

 下载:
下载:




-
二氧化氯(ClO2)是一种黄绿色的气体,易溶于水,在水中的溶解度约为Cl2的5倍,是一种有效的绿色氧化剂,可用于饮用水的消毒和纸浆漂白等[1-5]。近年来,不少研究报道将ClO2应用于烟气脱硫脱硝。李广培等[6]进行了ClO2对NO和Hg的气相氧化性能研究,发现在ClO2/NO为0.8时,NO的氧化效率高达82%;潘理黎等[7]对ClO2液相氧化协同氨法烟气脱硫脱硝进行了研究,发现其脱硝效率可达93.2%;孙淑君等[8]采用鼓泡反应器进行液相ClO2脱除烟气中NOx的实验研究,发现相比于其他氧化剂(KMnO4、NaClO2、H2O2),ClO2在较低的质量浓度下对NO转化效率即可保持100%。上述研究表明,ClO2应用于烟气脱硝可取得较好的效果,但由于实际中ClO2性质较为活泼,不易运输与存储,通常采取现场制备方式,而在制备过程中又常伴随着副产物氯气的逸出[1],具有一定的环境风险,此外,目前的ClO2制备工艺也具有较高的经济成本。
关于ClO2的制备,在1843年就有研究提出,氯酸钾与盐酸反应可生成ClO2和Cl2的混合气体,其原理为氯酸盐在酸性条件下被还原剂还原而生成ClO2[1],此后便陆续出现了以NaClO2[9]、NaClO3[10-11]为原料制备ClO2的方法。目前主流的方法是以NaClO3为原料,与还原剂在酸性条件下生成ClO2。使用NaClO3还原法制备ClO2,因使用的还原剂不同,此法又分为很多种,如HCl法、SO2法、NaCl法、Na2SO3、CH3OH法以及H2O2法[12-13]等。这些方法都各有优势,但同时也存在一定的弊端。HCl法、NaCl法在制备过程中,Cl−被氧化后,会有大量Cl2逸出,造成二次污染[1, 14];CH3OH法的副产物甲醛、甲酸会给工厂带来二次水污染[15]的问题;H2O2法中H2O2不稳定易分解且价格昂贵;SO2法虽然能避免Cl2逸出和二次水污染,但其具有制备过程繁杂、原料购置成本高等缺点,使其在工程应用中受到限制。因此,寻求一种绿色经济的ClO2制备方法将是ClO2应用于烟气脱硫脱硝领域的关键。
