


 下载:
下载:

 /(μmol·L− 1)
/(μmol·L− 1)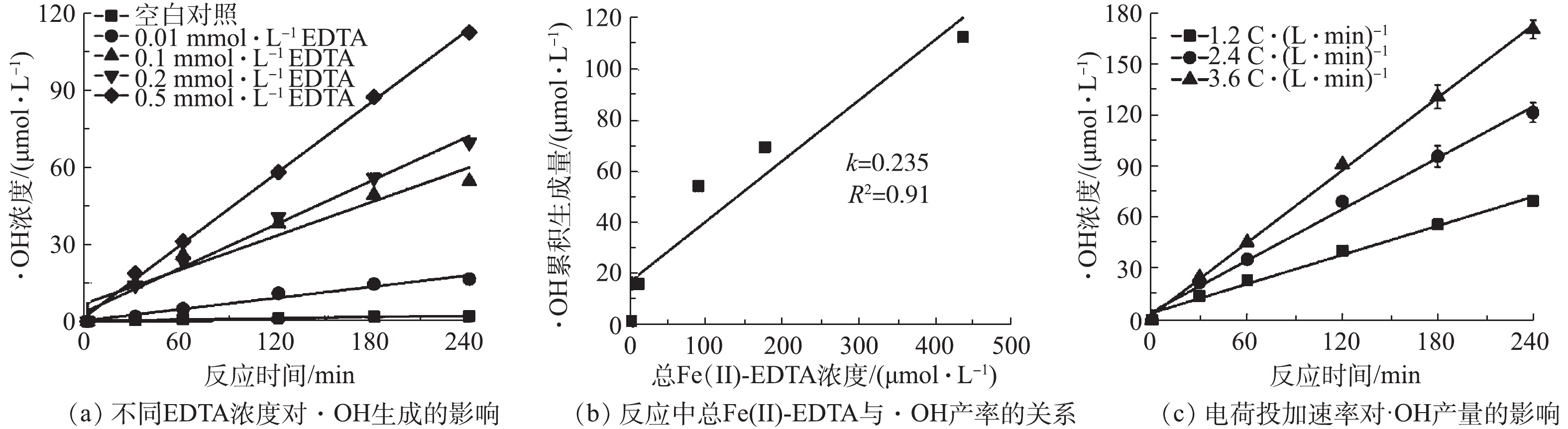

</td><td class="table_top_border" align="center" valign="middle">Fe(Ⅱ)-<inline-formula>$\left( { { {\rm{C} }_2}{ {\rm{O} }_4} } \right)_2^{2 - }$<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">草酸/(mmol·L<sup>− 1</sup>)</td><td class="table_top_border" align="center" valign="middle">Fe(Ⅱ)-<inline-formula>$\left( { { {\rm{C} }_2}{ {\rm{O} }_4} } \right)_2^{2 - }$<alternatives><img class="graphic" src="201906020_M9.jpg"><img class="graphic" src="201906020_M9.png"></alternatives></inline-formula>/(μmol·L<sup>− 1</sup>)</td><td class="table_top_border" align="center" valign="middle">Fe(Ⅱ)-(C<sub>2</sub>O<sub>4</sub>)<sup>0</sup>/(μmol·L<sup>− 1</sup>)</td><td class="table_top_border" align="center" valign="middle">总Fe(Ⅱ)-C<sub>2</sub>O<sub>4</sub>/(μmol·L<sup>− 1</sup>)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">10</td><td class="table_top_border2" align="center" valign="middle">2.04</td><td class="table_top_border2" align="center" valign="middle">2.45</td><td class="table_top_border2" align="center" valign="middle">4.49</td></tr><tr><td align="center" valign="middle">20</td><td align="center" valign="middle">3.39</td><td align="center" valign="middle">2.02</td><td align="center" valign="middle">5.42</td></tr><tr><td align="center" valign="middle">30</td><td align="center" valign="middle">4.13</td><td align="center" valign="middle">1.63</td><td