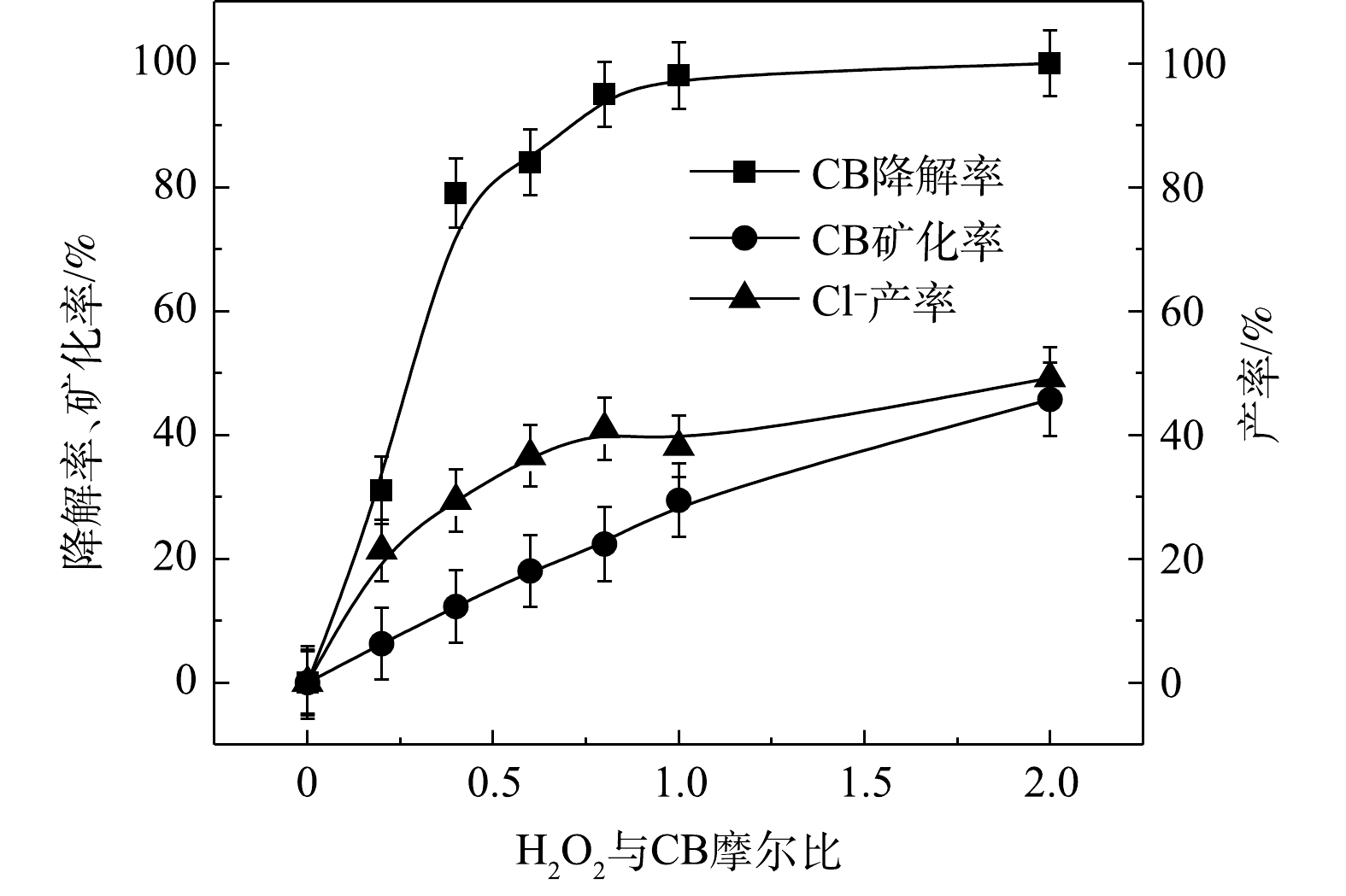
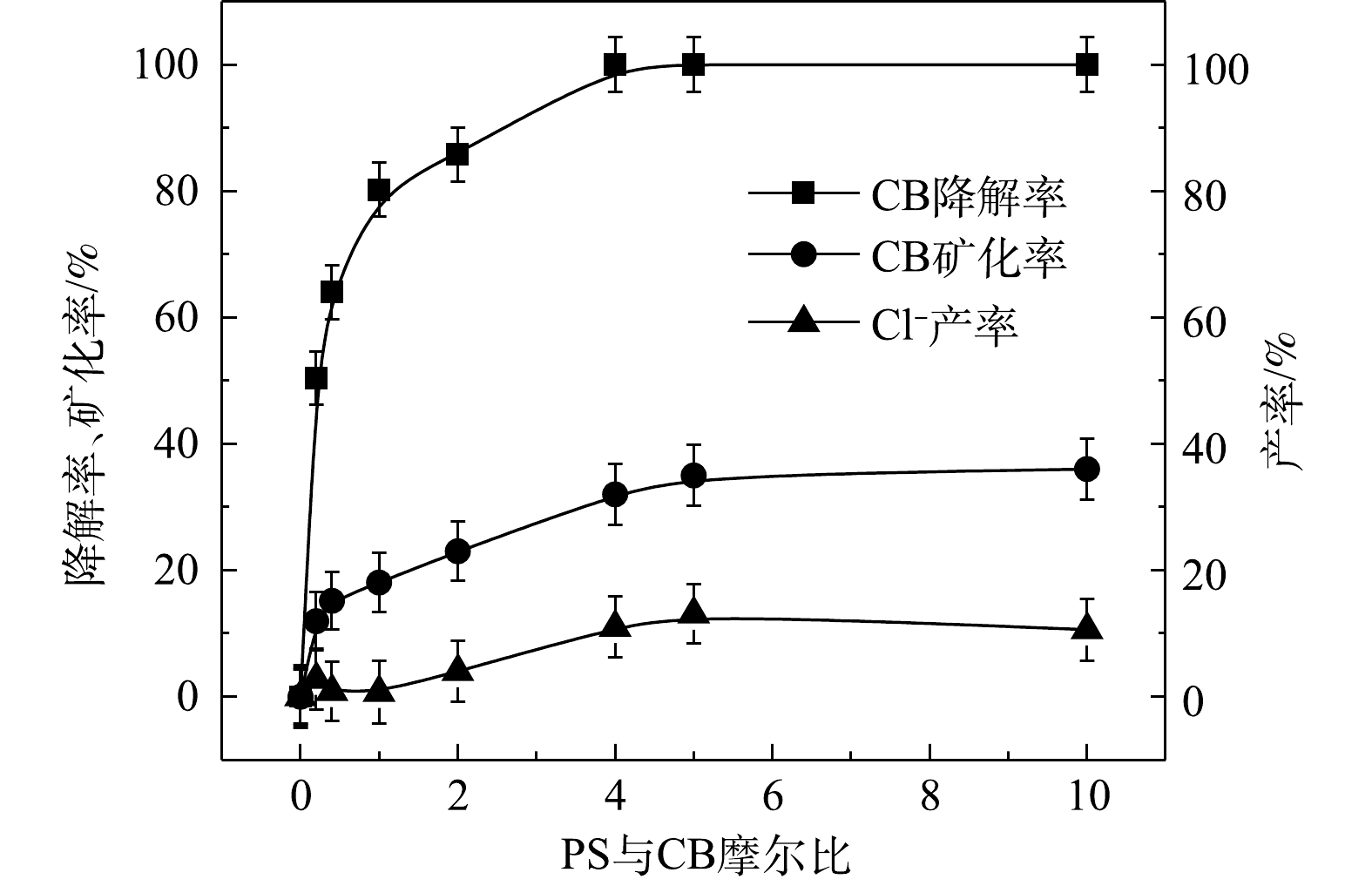
1) Fe2+/H2O2、Fe2+/PS、NaOH/PS体系氧化降解CB。Fe2+/H2O2、Fe2+/PS、NaOH/PS中不同氧化剂用量对CB的降解效果如图2所示。在Fe2+/H2O2氧化体系中,随着H2O2与CB摩尔比的增大,CB浓度明显降低,ηCB逐渐增大(图2(a))。当H2O2与CB的摩尔比为0.2和0.4时,ηCB分别为31%和79%,CB未能被完全降解,这表明较少量的H2O2不能完全氧化CB;当H2O2与CB摩尔比继续增大至1.0时,在反应420 min后,Fe2+/H2O2对CB的降解率高达98%。与Fe2+/H2O2类似,在Fe2+/PS体系中,随着PS与CB摩尔比的增大,CB的浓度降低较为明显(图2(b))。当PS与CB的摩尔比为0.2、0.4、0.8、1.0时,ηCB分别为50%、64%、80%、86%,CB未能被完全降解,这表明PS用量偏少,CB未能完全氧化分解;当PS与CB摩尔比继续增大至5.0、10.0时,CB被完全降解。
由图2(c)可知,当PS与CB的摩尔比为10.0时,NaOH/PS对CB的降解效果最佳,ηCB达到100%;但当PS与CB的摩尔比为20时,CB的降解效果反而变差。这可能是由于氧化剂浓度过高,自由基的自淬灭作用[20-21]显著(式(4)~式(7))。