氨法脱硫使用氨水吸收烟气中的SO2气体,先进行酸碱中和反应,即氨和SO2反应生成(NH4)2SO3,因具有脱硫速度快、效率高、能耗低等优点而广受企业欢迎。但由于氨法脱硫在脱硫过程中生成的(NH4)2SO3性能不稳定,会重新分解为SO2,造成二次污染,因此,将(NH4)2SO3氧化为更稳定的(NH4)2SO4,使其能够直接用作氮肥以实现资源化利用,是目前氨法脱硫中的研究热点。
本研究综合考虑ClO2的制备原理,在Na2SO3法和SO2法制备ClO2的基础上,探索将氨法脱硫的产物(NH4)2SO3应用于脱硝剂ClO2制备的可行性和影响因素,考察H2SO4浓度、(NH4)2SO3浓度、反应温度及反应时间对(NH4)2SO3法制备脱硝中氧化剂ClO2的影响;在研究过程中,将氨法脱硫产物(NH4)2SO3用于ClO2烟气氧化脱硝中氧化剂ClO2的制备,为后续烟气脱硝的脱硝剂ClO2的制备提供廉价还原剂,降低了ClO2制备过程的成本,同时也解决了氨法脱硫中(NH4)2SO3的氧化问题,为氨法脱硫和ClO2氧化脱硝工艺的组合运行提供了参考。
全文HTML
-
试剂包括氯酸钠(AR)、亚硫酸铵(AR)、硫酸(98%)、碘化钾(AR)、硫代硫酸钠(AR)、淀粉(5%)、盐酸(37%)、pH=7的磷酸盐缓冲液;仪器包括四口烧瓶(500 mL)、恒温加热磁力搅拌器(DF-101S)、恒温低温槽(DC-1006)、抽气泵(压力为−68~−92 kPa)。
实验装置如图1所示。实验系统由反应发生装置、ClO2吸收装置及末端气体吸收装置构成。反应在四口烧瓶中进行,通过恒温加热磁力搅拌器来控制反应温度,H2SO4经分液漏斗缓慢滴加入反应器中[16-17];制得的ClO2气体用冷藏的−5~0 ℃去离子水吸收,尾气用10%的NaOH吸收。系统末端采用气泵抽气,使得系统处于负压状态(−80~−72 kPa),从而促进ClO2气体的吸收,同时防止反应中气体的逸出而影响实验结果。
