






 下载:
下载:


</td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border2" align="center" valign="middle"><i>q</i><sub>m</sub>/(mg·g<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">FeOH</td><td class="table_top_border2" align="center" valign="middle">34.7</td><td class="table_top_border2" align="center" valign="middle">0.967</td><td class="table_top_border2" style="width:0.5%" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">66.7</td><td class="table_top_border2" align="center" valign="middle">0.995</td></tr><tr><td align="center" valign="middle">FN31</td><td align="center" valign="middle">35.0</td><td align="center" valign="middle">0.959</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">69.4</td><td align="center" valign="middle">0.971</td></tr><tr><td align="center" valign="middle">FN11</td><td align="center" valign="middle">36.8</td><td align="center" valign="middle">0.891</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">87.7</td><td align="center" valign="middle">0.963</td></tr><tr><td align="center" valign="middle">FN13</td><td align="center" valign="middle">47.0</td><td align="center" valign="middle">0.930</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">80.0</td><td align="center" valign="middle">0.961</td></tr><tr><td align="center" valign="middle">NiOH</td><td align="center" valign="middle">34.2</td><td align="center" valign="middle">0.969</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">78.5</td><td align="center" valign="middle">0.986</td></tr><tr><td align="center" valign="middle">FZ31</td><td align="center" valign="middle">59.9</td><td align="center" valign="middle">0.990</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">84.0</td><td align="center" valign="middle">0.958</td></tr><tr><td align="center" valign="middle">FZ11</td><td align="center" valign="middle">68.0</td><td align="center" valign="middle">0.992</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">87.0</td><td align="center" valign="middle">0.971</td></tr><tr><td align="center" valign="middle">FZ13</td><td align="center" valign="middle">70.92</td><td align="center" valign="middle">0.986</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">92.6</td><td