


 下载:
下载:






-
近年来,由于V/W/Ti系催化剂的高催化活性与热稳定性,其被广泛地应用于燃煤电厂的选择性催化还原(SCR)脱硝单元[1-3]。对于负载了钒氧化物的催化剂,在SCR反应过程中会发生SO2催化氧化生成SO3的副反应,其转化率为SO2浓度的0.5%~2%[4-6]。SO3是硫酸液滴和硫酸盐气溶胶等细微颗粒物(PM2.5)的来源之一,此外,其会增加烟囱中排烟的不透明度,并对锅炉运行产生不利影响[7-10]。现有技术尽管可以从SCR下游通过喷射碱性物质来控制SO3的排放,但由于运行成本太高且不能避免SO3生成对催化剂活性的影响,因此,应从催化剂入手最大限度地降低SO2的氧化率。为减少SO3的形成,应尽可能降低燃煤电厂SCR催化剂中V2O5的添加量[11];然而,为实现高NOx脱除效率,催化剂需要负载较高含量的V2O5[12]。因此,需要同时兼顾高NOx脱除效率和低SO3生成量之间的平衡。
为减少SO2氧化并减轻SO2对催化剂的毒性作用,研究人员探讨了SO2在不同催化剂上的氧化特性,并尝试使用不同的活性成分代替V2O5或添加助剂以减少SO2引起的副作用。WOJAYANTI等[13]发现,铜和铁基沸石催化剂比钒催化剂具有更高的氨储存能力。YU等[14]在SO2和H2O对催化剂活性影响的研究中发现,MnOx/SAPO-34催化剂比其他Mn基催化剂具有更好的抗SO2中毒性。在NH3-SCR中加入的SO2会在催化剂的表面以酸性硫酸铁的形式对Fe/CNTs产生不可逆的影响[15]。WOJAYANTI等[13]报道了Cu-SSZ-13催化剂在SO2存在下的催化活性,发现SO2会造成催化剂中毒而使其失活。有研究[16]发现,SO2对催化剂活性的影响更为温和且与物种无关,而SO3的存在对催化剂性能影响更为显著且更难逆转。HUANG等[17]在WO3(HWO)上添加V2O5以开发V2O5/HWO催化剂,发现其在SCR反应中表现出抗碱和抗硫中毒的特性。LIN等[18]发现CeO2-MnO2催化剂在SO2存在下会完全失活。此外,也有部分研究者[19-21]探究了新型低温催化剂配方,但研究的催化剂种类多样且并未实现工业应用,V/W/Ti系催化剂仍是目前使用的主流催化剂,因此,有必要深入研究V/W/Ti系催化剂表面SO3的生成特性。
