



 下载:
下载:




-
随着人口增长和工业发展对水的需求量越来越大,水资源短缺是全球面临的最严峻的问题及挑战。污水回用、苦咸水及海水淡化是缓解水资源短缺的有效方法[1],其中的关键环节就是除盐。传统除盐方法主要包括蒸馏法、反渗透法、电渗析法、离子交换法等[2-5],但均存在能耗大、成本高、二次污染等问题[6-9]。 电吸附除盐技术是利用外接直流电源产生的电场,使在电极间流动溶液中的带电离子向带有与自身电荷相反的电极移动,从而被电极表面产生的双电层吸附达到净化原水效果[10-13]。相比于传统除盐方法,电吸附除盐主要优势为能耗小、寿命长、无二次污染和电极易再生[14-16]。
但是,电吸附除盐技术存在吸附-脱附效率不稳定,循环除盐吸附效率衰减及电极再生性差等问题,这极大影响电吸附除盐技术的应用前景。电吸附效率的衰减及电极再生性差的主要原因为电极脱附不完全、电极的炭颗粒被水流冲刷流失及电极发生电化学反应损耗等。
LIANG等[17]采用添加炭黑至流动电极中增强其导电性,发现随着电极电压增加,吸附效率有所提高,但电荷效率明显下降。杨宏艳等[18]的研究发现,当电压为1.4 V、流速为2.5 mL·min−1、进料室盐溶液流速与阴阳电极室中电极浆料流速比为1∶2∶2时,流动电极电吸附除盐率达到70.38%。莫恒亮等[19]采用流动电极与电渗析耦合实现连续脱盐,其中电渗析为间歇性工作,整套装置水效达到95%。
基于上述研究,为解决传统电吸附的吸附效率衰减及电极再生难等问题,本研究采用流动电极代替传统的固定电极,对流动电极电吸附除盐的效率及出水水质的稳定性进行了优化,分别考察了电压、流速等对除盐率的影响,并探讨了流动电极中盐分的浓缩度和电极的电吸附适用性,本研究可为流动电极电吸附技术的工程应用提供参考。