align="center" valign="middle">0.942</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">ZrOH</td><td class="table_bottom_border" align="center" valign="middle">73.0</td><td class="table_bottom_border" align="center" valign="middle">0.983</td><td class="table_bottom_border" style="width:0.5%" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">104.2</td><td class="table_bottom_border" align="center" valign="middle">0.951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td rowspan="2" class="table_top_border" style="width:0.5%" align="center" valign="middle"></td><td colspan="2" class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{AsO}}_4^{3 - }$<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td rowspan="2" class="table_top_border" align="center" valign="middle">材料</td><td colspan="2" class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{PO}}_4^{3 - }$<alternatives><img class="graphic" src="202003074_M22.jpg"><img class="graphic" src="202003074_M22.png"></alternatives></inline-formula></td><td rowspan="2" class="table_top_border" style="width:0.5%" align="center" valign="middle"></td><td colspan="2" class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{AsO}}_4^{3 - }$<alternatives><img class="graphic" src="202003074_M23.jpg"><img class="graphic" src="202003074_M23.png"></alternatives></inline-formula></td></tr><tr><td class="table_top_border2" align="center" valign="middle"><i>q</i><sub>m</sub>/(mg·g<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border2" align="center" valign="middle"><i>q</i><sub>m</sub>/(mg·g<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">FeOH</td><td class="table_top_border2" align="center" valign="middle">34.7</td><td class="table_top_border2" align="center" valign="middle">0.967</td><td class="table_top_border2" style="width:0.5%" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">66.7</td><td class="table_top_border2" align="center" valign="middle">0.995</td></tr><tr><td align="center" valign="middle">FN31</td><td align="center" valign="middle">35.0</td><td align="center" valign="middle">0.959</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">69.4</td><td