由上述结果可知,相较于Fe2+/PS、NaOH/PS,Fe2+/H2O2对CB的降解效率最高,且氧化剂使用量最少。因此,在实验室的小试实验中,Fe2+/H2O2是去除污染场地地下水中CB的最佳氧化体系。
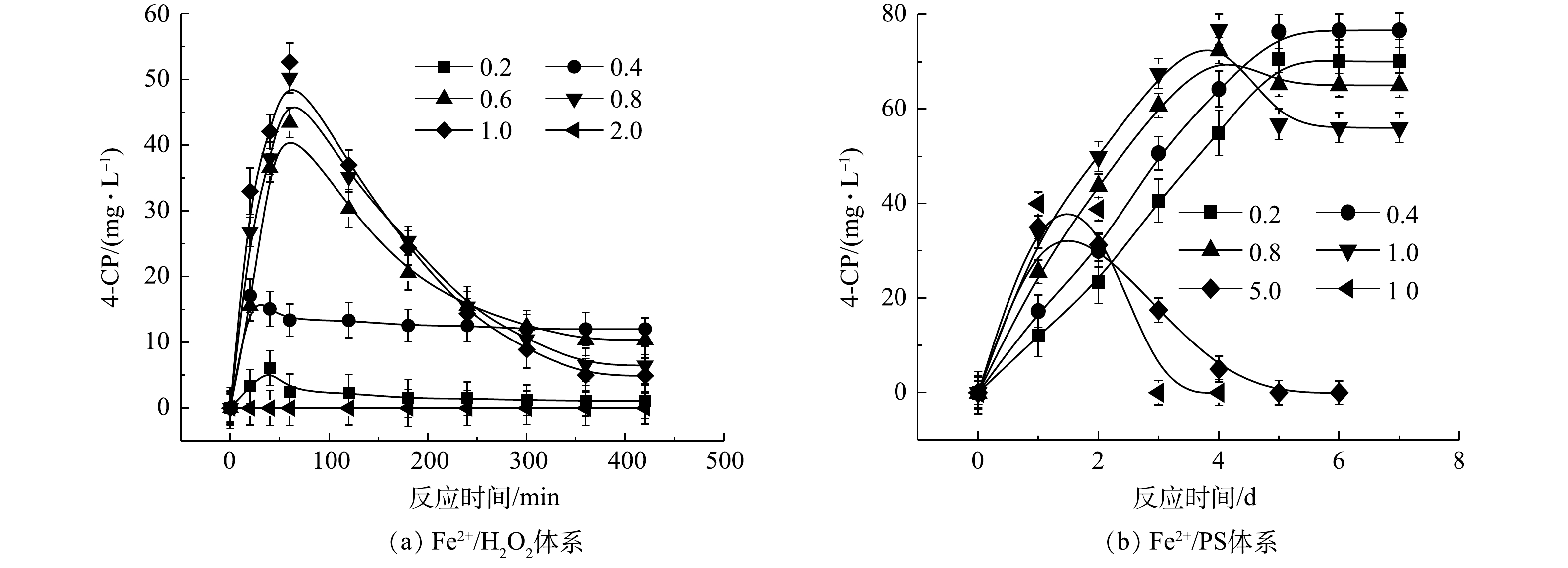
2) Fe2+/H2O2、Fe2+/PS、NaOH/PS体系中4-CP中间体的产量变化。在氧化CB的过程中,Fe2+/H2O2、Fe2+/PS、NaOH/PS 3种体系中的4-CP中间体浓度变化如图3所示。在Fe2+/H2O2氧化体系中:当H2O2与CB的摩尔比为0.2和0.4时,生成了浓度分别为1.09 mg·L−1和12.03 mg·L−1的4-CP中间体(图3(a));当H2O2与CB的摩尔比进一步增大时,4-CP中间体产物在反应60 min时达到最高浓度,但随着反应时间的增加,其浓度逐渐减少到较低的水平;当H2O2与CB摩尔比继续增大至1.0时,此时4-CP中间体浓度较低,为4.89 mg·L−1;而当H2O2与CB摩尔比为2.0时,未检出4-CP中间体产物。与Fe2+/H2O2不同,在Fe2+/PS中,当PS与CB的摩尔比分别为0.2、0.4、0.8、1.0时,生成了浓度分别为70.09、76.63、65.03、56.04 mg·L−1 