MA等[22]报道了V2O5在V2O5/AC上去除SO2的主要作用,表明V2O5将通过类似VOSO4的中间物质催化SO2氧化,其会与O2反应生成SO3和H2SO4。SHANG等[23]研究了SO2在TiO2颗粒上的非均相反应,发现SO2在TiO2颗粒上非均相反应的主要产物是硫酸盐。这些研究仅用于单一反应,并且该反应中使用的催化剂仅具有单一组分,不能代表电厂使用的商业催化剂。此外,SO3是一种高活性物质,其测量非常困难[24]。SCHWAMMLE等[25]与ZHENG等[26]研究了SO2/SO3的转化,发现SO2氧化随壁厚几乎呈线性增加,受NH3的抑制程度较大。LI等[27]发现NO促进了SO2向SO3的转化。本团队的前期研究中也发现,烟气中的H2O与CO2等也会影响SO3的生成[2]。实际烟气的成分很复杂,各气体组分对SO2氧化的影响尚不清楚,其机理尚未得出明确的结论,因此,需要了解多组分复合气体气氛下催化剂表面SO2催化氧化成SO3的特性。
本研究旨在了解商用V/Ti/W系SCR催化剂上的SO2催化氧化生成SO3的特性,利用BET、XRF、XRD等仪器对催化剂的物化特性进行分析,研究了SO2对商用V/Ti/W系SCR催化剂脱硝性能的影响,并在固定床反应系统上研究了催化剂组分(V含量)、操作条件(温度、空速)和进气组分(O2、SO2和NH3与NO的体积比(NH3∶NO))等因素对催化剂表面SO3生成反应的影响特性,以期为实现燃煤电厂排放烟气中SO3的有效控制及污染物超低排放提供参考。
全文HTML
-
本研究选取5种商用的蜂窝式催化剂,截距8.27 mm,内壁厚1.00 mm。因催化剂为方形,为方便实验操作,将催化剂样品破碎、筛分为平均粒径为20~40目的颗粒,以减少因催化剂形态导致的反应器设计以及气路短路等一系列其他因素对实验结果的影响,最终制得的催化剂为淡黄色小颗粒。
-