align="center" valign="middle">0.971</td></tr><tr><td align="center" valign="middle">FN11</td><td align="center" valign="middle">36.8</td><td align="center" valign="middle">0.891</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">87.7</td><td align="center" valign="middle">0.963</td></tr><tr><td align="center" valign="middle">FN13</td><td align="center" valign="middle">47.0</td><td align="center" valign="middle">0.930</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">80.0</td><td align="center" valign="middle">0.961</td></tr><tr><td align="center" valign="middle">NiOH</td><td align="center" valign="middle">34.2</td><td align="center" valign="middle">0.969</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">78.5</td><td align="center" valign="middle">0.986</td></tr><tr><td align="center" valign="middle">FZ31</td><td align="center" valign="middle">59.9</td><td align="center" valign="middle">0.990</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">84.0</td><td align="center" valign="middle">0.958</td></tr><tr><td align="center" valign="middle">FZ11</td><td align="center" valign="middle">68.0</td><td align="center" valign="middle">0.992</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">87.0</td><td align="center" valign="middle">0.971</td></tr><tr><td align="center" valign="middle">FZ13</td><td align="center" valign="middle">70.92</td><td align="center" valign="middle">0.986</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">92.6</td><td align="center" valign="middle">0.942</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">ZrOH</td><td class="table_bottom_border" align="center" valign="middle">73.0</td><td class="table_bottom_border" align="center" valign="middle">0.983</td><td class="table_bottom_border" style="width:0.5%" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">104.2</td><td class="table_bottom_border" align="center" valign="middle">0.951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td></tr><tr><td class="table_top_border2" align="center" valign="middle"><i>q</i><sub>m</sub>/(mg·g<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border2" align="center" valign="middle"><i>q</i><sub>m</sub>/(mg·g<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">FeOH</td><td