align="center" valign="middle">5.75</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">40</td><td class="table_bottom_border" align="center" valign="middle">4.86</td><td class="table_bottom_border" align="center" valign="middle">1.14</td><td class="table_bottom_border" align="center" valign="middle">6.00</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula>/(μmol·L<sup>− 1</sup>)</td><td class="table_top_border" align="center" valign="middle">Fe(Ⅱ)-(C<sub>2</sub>O<sub>4</sub>)<sup>0</sup>/(μmol·L<sup>− 1</sup>)</td><td class="table_top_border" align="center" valign="middle">总Fe(Ⅱ)-C<sub>2</sub>O<sub>4</sub>/(μmol·L<sup>− 1</sup>)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">10</td><td class="table_top_border2" align="center" valign="middle">2.04</td><td class="table_top_border2" align="center" valign="middle">2.45</td><td class="table_top_border2" align="center" valign="middle">4.49</td></tr><tr><td align="center" valign="middle">20</td><td align="center" valign="middle">3.39</td><td align="center" valign="middle">2.02</td><td align="center" valign="middle">5.42</td></tr><tr><td align="center" valign="middle">30</td><td align="center" valign="middle">4.13</td><td align="center" valign="middle">1.63</td><td align="center" valign="middle">5.75</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">40</td><td class="table_bottom_border" align="center" valign="middle">4.86</td><td class="table_bottom_border" align="center" valign="middle">1.14</td><td class="table_bottom_border" align="center" valign="middle">6.00</td></tr></tbody>