的4-CP中间体(图3(b)),且随反应时间的增加,其仍保持较高的浓度;当PS与CB摩尔比继续增大至5、10时,4-CP浓度在反应2 d后达到峰值,且随着反应时间增加逐渐减小,直至完全降解,这与SAI等[12]的研究结果一致。与Fe2+/H2O2、Fe2+/PS体系不同,NaOH/PS体系在氧化CB过程中不产生4-CP中间体(图1(c))。因此,在实际污染场地地下水修复时,针对局部区域存在氧化剂不过量的问题,宜采用NaOH/PS的氧化体系降解CB,可避免高毒性中间体4-CP的产生,从而有利于次生污染的防治。
-
通常,H2O2和PS通过活化剂产生活性自由基来氧化有机污染物[22]。在添加自由基捕获剂后,目标污染物的降解会受到显著抑制,由此可以利用自由基捕获实验来研究Fe2+/H2O2、Fe2+/PS和NaOH/PS三种氧化体系降解CB的反应机制。
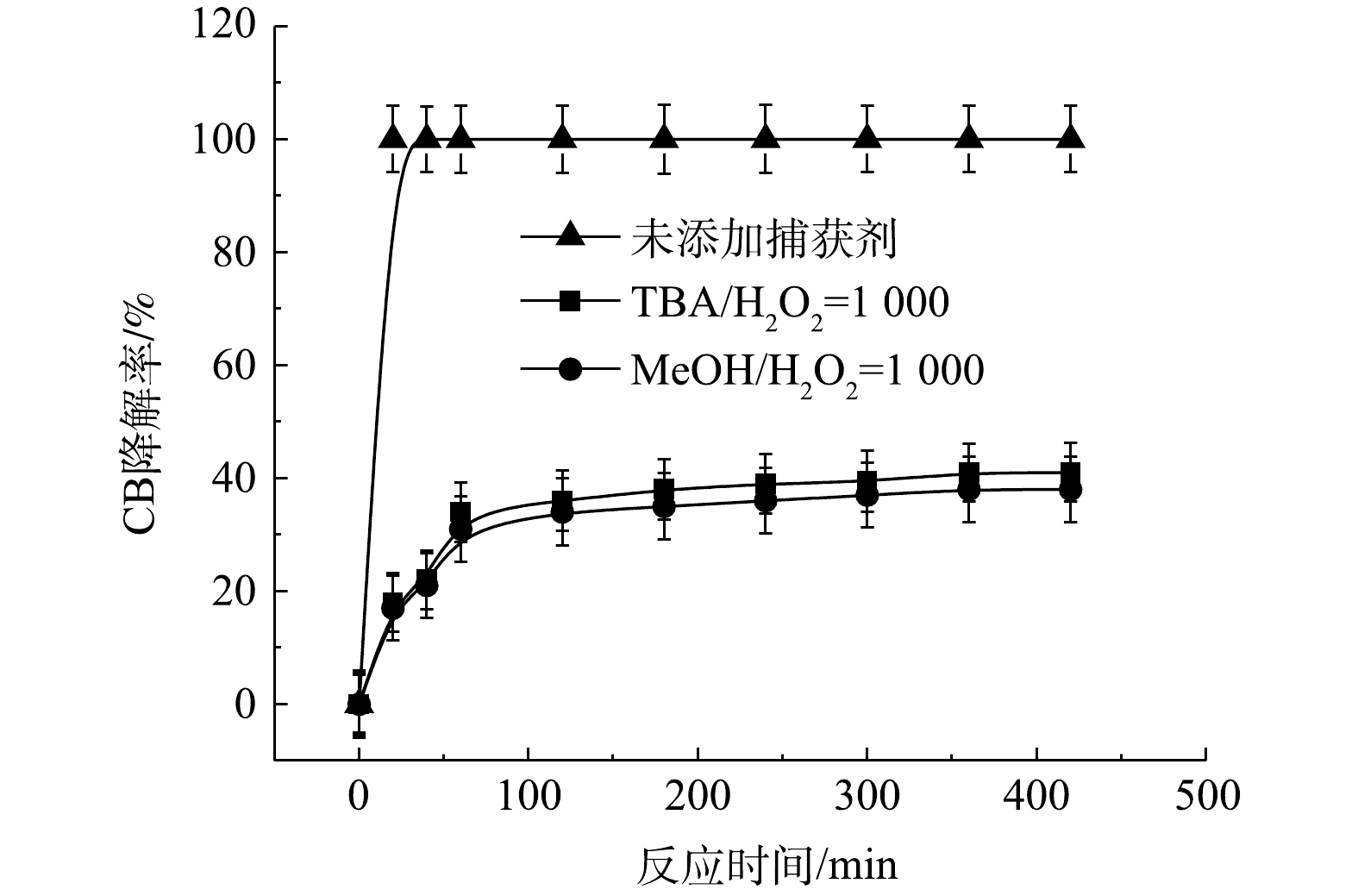
$ \cdot {\rm{SO}}_4^ - $ 和·OH通常被认为是活化H2O2或PS降解污染物的活性物种。TBA可以与·OH迅速反应(k = 6.0×108 L·(mol·s)−1),而MeOH与$ \cdot {\rm{SO}}_4^ - $ 和·OH的反应速率k分别为1.1×107 L·(mol·s)−1和9.7×108 L·(mol·s)−1[23]。可利用TBA和MeOH与2种自由基反应速率的差异来区分他们在3种氧化体系中的主要作用。