


 下载:
下载:











-
防毒面具被用于保障在有毒有害化学物质污染环境中作战及应急处置人员的呼吸安全。由于释放至环境中的污染物可能同时包含多种有毒有害物质,因此,要求防毒面具必须能够同时对多种污染物进行广谱高效的净化。空气净化材料作为防毒面具的核心组成,直接决定面具对受污染空气的净化能力。活性炭因具有价格低廉、制备工艺简便、吸附性能良好等优点,是目前防毒面具中广泛使用的空气净化材料。活性炭材料对沙林、苯与甲苯等高沸点有毒物质具有较好的物理吸附性能。然而,对于Cl2、SO2、NO2和NH3等难以通过物理吸附消除的低沸点有毒物质,活性炭的净化能力则较为有限。有研究[1]表明,目前各国军队现役防毒面具中广泛使用的、负载金属与有机胺作为活性组分的ASZM-TEDA型活性炭材料对上述多种有毒物质的防护时间均低于规定的最低时间要求(15 min)。近年来,针对活性炭材料性能的不足,设计开发能够对多种低沸点有毒物质进行广谱高效脱除的新型空气净化材料,已逐渐成为新的研究热点[2-6]。
氢氧化锆(Zr(OH)4)是一种无定形纳米多孔材料,表面分布有羟基基团、配位不饱和金属阳离子(Zrδ+)和氧空位等多种活性中心[7-9],是已知的同时具有酸碱性和氧化还原性的金属氢氧化物材料之一。以往大量的研究工作仅将Zr(OH)4作为氧化锆(ZrO2)合成过程的中间体,直到最近才由研究人员[10-15]发现并报道了其优良的气体吸附性能。这些研究[10-15]表明,负载Zn、Co和Ag等多种活性金属组分及有机胺三乙烯二胺(triethylenediamine,TEDA)制备的Zr(OH)4基空气净化材料,对低沸点有毒物质具有良好的脱除能力,对CNCl、HCN、SO2、NO2和NH3等多种有毒物质的脱除性能均优于ASZM-TEDA型活性炭材料,对酸性有毒气体NO2的脱除性能提升最为显著。在相同实验条件下,其对NO2的防护时间可达活性炭材料的4倍。然而,针对这一现象,即金属-有机胺改性的Zr(OH)4材料对NO2具有高效净化能力的机理解释,目前尚鲜见报道。
氮氧化物(NO2和NO)对人体有较大的危害,主要通过呼吸道吸入体内,造成急性或慢性中毒,进而对心脏、肾脏等重要人体器官造成损伤,尤其是NO2对人体的毒性可达NO的4~5倍。NO2属于酸性有毒气体中较难消除的目标化学物质,与其他酸性气体不同,其脱除难点主要在于NO2往往会发生还原反应,生成难以被吸附的NO有毒气体,造成材料床层穿透失效。因此,对氮氧化物的净化机理进行研究,有助于指导新型空气净化材料的设计,从而实现延缓甚至抑制NO释放的目的。本研究选取NO2作为目标污染物,首先制备得到负载活性组分Zn和/或TEDA的Zr(OH)4成型颗粒,通过比较负载活性组分前后的NO2和NO在材料床层内部的穿透行为,结合表征分析结果,探讨了不同活性组分负载条件下材料的氮氧化物净化机理,考察了基体、金属Zn和有机胺TEDA在NO2净化过程中各自的作用机制,并分析了“基体-金属-有机胺”三者之间存在的相互协同效应,为新型氮氧化物净化材料的设计开发提供参考。