采用化学法测量SO3的生成量,参照ASTM D-3226-73T,SO3取样采用控制冷凝法,在温度为65~100 ℃时将SO3从烟气流中分离并取样,同时计量收集时间。采用离子色谱仪(ThermoFisher ICS1100)分析试样中SO3的含量,并计算相应的SO3摩尔量。采用NaOH溶液收集SO2,在NaOH溶液中添加5 mL 30%的H2O2溶液,使得吸收的SO2完全氧化为SO3,随后采用离子色谱仪测量吸收液中SO2的含量,根据烟气流量计算相应的SO2摩尔量。实验中SO3生成率的计算方法见式(1)。
式中:
η 为SO3生成率;CSO3 为SO3摩尔量;CSO2 为SO2摩尔量。 -
SO3生成实验系统如图1所示。整个系统由配气混气系统、水蒸气发生器、反应系统、烟气收集以及测量记录部分组成。由于实验条件有限,不考虑实际使用中飞灰的影响,混气由NH3、NO、O2、SO2和N2组成,其中N2为平衡气。实验过程中总的气流量为1 L·min−1,通过质量流量计控制各组分气体的浓度。为模拟催化剂在燃煤锅炉中的使用情况,同时简化气路流程,实验时将5种气体先在混气瓶内混合均匀,然后通入水蒸气发生器的加热炉中预热,最后进入反应器中进行反应。水蒸气发生器出口与反应炉之间的管道缠绕有伴热带,以防止烟气在中间冷却影响实验结果。
反应炉为一段式加热的立式炉,脱硝反应时将反应器插入炉内,气流上进下出。反应器为内径20 mm、长590 mm的石英管。为了固定催化剂颗粒,石英管中间靠近反应炉热电偶的位置设计有一个80目筛板。为了使加热炉实际温度与设定的催化剂反应温度接近。实验时在石英管内部插入一个K型热电偶,并连接至烟气分析仪,同时记录实际反应温度和气体中各组分的浓度。通过调节加热炉的温度,使反应管内催化剂的温度与实验设计值相符合。
-
催化剂的比表面积和孔结构采用BET方法测试,在美国麦克公司的快速比表面积与孔隙度分析仪上进行测试,仪器型号为ASAP2020。测试前,样品在350 ℃的条件下进行脱气处理,处理时间为5 h。使用EDAX公司的XRF分析仪测定催化剂样品上的元素组成,仪器型号Eagle III,分析仪在4 kW下操作并使用RhKα辐射作为X射线源。晶体的结构通过X射线衍射(XRD)测试,采用荷兰帕纳科公司生产的型号为Empyrean的仪器,测试辐射源为CuKα,射线发生器的最大管压和最大管流分别为40 kV和40 mA。XRD测试的扫描角度2θ为20°~80°,扫描步长为0.017°。