全文HTML
-
本研究中原水采用去离子水和化学试剂配制而成,化学试剂Na2SO4、Fe2(SO4)3及CaSO4,均为分析纯,来自于天津光复精细化工研究所;去离子水为实验室自制。配制的原水模拟实际工程中某脱硫废水,其主要的成分为Na2SO4、Fe3+及Ca2+等,其中Na2SO4浓度为(1 250±100) mg·L−1、Fe3+浓度为 4~9 mg·L−1、Ca2+浓度为(200±50) mg·L−1、pH为7±0.3。
-
流动电极采用炭材料、石墨粉和蒸馏水(Na2SO4溶液) 混合配置,混合比例为质量比,其中炭材料的比表面积为1 500 m2·g−1,孔径为1.22 nm,比电容量为150 F·g−1,灰分为0.3%。取200 mL溶液按比例添加炭材料和石墨粉,搅拌器下搅拌1 h混合均匀备用。
-
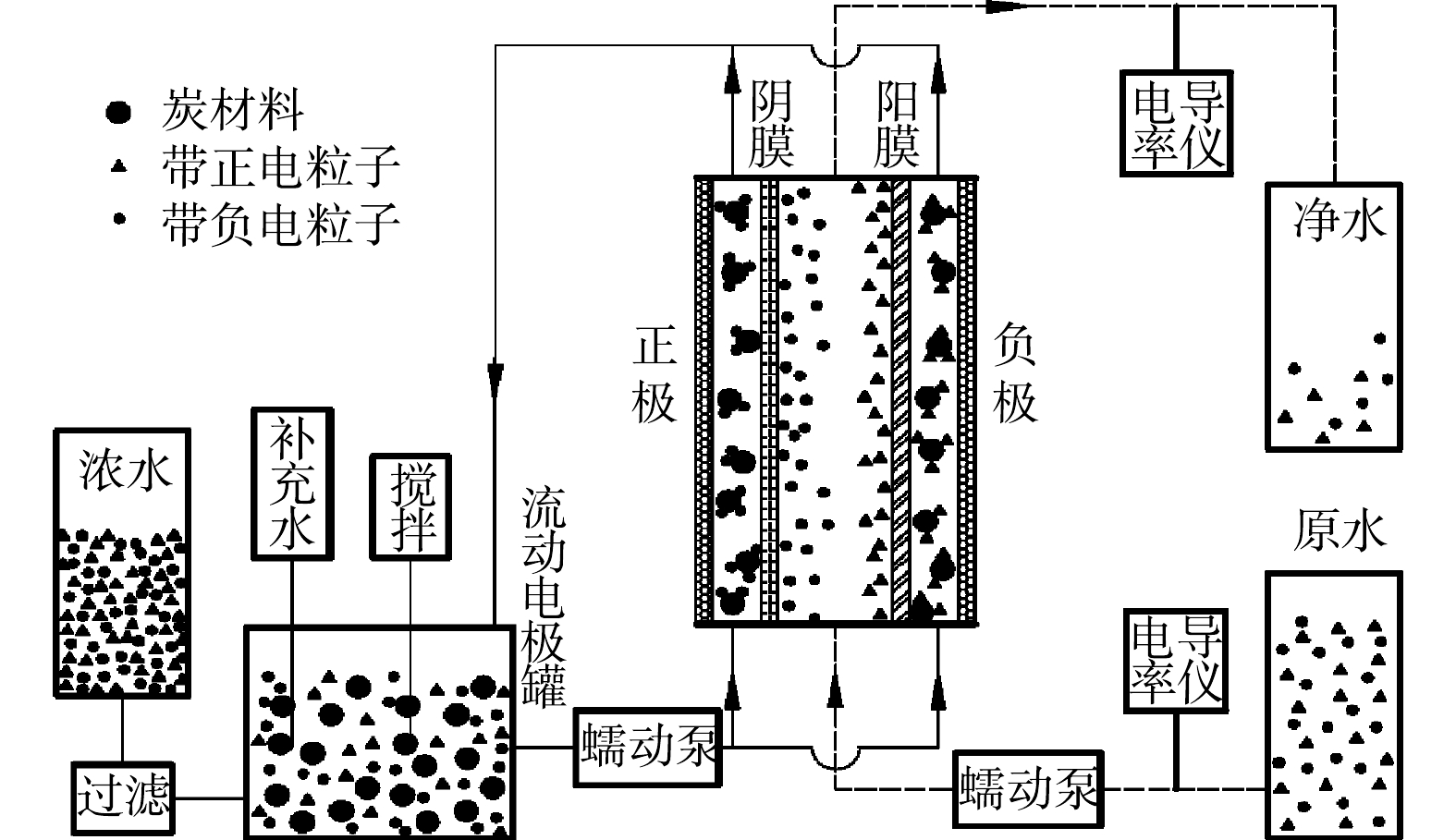
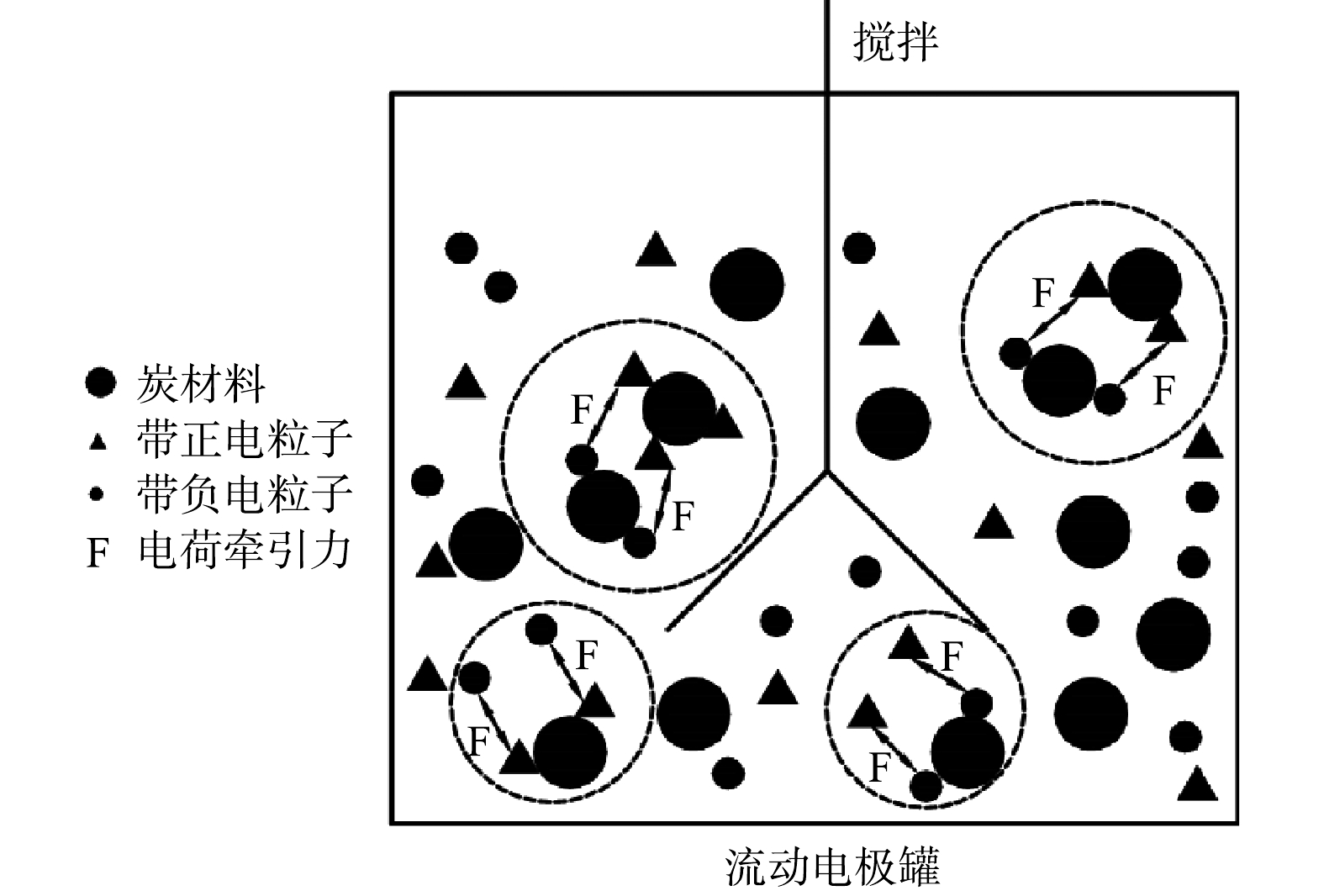
流动电极电吸附实验流程如图1所示。正负两电极分别贴附阴阳离子交换膜,在两电极间施加电压后,原水中带负电离子和带正电离子在电极电势驱动下分别透过阴阳离子交换膜,电吸附到流动电极中的多孔炭材料表面,随着流动电极循环带出,在流动电极罐中脱附,分别检测进水及出水溶液的电导率。流动电极中盐分过大会影响流动电极电吸附效率,故需要进行流动电极除盐,本研究采用过滤方法去除流动电极中的高盐,测试浓水电导率。
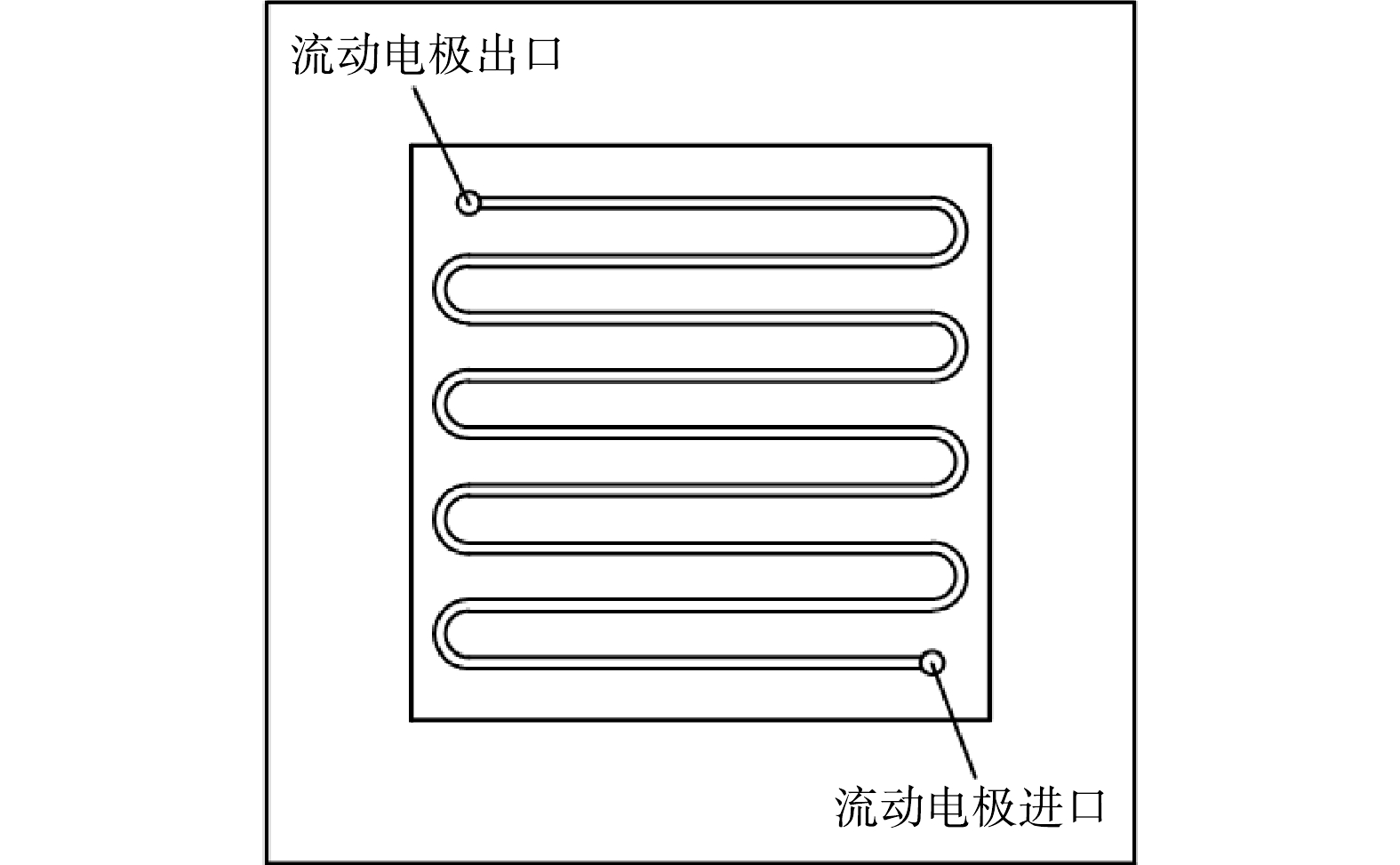