-
在实验中,反应发生器为500 mL的四口烧瓶,发生器中反应原液的体积为50 mL(其中NaClO3 和(NH4)2SO3混合液25 mL, H2SO4 溶液25 mL)。ClO2吸收液及末端尾气吸收液的体积均为500 mL,反应温度为50 ℃,反应时间为30 min,抽气泵的压力为−68~−92 kPa。反应的理论方程式[12]见式(1)和式(2),总反应见式(3)。
实验在固定其他因素不变的情况下,分别考察H2SO4浓度、反应温度、(NH4)2SO3浓度、反应时间对制备ClO2的影响,具体工况如表1所示。
通过五步碘量法[18]测定ClO2吸收液中ClO2及Cl2的浓度,ClO2的纯度和收率由式(4)和式(5)计算。
式中:
μ 为ClO2的纯度;CClO2 为吸收液中ClO2的浓度,mg·L−1;CCl2 为吸收液中Cl2的浓度,mg·L−1;μ1 为 ClO2的收率;mClO2实际 为实验条件下实际ClO2的产量,mg·L−1;mClO2理论 为实验条件下理论上ClO2的产量,mg·L−1。
1.1. 试剂、仪器及装置
1.2. 实验及分析方法
-
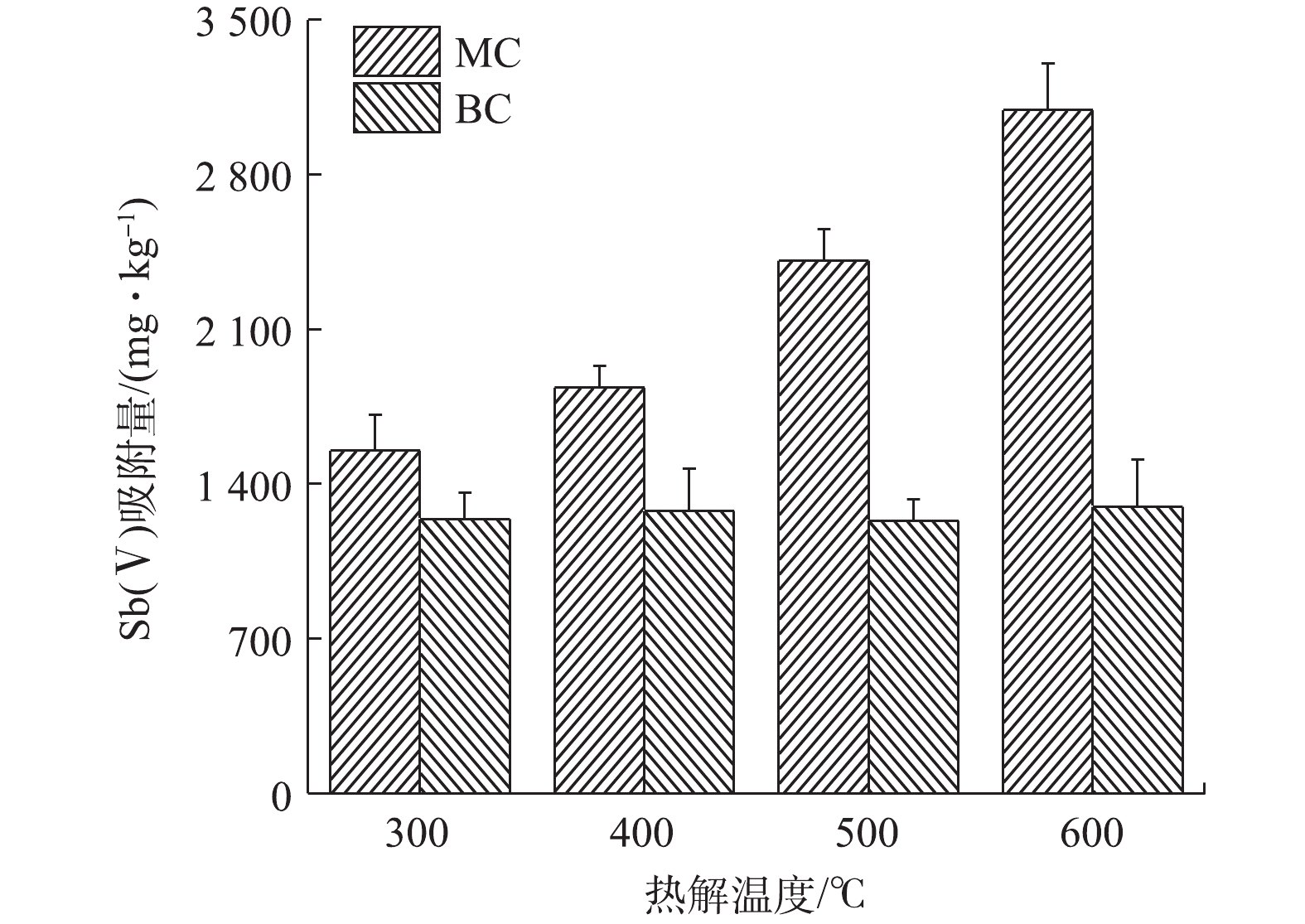
实验固定NaClO3浓度为2 mol·L−1,(NH4)2SO3浓度为1.25 mol·L−1,反应温度为50 ℃,分别在H2SO4浓度为6、7、8、9、10、11、12 mol·L−1的条件下,考察了H2SO4浓度对ClO2制备的影响,结果如图2所示。由图2(a)可看出,ClO2吸收液中的ClO2浓度随着硫酸浓度的增加呈现先上升后下降的趋势,其中硫酸浓度在10 mol·L−1时的效果最好,制备的ClO2浓度最高可达1 336 mg·L−1。