</table></div></foreignObject></svg>)
 百度学术
百度学术


-
铁电絮凝(Fe-EC)是在电场作用下,使金属铁阳极溶解产生铁离子,并在水中发生一系列物理化学反应,从而使水得到净化的技术。目前,铁电絮凝已被用于生活污水、工业废水以及饮用水等的净化和再生,对水中悬浮物、部分溶解性有机物以及重金属离子有较好的去除效果[1]。但铁电絮凝中污染物去除过程复杂,净化机理尚不明晰。以有机废水为例,铁电絮凝技术的主要作用被认为是吸附、沉淀和气浮作用[2]。除此之外,在铁电絮凝过程中,部分有机污染物可被氧化降解,其作用机制被认为有2种:其一,在含氯电解质中,Cl−在铁阳极表面氧化,形成活性氯(Cl2,HClO/ClO−),导致有机污染物氧化[3];其二,有机污染物在阳极表面直接失电子氧化[4-5]。但这2种氧化机制都是在电荷效率降低时阳极表面发生的副反应导致的。本课题组前期研究发现,铁电絮凝中存在Fe(Ⅱ)活化氧气产生羟基自由基而导致的有机污染物氧化机制[6],但这一过程的氧化效果较弱。
在工业生产中,许多有机配体(如草酸,柠檬酸和EDTA)被广泛用作金属螯合剂、食品添加剂等,最后这些物质通过各种途径进入污水处理厂或自然水体[7]。在天然水体及大气液相中,溶解态铁多以络合配体的形式存在,是水体净化中重要的氧化剂和还原剂[8]。近几年,铁-有机络合物在水处理中的应用也越来越多,Fe(Ⅱ)-有机络合物(如草酸盐,柠檬酸盐和丙酮酸盐)可以在有氧条件下还原氧气来产生活性氧物质(ROS),如·OH、H2O2和
O−2 ,且产生的ROS对多种有机物(如阿特拉津、4-氯苯酚和普萘洛尔)的氧化作用已经得到了证实[9]。YI等[10]也通过电子顺磁共振实验证明,EDTA有助于Fe(Ⅲ)/H2O2体系中产生·OH。因此,在使用铁电絮凝处理含有有机配体的有机污染废水或自然水体时,有机配体的存在可能会促进体系中·OH的产生,从而影响目标污染物的降解或转化。苯胺是化工生产中的原料和中间产物,在污废水中广泛存在。本研究采用苯胺作为目标污染物,在有机配体存在的铁电絮凝体系中证明这一作用机制的存在,继而以苯甲酸与·OH反应,生成对羟基苯甲酸为体系中·OH的定量手段[11],研究了有机配体对体系产·OH效率的影响以及不同有机配体含量时铁电絮凝体系中目标污染物的降解规律,研究结果为人们进一步了解铁电絮凝中污染物去除机制提供参考。
全文HTML
-
苯胺(C6H7N)、碳酸氢钠(NaHCO3)、甲酸(CH2O2)、四水合酒石酸钾钠(C4H12KNaO10)、丙酸(CH3CH2COOH)、二水合柠檬酸钠(Na3C6H5O7·2H2O)、乙二胺四乙酸二钠盐(C10H14N2Na2O8·2H2O)、苯甲酸钠(C7H5NaO2)、5,5-二甲基-1-吡咯啉-N-氧化物DMPO(C6H11NO)、三氟乙酸(CF3COOH)、乙醇等药品均为分析纯;甲醇(CH3OH)为色谱纯;铁电絮凝实验中电极材质为纯铁片(含铁量大于99.5%),尺寸为1.5 cm×8 cm;实验用水均为电阻率为18.25 MΩ·cm−1的超纯水。
-