1.1. 催化剂制备
1.2. SOx检测方法
1.3. 催化剂SO3生成性能实验
1.4. 催化剂表征
-
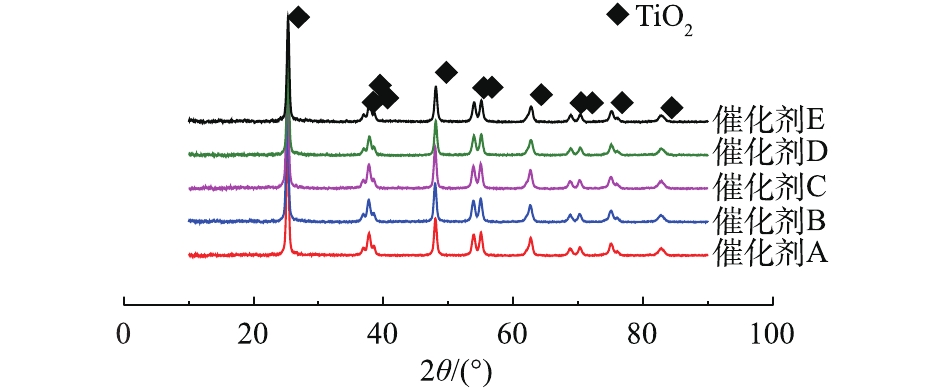
催化剂的活性成分及其他表面沉淀物含量如表1所示。可以看出,TiO2作为载体,在催化剂中占主要部分,V和W作为催化剂的活性物质和助剂,在催化剂中分别占1%~2%和2%~10%。除此之外,还有Si和Ca等元素。同时可以发现,在5种催化剂的表面均存在不同含量的S,主要是因为催化剂在运行一段时间后,烟气中的S会部分沉积在催化剂表面,这也是造成催化剂失活的原因之一。表2给出了实验中催化剂样品的物理特性。几种催化剂样品的比表面积、孔容与孔径均存在较大的差别,但是相对应的比表面积均较大。图2为催化剂的XRD谱图。结果表明,催化剂表面的衍射峰均为锐钛矿型的TiO2,并且没有在表面发现WO3和V2O5的晶体和金红石化的TiO2,说明活性物质在催化剂表面分散度很好。
-
图3给出了催化剂A~催化剂E的SO3生成率,反应温度为360 ℃,空速为10 000 h−1,600×10−6 SO2,3% O2。可以看出,不同催化剂的SO3生成速率在相同的反应条件下表现出明显的差异,这主要是由于不同催化剂的化学组成和物理结构的影响。通过比较催化剂A、B和C的化学组分发现,3种催化剂中各组分含量差异并不显著。通常,催化剂中V组分为1%~5%,V含量的高低会显著影响催化剂的脱硝与氧化活性。对比3种催化剂表面的SO3生成率,可以发现催化剂上的SO3生成率按C˃A˃B的顺序降低,这与催化剂上V含量降低的顺序相同而与催化剂的比表面积与孔容的降低顺序相反。这些结果表明,V负载量是决定SO3生成的重要参数,物理特性对SO3生成的影响低于化学成分性质。催化剂D中V含量最高,催化剂A与E中V含量基本一致,但比较催化剂A、D和E上SO3的生成速率发现,其与催化剂上V相应的含量变化并不一致,催化剂上SO3的生成率按A>D>E的顺序降低,说明催化剂中其余组分影响了SO3的生成。