此外,各种硫酸浓度下吸收液中的ClO2浓度均随反应时间呈逐渐上升的趋势,大部分在30 min时浓度最高。硫酸浓度为8 mol·L−1时,在反应时间由5 min延长至30 min时,ClO2吸收液中ClO2浓度由74 mg·L−1上升至1187 mg·L−1,升高了93.77%。
由图2(b)可以看出,ClO2的收率及纯度均随着H2SO4浓度的增加呈先上升后下降的趋势。H2SO4浓度由6 mol·L−1增至9 mol·L−1时,吸收液中ClO2的收率由3.3%增至19.9%,纯度由85.09%增至96.4%,说明硫酸对此反应的影响很大,这与DESHWAL等[1]的研究结果一致。但H2SO4浓度大于9 mol·L−1后,ClO2的收率和纯度便开始下降。这可能是因为:随着H2SO4投加量的逐渐增大,产生的SO2逐渐增多,继而产生的ClO2逐渐增多;而当H2SO4投加量继续增大时,(NH4)2SO3与H2SO4反应速度加快,产生大量SO2,SO2一部分未来得及参与NaClO3的液相反应而从液相中逃逸,另一部分则与ClO2发生副反应,生成H2SO4和HCl[19-21],其反应方程见式(6)。
-
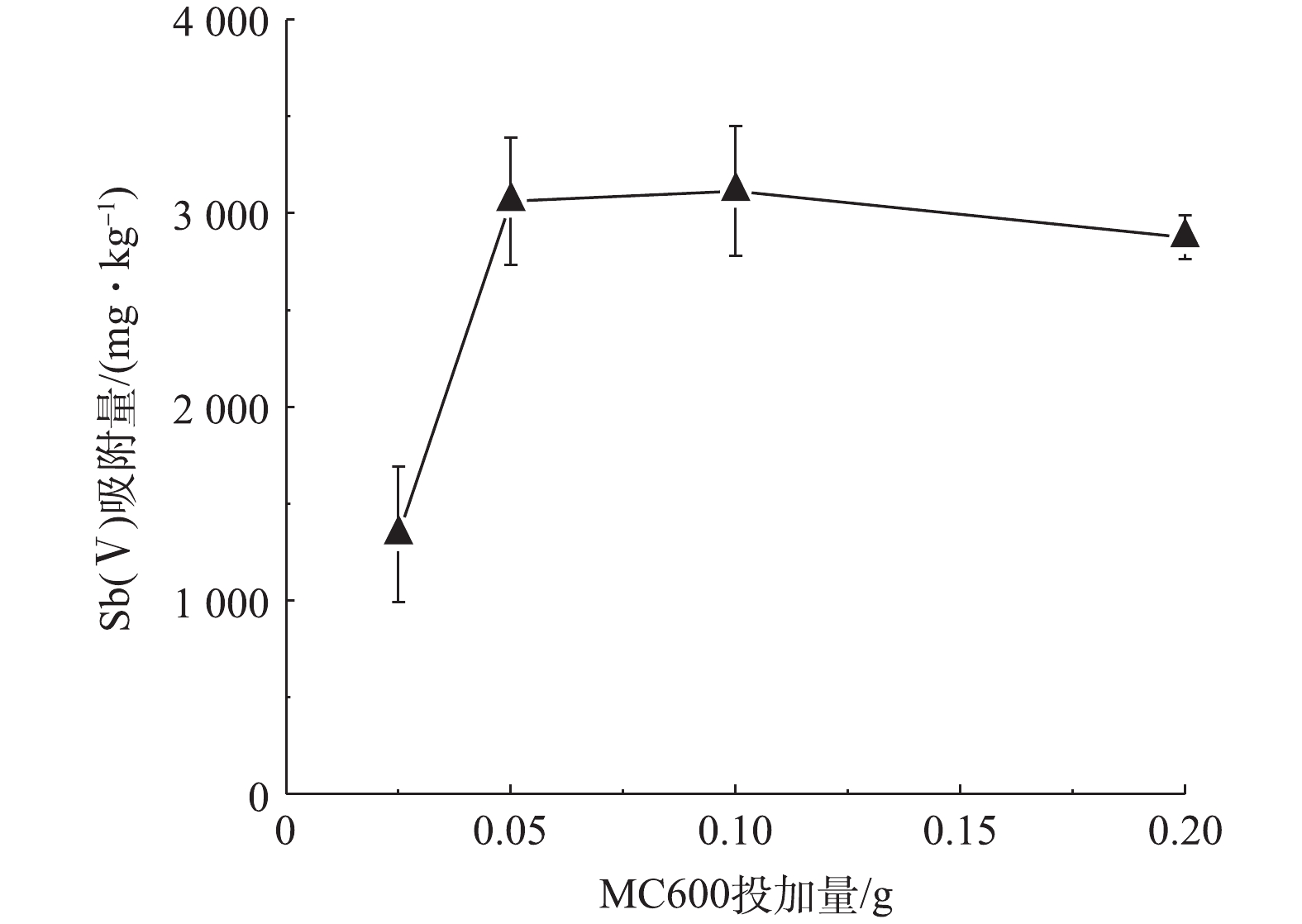
在实验中,设置NaClO3浓度为2 mol·L−1,(NH4)2SO3浓度为1.25 mol·L−1,H2SO4浓度为9 mol·L−1,分别在反应温度为10、20、30、40、50 ℃的条件下,考察了反应温度对ClO2制备的影响,结果如图3所示。