class="table_top_border2" align="center" valign="middle">34.7</td><td class="table_top_border2" align="center" valign="middle">0.967</td><td class="table_top_border2" style="width:0.5%" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">66.7</td><td class="table_top_border2" align="center" valign="middle">0.995</td></tr><tr><td align="center" valign="middle">FN31</td><td align="center" valign="middle">35.0</td><td align="center" valign="middle">0.959</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">69.4</td><td align="center" valign="middle">0.971</td></tr><tr><td align="center" valign="middle">FN11</td><td align="center" valign="middle">36.8</td><td align="center" valign="middle">0.891</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">87.7</td><td align="center" valign="middle">0.963</td></tr><tr><td align="center" valign="middle">FN13</td><td align="center" valign="middle">47.0</td><td align="center" valign="middle">0.930</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">80.0</td><td align="center" valign="middle">0.961</td></tr><tr><td align="center" valign="middle">NiOH</td><td align="center" valign="middle">34.2</td><td align="center" valign="middle">0.969</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">78.5</td><td align="center" valign="middle">0.986</td></tr><tr><td align="center" valign="middle">FZ31</td><td align="center" valign="middle">59.9</td><td align="center" valign="middle">0.990</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">84.0</td><td align="center" valign="middle">0.958</td></tr><tr><td align="center" valign="middle">FZ11</td><td align="center" valign="middle">68.0</td><td align="center" valign="middle">0.992</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">87.0</td><td align="center" valign="middle">0.971</td></tr><tr><td align="center" valign="middle">FZ13</td><td align="center" valign="middle">70.92</td><td align="center" valign="middle">0.986</td><td style="width:0.5%" align="center" valign="middle"></td><td align="center" valign="middle">92.6</td><td align="center" valign="middle">0.942</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">ZrOH</td><td