图1中正负电极选用不锈钢集流体,尺寸为125 mm×125 mm,在不锈钢上开2 mm×2 mm的流动电极流动槽,流动电极槽总长为1.1 m (图2)。原水通道由有机玻璃板形成,原水通道与流动电极槽一致,有机玻璃厚度为2 mm(阴膜和阳膜间距为2 mm)。
1.1. 原水及试剂
1.2. 流动电极的制备
1.3. 实验流程及方法
-
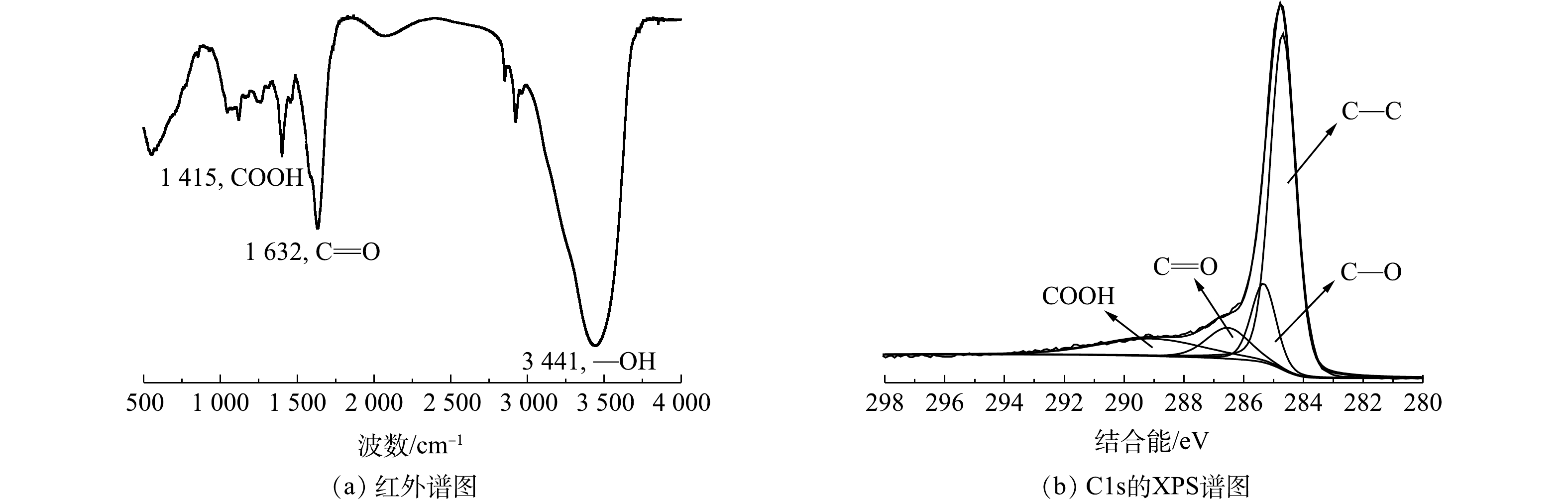
传统电吸附装置易出现吸附效率衰减,主要原因为吸附的离子未能及时完成有效脱附,尤其是重金属离子及结垢离子等,而此类离子易与炭材料表面基团发生化学反映,该类吸附比电吸附离子更难脱附,如活性炭吸附的重金属离子需要强酸才能达到有效脱附[20-21]。
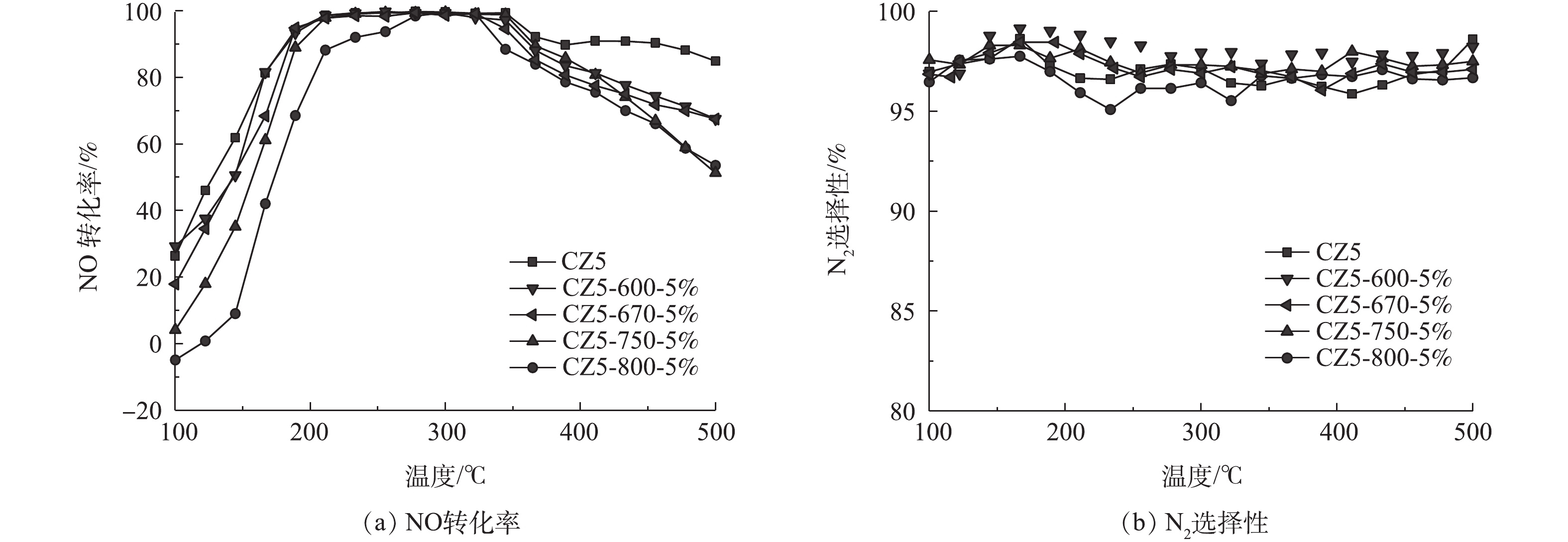
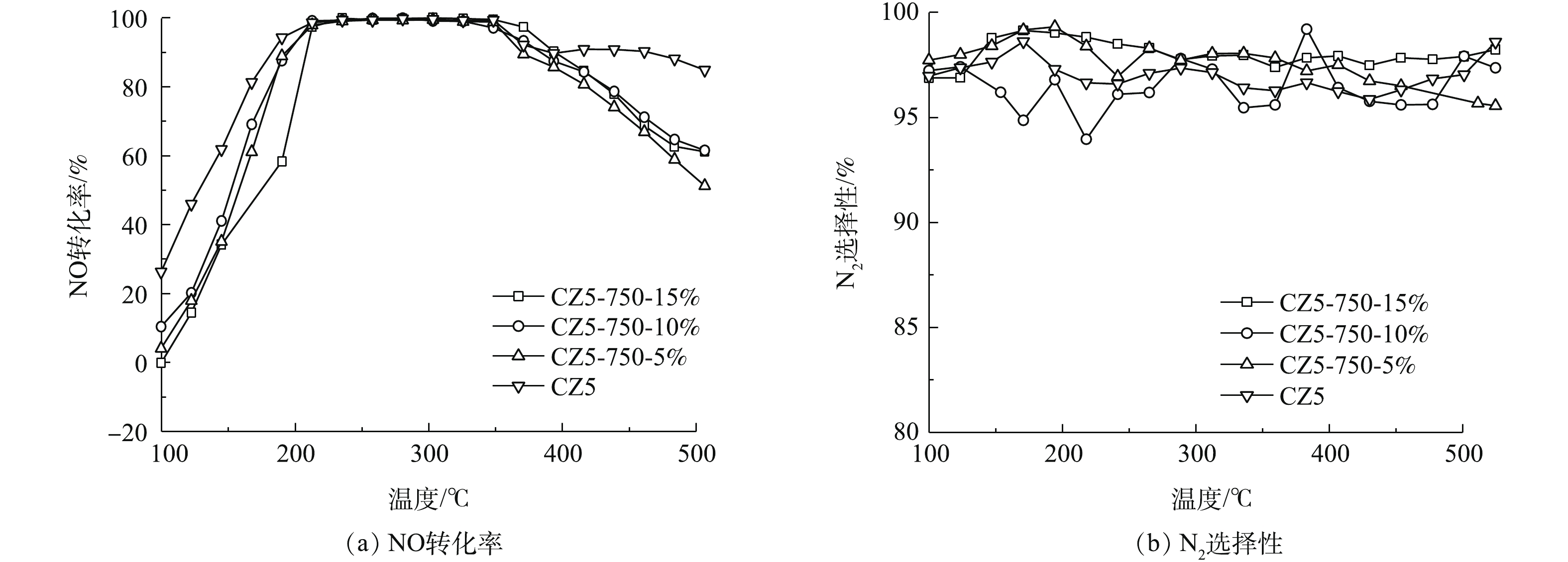
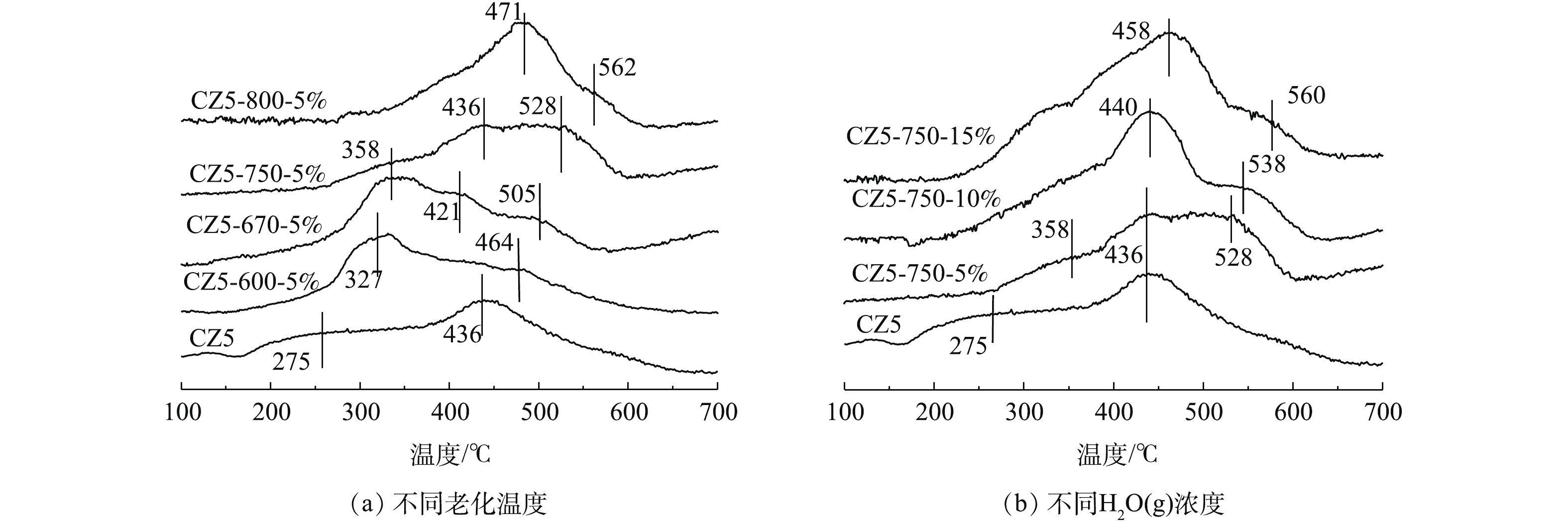
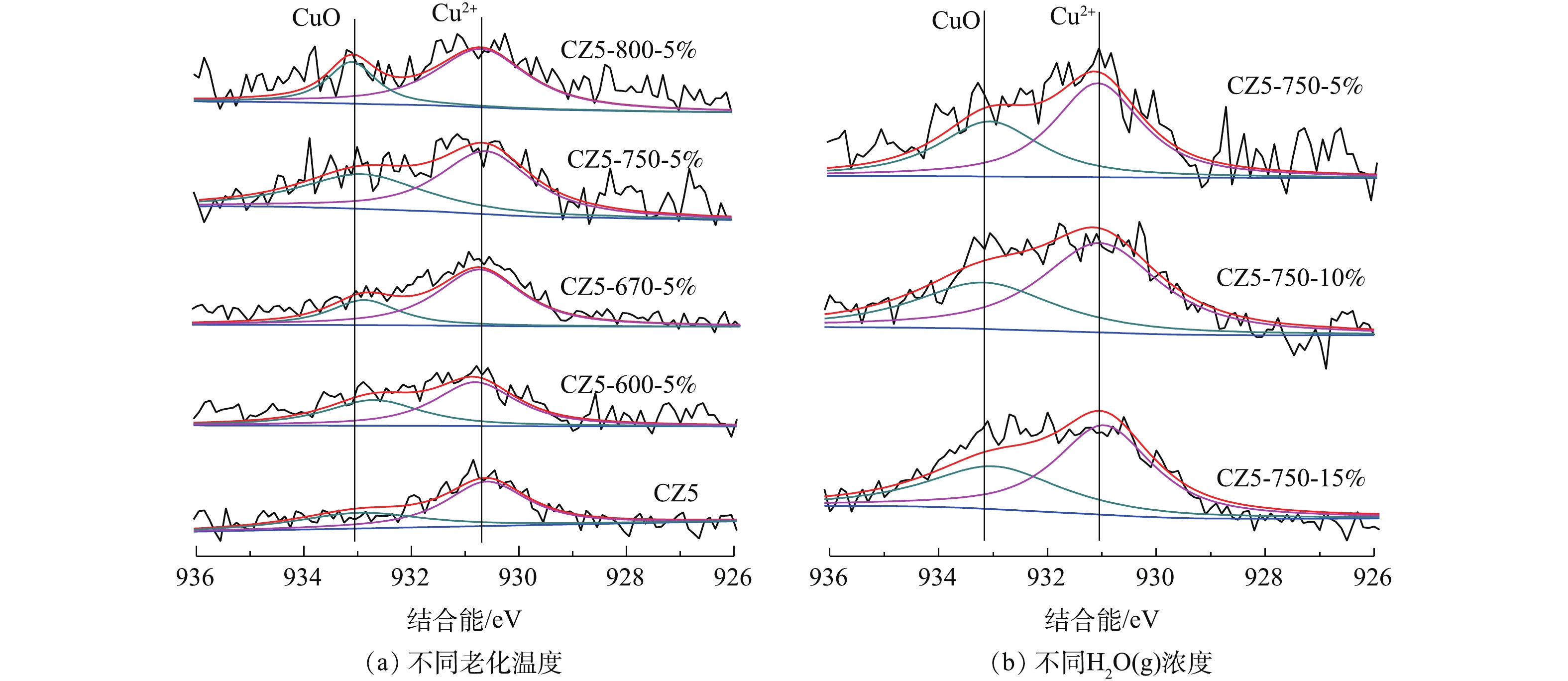
图3是对电极炭材料红外图谱及XPS分析。由图3可知,在波数1 415、1 632和3 441 cm−1处有明显的峰,分别对应的官能团为羧基(—COOH)、羰基(C=O) 及羟基(—OH)[22-23]。将活性炭中C1s的XPS谱图采用XPS Peak V4.1进行分峰处理[24-25],由图3可知,吸附材料活性炭表面的官能团同样主要以羧基、羰基及羟基为主。这类官能团易与重金属离子发生化学络合[26],且难脱附,本文采用流动电极电吸附法连续处理重金属离子Fe3+及结垢离子Ca2+,分析吸附-脱附循环。