对比催化剂中其余组分发现,3种催化剂中W与Si的差异较为显著,催化剂D中W的相对含量较高,而催化剂E中的Si相对含量较高。已有研究[27-28]同样发现,催化剂中的W与Si会抑制SO3的生成,这与本研究中催化剂D与E上相应的SO3生成率降低相一致。因此,可以得出结论,催化剂中W与Si的添加在SO3生成中起着显著的抑制作用。
在实际应用中,一方面应保证足够的V含量以提高催化剂脱硝效率;另一方面,不能单一提高V含量,也应控制相应的SO3生成量。催化剂中组分W与Si等对SO3的生成起抑制作用。在催化剂的制备过程中,除控制V含量以外,也可添加相应的活性组分(如W与Si等)控制SO3的生成。在V/W/Ti系催化剂中添加Si可以有效地抑制锐钛矿向金红石TiO2的转变,抑制晶粒的生长与催化剂比表面积的收缩。此外,添加Si后,V在催化剂表面高度分散,催化剂的水热稳定性显著提高[28-29]。在实际运行过程中,催化剂的选择也应综合考虑脱硝效率与SO3生成率2个指标。
-
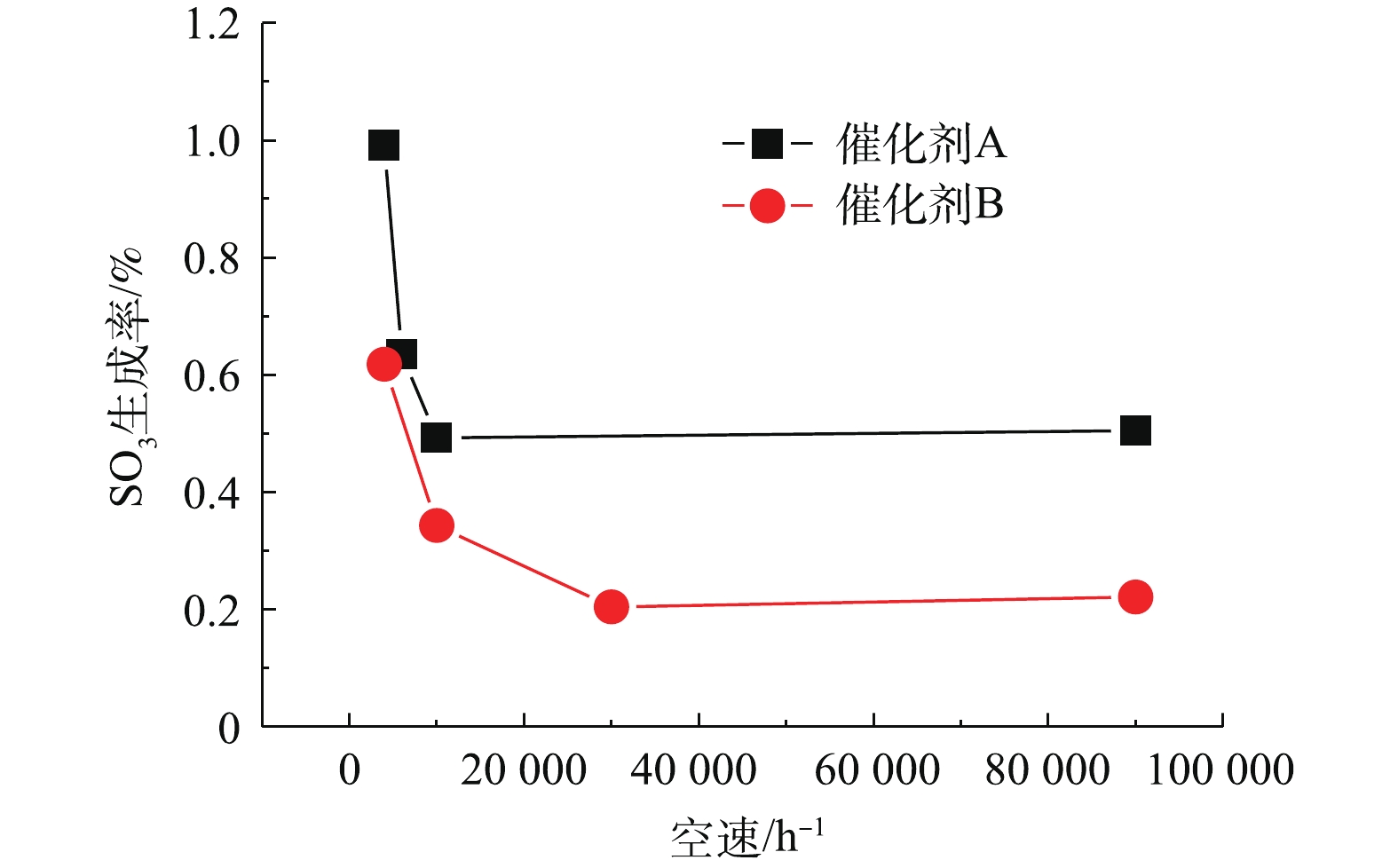
本研究考察了空速对催化剂A和催化剂B SO3生成率的影响。在本实验中,反应温度为360 ℃,600×10−6 SO2,3% O2,实验过程保持总气体流量始终为1 L·min−1,空速的变化通过改变催化剂床层的体积获得,这也会导致气体停留时间的变化。由于SO2氧化速率受SO2氧化反应控制,停留时间越短,意味着反应时间越短。空速对催化剂A和B上SO3生成的影响如图4所示。当空速从4 000 h−1增加到10 000 h−1时,催化剂A的SO3生成率明显降低;但当空速进一步增加到90 000 h−1时,SO3生成率几乎保持不变。当空速从4 000 h−1增加到30 000 h−1时,催化剂B的SO3生成率也明显下降;当空速进一步增加到90 000 h−1时,几乎不再变化。这主要是由于在低空速条件下,SO3的生成受气体扩散与化学反应量2个方面的影响,当空速增加到一定程度后,外扩散影响基本消失,SO3的生成主要由化学反应控制,因此,不再随着空速的增加而变化。由于催化剂A上的V含量高于催化剂B,因此,当SO3的生成主要受化学反应控制时,催化剂A上的SO3生成会高于催化剂B。