高效液相色谱仪(浙江福立,FL2200),配套C18色谱柱(岛津,4.6 mm×250 mm,5 μm)。电子自旋共振谱仪(日本JEOL,JES-FA200),pH计(奥力龙,MODEL818),超纯水机(圣德利,SDLA-B-0501-P),电子天平(梅特勒-托利多,AL204),直流电源(台湾固纬,GPS-2303C),电流表(FLUKE,F15B+)等。
-
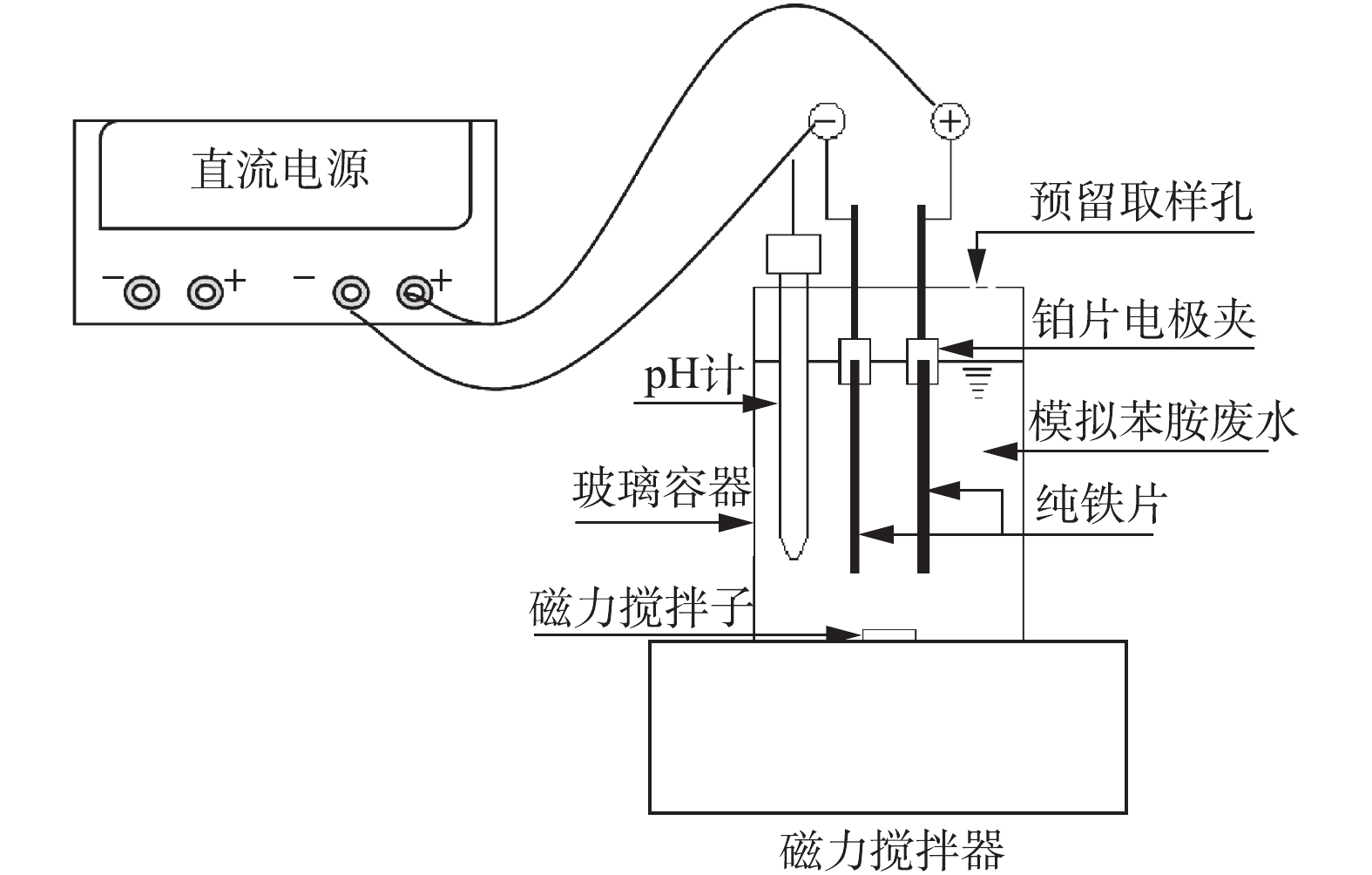
实验装置为600 mL圆柱形玻璃反应器,装有500 mL电解液,内置2片纯铁电极(1.5 cm×8 cm),电极间距为2 cm,实验装置如图1所示。由于碳酸氢根在地下水和地表水体中的广泛存在,本实验选择10 mmol·L−1 NaHCO3溶液为电解液,并在开始电解前搅拌30 min,使溶解氧饱和。实验初始电解液pH为8.5,4 h电解结束时上升至9.0。电解过程中采用磁力搅拌器使溶液保持完全混合,反应器开口与大气连通。采用稳压直流电源供电,并设置为恒电流模式,电流采用电流表进行监测,调节工作电流以达到所需的电荷投加速率。 根据法拉第电解定律计算相关指标,计算方法如式(1)所示。
式中:D是电解过程中铁投加速率,mol·(L·min)−1;Q是电荷投加率,C·(L·min)−1;Z是所涉及的电子数;F是法拉第常数,取值96 485 C·mol−1;V为电解液体积,L;I为电流强度,A。电解过程中实验装置用锡纸包裹以避光,实验分别在0、30、60、120、180、240 min用注射器吸取一定量水样,并用0.22 μm尼龙滤膜过滤,用于溶液中苯胺或对羟基苯甲酸浓度分析。所有实验至少重复2次。
在苯胺初始浓度为1 mg·L−1、电解质采用10 mmol·L−1 NaHCO3溶液、初始pH为8.5、电流强度为30 mA(即电荷投加速率为3.6 C·(L·min)−1)、电解时间为240 min时,分别投加以下有机配体:10 mg·L−1的甲酸、丙酸、柠檬酸和酒石酸钾钠;30 mmol·L−1草酸钠;0.2 mmol·L−1 EDTA。在此条件下,研究各种有机配体对Fe-EC降解苯胺的影响。
在铁电絮凝实验中,电解质为10 mL的10 mmol·L−1 NaHCO3,加入20 mmol·L−1 DMPO和0.05 mmol·L−1 EDTA,电解反应5 min后取样并过滤,加入DMPO捕获羟基自由基,进行ESR测试,开展DMPO捕获羟基自由基实验。
为了测量铁电絮凝中羟基自由基产率,本研究选择10 mmol·L−1苯甲酸钠作为羟基自由基的捕获剂,通过检测氧化产物对羟基苯甲酸的浓度而换算成羟基自由基产量(C·OH=Cp-HBA×5.87)[11]。电解质采用5 mmol·L−1 NaHCO3溶液,初始pH为8.5,电流强度为10 mA(即电荷投加速率为1.2 C·(L·min)−1)。在此条件下,研究草酸钠、EDTA的浓度对铁电絮凝中产·OH的作用。
采用10 mmol·L−1苯甲酸钠为·OH捕获剂,电解质采用5 mmol·L−1 NaHCO3溶液,初始pH为8.5,分别在反应开始时向溶液内投加30 mmol·L−1草酸钠和0.2 mmol·L−1 EDTA。以此为基础,研究不同电荷投加速率对铁电絮凝中产·OH的作用。
苯胺初始浓度为5 mg·L−1,电解质采用5 mmol·L−1 NaHCO3溶液,初始pH为8.5,电流强度为20 mA(即电荷投加速率为2.4 C·(L·min)−1)。在此条件下,研究EDTA浓度对铁电絮凝中苯胺降解的影响。
-