由图3(a)可看出,反应温度由10 ℃增至40 ℃时,吸收液中的ClO2浓度由1 262 mg·L−1降至965 mg·L−1,而温度继续增至50 ℃时,吸收液中的ClO2浓度会再次呈现升高趋势。由图3(b)可看出,随反应温度的升高,ClO2收率呈现逐渐下降的趋势。当温度由10 ℃升高至50 ℃时,ClO2的收率则由18.8 %下降至14.4 %,下降了4.4 %。这可能是由于反应温度过高,反应比较剧烈,瞬时产生的ClO2量过大,ClO2气体来不及进入后续吸收装置,从而导致分解[22]。
-
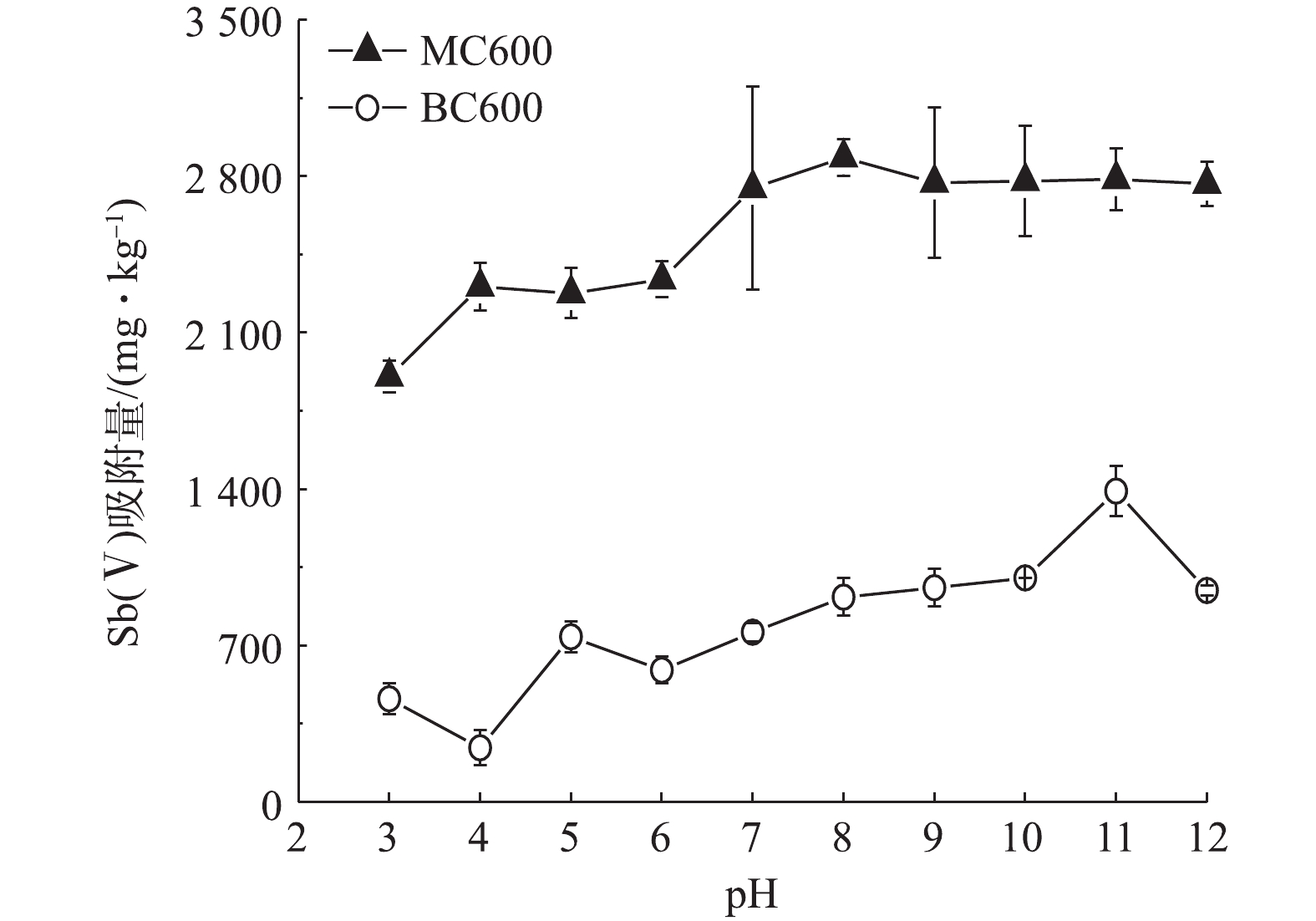
在实验中,设置NaClO3浓度为2 mol·L−1,H2SO4浓度为9 mol·L−1,反应温度为50 ℃,分别在(NH4)2SO3浓度为0.5、0.75、1.0、1.25 mol·L−1的条件下,考察了(NH4)2SO3浓度对ClO2制备的影响,结果如图4所示。
由图4(a)可看出,随着反应器中(NH4)2SO3浓度的不断增大,吸收液中的ClO2浓度越来越高,反应器中(NH4)2SO3的浓度由0.5 mol·L−1增至1.25 mol·L−1时,吸收液中ClO2的浓度由519.38 mg·L−1上升至1 336 mg·L−1,升高了61%。由图4(a)中的数据趋势可以看出,(NH4)2SO3浓度由0.75 mol·L−1增至1 mol·L−1时,吸收液中的ClO2浓度变化最为明显。图4(b)显示,随着(NH4)2SO3浓度的增高,ClO2的收率逐渐增高,此外(NH4)2SO3的浓度由0.5 mol·L−1增至1.25 mol·L−1时,ClO2的纯度可由60%增加至84%。这是因为(NH4)2SO3的增加会使
SO2−3 的浓度增加,从而与H2SO4反应,产生更多的SO2,SO2与NaClO3反应,产生更多的ClO2气体。 -
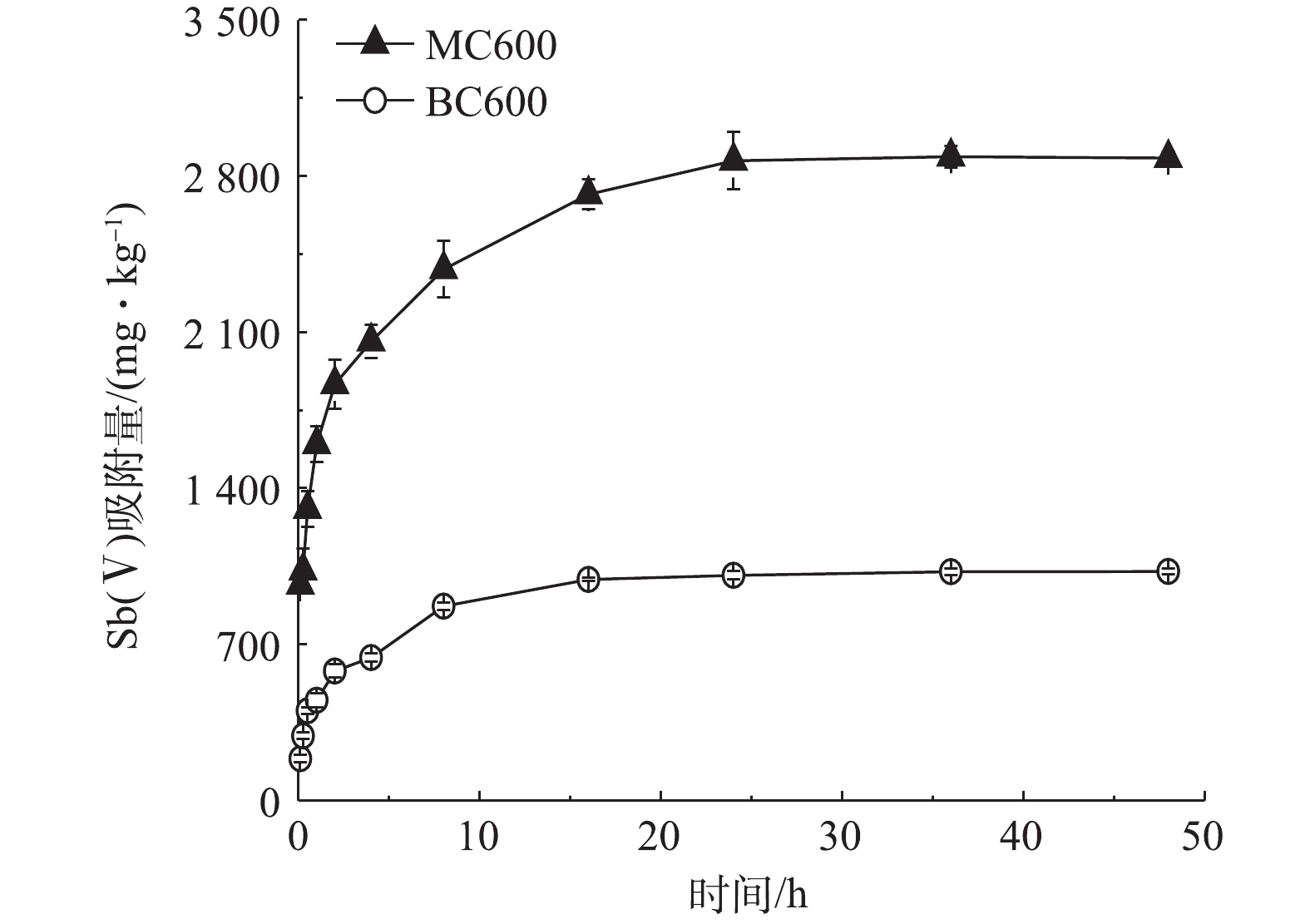
在实验中,设置NaClO3浓度为2 mol·L−1,(NH4)2SO3浓度为1.25 mol·L−1,H2SO4浓度为9 mol·L−1,反应温度为50 ℃,分别使反应原料在发生器中反应20、40、60、80、100、120 min,测定反应后吸收液中ClO2的浓度,考察反应时间对ClO2制备的影响,结果如图5所示。