-
图5给出了催化剂A和催化剂B的SO3生成率与反应温度的关系,空速为10 000 h−1,600×10−6 SO2,3% O2。结果表明,随着反应温度的升高,催化剂A和催化剂B的SO3生成率逐渐增加。这表明,随着温度的升高,催化剂的氧化活性增加。而从实验结果可以看出,即使反应温度降至280 ℃,仍然会产生SO3。如果SCR装置长期在280 ℃以下运行,则存在由NH4HSO4的生成引起的催化剂堵塞的风险。
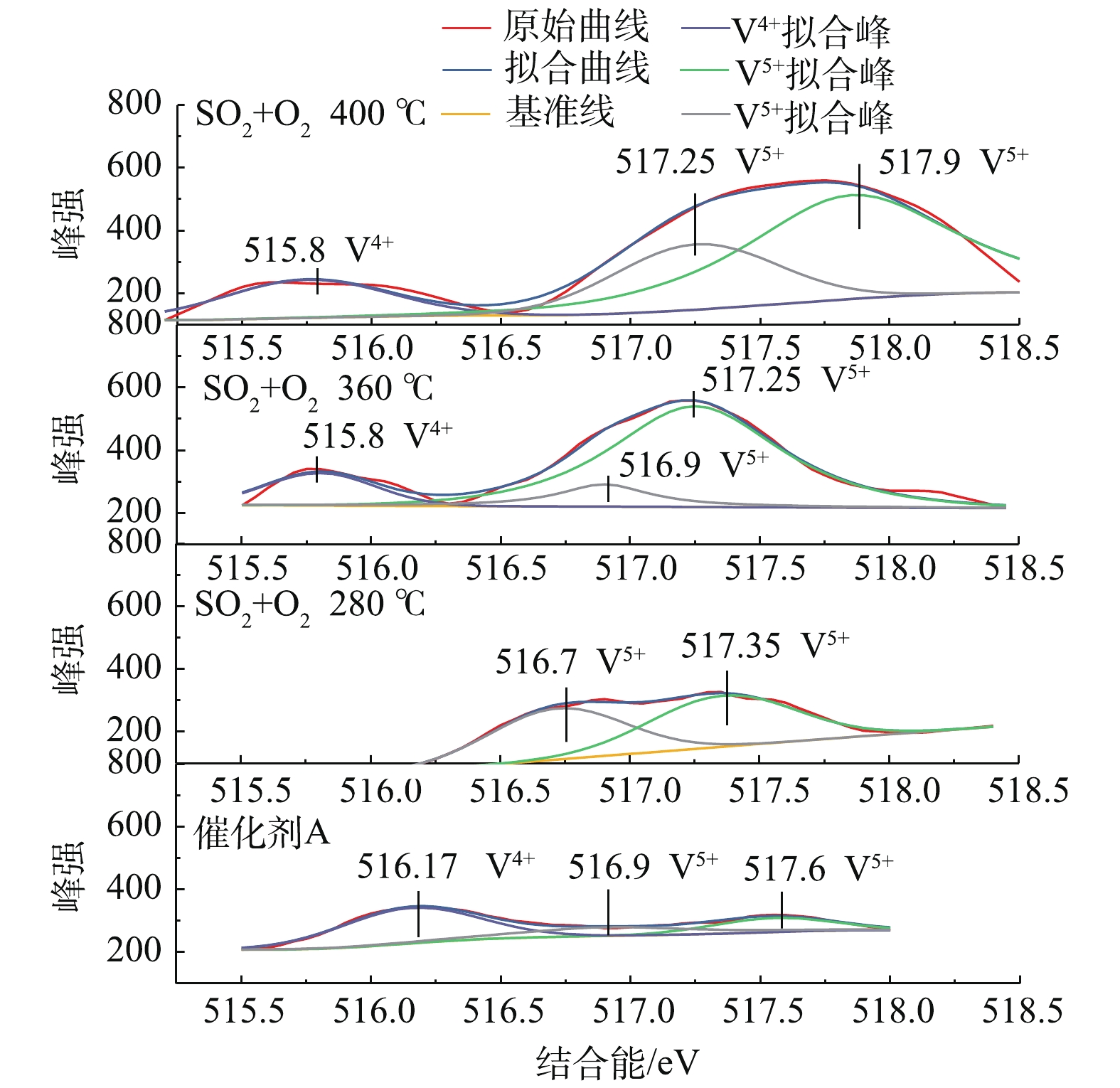
V在催化剂的氧化活性中起着至关重要的作用,催化剂A在不同温度下反应后的V2p XPS光谱如图6所示。可以看出,可以分配给V4+的位于516.17 eV的初始峰随着反应温度的升高而消失。此外,在位于515.25~516.25 eV的360 ℃和400 ℃处形成V4+的新峰,这也表明形成了新的钒化合物。许多研究证明,活性组分中间价的存在有利于提高反应活性[30],SO2氧化反应中新的钒化合物可能是类VOSO4中间体[31]。另外,如图6所示,随着温度的升高,分配给V5+的516.5~518.5 eV的另一个峰的结合能转移到更高的值,并且其相对量逐渐增加,这是由于V5+比例随温度的升高而增加的原因造成的[32]。在此过程中,SO3生成的催化反应可表示为式(2)和式(3)。
在催化反应中,V4+中间体的产生在促进SO3生成中起着至关重要的作用[33-34]。催化剂的氧化活性也依赖于V5+和V4+之间的转化,而温度的升高有利于这一反应的进行。
-
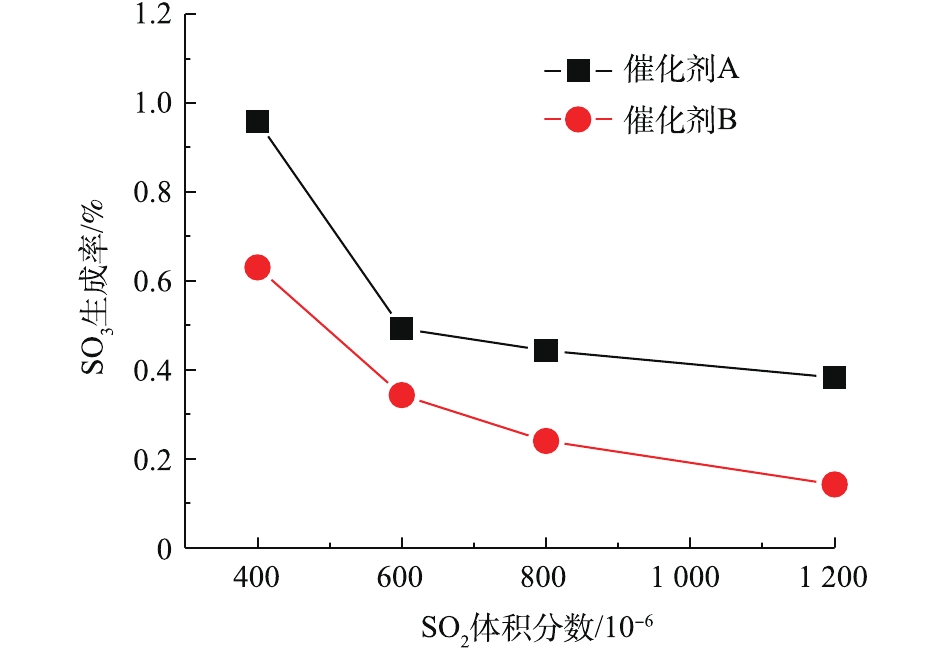