苯胺和对羟基苯甲酸浓度均采用高效液相色谱法(HPLC)测定;羟基自由基采用电子自旋共振谱仪(ESR)测定。
1.1. 实验试剂
1.2. 实验仪器
1.3. 实验方法
1.4. 分析方法
-
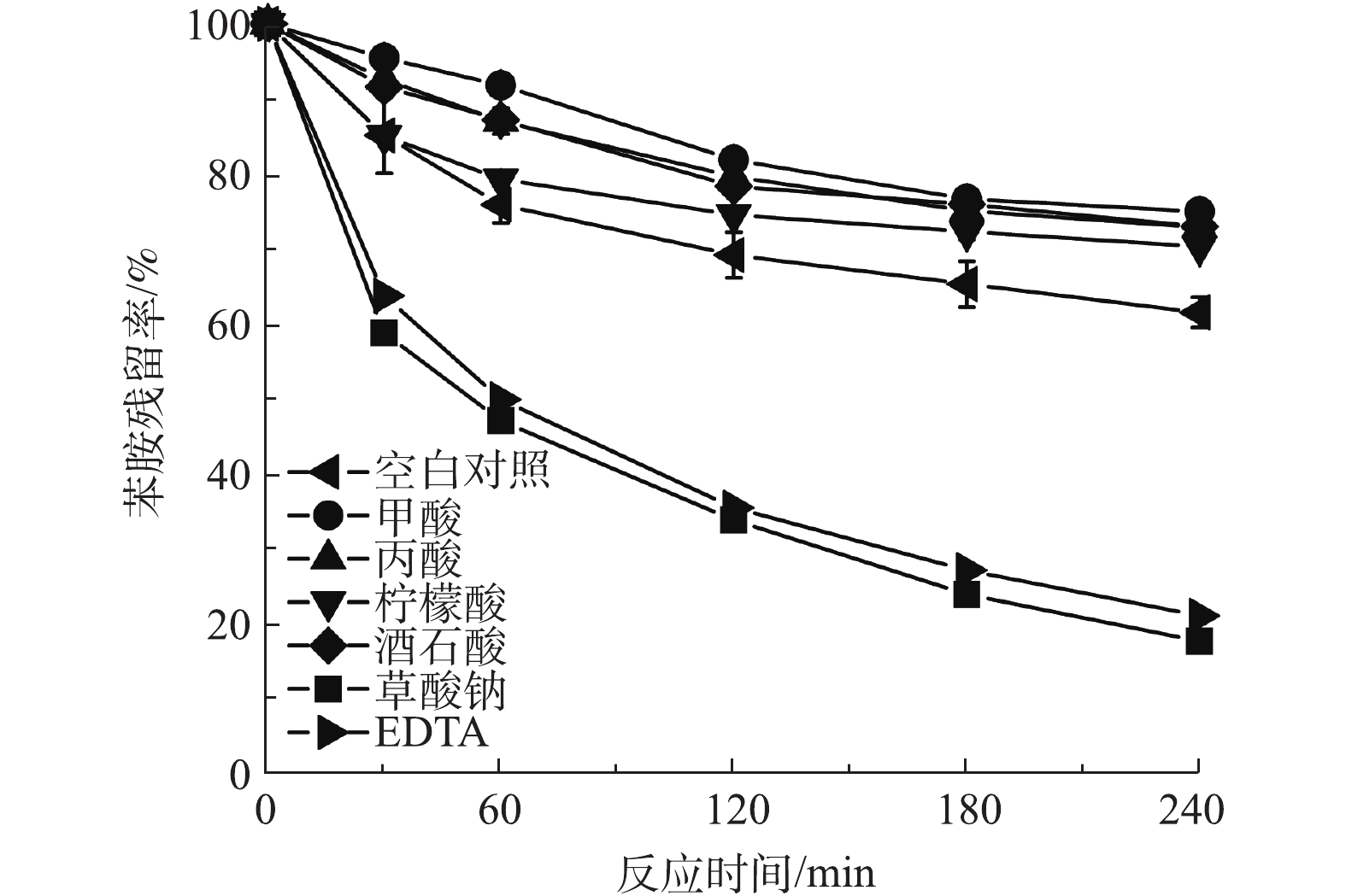
自然界中溶解态铁多以络合配体的形式存在,为研究有机配体在铁电絮凝中对有机污染降解的影响,实验中选取了几种水体中常见的有机酸,观察其在铁电絮凝中对有机污染物降解情况。 图2反映了溶液中存在不同有机配体时苯胺在铁电絮凝体系中的降解情况。在铁电絮凝中分别加入甲酸、丙酸、柠檬酸、酒石酸钾钠时,苯胺的残留率分别为74.9%、72.8%、70.3%、72.9%;在铁电絮凝中分别加入草酸钠和EDTA时,苯胺的残留率为17.5%和20.9%;不含有机配体时的对照实验中苯胺的残留率为61.4%。因此,甲酸、丙酸、柠檬酸和酒石酸钾钠在铁电絮凝体系中可使苯胺的降解率降低,而草酸钠和EDTA却对苯胺的去除有明显的促进作用。原因可能为:一方面,甲酸、丙酸、柠檬酸和酒石酸与Fe(Ⅱ)络合之后还原电位较高[12],没有明显提高其还原氧气的能力;另一方面,甲酸、丙酸、柠檬酸和酒石酸也能够与活性氧物质反应,进而影响苯胺的去除。
CHEN等[13]发现,Fe(Ⅱ)-草酸络合物在氧气参与下可以发生一系列氧化还原反应生成·OH,这一过程和铁电絮凝生成·OH的机理类似。NORADOVN等[14]发现,Fe(Ⅱ)-EDTA同样能够与O2反应,生成·OH并导致有机物的氧化降解。可能发生的反应如式(1)~式(4)所示(L表示草酸和EDTA配体)。
因此,在草酸钠和EDTA体系中,能够生成还原电位较低的Fe(Ⅱ)-C2O4和Fe(Ⅱ)-EDTA络合物[12],极大地促进了活性氧物质的生成,虽然草酸钠和EDTA也会消耗一部分产生的活性氧物质,但总体效果表现为促进苯胺的氧化降解。
-
在上述实验中,草酸和EDTA能够促进苯胺的降解,主要由于生成的活性氧物质所致。为进一步确认活性氧物质的主导作用,探究铁电絮凝体系中存在草酸钠和EDTA时苯胺的氧化降解机制,本研究做了乙醇淬灭实验和ESR测试。如图3(a)所示,在铁电絮凝体系中投加30 mmol·L−1草酸钠和0.2 mmol·L−1 EDTA时,反应240 min后,苯胺的残留率分别为17.5%和20.9%。乙醇与·OH的反应速率常数为1.9×109 L·(mol·s)−1,高浓度乙醇在铁电絮凝体系中可作为·OH的淬灭剂[15]。在上述实验体系中加入100 mmol·L−1乙醇后,苯胺的降解被极大地抑制,240 min时,苯胺的残留率为83.1%和93.6%。因此,淬灭实验结果证明了体系中导致苯胺氧化降解且起主导作用的活性中间氧化物种可能为·OH。
为进一步验证这一猜测,本研究采用DMPO作为·OH捕获剂,对含有EDTA的铁电絮凝体系进行了自由基检测(图3(b))。当只有DMPO存在时,只观察到较低的DMPO杂质波。当电絮凝反应体系中加入0.05 mmol·L−1 EDTA后,观察到相对较强的ESR信号,且这一信号与Fenton反应体系中检测到的信号峰相符。作为中间产物的·OH存在时间极短,浓度过低,很难被直接检测到,但它很容易与反磁性硝酮自旋捕获剂DMPO反应,形成稳定自旋加合物,因此,可以根据ESR光谱的磁参数进行识别[16]。波谱图包含4重峰的ESR信号,其具有1∶2∶2∶1的峰高模式和各向同性的超精细耦合常数,这是DMPO-OH加合物的特征。在图3(b)中,可观察到明显的DMPO-OH特征峰,从而证明体系中存在·OH。