-
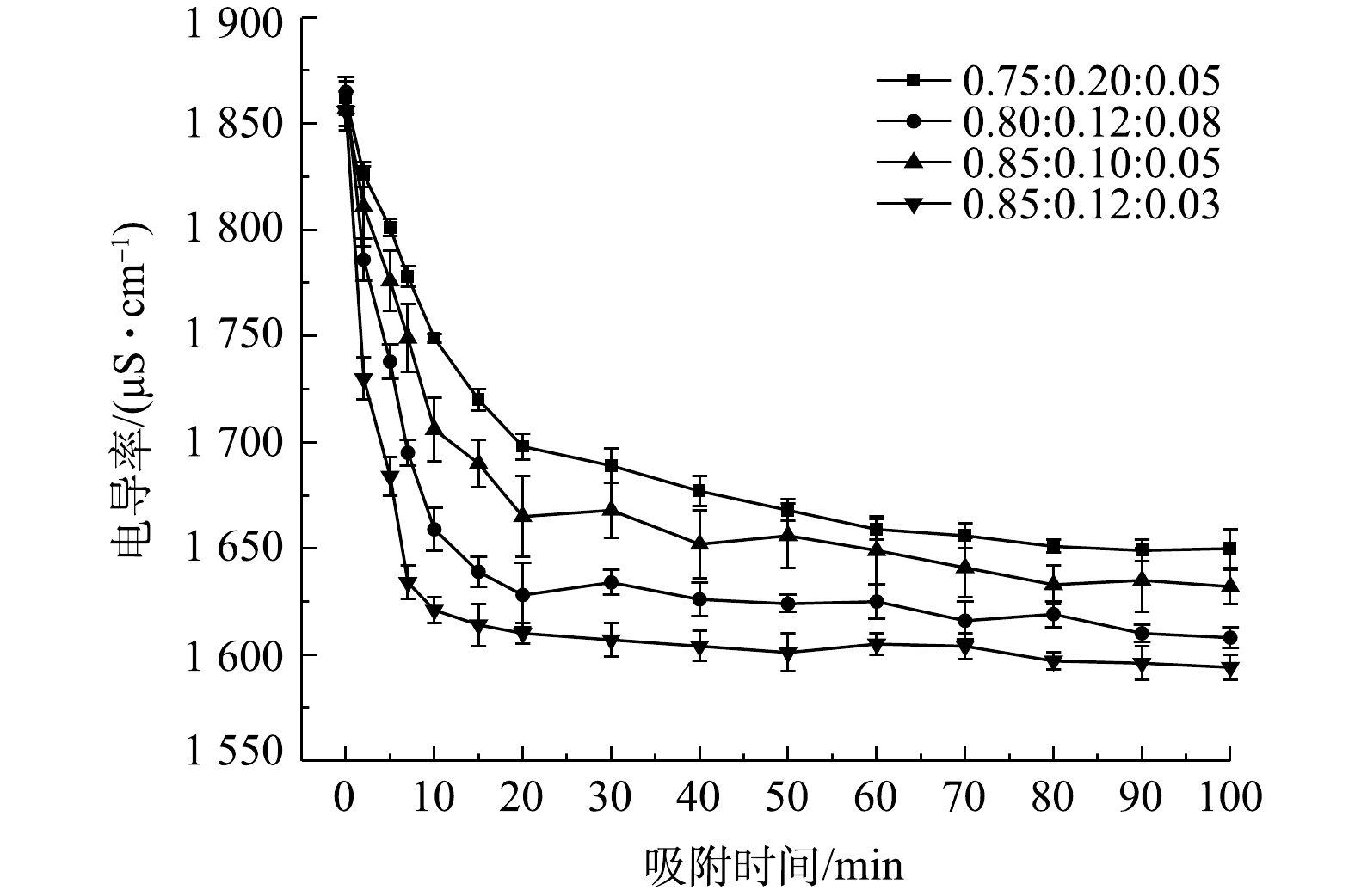
图4为溶液、炭材料及石墨粉的不同混合比例构成的流动电极电吸附Na2SO4。实验条件为初始电导率为(1 850±10) μS·cm−1、流动电极浆体为200 mL、Na2SO4溶液流速为5 mL·min−1、流动电极流速为10 mL·min−1、电压为1.5 V。由图4可知,当流动电极中固体比率较高时,电吸附效率较差。这是因为流动电极中固体率较高可导致流动电极的流动性变差,且影响流动电极浆体的分散性。而当流动电极固液比率恒定时,流动电极中石墨粉含量增加,导致炭材料比率降低,尽管流动电极浆体的导电性增加,但流动电极的比电容明显降低,从而导致吸附效率下降。因此,本研究选用的流动电极中溶液、炭材料及石墨粉的最佳混合比为0.85∶0.12∶0.03。
-
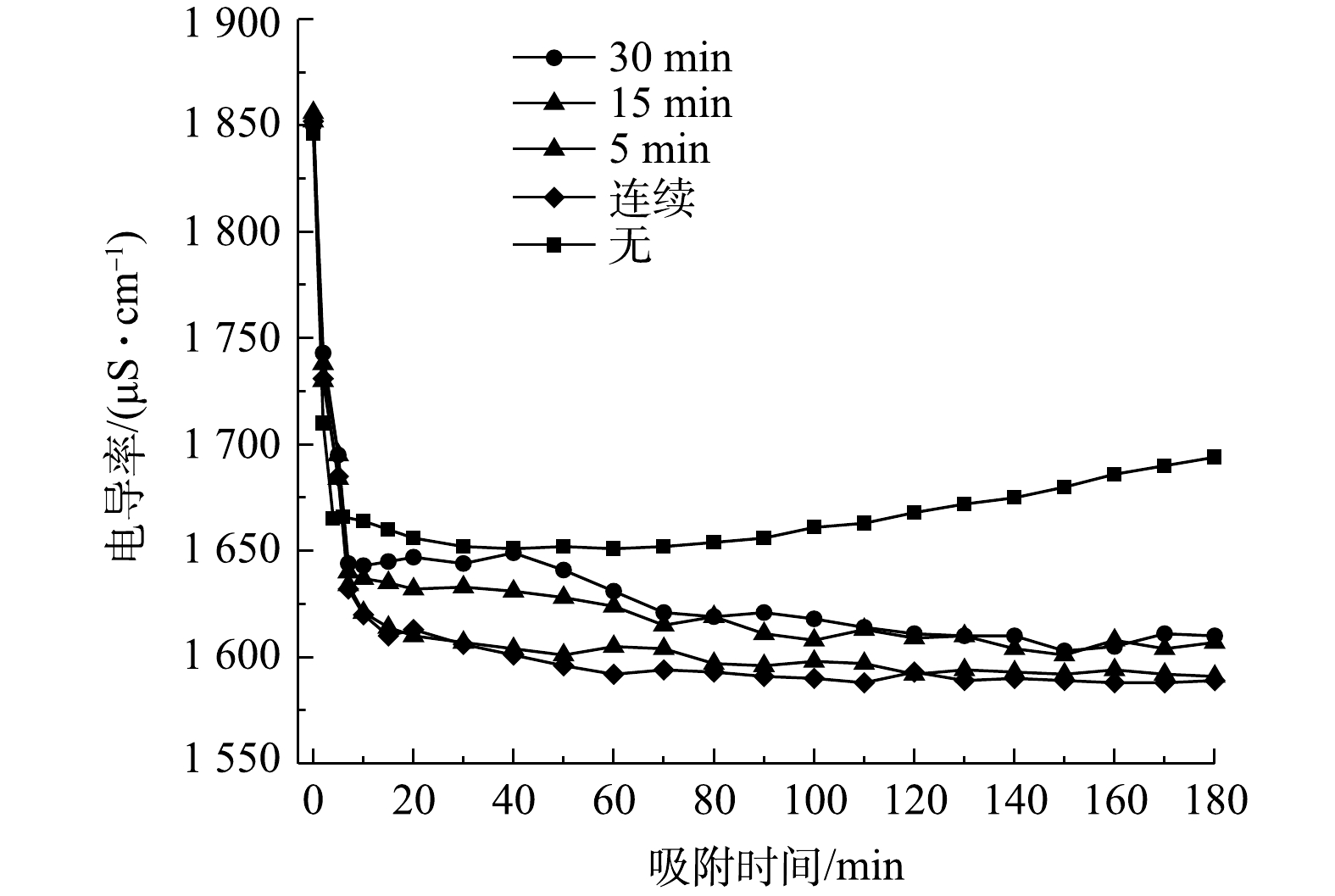
传统的电吸附除盐技术为净水和浓水交替出水,且净水出水水质波动较大[27]。流动电极电吸附实现了吸附和脱附分离,可优化出水水质的稳定性。但流动电极易出现固液不均匀,导致流动电极电阻增大,从而减弱电吸附效率。本研究采用流动电极搅拌解决流动电极分布不均匀及分层问题,流动电极搅拌用磁力搅拌器实现,每次搅拌设置为300 r·min−1,持续1 min。图5为流动电极的搅拌对电吸附Na2SO4溶液的影响,实验条件为初始电导率为(1 850±10) μS·cm−1,流动电极浆体为200 mL,Na2SO4溶液流速为5 mL·min−1,流动电极流速为10 mL·min−1,电压为1.5 V。