SO2浓度对SO3生成率的影响如图7所示,反应温度为360 ℃,空速为10 000 h−1,3% O2。当SO2浓度从400×10−6 增加到1 200×10−6时,催化剂A和B的SO3生成速率都降低。由于SO2氧化反应比催化剂孔内的SO2扩散慢得多,因此,SO3生成率主要受SO2氧化反应的控制而不是催化剂孔内的SO2扩散。SO2浓度对SO3生成的影响非常复杂。一方面,SO2分压的增加将导致反应式(4)向正反应方向移动,并且SO2的氧化将加速。
另一方面,当方程式(4)向正反应方向移动时,SO3分压将增加,这可能与SO2竞争吸附在催化剂上有关。SO3将对V—O—Ti键上的桥接氧电子产生更大的吸引力,从而优先吸附SO3。因此,随着SO2浓度的增加,SO3的生成率相对降低。
O2的存在是SO2氧化的先决条件,催化剂表面的O物种有助于SO2氧化。氧气浓度对催化剂A和B上SO3生成的影响如图8所示,反应温度为360 ℃,空速为10 000 h−1,600×10−6 SO2。可以看出,随着氧气浓度的增加,SO3的生成率略有增加。这表明在该实验条件下,O2浓度在催化剂表面上未完全平衡。O2浓度的增加将促进O2在催化剂表面的吸附,从而加速SO2的氧化。然而,DUNN等[6]研究了O2体积分数为1%~18%时的SO2氧化情况,发现当O2为2%~6%时,催化剂表面吸附的O2达到饱和,随着O2的增加,SO2的转化率保持不变,这说明不同催化剂其氧饱和度和活化能力是不同的。在本实验条件下,O2的吸附和活化能力未达到平衡,而且随着O2体积分数的增加而增加,但是增加到5%时,SO3生成率仍然低于0.8%,表明O2体积分数对氧化反应的影响远小于其他因素[22]。
以NH3作为还原剂,可将NO在催化剂表面选择性催化还原为N2,反应见式(5)。
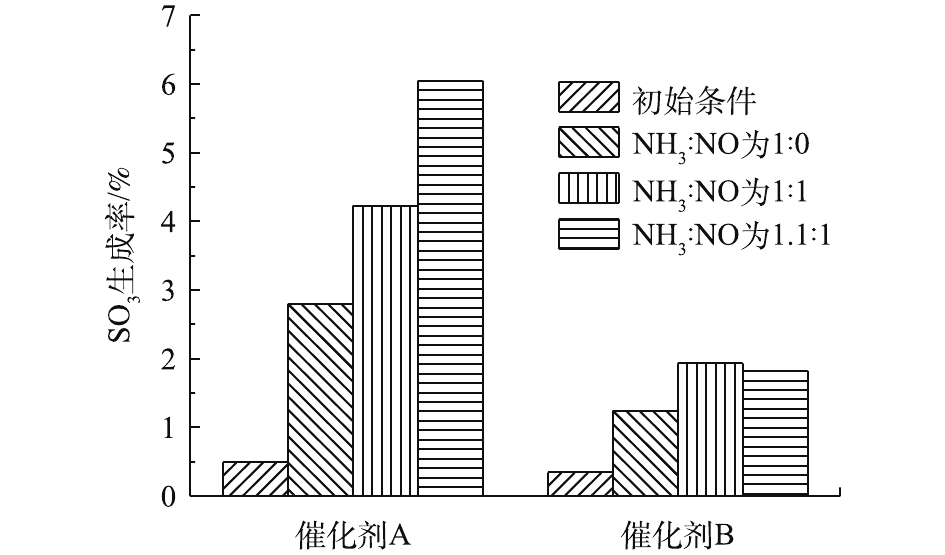
由于NH3还可以与SO3反应生成硫酸铵盐,因此,当NH3和NO同时存在于反应气氛中时,它对SO2氧化的影响仍然是未知的。NH3∶NO对催化剂A和B上SO3生成的影响如图9所示,反应温度为360 ℃, 空速为10 000 h−1,600×10−6 SO2,3% O2。在NH3存在下,加入500×10−6 NO,保持NH3∶NO为1∶1时,催化剂A和B的SO3生成率都进一步增加。