-
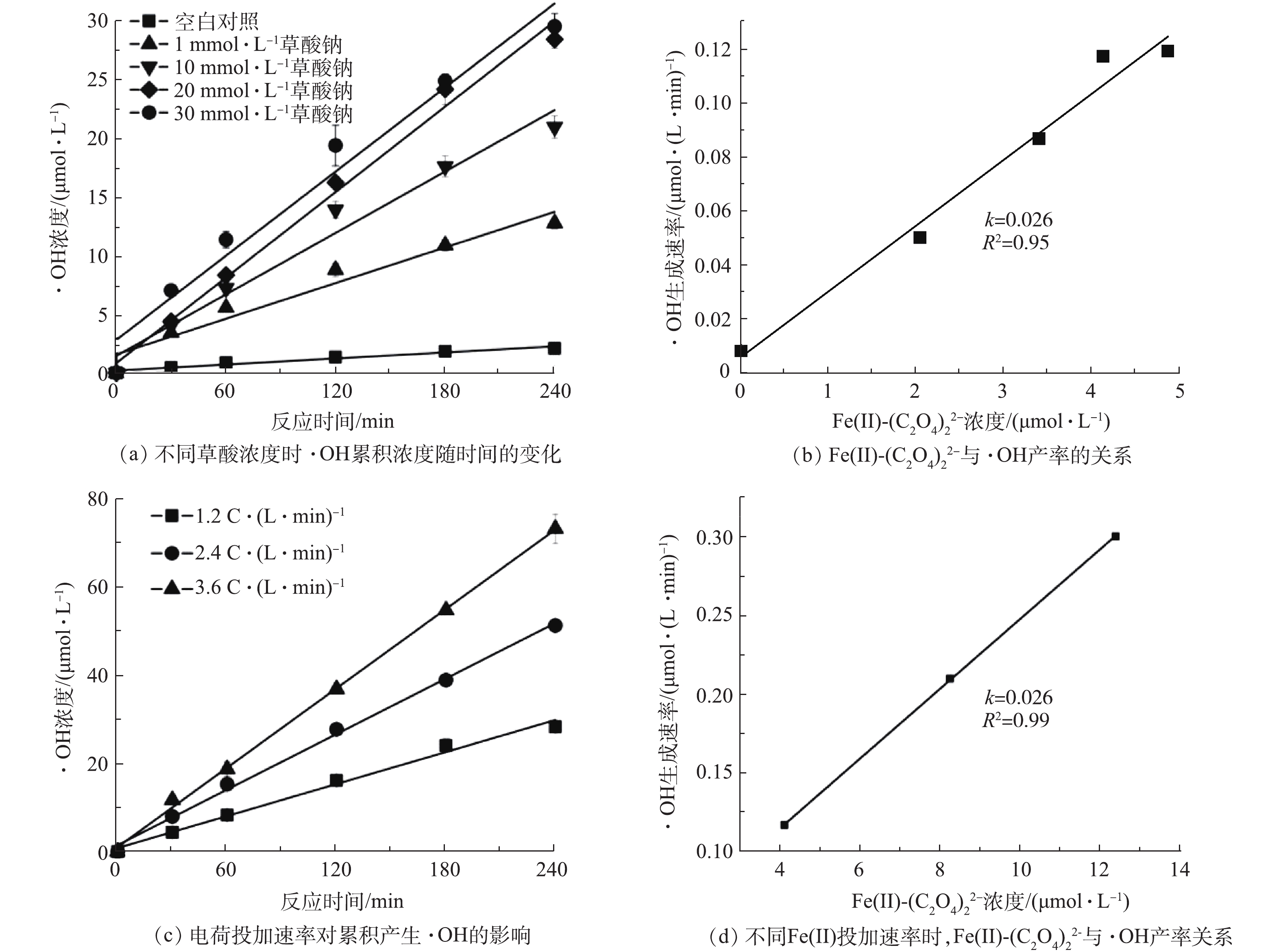
为了进一步探究有机配体存在时铁电絮凝体系中·OH的产生规律,本研究采用苯甲酸与·OH反应生成对羟基苯甲酸,作为·OH的定量分析手段[11],考察了不同条件下的·OH生成速率(见图4)。如图4(a)所示,铁电絮凝体系中·OH累积浓度随电解时间而增加,且符合拟零级动力学过程。当不存在有机配体时,铁电絮凝体系中的产生速率为0.008 6 μmol·min−1。当铁电絮凝中草酸浓度从10 mmol·L−1增加到50 mmol·L−1时,·OH产生速率从0.05 μmol·min−1增加至0.12 μmol·min−1。
根据法拉第电解定律[17],铁电絮凝中电荷投加速率为1.2 C·(L·min)−1时,铁阳极电解产生Fe(Ⅱ)的速率为6.22 μmol·(L·min)−1,反应240 min,产生的累积Fe(Ⅱ)浓度为1.5 mmol·L−1,按照Fe(Ⅱ)与草酸络合比1∶2计,10 mmol·L−1草酸是过量的。因此,当提高体系中草酸浓度时,Fe(Ⅱ)-草酸络合物的总量不变。但由图4(a)中的结果可知,体系中·OH产生速率随草酸浓度的增加而增加,可能是Fe(Ⅱ)-草酸络合物的形态发生变化所导致。为验证这一猜测,本研究采用Visual Minteq V3.1对反应体系中可能存在的Fe(Ⅱ)-草酸络合形态进行了计算,在反应体系中,Fe(Ⅱ)-草酸络合物的主要存在形式为Fe(Ⅱ)-(C2O4)0和Fe(Ⅱ)-
(C2O4)2−2 。随着草酸浓度的升高,Fe(Ⅱ)-草酸络合物的总含量几乎没有发生变化,而Fe(Ⅱ)-(C2O4)0含量降低,Fe(Ⅱ)-(C2O4)2−2 含量升高(表1)。利用计算得到的Fe(Ⅱ)-(C2O4)2−2 产生速率与实验结果中·OH的产生速率进行作图,二者呈现良好的线性关系(图4(b)),其比值为0.026。这一结果说明,体系中对·OH的产生起主要作用的是Fe(Ⅱ)-(C2O4)2−2 ,与已有研究结果[8, 12]一致,因为与Fe(Ⅱ)-(C2O4)0 相比,Fe(Ⅱ)-(C2O4)2−2 的还原电位更低。为了进一步验证Fe(Ⅱ)-
(C2O4)2−2 产生速率与体系中·OH产生速率之间的关系,本研究在草酸钠浓度为30 mmol·L−1条件下,对电荷投加速率与·OH生成的关系进行了研究。如图4(c)所示,当电荷投加速率分别为1.2、2.4、3.6 C·(L·min)−1时,体系中·OH 浓度的增长都符合拟零级动力学过程,这是因为体系中投加的捕获剂苯甲酸钠对·OH而言是过量的。通过Visual Minteq V3.1计算得到的体系中Fe(Ⅱ)-(C2O4)2−2 产生速率与·OH产生速率的关系如图4(d)所示,二者同样符合线性关系,且比值为0.026,与图4(b)中的比值一致。需要指出的是,即使当电荷投加速率增加到3.6 C·(L·min)−1时,10 mmol·L−1草酸相对于电解产生的Fe(Ⅱ)依然是过量的。因此,可以认为在草酸过量且溶液pH不变时,铁电絮凝体系中·OH的生成只取决于体系中Fe(Ⅱ)-
(C2O4)2−2 含量,二者存在一个固定的比值,约为0.026,即每氧化1 mol Fe(Ⅱ)-(C2O4)2−2 所产生的·OH量为26 mmol。有研究[18]显示,在中性条件下,对于还原性矿物质,氧化1 mol Fe(Ⅱ)时,·OH的产量为20 mmol[19],对于1 mol FeS为7.420 mmol。因此,Fe(Ⅱ)-(C2O4)2−2 相对于前2个体系中有明显的提升。 -