由图5可知,流动电极的搅拌对电吸附Na2SO4出水电导率的稳定性影响较大,当流动电极未搅拌时,出水Na2SO4溶液的电导率越来越高,电吸附效率越来越低,同时会引发流动电极浆体堵塞通道。随着搅拌频率的降低,出水电导率的波动减小,且电吸附效率增加。流动电极的搅拌一方面保证流动电极浆体的均匀性,防止出现固液分层以及循环回流动电极罐的浆体未经充分脱附便再次循环回电吸附装置的现象;另一方面,流动电极的搅拌可促进炭材料中吸附的离子在流动电极罐中快速脱附,如图6所示,流动浆体电极搅拌过程中,吸附在炭材料表面的离子会受到溶液或炭材料中相反电荷离子的牵引而脱落。
分析流动电极搅拌后浓水电导率变化,当连续吸附10 min后连续搅拌得到的浓水电导率为1 765 μS·cm−1,而无搅拌得到浓水电导率仅为876 μS·cm−1;当连续吸附30 min后连续搅拌得到的浓水电导率达到5 963 μS·cm−1,而无搅拌得到浓水电导率仅为3 459 μS·cm−1。由此可知,经过搅拌后得到的浓水电导率比未搅拌更高,这说明在搅拌作用下,炭材料表面电吸附的离子脱附率更高。
-
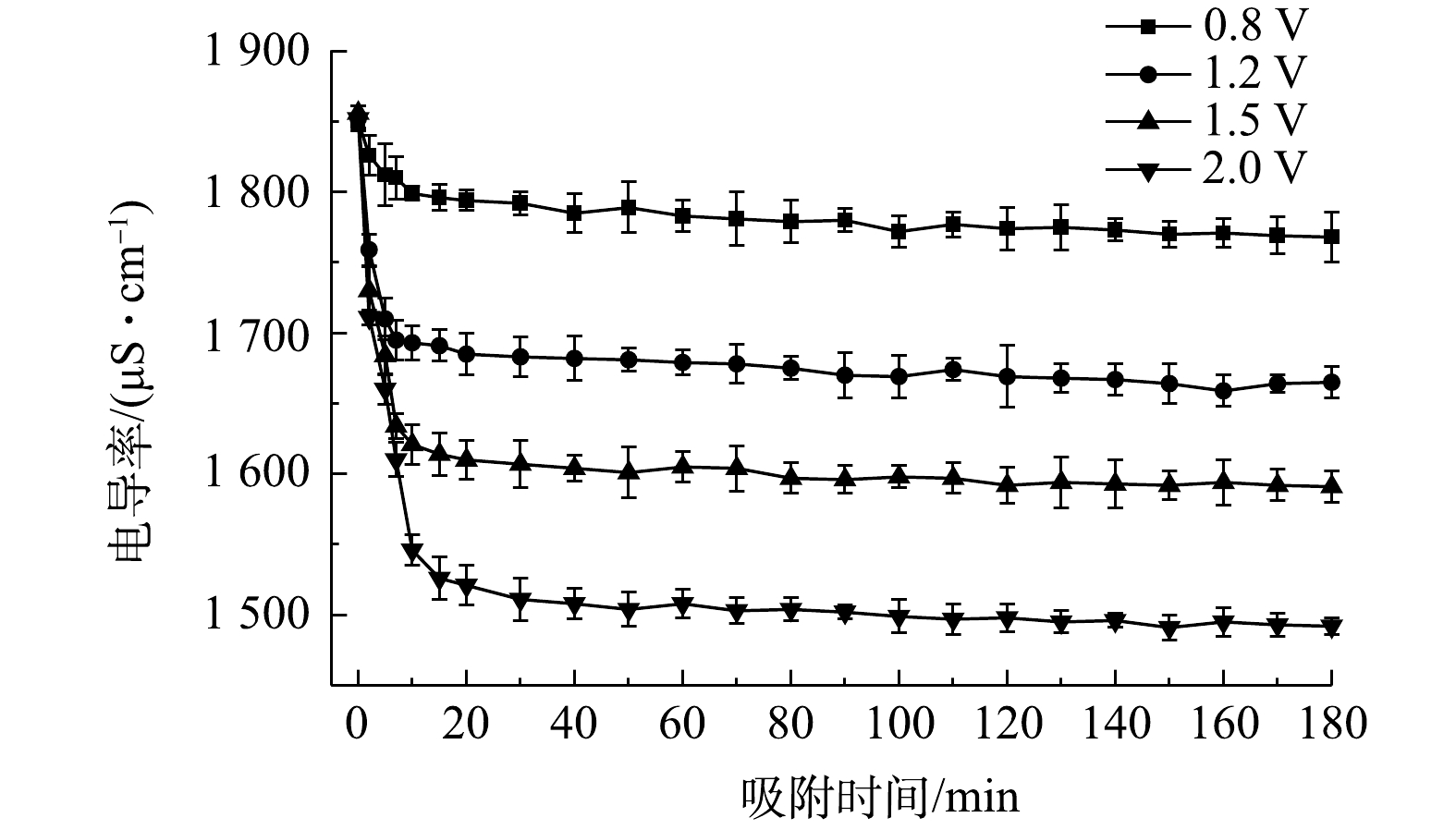
图7是电压对流动电极电吸附的Na2SO4溶液的影响,实验条件初始电导率为(1 850±10) μS·cm−1,流动电极浆体200 mL,Na2SO4溶液流速5 mL·min−1,流动电极流速10 mL·min−1,搅拌频率为5 min。如图7中所示,电压对电吸附Na2SO4溶液的处理效率影响很大,当电压为0.8、1.2、1.5和2 V时,电吸附Na2SO4的去除率为4.4%、10.1%、14.3%和19.4%。