由图5(a)可看出,吸收液中ClO2的浓度随反应的进行呈现先升高而后降低的趋势。在反应开始至60 min时,吸收液中ClO2浓度由965 mg·L−1迅速上升至1 113 mg·L−1,而当反应时间由60 min继续延长至120 min时,吸收液中ClO2浓度由1 113 mg·L−1下降至742 mg·L−1。这可能是因为,随反应时间的延长,反应物料逐渐消耗,反应速率逐渐降低,从而产生的气体量逐渐减少,但系统仍处于负压状态,产生的气体被抽出,溶解于吸收液中的ClO2也有部分随之逸出;也有可能是因为产生的ClO2与SO2发生了化学反应。此外,Cl2在水中的溶解度远小于ClO2,因此,随着反应系统在负压条件下的运行,Cl2更容易由水溶液中逸出,故ClO2的纯度会呈现上升趋势。
-
本研究以(NH4)2SO3为还原剂,在酸性条件下 (NH4)2SO3与NaClO3发生间接的氧化还原反应,从而制备ClO2,研究是在SO2以还原剂制备ClO2的基础上进行的。根据LENZI等[23]给出的还原剂在酸性条件下还原氯酸钠制备的普遍化机理,可知本研究的反应机理,反应见式(7)~式(12)。
为验证此反应机理与本实验反应机理的一致性,实验中将NaClO3溶液与(NH4)2SO3溶液混合加入反应器,在不加入H2SO4的情况下,观察到反应器内无黄绿色气体产生,此时在对应的ClO2吸收液中也并未测得ClO2;在不加入(NH4)2SO3的情况下,仅在反应器中加入NaClO3溶液,再缓慢加入H2SO4,也并未在吸收液中测得ClO2;而将NaClO3溶液与(NH4)2SO3溶液混合加入反应器,再逐渐加入H2SO4,反应器内便会开始产生黄绿色气体,对ClO2吸收液进行测定也发现其中含较高浓度的ClO2和少量的Cl2。