全文HTML
-
八水合氧氯化锆(ZrOCl2·8H2O)、硫酸、氢氧化钠、氨水、碳酸氢铵、碱式碳酸锌((ZnCO3)2·(Zn(OH)2)3)为分析纯,六水合三乙烯二胺(C6H12N2·6H2O)纯度为96%,均购自国药集团化学试剂有限公司。以上药品均未经处理直接使用。
-
首先通过氢氧化物沉淀结合水热改性的方法[16]制备得到Zr(OH)4粉体。之后采用“压片-破碎-筛分”的工艺使其成型为粒度在12~30目的片状颗粒,作为空气净化材料使用。将制得的Zr(OH)4成型颗粒于105 °C干燥12 h,以除去表面吸附的水分子,留待后续使用。
采用等体积浸渍法负载活性金属组分Zn,负载量以
mZn/mZr(OH)4 计算。制备单一负载Zn的Zr(OH)4时,首先将一定量的前驱体碱式碳酸锌加热溶解至氨水-碳酸氢铵溶液中,随后将上述溶液与Zr(OH)4混合均匀,待浸渍后的Zr(OH)4颗粒表观干燥后,放入烘箱中于130 °C活化6 h。TEDA的熔沸点较低,低温加热可使其快速升华。因此,可以通过升华沉积的方式进行负载,负载量以
mTEDA/mZr(OH)4 计算。制备单一负载TEDA的Zr(OH)4时,将一定量的TEDA与Zr(OH)4颗粒于广口瓶中混合均匀后,置于烘箱中60 °C恒温加热5 h,之后将广口瓶取出,使其自然冷却至室温。制备负载Zn和TEDA的Zr(OH)4时,为防止负载金属组分后干燥活化的过程中TEDA升华,采取先负载Zn后负载TEDA的顺序,分别依照二者单一负载时的方法进行制备。
-
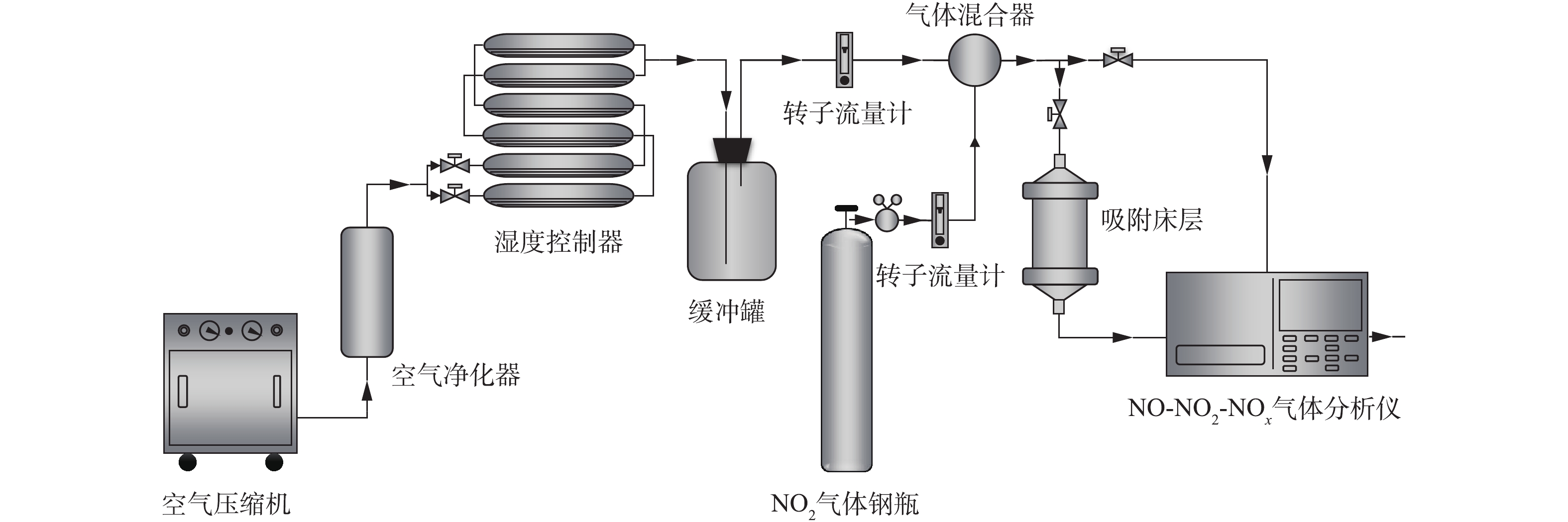
NO2脱除性能评价在如图1所示的装置中进行。将待评价的材料颗粒装填于固定床吸附装置内,目标气体由标准气体钢瓶产生,载气为空气,NO2和NO气体浓度采用Thermo Fisher Scientific 42i-HL NO-NO2-NOx分析仪进行检测。具体实验条件如下:固定床吸附器床层内径为1.0 cm,床层高度为2.0 cm,气流线速度为9.6 cm·s−1,温度为室温,气流相对湿度为50%,NO2初始浓度为1 000 mg·m−3。参考美国职业安全与健康管理局对于环境中NO2和NO容许暴露限值的规定[17],将尾气中NO2浓度到达10.5 mg·m−3或NO浓度到达33.5 mg·m−3的时间确定为穿透时间,二者任一个达到设定浓度时即认为材料被穿透。当尾气中NO2浓度进一步升高至21 mg·m−3(NO2容许暴露限值浓度的2倍)时,停止通入NO2。在停止通入NO2后,室温下继续用洁净空气对材料进行吹扫,同时记录尾气中NO2和NO浓度随时间的变化情况,以评价材料与NO2和NO吸附作用的强弱。