提高电极电压一方面可增加离子的迁移速率,另一方面增加了流动电极的电容以及电吸附过程中双电层厚度,可吸附更多带电离子[28]。但随着电压的升高,会发生水解反应及电极氧化反应 (式(1)~式(3)),即损坏了电极材料,又降低了电荷效率,故电吸附电压不宜过高,本研究选取电压为1.5 V。
-
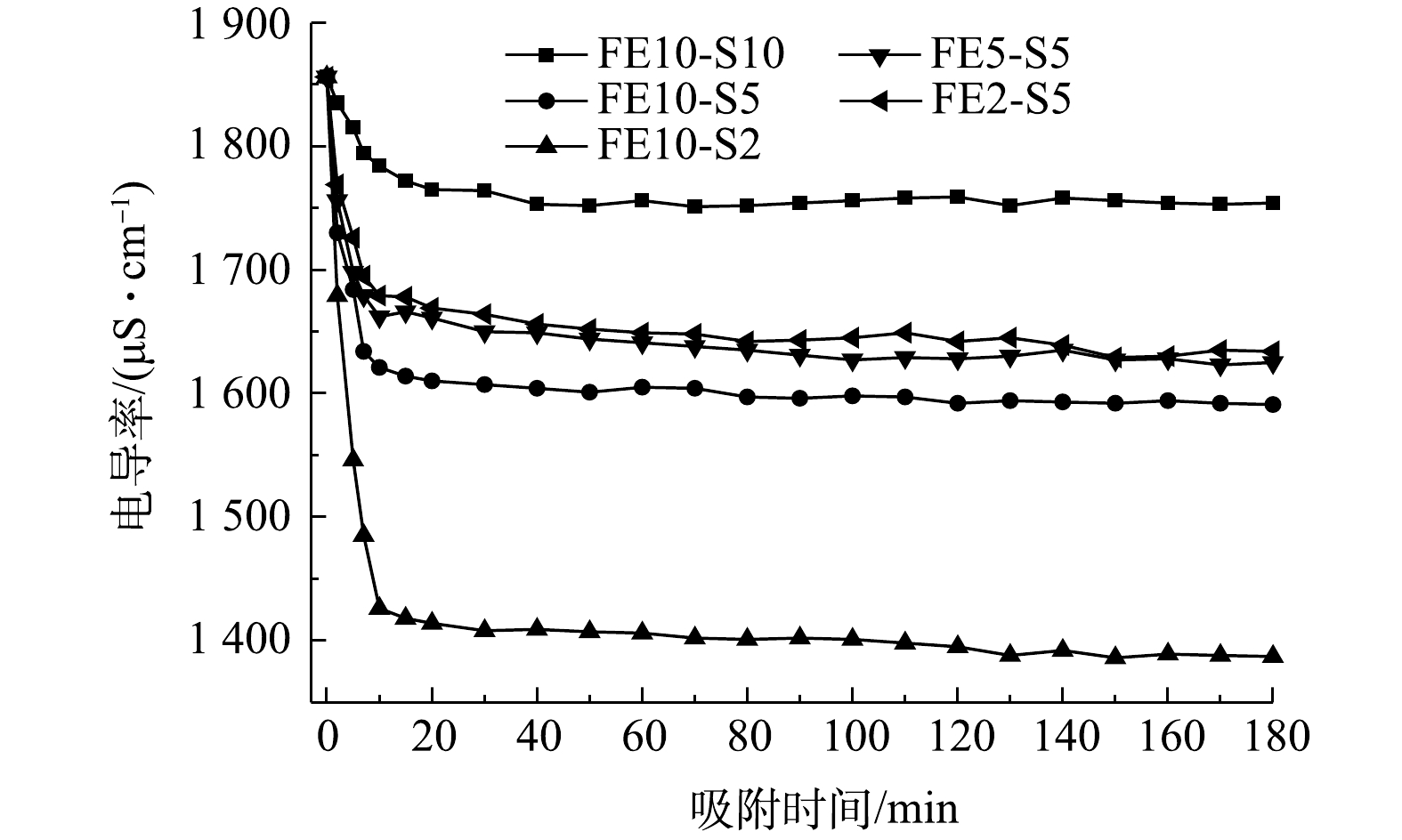
为了简便,将流动电极记作“FE”,Na2SO4溶液记作“S”,其中FE10-S5为流动电极流速为10 mL·min−1,Na2SO4溶液流速为5 mL·min−1。图8为Na2SO4溶液流速和流动电极流速对电吸附Na2SO4溶液的影响。由图8可知,Na2SO4溶液的流速对电吸附Na2SO4溶液的去除率影响较大,当Na2SO4溶液流速由5 mL·min−1降至2 mL·min−1,Na2SO4的去除率由14.3%增加至25.3%。Na2SO4溶液流速降低,溶液在吸附模块中停留时间增长,对应的吸附量增加,但随着流速降低,设备的处理能力会严重削弱。此外,由图8可知,流动电极的流速对电吸附Na2SO4溶液的影响较小,其中FE10-S5组分电吸附能力最大。当流动电极流速高于溶液速度时,可以保证流动电极不会因吸附饱和而降低在停留时间内的吸附率。但流动电极流速与溶液流速相差过大导致离子膜两侧压差大,不仅损害膜组件,而且增加水分子的迁移,因此,流动电极流速为10 mL·min−1和Na2SO4溶液流速为5 mL·min−1适宜。
-
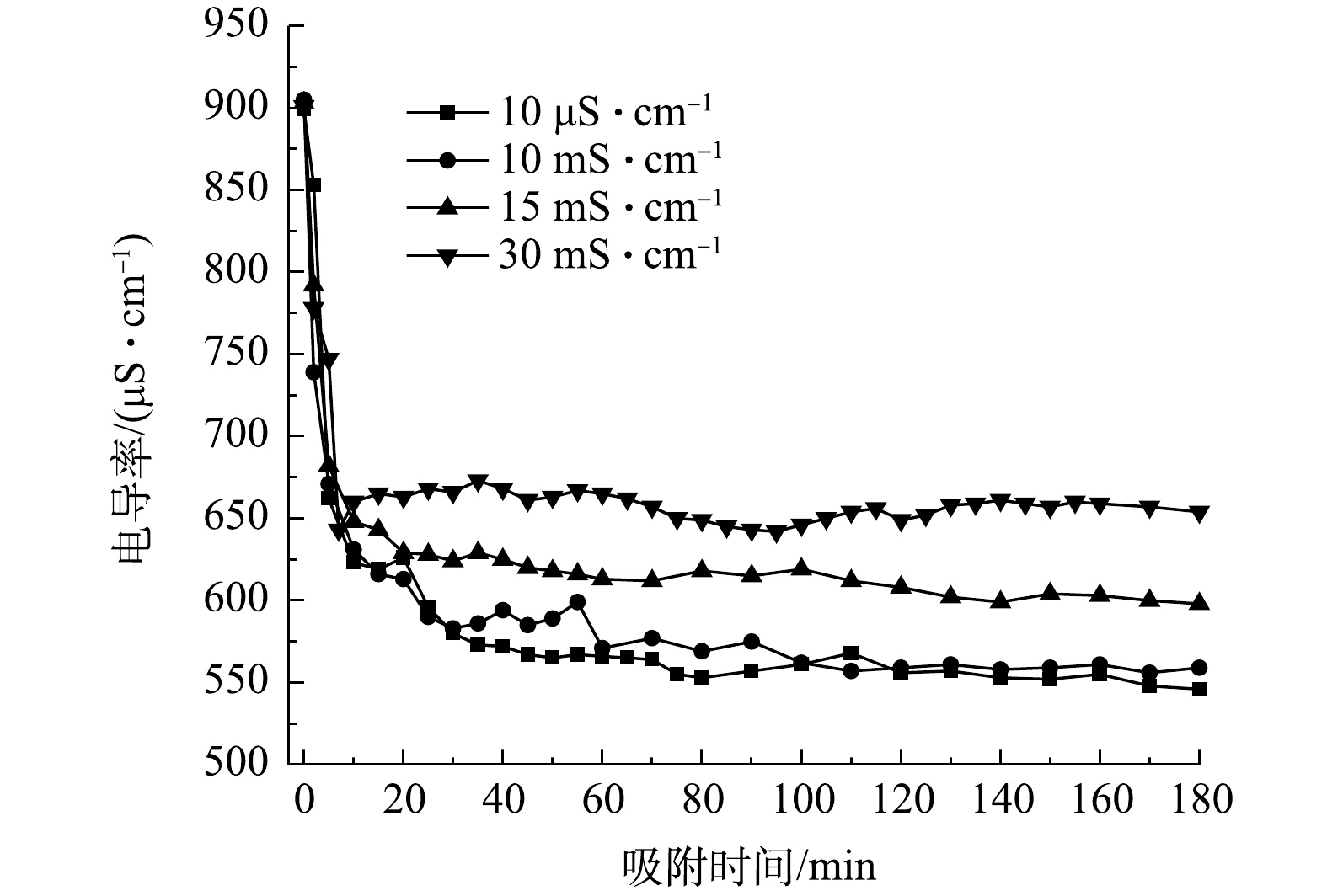