class="table_bottom_border" align="center" valign="middle">73.0</td><td class="table_bottom_border" align="center" valign="middle">0.983</td><td class="table_bottom_border" style="width:0.5%" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">104.2</td><td class="table_bottom_border" align="center" valign="middle">0.951</td></tr></tbody>
</table></div></foreignObject></svg>)


-
含氧阴离子是水环境中广泛存在的对水生动植物及水生态产生重要影响的一些非金属离子及一些重金属离子的总称,其统一的化学表示为
XOm−n ,其中PO3−4 和AsO3−4 是水环境中典型的含氧阴离子[1-4]。金属水合氧化物吸附剂材料,如FeOOH[5-7]、Al(OH)3[8-10]、TiO2[11]、ZrO2[12]、Zr(OH)4[13-14]和La(OH)3[15]等,因其具有高效的含氧酸阴离子吸附性能而受到广泛关注,特别是近年来金属复合材料的增强吸附机制已成为环境材料领域的研究热点。与单一组分金属水合氧化物相比,某些双金属及多金属复合材料拥有更好的含氧酸阴离子吸附性能[2, 16-27]。有研究[16, 20, 23, 27]表明,这些金属复合材料对含氧阴离子吸附性能具有特定的最优金属比,或通过物理混合物对比证实了金属复合材料的吸附增强特性[18]。前期对镧锆复合金属材料吸附研究提出金属离子掺杂电子结构调控增强吸附[28],但是金属复合材料的吸附增强机制尚待深入研究。另外,通过对比研究发现,上述材料中的镧(La)和锆(Zr)等前过渡金属材料表现出比铁(Fe)等后过渡金属更为突出的含氧阴离子吸附性能,而且这些过渡金属材料的含氧阴离子高效吸附特性在一些特定反应过程中蕴藏着潜在的规律,如析氧反应(OER)和氧还原反应(ORR)等[29-31]。这些研究[29-31]提出,过渡金属3d与O2p的σ*反键轨道电子填充度,即eg描述符,用以揭示过渡金属材料的OER和ORR电催化活性,但过渡金属水合物的形成机制及其对含氧阴离子的作用机制仍待深入研究。本研究的主要目标为:采用统一合成方法制备系列过渡金属水合氧化物及其复合材料;研究过渡金属水合氧化物对典型含氧酸阴离子的吸附效能;探讨过渡金属水合氧化物的形成机制和过渡金属水合氧化物及其复合材料对含氧阴离子的吸附机制,以期为后续过渡金属水合氧化物材料开发及其对含氧阴离子吸附过程及调控机制等研究提供理论支持。
全文HTML
-
过渡金属水合氧化物吸附材料是通过水热法制备的,具体制备过程如下。选择Fe(NO3)3·9H2O、Ni(NO3)3·6H2O和Zr(NO3)4·5H2O作为起始原料,使用乙醇作为溶剂,并加入环氧丙烷引发胶凝作用。在60 ℃磁力搅拌下,分别将12 mmol 金属硝酸盐溶解在80 mL乙醇中,用于获得金属硝酸盐溶液。澄清后,将前体溶液冷却至室温,定量吸取0.024 mol环氧丙烷溶液并在剧烈搅拌下缓慢加入所得到的金属硝酸盐溶液中。2 min后,将形成的透明溶液倒入100 mL聚四氟乙烯(PTFE)水热反应釜内胆中,并转移到不锈钢高压釜中,在自生压力下于200 ℃进行溶剂热处理。20 h后,将高压釜缓慢冷却至环境温度。将所得的湿凝胶在60 ℃下蒸发48 h,得到干凝胶,然后先后分别用去离子水和无水乙醇反复洗涤3次。将获得的沉淀物在60 ℃下干燥48 h,然后研磨成细粉。根据样品合成过程中使用的金属盐命名样品,即FeOH、NiOH和ZrOH。Fe-Ni和Fe-Zr复合金属水合氧化物由相同方法制备,其样品的命名是根据样品合成过程中的Fe:Ni或Fe:Zr的物质的量之比,即FN31、FN11、FN13或FZ31、FZ11、FZ13。材料制备及实验中所用试剂均为国药分析纯试剂。
-
吸附实验是通过将0.01 g吸附剂悬浮在50 mL去离子水中而进行的,P浓度为10~100 mg·L−1和As浓度为5~50 mg·L−1。在实验前,使用1 mol·L−1 HCl或KOH调节液将溶液pH 调至7.0±0.1,然后将上述悬浊液放置在(25±1) ℃恒温摇床中,以200 r·min−1转速摇动24 h。到达预定时间后,通过电感耦合等离子体发射光谱法(ICP-OES)测定上清液中磷或砷的浓度。每个数据点分别做3次平行实验,根据Langmuir吸附模型对所得实验数据进行拟合,如式(1)所示。
式中:qe为平衡吸附量,mg·g−1;qm为吸附材料的吸附容量,mg·g−1;KL为吸附参数,L·mg−1;Ce为平衡时体系中吸附质的浓度,mg·L−1。
-