-
采用Nicolet 600型傅里叶变换红外光谱仪(FT-IR)对样品进行红外光谱分析。采用KBr压片法测试,测试波数为2 000~600 cm−1。采用Thermo Fisher Scientific ESCALAB 250 Xi型X-射线光电子能谱仪(XPS)对样品的表面元素化学态以及各元素相对含量进行分析。X-射线源为单色化的Al的Kα谱线(1 486.6 eV),以样品表面外来污染C1s结合能(284.8 eV)作为参考结合能进行荷电校正。
1.1. 实验试剂
1.2. 材料制备
1.3. NO2脱除性能评价
1.4. 表征分析方法
-
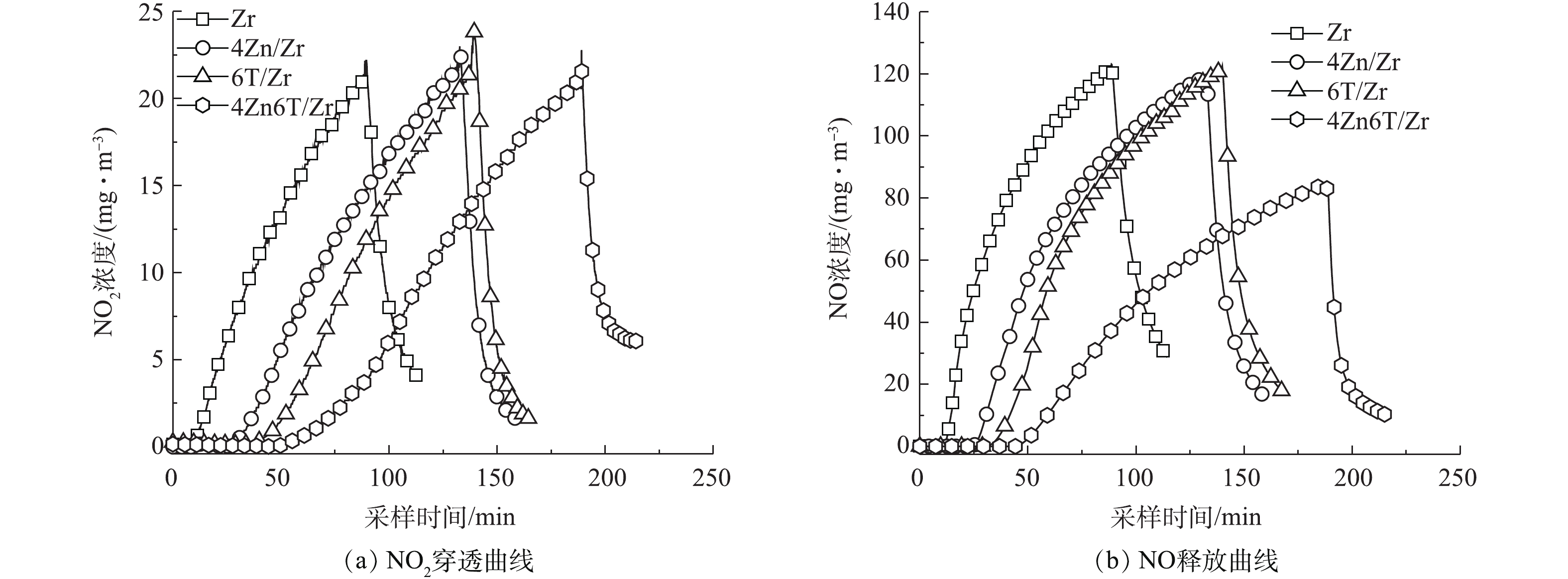
Zn和TEDA的负载量对材料穿透时间影响的实验结果(表1)表明,在同时负载Zn和TEDA的情况下,当二者负载的质量分数分别为4%和6%时,穿透时间达到最长。因此,在后续对不同活性组分负载对材料NO2净化性能和机理影响的研究中,选取Zr(OH)4基体(记为Zr)、单一负载4% Zn的样品(4Zn/Zr)、单一负载6% TEDA的样品(6T/Zr)以及同时负载4% Zn和6% TEDA的样品(4Zn6T/Zr)的NO2穿透曲线以及NO释放曲线进行对比,结果分别如图2(a)和图2(b)所示。
通过对图2(a)中NO2穿透曲线上方的面积进行积分,获得了穿透时各样品的NO2吸附量,结果如表2所示。由于材料在实际使用时是按体积而非质量对防毒面具滤毒罐进行装填,因此,在表2中同时列出了更具有实际意义的按体积平均的NO2穿透吸附量。可以看出,同时负载Zn和TDEA使得材料的NO2穿透吸附量由5.5 mg·cm−3增加至24.8 mg·cm−3,穿透吸附量增加了351%,对NO2的吸附性能得到大幅提升。其次,由图2(a)可见,负载活性组分前后,NO2穿透曲线的位置和形状存在明显差异。NO2在Zr基体上的穿透呈较为陡峭的凸形曲线,负载活性组分使得NO2的穿透逐步得到延缓,其穿透曲线逐渐向右移动并趋向于平缓的凹形曲线。4Zn/Zr的穿透曲线为与Zr基体形状相似的凸形曲线,表明单一负载金属Zn并不能改变净化机理,Zn在吸附过程中很可能直接与NO2作用或作为酸性中间产物的反应位点,主要为Zr基体表面碱性吸附位点提供辅助作用。而6T/Zr、4Zn6T/Zr的穿透曲线为形状不同的凹形曲线,说明有机胺TEDA的加入使净化机理发生了改变,其中以同时负载Zn和TEDA的4Zn6T/Zr曲线形状(即净化机理)变化最为明显。此外,洁净空气吹扫使得尾气中NO2浓度迅速下降,表明各材料对NO2的吸附作用均主要为化学吸附,因物理吸附而残留在材料孔道内的NO2较少,吸附质与表面吸附活性中心较强的化学相互作用使得有毒物质不易因空气吹扫而发生脱附。