图5反映了EDTA对·OH产量的影响。图5(a)反映了不同EDTA浓度条件下铁电絮凝体系中·OH的生成量,其生成过程符合拟零级动力学过程。当电荷投加速率为1.2 C·(L·min)−1时,EDTA从0.01 mmol·L−1增加到0.5 mmol·L−1,反应进行240 min后,铁电絮凝反应中·OH的生成速率从0.07 μmol·(L·min)−1增加到0.47 μmol·(L·min)−1,相比不加配体时·OH生成速率有明显的提升。当电荷投加速率为1.2 C·(L·min)−1时,铁阳极电解产生Fe(Ⅱ)的速率为6.22 μmol·(L·min)−1,按EDTA最大浓度0.5 mmol·L−1且EDTA与Fe的络合比为1∶1计(图5(b)),80 min内,EDTA被完全络合,故与草酸钠体系不同,EDTA相对于Fe有所不足。为了解EDTA存在时铁电絮凝中主要起作用的铁络合物,本研究对反应体系中可能存在的铁络合形态进行了计算。由Visual Minteq V3.1拟合出EDTA在铁电絮凝中的络合形态,主要有Fe(Ⅱ)-EDTA2−、Fe(Ⅱ)-OHEDTA3−和Fe(Ⅱ)-HEDTA−,其中Fe(Ⅱ)-EDTA2−占87%左右(表2)。同时,在铁电絮凝反应中电荷投加速率为1.2 C·(L·min)−1时,单位时间形成的Fe(Ⅱ)-EDTA2−络合物含量极少(约为5.40 μmol·(L·min)−1),且EDTA浓度升高,其含量无明显变化。由表2可知,随着EDTA浓度升高,Fe(Ⅱ)-EDTA2−含量明显升高,Fe(Ⅱ)-OHEDTA3−含量缓慢变化,反应中起主要影响的物质是Fe(Ⅱ)-EDTA2−。这说明单位时间内反应中的EDTA浓度较低,难以定量Fe(Ⅱ)-EDTA2−对·OH影响,所以在研究EDTA体系时,以反应中累积生成的Fe(Ⅱ)-EDTA2−总量来衡量·OH生成速率。由图5(b)可知,1 mol Fe(Ⅱ)-EDTA2− 反应产生的·OH为235 mmol,这一产率为草酸体系的9倍。其主要原因是:Fe(Ⅱ)-EDTA还原电位(E0= 0.131 V)低于Fe(Ⅱ)-草酸络合物(E0=0.256 V)[12]。还原电位越低,还原能力越强,Fe(Ⅱ)-EDTA更容易与氧气发生系列反应生成·OH。
由图5(c)可知,·OH的产生速率随电荷投加速率的增加而增加,但由Fe(Ⅱ)与EDTA络合比1∶1可知,当电荷投加速率为3.6 C·(L·min)−1时,0.2 mmol·L−1 EDTA在 90 min左右能够被络合完全,因此,当EDTA量固定时,反应过程中产生的Fe(Ⅱ)-EDTA总量是不变的。而·OH的产生速率随电荷投加速率增加的原因可能是:一方面,电荷投加速率增加时Fe(Ⅱ)产生速率增加,降低了溶液的氧化还原电位[20];另一方面,电荷投加速率增加导致电压升高,促进Fe(Ⅲ)-EDTA在阴极表面还原为Fe(Ⅱ)-EDTA。
上述结果说明在铁电絮凝体系中EDTA浓度和Fe(Ⅱ)投加速率的增加均有利于·OH的生成,且·OH与形成的总Fe(Ⅱ)-EDTA含量呈正相关。
-