1) Fe2+/H2O2反应机理及CB的降解机制。MeOH、TBA对Fe2+/H2O2降解CB的影响如图4所示。TBA抑制了Fe2+/H2O2体系对CB的降解,ηCB由100%降低至41%(n[TBA/H2O2]为1 000),这表明Fe2+/H2O2体系中存在·OH。当MeOH以MeOH/H2O2=1 000的摩尔比添加到反应中,ηCB由100%降低至38%(TBA添加至反应中,ηCB由100%降低至41%)。相较于TBA,MeOH对CB降解抑制作用仅略微增强,这表明在Fe2+/H2O2的氧化体系中以·OH为该反氧化体系的主导自由基(式(8)),且除了·OH,可能存在其他活性物质[24]。通过对比Fe2+/H2O2体系中H2O2与CB不同摩尔比下ηCB、ηTOC及ηCl−,进一步确认Fe2+/H2O2在氧化CB过程中稳定中间体的产生情况,其结果如图5所示。由图5可知,随着H2O2与CB摩尔比的增大,Fe2+/H2O2对ηCB、ηTOC及ηCl−均增大,且ηCl−始终大于ηTOC。这表明CB在被·OH(已证明Fe2+/H2O2的主要自由基为·OH)氧化的过程中发生了脱氯反应,生成了盐酸,这是一种典型的基于·OH链反应的高级氧化过程[16,25-26]。