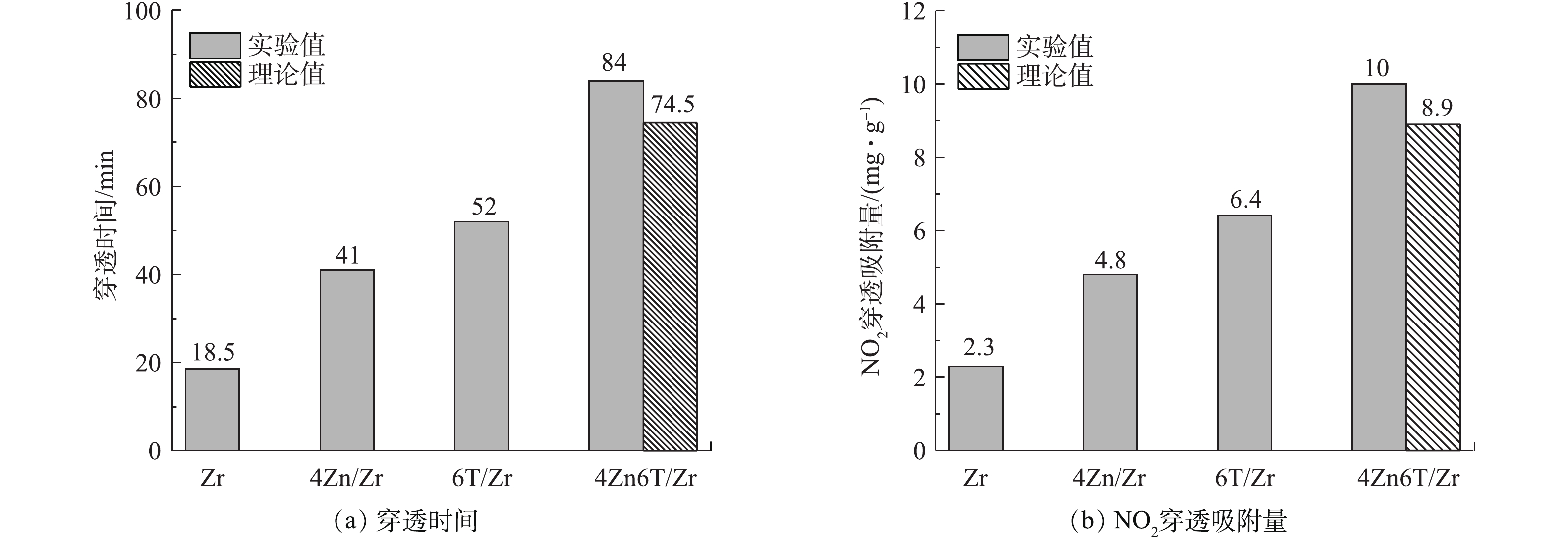
为了使基体、金属和有机胺三者之间可能存在的相互作用可视化,我们将4Zn6T/Zr样品穿透时间和NO2穿透吸附量的实验值与理论计算值进行了比较,结果如图3所示。理论计算值是根据单一负载Zn和TEDA时相应值的增量分别进行简单的线性加和求得。由图3可见,同时负载Zn和TEDA使得Zr(OH)4的穿透时间和NO2穿透吸附量均高于理论计算值,这表明“基体-金属-有机胺”三者之间可能存在相互协同效应,同时添加活性金属组分Zn和有机胺TEDA,对材料NO2净化性能产生了“1+1+1>3”的提升效果。
NO2脱除的另一个重要方面是控制过程中NO的释放。在实验条件下,所有样品首先达到穿透浓度的物质均为NO。如图2(b)和表2所示,负载活性组分能够增强材料表面对NO的截留能力,延缓NO的释放并减少过程中NO的产生。值得注意的是,虽然单一负载Zn或TEDA的材料能够延缓NO的释放,但对于NO2转化为NO的抑制作用并不明显,过程中释放的NO与Zr基体接近。而负载Zn和TEDA则能够在延缓NO释放的同时,又可有效抑制NO2转化为NO,使NO释放百分比相比于Zr基体降低约60%。这一结果再次证明了“基体-金属-有机胺”三者之间协同效应的存在。
-
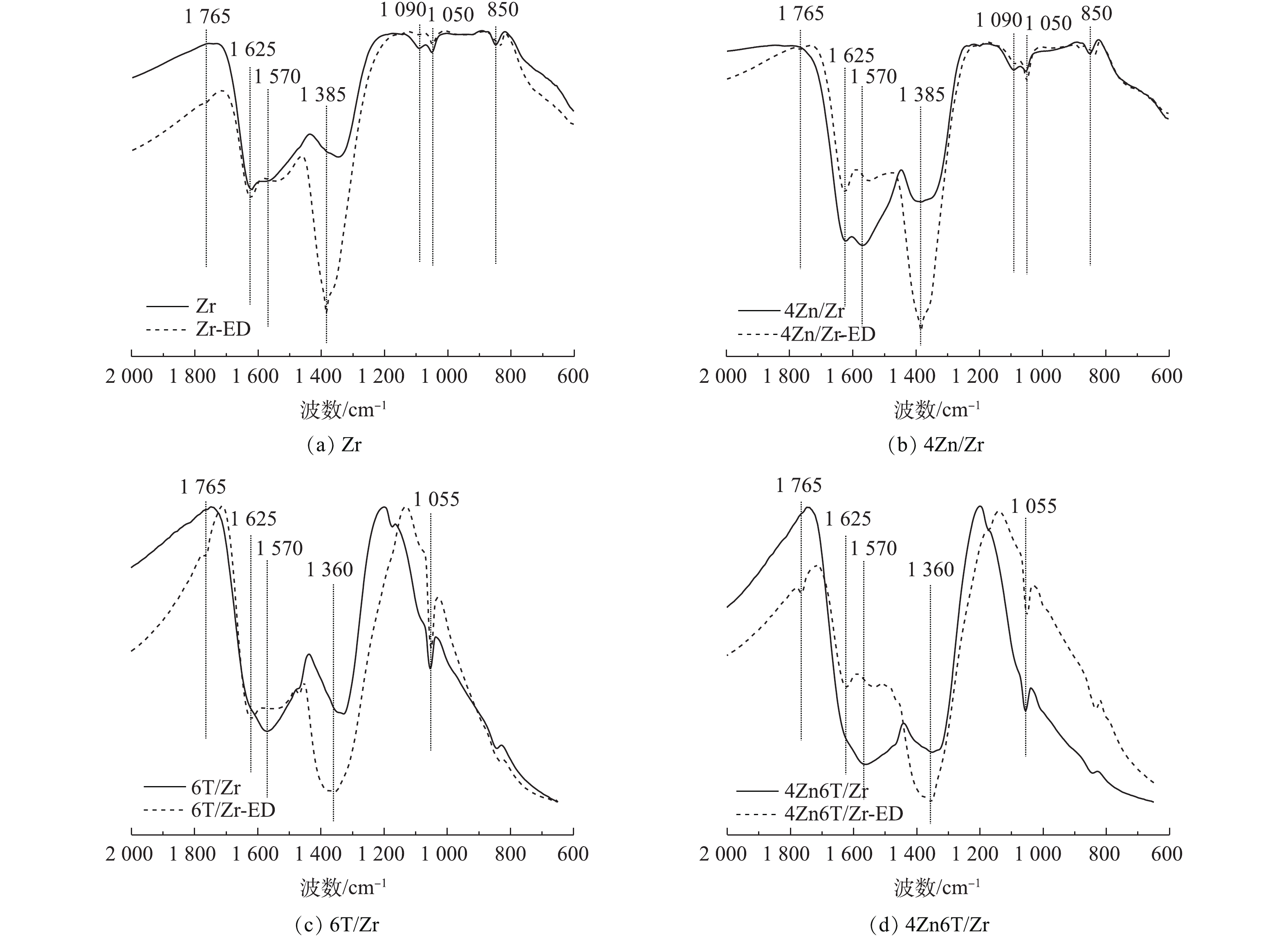
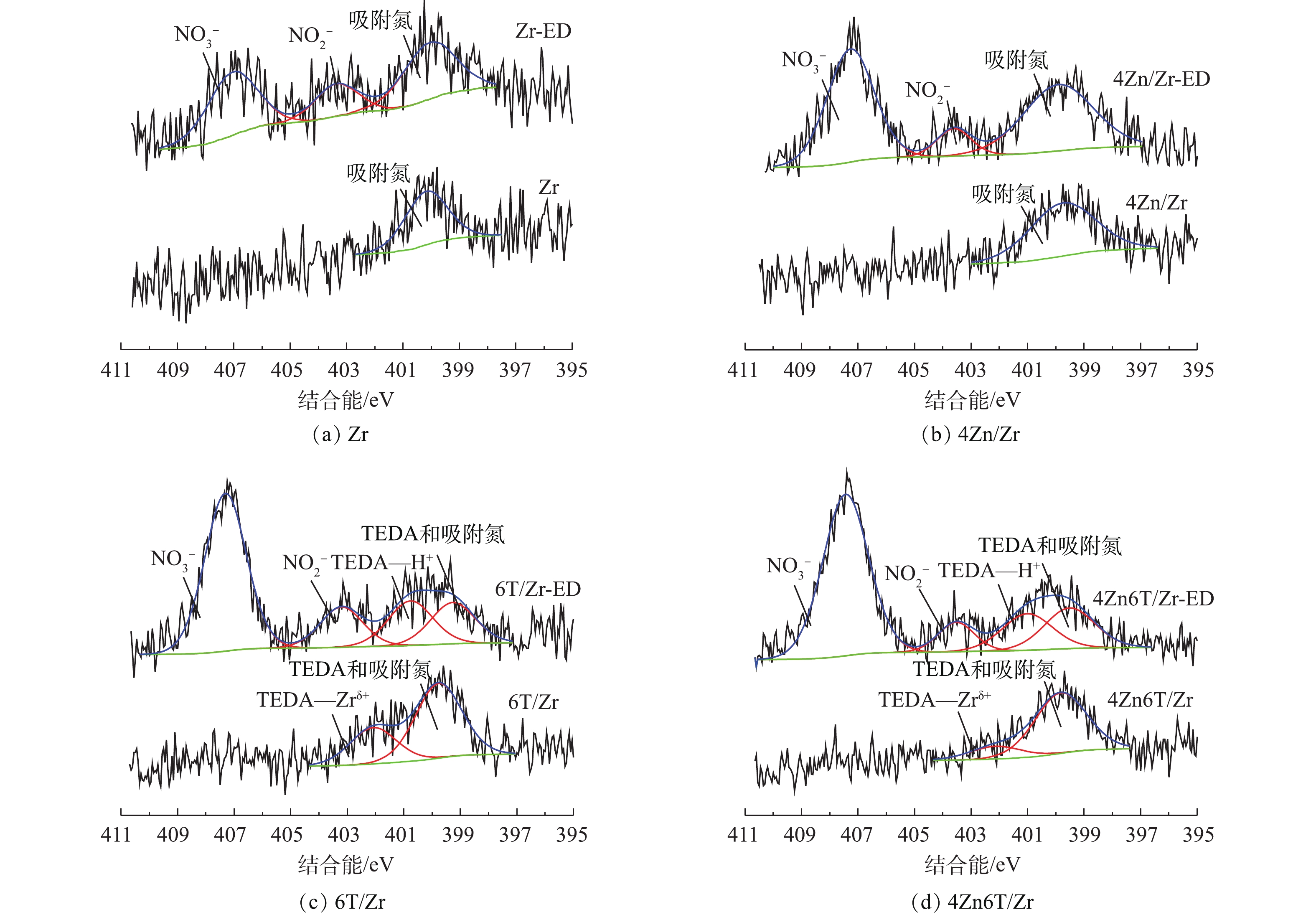
分别对吸附NO2前后的Zr、4Zn/Zr、6T/Zr和4Zn6T/Zr 4种材料样品进行红外光谱分析,通过材料表面官能团的变化,研究NO2吸附过程中所涉及的表面化学相互作用。吸附NO2后的样品通过在其名称后添加后缀“-ED”进行表示。红外光谱的表征结果如图4所示。对于未吸附NO2的Zr基体,1 625、1 570和1 340 cm−1处吸收峰归属于材料表面羟基基团以及配位水分子的弯曲振动[18-21]。1 090 cm−1和1 050 cm−1处的双峰以及850 cm−1处的吸收峰分别归属于锆酰基团(Zr=O)与Zr—O键的晶格振动[18,20]。在负载Zn后,1 625 cm−1与1 570 cm−1处双峰的相对强度和半峰宽均有所增加,这是由于前驱体碱式碳酸锌中CO32-的伸缩振动峰与羟基弯曲振动峰叠加所致[18]。对于6T/Zr、4Zn6T/Zr样品,水分子的干扰以及TEDA负载使得材料的红外光谱与Zr、4Zn/Zr样品有所不同,1 625 cm−1与1 570 cm−1处表面羟基基团以及水分子弯曲振动引起的双峰合并为1 570 cm−1处较强而宽的吸收峰,1 340 cm−1处羟基基团的吸收峰红移至1 330 cm−1处,并在1 055 cm−1处出现了C—N键骨架振动峰[22-23]。