采用SU8000扫描电子显微镜(SEM,日本,日立公司)、X’ Pert PRO MPDX射线衍射仪(XRD,德国,布鲁克公司)、PHI 5600 X射线光电子能谱仪(XPS,美国,Physical Electronics公司)对反应前后的材料样品进行形貌、物相和表面元素形态分析。采用中科院高能物理研究所北京同步加速器辐射设施(BSRF)的4B7B线站对材料样品表面氧元素进行氧近边光吸收谱(XAS)分析。采用iCAP8000电感耦合等离子体光谱仪(美国,赛默飞公司)对溶液中的磷和砷的浓度进行分析。
1.1. 材料制备
1.2. 吸附实验
1.3. 表征方法
-
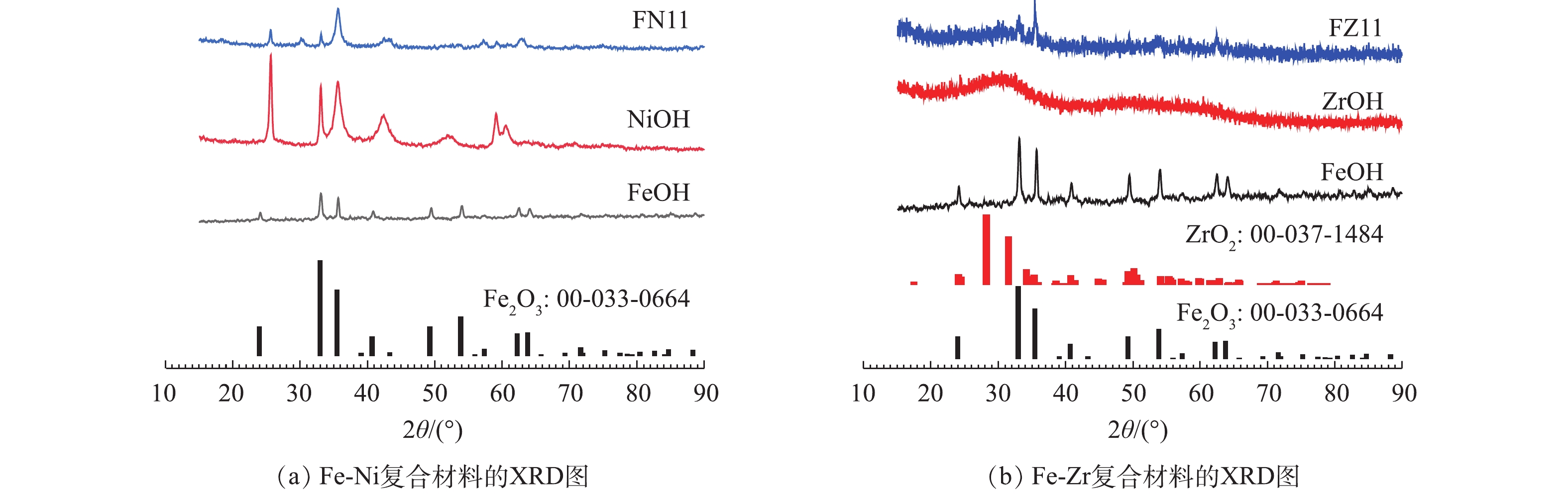
合成的FeOH、NiOH、ZrOH及Fe-Ni、Fe-Zr复合材料的SEM和XRD图谱如图1和图2所示。FeOH材料呈纳米颗粒团簇状,颗粒尺寸约为100 nm,颗粒聚集体尺寸可达数微米,其结构松散、易脱落。FeOH材料的XRD谱图与三方相Fe2O3(00-033-00664)卡片比对吻合,这说明所合成的FeOH材料为颗粒均匀且结构松散的铁氧化物。NiOH材料呈纳米花瓣状,其球状尺寸约为1 μm,整体花簇状尺寸可达5 μm以上。NiOH材料的XRD图谱显示,其各衍射峰峰位置与FeOH材料(Fe2O3)相近,这说明NiOH材料为镍的氧化物。前3个强衍射峰的位置相对于晶体相Fe2O3后移,这进一步说明在相同晶格中Fe和Ni原子的电荷的差别[32-33]。此外,NiOH和FeOH材料的XRD衍射模式,即衍射峰强度比存在显著差异。在NiOH材料的XRD谱图中,位于24°附近的峰最强,而在FeOH材料中此峰的强度远远小于在33°附近的峰。XRD衍射模式的差异与晶体的晶面取向相关,这正是NiOH与FeOH材料的形貌不同的表现。ZrOH材料呈块状,结构密实,块形尺寸为5 μm以上。合成所得ZrOH材料的XRD谱图与ZrO2(00-037-1484)卡片比对吻合,其在大约30°及50°~60°处的大峰包,表明ZrOH材料为非晶态ZrO2相材料。
另外,由图1可知,复合金属水合氧化物的形貌结构存在一定的变化趋势。Fe-Ni复合金属水合氧化物的形貌从FN31的类似FeOH松散团簇状结构随着镍含量的升高发展为FN13的类似NiOH材料花簇状结构。与之类似,Fe-Zr复合金属水合氧化物的形貌随着锆含量的升高从FZ31的类似FeOH松散团簇状结构发展为FZ13的类似ZrOH材料块状密实结构。结合上述对其单一金属水合氧化物的物相分析,Fe-Ni、Fe-Zr复合金属水合氧化物材料中存在FeOH-NiOH和FeOH-ZrOH间晶体物相的竞争。由XRD模式(图2)的差异可知,复合金属水合氧化物材料的晶面取向有所不同,说明在形成复合金属水合氧化物材料过程中存在晶体物相的相互干扰。如图2(a)所示,FN11的XRD图谱中无明显其他新衍射峰的出现,由此可知FN11材料中无其他物相形成。Fe-Ni复合金属水合氧化物的XRD峰强度的相对变化,表明FN11材料中仅为FeOH-NiOH间金属离子的互混,最近LEE等[34]采用Raman谱也证实了镍铁氢氧化物中Fe和Ni原子的掺杂。然而,FZ11的XRD的图谱中在对应位置处均出现显著的FeOH的特征衍射峰和30°附近对应的ZrO2峰包,如图2(b)所示。FZ11材料中包含这2种晶体相成分,确认了Fe-Zr复合材料为FeOH和ZrOH混合晶体颗粒。同时,Fe-Ni、Fe-Zr复合金属水合氧化物材料所对应晶体衍射峰强度均显著有所降低,这表明复合材料中FeOH、NiOH和ZrOH晶粒尺寸的减小,暗示材料中晶体物相间存在竞争干扰。
-
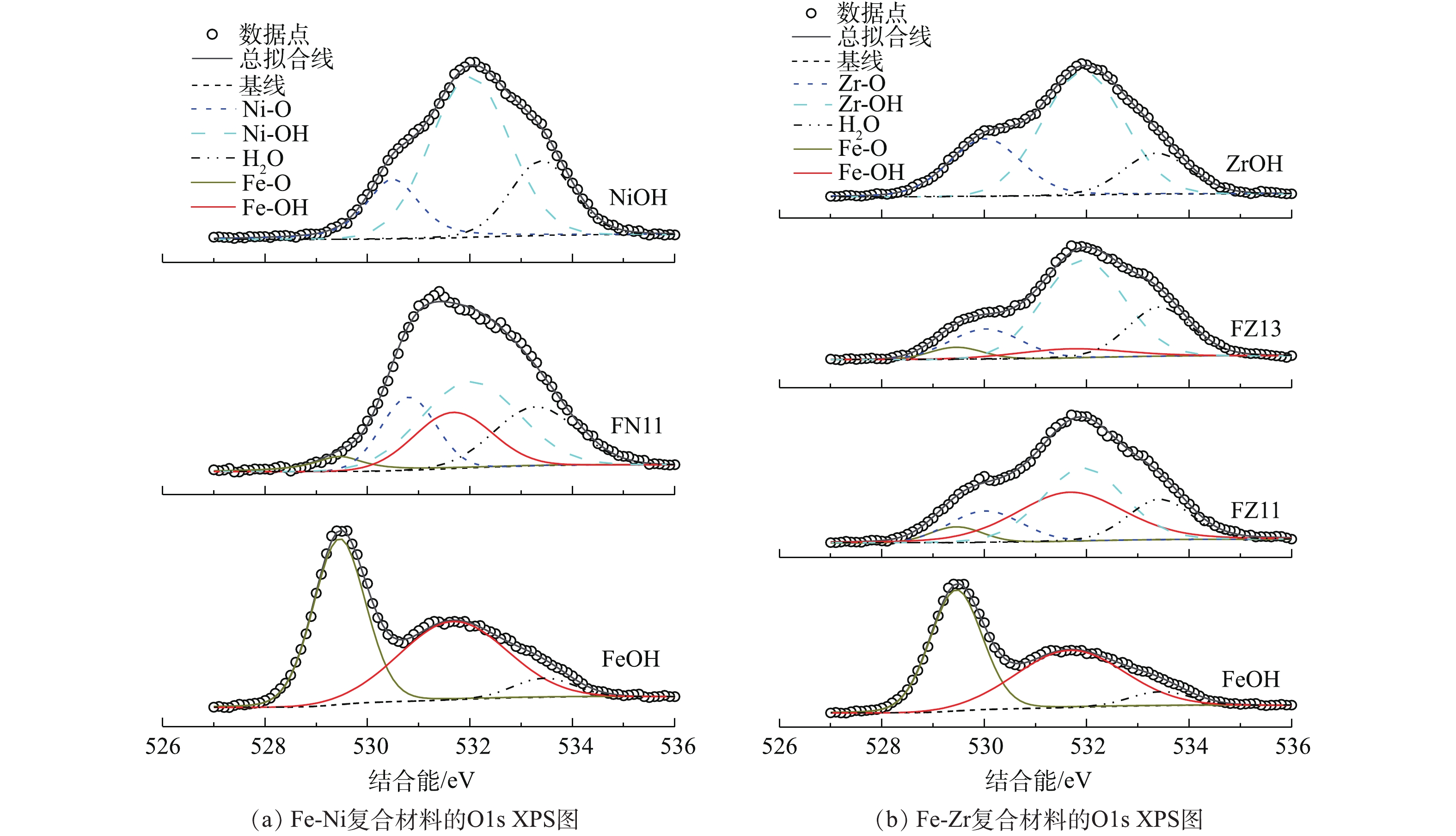
通过O1s XPS表征来进一步确认各金属水合氧化物表面的氧物种形态。由图3可以看出,在各金属水合氧化物中的氧物种形态主要以形成晶体的晶格氧M—O、表面金属羟基M—OH及其结合水形式存在[35-37]。对这些材料中O1s XPS谱的峰形分析表明,FeOH、NiOH、ZrOH金属水合氧化物材料表面氧形态组成存在显著差异:FeOH材料以晶格氧M—O为主,而NiOH和ZrOH 材料以M—OH为主;另外表面结合水含量也存在差异:FeOH材料结合水含量较低,而NiOH和ZrOH材料结合水含量较高。由以上分析可知,此合成方法制得的FeOH中铁离子易与体系氧物种缩合形成氧化物晶体,而NiOH和ZrOH分别形成水合态氧化物晶体NiOH和非晶水合氧化物ZrOH。比较Fe-Ni和Fe-Zr复合材料的O1s XPS图谱发现,FeOH在形成氧化物晶体的结晶过程中,易受到体系中Ni3+和Zr4+金属离子的干扰,致使Fe-Ni、Fe-Zr复合材料中M—OH和结合水含量显著增加。一方面,Fe-Ni和Fe-Zr复合材料中同时分别含有Fe—O、Ni—O、Fe—OH、Ni—OH和Fe—O、Zr—O、Fe—OH、Zr—OH,这表明复合金属水合氧化物材料中氧物种组成成分没有发生变化。另一方面,Fe-Ni复合材料的XRD图谱已经确认了材料中Fe和Ni在相近的晶体晶格中的相互掺杂,并且Fe3+和Ni3+离子半径相近,满足相互掺杂的必要条件(与6个氧配位的高和低自旋的Fe3+离子半径分别为0.064 5 nm和0.055 0 nm,与6个氧配位的高和低自旋的Ni3+离子半径分别为0.060 0 nm和0.056 0 nm[38]);对于Fe-Zr复合材料而言,由于非晶态的氧化锆与Fe2O3晶体相的显著差别,以及Zr4+离子半径较大(与6个和7个氧配位的Zr4+离子半径分别为0.072 0 nm和0.078 0 nm[38]),Fe3+和Zr4+不容易相互掺杂,Fe-Zr复合材料而是形成混合的FeOH和ZrOH晶粒。