2) Fe2+/PS反应机理及CB的降解机制。MeOH、TBA对Fe2+/PS体系降解CB的影响如图6所示。与Fe2+/H2O2体系不同,TBA对CB降解的抑制作用显著低于MeOH,这表明在Fe2+/PS的氧化体系中·OH和
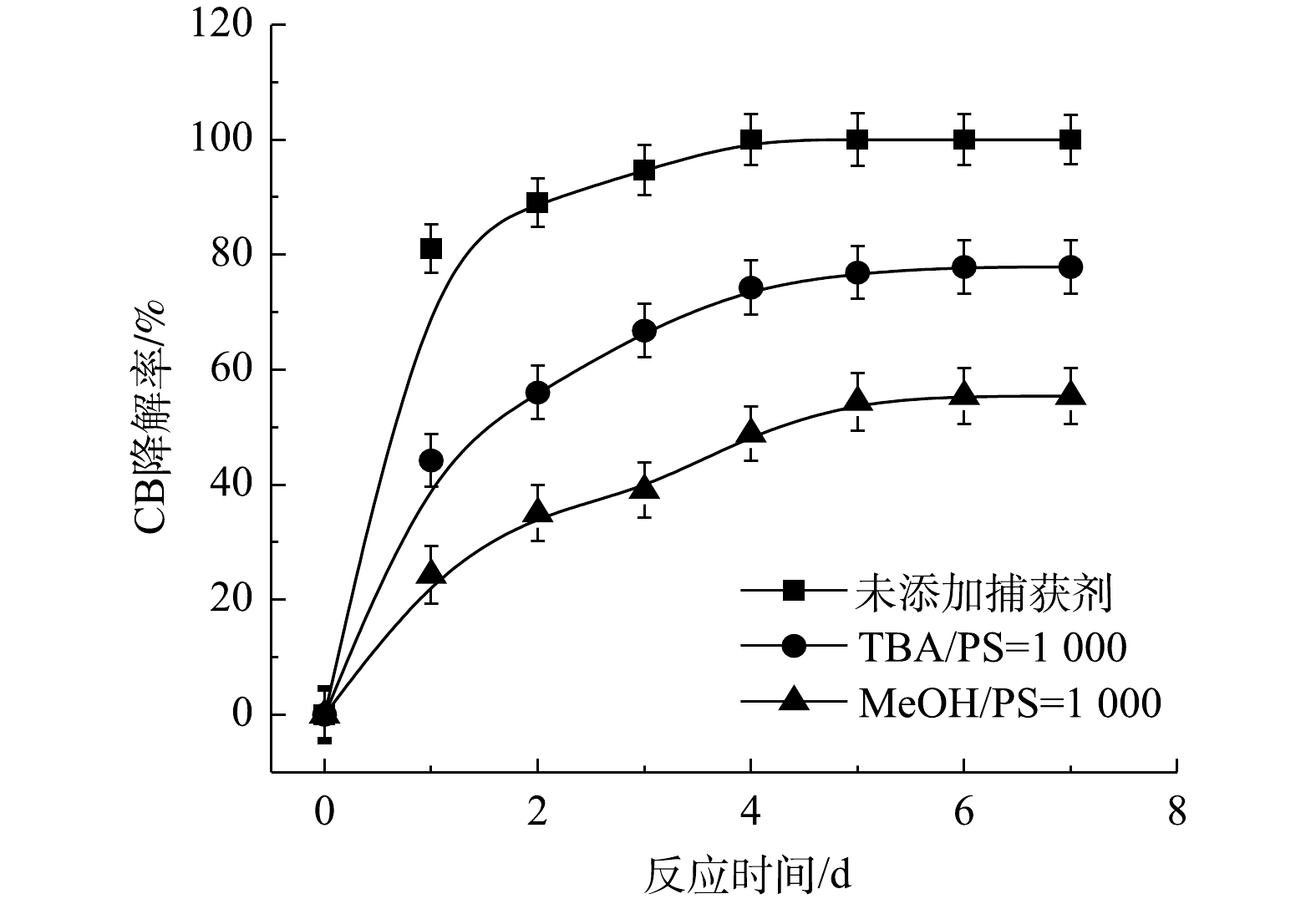
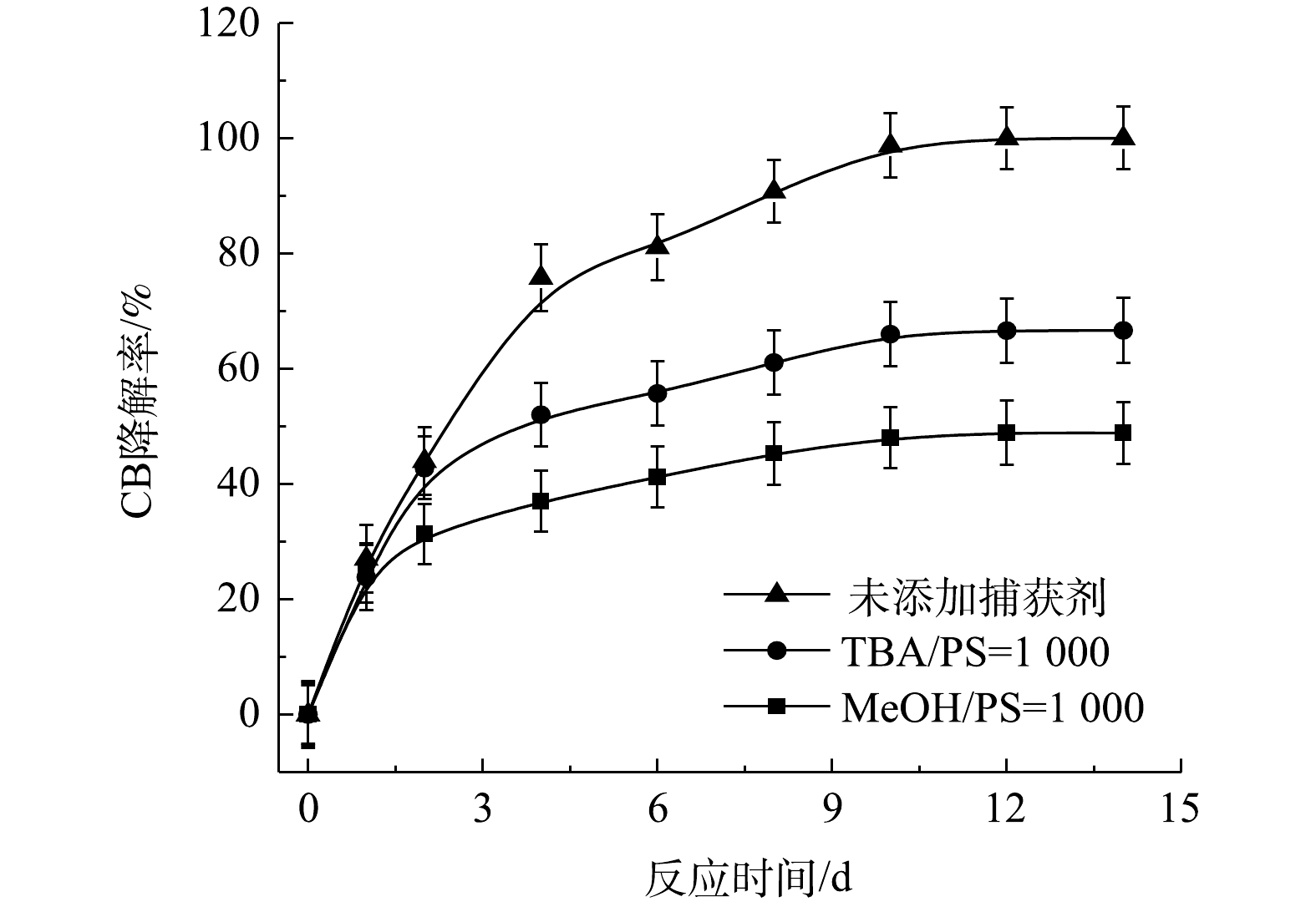
$ \cdot {\rm{SO}}_4^ - $ 共存[27-28](式(9)和式(10))。Fe2+/PS体系中PS与CB不同摩尔比下ηCB、ηTOC及ηCl−的结果如图7所示。由图7可知,Fe2+/PS对CB的矿化率始终大于ηCl−,且ηTOC和ηCl−均小于ηCB,这说明Fe2+/PS在降解CB的过程中生成较多的氯代中间体产物,这与2.2章节2)中Fe2+/PS在氧化CB的过程中生成较多的氯代酚类中间体的结论一致。3) NaOH/PS反应机理及CB降解机制。MeOH、TBA对NaOH/PS降解CB的影响如图8所示。TBA抑制了NaOH/PS氧化体系对CB的降解,ηCB由100%降低至66%(n[TBA/PS] = 1 000),这表明NaOH/PS体系中存在·OH。与TBA相比,MeOH对NaOH/PS降解CB的抑制作用显著,NaOH/PS体系对CB的降解率由100%降低至48%(n[MeOH/PS = 1 000)。这说明NaOH/PS体系中·OH和
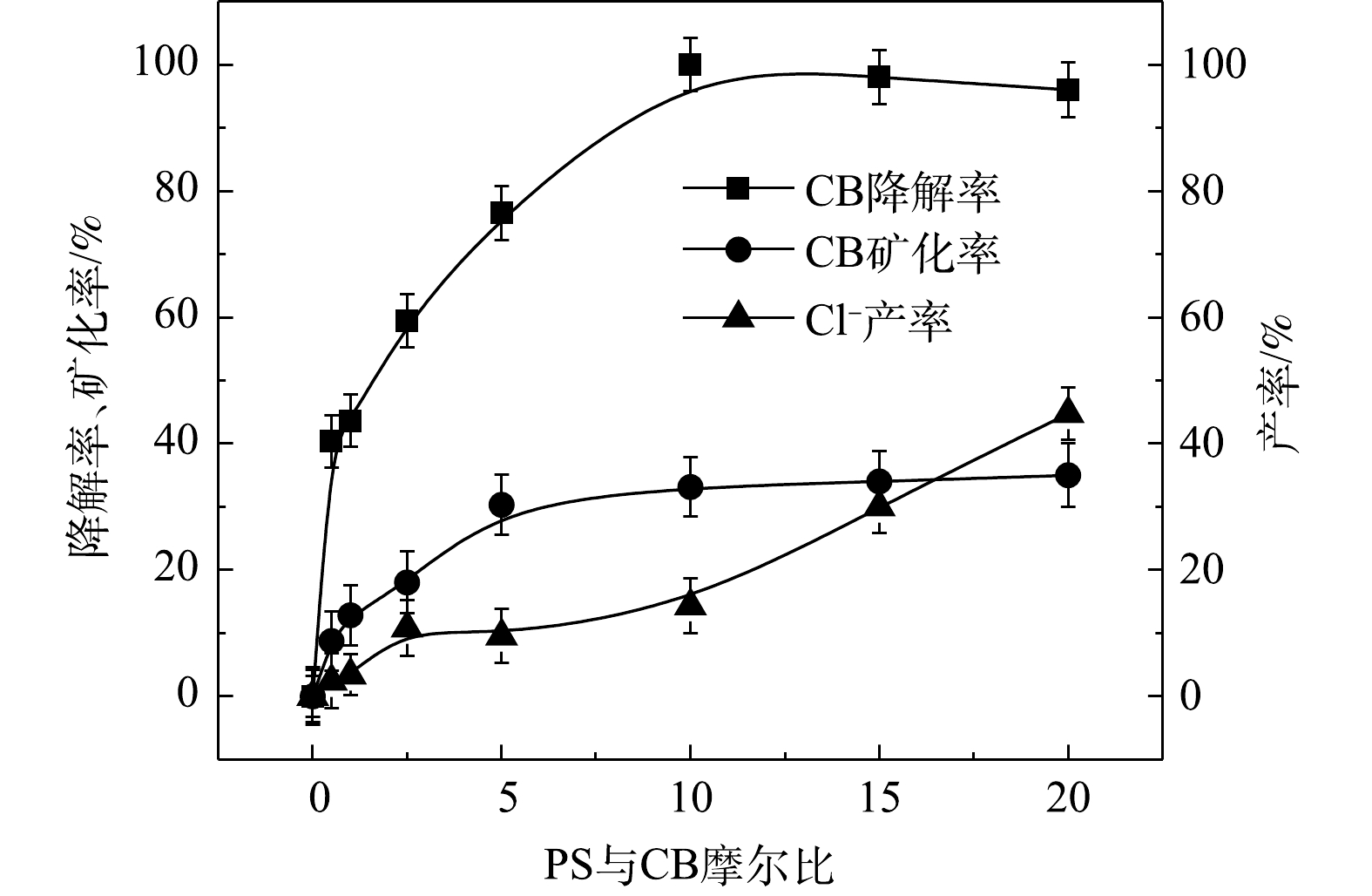