在吸附NO2后,各吸附材料在1 570 cm−1附近对应于羟基基团的吸收峰强度均有所降低。这表明在负载活性组分前后,表面羟基基团均参与了NO2的吸附过程。在各样品对应的谱图中,在1 385 cm−1或1 360 cm−1处均出现了新的强吸收峰,该峰归属于游离
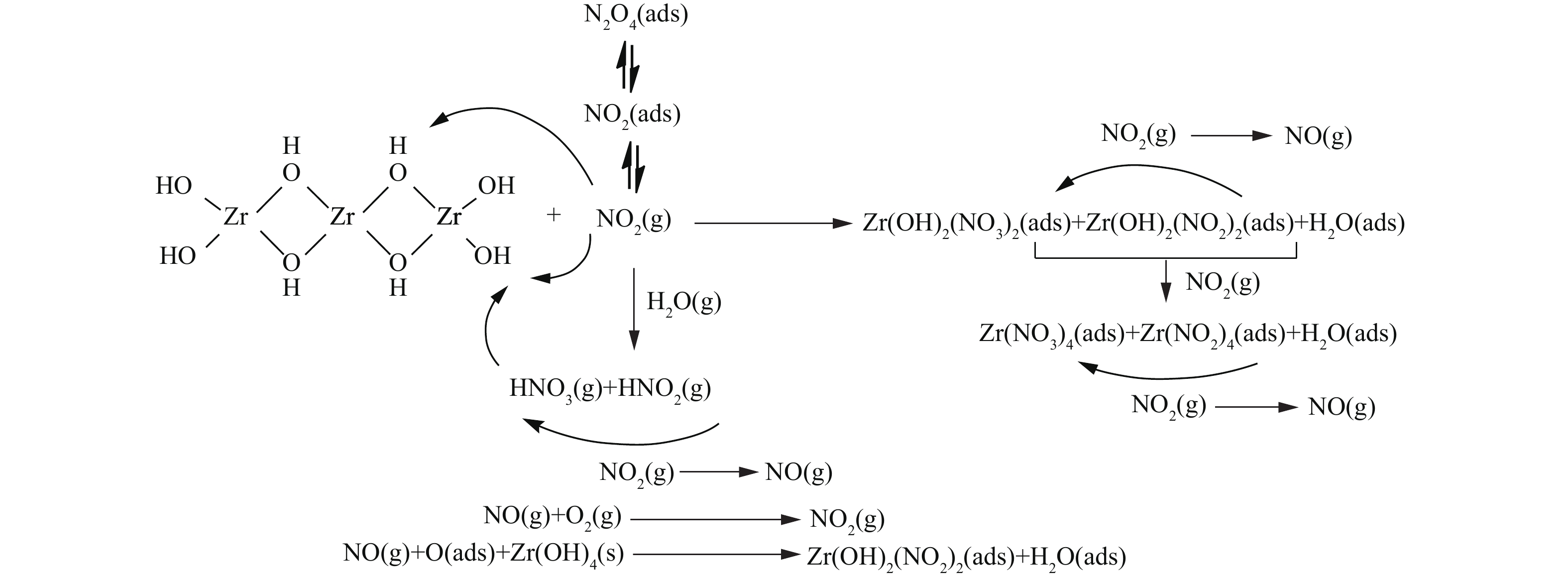
NO−3 离子中N=O键的不对称伸缩振动[23-25]。这表明在实验条件下,NO2在各材料表面吸附最终产物均主要为硝酸盐化合物。此外,各样品谱图中在1 765 cm−1处均出现微弱的N2O4振动吸收峰[24-25],并且峰强度随穿透时间的延长而逐渐增大。这表明除与材料表面活性中心反应发生化学吸附外,少量NO2分子还可通过物理作用吸附在材料表面和孔隙中,并与其二聚体形式(N2O4)混合存在,过程如式(1)所示。 -
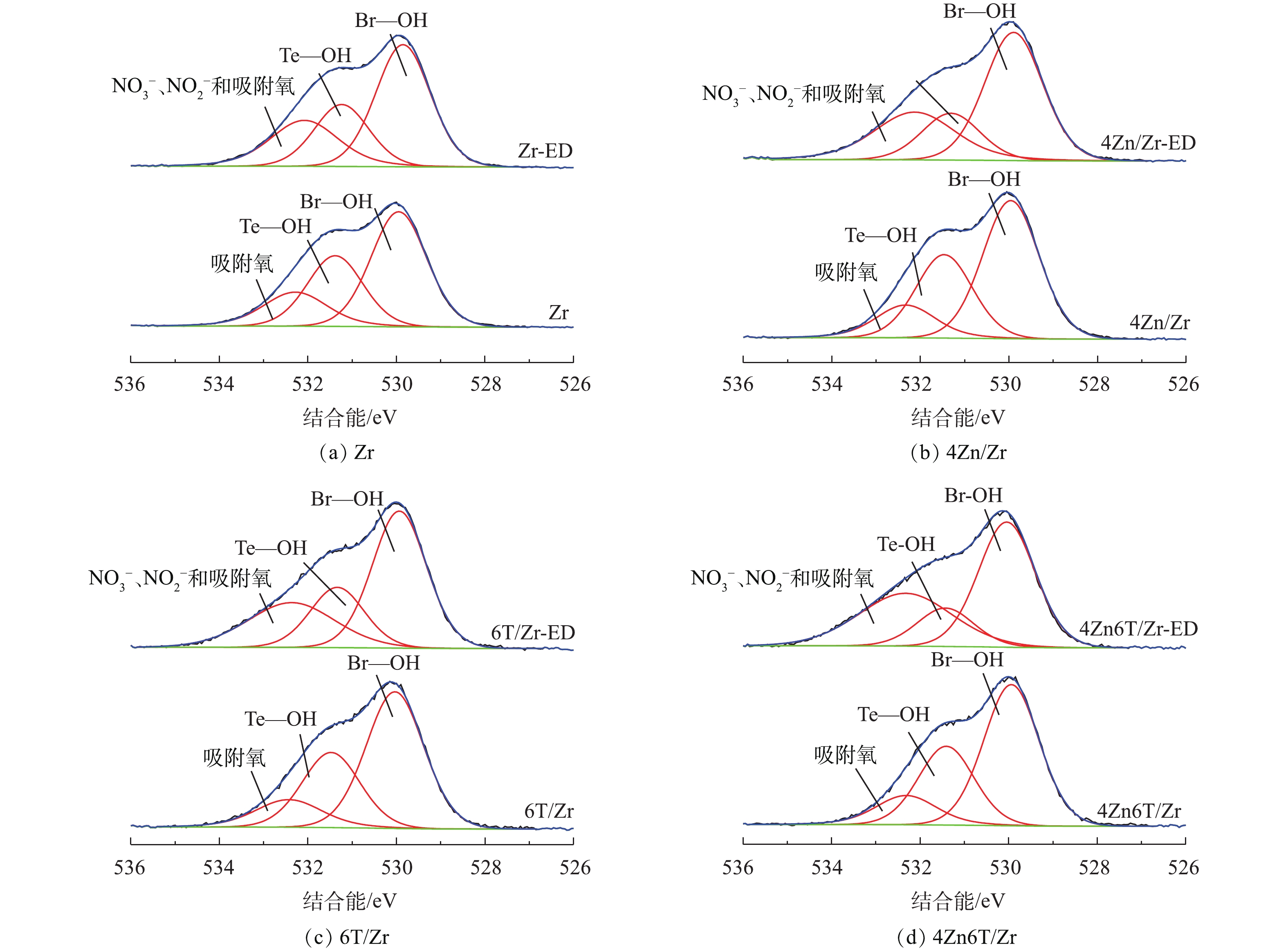
利用XPS进一步对吸附NO2前后Zr、4Zn/Zr、6T/Zr以及4Zn6T/Zr 4种材料样品进行了分析,以研究吸附前后材料表面元素组成和化学态的变化。图5为吸附NO2前后4种材料样品的O1s的XPS谱图。对于未吸附NO2的样品,O1s能谱图中存在3种类型的光电子峰,分别对应3种官能团或表面物种:约529.9 eV和531.4 eV处的光电子峰分别归属于Zr(OH)4基体表面桥式羟基(Br—OH)和端式羟基(Te—OH)基团[9,12,26],约532.3 eV处的光电子峰则归属于样品在环境中暴露所引入的表面吸附氧物种。桥式羟基和端式羟基的结合能不同,具有不同的配位环境,因而具有不同的酸碱性质及反应活性。有研究[9,12,26]表明,通常Zr(OH)4基体表面的桥式羟基呈Brönsted酸性,而端式羟基则呈Brönsted碱性。在吸附NO2后,各样品O1s能谱图中对应于端式羟基的光电子峰强度降低,而对应于桥式羟基的光电子峰强度则无明显变化。约532.3 eV处的光电子峰强度和峰宽在吸附NO2后均有明显增加,这是由于吸附生成的硝酸盐(