综上所述,本实验的反应机理与式(7)~式(11)保持一致,(NH4)2SO3在有H2SO4存在的情况下被氧化为(NH4)2SO4,同时产生了SO2。产生的SO2作为有效还原剂,与NaClO3反应,生成(NH4)2SO4和ClO2(式(13)和式(14)),总反应见式(15)。
2.1. H2SO4浓度对ClO2制备的影响
2.2. 反应温度对ClO2制备的影响
2.3. (NH4)2SO3浓度对ClO2制备的影响
2.4. 反应时间对ClO2制备的影响
2.5. 反应机理分析
-
1)实验结果表明,使用NaClO3、H2SO4和(NH4)2SO3制备脱硝氧化剂ClO2的方法是可行的。在NaClO3浓度为2 mol·L−1,H2SO4浓度为9 mol·L−1,(NH4)2SO3浓度为1.25 mol·L−1,反应温度为50 ℃的条件下,ClO2吸收液中的ClO2浓度可达1 336 mg·L−1,满足ClO2氧化脱硝要求。
2)在反应过程中,吸收液中ClO2的浓度随着反应器中H2SO4的加入量、反应时间的升高呈现先升高后降低的趋势,随(NH4)2SO3浓度的增加呈现不断上升状态,随反应温度的增高呈波动变化状态;在20 ℃之前呈上升趋势,20~50 ℃呈下降趋势,之后再回升,20 ℃和50 ℃时均具有较好的实验效果。为避免温度过高导致(NH4)2SO3的分解,因此,选择温度为20 ℃。
3)实验中硫酸浓度对制备ClO2的反应有很大的影响。H2SO4浓度由6 mol·L−1升至9 mol·L−1时,吸收瓶中ClO2浓度由222.59 mg·L−1上升至1 335.55 mg·L−1,升高了83.33%;反应器中(NH4)2SO3的浓度由0.5 mol·L−1增至1.25 mol·L−1时,吸收液中ClO2的浓度可由519.38 mg·L−1上升至1 298.45 mg·L−1,升高了60%。