$ \cdot {\rm{SO}}_4^ - $ 共存(式(10)和式(11))[20,29-30]。NaOH/PS体系中PS与CB不同摩尔比下ηCB、ηTOC及ηCl−的结果如图9所示。由图9可知,与Fe2+/H2O2、Fe2+/PS体系均不相同,随着PS与CB摩尔比的增大,NaOH/PS体系对CB的矿化率先大于而后又小于ηCl−,这说明PS与CB摩尔比较小时,由于PS投加量较少时,NaOH/PS体系氧化能力不强,不能实现对氯代有机物的脱氯反应,而随着PS与CB摩尔比进一步增大,PS投加量增多,NaOH/PS体系中产生了大量的自由基活性物质,且在强碱性环境中,实现了氯代有机物的消除反应[7,10,29],从而使得ηCl−大于ηTOC。上述结果表明,相较于Fe2+/PS,Fe2+/H2O2和NaOH/PS更适于氯苯的降解,且不会生成大量的氯代酚类中间体。然而,在Fe2+/H2O2体系中的·OH是瞬间释放和消灭的[31]。因此,在实际的CB污染场地地下水修复中,Fe2+/H2O2对CB的氧化作用时间较短,实际修复效果较差。NaOH/PS释放自由基则相对缓慢,氧化作用持续时间长,有利于污染物的降解。在实际的CB污染场地地下水修复中,NaOH/PS具有更好地应用前景。
-
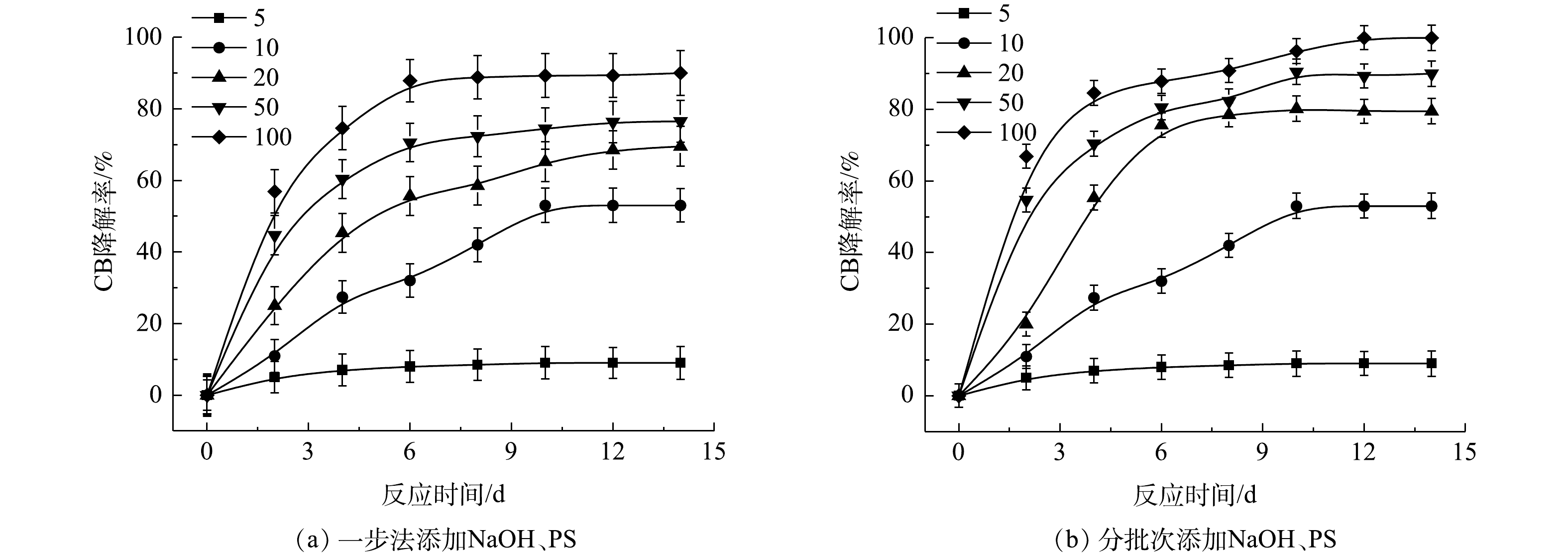