NO−3 )、亚硝酸盐(NO−2 )等产物的光电子峰叠加所致[27]。表3总结了吸附NO2前后4种材料中不同含氧官能团或表面物种所占相对原子百分比以及桥式羟基基团与表面锆原子比例(Br—OH/Zr)的变化。由表3可知,在吸附NO2后,端式羟基所占百分比显著下降,并且随着穿透时间的延长,下降幅度逐步增大;而桥式羟基所占比例则几乎不变,Br—OH/Zr在各种条件下均稳定在1.8~2.0。由图5和表3可知,在NO2净化过程中,仅有端式羟基参与了表面吸附反应,而桥式羟基则在整个过程中较为稳定,对NO2的脱除没有贡献。
图6对比了吸附NO2前后4种材料的N1s XPS谱图。对于未吸附NO2的Zr、4Zn/Zr样品,N1s能谱图中约400 eV处存在一宽而弱的光电子峰,该峰可能归属于样品在环境中暴露所引入的某些表面吸附氮物种。当负载有机胺TEDA后,材料的N1s能谱图中出现了2种类型的光电子谱峰。参考LIU等[28]对于Cu-TEDA改性分子筛N1s能谱图的分析结果,位于约399.8 eV处较强的光电子峰应归属于TEDA中裸露的N原子以及吸附氮物种;而位于约402.1 eV处相对较弱的光电子峰则表明部分TEDA中N原子与材料表面金属阳离子间存在配位相互作用,这种配位相互作用要强于一般浸渍负载过程中活性组分与基体间的物理相互作用,一定程度上有利于提升活性组分的稳定性,抑制其在材料使用和贮存过程中的流失。据报道[7-9],Zr(OH)4表面存在多种配位不饱和金属阳离子位点Zrδ+ (2类Zr4+离子和1类Zr3+离子)。无论材料表面是否有Zn原子存在,6T/Zr以及4Zn6T/Zr样品的N1s能谱图中约402.1 eV处均存在光电子峰,故部分TEDA分子很可能与金属阳离子Zrδ+形成了配合物。值得注意的是,活性金属Zn(碱式碳酸锌)的负载使得TEDA与Zrδ+间配位相互作用减弱,光电子峰强度降低。这可能是由于碱式碳酸锌预先占据了部分不饱和Zrδ+位点所致,表明碱式碳酸锌与Zr基体之间可能也存在着某些化学作用,同时也从侧面证明了4Zn6T/Zr中“基体-金属-有机胺”三者间化学协同作用的存在。
在吸附NO2后,在各材料的N1s能谱图中,位于407.3 eV和403.5 eV附近出现了2个新的光电子峰,分别归属于吸附生成产物硝酸盐(
NO−3 )和亚硝酸盐(NO−2 )[27,29],与O1s能谱分析结果相符。此外,对于负载有机胺TEDA的样品6T/Zr以及4Zn6T/Zr,在N1s能谱图中约402.1 eV处,TEDA与Zrδ+配位形成的光电子峰在吸附NO2后消失,而在约401.2 eV处则出现了新的光电子峰,根据PETERSON等[12]的报道,该光电子峰应归属于NO2吸附过程中表面H+离子与TEDA结合所形成的酸碱配合物。由于H+离子的L-酸性强于金属阳离子Zrδ+,因此,H+离子能够将N原子从吸附前TEDA与Zrδ+形成的配合物中夺去。TEDA—H+配合物主要由TEDA的水合质子化过程或与HNO3等酸性中间产物反应形成。TEDA的水合质子化过程能够产生OH−离子,有利于促进NO2水解;而吸附过程中生成的HNO3等酸性中间产物与TEDA反应,则使得N原子裸露的TEDA比例逐渐减少,导致TEDA逐步丧失促进NO2水解的能力。表4总结了吸附NO2前后表面含氮物种的相对含量变化情况。可以看出,对于4种不同的吸附材料而言,硝酸盐(