由于·OH与形成的Fe(Ⅱ)-EDTA含量呈正相关,电荷投加速率一定时,为探究不同有机配体浓度在铁电絮凝降解有机污染物时的影响,本实验采用苯胺作为目标污染物,以EDTA作有机配体进行了研究。图6(a)反映了在铁电絮凝过程中加入不同浓度的EDTA后苯胺的降解情况,苯胺的残留率分别为41%、20%、28%、42%;不加EDTA作为对照时,苯胺残留率为71%。随EDTA浓度的增加,铁电絮凝体系对苯胺的降解效果呈现先升高后降低的趋势。由图6(b)可知,当EDTA从0增加到0.5 mmol·L−1时,苯胺的氧化反应速率常数由0.002 8 min−1增加至0.010 7 min−1,而后降低至0.005 9 min−1;当EDTA为0.05 mmol·L−1时,苯胺的氧化反应速率常数达到最大值。苯胺、EDTA与·OH的反应速率常数分别为1.4×1010 L·(mol·s)−1[21]和5×108 L·(mol·s)−1[22]。因此,在二者含量相近时,反应中产生的Fe(Ⅱ)-EDTA和Fe(Ⅲ)-EDTA均能够与苯胺竞争·OH。EDTA与苯胺对·OH的竞争效率[23]可根据式(2)进行计算。
式中:F为竞争效率;CEDTA为EDTA浓度,mmol·L−1;KEDTA为EDTA与·OH反应速率常数,L·(mol·s)−1;CAN为苯胺浓度,mmol·L−1;KAN为苯胺与·OH反应速率常数,L·(mol·s)−1。当苯胺浓度为5 mg·L−1(即0.055 mmol·L−1),EDTA浓度为0.05 mmol·L−1时,其竞争效率为3.1%,即EDTA的竞争作用可忽略。但是当EDTA含量升高至0.5 mmol·L−1时,竞争效率达31%,影响显著,这一预测与以上实验结果(图6(a))相符。
为进一步证明EDTA对苯胺的竞争作用,本研究对体系中Fe(Ⅲ)-EDTA含量进行了检测,其结果如图6(c)所示。当Fe(Ⅲ)与EDTA按1∶1络合时,Fe(Ⅲ)-EDTA的最大含量为0.05 mmol·L−1,由于电荷投加速率为2.4 C·(L·min)−1时Fe的投加速率为12.44 μmol·(L·min)−1,因此,30 min内,EDTA会被络合完全,Fe(Ⅲ)-EDTA含量达到最大值。其后,Fe(Ⅲ)-EDTA含量随时间延长逐渐降低,验证了Fe(Ⅲ)-EDTA在铁电絮凝过程中被氧化,即Fe(Ⅲ)-EDTA与苯胺竞争·OH。
因此,虽然EDTA含量的升高促进了铁电絮凝中·OH的产生,但由于其对·OH的竞争作用,使得EDTA含量超过0.05 mmol·L−1时苯胺的降解效果减弱。
2.1. 不同有机配体对电絮凝降解苯胺的影响
2.2. 羟基自由基介导的氧化降解机制
2.3. 草酸存在时铁电絮凝体系中·OH的产生机制
2.4. EDTA对铁电絮凝体系产·OH的作用
2.5. 铁电絮凝中EDTA浓度对苯胺降解的影响
-
1)在铁电絮凝体系中甲酸、丙酸、柠檬酸和酒石酸钾钠会抑制苯胺的降解,而EDTA和草酸则能促进苯胺的降解。
2) EDTA存在时促进了铁电絮凝体系中·OH的产生,且·OH是导致苯胺降解的一种主要活性氧化物种。
3)铁电絮凝-草酸体系中产生的Fe(Ⅱ)-
(C2O4)2−2 是促进·OH产生的主要Fe(Ⅱ)物种,且1 mol Fe(Ⅱ)-(C2O4)2−2 反应可以产生26 mmol ·OH;而铁电絮凝-EDTA体系中具有活性的主要Fe(Ⅱ)物种为Fe(Ⅱ)-EDTA2−,1 mol Fe(Ⅱ)-EDTA2−反应可以产生235 mmol ·OH。4) EDTA对铁电絮凝体系中苯胺降解既有促进又有竞争作用,在其浓度为0.05 mmol·L−1时苯胺降解效率最佳。