图9为不同流动电极电导率对电吸附影响,流动电极的初始电导率分别选取10 μS·cm−1蒸馏水、10、15和30 mS·cm−1 Na2SO4溶液,实验条件为流动电极浆体200 mL、Na2SO4溶液流速5 mL·min−1、流动电极流速10 mL·min−1、电压为1.5 V、搅拌间隔5 min。由图9可知,当流动电极中电导率由10 μS·cm−1升至10 mS·cm−1时,电吸附效率并无明显下降。由此可知,当流动电极电吸附处理Na2SO4溶液时,流动电极中吸附的盐分可浓缩11倍(相比于原水),可极大降低浓水量,从而增加得水率。当流动电极中盐分继续增加到15 mS·cm−1和30 mS·cm−1时,除盐率出现下降,太多的盐分存在流动电极溶液中会降低吸附在炭材料中离子的脱附率。同时,流动电极溶液中盐分浓度过大会发生浓差渗透现象,流动电极溶液中离子所受的浓差扩散力大于其电场力而反渗到原水中,进而降低了除盐率。
-
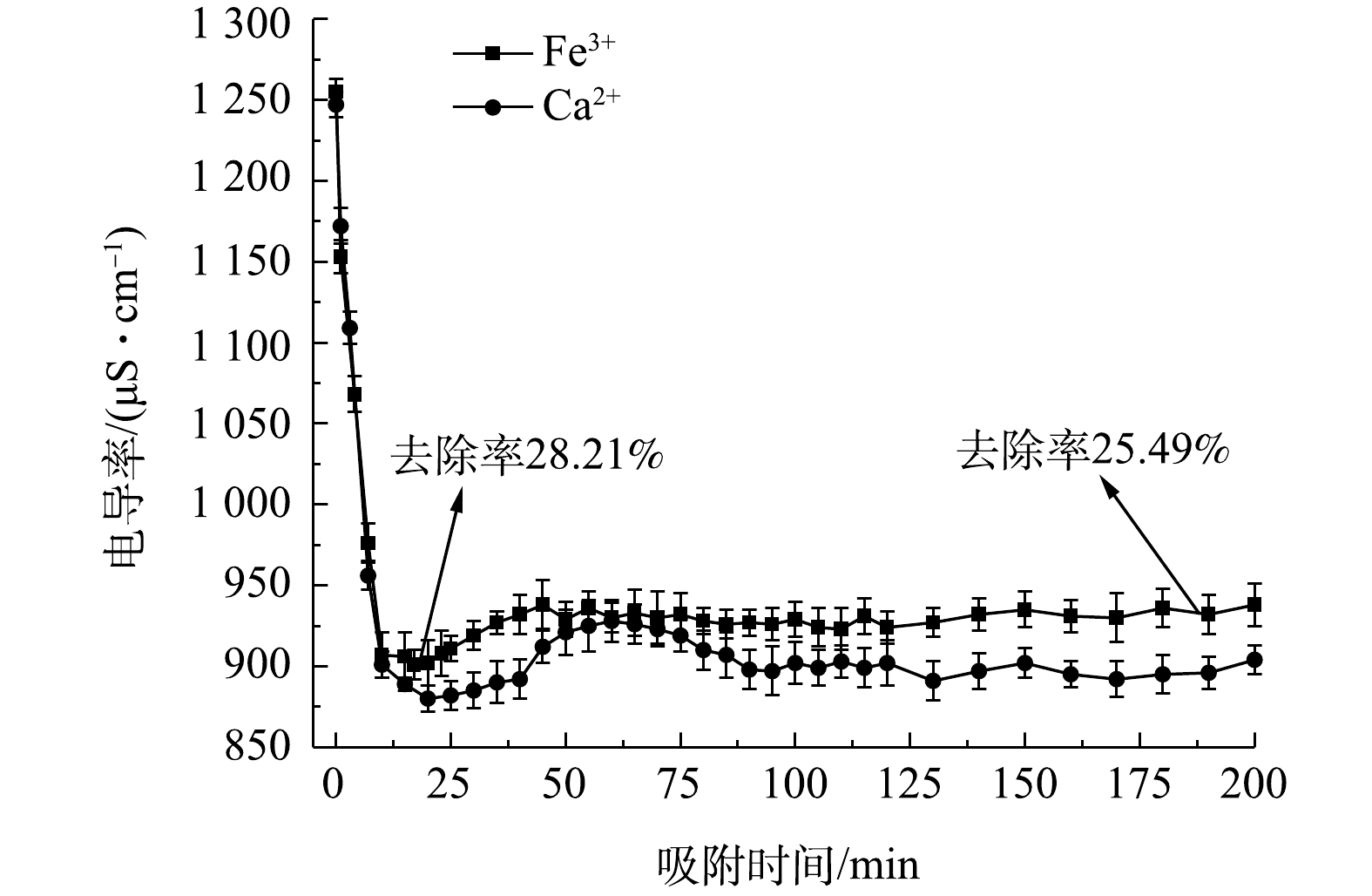
图10为流动电极电吸附处理Fe3+和结垢离子Ca2+,实验条件为流动电极浆体200 mL、原水溶液流速5 mL·min−1、流动电极流速10 mL·min−1、电压为1.5 V,搅拌间隔5 min。由图10可知,电吸附初期的出水电导率较低,电吸附效率较高,电吸附前期Fe3+的去除率达到28.21%。由FT-IR和XPS分析结果可知,炭材料表面以羧基、羰基及羟基基团为主,这类官能团易与重金属离子及结垢离子发生化学吸附[20-21, 26],即使添加搅拌辅助,仍难以有效脱附。由图10可知,在连续电吸附50 min后,流动电极吸附Fe3+和Ca2+趋于稳定,但去除率比前期低,其中Fe3+的去除率为25.49%。