表2为污染场地地下水与实验CB溶液的参数比较。由表2可知,实际的污染场地地下水COD远高于实验CB溶液,这是由于其存在大量的其他有机物。在相同CB浓度下,为了达到与实验CB溶液相当的降解效果,需增加氧化剂PS的投加量。为此,以常州市某污染场地地下水为研究对象,考察PS投加量以及添加方式对NaOH/PS降解实际污染地下水中CB的影响,其结果如图10所示。由图10(a)可知,随着PS投加量的增大,ηCB增大,当CB与PS的摩尔比为100时,ηCB接近90%。而在相同反应条件下,将氧化药剂添加方式由一步法改为分批次添加,此时ηCB达到100%(图10(b))。这表明在活化剂与氧化剂使用量较大的情况下,采用分批次添加,逐级氧化的方式,可以实现CB污染物的高效降解。此外,上文2.2节1)中结论表明PS与CB的摩尔比超过10,NaOH/PS体系自由基的淬灭作用增强,导致降解效果变差。因此,采用分批次添加,逐级氧化的方式,可以减弱自由基的自淬灭作用,达到高效降解CB的目标。这为采用原位化学氧化法进行污染场地地下水修复提供了参考。此外,由上文讨论的结果可知,Fe2+/H2O2、Fe2+/PS体系在氧化剂不过量的条件下,氧化CB的过程中会生成毒性更强的氯代酚类中间体。与Fe2+/H2O2、Fe2+/PS不同,NaOH/PS不会出现此现象。在实际污染地下水修复中,氧化剂在地下水中释放缓慢,并且氧化剂在修复过程中扩散不均匀和自分解,致使地下水中的有效氧化剂量较小,在以Fe2+/H2O2、Fe2+/PS 2种氧化体系进行原位化学氧化修复过程中会生成毒性较强的中间体产物,产生二次污染,使得不能完全达到修复的目标。