当NH3∶NO增加到1.1∶1时,催化剂A的SO3生成率明显增加,而催化剂B的SO3生成率却略有下降。NH3和NO的存在将大大加速SO2的氧化。NH3的加入进一步促进了NO的反应,这可能是由于3个原因引起的[35]:1)如式(5)所示,NH3选择性催化还原NO会产生H2O,这会通过OH官能团的形成促进SO2的氧化,并且在脱硝反应时,吸附的NO被还原为N2,伴随着活化O原子的损失,其可能被SO2氧化消耗;2)当催化剂表面的SO2吸附达到一定浓度时,络合物将电离,最终抑制SO2的吸附和氧化。NH3与硫酸盐反应,使SO2吸附平衡向正方向移动,从而促进SO2的表面吸附;3)NH3与SO2氧化产物反应生成硫酸铵盐,这将减少气体中SO3的含量,如前所述,SO3还将抢占V活性位点的SO2吸附,因此,SO3的还原将促进SO2的吸附。
但是,催化剂A和催化剂B之间的变化趋势有一些差别。当NH3∶NO从1∶1增加到1.1∶1时,催化剂B上SO3的生成率会略微降低。这可能由于V活性位点的减少,导致催化剂B表面上V活性位点的相对含量不足以进行所有反应,因此,与NH3∶NO为1∶1相比,SO3生成率不会增加。
2.1. 催化剂物化性质
2.2. 催化剂化学组分对SO3生成率的影响
2.3. 空速对催化剂SO3生成率的影响
2.4. 反应温度对催化剂SO3生成率的影响
2.5. 进气组分对催化剂SO3生成率的影响
-
1)催化剂的化学组成对SO3的生成影响明显高于物理特性的影响,V负载量是决定SO3生成重要参数,V负载的增加对SO3的形成有积极作用,而Si的存在对SO2的氧化起到显著的抑制作用。
2)当空速增加到一定值时,空速的增加将不再影响SO2的氧化,但对于不同的催化剂,这个值是不同的。随着反应温度的升高,催化剂的SO3生成速率逐渐增加,这表明随着温度的升高,催化剂的氧化活性会增加。温度的升高有利于V5+和V4+之间的转化反应,从而提高催化剂的氧化活性。即使反应温度降至280 ℃,仍然会产生SO3。
3)随SO2浓度的增加,生成的SO3通过竞争吸附抑制SO2的氧化,导致SO3生成率降低。总之,O2、NH3、NO的存在将促进SO2的转化,浓度越高,促进作用越大。