这是因为随着前期在炭材料表面化学吸附点位饱和后,流动电极后续吸附Fe3+和Ca2+以双电层吸附为主,电吸附在炭材料表面的各类离子会随着流动电极浆体的循环带出电吸附装置,进入流动电极罐,与传统固定电极电吸附相比,流动电极电吸附在炭材料表面的离子有充足的时间进行有效脱附。因此,后续流动电极吸附处理Fe3+和Ca2+时,出水电导率稳定,可实现稳定的吸附-脱附循环。
2.1. 流动电极电吸附处理金属离子及结垢离子
2.2. 流动电极浆体中溶液、炭材料及石墨粉混合比率对电吸附Na2SO4溶液的影响
2.3. 流动电极搅拌对电吸附Na2SO4溶液的影响
2.4. 电压对电吸附Na2SO4溶液的影响
2.5. 流动电极流速和原水流速对电吸附Na2SO4溶液的影响
2.6. 流动电极电吸附浓水的浓缩度分析
2.7. 流动电极电吸附处理Fe3+和Ca2+
-
1)流动电极浆体的搅拌可提高电吸附出水电导率的稳定性,减少电吸附效率的衰减。随着搅拌频率的降低,出水电导率的波动减小,且电吸附效率增加。
2)电压的增加可显著提高电吸附去除率,当电压为1.5 V时,电吸附Na2SO4溶液的去除率达到14.3%。
3) Na2SO4溶液的流速对电吸附Na2SO4溶液的去除率影响显著,当Na2SO4溶液流速由5 mL·min−1降至2 mL·min−1时,Na2SO4的去除率由14.3%增加至25.3%,但流动电极的流速影响相对较小。
4)流动电极中吸附的盐分浓缩度高,达到11倍以上,降低了电吸附过程中的浓水量。
5)使用流动电极电吸附技术处理Fe3+及Ca2+时,均可实现稳定的吸附-脱附循环。