而采用NaOH/PS方式降解污染场地地下水中的CB,则可以避免次生污染物的产生,达到地下水修复效果评估的要求
2.1. 氯苯氧化的中间产物确认
2.2. 不同体系氧化降解CB
2.3. 反应机理和分解物的讨论
2.4. NaOH/PS降解实际污染场地地下水中的CB
-
1)当H2O2浓度为1 mmol·L−1、反应7 h时,Fe2+/H2O2对CB的降解率为98%,且在氧化过程中生成了4-CP,其平衡浓度为4.89 mg·L−1;当PS浓度为1.0 mmol·L−1、反应7 d时,Fe2+/PS对CB的降解率为86%,且在降解过程中会生成4-CP,其平衡浓度可高达56.04 mg·L−1。与Fe2+/H2O2、Fe2+/PS不同,NaOH/PS在氧化CB的过程始终不会生成4-CP。但当PS与CB的摩尔比超过10时,NaOH/PS体系存在自由基的淬灭作用。
2)游离氯离子和总有机碳的测定结果表明,与Fe2+/PS体系氧化过程不同,NaOH/PS、Fe2+/H2O2体系在氧化CB过程中会发生脱氯反应。
3)在实际的污染场地地下水修复中,宜采用NaOH/PS氧化体系,以分批次添加PS、逐级氧化的方式进行CB污染物的去除,可实现CB污染物的高效降解,同时可避免有害中间体4-CP的产生。