NO−3 )均为吸附NO2后的主要产物,相对含量随着穿透时间的延长而逐步增大;而亚硝酸盐(NO−2 )在吸附产物中的含量则相对较少,相对含量和穿透时间之间并没有明显的关系。根据反应前后氧化数的变化,吸附产物中硝酸盐和亚硝酸盐的理论含量比例应为1∶1。实验测得的亚硝酸盐含量低于理论值,这表明在吸附过程中,亚硝酸盐能够被进一步氧化为硝酸盐,从而导致硝酸盐最终成为主要的吸附产物。值得注意的是,负载活性组分后,材料表面吸附产物中亚硝酸盐的含量相比于Zr基体均有不同程度的降低,其中以添加金属Zn的样品4Zn/Zr以及4Zn6T/Zr亚硝酸盐含量降低最为显著。这意味着负载活性金属组分Zn能够有效促进吸附产物中亚硝酸盐向硝酸盐的转化。 -
基于上述性能评价与表征分析结果,我们提出了Zr、4Zn/Zr、6T/Zr以及4Zn6T/Zr 4种材料对氮氧化物的净化机理。如图7所示,Zr(OH)4表面羟基中具有Brönsted碱性的端式羟基可与气流中的NO2直接作用,形成硝酸盐和亚硝酸盐而将其吸附在表面,而具有Brönsted酸性的桥式羟基则不参与NO2的吸附过程。此外,NO2还可通过与气流中存在的水分子发生水解反应,从而将其转变为HNO3和HNO2,间接与碱性端式羟基形成硝酸盐和亚硝酸盐产物。随着NO2吸附的不断进行,表面生成的吸附态亚硝酸盐被NO2进一步氧化为硝酸盐,并释放出NO气体,最终导致硝酸盐成为主要吸附产物。在亚硝酸盐氧化过程中,释放出的NO气体可以自发与载气(空气)中的O2分子反应重新转变为NO2,或被Zr(OH)4表面含氧物种氧化重新转变为亚硝酸盐,进而返回至上述表面吸附反应中。随着NO2吸附过程的不断进行,整个过程中NO的生成速率逐步超过其消耗速率,致使材料床层最终被NO穿透。
单一负载金属Zn并不改变净化机理,4Zn/Zr对NO2的吸附仍主要按照上述机制进行。Zn在吸附过程中很可能直接与NO2作用或作为酸性中间产物的反应位点,主要为Zr基体表面碱性吸附位点端式羟基提供辅助作用。此外,Zn的存在还能够有效促进吸附产物中亚硝酸盐向硝酸盐的转化,加快NO2吸附过程中生成的NO返回到表面反应体系中,从而延缓NO的释放。Zn在氮氧化物净化过程中的作用机理如式(2)~式(5)所示。