-
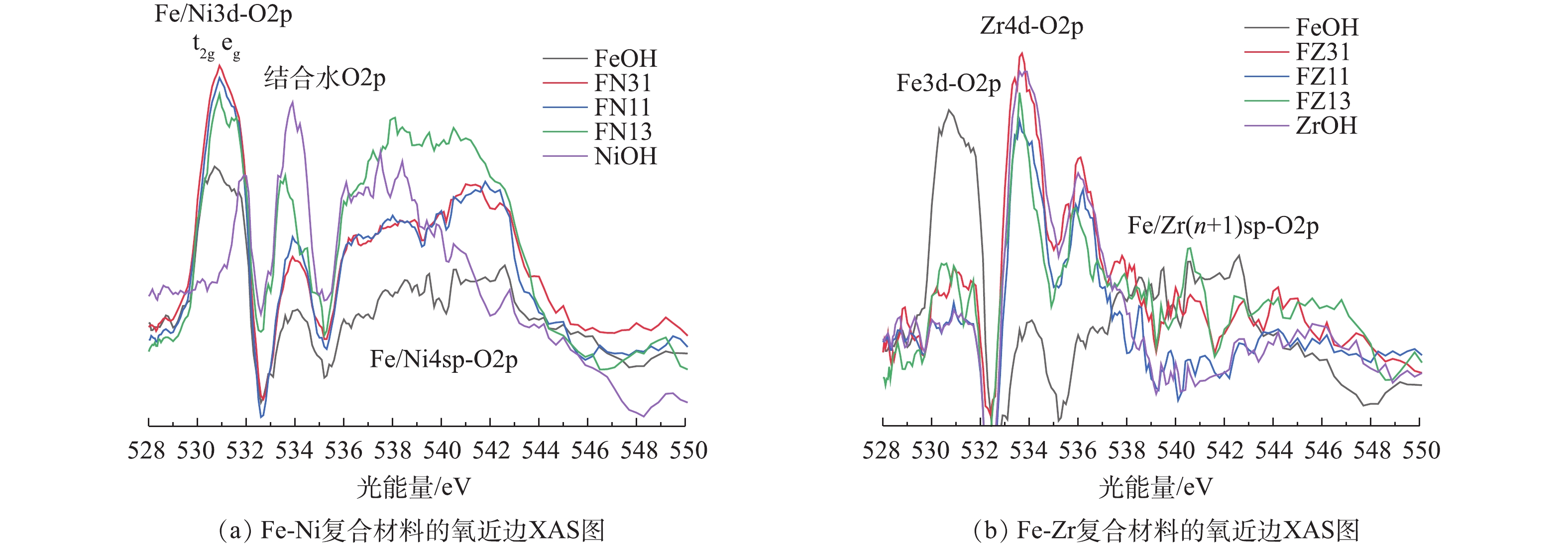
为进一步确定各金属水合氧化物材料表面的氧物种形态及物理化学特征,我们分别调查了Fe-Ni、Fe-Zr及其复合材料的氧近边XAS谱。氧近边XAS谱表现的是10 nm附近的材料表面的物理化学特征,O1s轨道电子到费米能级及费米能级以上空域的激发态[39]。过渡金属水合氧化物复杂的电子结构与局域化的金属d电子态和d电子相互作用有关。通过揭示具有d轨道的O2p杂化状态,氧近边XAS分析为研究由不同金属的存在引起的化学状态变化的灵敏方法,这有助于进一步了解复合材料对含氧阴离子的吸附机理。
图4为Fe-Ni及Fe-Zr复合材料的氧近边XAS图。Fe和Ni的水合氧化物及其复合材料的氧近边XAS谱如图4(a)所示,其主要由530~532、533~535及536~543 eV的3个谱带组成。为了阐明能量顺序,进一步分析了金属-氧配体的成键或反键特性。有研究[39-40]表明,可以将谱峰分配给金属和氧配体的t2g、eg和4sp杂化轨道。此外,结合水的谱峰位置大约在534~537 eV。氧近边XAS谱中位于530~532 eV的谱峰表明过渡金属Fe/Ni与氧配体间的成键或反键作用,即t2g和eg杂化轨道,其电子分布表征了过渡金属d电子间的相互作用。对于FeOH金属水合氧化物材料,Fe3+ 电子态3d5采取的是
t32ge2g ,氧近边XAS谱图在相应位置表现为两峰的交叠。对于NiOH金属水合氧化物材料,Ni3+电子态3d7采取的是t62ge1g ,氧近边XAS谱图在相应位置表现为孤立的eg峰。进一步比较Fe-Ni复合材料谱图发现,在复合材料形成过程中,少量的掺杂便可显著的调控过渡金属d电子态;比较结合水的峰强度,发现少量的镍掺杂可大幅提升材料的结合水含量,使得Fe-Ni复合材料的水合程度增加,这符合XRD和XPS的相关结论。Fe和Zr的水合氧化物及其复合材料的氧近边XAS谱如图4(b)所示,主要由530~532、533~535、535~537和538~543 eV处的4个峰组成。同理,可以用金属-氧配体间的成键或反键相互作用进行谱分析。对于FeOH金属水合氧化物材料,Fe3+电子态3d5采取的是
t32ge2g 。对于ZrOH金属水合氧化物材料,Zr4+电子态4d0采取的是t02ge0g ,533 eV和536 eV 处2个峰与之相对应。进一步比较Fe-Zr复合材料图谱发现,在复合材料形成的过程中,少量锆的掺杂便可显著的调控过渡金属d电子态;比较结合水的峰强度发现,少量的锆掺杂就可大幅提升材料的结合水含量,使得Fe-Zr复合材料的水合程度增加,这与XPS的相关结论一致。 -
图5为FeOH、NiOH、ZrOH、Fe-Ni和Fe-Zr复合材料的
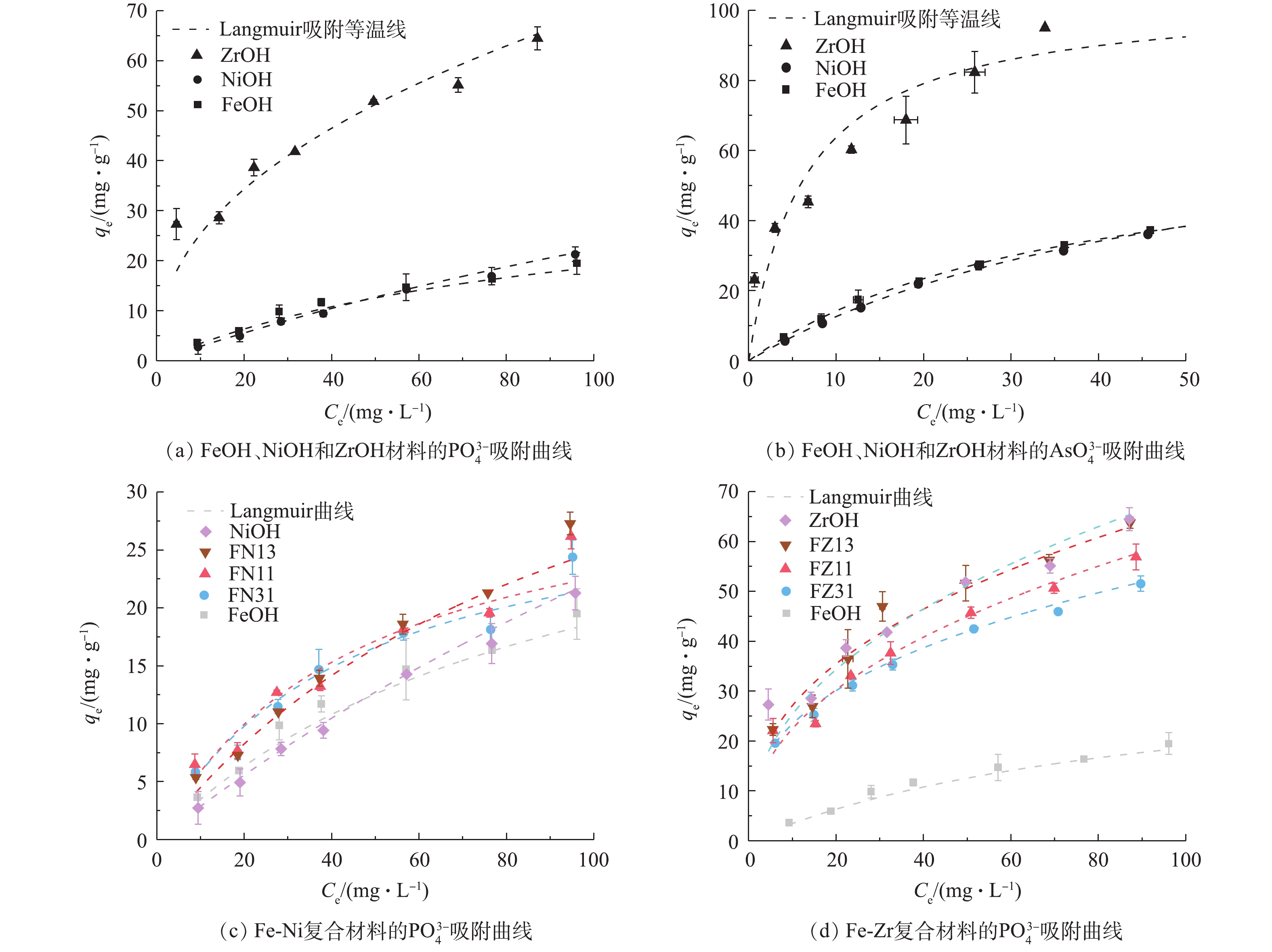
PO3−4 和AsO3−4 吸附曲线。考虑到磷酸根及砷酸根具有相近的pKa(分别为2.12、7.2、12.36和2.2、7.00、11.50),本研究选定在pH=7.0的条件来评价所选材料的吸附容量,以此评价各吸附材料的吸附性能,从而构建材料特征与吸附性能的关系。Fe、Ni和Zr金属水合氧化物的磷酸根和砷酸根吸附性能如图5(a)和图5(b)所示。在pH=7.0、温度为25 ℃条件下,Fe、Ni和Zr金属水合氧化物的磷酸根和砷酸根的拟合吸附容量分别约为34.7、34.2、73.0 mg·g−1和66.7、78.5、104.2 mg·g−1。在相同条件下,ZrOH材料对磷和砷的吸附性能也远大于FeOH和NiOH,吸附能力顺序为FeOH、NiOH<ZrOH。Fe-Ni和Fe-Zr复合金属水合氧化物的磷酸根和砷酸根吸附性能分别如图5(c)和图5(d)所示,其吸附容量拟合值见表1。首先,Fe-Ni和Fe-Zr复合金属水合氧化物的磷酸根吸附性能趋势显著不同:Fe-Ni复合金属水合氧化物的磷酸根吸附性能要优于单一金属水合氧化物FeOH和NiOH,而Fe-Zr复合金属水合氧化物的磷酸根吸附性能随吸附材料锆含量的升高而显著增加。另外,Fe-Ni和Fe-Zr复合金属水合氧化物的砷酸根吸附性能具有类似的趋势。Fe-Ni复合金属水合氧化物的含氧阴离子吸附性能有所增强的原因可能为Fe-Ni复合材料形成过程中的铁镍水合干扰引起的形貌改变,使得吸附材料表面暴露的有效吸附位点的增多,也可能是材料中铁镍掺杂改变了过渡金属3d电子的状态,从而影响了金属铁镍的本征吸附活性。Fe-Zr复合金属水合氧化物的锆依赖吸附活性可归因于Zr金属的本征吸附活性。
2.1. 材料的物相及形貌
2.2. XPS分析
2.3. XAS分析
2.4. 对磷酸根和砷酸根的吸附效能
-