作为环状双叔胺,TEDA在过程中并不直接与NO2发生反应,一般认为其作用机制在于能够有效催化NO2的表面水解反应[11-12],将NO2转变为酸性更强的HNO3和HNO2,使其与Zr基体表面端式羟基反应的速率加快,进而使得NO2和NO的穿透延缓。随着NO2吸附过程的进行,材料表面端式羟基逐渐被消耗,水解产物HNO3和HNO2开始与TEDA反应,形成酸碱络合物TEDA—H+,致使TEDA中毒,NO2催化水解速率逐步降低,造成材料床层逐渐被穿透。TEDA在氮氧化物净化过程中的作用机理如式(6)~式(9)所示。
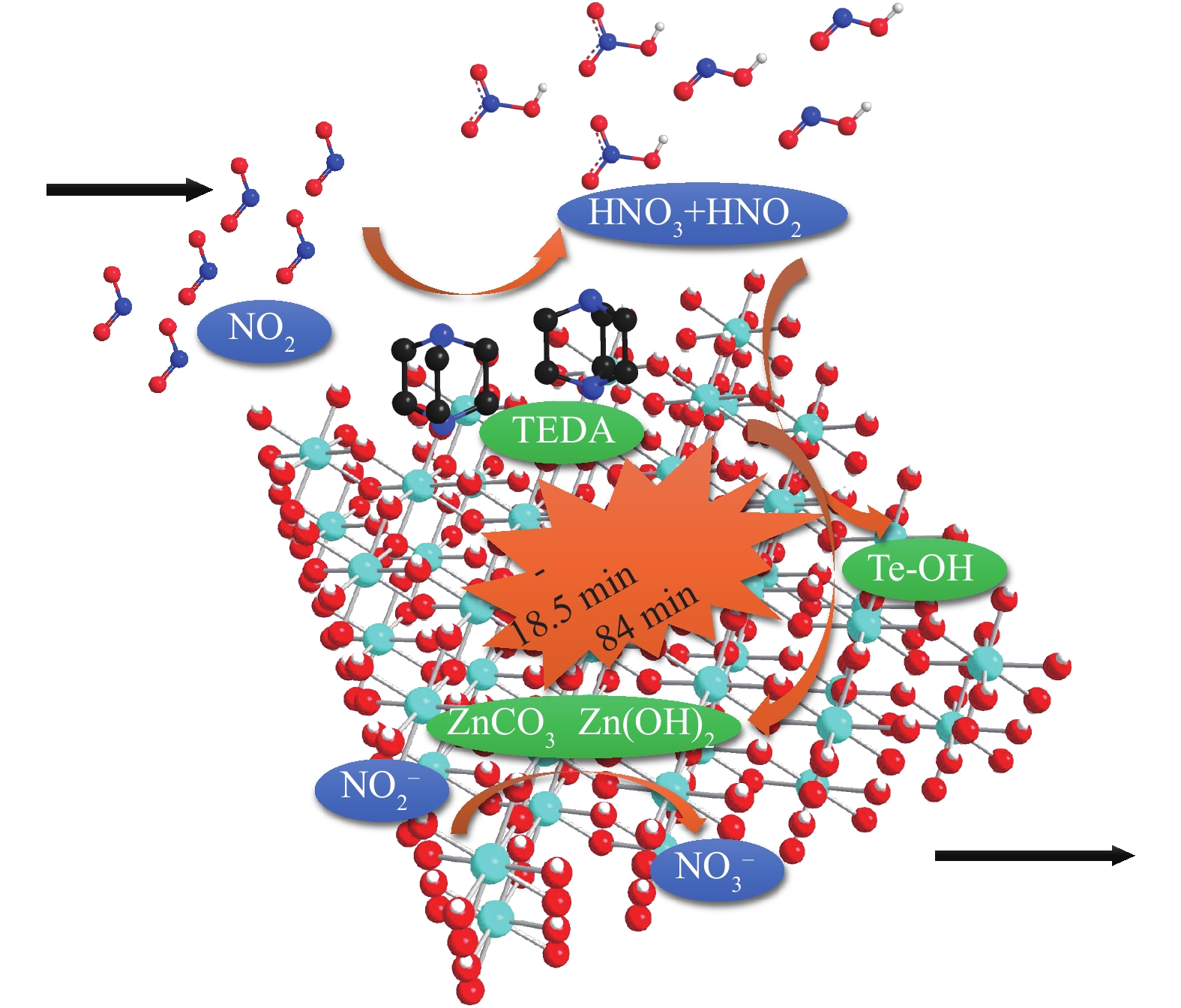
当基体同时负载Zn和有机胺TEDA时,基体、金属和有机胺不仅能够实现其单独存在时对NO2的作用机制,三者之间还能够相互“分工”,有效地进行协同配合。如图8所示,TEDA通过催化NO2的水解反应,使NO2转变为酸性更强的HNO3和HNO2,二者能够同时与端式羟基基团以及碱式碳酸锌等多种表面碱性中心反应,快速得到消耗,从而减少NO2和NO的释放,并有效延缓TEDA中毒,使得材料的穿透时间显著延长。此外,Zn还能够有效促进吸附产物中亚硝酸盐向硝酸盐的转化,加速NO2吸附过程中生成的NO返回表面反应体系中,进一步减少并延缓了NO的释放。上述“基体-金属-有机胺”三者之间相互协同效应的存在,使得同时添加Zn和TEDA对材料的NO2脱除性能产生了“1+1+1 > 3”的增益提升效果,显著提高了穿透时间和穿透吸附量,有效降低了NO释放速率和生成比例。
2.1. 负载活性组分前后氮氧化物床层穿透行为比较
2.2. FT-IR分析
2.3. XPS分析
2.4. 氮氧化物净化机理分析
-
1) 同时负载质量分数为4%的Zn和6%的TEDA使得材料对NO2的净化能力获得显著提升,与Zr(OH)4基体相比较,穿透时间由18.5 min延长至84 min,NO2穿透吸附量由5.5 mg·cm−3增加至24.8 mg·cm−3,NO释放比例由13%降低至6%。
2) Zr(OH)4基体主要通过表面的端式羟基与NO2直接或间接作用,从而形成硝酸盐和亚硝酸盐将其脱除,硝酸盐为主要吸附产物。
3) 活性金属组分Zn主要为端式羟基提供辅助作用,在脱除过程中,为NO2或酸性中间产物提供新的表面碱性吸附位点,并能够促进吸附产物中亚硝酸盐向硝酸盐的转化,延缓NO的释放。
4) 有机胺TEDA通过催化NO2的表面水解反应,将NO2转变为酸性更强的HNO3和HNO2,使其与表面碱性位点的反应速率加快,进而延缓NO2和NO的穿透。
5) 当负载Zn和TEDA进行改性时,基体、金属和有机胺可有效地进行相互配合,三者之间的协同效应对氢氧化锆材料NO2净化性能可产生“1+1+1 > 3”的增益提升效果,在延缓NO释放的同时亦显著降低了NO2净化过程中NO的生成比例。