图6为Fe-Ni和Fe-Zr复合材料的形成及其含氧阴离子吸附机理示意图。Fe、Ni、Zr金属水合氧化物的形成过程如图6(a)所示。在金属离子水解过程中,金属离子结合羟基,在水热合成中环氧丙烷的氧结合质子,可加速金属离子结合羟基并相互缩合,使之发生进一步晶体化。当混有不同金属离子时,根据金属离子水解能力的不同,结合羟基能力强的金属离子先结合,在到达一定程度后,结合羟基能力弱的金属离子也开始水解,并参与到金属羟基团簇的缩合反应中。在本研究中,根据Fe(OH)3和Zr(OH)4的溶度积(Ksp分别为1.6×10−36和3.0×10−49)可知,离子形成氢氧化物沉淀的能力为Zr4+>Fe3+,Zr4+先于Fe3+离子水解形成沉淀[41-42],这也是Fe-Zr复合材料的形貌以ZrOH材料为基础而变化的原因;由XAS分析结果可知,Fe3+与Ni3+离子的电子构型分别为
t32ge2g 和t62ge1g ,根据eg轨道填充规则,Ni—O比Fe—O结合略强,其形成氢氧化物沉淀的能力相近,Fe-Ni复合材料形成过程中Fe3+与Ni3+离子同步水解沉淀,这为Fe3+与Ni3+离子掺杂提供了必要条件。根据XRD分析,Fe-Ni复合材料中Fe和Ni在晶体晶格中相互掺杂形成Fe-Ni复合氧化物,Fe-Zr复合材料中,由于ZrO2晶型不同以及Zr4+离子的离子半径较大,Fe3+不容易掺杂其中,Fe-Zr复合材料以混合的FeOH和ZrOH晶粒形式存在。同时XPS与XAS分析表明,少量的镍和锆离子的掺杂可大幅提升复合材料的结合水含量,表明Fe-Ni和Fe-Zr复合材料为水合氧化物。 -
金属水合氧化物的含氧阴离子吸附作用包括物理吸附和化学吸附[43]。物理吸附发生在吸附剂与吸附质相距较远的空间,吸附剂与吸附质之间的作用力不是很大,由于分子热运动的存在,不足以将其牢牢固定在吸附剂的表面。当这些被物理吸附在表面附近的吸附质由于不规则热运动更加靠近吸附剂表面的吸附位点时,由于其碰撞方向匹配及间距足够小,此时吸附位点上被物理吸附的水分子被这个吸附质分子挤开,吸附质分子通过离子键或共价键的形式牢牢固定在吸附剂表面,此过程为化学吸附作用,如图6(b)所示。
对于复合金属水合氧化物的吸附机制,需要从复合材料的形成过程加以分析。就Fe-Ni复合金属水合氧化物的物质结构和磷酸根吸附性能的综合分析来看,Fe-Ni复合金属水合氧化物的除磷性能有所增强。其吸附增强的可能机制是金属的掺杂调控了化学吸附作用。如前所述,Fe-Ni复合材料形成过程的铁镍水合干扰引起的形貌改变,使得吸附表面的增加或暴露的吸附位点增多,也可能是材料中铁镍掺杂改变了过渡金属3d电子的状态,从而影响了金属铁镍的本征吸附活性。根据氧近边 XAS表征结果分析,可能是两者共同的结果。Fe-Ni复合材料形成过程的铁镍水合干扰,引起了金属氧化物Fe2O3结晶度的降低,金属水合位点增多,这也是引起其形貌改变的原因,形貌的改变并不是吸附性能增强的原因,其真正原因是金属水合位点的增多,即有效化学吸附活性位点的增多。一方面,XPS和XAS分析结果表明,少量的镍掺杂可大幅提升Fe-Ni复合材料的结合水含量。如图6(c)所示,根据eg轨道填充规则[31], Ni3+离子的
t62ge1g 电子构型更易容纳水分子中氧的2p电子,导致含镍材料中结合水含量增加。Fe-Ni复合材料的铁镍掺杂调控了eg轨道电子态,削弱了金属与羟基及结合水的结合强度,使得有效的含氧阴离子吸附活性位点增多。另一方面,Fe-Ni复合材料的铁镍掺杂调控也能增加其化学吸附物种的电子状态,使其以相对稳定的状态固定在其表面上。对于Fe-Zr复合金属水合氧化物,其物质结构是Fe与Zr水合氧化物晶粒的混合。非晶态锆氧化物ZrOH材料拥有大量的结合水,而FeOH材料中的结合水含量相对较少。与Fe-Ni复合材料类似地,Zr4+离子的t02ge0g 电子构型使其更易容纳水分子中氧的2p电子,导致含锆材料中结合水含量增加。Fe-Zr金属掺杂调控带来的金属水合位点的增多远不及因锆含量降低带来的金属水合位点的减少,且不管Fe与Zr水合氧化物团簇的混合提供的电子调控能力。所以Fe-Zr复合金属水合氧化物的除磷除砷性能随吸附材料锆含量的升高而显著增加,与Fe-Ni复合金属水合氧化物形成对比。Fe-Zr复合金属水合氧化物的锆依赖吸附活性可归因于Zr4+离子的t02ge0g 电子构型以及其高配位性质导致的高本征吸附活性。
3.1. 金属水合氧化物的形成过程及掺杂特征
3.2. 对磷酸根和砷酸根吸附机理的差异
-
1)复合金属水合氧化物材料的形成机制的差异:Fe和Ni的水合氧化物由于其离子半径和形成的晶体结构相近,Fe和Ni元素掺杂形成Fe-Ni复合水合氧化物材料;而Fe和Zr的水合氧化物由于其离子半径和形成的晶体结构差异显著,Fe和Zr离子掺杂仅形成FeOH和ZrOH微小晶体颗粒的混合团簇。
2)过渡金属水合氧化物材料的含氧阴离子吸附性能顺序为FeOH、NiOH<Fe-Ni<Fe-Zr<ZrOH。复合水合氧化物吸附材料的含氧阴离子的吸附机制的差异为:Fe-Ni复合水合氧化物材料中Fe和Ni元素掺杂可调控材料3d电子态,通过电子态调控强化吸附性能;而Fe-Zr复合金属水合氧化物的锆依赖吸附活性可归因于Zr4+离子的
t02ge0g 电子构型以及其高配位性质导致的高本征吸附活性。
